

Abstract
The cellular response to alkylation damage is complex, involving multiple DNA repair pathways and checkpoint proteins, depending on the DNA lesion, the cell type, and the cellular proliferation state. The repair of and response to O-alkylation damage, primarily O6-methylguaine DNA adducts (O6-mG), is the purview of O6-methylguanine-DNA methyltransferase (MGMT). Alternatively, this lesion, if left un-repaired, induces replication-dependent formation of the O6-mG:T mis-pair and recognition of this mis-pair by the post-replication mismatch DNA repair pathway (MMR). Two models have been suggested to account for MMR and O6-mG DNA lesion dependent formation of DNA double-strand breaks (DSBs) and the resulting cytotoxicity – futile cycling and direct DNA damage signaling. While there have been hints at crosstalk between the MMR and base excision repair (BER) pathways, clear mechanistic evidence for such pathway coordination in the formation of DSBs has remained elusive. However, using a novel protein capture approach, Fuchs and colleagues have demonstrated that DSBs result from an encounter between MMR-induced gaps initiated at alkylation induced O6-mG:C sites and BER-induced nicks at nearby N-alkylation adducts in the opposite strand. The accidental encounter between these two repair events is causal in the formation of DSBs and the resulting cellular response, documenting a third model to account for O6-mG induced cell death in non-replicating cells. This graphical review highlights the details of this Repair Accident model, as compared to current models, and we discuss potential strategies to improve clinical use of alkylating agents such as temozolomide, that can be inferred from the Repair Accident model.
Keywords: MGMT, alkylation, base excision repair, mismatch repair, futile cycle, cell death
1. Introduction
The cellular response to and the repair of DNA damage induced by alkylating agents is complex, involving at least three DNA repair pathways: direct repair, base excision repair (BER) and mismatch repair (MMR). Specifically, this includes direct repair of the O6-methylguaine (O6-mG) lesion by O6-methylguanine-DNA methyltransferase (MGMT), direct repair of 1-methyladenine (1-mA) and 3-methylcytosine (3-mC) lesions by ALKBH proteins and BER of the remaining lesions such as N3-methyladenine (N3-mA) and N7-methylguanine (N7-mG) [1–3]. Although not directly cytotoxic, the O6-mG lesion induces cellular toxicity in response to MMR recognition and processing [4, 5]. An active MMR pathway is required for cytotoxicity of the O6-mG lesion [6], with the mechanism of cell death characterized by two complementary models (Fig. 1). However, questions regarding O6-mG induced cell death have remained unanswered, including the observation that cytotoxicity of the O6-mG/C or O6-mG/T base pair is associated with DNA strand breaks [7] or nicks suggested to result from BER intermediates (abasic sites) [8]. While the mechanisms of the individual repair pathways have been characterized in detail, there has been continued debate regarding pathway crosstalk in response to alkylation damage [3]. The model proposed by Fuchs et al [9] adds a further dimension to the debate, promoting essential BER/MMR pathway functional interaction in the cellular response to and repair of the O6-mG lesion, with emphasis on the pathways involved in the response in non-replicating cells.
Fig. 1 –
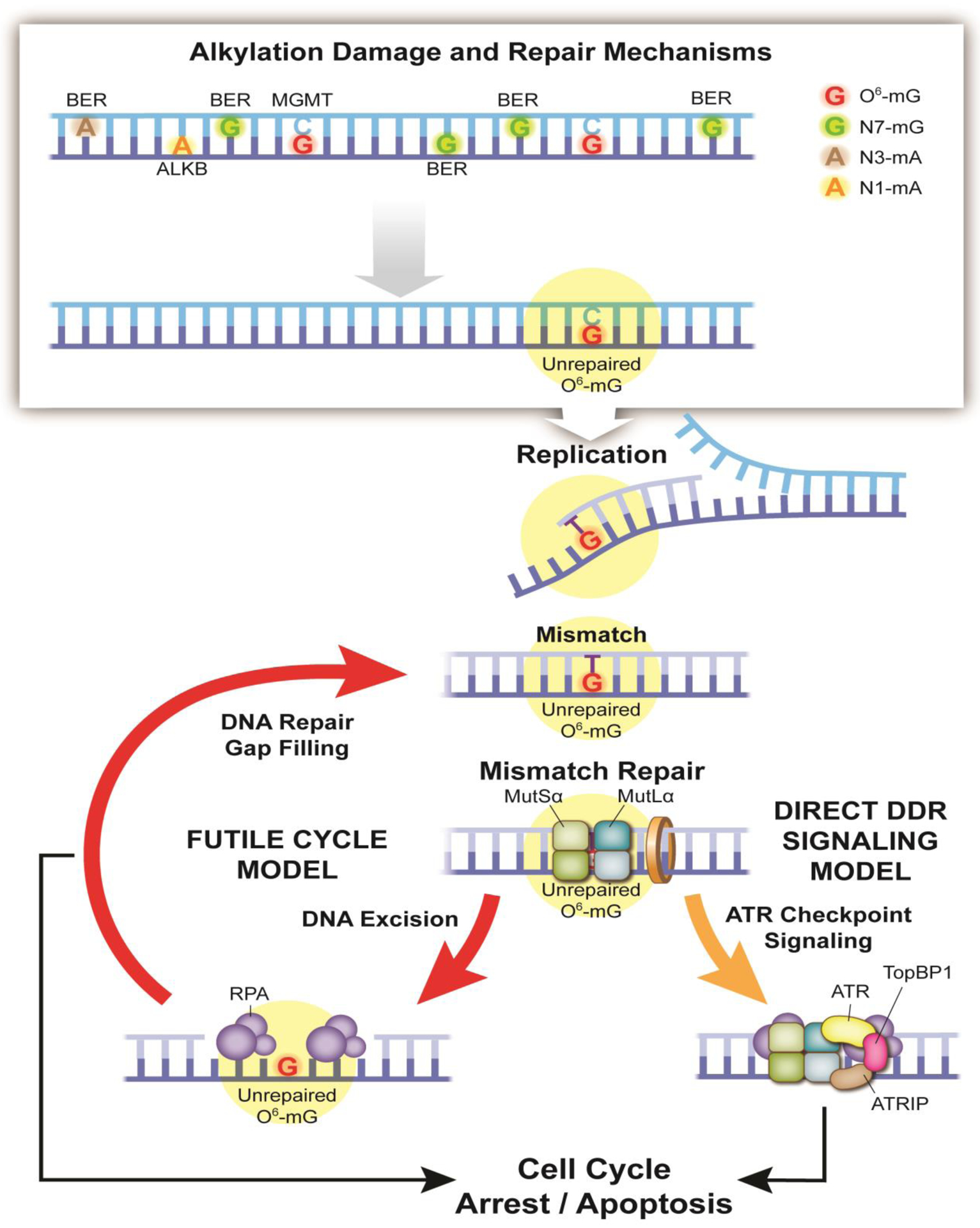
Classic models of the cellular response to alkylation damage. Alkylating agents such as temozolomide induce a spectrum of adducts/lesions, requiring several DNA repair pathways to maintain genome integrity [1, 3, 15, 28]. These include the Direct Reversal protein MGMT for repair of the O6-mG adduct, the ALKB family of Direct Reversal proteins for repair of the 1-methyladenine (1-mA) and 3-methylcytosine (3-mC) adducts, and proteins of the BER pathway for repair of the N3-methyladenine (N3-mA) and N7-methylguanine (N7-mG) adducts. If left un-repaired, a T base is preferentially inserted opposite the O6 modified base (O6-mG), forming the O6-mG:T mis-pair. This mismatch then initiates MutSα/MutLα recognition and repair. Iterative rounds of DNA excision and DNA repair gap filling ultimately lead to cell cycle arrest and apoptosis, as shown in the Futile Cycle model (left). Concurrently, the MutSα and MutLα proteins directly recruit the ATR checkpoint proteins (right), initiating the Direct DDR Signaling model.
2. MMR mediated response to alkylation damage – futile cycling or direct signaling
Genomic DNA is subject to base alkylation resulting from both endogenous and exogenous sources. These include cellular metabolic products (endogenous) and environmental or exogenous genotoxins such as nitroso-compounds and chemotherapeutic agents [1, 3]. Temozolomide (TMZ) is the predominant mono-functional DNA alkylating agent used in the treatment of glioma, among other cancers. This orally administered, bioavailable chemotherapeutic rapidly breaks down to yield the active metabolite MTIC [5-(3-methyl-1-triazeno) imidazole-4-carboxamide]. MTIC then spontaneously breaks down to the methyl diazonium ion, which methylates DNA in the N7 position of guanine (N7-mG), the N3 position of adenine (N3-mA), and the O6 atom of guanine (O6-mG) as well as minor fractions at the N1 position of adenine (1-mA) and the N3 position of cytosine (3-mC). Cellular protection from TMZ and other alkylating agents requires at least three DNA repair processes, including BER, MMR and direct reversal repair proteins such as MGMT [10] and the ALKBH proteins [2, 3] (Fig 1). While the O6-mG lesion is a minor fraction of the TMZ-induced lesions, it is the most cytotoxic and mutagenic. High expression of MGMT blocks alkylating agent induced cell death by directly reverting O6-mG to G. Conversely, cells are highly sensitive to alkylating agents upon loss of MGMT expression [7, 11]. Thus, to improve cancer therapy, numerous strategies to limit O6-mG repair, by depleting or inhibiting MGMT, have been developed [11].
The O6-mG lesion, when not repaired by the direct reversal protein MGMT, is stable. During replication, predominantly thymine and, to a lesser extent, cytosine are inserted opposite the O6-mG lesion. As both of these insertion events evade proofreading, insertion of T is highly mutagenic [12]. Thus, while the lesion itself is not inherently cytotoxic, O6-mG induced cell death depends on replication dependent formation of the O6-mG:T mis-pair and recognition of this mis-pair or mismatch by the post-replication mismatch DNA repair (MMR) pathway (Fig. 1). The mis-pair is recognized by the MSH2/MSH6 heterodimer (the MutSα complex) that in turn recruits the MLH1/PMS2 heterodimer (the MutLα complex). At this juncture, two complementary models have been proposed to explain the resulting onset of apoptosis. As shown (Fig. 1, left), recognition of the mismatch leads to MMR-induced exonuclease activity and the formation of a large DNA gap, followed by MMR-induced DNA synthesis. During gap-filing, T is again inserted opposite O6-mG [12], thus forming again the O6-mG:T mismatch which re-initiates MutSα/MutLα recognition and repair, leading to repeated (futile) cycles of mismatch recognition-DNA excision-DNA synthesis [5, 13]. In a crucial observation made 40 years ago, Karran and Marinus [14], recognized that owing to their location in the parental strand, O6-mG lesions are not removed during the multiple MMR-mediated repair attempts. This unique feature distinguishes O6-mG lesions from base analogs such as 2-AP and BrdU which both reside in the daughter strand and are thus efficiently repaired by MMR. Repeated rounds of excision and re-synthesis will eventually lead to the collapse of the replication fork, ATR/CHK1 signaling and the onset of apoptosis [1, 3, 15].
However, an elegant study by Hsieh and colleagues proposed a direct signaling model (Fig. 1, right). Here, the O6-mG:T mis-pair is recognized and bound by the MutSα and MutLα complexes that in turn recruit the DNA damage response proteins ATR, ATRIP and TopBP1 to initiate DNA damage response (DDR) checkpoint activation [16, 17]. Subsequently, it was found that MutSα directly interacts with ATR, TopBP1, and Chk1 while MutLα interacts with TopBP1 [18]. This DDR checkpoint triggers the onset of cell cycle arrest and apoptosis. While signaling in response to the O6-mG:T mismatch in cancer cells requires two rounds of replication, normal stem cells show the same MMR-dependent signaling in the first S-phase [19]. Overall, these two models (Fig. 1) of O6-mG induced cell death are consistent with the early observations of several groups documenting the requirement for the MMR pathway [6], the absence of MGMT [10] and a correlation with DNA strand breaks [7] for the onset of O6-mG induced cell death.
3. DNA Repair Accident model
The clinically important alkylating agent TMZ is regarded as the first-line therapy, combined with surgery and radiation, for the treatment of glioblastoma, while its mechanism of action leading to cytotoxic effects is still under debate. As defined above, TMZ mainly induces a spectrum of DNA lesions, including N7-mG (70–75%), N3-mA (8–12%), and O6-mG (8–9%). These damaged DNA bases trigger activation of several DNA repair systems, including BER for N7-mG and N3-mA and MGMT or MMR for the O6-mG lesion. It has long been held that the cytotoxic effect of TMZ depends on a DNA damage response signal or a lethal by-product of O6-mG-induced MMR processing following DNA replication (direct signaling or the futile cycle models, Fig 1). In addition to processing by the MMR pathway, BER intermediates have also been proposed to contribute to the cytotoxic effects of TMZ [8, 20, 21]. Further, it has been suggested that DSBs leading to cytotoxicity might be produced by crosstalk between BER and MMR [22] or accumulation of BER intermediates in addition to the O6-mG lesion [8].
Since glioblastoma tumors are composed of a large fraction of non-dividing, quiescent, cells [23], it was deemed essential to investigate the mode-of-action of TMZ in resting cells. To address this issue, we utilized a newly developed approach aimed at capturing nucleoprotein complexes from nuclear extracts (termed IDAP; Isolation of DNA Associated Proteins), an approach that turned out to be efficient and versatile [24, 25]. Briefly, the core aspect of the IDAP methodology centers around the capture of a DNA-fragment-of-interest on a magnetic bead: a specific oligonucleotide (TFO probe) forms a triple helix with a cognate dsDNA sequence while the other extremity of the TFO probe carries a biotin moiety that interacts with a streptavidin-conjugated magnetic bead. To implement the IDAP approach, we incubated plasmid DNA (damaged by MNU, a TMZ mimic), under non-replicating conditions, with protein extracts of Xenopus laevis eggs. Many proteins, specifically recruited by the presence of MNU-induced DNA damage, were captured by the probe, and identified by mass spectrometry (MS) analysis. Of particular interest, the core MMR proteins were highly enriched. Through subsequent biochemical assays, it was revealed that both MMR and BER proteins are active on MNU-treated DNA. We found that concurrent BER and MMR processes on the same DNA molecule could accidentally lead to DSB formation when repair intermediates of BER and MMR encounter each other, an event we will refer to as a “Repair Accident” (RA) (Fig. 2) [9]. Future studies should therefore be considered that would further evaluate the role of BER proteins in this model. This may be achieved by probing the impact of BER defects (loss of expression for example of APE1, PARP1 or other BER proteins) or BER inhibition in resting cells and evaluating the contribution of the RA model in the formation of DSBs. Overall, we propose that DSB‟s generated via concurrent BER, and MMR processing represents an additional mechanism for TMZ-induced cytotoxicity in non-dividing or quiescent cells.
Fig. 2 –
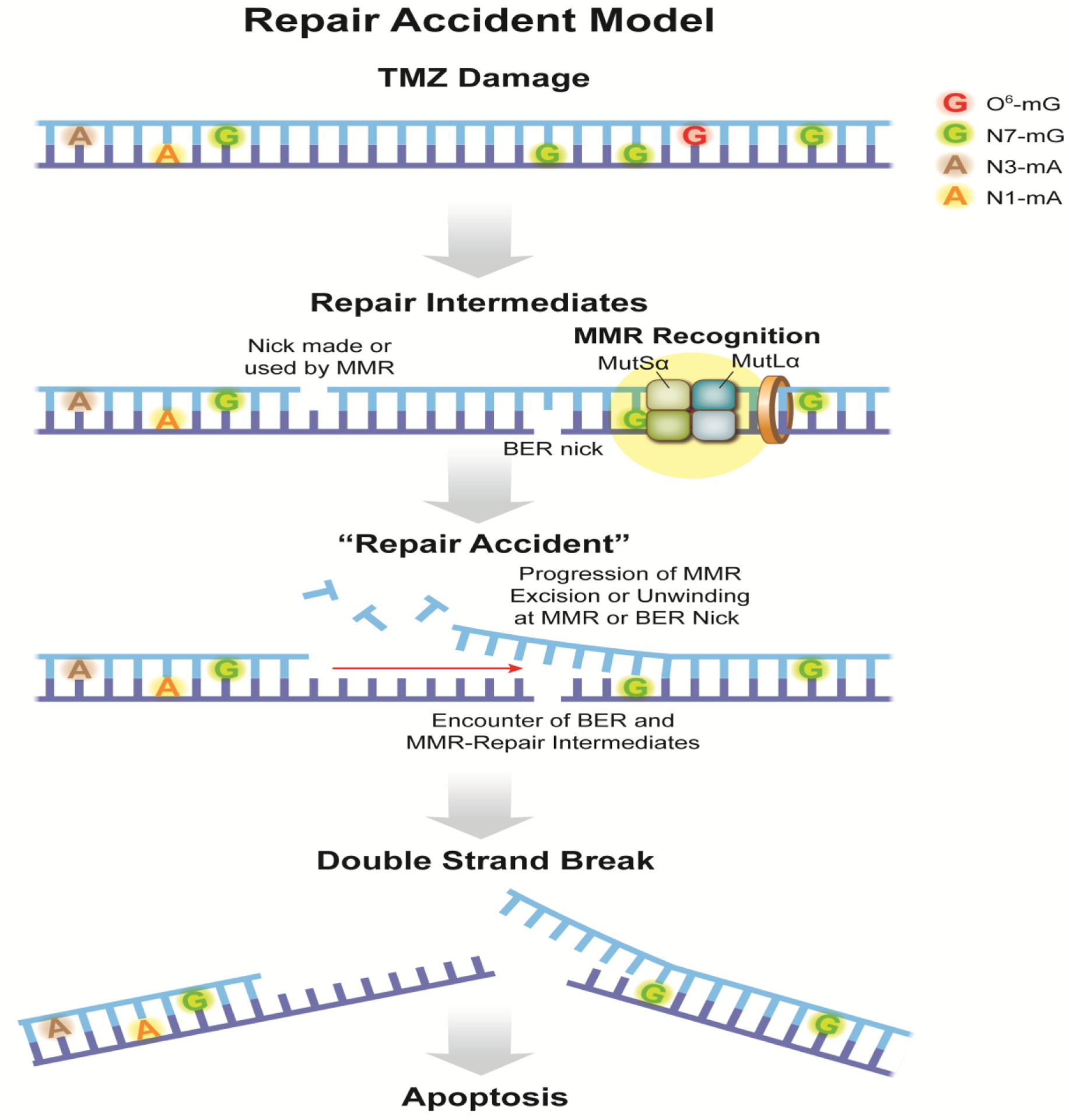
Repair Accident (RA) model. When temozolomide (TMZ) reacts with DNA it produces a variety of adducts among which the N-alkyl adducts, N7-mG and N3-mA, represent >80% of all alkylation events. Importantly, 8–9% of the lesions induced by TMZ include O6-mG. O-alkylation adducts are a hallmark of SN1 alkylating agents. The BER pathway acts at N-alkyl adducts, while the core MMR proteins recognize O6-mG:C base pairs. In TMZ treated DNA, initiation of MMR involves recognition by MutSα of the O6-mG:C base pair. MMR-mediated gap formation starts at a nick made by MutLα or at a nick produced by BER during repair of a nearby N-alkylation adduct. In contrast to MMR at the replication fork, lack of an instruction signal makes it equally likely that the initiating nick is in either strand. In any case, Exo1-mediated strand degradation or helicase unwinding proceeds towards the initiating O6-mG:C lesion. With an average MMR excision track length of several hundred bases, the accidental occurrence of another nicked BER intermediate in the opposite strand will give rise to a DSB. We suggest naming such a circumstance a “Repair Accident” [9].
4. Discussion/Summary
Over the last 20 years, numerous investigations have concluded that the cellular toxicity of SN1 alkylating agents is due to the minor O6-mG adduct with an obligatory involvement of the MMR pathway. In all these studies, the target for MMR is not O6-mG per se but the O6-mG:T mis-pair or mismatch that forms during replication; this mismatch was suggested either to trigger multiple MMR repair attempts (futile processing) or to act as a checkpoint signal (direct signaling) (Fig.1).
The prevailing models for the cellular response to alkylation damage such as that induced by TMZ (futile cycling and direct DNA damage signaling) have been extensively tested in numerous cellular and animal models that highlight the requirement for cell replication as a prerequisite for apoptotic signaling [5]. However, there has been continued debate and evaluation of the mechanisms that connect MMR pathway proteins and activity to apoptosis. Neither model can explain all the observations, suggestive of a missing piece to the puzzle. Further, different cell types appear to respond with modified mechanisms of response. For example, while most cell types show a requirement for two rounds of replication for activation of the ‘futile cycle’ model [5], colon cancer stem cells activate the signal in an MMR-dependent manner in the first cell cycle [19]. Conversely, the direct signaling model has been supported both in cellular models [17] and in animal models, as reviewed in [5]. However, in both cases, a role for replication is essential and therefore highlights aspects of alkylation-induced cell death that cannot be explained for non-dividing or quiescent cells. There have been numerous suggestions of crosstalk between MMR and BER in the response to alkylating agents [20, 22]. For example, it was shown that the protein ASCIZ rapidly forms MLH1-dependent foci in response to methyl methane sulfonate (MMS) treatment. It was suggested that alkylation induced ASCIZ foci is dependent on activity of the BER pathway but does not depend on DNA replication or the formation of DSBs [21]. Further analysis would be required to determine if this signaling model via ASCIZ is related to the ‘Repair Accident’ model we highlight herein and below.
In a recent study, Fuchs et.al. propose that TMZ treatment can lead to DSB‟s in the absence of replication by virtue of an accidental encounter between an MMR event initiated at an O6-mG:C base pair and a nearby BER intermediate from processing of an N-alkylation site (Repair Accident (RA) model) (Fig. 2). In contrast to the previous models that can be qualified as late events, since they involve replication and cell cycle(s), induction of DSBs within the framework of the RA model occurs soon after TMZ exposure and represents an early response.
During glioblastoma treatment, a dose of TMZ is delivered daily, concomitantly with a radiotherapy session, for 6 weeks (for a recent review, see [26]). In this context, triggering DSBs by TMZ treatment in non-dividing cells via the RA mechanism might be highly effective since most cells in a glioblastoma tumor are not proliferating and not subject to the effects of most chemotherapeutic agents [23]. Extrapolation of our experimental data to the concentration of TMZ achieved in serum following a single dose suggests that about 10 DSBs/cell can form via the Repair Accident model each day, a number comparable to the DSBs induced by 0.5–1 Gy of ionizing radiation (IR). In addition, it was empirically established that treatment (TMZ plus radiotherapy) exhibits supra-additive cytotoxicity as long as TMZ administration precedes radiotherapy [27]. Our data may provide some rationale for this observation. Indeed, the single-strand DNA (ssDNA) gaps, that are formed at early time points during MMR processing at O6-mG:C sites (Fig. 2), constitute preferential targets for IR-induced single-strand breaks (SSBs), leading to DSBs. Such events provide a plausible explanation for the observed supra-additivity when TMZ precedes IR.
Potential improvements for the clinical use of TMZ might be considered based on the Repair Accident model. In this model, DSBs are formed as a consequence of concomitant processing of lesions by proteins of both the MMR and BER pathways. Processing of O6-mG:C sites by MMR entails the formation of ssDNA gaps that are several hundred bases in length; these gaps are either produced by the action of an exonuclease (Exo1) or via helicase unwinding. If these excision tracks encounter a BER intermediate (nicks or abasic sites), then a DSB will likely occur (Fig. 2). Slowing down or inhibiting the latter steps of BER, i.e., the steps that occur between incision and ligation, will potentially increase DSB occurrence. Thus, inhibitors of the downstream BER proteins Polβ, PARP1, PARP2 and DNA ligase III, or defects in expression of XRCC1, would increase the half-life of strand discontinuities and consequently promote DSB formation in resting cells via this model. Our work may also suggest that, in addition to brain tumors, TMZ could be instrumental in the treatment of any cancer with a high index of non-dividing cells. The RA model, a proposed third mechanism of DSB generation by alkylating agents such as TMZ, is unique from the prevailing models (futile cycling and direct DNA damage signaling) since the RA model would preclude the requirement for replication/cell cycle dependence for cytotoxicity.
Acknowledgements
RWS is an Abraham A. Mitchell Distinguished Investigator. Research in the Sobol lab on DNA repair, the analysis of DNA damage and the impact of genotoxic exposure is funded by grants from the National Institutes of Health (NIH) [CA148629, ES014811, ES029518, ES028949 and CA238061] and from the National Science Foundation (NSF) [NSF-1841811]. Support for RWS is also provided from the Abraham A. Mitchell Distinguished Investigator Fund and from the Mitchell Cancer Institute Molecular & Metabolic Oncology Program Development fund. We thank Dr. Aishwarya Prakash (University of South Alabama) for critically evaluating the figures during the preparation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
RWS is a scientific consultant for Canal House Biosciences, LLC. The authors state that there is no conflict of interest.
References
- [1].Fu D, Calvo JA, Samson LD, Balancing repair and tolerance of DNA damage caused by alkylating agents, Nat Rev Cancer, 12 (2012) 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Fedeles BI, Singh V, Delaney JC, Li D, Essigmann JM, The AlkB Family of Fe(II)/alpha-Ketoglutarate-dependent Dioxygenases: Repairing Nucleic Acid Alkylation Damage and Beyond, J Biol Chem, 290 (2015) 20734–20742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Soll JM, Sobol RW, Mosammaparast N, Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities, Trends Biochem Sci, 42 (2017) 206–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Gerson SL, MGMT: its role in cancer aetiology and cancer therapeutics, Nat Rev Cancer, 4 (2004) 296–307. [DOI] [PubMed] [Google Scholar]
- [5].Jiricny J, The multifaceted mismatch-repair system, Nat Rev Mol Cell Biol, 7 (2006) 335–346. [DOI] [PubMed] [Google Scholar]
- [6].Kat A, Thilly WG, Fang WH, Longley MJ, Li GM, Modrich P, An alkylation-tolerant, mutator human cell line is deficient in strand-specific mismatch repair, Proc Natl Acad Sci U S A, 90 (1993) 6424–6428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Kalamegham R, Warmels-Rodenhiser S, MacDonald H, Ebisuzaki K, O6-methylguanine-DNA methyltransferase-defective human cell mutant: O6-methylguanine, DNA strand breaks and cytotoxicity, Carcinogenesis, 9 (1988) 1749–1753. [DOI] [PubMed] [Google Scholar]
- [8].Fortini P, Rosa S, Zijno A, Calcagnile A, Bignami M, Dogliotti E, Methoxyamine modification of abasic sites protects CHO cells from the cytotoxic and mutagenic effects of oxygen alkylation, Carcinogenesis, 13 (1992) 87–93. [DOI] [PubMed] [Google Scholar]
- [9].Fuchs RP, Isogawa A, Paulo JA, Onizuka K, Takahashi T, Amunugama R, Duxin JP, Fujii S, Crosstalk between repair pathways elicits double-strand breaks in alkylated DNA and implications for the action of temozolomide, Elife, 10 (2021) e69544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Karran P, Bignami M, Self-destruction and tolerance in resistance of mammalian cells to alkylation damage, Nucleic Acids Res, 20 (1992) 2933–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Dolan ME, Moschel RC, Pegg AE, Depletion of mammalian O6-alkylguanine-DNA alkyltransferase activity by O6-benzylguanine provides a means to evaluate the role of this protein in protection against carcinogenic and therapeutic alkylating agents, Proc Natl Acad Sci U S A, 87 (1990) 5368–5372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Warren JJ, Forsberg LJ, Beese LS, The structural basis for the mutagenicity of O(6)-methyl-guanine lesions, Proc Natl Acad Sci U S A, 103 (2006) 19701–19706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wang JY, Edelmann W, Mismatch repair proteins as sensors of alkylation DNA damage, Cancer Cell, 9 (2006) 417–418. [DOI] [PubMed] [Google Scholar]
- [14].Karran P, Marinus MG, Mismatch correction at O6-methylguanine residues in E. coli DNA, Nature, 296 (1982) 868–869. [DOI] [PubMed] [Google Scholar]
- [15].Li GM, Mechanisms and functions of DNA mismatch repair, Cell research, 18 (2008) 85–98. [DOI] [PubMed] [Google Scholar]
- [16].Li Z, Pearlman AH, Hsieh P, DNA mismatch repair and the DNA damage response, DNA Repair (Amst), 38 (2016) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Yoshioka K, Yoshioka Y, Hsieh P, ATR kinase activation mediated by MutSalpha and MutLalpha in response to cytotoxic O6-methylguanine adducts, Mol Cell, 22 (2006) 501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Liu Y, Fang Y, Shao H, Lindsey-Boltz L, Sancar A, Modrich P, Interactions of human mismatch repair proteins MutSalpha and MutLalpha with proteins of the ATR-Chk1 pathway, J Biol Chem, 285 (2010) 5974–5982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gupta D, Lin B, Cowan A, Heinen CD, ATR-Chk1 activation mitigates replication stress caused by mismatch repair-dependent processing of DNA damage, Proc Natl Acad Sci U S A, 115 (2018) 1523–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Agnihotri S, Gajadhar AS, Ternamian C, Gorlia T, Diefes KL, Mischel PS, Kelly J, McGown G, Thorncroft M, Carlson BL, Sarkaria JN, Margison GP, Aldape K, Hawkins C, Hegi M, Guha A, Alkylpurine-DNA-N-glycosylase confers resistance to temozolomide in xenograft models of glioblastoma multiforme and is associated with poor survival in patients, J Clin Invest, 122 (2012) 253–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McNees CJ, Conlan LA, Tenis N, Heierhorst J, ASCIZ regulates lesion-specific Rad51 focus formation and apoptosis after methylating DNA damage, EMBO J, 24 (2005) 2447–2457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Simonelli V, Leuzzi G, Basile G, D’Errico M, Fortini P, Franchitto A, Viti V, Brown AR, Parlanti E, Pascucci B, Palli D, Giuliani A, Palombo F, Sobol RW, Dogliotti E, Crosstalk between mismatch repair and base excision repair in human gastric cancer, Oncotarget, 8 (2017) 84827–84840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Das S, Srikanth M, Kessler JA, Cancer stem cells and glioma, Nat Clin Pract Neurol, 4 (2008) 427–435. [DOI] [PubMed] [Google Scholar]
- [24].Isogawa A, Fuchs RP, Fujii S, Versatile and efficient chromatin pull-down methodology based on DNA triple helix formation, Sci Rep, 8 (2018) 5925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Isogawa A, Fuchs RP, Fujii S, Chromatin Pull-Down Methodology Based on DNA Triple Helix Formation, Methods Mol Biol, 2119 (2020) 183–199. [DOI] [PubMed] [Google Scholar]
- [26].Strobel H, Baisch T, Fitzel R, Schilberg K, Siegelin MD, Karpel-Massler G, Debatin KM, Westhoff MA, Temozolomide and Other Alkylating Agents in Glioblastoma Therapy, Biomedicines, 7 (2019) 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Bobola MS, Kolstoe DD, Blank A, Silber JR, Minimally cytotoxic doses of temozolomide produce radiosensitization in human glioblastoma cells regardless of MGMT expression, Mol Cancer Ther, 9 (2010) 1208–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Gupta D, Heinen CD, The mismatch repair-dependent DNA damage response: Mechanisms and implications, DNA Repair (Amst), 78 (2019) 60–69. [DOI] [PubMed] [Google Scholar]