

Abstract
Purpose:
Programmed cell death protein 1 (PD-1) blockade can mediate objective responses in advanced sarcomas, but their durability has not been established and it is unclear if hyperprogressive disease (HPD) occurs in sarcomas treated with PD-1 inhibitors.
Experimental Design:
We pooled patients who were treated prospectively with nivolumab or pembrolizumab as monotherapy or with bempegaldesleukin, epacadostat, ipilimumab, or talimogene laherparepvec. We did a new independent assessment for HPD and analyzed clinical, pathologic, and genomic data from baseline tumor biopsies. Our primary endpoint was the incidence of HPD; secondary endpoints were clinical or genomic correlates of response or HPD.
Results:
We treated 134 patients with advanced sarcoma from 2015 to 2019. Twenty-one patients (16%) had a complete or partial response (CR/PR), and 30% of responses were durable for over 2 years. Forty-eight (36%) patients had stable disease (SD), 45 (34%) had progressive disease without HPD (PD), and 15 (11%) had HPD. Five patients (4%) were not evaluable for HPD. The sarcoma subtypes, sites of metastasis, clinical course, and genomic alterations in patients with PD and HPD were similar, except HPD tumors were smaller at baseline.
Conclusions:
In patients with advanced sarcoma, PD-1 blockade can mediate durable responses. HPD occurs in sarcoma at an incidence that is similar to what has been reported in other solid tumors, but patients with HPD were clinically and biologically similar to those who had PD. Further research is required to establish whether HPD is a biologically distinct phenomenon and whether a theoretical risk of HPD should influence patient management.
Translational Relevance.
Programmed cell death protein 1 (PD-1) blockade is an effective treatment for some patients with advanced sarcomas, but there are few known predictors of response and it is unclear if hyperprogressive disease (HPD) occurs in this setting. We did a pooled analysis of patients who were enrolled on prospective trials evaluating PD-1 inhibitors, did whole-exome and RNA sequencing of baseline tumor biopsies, and performed a new radiographic assessment for HPD. Response to PD-1 blockade was associated with sarcoma subtype, disease burden, and patterns of metastatic spread, but not with tumor mutational burden or any specific genomic alterations. HPD occurred in 11% of patients, but patients with HPD were clinically and genotypically comparable with those with non-HPD progressive disease. Moreover, while these two groups had similar outcomes after treatment, we unexpectedly found their disease burden at baseline was unevenly distributed. Further research will be required to determine if HPD is biologically distinct and clinically meaningful.
Introduction
Sarcomas are a heterogeneous group of mesenchymal neoplasms of bone and soft tissue origin and include more than 70 distinct subtypes. Approximately 15,000 cases of soft tissue and bone sarcomas are diagnosed annually in the United States. Despite primary combined modality therapy, 25% to 50% of patients develop recurrent and/or metastatic disease (1, 2). For patients with unresectable advanced disease, chemotherapy remains the standard of care. However, complete responses (CR) are rare, and the median overall survival (OS) in the metastatic setting is 15 to 20 months (3–6).
Standard cytotoxic chemotherapy agents such as doxorubicin alone or gemcitabine with docetaxel lead to responses in about 20% of patients in the front line setting (4, 5). In the second line, chemotherapy response rates are even lower (7, 8). With pazopanib, despite improved progression-free survival (PFS) over placebo, the median was 4 months without an improvement in OS (9). For most patients, with cytotoxic chemotherapy the duration of benefit is short-lived without any potential for cure. Therefore, novel and effective systemic therapies are desperately needed for patients with unresectable and metastatic sarcomas.
Early trials evaluating anti–programmed cell death protein 1 (anti–PD-1) monotherapy in sarcoma reported low objective response rates (ORR) and brief PFS in an unselected sarcoma population. In SARC028, the ORR to pembrolizumab in the entire population was 11% (10). A study of pembrolizumab plus metronomic cyclophosphamide reported an ORR of 2% (11). A study of pembrolizumab plus axitinib reported an ORR of 10% in sarcomas other than alveolar soft-part sarcoma (ASPS), which can be uniquely responsive (12).
The literature to date is limited by the rarity of individual subtypes of sarcomas, lack of controls in clinical trials, and short follow-up. Consequently, it is not clear whether responses to immune checkpoint inhibitors (ICI) are durable, and heterogeneity among sarcoma subtypes has diluted the perceived response rate of “sarcomas” as a group. For example, the ORR for Ewing's sarcoma, osteosarcoma, gastrointestinal stromal tumors (GIST), and leiomyosarcoma has been below 10% (13). In contrast, the ORR for angiosarcoma and undifferentiated pleomorphic sarcoma (UPS) may exceed 20%, and in ASPS it could be closer to 50%.
Other than histology, there are no reliable clinical, biochemical, or genomic predictors of response in sarcoma. In melanoma, there is a correlation between disease burden at baseline and outcomes after checkpoint inhibitors, although no proof of a causal relationship (14, 15). Also, while sarcomas classically metastasize to lungs, some like leiomyosarcoma have a predilection for liver metastases and others including angiosarcoma and epithelioid sarcoma metastasize to lymph nodes (16). The experience in other solid tumors is that liver metastases tend to be less responsive to immunotherapy, while lymph node metastases are particularly responsive (17, 18). Whether any of these factors correlate with response in sarcoma has not been evaluated.
Furthermore, there is increasing awareness that some patients experience hyperprogressive disease (HPD) after treatment with PD-1 inhibitors. HPD is a response pattern in which tumor growth is accelerated by treatment with checkpoint inhibitors (19, 20). The incidence of HPD in other solid tumors treated with immunotherapy is reported to range from 4% to 29%. In diverse cancers, HPD was reported to be associated with alterations in MDM2, MDM4, and EGFR (21). To our knowledge there has never been an evaluation of HPD in a controlled trial, but HPD was not reported to occur in prior chemotherapy trials. It has not been explored if HPD occurs in an exclusive sarcoma population after PD-1 blockade.
Given these outstanding questions we undertook to evaluate patients with advanced sarcoma who were treated with PD-1 blockade at our institution under the auspices of clinical trials (22–25). All patients had prospective response evaluation, and for this study we did a new independent assessment for HPD. We evaluated the clinical and biological characteristics of patients who experienced HPD to better understand the significance of this response pattern.
Materials and Methods
Treatment with anti–PD-1
All patients had unresectable or metastatic sarcoma and were treated prospectively on trial at Memorial Sloan Kettering Cancer Center (MSKCC) with antibodies against PD-1, with or without other immunomodulatory agents. Patients who were enrolled on more than one trial at MSKCC were only included in this study once and were analyzed with the first trial for which they were enrolled. The results of these four prospective clinical trials have been reported elsewhere: (i) nivolumab with or without ipilimumab (22), (ii) pembrolizumab with talimogene laherparepvec (T-VEC; ref. 23), (iii) nivolumab with bempegaldesleukin (24), and (iv) pembrolizumab with epacadostat (25).
Study investigators obtained written informed consent from all patients enrolled. These studies were conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, and US Common Rule) and were approved by the MSKCC Institutional Review Board.
Data collection
Patient demographic information including age, tumor histology, and lines of prior systemic treatment before PD-1 blockade were prospectively recorded. Disease burden refers to the sum of target lesions at baseline in millimeters, taken from prospectively recorded RECIST measurements. Site(s) of metastatic involvement were determined retrospectively by clinical, radiographic, and/or pathologic review. Equivocal lymph node findings required pathologic confirmation.
Outcomes
Time to treatment refers to the interval between the diagnosis of metastatic or unresectable sarcoma and the first dose of the study drug(s). PFS was prospectively recorded and measured after the first dose of the study drug(s). We counted death without disease as a progression event. OS was also measured after the first dose of the study drug.
HPD assessment
We evaluated patients for HPD as previously described (19). Briefly, this required new tumor response evaluations (performed using RECIST v1.1) starting with a pre-baseline set of images with at least two subsequent time points (immediately before PD-1 blockade and at least one time point after). These new response evaluations were performed in a blinded fashion by a single radiologist (S. Hwang, 15 years of experience) using an independent software application dedicated for the RECIST v1.1 assessment. Using these measurements we calculated tumor growth rate (TGR) before and after PD-1 blockade. HPD was defined as an increase in TGR of 50% or more in target lesions. Patients without adequate pre-baseline imaging, or at least one follow-up imaging study, were not eligible for HPD assessment.
Statistics
Continuous variables are reported as median and range, discrete variables as frequency. We compared demographic data using a χ2 or Fisher exact test. We calculated median follow-up from surviving patients. Time to event and survival curves are shown using the Kaplan–Meier method with univariate comparisons by the log-rank test. Disease burden, tumor mutational burden (TMB), and fraction genome altered (FGA) were compared using an unpaired Mann–Whitney test. We used GraphPad Prism to perform statistics and generate figures.
Whole-exome and RNA sequencing
Details on nucleic acid extraction, whole-exome sequencing (WES), and RNA sequencing (RNA-seq) are provided in the Supplementary Methods document. WES was performed on 96 samples [17 complete or partial response (CR/PR), 32 stable disease (SD), 33 progressive disease without HPD (PD) and 14 HPD]. DNA was extracted from the tumor and matched normal DNA samples and was aligned to GRCh37 using the TEMPO pipeline (https://ccstempo.netlify.app/). The same pipeline was used to determine somatic mutations (substitutions, small insertions, and deletions) and gene-level focal copy-number alterations. Detected variants were annotated for truncating and nontruncating driver mutations using OncoKB annotation api (https://api.oncokb.org/oncokb-website/api). See the Supplementary Methods document for further details regarding RNA library preparation and sequencing.
Differential expression analysis
We used Kallisto (v.0.46.1) to align paired-end FASTQ raw sequencing reads to GRCh37.75 human reference, and to quantify transcript abundances from baseline tumor samples (26). A total of 71 samples were transcriptionally profiled including 12 CR/PR, 21 SD, 28 PD, and 10 HPD. To define sequencing and alignment quality we used CollectRnaSeqMetrics from Picard tools (http://broadinstitute.github.io/picard/), and MultiQC v.1.9 (27).
Transcript level abundances obtained by Kallisto were further aggregated to gene-level expression using sleuth v0.30 R package (28), and we obtained transcript to gene mapping using biomaRt (v.3.12) package in R, and gene symbols defined by Ensembl GRCh37.75 (29). Sleuth R package was also used to normalize gene counts across samples and for linear models to determine differential expression analysis. Models were corrected for batch effects due to sequencing and for sarcoma subtypes.
Gene set enrichment analysis (GSEA) of pathways associated with response (HPD vs. PD) was performed using the clusterProfiler package and GSEA function in R (30, 31). We used 50 hallmark pathways with well-defined biological processes, downloaded from MSigDB (https://www.gsea-msigdb.org/gsea/msigdb/). P value based on comparison between two conditions (HPD and PD) was multiplied with the sign of log-fold change and used for the ranking of the genes. Weighted enrichment statistics and 100,000 gene set permutations were then calculated and used for detection of significantly enriched pathways.
Immune cell deconvolution
Immune cell proportion estimation for 10 cell types was computed from normalized gene expression using the quanTIseq deconvolution method in immunedeconv R package v.2.0.2–1 (32, 33). To allow for enhanced visualization of cellular fraction across all samples we calculated z-score (z = (x-μ)/σ) of each cell type, where x is the raw cell fraction, μ is the all samples mean, and σ is the standard deviation for all samples. We then used the hierarchical clustering method (i.e., maximum clustering distance) in complex heatmap package v.3.4 in R to group and visualize samples (34).
Availability of data and material
The sequencing data generated in this study are not yet publicly available due to patient privacy requirements that are pending approval but will be available upon request from the corresponding author.
Results
We treated 134 patients with advanced sarcoma from 2015 to 2019 (Fig. 1A). For the entire cohort, median follow-up was 33 months [95% confidence interval (CI), 31.3–35.1 months], median PFS was 2 months (95% CI, 1.9–3.4 months), and median OS was 16 months (95% CI, 10.8–19.6 months). The number of patients that received each regimen included: 20 (15%) nivolumab ± ipilimumab, 18 (13%) pembrolizumab plus T-VEC, 68 (51%) nivolumab plus bempegaldesleukin, and 28 (21%) pembrolizumab plus epacadostat.
Figure 1.
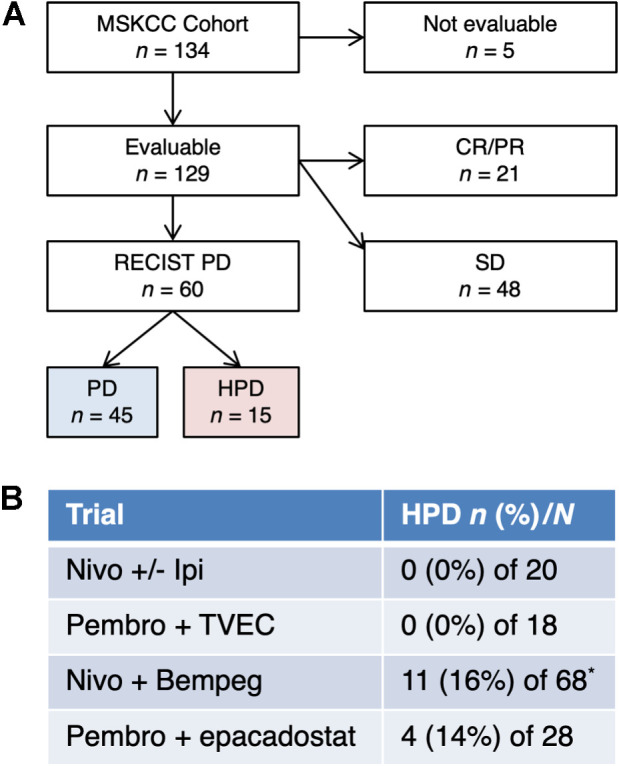
HPD in sarcoma. A, CONSORT diagram for comparison. Five patients were not evaluable. B, Incidence of HPD in each protocol (P = 0.03 by χ2 test). The number of patients with HPD (n, %) and the total number treated on protocol (N) are indicated. Patients who were enrolled on multiple trials were only included once, on the first study. *Five patients in the nivolumab plus bempegaldesleukin trial were not evaluable.
Enrollment in the four protocols was not randomized, leading to unequal distributions of patient demographic variables, sarcoma subtypes and pretreatment characteristics (Supplementary Fig. S1). For example, the median time to treatment in the nivolumab plus bempegaldesleukin trial was 12 months but it was 29 months in the pembrolizumab plus T-VEC trial (P = 0.01; Supplementary Fig. S2). These baseline differences preclude meaningful between-studies comparisons, but there were nonetheless no significant differences in PFS or OS between studies (Supplementary Fig. S3).
Twenty-one (16%) of 134 patients had a CR/PR, 48 (36%) had SD, 45 (34%) had PD, and 15 (11%) had HPD (Fig. 1A). Five patients were not evaluable due to inadequate imaging (all were in the nivolumab plus bempegaldesleukin trial). Eleven instances of HPD occurred in the nivolumab plus bempegaldesleukin trial and 4 occurred in the pembrolizumab plus epacadostat trial. No cases of HPD were identified in the nivolumab ± ipilimumab or pembrolizumab plus T-VEC trials (P = 0.03; Fig. 1B).
The median PFS of patients with a CR/PR, SD, PD, and HPD was 15.0 months (95% CI, 11.8–not reached months), 5.5 months (95% CI, 4.1–7.3 months), 1.6 months (95% CI, 1.5–1.8 months), and 1.6 months (95% CI, 1.0–1.8 months) respectively (Fig. 2A). For patients with a CR/PR, 2-year PFS was 30.4% (95% CI, 50.6%–12.0%). Also, PFS was the same for the PD and HPD groups because these patients all progressed at early time points. Median OS for patients with a CR/PR, SD, PD, and HPD was not reached (95% CI, not reached–20.6 months), 18.0 months (95% CI, 10.9–26.6 months), 7.7 months (95% CI, 5.0–13.8 months), and 5.9 months (95% CI, 3.1–9.9 months) respectively (PD vs. HPD, P = 0.34; Fig. 2B). For patients with a CR/PR, 2-year OS was 73.7% (95% CI, 87.9%–47.4%).
Figure 2.

Outcomes of patients stratified by best response. Kaplan–Meier curves show PFS (A) and OS (B) after the first dose of the study drug. ns, not statistically significant.
Demographic characteristics
Patients with a CR/PR, as compared with those who had SD, had a similar age and gender distribution (Table 1). Their mean number of prior systemic therapies was 2.0 and 2.9, respectively (P = 0.12), and median time to treatment was 12 months and 25 months (P = 0.019; Supplementary Fig. S4A). Patients with PD and HPD also had comparable age and gender distributions (Table 1). They were pretreated with a mean of 2.7 and 2.3 lines of systemic therapy (P = 0.79) and their time to treatment was 18 and 16 months (P = 0.67; Supplementary Fig. S4B).
Table 1.
Patient demographics.
| Evaluable | CR/PR | SD | PD | HPD | |
|---|---|---|---|---|---|
| Patients | 129 (96%)a | 21 (16%) | 48 (36%) | 45 (34%) | 15 (11%) |
| Median age (IQR) | 57 (44–66) | 65 (54–75) | 60 (53–67) | 47 (38–62) | 56 (44–59) |
| Gender (n female, %) | 66 (51%) | 10 (48%) | 25 (52%) | 22 (49%) | 9 (60%) |
| Prior systemic Rx (% of each response) | |||||
| None | 16 (12%) | 4 (19%) | 6 (13%) | 5 (7%) | 1 (12%) |
| One line | 27 (21%) | 6 (29%) | 12 (25%) | 6 (20%) | 3 (21%) |
| Two lines | 28 (22%) | 4 (19%) | 6 (13%) | 14 (27%) | 4 (22%) |
| Three or more lines | 58 (45%) | 7 (33%) | 24 (50%) | 20 (47%) | 7 (45%) |
| Time to PD-1 (95% CI), months | 18 (14–22) | 12 (3–14) | 25 (16–33) | 18 (10–20) | 16 (6–23) |
| Histology (% of each subtype) | |||||
| ASPS | 3 | 1 (33%) | 2 (67%) | 0 (0%) | 0 (0%) |
| Angiosarcoma | 12 | 5 (42%) | 4 (33%) | 2 (17%) | 1 (8%) |
| Chondrosarcoma | 10 | 1 (10%) | 2 (20%) | 6 (60%) | 1 (10%) |
| EHE | 5 | 0 (0%) | 4 (80%) | 0 (0%) | 1 (20%) |
| Epithelioid sarcoma | 5 | 2 (40%) | 0 (0%) | 3 (60%) | 0 (0%) |
| Leiomyosarcoma | 26 | 2 (8%) | 13 (50%) | 7 (27%) | 4 (15%) |
| LPS, dedifferentiated | 10 | 0 (0%) | 8 (80%) | 2 (20%) | 0 (0%) |
| LPS, myxoid/round cell | 4 | 1 (25%) | 1 (25%) | 0 (0%) | 2 (50%) |
| Myxofibrosarcoma | 5 | 2 (40%) | 2 (40%) | 1 (20%) | 0 (0%) |
| Osteosarcoma | 7 | 0 (0%) | 0 (0%) | 6 (86%) | 1 (14%) |
| Small blue round cell | 7 | 0 (0%) | 0 (0%) | 5e (71%) | 2d (29%) |
| Spindle cell HG NOS | 5 | 1 (20%) | 2 (40%) | 1 (20%) | 1 (20%) |
| UPS | 16 | 5 (31%) | 3 (19%) | 6 (38%) | 2 (13%) |
| Other | 14 | 1b (7%) | 7c (50%) | 6f (43%) | 0 (0%) |
Note: The characteristics of each response group are shown. Time to ICI refers to time in months between the diagnosis of stage IV or unresectable disease and the first dose of PD-1 blockade.
Abbreviations: IQR, interquartile range; LPS, liposarcoma; spindle cell HG NOS, spindle cell sarcoma high grade not otherwise specified.
aFive patients were not evaluable for response.
bPleomorphic LPS.
cOne follicular dendritic cell sarcoma, 2 GIST, 2 malignant solitary fibrous tumor, 1 pecoma, 1 rhabdomyosarcoma.
dOne Ewing's and 1 synovial sarcoma.
eOne Ewing's, 2 synovial, and 2 dermoplastic small round cell sarcomas.
fTwo GIST and 1 each of epithelioid sclerosing fibrosarcoma, malignant peripheral nerve sheath tumor, phyllodes tumor, and pleomorphic LPS.
Sarcoma subtypes
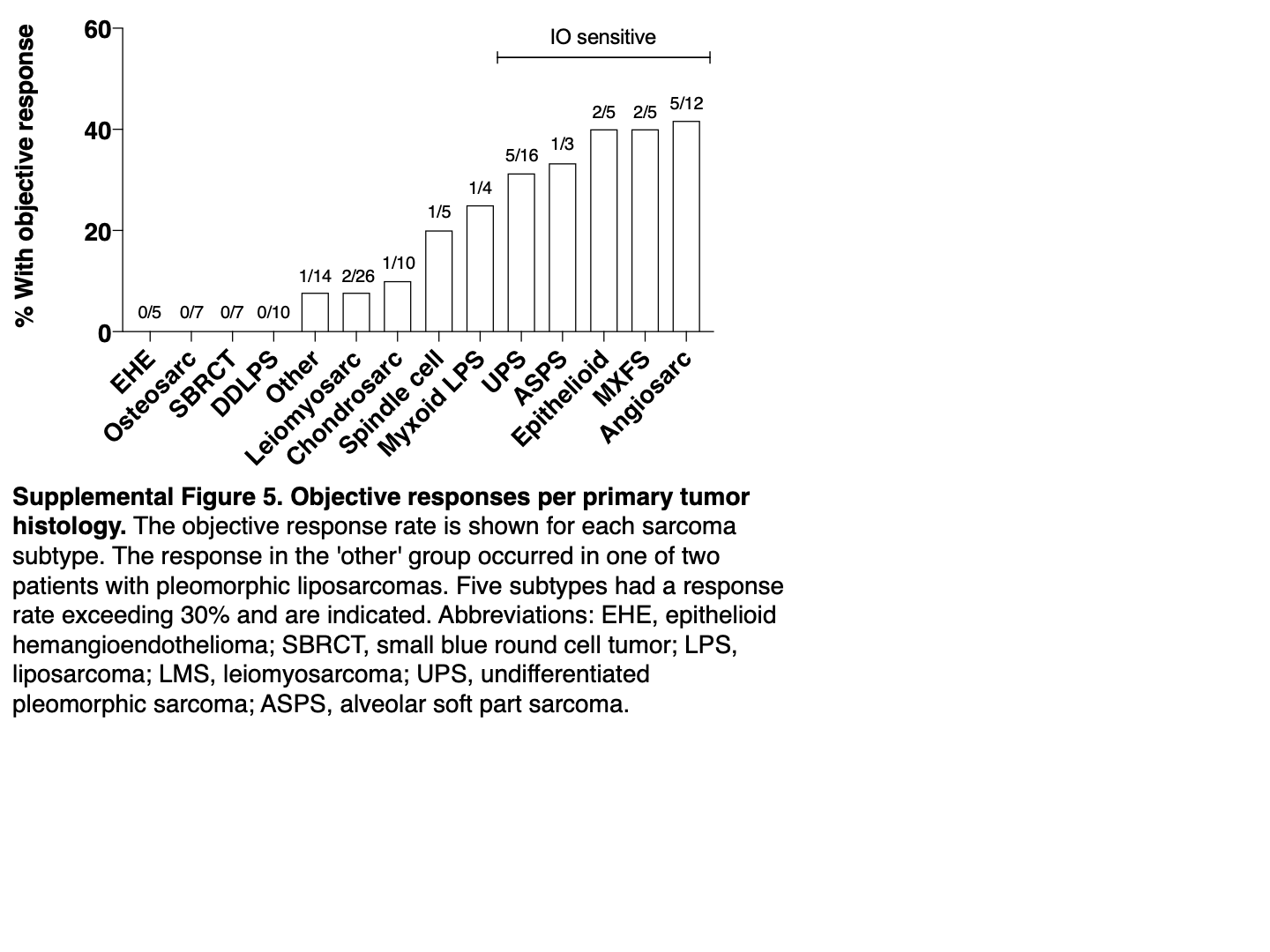
The ORR for each sarcoma subtype ranged from 0% to 42% (Table 1; Supplementary Fig. S5). Five histologies had an ORR that exceeded 30%. These immune-sensitive subtypes were angiosarcoma (2 CR and 3 PR in 12 patients), myxofibrosarcoma (1 CR and 1 PR in 5 patients), epithelioid sarcoma (2 PR in 5 patients), and UPS (5 PR in 16 patients). Also, 1 of 3 patients with ASPS had a response, consistent with other series showing exceptionally high ORR in ASPS (12, 35).
None of the 10 patients with dedifferentiated liposarcoma responded. However, a response was observed in 1 of 4 patients with myxoid liposarcoma, 1 of 2 patients with pleomorphic liposarcoma, and 1 of 5 with high-grade spindle cell sarcoma. Responses were rare or absent in other histologies including chondrosarcoma, leiomyosarcoma, osteosarcoma, and small blue round cell tumors (SBRCT). Finally, while none of the 5 patients with epithelioid hemangioendothelioma (EHE) had a response, 2 had prolonged SD (patients 109 and 94 on Supplementary Table S1).
The subtypes of the 15 patients with HPD were as follows: 1 angiosarcoma, 1 chondrosarcoma, 1 EHE, 4 leiomyosarcoma, 2 myxoid liposarcoma, 1 osteosarcoma, 1 Ewing's sarcoma, 1 synovial sarcoma, 1 high-grade spindle cell sarcoma, and 2 UPS (Table 1). There was no obvious difference in the distribution of sarcoma subtypes between patients with PD and HPD (Table 1). For example, “immune-sensitive” sarcoma subtypes (i.e., angiosarcoma, ASPS, epithelioid, myxofibrosarcoma, or UPS) were present in 12 (27%) of 45 patients with PD and 3 (20%) of 12 with HPD. Moreover, PD-L1 expression was similar in patients with PD and HPD (Supplementary Fig. S6).
Baseline disease burden
Patients with a CR/PR, as compared with those with SD, had a significantly lower baseline disease burden (62 mm vs. 109 mm, P = 0.011; Fig. 3). The disease burden of the 3 patients who had a CR was 14 mm, 29 mm, and 38 mm. Only 1 patient with a PR had a disease burden that was substantially higher than 100 mm; this was an individual with pleomorphic liposarcoma in the mediastinum who had a baseline burden of 202 mm. Unexpectedly, we found the baseline disease burden of patients with PD and HPD were not evenly distributed (104 mm vs. 59 mm, P = 0.035).
Figure 3.
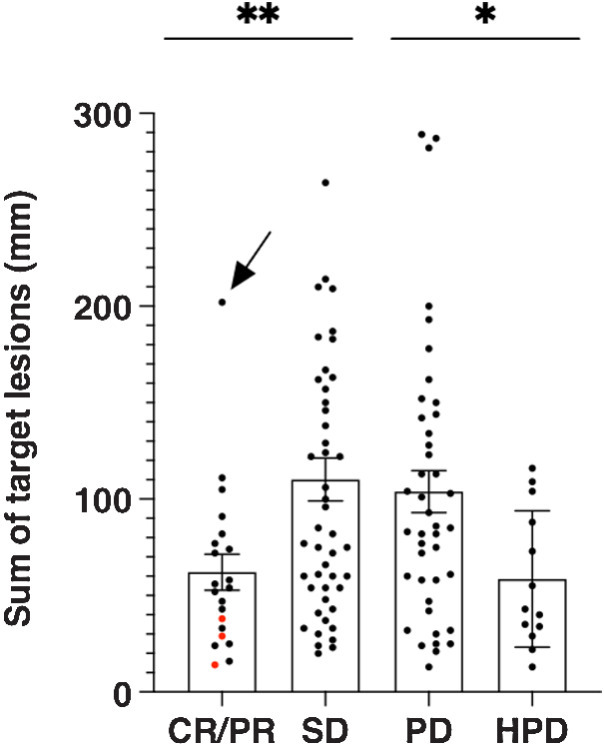
Response and tumor burden at baseline. Tumor burden is represented by the sum of prospectively recorded target lesions at baseline per RECIST. The 3 patients with complete responses are indicated by red dots. The patient indicated by the arrow had a pleomorphic liposarcoma in the mediastinum. *, P < 0.05; **, P < 0.01.
Site of metastasis
The most common site of metastasis was lung in 80 evaluable patients, followed by liver in 31, bone in 24, and lymph nodes in 18. In addition, 28 patients had “other” nonvisceral metastases in skin, soft tissue, peritoneum, or retroperitoneum (Fig. 4A). Some patients had metastases in multiple different organ systems. ORR of patients with metastases in lung, bone, and ‘other’ metastases was 14%, 13%, and 14% respectively.
Figure 4.

Patterns of metastatic spread and response rate. Bars indicate the percentage of patients with metastases in the indicated site that had an objective response. The numbers above each bar show patients with a response over patients evaluable. Some patients had metastases in multiple sites. “Other” metastases include skin and soft tissue, peritoneum, and retroperitoneum. A, Response rate of patients with metastases in the given site. B, Response rate is grouped by the presence or absence of LN metastases and whether the sarcoma subtype was considered immune-sensitive or not. Sensitive subtypes were ASPS, epithelioid sarcoma, myxofibrosarcoma, UPS, and angiosarcoma. C, Distribution of metastases in patients with PD and HPD. LN, lymph node; ns, not statistically significant; Sens, sensitive. *, P < 0.05; **, P < 0.01.
The ORR in patients with liver metastases was 3%, with a single PR occurring in a patient with angiosarcoma (patient 134 on Supplementary Table S1). Four patients with liver metastases had prolonged SD (patients 77, 109, 47, and 94 on Supplementary Table S1). Involvement of the liver varied by sarcoma subtype; for example, 11 (42%) of 26 patients with leiomyosarcoma and 5 (100%) of 5 with EHE had hepatic involvement. Of note, only 4 (13%) of 31 patients with liver metastases had an ‘immune-sensitive’ sarcoma subtype (1 had UPS and 3 had angiosarcoma).
Lymph node metastases were present in 18 (14%) of 129 evaluable patients with the following subtypes: 3 angiosarcoma, 3 epithelioid sarcoma, 3 UPS, 3 SBRCT, 2 chondrosarcoma, 2 leiomyosarcoma, 1 ASPS, and 1 EHE. Eight (44%) of 18 patients with nodal metastases had an objective response (Fig. 4A). Interestingly, all 8 nodal responders also had immune-sensitive sarcoma subtypes (Fig. 4B). Conversely, none of the patients who had nodal metastases with other subtypes (SBRCT, chondrosarcoma, leiomyosarcoma, EHE) had a response.
Patients with PD and HPD had similar patterns of metastatic spread (Fig. 4C). For example, 32 (71%) of 45 patients with PD had lung metastases, as did 11 (73%) of 15 patients with HPD. Metastases to the liver and lymph nodes were also comparably distributed between patients with PD and HPD.
Genomic analyses
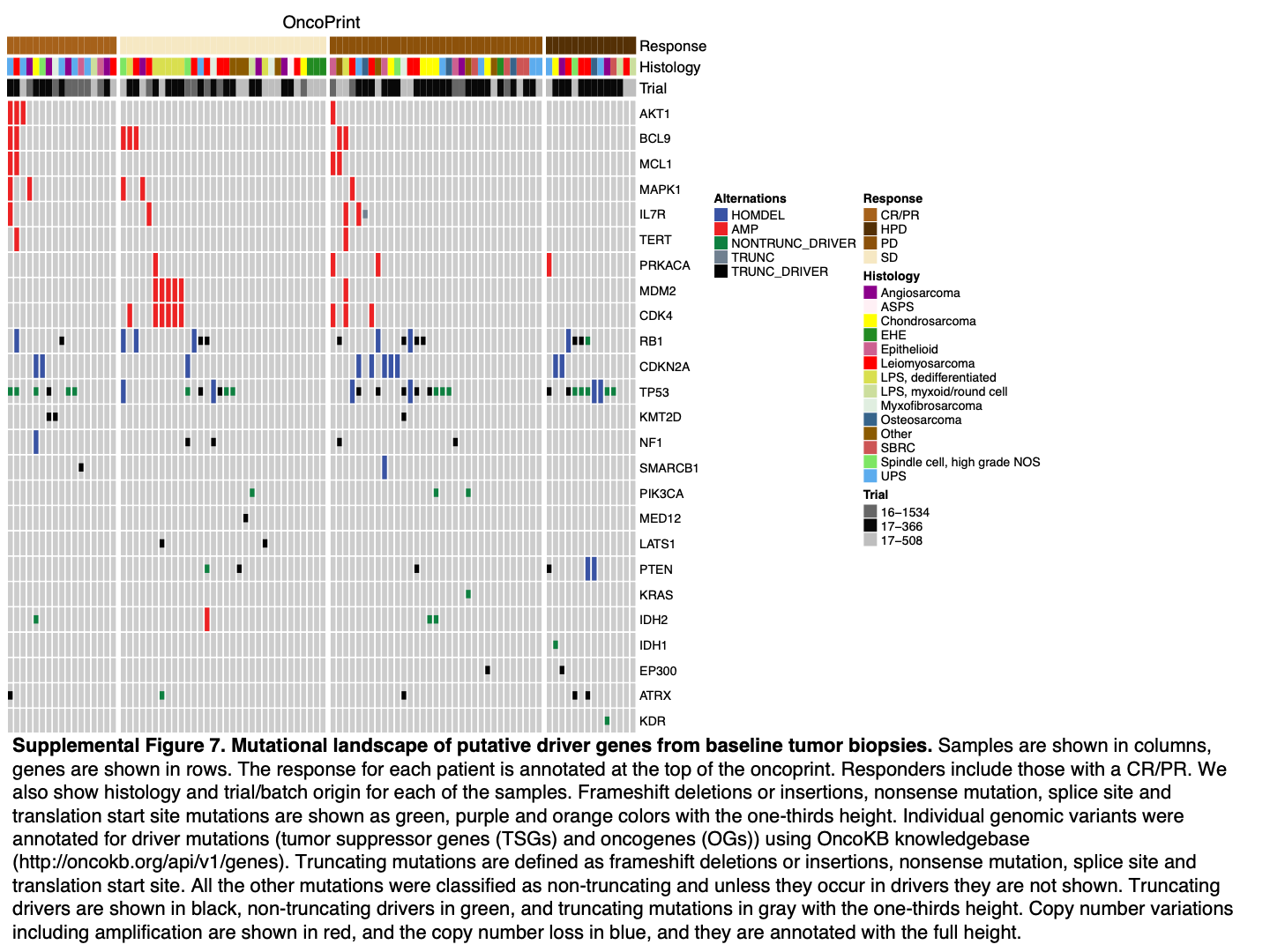
WES of baseline tumor specimens was performed in 113 of 129 evaluable patients (Supplementary Fig. S7). As expected, individual alterations were rare, with TP53 being by far the most common, and there was considerable heterogeneity among sarcoma subtypes. No patient with HPD had alterations in MDM2, MDM4, and EGFR, which were previously reported to be associated with HPD in other cancer types (21).
In order to determine genomics make-up of the faster tumor growth observed in HPD, we calculated tumor mutational burden (TMB) which is associated with response to PD-1 blockade across a wide variety of solid tumors (36). TMB was higher in patients with CR/PR compared with those with SD, with a median of 1.5 and 0.9 mutations per megabase (Mt/MB), respectively (P = 0.04; Fig. 5A). There was no difference in TMB between patients with PD and HPD (1.2 and 1.1 Mt/MB, respectively). Within the subtypes of UPS and angiosarcoma, TMB was not different between responders from nonresponders (Fig. 5B).
Figure 5.

Association between response and genomic alterations. Bars show median with 95% confidence intervals. A, TMB is shown for each patient with the given response. The median for CR/PR was 1.5 while for SD it was 0.93 (P = 0.04). Two patients with SD and PD had a TMB of 14.6 and 8.7, respectively. B, TMB is shown between responders (CR/PR) and nonresponders (NR) for UPS and angiosarcoma. The TMB for patients with epithelioid sarcoma, myxofibrosarcoma (MXFS), and myxoid LPS are also shown, with red circles indicating responders. C, FGA is shown as a percentage for each patient. LPS, liposarcoma; MXFS, myxofibrosarcoma.
We also assessed the fraction of genome altered (FGA) by copy-number gains or losses. FGA trended higher in patients with a CR/PR (median 72%) than SD (median 47%; P = 0.14; Fig. 5C). This might reflect the uneven distribution of subtypes in the different response groups because FGA varies considerably between sarcomas with simple and complex genotypes (37). Moreover, between patients with PD and HPD we observed similar FGA.
We used transcriptome data from baseline tumor biopsies to evaluate 50 hallmark pathways through GSEA and compared tumors exhibiting HPD with PD (Fig. 6; Supplementary Table S2). Several metabolic pathways were upregulated in HPD tumors relative to PD tumors including glycolysis, hypoxia, and oxidative phosphorylation. Also, HPD tumors had elevation of IFNα and IFNγ response pathways compared with PD tumors. In contrast, PD tumors had elevation of angiogenesis, epithelial–mesenchymal transition, and cell cycle pathways including E2F targets, G2M checkpoint, and mitotic spindle.
Figure 6.
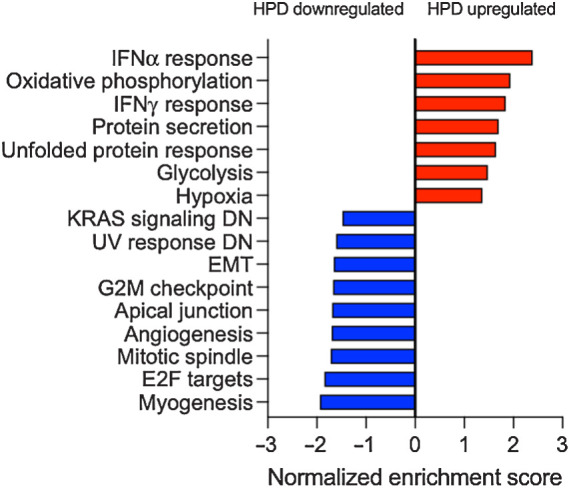
GSEA of pathways associated with HPD as compared with PD. RNA-seq was performed on baseline tumor biopsies. Bars are a summary of results from GSEA plots with the gene set names from Hallmark pathways and the normalized enrichment score. Positive enrichment score represents pathways that were correlated with HPD while negative enrichment score represents those that were correlated with PD. All gene sets shown had an adjusted P value < 0.05. IFN, interferon.
Finally, we used quanTIseq analysis to perform deconvolution of the tumor immune microenvironment using RNA-seq data (Supplementary Fig. S8). However, between PD and HPD tumors we observed no difference in B cells, M1 or M2 macrophages, CD4, CD8, or T-regulatory cells.
Discussion
Sarcomas are a heterogeneous group of solid tumors that, as a class, have been less responsive to immunotherapy than many other solid tumors. To our knowledge, this is the largest series to date evaluating patients with advanced sarcoma treated with PD-1 inhibitors and it has the longest follow-up. In several sarcoma subtypes, the response rate exceeded 30%, and some responses have been durable for years. However, we observed that HPD occurred in 15 (11%) patients. This is similar to what has been reported in other solid tumors (38). Of note, this was a pooled analysis of four prospective studies and between them the incidence of HPD was 0% to 16%. This difference was significant, but we doubt that bempegaldesleukin or epacadostat was the cause of HPD, because HPD occurred in the absence of these drugs in other series (38).
An important and unexpected finding is that patients with HPD had a significantly smaller disease burden at baseline than patients who had a best response of PD (but not HPD). This is problematic because HPD is defined radiographically. Because radiographically determined tumor size was not equal before treatment, it is difficult to interpret the significance of differential growth kinetics after treatment. It is unclear whether this was also the case in other series (19).
It has been suggested that HPD may simply reflect the natural history of disease in some patients (38). There was little that distinguished patients with PD and HPD other than baseline tumor size. Moreover, there were no genomic alterations that were associated with HPD. Others found that alterations in MDM2, MDM4, and EGFR were associated with HPD but we did not see this in our series (21). Two potential explanations for our findings is that HPD is a consequence of tumor growth kinetics and/or is an artifact of radiographic tumor measurements. Larger tumors, which were less likely to exhibit HPD, might be growth-constrained by lack of blood supply, an explanation supported by the fact that PD patients had upregulated angiogenesis pathways relative to HPD patients. Also, radiographic tumor measurements can be variable on CT scans. In absolute terms that variance should be constant, but in relative terms it would be of greater magnitude in small tumors (39). This might explain why HPD was more likely to be called in patients with a small disease burden—there are more outliers as variance increases.
Yet neither of these explanations fully accounts for why HPD may occur more frequently after PD-1 blockade than after chemotherapy in non–small cell lung cancer (NSCLC), though this has never been evaluated in sarcoma (19). We did observe that HPD tumors had upregulated IFNα and IFNγ responses at baseline relative to PD tumors, although our RNA-seq data was limited by small sample size and batch effects. But in colorectal cancer it was recently shown that ICI can induce tumor inflammation even in the absence of an effective antitumor immune response (40). It is possible that something similar occurs in sarcomas, leading to tumor swelling from inflammation which manifests radiographically as HPD.
In this series we identified several correlates of response although surprisingly TMB was not one of them. Responders had a slight elevation in TMB compared with those with SD, but not compared with those with PD or HPD. Thus, a low TMB in sarcoma does not necessarily predict a lack of response to immunotherapy. And the 2 patients with the highest TMB (14.6 and 8.7 Mt/MB) did not have an objective response. The limited utility of TMB as a biomarker in sarcoma is best illustrated by ASPS, which despite being a fusion-driven sarcoma with an extremely low TMB remains quite sensitive to PD-1 blockade (13).
In studies of patients with melanoma and NSCLC treated with PD-1 blocking antibodies, it was reported that the liver represents one of the least responsive sites of metastasis (17, 18). In the current series, the ORR for patients with liver metastases was indeed low, but sarcomas that metastasized to the liver tended to be subtypes that are not responsive even in the absence of hepatic involvement (i.e., leiomyosarcoma). Of the 4 patients with ‘immune-sensitive’ sarcoma subtypes who had liver metastases, 2 have done exceptionally well (Supplementary Table S1).
Lymph node metastases from other solid tumors are among the most responsive sites to PD-1 blockade (18). In our series, the presence of nodal metastases was associated with response, but only among ‘immune-sensitive’ sarcoma subtypes. Since response assessment includes nonnodal sites, this raises the possibility that tropism for lymph nodes in certain sarcomas is a favorable prognostic marker in the context of immunotherapy. This clinical observation would be consistent with recent data showing the importance of B cells for immunotherapy response and survival in sarcoma (41).
We also observed that disease burden at baseline was associated with response to PD-1 blockade, an observation that has been reported in other solid tumors but to our knowledge not in sarcoma (14). In fact, with a single exception there were no responders who had a baseline target lesion sum that was substantially more than 100 mm. Responders also tended to be more lightly pretreated and were earlier in their disease course.
It remains to be determined if the association between disease burden and response to PD-1 blockade is causal. After all, patients with immunogenic tumors and those with indolent biology might be expected to have smaller tumors and would be more likely to respond to immunotherapy. However, it is at least possible that tumor progression directly causes an immunosuppressive effect in the tumor microenvironment. The hypoxic and necrotic microenvironment of large tumors contain high concentrations of immunosuppressive metabolites (42–44). This possibility provides some rationale for attempting IO strategies earlier in the disease course. Differences in disease burden could explain some of the discordant response rates to anti–PD-1 observed between the SARC028 and Alliance trials.
Relatively small numbers of patients with each sarcoma subtype, short follow-up, and lack of control groups limits this study. This series includes data from four nonrandomized trials evaluating PD-1 blockade either alone or in combination with other agents that could have altered the efficacy of the treatment. Response and progression data were prospectively recorded but patterns of metastatic spread were recorded retrospectively. Also, the association between response and a tropism for lymph nodes was a retrospective observation made in a small number of patients. Independent validation is necessary before any conclusions can be drawn about the significance of lymph node metastases for immunotherapy in sarcoma. Finally, the power of gene expression data to evaluate the biology of HPD was greatly limited by small numbers of patients, batch effects, and heterogeneity from multiple sarcoma subtypes.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments
The four trials included in this pooled analysis were funded as previously reported (22–25). Additional funding was provided by the Cycle for Survival, Mary and Arden Witherwax Foundation, Sarcoma Foundation of America, NCI SPORE in Soft Tissue Sarcoma (P50 CA217694), and MSKCC Support Grant/Core Grant (P30 CA008748). We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology.
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
This article is featured in Highlights of This Issue, p. 811
Footnotes
Note: Supplementary data for this article are available at Clinical Cancer Research Online (http://clincancerres.aacrjournals.org/).
Authors' Disclosures
M.M. Gounder reports personal honoraria/advisory boards and/or associated research paid to institution from Athenex, Ayala Pharmaceuticals, Bayer, Boehringer Ingelheim, Daiichi Sankyo, Epizyme, Karyopharm Therapeutics, Rain Therapeutics, Springworks, TRACON Pharmaceuticals, and TYME; other support from Guidepoint, GLG Pharma Corporation, Third Bridge, and Flatiron Health; CME honoraria from Medscape, MORE Health, Physicians' Education Resource, and touchIME; royalties from Wolters Kluwer; patents with MSKCC (GODDESS PRO); uncompensated research with Foundation Medicine; and grants from FDA (R01 FD005105) and the NCI,NIH (P30CA008748)-core grant (Cancer Center Support Grant shared resources and core facility). C.M. Kelly reports other support from Amgen, Merck, and Incyte during the conduct of the study; C.M. Kelly also reports other support from Servier, Xencor, Exicure, and Kartos Therapeutics, as well as personal fees from Exicure and Kartos outside the submitted work. S. Movva reports grants from MSK Core and Sarcoma Spore during the conduct of the study; S. Movva also reports grants from Trillium Therapeutics, Hutchinson MediPharma, and Ascentage Pharma, as well as non-financial support from Merck, Bristol Myers Squibb, and Clovis Oncology outside the submitted work. K.A. Thornton reports personal fees from Epizyme and GlaxoSmithKline outside the submitted work. P. Chi reports grants and personal fees from Deciphera and Ningbo NewBay, personal fees from Zai Lab, and grants from Pfizer outside the submitted work. B.A. Nacev reports grants from the NCI, the Connective Tissue Oncology Society during the conduct of the study. W.D. Tap reports personal fees from Eli Lilly, EMD Serono, Mundipharma, C4 Therapeutics, Daiichi Sankyo, Blueprint Medicines, Agios Pharmaceuticals, NanoCarrier, Deciphera, Adcendo, Ayala Pharmaceuticals, Kowa, Servier, Bayer, Epizyme, Cogent, MedPacto, Foghorn Therapeutics, and Amgen outside the submitted work; in addition, W.D. Tap has a patent for Companion Diagnostic for CDK4 inhibitors - 14/854,329 pending to MSK/SKI, Enigma and CDH18 as companion Diagnostics for CDK4 inhibition - SKI2016-021-03 pending to MSK–SKI. W.D. Tap reports being on the Scientific Advisory Board of Certis Oncology Solutions with stock ownership, is a cofounder of Atropos Therapeutics with stock ownership, and is on the Scientific Advisory Board of Innova Therapeutics. S.P. D'Angelo reports other support from EMD Serono, Amgen, Nektar Therapeutics, Immune Design, GlaxoSmithKline, Incyte, Merck, Adaptimmune, Immunocore, Bristol Meyers Squibb, and Deciphera during the conduct of the study, as well as other support from EMD Serono, Amgen, Nektar Therapeutics, Immune Design, GlaxoSmithKline, Incyte, Merck, Adaptimmune, Immunocore, Bristol Meyers Squibb, and Deciphera outside the submitted work. No disclosures were reported by the other authors.
Authors' Contributions
N.D. Klemen: Conceptualization, data curation, formal analysis, investigation, writing–original draft. S. Hwang: Investigation, writing–review and editing. M. Bradic: Software, formal analysis, methodology. E. Rosenbaum: Investigation, writing–original draft. M.A. Dickson: Data curation, writing–review and editing. M.M. Gounder: Data curation, writing–review and editing. C.M. Kelly: Data curation, investigation, writing–original draft, writing–review and editing. M.L. Keohan: Data curation, writing–review and editing. S. Movva: Data curation, writing–review and editing. K.A. Thornton: Data curation, writing–review and editing. P. Chi: Data curation, writing–review and editing. B.A. Nacev: Data curation, writing–review and editing. J.E. Chan: Data curation, writing–review and editing. E.K. Bartlett: Data curation, investigation, writing–original draft, writing–review and editing. A.L. Richards: Software, formal analysis, investigation, methodology, writing–review and editing. S. Singer: Data curation, investigation, writing–review and editing. M.T.A. Donoghue: Formal analysis, methodology, writing–review and editing. W.D. Tap: Data curation, writing–review and editing. S.P. D'Angelo: Conceptualization, resources, data curation, formal analysis, supervision, investigation, methodology, writing–original draft, project administration, writing–review and editing.
References
- 1. Lindberg RD, Martin RG, Romsdahl MM, Barkley HT. Conservative surgery and postoperative radiotherapy in 300 adults with soft tissue sarcomas. Cancer 1981;47:2391–7. [DOI] [PubMed] [Google Scholar]
- 2. Weitz J, Antonescu CR, Brennan MF. Localized extremity soft tissue sarcoma: improved knowledge with unchanged survival over time. J Clin Oncol 2003;21:2719–25. [DOI] [PubMed] [Google Scholar]
- 3. Billingsley KG, Burt ME, Jara E, Ginsberg RJ, Woodruff JM, Leung DH, et al. . Pulmonary metastases from soft tissue sarcoma: analysis of patterns of diseases and postmetastasis survival. Ann Surg 1999;229:602–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tap WD, Wagner AJ, Schöffski P, Martin-Broto J, Krarup-Hansen A, Ganjoo KN, et al. . Effect of doxorubicin plus olaratumab vs doxorubicin plus placebo on survival in patients with advanced soft tissue sarcomas: The ANNOUNCE randomized clinical trial. JAMA 2020;323:1266–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Seddon B, Strauss SJ, Whelan J, Leahy M, Woll PJ, Cowie F, et al. . Gemcitabine and docetaxel versus doxorubicin as first-line treatment in previously untreated advanced unresectable or metastatic soft tissue sarcomas (GeDDiS): a randomized controlled phase III trial. Lancet Oncol 2017;18:1397–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Glabbeke MV, Oosterom ATv, Oosterhuis JW, Mouridsen H, Crowther D, Somers R, et al. . Prognostic factors for the outcome of chemotherapy in advanced soft tissue sarcoma: an analysis of 2,185 patients treated with anthracycline-containing first-line regimens–a European Organization for Research and Treatment of Cancer Soft Tissue and Bone Sarcoma Group Study. J Clin Oncol 1999;17:150–7. [DOI] [PubMed] [Google Scholar]
- 7. Demetri GD, von Mehren M, Jones RL, Hensley ML, Schuetze SM, Staddon A, et al. . Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016;34:786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schöffski P, Chawla S, Maki RG, Italiano A, Gelderblom H, Choy E, et al. . Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomized, open-label, multicenter, phase III trial. Lancet 2016;387:1629–37. [DOI] [PubMed] [Google Scholar]
- 9. van der Graaf WTA, Blay J-Y, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, et al. . Pazopanib for metastatic soft tissue sarcoma (PALETTE): a randomized, double-blind, placebo-controlled phase III trial. Lancet 2012;379:1879–86. [DOI] [PubMed] [Google Scholar]
- 10. Tawbi HA, Burgess M, Bolejack V, Tine BAV, Schuetze SM, Hu J, et al. . Pembrolizumab in advanced soft tissue sarcoma and bone sarcoma (SARC028): a multicenter, two-cohort, single-arm, open-label, phase II trial. Lancet Oncol 2017;18:1493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Toulmonde M, Penel N, Adam J, Chevreau C, Blay J-Y, Cesne AL, et al. . Use of PD-1 targeting, macrophage infiltration, and IDO pathway activation in sarcomas. JAMA Oncol 2018;4:93–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wilky BA, Trucco MM, Subhawong TK, Florou V, Park W, Kwon D, et al. . Axitinib plus pembrolizumab in patients with advanced sarcomas including alveolar soft-part sarcoma: a single-center, single-arm, phase II trial. Lancet Oncol 2019;20:837–48. [DOI] [PubMed] [Google Scholar]
- 13. Klemen ND, Kelly CM, Bartlett EK. The emerging role of immunotherapy for the treatment of sarcoma. J Surg Oncol 2020;1:435–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Joseph RW, Elassaiss-Schaap J, Kefford R, Hwu W-J, Wolchok JD, Joshua AM, et al. . Baseline tumor size is an independent prognostic factor for overall survival in patients with melanoma treated with pembrolizumab. Clin Cancer Res 2018;24:4960–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ribas A, Hamid O, Daud A, Hodi FS, Wolchok JD, Kefford R, et al. . Association of pembrolizumab with tumor response and survival among patients with advanced melanoma. JAMA 2016;315:1600–10. [DOI] [PubMed] [Google Scholar]
- 16. Brennan MF, Antonescu CR, Maki RG. Management of soft tissue sarcoma. New York: Springer Science + Business Media; 2013. [Google Scholar]
- 17. Tumeh PC, Hellmann MD, Hamid O, Tsai KK, Loo KL, Gubens MA, et al. . Liver metastasis and treatment outcome with anti–PD-1 monoclonal antibody in patients with melanoma and NSCLC. Cancer Immunol Res 2017;5:417–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Osorio JC, Arbour KC, Le DT, Durham JN, Plodkowski AJ, Halpenny DF, et al. . Lesion-level response dynamics to programmed cell death protein (PD-1) blockade. J Clin Oncol 2019;37:3546–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferrara R, Mezquita L, Texier M, Lahmar J, Audigier-Valette C, Tessonnier L, et al. . Hyperprogressive disease in patients with advanced non–small cell lung cancer treated with PD-1/PD-L1 inhibitors or with single-agent chemotherapy. JAMA Oncol 2018;4:1543–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Champiat S, Dercle L, Ammari S, Massard C, Hollebecque A, Postel-Vinay S, et al. . Hyperprogressive disease is a new pattern of progression in cancer patients treated by anti–PD-1/PD-L1. Clin Cancer Res 2017;23:1920–8. [DOI] [PubMed] [Google Scholar]
- 21. Forschner A, Hilke F-J, Bonzheim I, Gschwind A, Demidov G, Amaral T, et al. . MDM2, MDM4, and EGFR amplifications and hyperprogression in metastatic acral and mucosal melanoma. Cancers 2020;12:540–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. D'Angelo SP, Mahoney MR, Tine BAV, Atkins J, Milhem MM, Jahagirdar BN, et al. . Nivolumab with or without ipilimumab treatment for metastatic sarcoma (Alliance A091401): two open-label, noncomparative, randomized, phase II trials. Lancet Oncol 2018;19:416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kelly CM, Antonescu CR, Bowler T, Munhoz R, Chi P, Dickson MA, et al. . Objective response rate among patients with locally advanced or metastatic sarcoma treated with talimogene laherparepvec in combination with pembrolizumab. JAMA Oncol 2020;6:402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. D'Angelo SP, Conley AP, Kelly CM, Dickson MA, Gounder MM, Chi P, et al. . Pilot study of NKTR214 and nivolumab in patients with sarcomas. J Clin Oncol 2019;37:11010. [Google Scholar]
- 25. Kelly CM, Chi P, Dickson MA, Gounder MM, Keohan ML, Qin L-X, et al. . A phase II study of epacadostat and pembrolizumab in patients with advanced sarcoma. J Clin Oncol 2019;37:11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol 2016;34:525–7. [DOI] [PubMed] [Google Scholar]
- 27. Ewels P, Magnusson M, Lundin S, Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 2016;32:3047–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pimentel H, Bray NL, Puente S, Melsted P, Pachter L. Differential analysis of RNA-seq incorporating quantification uncertainty. Nat Methods 2017;14:687–90. [DOI] [PubMed] [Google Scholar]
- 29. Durinck S, Spellman PT, Birney E, Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 2009;4:1184–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. . Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Yu G, Wang L-G, Han Y, He Q-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 2012;16:284–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sturm G, Finotello F, Petitprez F, Zhang JD, Baumbach J, Fridman WH, et al. . Comprehensive evaluation of transcriptome-based cell-type quantification methods for immuno-oncology. Bioinformatics 2019;35:i436–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Finotello F, Mayer C, Plattner C, Laschober G, Rieder D, Hackl H, et al. . Molecular and pharmacologic modulators of the tumor immune contexture revealed by deconvolution of RNA-seq data. Genome Med 2019;11:34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 2016;32:2847–9. [DOI] [PubMed] [Google Scholar]
- 35. Somaiah N, Conley AP, Lin HY, Amini B, Sabir SH, Araujo DM, et al. . A phase II multi-arm study of durvalumab and tremelimumab for advanced or metastatic sarcomas. J Clin Oncol 2020;38:11509. [Google Scholar]
- 36. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blackhall F, DeVita VT, Hellman S, Rosenberg SA. Cancer: principles and practice of oncology, seventh edition. Surg Oncol 2005;14:195. [Google Scholar]
- 38. Adashek JJ, Subbiah IM, Matos I, Garralda E, Menta AK, Ganeshan DM, et al. . Hyperprogression and immunotherapy: fact, fiction, or alternative fact? Trends Cancer 2020;6:181–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Oxnard GR, Zhao B, Sima CS, Ginsberg MS, James LP, Lefkowitz RA, et al. . Variability of lung tumor measurements on repeat computed tomography scans taken within 15 minutes. J Clin Oncol 2011;29:3114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chalabi M, Fanchi LF, Dijkstra KK, Berg JG, Aalbers AG, Sikorska K, et al. . Neoadjuvant immunotherapy leads to pathologic responses in MMR-proficient and MMR-deficient early-stage colon cancers. Nat Med 2020;1–25. [DOI] [PubMed] [Google Scholar]
- 41. Petitprez F, de Reyniès A, Keung EZ, Chen TW-W, Sun C-M, Calderaro J, et al. . B cells are associated with survival and immunotherapy response in sarcoma. Nature 2020;577:556–60. [DOI] [PubMed] [Google Scholar]
- 42. Virgilio FD, Sarti AC, Falzoni S, Marchi ED, Adinolfi E. Extracellular ATP and P2 purinergic signaling in the tumor microenvironment. Nat Rev Cancer 2018;18:601–18. [DOI] [PubMed] [Google Scholar]
- 43. Eil R, Vodnala SK, Clever D, Klebanoff CA, Sukumar M, Pan JH, et al. . Ionic immune suppression within the tumor microenvironment limits T-cell effector function. Nature 2016;537:539–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Brand A, Singer K, Koehl GE, Kolitzus M, Schoenhammer G, Thiel A, et al. . LDHA-associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab 2016;24:657–71. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing data generated in this study are not yet publicly available due to patient privacy requirements that are pending approval but will be available upon request from the corresponding author.