

Assessment of tumor-infiltrating T-cell and B-cell diversity in renal cell carcinoma advances the understanding of tumor immune system interactions, linking tumor immune ecology with tumor burden, aggressiveness, and patient survival.
Abstract
Immune-modulating systemic therapies are often used to treat advanced cancer such as metastatic clear cell renal cell carcinoma (ccRCC). Used alone, sequence-based biomarkers neither accurately capture patient dynamics nor the tumor immune microenvironment. To better understand the tumor ecology of this immune microenvironment, we quantified tumor infiltration across three distinct ccRCC patient tumor cohorts using complementarity determining region-3 (CDR3) sequence recovery counts in tumor-infiltrating lymphocytes and a generalized diversity index (GDI) for CDR3 sequence distributions. GDI can be understood as a curve over a continuum of diversity scales that allows sensitive characterization of distributions to capture sample richness, evenness, and subsampling uncertainty, along with other important metrics that characterize tumor heterogeneity. For example, richness quantified the total unique sequence count, while evenness quantified similarities across sequence frequencies. Significant differences in receptor sequence diversity across gender and race revealed that patients with larger and more clinically aggressive tumors had increased richness of recovered tumoral CDR3 sequences, specifically in those from T-cell receptor alpha and B-cell immunoglobulin lambda light chain. The GDI inflection point (IP) allowed for a novel and robust measure of distribution evenness. High IP values were associated with improved overall survival, suggesting that normal-like sequence distributions lead to better outcomes. These results propose a new quantitative tool that can be used to better characterize patient-specific differences related to immune cell infiltration, and to identify unique characteristics of tumor-infiltrating lymphocyte heterogeneity in ccRCC and other malignancies.
Significance:
Assessment of tumor-infiltrating T-cell and B-cell diversity in renal cell carcinoma advances the understanding of tumor-immune system interactions, linking tumor immune ecology with tumor burden, aggressiveness, and patient survival.
See related commentary by Krishna and Hakimi, p. 764
Introduction
Renal cell carcinoma ranks seventh and tenth among the most diagnosed cancers among men and women in the United States, respectively, accounting for 3.8% of all cancer cases and 2.5% of all cancer deaths (1). The most common type of renal cell carcinoma is clear cell renal cell carcinoma (ccRCC). Historically, metastatic ccRCC has been one of the first malignancies successfully treated with immune-modulating systemic therapy, using IL2 and IFNα (2). Immune checkpoint inhibitors (ICI) such as nivolumab, ipilimumab, pembrolizumab, and avelumab, have emerged as the first-line therapy for metastatic ccRCC, typically administered in combination with each other or with a targeted therapeutic agent (3). The arrival of ICIs has precipitated a tremendous research effort aiming to accurately characterize the tumor immune microenvironment and explore potential biomarkers to predict ICI response, for which robust markers have been largely elusive. Most investigations have focused on tumor-centric variables including somatic mutations and gene expression. Fewer studies have been focused on host factors that contribute to the microenvironment or focused on how differences among these factors may affect clinical or therapeutic outcomes.
Across tumor types, response to ICI has been correlated with higher frequencies of somatic mutations that are believed to give rise to tumor-specific neoantigens, and to stimulate a robust antitumor immune response (4–6). In contrast, analyses of renal cell carcinomas have demonstrated a relatively low frequency of somatic mutations, yet very high levels of immune cell infiltration. These findings suggest that a high mutational burden is not solely responsible for inducing immune infiltration in ccRCC (7–11). In addition, recent work has demonstrated that CD8+ T-cell infiltration alone does not predict response to ICI. Refinements of characterizing immune cell populations are needed to understand the microenvironment and the biology underlying ICI response (12).
To initiate an antitumor immune response, tumor-specific neoantigens first require recognition by a T- or B-cell receptor (TCR, BCR) on a tumor-infiltrating lymphocyte. The tumor-infiltrating lymphocyte, complementarity determining region-3 (CDR3) is a highly variable region in the TCR/BCR that provides a complementary binding surface for antigens and largely determines the antigen specificity of the receptor. Investigations have shown the promise of CDR3 features as prognostic biomarkers for several malignancies (13–16), using sequencing and bioinformatic pipelines to recover single reads that represent the CDR3 amino acid sequence. These reads can be quantified as total recovered reads, potentially as a primary metric of immune infiltration (14–16). However, total count-based measures of CDR3 variability are unlikely to reflect the underlying complex biology of host adaptive immune response with the same accuracy as other measures of receptor diversity (17–19).
We hypothesized that immune cell receptor sequence diversity recapitulates important features of tumor biology such as origin, environment-driven evolution, and progression risk. We leverage properties of a generalized diversity index (GDI; refs. 20–22), a measure applied in ecology and evolution, to quantify CDR3 diversity, and assess whether this diversity is associated with important clinicopathologic outcomes in ccRCC. GDI is evaluated as a continuous function along a range of order of diversity (q) values (20–22). At low values of q (low-q GDI), the index is a measure of distribution richness, that is, the count of distinct types, sequences or clones, while the value at q = 1 is closely related to Shannon diversity index (23). At high values of q (high-q GDI), the index approaches a measure of evenness or dominance, that is, focusing on the dominant clone or sequence and its frequency. Here, we applied these diversity metrics to ccRCC tumor samples, and assessed the properties of GDI and their utility to serve as possible prognostic markers. We assessed tumor-infiltrating lymphocyte TCR and BCR CDR3 diversity across the range of q, and for isolated values that have direct statistical interpretations. We analyzed three independent cohorts of patients with ccRCC with bulk RNA-sequencing (RNA-seq) samples; the Moffitt Total Cancer Care (TCC) cohort (24), the Clinical Proteomic Tumor Analysis Consortium 3 (CPTAC-3) cohort (25), and The Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma (TCGA-KIRC) cohort (26, 27).
Materials and Methods
Clinical samples
Following Institutional Review Board (IRB) approval (H. Lee Moffitt Cancer Center's Total Cancer Care protocol MCC# 14690; approved by the Institutional Review Board; Advarra IRB Pro00014441), we retrospectively obtained clinicopathologic and bulk RNA-seq patient data from electronic medical records, where all patients had provided written consent under the institutional TCC Protocol. RNA was prepared using the Qiagen RNAeasy plus mini kit for RNA (frozen tissue) or the Qiagen All prep FFPE DNA/RNA kit (formalin-fixed paraffin-embedded tissue). RNA-seq libraries were prepared using the standard Illumina TruSeq RNA Access kit (now called TruSeq RNA Exome), according to manufacturer protocols. RNA-seq libraries were sequenced on an Illumina HiSeq 4000 according to manufacturer protocols. RNA-seq reads were aligned to the human reference genome (hs37d5) in an intron-aware manner with Spliced Transcripts Alignment to a Reference (STAR; ref. 28). Table 1 shows a summary of the clinical information obtained from individuals in the Moffitt TCC cohort. Relevant clinical and pathologic outcomes available from the Moffitt TCC cohort, including ranges of percentage of tumor with EGFR spice variant alpha are recorded in Table 1. Summaries of numbers of reads per samples in each of the cohorts are available in Supplementary Fig. S1.
Table 1.
Clinical and demographic summary of ccRCC cohorts.
| Variables | TCC ccRCC patients, no. (%) (n = 105) | CPTAC-3 ccRCC patients, no. (%) (n = 110) | TCGA-KIRC ccRCC patients, no. (%) (n = 441) |
|---|---|---|---|
| Sex | |||
| Female | 35 (33.3) | 30 (27.3) | 153 (34.7) |
| Male | 70 (66.7) | 80 (72.7) | 288 (65.3) |
| Race | |||
| Asian Indian or Pakistani | 2 (1.9) | 1 (0.01) | 7 (1.6) |
| Black | 3 (2.9) | 1 (0.01) | 30 (6.8) |
| Other | 7 (6.7) | — | — |
| White | 93 (88.5) | 61 (55.5) | 397 (90.0) |
| Not reported | — | 47 (42.7) | 7 (1.6) |
| Tumor laterality | |||
| Left | 62 (59.0) | NA | NA |
| Right | 43 (41.0) | NA | NA |
| Surgery type | |||
| Partial nephrectomy | 22 (21.0) | NA | NA |
| Radical nephrectomy | 65 (61.9) | NA | NA |
| Radical nephrectomy with thrombectomy | 18 (17.1) | NA | NA |
| Fuhrman nuclear grade | |||
| 1 | 0 | 7 (6.4) | 9 (2.0) |
| 2 | 30 (28.6) | 53 (48.2) | 182 (41.3) |
| 3 | 62 (59.0) | 41 (37.3) | 179 (40.6) |
| 4 | 13 (12.4) | 9 (8.2) | 68 (15.4) |
| Not reported | — | — | 3 (0.7) |
| pT | |||
| T1 | 31 (29.5) | 52 (47.3) | 211 (47.8) |
| T2 | 3 (2.9) | 13 (11.8) | 58 (13.2) |
| T3+T4 | 70 (66.7) | 45 (40.9) | 172 (39.0) |
| Not reported | 1 (1.0) | — | — |
| pN | |||
| N0 | 27 (25.7) | 16 (14.5) | 203 (46.0) |
| N1 | 5 (4.8) | 4 (3.6) | 13 (2.9) |
| NX | 73 (69.5) | 89 (81.9) | 225 (51.0) |
| pM | |||
| M0 | — | 34 (30.9) | 353 (80.0) |
| M1 | 23 (21.9) | 3 (2.7) | 73 (16.6) |
| MX | 82 (78.1) | — | 14 (3.2) |
| Not reported | — | 73 (66.4) | 1 (0.2) |
| Sarcomatoid status | |||
| No | 99 (94.3) | NA | NA |
| Yes | 6 (5.7) | NA | NA |
| Vitality | |||
| Alive | 82 (78.1) | 96 (87.3) | 118 (83.1) |
| Dead | 23 (21.9) | 14 (12.7) | 24 (16.9) |
| Age at surgery (yr) | Age at diagnosis (yr) | Age at diagnosis (yr) | |
| Median (range) | 65 (36–87) | 60 (30–89) | NA |
| Pathological tumor size (cm) | |||
| Median (range) | 6.0 (1.3–17.5) | 6.4 (1.0–16.0) | NA |
| % EGFR splice variant alpha | |||
| Median (range) | 1.01 (0.00–41.33) | NA | NA |
| BAP1 mutation | |||
| Wild type | 61 (58.1) | 93 (84.5) | 372 (84.4) |
| Alteration | 4 (3.8) | 17 (15.5) | 47 (10.6) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| KDM5C mutation | |||
| Wild type | 56 (53.3) | 91 (82.7) | 389 (88.2) |
| Alteration | 9 (8.6) | 19 (17.3) | 30 (6.8) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| MTOR mutation | |||
| Wild type | 62 (59.0) | 104 (94.5) | 392 (88.9) |
| Alteration | 3 (2.9) | 6 (5.5) | 27 (6.1) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| PBRM1 mutation | |||
| Wild type | 41 (39.0) | 66 (60.0) | 258 (58.5) |
| Alteration | 24 (22.9) | 44 (40.0) | 161 (36.5) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| PTEN mutation | |||
| Wild type | 60 (57.1) | 105 (95.5) | 399 (90.5) |
| Alteration | 5 (4.8) | 5 (4.5) | 20 (4.5) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| SETD2 mutation | |||
| Wild type | 55 (52.4) | 95 (86.4) | 360 (81.6) |
| Alteration | 10 (9.5) | 15 (13.6) | 59 (13.4) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| TP53 mutation | |||
| Wild type | 61 (58.1) | 104 (94.5) | 408 (92.5) |
| Alteration | 4 (3.8) | 6 (5.5) | 11 (2.5) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| VHL mutation | |||
| Wild type | 18 (17.1) | 28 (25.5) | 186 (42.2) |
| Alteration | 47 (44.8) | 82 (74.5) | 233 (52.8) |
| Not reported | 40 (38.1) | — | 22 (5.0) |
| Diabetes status | |||
| No | 76 (72.4) | NA | NA |
| Yes | 29 (27.6) | NA | NA |
| Not reported | — | 110 | 441 |
Abbreviations: ccRCC, clear cell renal cell carcinoma; CPTAC-3, Clinical Proteomic Tumor Analysis Consortium 3; NA, not available; TCC, Total Cancer Care Protocol; TCGA-KIRC, The Cancer Genome Atlas Kidney Renal Clear Cell Carcinoma; yr, years.
To further investigate the trends identified in point estimates of diversity from TCR and B-cell immunoglobulin recoveries the Moffitt TCC patient bulk RNA-seq, we validated trends identified in the Moffitt TCC cohort analysis with complementary analysis with the RNA-seq from the under CPTAC-3 cohort (written consent had been obtained under CPTAC guidelines). Table 1 shows a summary of the clinical information obtained from individuals in the CPTAC-3 cohort. TCGA-KIRC cohort RNA-seq–based TRA and TRB CRD3s were obtained from Thorsson and colleagues (26, 27) based on the dbGAaP-approved protocol number 6300. Relevant clinical and pathologic outcomes, aligning with outcomes available in the Moffitt TCC cohort, that are available in CPTAC-3 and TCGA-KIRC cohorts are reported in Table 1.
Recovery of immune receptor V(D)J recombination reads from bulk RNA-seq
Recovery of immune receptor V(D)J recombination reads was performed in two steps. First, RNA-seq binary alignment map (BAM) files were searched, as a straight string search, for 10-mer nucleotide sequences representing the 3′ ends of every human V-gene and 5′ end of every human J-gene, for all seven immune receptors. Next, the resulting reads were aligned to reference V and J regions obtained from the International Immunogenetics Information System. The quantitative parameters for the pairwise alignment were: (i) nucleotide match, +5, (ii) mismatch, −10, (iii) opening gap, −10, and (iv) extending gap, −10. The threshold for a V or J gene segment match was a score of ≥65. To ensure V and J read fidelity, only reads with a 20 nucleotide or greater match length for both V and J regions, and within the 20 nucleotides, a >90% nucleotide match fidelity for both V and J regions were considered as matches. In addition, and a productive CDR3 domain, defined as an in-frame junction without stop codons, was required for recombination read identification. Code for the method described above can be obtained at: https://github.com/bchobrut-USF/vdj under “Code Package A.” See also https://hub.docker.com/r/bchobrut/vdj for a container version of the code with a README file.
Generalized diversity index for patient quantifying CDR3 receptor diversity
The GDI can be viewed as a continuous, non-increasing function over a range of values described by the parameter q, called order of diversity. This parameter allows a consideration of multiple scales of diversity simultaneously or in combination. GDI is often used in ecology (20) and was more recently introduced to quantify intratumor heterogeneity and evolution (29–31). Formally, GDI is calculated as:
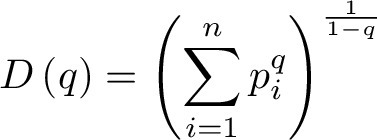 |
where 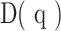 is called the diversity index at the given order of diversity
is called the diversity index at the given order of diversity 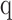 ,
, 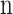 is the number of unique CDR3 sequences recovered across the entire cohort, and
is the number of unique CDR3 sequences recovered across the entire cohort, and 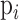 is the relative proportion of
is the relative proportion of 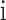 -th CDR3 sequence. We typically evaluated the diversity score,
-th CDR3 sequence. We typically evaluated the diversity score, 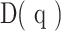 , for
, for 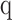 between 0.01 and 100 numerically for each patient, for each of the receptor and immunoglobulin types individually, as well as in biologically meaningful groups (TRA+TRB together, TRG+TRD together, and IGH+IGK+IGL together). Varying the value of q represents interpolating between richness and evenness: richness is weighted more at low values of q, and evenness is weighted more at higher values of q. High-q GDI scales inversely with dominance or clonality.
between 0.01 and 100 numerically for each patient, for each of the receptor and immunoglobulin types individually, as well as in biologically meaningful groups (TRA+TRB together, TRG+TRD together, and IGH+IGK+IGL together). Varying the value of q represents interpolating between richness and evenness: richness is weighted more at low values of q, and evenness is weighted more at higher values of q. High-q GDI scales inversely with dominance or clonality.
Point estimates derived from patient's GDI, D(q), can be used for easy of comparison of sequence distributions across patients and cohorts. These point estimates of interest include (i) low-q diversity (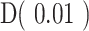 ), which describes the epitope richness of the patients, (ii) high-q diversity (
), which describes the epitope richness of the patients, (ii) high-q diversity (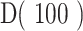 ), which describes dominance of the “main driving” epitope, and (iii) ΔD, which measures the difference between low-q and high-q diversity (
), which describes dominance of the “main driving” epitope, and (iii) ΔD, which measures the difference between low-q and high-q diversity (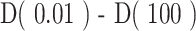 ). Furthermore, when visualizing the continuum of diversity measures D(q) with
). Furthermore, when visualizing the continuum of diversity measures D(q) with 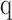 in log-scaling, the continuum of diversity measures appears to have an inflection point (IP), corresponding to a scale of diversity where small changes in the key parameter q can have large impact: the higher this value, the more even we expect a distribution to be, as the IP tends to infinity for perfectly even distributions (corresponding to n sequences all at frequency 1/n). Thus, two additional point estimates of interest that we used are (iv) the value q at which an IP occurs, and (v) the slope at the IP (denoted as IP slope). All code for calculating CDR3 diversity and its summary metrics has been implemented in Julia (version 1.4.0) and documented in the publicly available package OncoDiversity.jl.
in log-scaling, the continuum of diversity measures appears to have an inflection point (IP), corresponding to a scale of diversity where small changes in the key parameter q can have large impact: the higher this value, the more even we expect a distribution to be, as the IP tends to infinity for perfectly even distributions (corresponding to n sequences all at frequency 1/n). Thus, two additional point estimates of interest that we used are (iv) the value q at which an IP occurs, and (v) the slope at the IP (denoted as IP slope). All code for calculating CDR3 diversity and its summary metrics has been implemented in Julia (version 1.4.0) and documented in the publicly available package OncoDiversity.jl.
To determine the impact of all five point estimates of diversity, we ran a correlation analysis and determined that we could reduce our five point estimates of diversity down to three metrics for comparison across receptor groups and patients. The Spearman correlation coefficients were calculated between point-estimate metrics and comparisons between low-q diversity, ΔD diversity, and the IP slope all had significant and very strong Spearman correlation coefficients of 0.98 or greater, so moving forward, we just focused on one of those metrics as a measure of species richness diversity (Supplementary Fig. S2). There was not a strong correlation between high-q diversity and the IP q metrics and either metric compared with any of the three species richness diversity metrics (low-q diversity, ΔD diversity, and IP slope), so we continue to look at the high-q diversity and IP q separately and in additional to the single species richness measure.
Assessment of clinical and survival associations with CDR3 features
Clinical associations were evaluated for recoveries in TRA, TRB, TRG, TRD, IGH, IGK, and IGL separately as well as in combinations of TRA+TRB, TRG+TRD, and IGH+IGK+IGL. After point estimates of diversity were calculated for each patient and each receptor subtype/combination, the clinical parameter values were assessed to identify whether CDR3 receptor diversity could discriminate patients with ccRCC based on relevant clinical and pathologic outcomes, as well as the percentage of tumor with EGFR spice variant alpha previously found to be prognostic in ccRCC (32). Largest diameter size and age were the only two continuous variables, which were evaluated by dividing the cohort into above and below the median of the diversity and compared with unpaired t tests. All other categorial data types were divided by categories and the diversity metric was compared across the categories using unpaired t test for two categories and ANOVA for three or more categories.
Survival correlations for the above combinations were performed by separating the cohort into above and below the median based on point estimates of the generalized diversity index. In addition, the maximally selected rank statistical analysis was performed to estimate an optimal cut-off point in the quantitative point estimates as a binary classification rule regarding overall survival time (33). The Kaplan–Meier curve method was used to calculate survival probability and log-rank test was used to compare the above (high diversity) and below (low diversity) groups. GraphPad Prism software (version 8) and R version 3.6.1 were used for computing statistical comparisons and outputting figures.
xCell scores
From bulk RNA-seq for each patient, xCell scores were calculated for various T-cell and B-cell subtypes as well as the immune score, stroma score, and microenvironment score. Then for each xCell score calculated a Spearman correlation was calculated for the total and unique recoveries identified for TRA receptor, IGL receptor, and aggregate combination (TRs+IGs).
Patients in the Moffitt TCC cohort had previously undergone bulk RNA-seq of macrodissected tumor samples using the TruSeq RNA Exome kit (Illumina) for 50 million 100–bp paired-end reads. RNA-seq reads were aligned to the human reference genome in a splice-aware fashion using STAR (28), allowing for accurate alignments of sequences across introns. Aligned sequences were assigned to exons using the HTseq package (34) to generate initial counts by region. Normalization, expression modeling, and difference testing were performed using DESeq2 (35). For the CPTAC cohorts, detailed methodology regarding RNA-seq can be found at its source webpage (25).
RNA-seq data were analyzed for cell-type enrichment using the xCell bioinformatic pipeline (25). xCell uses a compendium of validated gene expression signatures for 64 individual cell types derived from thousands of expression profiles. Single-sample gene set enrichment analysis scores were adjusted for spillover compensation to generate an adjusted enrichment score for each cell type within the specimen, which is referred to as the xCell score. xCell scores were generated for each of the 64 cell types for each ccRCC tumor specimen.
Data and code availability
Code for VDJ epitope recoveries from patient sequencing data (BAM files) is publicly available on Docker at https://hub.docker.com/r/bchobrut/vdj and GitHub at https://github.com/bchobrut/vdj_recovery.
The code and documentation describing how to calculate patient CDR3 diversity from CDR3 sequence recoveries and run our pipeline and reproduce our results are open-source and publicly available through the OncoDiversity.jl GitHub repository (https://github.com/mcfefa/OncoDiversity.jl). A virtual machine producing the full diversity environment is available on Code Ocean (https://codeocean.com/capsule/9959428/tree/).
Results
GDI quantifies tumor-infiltrating lymphocyte receptor subtype diversity in the TCC cohort
For each patient, we measured individual receptor CDR3 diversities across the seven human adaptive immune receptor genes (TRA, TRB, TRG, TRD, IGH, IGK, IGL), as well as common combinations of these receptor subtypes (TRA+TRB, TRG+TRD, IGH+IGK+IGL, along with all seven together, denoted at TRs+IGs). In the TCC cohort (n = 105), CDR3 sequences were recovered from bulk RNA-seq of patient tumor tissue. GDI was then calculated for each subtype and group of subtypes (the landscape of recoveries across common groups are shown in Fig. 1A and B and Supplementary Fig. S3 and distribution of individual recoveries in Supplementary Fig. S4). The Moffitt TCC cohort of patients with ccRCC represented a cohort of clinically high-risk and advanced patients. Over two-thirds of the cohort contains patients with pathologic stages 3 or 4, including 6% of patients with highly aggressive sarcomatoid histology (Table 1). To quantify the GDI, we generated a continuum of diversity measures D(q) for each patient across values of the order of diversity, q. Then, we were able to compare clinical variables at specific point estimates of the continuum of diversity measures, as shown in Fig. 1C. We compared immune receptor subtype diversity across patients, and found that the point estimates ΔD diversity, high-q diversity, and IP of the GDI curve (see Materials and Methods) were unique summary metrics. The value of ΔD summarizes richness (total number of unique sequences) of receptor subtypes in a patient sample. High-q diversity focuses on the dominance (frequency of largest sequence) of a receptor subtype and deemphasizes a rare receptor subtype. IP is a measure of receptor subtype evenness, with high IP values indicating an overall more level distribution, largely independent of receptor subtype richness (29–31).
Figure 1.

Tumor-infiltrating lymphocyte receptor diversity as a marker in ccRCC. A, Overall workflow schematic of calculating tumor-infiltrating lymphocyte diversity across a cohort of patients. Patient tumors undergo bulk RNA-sequencing and then CDR3 sequences from TCRs and BCRs are recovered. Then for each patient, CDR3 sequences are segregated by receptor class (TRA, TRB, TRG, TRD, IGH, IGK, and IGL) and patient frequencies across the CDR3 landscape per receptor are calculated and used to quantify the individual patient's receptor diversity using the generalized diversity index from ecology. B, Receptor recovery distributions across the seven major receptor types in the Moffitt cohort; written consent was provided under the TCC protocol. C, Patient diversity curves can be distilled down to five point estimates of diversity: low-q (q = 0.01), high-q (q = 100), ΔD (D(0.01) − D(100)), IP, and IP slope.
Immune receptor subtype richness is associated with important pathologic features in the TCC cohort
Across individual receptor subtypes, TRA and IGL receptor diversity consistently showed increased richness (in Fig. 2 exemplified with ΔD diversity comparisons) in tumors with larger diameters, higher grade, sarcomatoid status, and tumors from the left side. TRA receptor diversity split the Moffitt TCC cohort at the median of ΔD diversity. Of these, patients with ΔD values below the cohort median had a mean largest diameter size of 6.1 cm, compared with those with above the median who had a mean largest diameter size of 7.6 cm (Fig. 2A, i; P: 0.0287). This same trend was reflected in IGL receptor diversity with the high ΔD diversity group (Fig. 2B, i; P: 0.0195).
Figure 2.
![Figure 2. Patients with tumors that are larger in diameter, higher grade, left laterality, and sarcomatoid status have increased diversity in TRA and IGL receptors. A, TRA receptor CDR3 sequence ΔD diversity across the Moffitt TCC cohort have increased diversity in (i) larger diameter tumors (low diversity had mean diameter of 6.1 cm and high diversity had a mean diameter for 7.6 cm; P: 0.0287), (ii) left laterality tumors (score of 37.55 vs. 16.39; P: 0.0097), (iii) with higher grade tumors (mean score from grade 2 was 12.96, mean score from grade 3 was 33.09, and mean score from grade 4 was 42.62; P: 0.0227), and (v) in patients with sarcomatoid status [sarcomatoid status evaluated as yes (at least 5%) or no, in TRA no had a mean ΔD diversity score of 26.47 vs. yes with a mean score of 61.32; P 0.0430]. B, IGL receptor CDR3 sequence ΔD diversity showed the same trends as TRA receptor CDR3 sequence diversity for (i) size (low diversity had mean diameter of 6.1 cm and high diversity had a mean diameter of 7.6 cm: P: 0.0195), (ii) laterality (score of 331.1 vs. 141.3; P: 0.0445), (iii) grade (mean score from grade 2 was 93.99, mean score from grade 3 was 281.4, and mean score from grade 4 was 465.5; P: 0.0459), and (iv) sarcomatoid status (no had a mean score of 223.9 vs. yes with a mean score of 704.4; P: 0.0152). Unpaired t tests were used to compare two group data and ANOVA was used to compare grade, three group data. *, P < 0.05; **, P < 0.01.](https://www.ncbi.nlm.nih.gov/core/lw/2.0/html/tileshop_pmc/tileshop_pmc_inline.html?title=Click%20on%20image%20to%20zoom&p=PMC3&id=9359723_929fig2.jpg)
Patients with tumors that are larger in diameter, higher grade, left laterality, and sarcomatoid status have increased diversity in TRA and IGL receptors. A, TRA receptor CDR3 sequence ΔD diversity across the Moffitt TCC cohort have increased diversity in (i) larger diameter tumors (low diversity had mean diameter of 6.1 cm and high diversity had a mean diameter for 7.6 cm; P: 0.0287), (ii) left laterality tumors (score of 37.55 vs. 16.39; P: 0.0097), (iii) with higher grade tumors (mean score from grade 2 was 12.96, mean score from grade 3 was 33.09, and mean score from grade 4 was 42.62; P: 0.0227), and (v) in patients with sarcomatoid status [sarcomatoid status evaluated as yes (at least 5%) or no, in TRA no had a mean ΔD diversity score of 26.47 vs. yes with a mean score of 61.32; P 0.0430]. B, IGL receptor CDR3 sequence ΔD diversity showed the same trends as TRA receptor CDR3 sequence diversity for (i) size (low diversity had mean diameter of 6.1 cm and high diversity had a mean diameter of 7.6 cm: P: 0.0195), (ii) laterality (score of 331.1 vs. 141.3; P: 0.0445), (iii) grade (mean score from grade 2 was 93.99, mean score from grade 3 was 281.4, and mean score from grade 4 was 465.5; P: 0.0459), and (iv) sarcomatoid status (no had a mean score of 223.9 vs. yes with a mean score of 704.4; P: 0.0152). Unpaired t tests were used to compare two group data and ANOVA was used to compare grade, three group data. *, P < 0.05; **, P < 0.01.
TRA receptor ΔD diversity in CDR3 amino acid sequences recovered from tumors with left laterality had an average diversity score 2.3-fold higher compared with those with right laterality tumors (Fig. 2A, ii; P: 0.0097), which was also reflected in IGL receptor ΔD diversity (Fig. 2B, ii; P: 0.0445). In addition, patients with high tumor grade had increased TRA receptor ΔD diversity (Fig. 2A, iii; P: 0.0227), which was also demonstrated in IGL receptor ΔD diversity (Fig. 2B, iii; P: 0.0459).
Overall, Moffitt TCC cohort tumors that were identified with sarcomatoid histology had increased overall lymphocyte receptor diversity compared with those individuals who did not have sarcomatoid histology (demonstrated in Fig. 2A and B, iv; P: 0.0430 in TRA and P: 0.0152 in IGL). This trend for increased diversity in sarcomatoid carcinoma tumors was statistically significant in all combinations except for TRG receptor diversity, which was one of the rarest CDR3 receptor subtypes recovered.
Our observations of increased lymphocyte receptor richness in larger diameter tumors, higher grade tumors, left laterality tumors, and sarcomatoid carcinomas were also discovered in other receptor subtypes, as well as in the combinations (Supplementary Fig. S5 shows size, laterality, grade, and sarcomatoid status across all combinations of receptors, Supplementary Fig. S6 shows the Shannon index of TRA and IGL across size, laterality, grade, and sarcomatoid status and Supplementary Data S1 contains statistics for all comparison combinations across all clinical features for the Moffitt TCC cohort).
Independent validation of GDI metrics
To validate the findings of increased diversity with poor pathologic features we first calculated xCell scores (36) for patients in the Moffitt TCC cohort to confirm that the detected recoveries came from tumor-infiltrating lymphocytes. TRA receptor total and unique recoveries had the highest Spearman correlation scores with T-cell subtype xCell score and were less correlated with the B-cell subtype xCell score (Fig. 3A and full correlation analysis of all xCell scores with total and unique recoveries of TRA is shown in Supplementary Fig. S7, with total and unique recoveries of IGL in shown in Supplementary Fig. S8, and with total and unique recoveries of all CDR3s recovered is shown in Supplementary Fig. S9). The strongest correlation associated with TRA receptor was with CD8+ Tcm (r = 0.819 with total recoveries; r = 0.714 with unique recoveries) and CD8+ T cells (r = 0.751 with total recoveries; r = 0.695 with unique recoveries), while the weakest correlation was with CD4+ Tcm (r = 0.177 with total recoveries; r = 0.185 with unique recoveries) and CD4+ naïve T cells (r = 0.243 with total recoveries; r = 0.097 with unique recoveries). These correlations also held true for IGL receptor recoveries and total (TRs+IGs) receptor recoveries, but the correlations were moderate in strength (most Spearman correlation coefficients between 0.2–0.5) compared with the Spearman correlation coefficient associated with TRA (most correlation coefficients between 0.3–0.7). Furthermore, the immune score strongly correlated positively with total and unique recoveries, compared with the microenvironment score and stroma score, which both showed weaker Spearman correlation coefficients.
Figure 3.

CDR3 sequence diversity trends were validated using xCell scores and a secondary RCC CPTAC3 cohort. A, Spearman correlation coefficient, r, was calculated between the total and unique number of TRA, IGL, and total (TRs+IGs) recoveries and the xCell scores for various T-cell and B-cell subtypes, immune score, stroma score, and microenvironment score. B, Grade, largest diameter size, and stage (pT) trends in TRA receptor and IGL receptor ΔD diversity in the CPTAC-3 cohort. For TRA recoveries, patients (n = 108) had increased ΔD diversity in (i) higher grade (mean score from grade 1 was 8.558, mean score from grade 2 was 27.94, mean score from grade 3 was 38.60, and mean score from grade 4 was 15.92; P: 0.0212), (ii) a higher mean ΔD diversity score in tumors with diameters greater than or equal to 7 cm (mean score in diameters ≥ 7 cm was 37.91, mean score in diameters < 7 cm was 28.12; P: 0.1850), and (iii) in more advanced pT stage (mean score from T1 was 25.01, mean score from T2 was 51.07, mean score from T3 was 29.24, mean score from T4 was 5.32; P: 0.0368). Similarly, for IGL recoveries, patients had increased ΔD diversity in (iv) higher grade (mean score from grade 1 was 50.14, mean score from grade 2 was 160.6, mean score from grade 3 was 156.9, and mean score from grade 4 was 84.79; P: 0.5268), (v) a higher mean ΔD diversity score in tumors with diameters greater than or equal to 7 cm (mean score in diameters ≥ 7 cm was 235.6, mean score in diameters < 7 cm was 126.6; P: 0.0545), and (vi) in more advanced pT stage (mean score from T1 was 147.0, mean score from T2 was 291.1, mean score from T3 was 97.58, mean score from T4 was 120.5; P: 0.0637). C, Grade and stage (pT) trends in TRA receptor ΔD diversity in the TCGA-KIRC cohort. Patients (n = 390) had increased ΔD diversity in (i) higher grade (mean score from grade 1 was 10.28, mean score from grade 2 was 16.62, mean score from grade 3 was 22.49, and mean score from grade 4 was 28.32; P: 0.0008), and (ii) in more advanced pT stage (mean score from T1 was 16.64, mean score from T2 was 24.25, mean score from T3 was 24.78, mean score from T4 was 17.90; P: 0.0027). Unpaired t tests were used to compare two group data and ANOVA was used to compare grade, three group data. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Once we confirmed that the diversity scores, we measured were attributable to the immune cell infiltration in the tumor with the xCell scores, we sought to independently validate our findings with a replicative study, using the ccRCC CPTAC-3 cohort (n = 110). The CDR3 recovery landscape of CPTAC-3 and Moffitt TCC differed slightly. CPTAC-3 had more recoveries from T cells; B cells accounted for an average of only 78.96%. However, we did not identify any significant trend of differences between the proportions of T cells and B cells recovered on the basis of stage (CPTAC-3 recovery landscape is shown in Supplementary Fig. S10 and fraction of BCRs and TCRs recovered per patient grouped by stage is detailed in Supplementary Fig. S11). Laterality and sarcomatoid status could not be evaluated, as these are not available in CPTAC-3.
We confirmed the observation that higher grade and larger size of tumors are associated with increases in TRA and IGL receptor ΔD diversity in the CPTAC-3 cohort (Fig. 3B compared with Supplementary Fig. S12, and Supplementary Data S2 contains statistics for all comparisons across all clinical features for the CPTAC-3 cohort)—which independently validated that rich, but rather uneven receptor subtype distribution is associated with larger and high-grade tumors. TRA receptor ΔD diversity was significantly different between low and high grade (Fig. 3B, i; P: 0.0212) and pT stages (Fig. 3B, iii; P: 0.0368). In both TRA and IGL receptor distributions, the ΔD diversity score showed that large tumors (described pathologically as tumors with the largest diameter of 7 cm or greater) have increased receptor subtype diversity compared with smaller tumors (those with largest diameters below 7 cm). Of note, the CPTAC-3 had a significantly different distribution of tumor sizes, with many small tumors (largest diameter < 7 cm), compared with the Moffitt TCC cohort (Fig. 3B, ii and v; Supplementary Fig. S13 compares the Moffitt TCC and CPTAC-3 cohorts based on distribution of tumor sizes).
Furthermore, the differences in grade and stage (size data not available) could be replicated in the TRA recoveries from previously obtained TCR CDR3s (26, 27) from TCGA-KIRC Cohort. As demonstrated in the Moffitt TCC cohort (Fig. 2A, iii; Supplementary Fig. S12A, iii) and CPTAC-3 cohort (Fig. 3B, i and iii, TCGA-KIRC cohort patients with higher grade (Fig. 3C, i; P: 0.0008) and higher pT stage (Fig. 3C, ii; P: 0.0027) had significantly higher TRA receptor ΔD diversity.
In addition to tumor sample differences, CPTAC-3 cohort had a subset of 75 patients with matched normal tissue samples (matched normal tissue recovery landscape described in Supplementary Fig. S14). Lymphocyte receptor richness was increased in tumor samples compared with normal tissue, which was observed across all receptor subtypes and combinations (Fig. 4A; Supplementary Fig. S15; Supplementary Data S2). In the TRs+IGs combination, tumor tissues had on average, at least 2.6-fold increase in richness of CDR3 sequences recovered, compared with the matched patient's normal tissue (mean score of 144.0 in normal tissue compared with a mean score of 377.1 in matched tumor). Furthermore, sequence dominance (measured by low values of high-q diversity) was decreased in all receptor subtypes except for the IGL receptor and IGH+IGK+IGL receptor combination (Fig. 4B; Supplementary Fig. S15; Supplementary Data S2). In the TRs+IGs combination, the tumor tissue had on average, at least 1.3-fold increase in high-q diversity compared with normal tissue, which indicates that the most abundance CDR3 sequence in the sample was about 2% lower, thus less dominant, in the tumor (mean score of 14.27 in the normal tissue compared with a mean score of 19.17 in the tumor tissue). Analyzing the IP q-metric of evenness in TRs+IGs combinations in the normal-tumor matched patients showed that normal samples have an almost 10% mean increase evenness compared with their matched tumor samples (Fig. 4C; P 0.0857). These data supported the hypothesis that normal tissues are expected to show very even CDR3 sequence distributions, and that better outcomes are to be expected in tumors that appear more normal in this context.
Figure 4.

Novel measures of diversity and overall survival. A and B, Tumor samples have increased TRs+IGs (all receptor combination) species richness (A) and evenness (B) of CDR3 receptor sequences compared with patient-matched normal tissue. C, However, tumor samples have reduced TRs+IGs IP q diversity compared with normal tissue (mean of normal 1.602 vs. mean of tumor 1.465; P 0.0857). D, Individuals in the Moffitt TCC cohort with larger TRA distribution IP (see Materials and Methods) had significantly improved overall survival (HR: 0.526, Cox P: 0.036), with a median survival of 115 months compared with those with lower IP. E, TCGA-KIRC overall survival supports the trend of lower IP has reduced median survival (92.13 months compared with undefined in high IP group; log-rank P: 0.6104). F–H, Four examples of patients of the Moffitt TCC cohort with fundamentally different characteristics. While ΔD assesses mainly sequence richness, the inflection IP, clearly visible in G, is a robust measure of evenness; high IP distributions are more even. **, P < 0.01; ***, P < 0.001.
Immune receptor subtype evenness, measured by IP, is associated with survival
We first analyzed associations between immune receptors and overall survival in the Moffitt TCC cohort. Each of our chosen diversity metrics (Materials and Methods) was compared for each of the seven immune receptor types. We used the maximally selected rank statistics (maxstat) approach (Materials and Methods), with a cut-off point that yielded a maximal survival difference, together with a multivariate Cox regression analysis. We found larger TRA sequence distribution IP, which measures distribution evenness, was significantly associated with longer overall survival (Fig. 4D). The IP value of the cut-off point in the cohort that yielded a maximal survival difference was 0.826. Using this optimal cut-off point resulted in a HR of 0.526 (log-rank P: 0.049, Cox P: 0.036) with the low IP group (IP < 0.826, n = 15) having a median overall survival of 80 months, and the high diversity group (IP > 0.826, n = 88) having a median overall survival of 115 months. This trend could not be confirmed with the CPTAC-3 cohort due to the lack of survival information, that is, overall survival data information was censored for 85 of the 98 CPTAC-3 cases (Supplementary Fig. S16). This trend was supported (although not statistically significantly) with the TCGA-KIRC cohort with individual with IP above the median produced a low IP group (IP < 2.86, n = 192) having a median overall survival of 92.1 months, and the high IP group (IP > 2.86, n = 197) having an undefined median overall survival due to survival fraction not falling below 50% in this group, which is likely due to this cohort also being comprised of lower grade and stage tumors compared with the Moffitt TCC cohort (Fig. 4E; log-rank P: 0.6104).
To further illustrate the utility of our diversity metrics for receptor subtype heterogeneity estimation, we show in Fig. 4F–H four Moffitt TCC cohort examples of characteristic differences in richness versus evenness space. In both examples of low evenness, dominance (low high-q diversity) is high (Fig. 4G). Interestingly, the survival difference unveiled by IP distribution evenness comparison does not necessarily coincide with tumor size, as size rather correlated with richness (ΔD, Fig. 4H). Taken together, these findings highlight the ability of these metrics to assess immune sequence distribution heterogeneity via GDI-derived point estimates.
Demographic and aberrant splicing differences in tumor-infiltrating lymphocyte receptor diversity
In addition to capturing differences in tumor pathology and survival, the GDI was also able to discriminate patients based on demographic differences. BCR high-q diversity is an estimate of dominance by the most abundant sequence: the lower this value, the more dominant the most abundant sequence. High-q diversity demonstrated differences in immune infiltration of White versus non-White patients with ccRCC in the TCC cohort (Supplementary Fig. S17). IGH and IGL receptor high-q diversity alone, as well as the IGH+IGK+IGL and total recovery (TRs+IGs) showed at least a 2.1-fold increase in non-White patients compared with White patients (Supplementary Fig. S17A and S17B; P < 0.01). This trend was also observed in the IGK receptor high-q diversity, but only with a 1.6-fold increase in non-White individuals (Supplementary Fig. S17C; P: 0.0561).
Furthermore, diversity metrics from TCRs TRG and TRD discriminate patients based on gender differences in the TCC cohort. TRG and TRD receptor IP q was at least 1.7-fold higher in female patients compared with male patients (TRG: score of 2.503 in females vs. a score of 1.442 in males, P: 0.0033; TRD: score of 2.883 in females vs. a score of 1.454 in males, P: 0.0003; Supplementary Fig. S18). Moreover, when high-q diversity (dominance of the most abundant sequence) was compared between female and male patients with respect to TRG and TRD diversity, in both the Moffitt TCC and CPTAC-3 cohorts, female patients had increased high-q diversity compared with male patients (Supplementary Fig. S19).
We evaluated association of GDI with a recently described aberrant EGFR splice variant in ccRCC (36). The Moffitt TCC cohort of patients were profiled for the presence of this EGFR variant (reported as % EGFR variant), and we found a positive correlation between the percentage of tumors expressing the EGFR variant and the evenness (IP q) scores from BCRs (Supplementary Fig. S20). This correlation was most significant in IGL receptor diversity, with a Spearman correlation coefficient of r = 0.3430 (P: 0.004, BCR IP vs. EGFR variant correlation analysis is demonstrated in Supplementary Fig. S20 and TCR IP vs. EGFR variant correlation analysis is demonstrated in Supplementary Fig. S21), indicating more even (high IP) distributions have higher proportion of cells with that EGFR variant. This positive correlation between IP and percentage of EGFR variant was not as strong nor significant in the TCRs (Supplementary Fig. S21).
To determine whether preexisting host inflammatory environment contributed to differences in patient CDR3 sequence diversity, we identified patients in the Moffitt TCC cohort who were diagnosed with diabetes and investigate whether there were any associations with the diversity metrics and diabetes status. We found that none of the 11 receptor combinations explored had a significant difference in any of the metrics of CDR3 diversity (individual comparisons are reported in Supplementary Data S1 and TRA, IGL, and TRs+IGs ΔD and IP comparisons are demonstrated in Supplementary Fig. S22).
Finally, we evaluated associations of GDI across the mutational landscape across all three cohorts. For each of the cohorts, we had mutational status on a subset of patients for common driver mutations in ccRCC including: BAP1, SETD2, KDM5C, MTOR, PBRM1, PTEN, TP53, and VHL. Across all three cohorts, the number of mutations in this shortlist of driver mutations was negatively correlated with richness (Fig. 5A). Interestingly, the Moffitt TCC cohort, generally considered the more aggressive cohort, had the fewest number of associations between diversity metrics and mutation status, while the CPTAC-3 and TCGA-KIRC cohorts had more significant associations (detailed for CPTAC-3 in Supplementary Data S2 and TCGA-KIRC in Supplementary Data S3). Furthermore, BCRs (IGH, IGK, IGL) had more significant associations with mutational status than TCRs (TRA, TRB, TRG, TRD; compared between the Moffitt TCC cohort described in Supplementary Data S1 and CPTAC-3 cohort in Supplementary Data S2). Specifically, with IGL recoveries in the CPTAC-3 cohort in patients with mutations in KDM5C, PBRM1, and VHL all had reduced richness (Fig. 5B, i–iii) and mutation in PTEN was associated with increased evenness (Fig. 5B, iv). These trends were confirmed in the total (TRs+IGs) recoveries from the CPTAC-3 cohort (Supplementary Fig. S23A) and the trends were supported by the Moffitt TCC cohort in direction, however they were not statistically significant (Fig. 5C for IGL trends and Supplementary Fig. S23B for TRs+IGs trends). In T-cell recoveries, both the CPTAC-3 and TCGA-KIRC cohorts showed reduced richness was associated with a mutation in PBRM1 (Fig. 5D); however, this trend was not observed in the Moffitt TCC cohort.
Figure 5.

Associations of diversity metrics with mutational landscape in ccRCC. A, Number of mutations were negatively correlated with CDR3 recovery richness. Spearman correlation coefficients between number of mutations and ΔD diversity were calculated for the (i) Moffitt TCC total (TRs+IGs) recoveries (Spearman r: −0.3007, P: 0.0167; ANOVA, P: 0.0025); CPTAC-3 total recoveries (Spearman r: −0.2452, P: 0.0098; ANOVA, P: 0.0213), and (iii) TCGA-KIRC total available (TRA+TRB) recoveries (Spearman r: −0.07348, P: 0.1332; ANOVA, P: 0.3905). B, IGL recoveries had reduced richness in CPTAC-3 patients with (i) KDM5C mutations (mean score of wild type was 167.9 and mutant was 44.41; P: 0.0298), (ii) PBRM1 mutations (mean score of wild type was 182.2 and mutant was 92.06; P: 0.0421), and (iii) VHL mutations (mean score of wild type was 247.7 and mutant was 115.0; P: 0.0094) and increased evenness in patients with (iv) PTEN mutations (mean score of wild type was 10.61 and mutant was 35.35; P: 0.0001). C, IGL recoveries had reduced richness in Moffitt TCC patients with (i) KDM5C mutations (mean score of wild type was 248.1 and mutant was 42.96; P: 0.0906), (ii) PBRM1 mutations (mean score of wild type was 259.8 and mutant was 156.4; P: 0.2200), and (iii) VHL mutations (mean score of wild type was 307.7 and mutant was 190.4; P: 0.1993) and increased evenness in patients with (iv) PTEN mutations (mean score of wild type was 0.7323 and mutant was 1.427; P: 0.0083). D, TRA receptor richness was (i) not different in Moffitt TCC patients with PBRM1 mutations (mean score of wild type was 32.03 and mutant was 24.59; P: 0.5397), (ii) decreased in CPTAC-3 patients with PBRM1 mutations (mean score of wild type was 33.63 and mutant was 22.53; P: 0.0470), and (iii) decreased in TCGA-KIRC patients with PBRM1 mutations (mean score of wild type was 23.84 and mutant was 17.06; P: 0.0032). Unpaired t tests were used to compare two group data and ANOVA was used to compare grade, three group data. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Discussion
Here, we demonstrate how a GDI, often used in ecology and evolution (31, 37, 38), can be applied to quantitatively characterize tumor-infiltrating lymphocyte receptor subtype diversity in ccRCC. We identified point estimates of this index that are associated with important differences in patient demographics, tumor pathology, and survival. These metrics can help objectively characterize host differences in immune receptor subtypes in patients with ccRCC. These novel objective metrics can provide insight into underlying tumor and host immune relationships by defining differences within and across patients. We used bulk sequencing data from ccRCC tumors to better understand these differences in patient immune receptor subtypes and these metrics can be replicated in other similar cohorts with available sequencing data. These host diversity metrics could be especially helpful in elucidating the ideal tumor microenvironment for response to immunotherapy agents in patients with metastatic ccRCC (39).
The recovery of adaptive immune receptor recombination reads from RNA-seq files is obtained via PCR amplification of adaptive immune receptors. This procedure is also called the immune repertoire approach (40). Our work here strongly indicates the specific value of the TRA and IGL GDI, discussed in more detail below. Thus, it has made sense to apply the immune repertoire approach to increase the number of recombination reads for all adaptive immune receptors. We expect that other immune receptor genes may have prognostic value, with more recombination reads to evaluate. Immune repertoire approaches, particularly when applied to cancer samples, often result in a majority of reads that represent relatively few clonotypes. In addition, human aging substantially reduces clonotype diversity (41, 42), particularly Fig. 2C and D therein. Thus “sampling of the repertoire,” by mining genomics files over large patient databases, can generate conclusions regarding clonotype associations with clinical features. More recent preparations of RNA-seq files, including those we use here, have become much more robust over the last several years, both in terms of read quantity and lengths (27). Recovery of adaptive immune receptors from those files also has become more robust. In summary, our results from adaptive immune receptor read recoveries from the RNA-seq files are informative. Further studies using the immune repertoire approach, representing a more comprehensive clonotype collection, should be applied in the future.
Our findings suggest that individuals with more advanced disease have increased richness in tumor recovered CDR3 sequences. In the Moffitt TCC cohort, we detected statistically significant differences in TRA and IGL diversity with increased richness in tumors with larger diameter and higher grade. Furthermore, we identified tumors with sarcomatoid carcinoma pathology that represent a rare and aggressive histology and showed significant increases in different diversity metrics and receptor subtypes, in particular increased CDR3 sequence richness. The immune receptor profiles of these tumors are particularly interesting because sarcomatoid histology has also been associated with very favorable response to checkpoint inhibitors. We postulate that a similar immune receptor profile in other patient tumors may portend a favorable response to checkpoint inhibition. Also, we found a significant increase in richness in left-sided tumors. This difference may explain some of the host-related factors associated with left-sided tumors that have a poorer clinical outcome than right-sided tumors (43). Many of these associations were able to be validated in similar comparisons with the CPTAC-3 cohort studies.
In the TCC cohort, BCRs with IGL had the most recoveries and TRA had the most T-cell CDR3 sequences recovered (Supplementary Fig. S3). It should be noted that point estimates of diversity or heterogeneity are only comparable within the subtype of interest, within a cohort. However, trends can be compared between point estimates and patient cohorts. Furthermore, different cohorts based on their clinical context may show differences in immune cell infiltrates, even when considering simple cell marker differences between T cells and B cells. Interestingly, B-cell CDR3 recoveries dominated in the TCC cohort, accounting for an average of 93.41% (ranging from at least 53.24% to 99.95%). This contrasted with the CPTAC-3 renal cell carcinoma cohort, which contains generally less aggressive tumors, with only 40.9% of the cohort respectively comprised of stage 3 or 4 tumors.
Our results demonstrate that increased richness is indicative of larger and more advanced ccRCC tumors, which may be related to differences in underlying tumor biology. Evenness, as measured by the IP q, segregated patients based on survival. In a cross-validation cut-off point analysis, patients with higher TRA evenness had a significantly improved overall survival compared with individuals with lower TRA evenness. These results indicate that patients' TRA evenness, not richness, may be a possible prognostic biomarker and could have direct therapeutic consequences for response to systemic agents that elicit their effect in the tumor microenvironment (39). Furthermore, this evenness metric could be extended to other solid tumor types, as a quantitative metric of host contributions to immune infiltration that is a result of tumor evolution. These characterizations might become especially interesting in the context of terminally exhausted CD8+ T cells, which were recently shown to be enriched in advanced renal cell carcinoma, interacting with M2-like tumor-associated macrophages, leading to immune dysfunction and poorer prognosis (44).
We identified demographic differences based on the CDR3 diversity of BCRs, both individually and in combination, showing increased high-q diversity. This amounted to decreased dominance of the most abundant sequence, in non-White individuals compared with White individuals in the Moffitt TCC cohort. However, in this cohort, 88.5% of the patients were White (Table 1) and we were unable to confirm these results in the CPTAC-3 cohort due to missing data. Nevertheless, previously found race-related differences in BCR pathway activation in African Americans compared with European Americans lends support to our finding in differences in BCR dominance/clonality diversity (45). Furthermore, high-q diversity/sequence dominance may reveal gender-based differences in the ccRCC microenvironment. We found that females had higher clonality compared with male patients, which persists in the CPTAC-3 cohort. These demographic differences need to be further investigated, but our results suggest underlying race and gender differences in the heterogeneity of ccRCC microenvironments as reflected in tumor immune infiltration differences.
Biodiversity has historically been summarized into: alpha-diversity, which measures a single community's diversity; beta-diversity, which quantifies the relative change of species between communities; and gamma diversity, which measures the total diversity in ecology (46). Diversity at the individual receptor subtype level relates closest to alpha-diversity, which is then compared across the patients in a cohort. In a beta-diversity context, we see that many of the trends hold true between the Moffitt TCC and CPTAC-3 cohorts. The most commonly used diversity measures applied to cancer systems have been Shannon and Simpson indices (47, 48), which are special cases of the GDI at intermediate values of the parameter q (49, 50). Our analysis found that it is at the extremes of the continuum of diversity measures (low-q and high-q values) that we can stratify patients in clinically meaningful ways (e.g., Fig. 2 vs. Supplementary Fig. S6). In this sense, novel properties of the GDI that are discussed here may allow a more nuanced, and thus more clinically comprehensive characterization of sequence heterogeneity. These novel objective diversity scales could have important applications for other systems in which tumor heterogeneity with its ecological and evolutionary impact is quantified.
Different point estimates based on generalized diversity give unique information about the tumor. Increased richness in TRA and IGL diversity informs the size and aggressiveness of a tumor. Dominance of the most abundant sequence segregates patients based on prognosis. We identified a novel measure of evenness among immune receptor subtypes that could accurately classify patients' overall survival. We also found important differences in receptor subtype contributions based on patient demographics such as race and gender. Using these diversity metrics, we identify a new statistical approach to stratify ccRCC patients based on differences in immune infiltration diversity and further guide precision oncology.
Authors' Disclosures
M.C. Ferrall-Fairbanks reports other support from UF Foundation during the conduct of the study. J.K. Teer reports grants from Department of Defense and NIH during the conduct of the study; in addition, J.K. Teer has a patent for Large Data Set Negative Information Storage Model pending. E.M. Siegel reports grants from NCI/NIH during the conduct of the study. B.J. Manley reports other support from M2GEN during the conduct of the study. P.M. Altrock reports grants from NIH/NCI and ACR during the conduct of the study and grants from KITE Pharma/Gilead outside the submitted work. No disclosures were reported by the other authors.
Supplementary Material
Acknowledgments
This work was supported in part by the Biostatistics and Bioinformatics Shared Resource at the H. Lee Moffitt Cancer Center & Research Institute, a NCI-designated Comprehensive Cancer Center (P30-CA076292), and TCC Protocol at Moffitt Cancer Center, which was enabled in part by the generous support of the DeBartolo Family. Some data used in this publication were generated by the NCI Clinical Proteomic Tumor Analysis Consortium (CPTAC), available to author via dbGaP project approval number 6757. Editorial assistance was provided by the Moffitt Cancer Center's Scientific Editing Department by Dr. Paul Fletcher and Daley Drucker (no compensation was given beyond their regular salaries). The authors also thank Gregory J. Kimmel, PhD for valuable comments.
This work was supported by U.S. Army Medical Research Acquisition Activity Department of Defense (KC180139 to B.J. Manley). P.M. Altrock was supported by an American Cancer Society Moffitt IRG award, the Richard O. Jacobson Foundation (Evolutionary Therapy Center of Excellence at Moffitt Cancer Center), and the William G. ‘Bill’ Bankhead Jr and David Coley Cancer Research Program (20B06).
The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked advertisement in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
Note: Supplementary data for this article are available at Cancer Research Online (http://cancerres.aacrjournals.org/).
Authors' Contributions
M.C. Ferrall-Fairbanks: Conceptualization, data curation, software, formal analysis, validation, investigation, visualization, methodology, writing–original draft, writing–review and editing. N.H. Chakiryan: Conceptualization, data curation, investigation, writing–review and editing. B.I. Chobrutskiy: Data curation, adaptive immune receptor recombination read extraction, moffitt cohort. Y. Kim: Validation, statistical analysis, critical review. J.K. Teer: Data curation, statistical analysis, critical review, logistic support. A. Berglund: Data curation, statistical analysis, critical review, logistic support. J.J. Mulé: Investigation, critical review. M. Fournier: Data curation, logistic support. E.M. Siegel: Data curation. J. Dhillon: Investigation, critical review. S.S.A. Falasiri: Data curation, adaptive immune receptor recombination read extraction, moffitt cohort. J.F. Arturo: Data curation, adaptive immune receptor recombination read extraction, moffitt cohort. E.N. Katende: Data curation, logistic support. G. Blanck: Supervision, writing–original draft, writing–review and editing. B.J. Manley: Conceptualization, resources, supervision, investigation, methodology, writing–original draft, project administration, writing–review and editing. P.M. Altrock: Conceptualization, resources, software, supervision, funding acquisition, investigation, methodology, writing–original draft, writing–review and editing.
References
- 1. Cronin KA, Lake AJ, Scott S, Sherman RL, Noone AM, Howlader N, et al. Annual report to the nation on the status of cancer, part I: national cancer statistics. Cancer 2018;124:2785–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Motzer RJ, Russo P. Systemic therapy for renal cell carcinoma. J Urol 2000;163:408–17. [PubMed] [Google Scholar]
- 3. Escudier B. Combination therapy as first-line treatment in metastatic renal-cell carcinoma. N Engl J Med 2019;380:1176–8. [DOI] [PubMed] [Google Scholar]
- 4. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 2014;371:2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 2015;348:124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yarchoan M, Hopkins A, Jaffee EM. Tumor mutational burden and response rate to PD-1 inhibition. N Engl J Med 2017;377:2500–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Maleki Vareki S. High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. J Immunother Cancer 2018;6:157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Miao D, Margolis CA, Gao W, Voss MH, Li W, Martini DJ, et al. Genomic correlates of response to immune checkpoint therapies in clear cell renal cell carcinoma. Science 2018;359:801–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Braun DA, Hou Y, Bakouny Z, Ficial M, Sant' Angelo M, Forman J, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 2020;26:909–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hajiran A, Chakiryan N, Aydin AM, Zemp L, Nguyen J, Laborde JM, et al. Reconnaissance of tumor immune microenvironment spatial heterogeneity in metastatic renal cell carcinoma and correlation with immunotherapy response. Clin Exp Immunol 2021;204:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chobrutskiy BI, Zaman S, Diviney A, Mihyu MM, Blanck G. T-cell receptor-alpha CDR3 domain chemical features correlate with survival rates in bladder cancer. J Cancer Res Clin Oncol 2019;145:615–23. [DOI] [PubMed] [Google Scholar]
- 14. Roca AM, Chobrutskiy BI, Callahan BM, Blanck G. T-cell receptor V and J usage paired with specific HLA alleles associates with distinct cervical cancer survival rates. Hum Immunol 2019;80:237–42. [DOI] [PubMed] [Google Scholar]
- 15. Chobrutskiy BI, Zaman S, Tong WL, Diviney A, Blanck G. Recovery of T-cell receptor V(D)J recombination reads from lower grade glioma exome files correlates with reduced survival and advanced cancer grade. J Neurooncol 2018;140:697–704. [DOI] [PubMed] [Google Scholar]
- 16. Callahan BM, Yavorski JM, Tu YN, Tong WL, Kinskey JC, Clark KR, et al. T-cell receptor-beta V and J usage, in combination with particular HLA class I and class II alleles, correlates with cancer survival patterns. Cancer Immunol Immunother 2018;67:885–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zarnitsyna VI, Evavold BD, Schoettle LN, Blattman JN, Antia R. Estimating the diversity, completeness, and cross-reactivity of the T cell repertoire. Front Immunol 2013;4:485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nikolich-Zugich J, Slifka MK, Messaoudi I. The many important facets of T-cell repertoire diversity. Nat Rev Immunol 2004;4:123–32. [DOI] [PubMed] [Google Scholar]
- 19. Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A 2009;106:20216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jost L. Entropy and diversity. Oikos 2006;113:363–75. [Google Scholar]
- 21. Hill MO. Diversity and evenness: a unifying notation and its consequences. Ecology 1973;54:427–32. [Google Scholar]
- 22. MacArthur RH. Patterns of species diversity. Biol Rev Camb Philos Soc 1965;40:510–33. [Google Scholar]
- 23. Kaplinsky J, Arnaout R. Robust estimates of overall immune-repertoire diversity from high-throughput measurements on samples. Nat Commun 2016;7:11881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Moffitt Cancer Center. Total Cancer Care. Available from: https://moffitt.org/research-science/total-cancer-care/.
- 25. NCI Office of Cancer Clinical Proteomics Research. Clinical Proteomic Tumor Analysis Consortium (CPTAC). Available from: https://cptac-data-portal.georgetown.edu/cptac/public?scope=Phase+III.
- 26. Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Ou Yang TH, et al. The immune landscape of cancer. Immunity 2018;48:812–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Patel DN, Yeagley M, Arturo JF, Falasiri S, Chobrutskiy BI, Gozlan EC, et al. A comparison of immune receptor recombination databases sourced from tumour exome or RNAseq files: verifications of immunological distinctions between primary and metastatic melanoma. Int J Immunogenet 2021;48:409–18. [DOI] [PubMed] [Google Scholar]
- 28. Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ferrall-Fairbanks MC, Ball M, Padron E, Altrock PM. Leveraging single-cell RNA sequencing experiments to model intratumor heterogeneity. JCO Clin Cancer Inform 2019;3:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ferrall-Fairbanks MC, Altrock PM. Investigating inter- and intrasample diversity of single-cell RNA sequencing datasets. In: Markowitz J, editor. Translational bioinformatics for therapeutic development: methods and protocols. Springer; 2020. [DOI] [PubMed] [Google Scholar]
- 31. Miroshnychenko D, Baratchart E, Ferrall-Fairbanks MC, Vander Velde R, Laurie MA, Bui MM, et al. Spontaneous cell fusions as a mechanism of parasexual recombination in tumor cell populations. Nat Ecol Evol 2021;5:379–91. [DOI] [PubMed] [Google Scholar]
- 32. Zaman S, Hajiran A, Coba GA, Robinson T, Madanayake TW, Segarra DT, et al. Aberrant epidermal growth factor receptor RNA splice products are among the most frequent somatic alterations in clear cell renal cell carcinoma and are associated with a poor response to immunotherapy. Eur Urol Focus 2021;7:373–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hothorn T, Lausen B. On the exact distribution of maximally selected rank statistics. Comput Stat Data Anal 2003;43:121–37. [Google Scholar]
- 34. Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Aran D, Hu Z, Butte AJ. xCell: digitally portraying the tissue cellular heterogeneity landscape. Genome Biol 2017;18:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tuomisto H. A consistent terminology for quantifying species diversity? Yes, it does exist. Oecologia 2010;164:853–60. [DOI] [PubMed] [Google Scholar]
- 38. Cazzolla Gatti R, Amoroso N, Monaco A. Estimating and comparing biodiversity with a single universal metric. Ecol Modell 2020;424:109020. [Google Scholar]
- 39. Diaz-Montero CM, Rini BI, Finke JH. The immunology of renal cell carcinoma. Nat Rev Nephrol 2020;16:721–35. [DOI] [PubMed] [Google Scholar]
- 40. Rosati E, Dowds CM, Liaskou E, Henriksen EKK, Karlsen TH, Franke A. Overview of methodologies for T-cell receptor repertoire analysis. BMC Biotechnol 2017;17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Egorov ES, Kasatskaya SA, Zubov VN, Izraelson M, Nakonechnaya TO, Staroverov DB, et al. The changing landscape of naive T cell receptor repertoire with human aging. Front Immunol 2018;9:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Britanova OV, Shugay M, Merzlyak EM, Staroverov DB, Putintseva EV, Turchaninova MA, et al. Dynamics of individual T cell repertoires: from cord blood to centenarians. J Immunol 2016;196:5005–13. [DOI] [PubMed] [Google Scholar]
- 43. Guo S, Yao K, He X, Wu S, Ye Y, Chen J, et al. Prognostic significance of laterality in renal cell carcinoma: a population-based study from the surveillance, epidemiology, and end results (SEER) database. Cancer Med 2019;8:5629–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell 2021;39:632–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Longo DM, Louie B, Mathi K, Pos Z, Wang E, Hawtin RE, et al. Racial differences in B cell receptor signaling pathway activation. J Transl Med 2012;10:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jost L. Partitioning diversity into independent alpha and beta components. Ecology 2007;88:2427–39. [DOI] [PubMed] [Google Scholar]
- 47. Almendro V, Cheng YK, Randles A, Itzkovitz S, Marusyk A, Ametller E, et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep 2014;6:514–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Marusyk A, Tabassum DP, Altrock PM, Almendro V, Michor F, Polyak K. Non-cell-autonomous driving of tumour growth supports sub-clonal heterogeneity. Nature 2014;514:54–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Shannon CE. A mathematical theory of communication. Bell Syst Tech J 1948;27:379–423. [Google Scholar]
- 50. Simpson EH. Measurement of diversity. Nature 1949;163:688. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code for VDJ epitope recoveries from patient sequencing data (BAM files) is publicly available on Docker at https://hub.docker.com/r/bchobrut/vdj and GitHub at https://github.com/bchobrut/vdj_recovery.
The code and documentation describing how to calculate patient CDR3 diversity from CDR3 sequence recoveries and run our pipeline and reproduce our results are open-source and publicly available through the OncoDiversity.jl GitHub repository (https://github.com/mcfefa/OncoDiversity.jl). A virtual machine producing the full diversity environment is available on Code Ocean (https://codeocean.com/capsule/9959428/tree/).