

Abstract
Renal cell carcinomas associated with hereditary leiomyomatosis and renal cell cancer (HLRCC) are notoriously aggressive and represent the leading cause of death among HLRCC patients. To date, a safe and effective standardized therapy for this tumor type is lacking. Here we show that the engineered synthetic therapeutic enzyme, Cyst(e)inase, when combined with rapamycin, can effectively induce ferroptosis in HLRCC cells in vivo. The drug combination promotes lipid peroxidation to a greater degree than cysteine deprivation or Cyst(e)inase treatment alone, while rapamycin treatment alone does not induce ferroptosis. Mechanistically, Cyst(e)inase induces ferroptosis by depleting the exogenous cysteine/cystine supply, while rapamycin reduces cellular ferritin level by promoting ferritins’ destruction via ferritinophagy. Since both Cyst(e)inase and rapamycin are well-tolerated clinically, the combination represents an opportunity to exploit ferroptosis induction as a cancer management strategy. Accordingly, using a xenograft mouse model, we showed that the combination treatment resulted in tumor growth suppression without any notable side effects. In contrast, both Cyst(e)inase only and rapamycin only treatment groups failed to induce a significant change when compared to the vehicle control group. Our results demonstrated the effectiveness of Cyst(e)inase-rapamycin combination in inducing ferroptotic cell death in vivo, supporting the potential translation of the combination therapy into clinical HLRCC management.
Keywords: HLRCC, Ferroptosis, Cysteinase, Rapamycin, FH
Introduction.
Hereditary leiomyomatosis and renal cell cancer (HLRCC) is an autosomal dominant hereditary cancer syndrome with incomplete penetrance.(1) HLRCC patients have a high risk of developing skin leiomyomas, uterine fibroids, and type 2 papillary renal cell carcinoma.(1) Importantly, HLRCC associated renal cell carcinoma is particularly aggressive and is resistant to all known therapies.(2) Moreover, the tumor often metastasizes early, limiting the effectiveness of surgical interventions.(2,3) Thus, there is a critical need for a safe and effective targeted treatment option for HLRCC associated renal tumors.
Biallelic inactivation of the fumarate hydratase gene, FH, underlies the biology of HLRCC.(4) FH encodes the citric acid cycle enzyme fumarase. Consequently, one of the most direct results of FH inactivation is intracellular fumarate accumulation. In HLRCC cells, fumarate can accumulate to millimolar concentrations. This accumulated fumarate can covalently modify cysteine residues on proteins to form succinyl adducts in a post-translational modification process known as succination.(5) One of the earliest reported consequences of succination in HLRCC is the sustained activation of the NRF2 transcription factor.(5) Subsequently, succination has been shown to alter cellular signaling and metabolism, including iron signaling, and enhancing susceptibility of HLRCC cells to an iron-dependent cell death mechanism known as ferroptosis.(6,7)
HLRCC cells’ susceptibility to ferroptotic cell death presents an opportunity to develop a safe and effective treatment strategy for this intractable tumor type. Mechanistically, lipid peroxide accumulation precedes ferroptotic cell death.(8) Specifically, the intracellular labile iron pool catalyzes Fenton’s reaction and generates lipid peroxides. Under normal cellular conditions, these lipid peroxides are neutralized by glutathione peroxidase 4 (GPX4), leaving the cells unharmed. Ferroptosis is induced by either directly or indirectly inhibiting GPX4, leading to lipid peroxide accumulation and subsequent oxidative cell death. To date, GPX4 has been directly inhibited by utilizing a GPX4 inhibitor such as RSL3. In addition, strategies that indirectly inhibit GPX4 include the depletion of cellular glutathione by suppressing the glutamate-cystine antiporter system (system Xc) using inhibitors such as erastin and glutamate.(8) Here, we seek to explore a strategy to induce ferroptosis in vivo using therapeutics with favorable safety and pharmacological characteristics. To do this, we utilize an engineered synthetic enzyme known as Cyst(e)inase. This enzyme is designed to degrade circulatory cysteine and cystine, and it is a biologic currently under clinical trial for treating cystinuria (9). We compared Cyst(e)inase to rapamycin, a drug approved for advanced renal cell carcinoma treatment.(10–12) Moreover, we also explored the efficacy of Cyst(e)inase and rapamycin combination for HLRCC treatment.
Materials and Methods
Cell lines and culture conditions.
UOK262 cell lines were a gift from Dr. Marston Linehan, National Cancer Center, National Institute of Health.(13) NCCFH1 was previously developed from the pleural fluid of a patient with metastatic HLRCC.(14) UOK262 cell line was authenticated through Sanger’s sequencing to confirm the presence of c.1316A>C FH mutation. NCCFH1 cell line was authenticated in the same way to confirm the presence of c.1162 delA FH mutation. All HLRCC cells were maintained in RPMI1640 media (Gibco, cat # 11875–093) with 10% Fetal Bovine Serum (Genesee Scientific). Both cell lines have been tested negative for mycoplasma contamination using a QPCR based method.
For cysteine deprivation experiments, L-glutamine, L-methionine and L-cystine free RPMI1640 media (Sigma, Cat # R7513) were used instead. The media was back supplemented with L-glutamine (Sigma, Cat # G7513) to a final concentration of 2mM, L-methionine (Sigma, Cat # M5308) to a final concentration of 0.1mM, and either without L-cystine supplementation for cysteine deprivation group, or with L-cystine hydrochloride (Sigma, Cat # 57579) supplementation to a final concentration of 0.2mM for the control group. Cysteine deprivation experiments were carried out in the presence of 5% instead of 10% FBS to minimize the effects of cysteine present in the fetal bovine serum.
Antibodies and chemicals
The following antibodies were used: Actin (Sigma), FTH1 (Santa Cruz Biotechnology, SC-376594), IRP2 (Santa Cruz Biotechnolog, SC-33682), S6 (Cell Signaling Technology, 54D2), p-S6 (Cell Signaling Technology, 91B2), 2-SC (Discovery Antibodies, crb2005017e/6773). Rapamycin and Deferoxamine were purchased from Sigma. Ferrostatin was purchased from Tocris. Cyst(e)inase was prepared as previously described.(9) the following dosages were used in in vitro experiments: Deferoxamine = 100μM, ferrostatin = 1μM, Cyst(e)inase = 200nM, Rapamycin = 1μM. Heat inactivated Cyst(e)inase was prepared by incubating Cyst(e)inase at 100°C for 10 minutes. Heat inactivated Cyst(e)inase was used at a final concentration of 200nM.
C11-Bodipy assay
Lipid peroxidation sensor, Bodipy™ 581/591 C11 (denoted as C11-Bodipy) was purchased from Thermo Scientific. The assay was carried out according to a procedure previously described by Dixon and co-workers.(15) The time point by which ferroptosis occurs is dependent on the cell lines and ferroptosis inducers. Thus, Bodipy staining, and measurement were done at different time points post treatment for different cell lines. For experiments not involving rapamycin cotreatment, Bodipy staining were done at 48-hour post treatment for UOK262, and 24-hour post treatment for NCCFH1. For experiments involving rapamycin co-treatments, Bodipy staining were done at 42-hour post treatment for UOK262, and at 19-hour post treatment for NCCFH1.
Immunohistochemistry
Immunohistochemistry (IHC) staining was performed using DAKO EnVision+ kit according to a manufacturer-recommended protocol. We used a citrate buffer pH6.0 heat-mediated antigen retrieval procedure to reveal the antigens in the paraffin-embedded tissues, and Vector® NovaRed® HRP substrate for color development. IHC slides were counterstained with Hematoxylin QS (Vector Laboratory).
Animal Experiments.
All animal experiments were performed by the University of Arizona Cancer Center’s Experimental Mouse Shared Resource (EMSR) and were approved by the university’s Institutional Animal Care and Use Committee (IACUC, Protocol No. :07-029). Briefly, the experiment was performed using 20 females and 20 males of NOD-scid IL2Rgammanull (NSG™) mice (Source of the mice: University of Arizona Cancer Center breeding colonies). Ten million NCCFH1 cells suspended in a 1:1 ratio of PBS:Matrigel were injected into the right flank of each animal. Tumors were measured twice a week using a digital caliper, and tumor volumes were calculated according to the volume of an ellipsoid. When mean tumor volumes reached 100mm3, an equal number of male and female mice were randomly assigned into four different groups. Thirty three of the 40 mice used in the experiment developed tumors. Thus, the final group assignment was as follow: vehicle (control group, n=6, 3 female & 3 male), rapamycin (rapamycin only group, n=7, 4 female & 3 male), Cyst(e)inase (Cyst(e)inase only group, n=10, 5 female & 5 male), rapamycin+Cyst(e)inase (combination group, n=10, 5 female & 5 male). Treatments were administered once every three days via intraperitoneal injection of either vehicle (Phosphate buffered saline containing 30% PEG300), rapamycin (0.6mg/ml suspended in vehicle, injection dose = 0.6mg per mouse), Cyst(e)inase (7.1mg/ml suspended in vehicle, injection dose = 7.1mg per mouse), and rapamycin+Cyst(e)inase combination (0.6mg/ml rapamycin, 7.1mg/ml Cyst(e)inase suspended in vehicle. Injection dose = 1ml per mouse). The tumor growth ratio of each time point, for each mouse, was calculated as the tumor volume of that time point divided by the tumor volume from the same mouse at 21-day post xenografting. Data was presented at mean ± standard error.
Qualitative and qualitative assessment of cell density and cell growth studies
Incucyte® Live-Cell Imaging System and crystal violet staining followed by digital imaging were used as qualitative cell density assessment methods. Quantitative assessments of cell density were performed by dissolving crystal violet stain cells in 1% SDS followed by spectrophotometry measurement at 590 nm.
Quantitative PCR of PTGS2
Total RNA from cell lines and xenograft tumors were isolated using a Direct-zol RNA isolation kit (Zymo Research). Complimentary DNAs (cDNA) were generated using large scale cDNA synthesis kit (Thermo Scientific) according to the manufacturers recommended protocol. The following Taqman gene expression assay probes were used: PTGS2 [IDT Assay ID: Hs.PT.58.77266], Actin [Thermo Scientific Taqman gene expression assay. Assay ID: Hs01060665_g1].
Statistical analysis
All statistical analyses were performed in R statistical environment. For animal experiment, significant difference between groups were determined by analysis of variance (ANOVA).
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.
Results
Cysteine deprivation induces ferroptotic cell death in HLRCC cells.
We previously showed that erastin induces ferroptosis in FH−/− cells.(6) Since erastin is a system Xc inhibitor that inhibits cells’ ability to uptake exogenous cystine, it depletes cellular glutathione and lead to GPX4 inhibition.(15) We reasoned that cysteine deprivation could also induce ferroptosis in HLRCC cells in a similar manner. Using two human HLRCC cell lines, UOK262 and NCCFH1, cultured in control and cysteine deprived media, we showed that cysteine deprivation-induced cell death in both cell lines. The cell death could be inhibited by either an iron chelator, deferoxamine (DFO), or a ferroptosis inhibitor, ferrostatin, indicating that both cell lines were undergoing ferroptotic cell death (Figure 1A–D). Consistently, C11-Bodipy measurement indicated that cysteine deprivation increases lipid peroxidation in both cell lines, and DFO could abrogate the effects of cysteine deprivation (Figure 1E–F).
Figure 1. Cysteine deprivation induces ferroptosis in HLRCC cells.
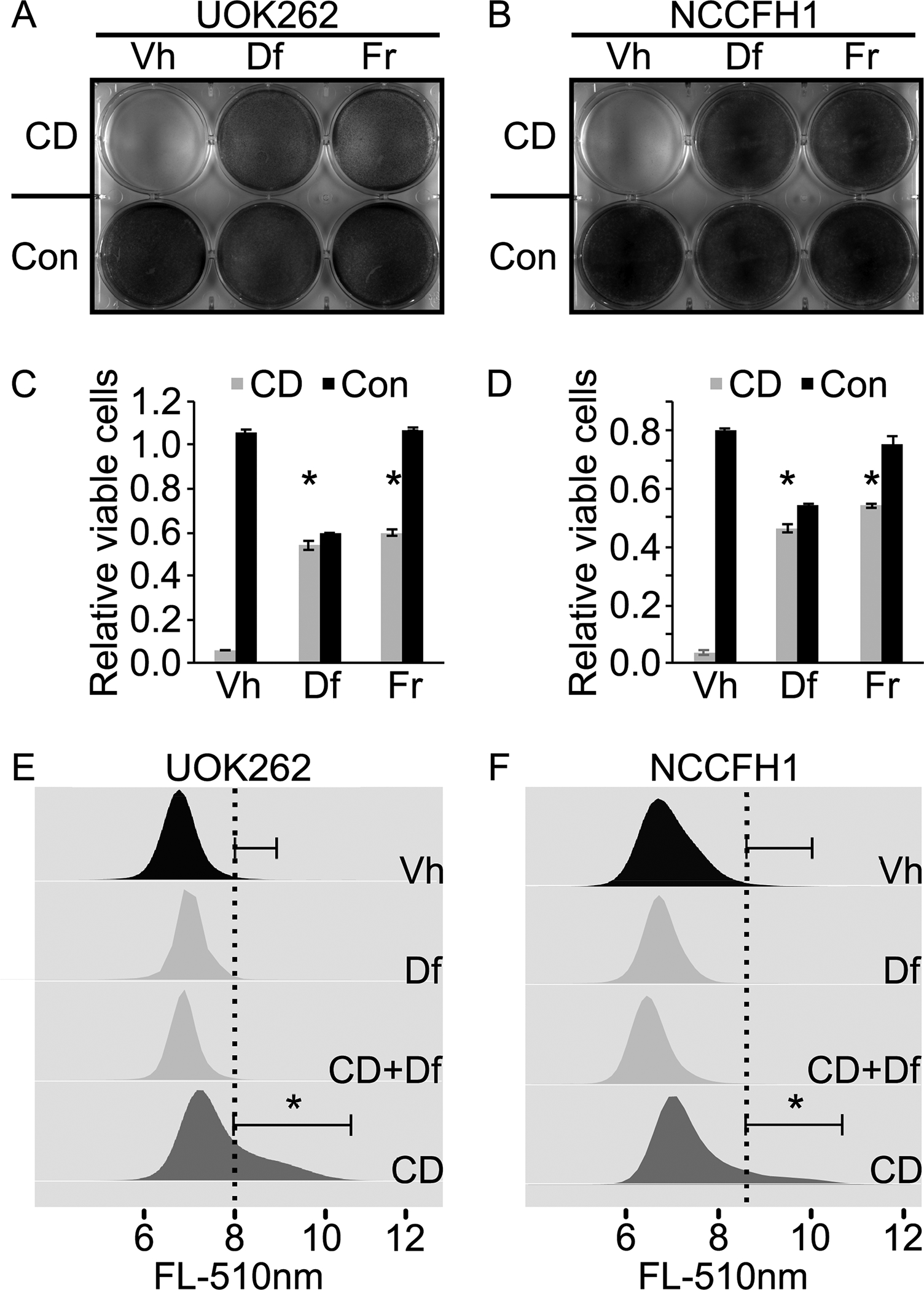
A. Cysteine deprivation (CD) induces UOK262 cell death. Deferroxamine (Df) and ferrostatin (Fr) abrogate the cell killing effects of CD, indicating ferroptosis as the cell death mechanism. Equal number of UOK262 cells were seeded onto a 6-well plate. At 24 hours post seeding, media in the top row (3 wells) were replaced with cysteine free RPMI1640 supplemented with 5% FBS. Media in the bottom row (3 wells) were replaced with complete RPMI1640 supplemented with 5% FBS. Deferroxamine (Df) was spiked into the wells in the middle column (middle two wells) to a final concentration of 100μM. Ferrostatin (Fr) was spiked into the last column (last two wells) to a final concentration of 1μM. Vehicle (Vh) consisting of DMSO was spiked into the wells in the first column to a final concentration of 0.1%. Cells were fixed and stained with crystal violet at 24 hours post treatment. B. CD induces ferroptotic cell death in NCCFH1 cells. Experiment described in A was repeated with a different HLRCC cell line, NCCFH1. C and D. Quantitative measurement of viable cells following experiments similar to those described in A and B. Cells remain adhered to the 6-well plate were stained with crystal violet. After that, the stained cells were dissolved in 1% SDS and subjected to absorbance measurement at 590nm. Bar heights and error bars indicate means and standard deviations respectively. These statistics were calculated from two independent experiments. Each of those two independent experiments contained three technical replicates. Statistical significance was determined through two-way ANOVA followed by Tukey Honest Significant Differences. An asterisk, *, indicates a p-value < 0.05 when compared to the corresponding Vh control group. E and F. Cysteine deprivation increases lipid peroxidation rate in HLRCC cells. Df rescues the CD-induced increase in lipid peroxidation. Experiments described in A and B were repeated. Instead of measuring the remaining viable cells as the end point, lipid peroxidation rates were measured using a C11-Bodipy-flow cytometry method. Flow cytometry data generated were analyzed and visualized using flowCore and flowWiz packages in R statistical environment. Statistical significance was determined through Student’s t-test. An asterisk, *, indicates a p-value < 0.05.
Cyst(e)inase induces ferroptotic cell death in HLRCC cells.
Cyst(e)inase is an engineered synthetic enzyme that degrades both cystine and cysteine and can effectively deplete circulatory cystine and cysteine in mice and in human with no observable side effects.(9) Thus, it represents a viable way to induce cysteine deprivation in vivo. Previous studies showed that Cyst(e)inase can effectively suppress proliferation of various cell lines in vitro at 200nM.(9) We utilized Cyst(e)inase in place of the cysteine deprivation to induce ferroptosis in UOK262 and NCCFH1 cell lines. We found that consistent with cysteine deprivation, 200nM Cyst(e)inase induced cell death in both cell lines. The cell death could be inhibited by either DFO or ferrostatin (Figure 2A–D), supporting that those cells were undergoing ferroptosis. Similarly, C11-Bodipy measurement also indicated that Cyst(e)inase increased lipid peroxidation in both cell lines (Figure 2E–F). Furthermore, heat-inactivated Cyst(e)inase was unable to induce cell death, indicating that it is the enzymatic activity of Cyst(e)inase is mediating the effect (Figure 2A–D).
Figure 2. Cyst(e)inase induces ferroptosis in HLRCC cells.
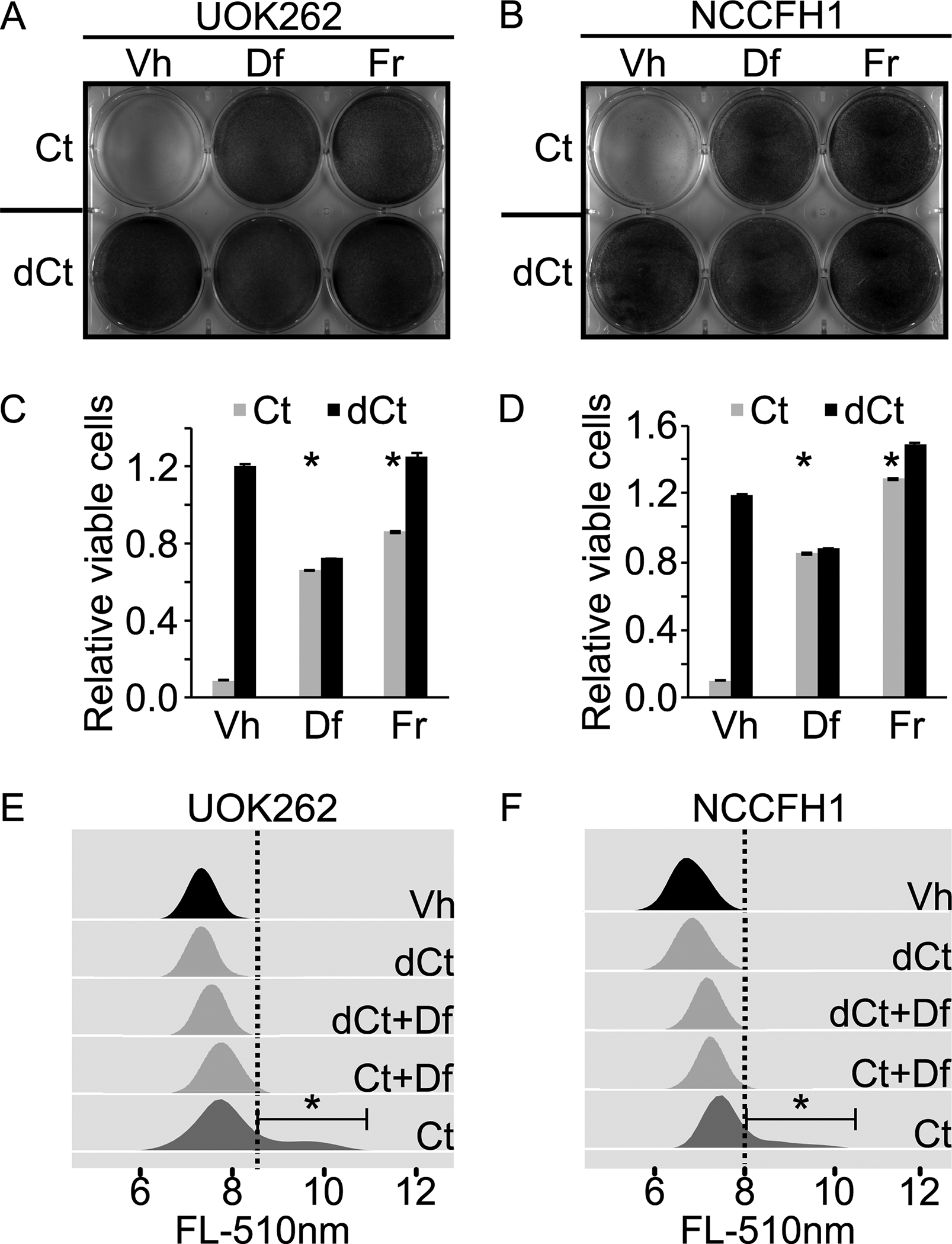
A. and B. Enzymatic activity of Cyst(e)inase induces ferroptosis in HLRCC cells. HLRCC cell lines, UOK262 and NCCFH1 each were seeded evenly onto a 6-well plate. Wells in the top row (3 wells) were treated with Cyst(e)inase (Ct)[final concentration = 200nM], while wells in the bottom row (3 wells) were treated with heat inactivated Cyst(e)inase (dCt) [200nM, heat inactivated at 100°C for 10 minutes]. Deferroxamine (Df) was spiked into the two wells in the middle column to a final concentration of 100μM, and ferrostatin (Fr) was spiked into the two wells in the third column to a final concentration of 1μM. Vehicle (Vh) consists of DMSO was spiked into the wells in the first column to a final concentration of 0.1%. Cells were fixed and stained with crystal violet at 24 hours post treatment. C and D. Quantitative measurement of viable cells following experiments similar to those described in A and B. Cells remain adhered to the 6-well plate were stained with crystal violet. After that, the stained cells were dissolved in 1% SDS and subjected to absorbance measurement at 590nm. Bar heights and error bars indicate means and standard deviations respectively. These statistics were calculated from two independent experiments. Each of those two independent experiments contained three technical replicates. Statistical significance was determined through two-way ANOVA followed by Tukey Honest Significant Differences. An asterisk, *, indicates a p-value < 0.05 when compared to the corresponding Vh control group. E and F. Enzymatic activity of Cyst(e)inase increases lipid peroxidation rate in HLRCC cells. Experiments described in A and B were repeated. Instead of measuring the relative number of cells remain viable as the end point, cells were stained with C11-Bodipy and then measured using flow cytometry. Data were analyzed and visualized using flowCore and flowWiz packages in R statistical environment. Statistical significance was determined through Student’s t-test. An asterisk, *, indicates a p-value < 0.05.
Rapamycin enhances sensitivity to ferroptotic cell death.
We previously showed that because of sustained NRF2 activation and the succination of iron regulatory proteins, HLRCC cells express high level of ferritin. Since ferritin suppresses ferroptosis by sequestering iron, releasing the ferritin stored iron may further enhance HLRCC cells’ susceptibility to ferroptosis. Studies have shown that increased autophagy will lead to the destruction of intracellular ferritin and the release of its stored iron.(16) Among the different autophagy induction methods, we chose to explore mammalian target of rapamycin (MTOR) inhibition by rapamycin because its analogs are approved by the FDA for RCC treatment and demonstrated favorable clinical and pharmacological characteristics.(11) As expected, rapamycin treatment inhibited protein S6 phosphorylation and reduced intracellular ferritin level in both UOK262 and NCCFH1 cells (Figure 3A). Cellular labile iron pool dictates intracellular iron regulatory proteins (IRPs, IRP1/2) levels. Increased labile iron pool stabilizes cellular iron sensor protein F-Box and Leucine-Rich Repeat Protein 5 (FBXL5), which mediates IRPs degradation.(17,18) Accordingly, rapamycin treatment resulted in decreased IRP2 level, indicating an increase in the labile iron pool (Figure 3A). However, rapamycin treatment alone was not sufficient to induce ferroptosis (Figure S1). Hence, we explored rapamycin-cysteine deprivation combination treatment and found that rapamycin enhances cysteine deprivation-induced ferroptosis in HLRCC cells. To investigate the effects of Cyst(e)inase-rapamycin combination in vitro, we performed time point studies using Incucyte® live cell imaging system. We found that Cyst(e)inase-rapamycin combination induced ferroptosis at an earlier time point. Specifically, we found that Cyst(e)inase-rapamycin combination induces ferroptotic cell death at 42 and 24 hours in UOK262 and NCCFH1 cells respectively (Figure 3B and C). This is earlier than Cyst(e)inase alone treated groups where ferroptosis occurred at 54 and 27 hours in UOK262 and NCCFH1 cells respectively (Figure 3B and 3C). Consistently, rapamycin alone treated group failed to induce ferroptosis in both HLRCC cell lines (Figure 3B and C). We repeated the experiment shown in Figure 3B and 3C using cysteine deprivation (Cd) in place of Cyst(e)inase treatment and measured the percentage of cells that showed increased lipid peroxidation at 42-hour post-treatment initiation for UOK262 and at 19 hour post treatment initiation for NCCFH1. C11-Bodipy measurements showed that rapamycin-cysteine deprivation combination increased intracellular lipid peroxidation in a larger subpopulation of cells as compared to cysteine deprivation alone, and rapamycin treatment alone did not significantly affect lipid peroxidation (Figure 3D and E).
Figure 3. Prolonged rapamycin treatment leads to decrease intracellular ferritin level, enhancing ferroptosis susceptibility of HLRCC cells.
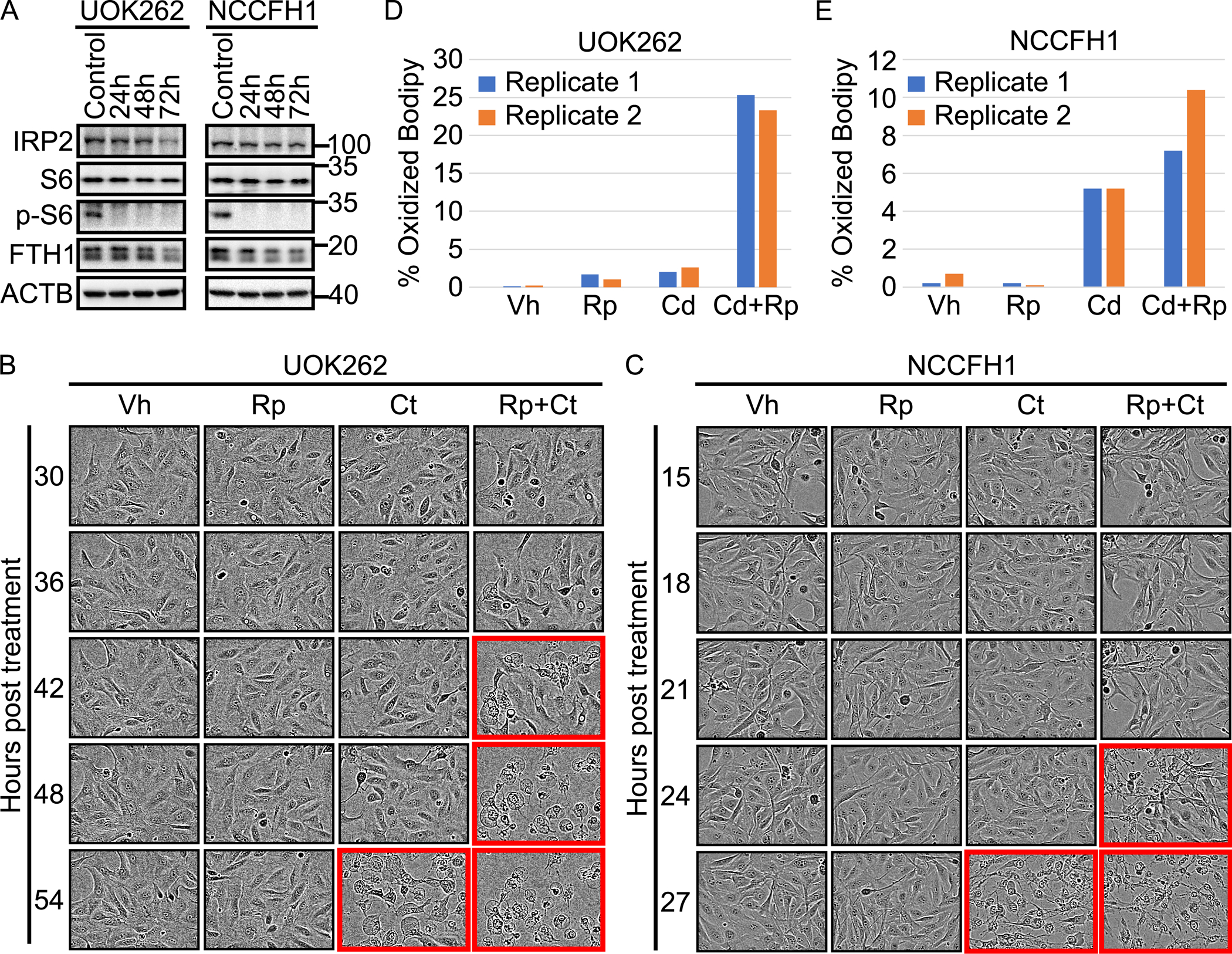
A. Prolonged rapamycin treatment decreased intracellular ferritin levels. HLRCC cell lines: UOK262 and NCCFH1 were treated with 1μM rapamycin for the indicated durations. Immunoblot for phospho-S6 showed that the rapamycin treatment inhibited MTOR signaling. Immunoblotting for ferritin heavy chain showed decreased protein levels following rapamycin treatment. There was also a moderate decrease in IRP2 level, suggesting an increase in labile iron pool. B and C. Cyst(e)inase-Rapamycin combination induces ferroptotic cell death at earlier time points than Cyst(e)inase treatment alone. HLRCC cells: UOK262 and NCCFH1 were treated with either vehible (Vh), Rapamycin (Rp)[1μM], Cyst(e)inase (Ct)[200nM], or Rapamycin+Cyst(e)inase (Rp+Ct)[1μM Rp + 200nM Ct]. Images were taken at the indicated time points using the Incucyte® live cell imaging system. Panels with red colored outline show cells undergoing ferroptotic cell death. The images shown were from a representative experiment. The entire experiment, for both cell lines, were repeated once and the repeat experiment showed consistent results. D and E. C11-Bodipy experiment showed that rapamycin enhances Cyst(e)inase-induced lipid peroxidation. Cells were treated with indicated drugs, C11-Bodipy staining were measured by flow cytometry. Data generated were analyzed and visualized using flowCore and flowWiz packages in R statistical environment. Data from two biological replicates are shown as different colored bar.
Cyst(e)inase-rapamycin combination suppressed HLRCC tumor growth in vivo
To evaluate the efficacy of rapamycin, Cyst(e)inase, and Cyst(e)inase-Rapamycin combination in vivo, we performed a xenograft study by establishing subcutaneous HLRCC tumors in the flank regions of NOD-SCID Gamma (NSG™) mice. First, we performed a pilot study to evaluate the tumorigenic potential of HLRCC cell lines, UOK262 and NCCFH1 and found that only NCCFH1 cells were tumorigenic. Due to intracellular fumarate accumulation, many proteins in HLRCC cells are highly succinated. Hence, 2-succinylcysteine (2-SC) staining is a reliable diagnostic tool to detect HLRCC tumors. (19) Accordingly, NCCFH1 derived xenograft tumors were also positively stained for 2-SC (Figure 4A, 4B). Unlike NCCFH1, UOK262 failed to grow a tumor even after 100 days post-injection (Figure S2). Therefore, we proceeded to perform a xenograft study using only NCCFH1 cells. The study consisted of 4 groups of NSG mice carrying NCCFH1 derived xenograft tumors. A total of 20 female and 20 male NSG mice were xenografted with NCCFH1 cells. Thirty three out of the 40 mice developed tumors. When the mean tumor volume reached 100mm3, the mice were randomly assigned into four different groups: vehicle (control group, n=6, 3 female & 3 male), rapamycin (rapamycin only group, n=7, 4 female & 3 male), Cyst(e)inase (Cyst(e)inase only group, n=10, 5 female & 5 male), rapamycin+Cyst(e)inase (combination group, n=10, 5 female & 5 male). Once every 3 days, mice in each group was intraperitoneally injected with either vehicle (control group), rapamycin (rapamycin only group), Cyst(e)inase (Cyst(e)inase only group), or rapamycin+Cyst(e)inase (combination group). Overall, mice in all four treatment groups did not show any adverse reaction to the treatment. However, one mouse in the vehicle control group unexpectedly passed away on day 111 post-xenografting (Supplemental Table). There was no significant change in body weight among mice in all four treatment groups (Figure S3). The xenograft study indicated that only the combination treatment group resulted in a significant tumor growth suppression when compared to control (Figure 4C). Small changes in tumor growth rate were also observed in rapamycin only (Figure 4D) and Cyst(e)inase only treatment groups (Figure 4E) when compared to the control group. However, these changes were not statistically significant. Upon terminating the study, gross necropsy of the animals found visible lung metastases in four mice: two from the control group, one from the rapamycin only group, and one from the Cyst(e)inase only group. There was no observable metastasis in the combination group. Representative lung metastases (Hematoxylin and Eosin, and 2-SC stained images) from the control group, the Cysteinase only group, and the rapamycin only group are shown in Figure S4. We did not observe any sex-dependency in the results.
Figure 4. Xenograft model shows that HLRCC tumors are more sensitive to Cyst(e)inase-Rapamycin combination than Cyst(e)inase or Rapamycin alone.
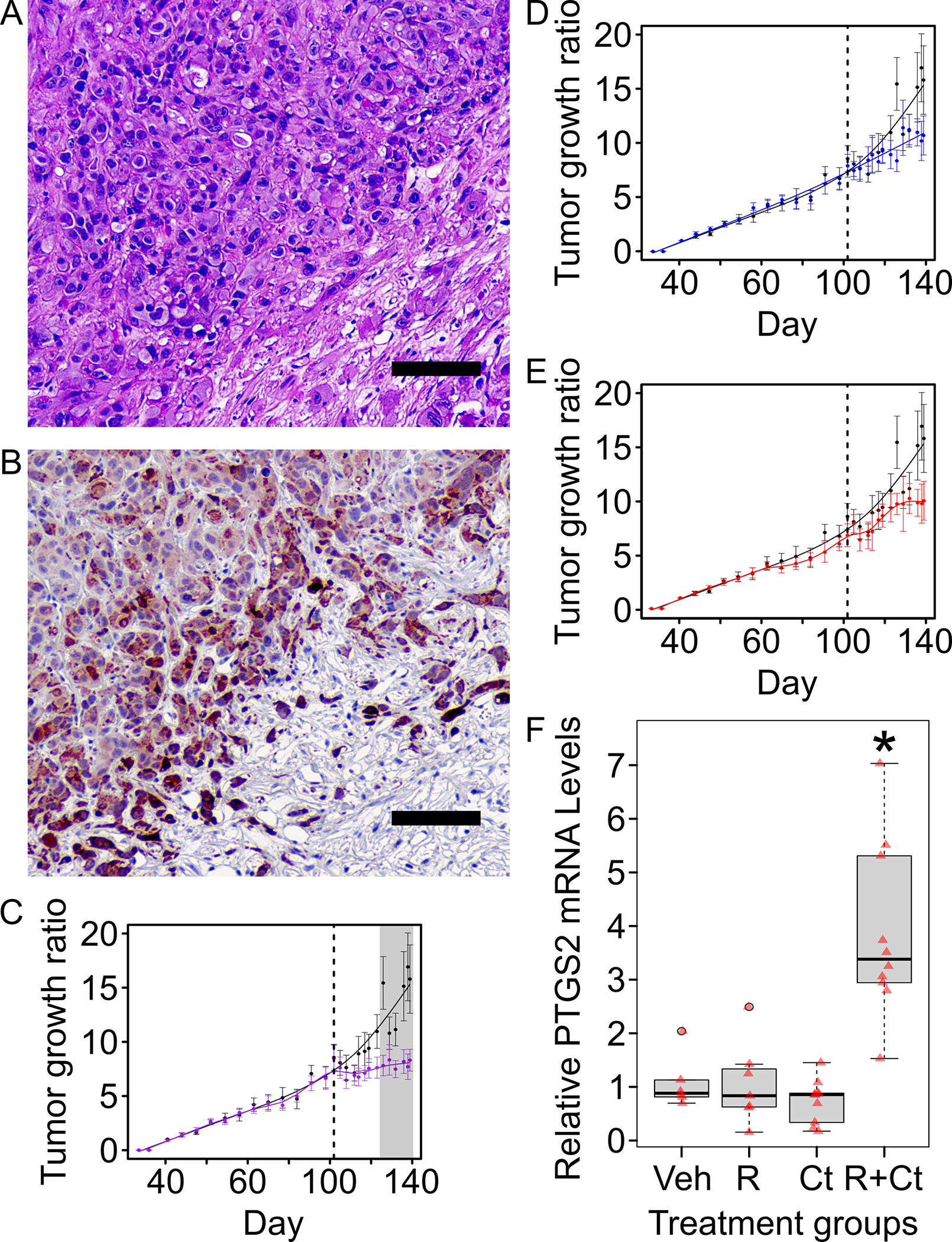
A. Hematoxylin and Eosin (H&E) staining of a representative xenograft tumor from the control group. (scale bar = 100μm) B. Immunohistochemistry staining of the same representative xenograft tumor shown in “A” for proteins with 2-succinyl-cysteine (2-SC) modification; 2-SC is a reliable marker for FH inactivation and is a marker for HLRCC tumors. (scale bar = 100μm) C. D. & E. Xenograft mouse model studies showed that Rapamycin-Cyst(e)inase combination treatment suppresses tumor growth in vivo. Ten million NCCFH1 cells suspended in a 1:1 ratio of PBS:Matrigel were injected into the right flank of each NSG mouse (equal number of male-female mice per group). Tumors were allowed to grow to an average tumor volume of 100mm3 before proceeding with the treatments. Treatment start day is indicated with a dotted vertical line. Error bars represent standard error of mean (SEM). Statistical significance was determined using analysis of variance (ANOVA) followed by Tukey Honest Significant Difference (Tukey-HSD). Grey shaded area indicate data points where treatment group is significantly different (p-value <0.05) from control group (black line). Panels C, D, and E share the same control group. C. Rapamycin-Cyst(e)inase combination treatment group (purple line) showed growth suppression when compared to the control group (black line). D. Rapamycin alone treatment group (blue line) did not show a significant growth suppression when compared to control group (black line). E. Cyst(e)inase alone treatment group (red line) did not show a statistically significant growth suppression when compared to the control group (black line). F. Rapamycin-Cyst(e)inase combination group (R+Ct) showed significant upregulation of ferroptosis marker, PTGS2 when compared to vehicle (Veh), rapamycin (R), or Cyst(e)inase (Ct) only groups. PTGS2 mRNA levels in xenograft tumors were determined using a Taqman® gene expression qPCR assay. Taqman® probe for beta-actin was used as internal loading control. Results from the qPCR was analyzed using the delta-delta-ct method with samples in the Veh group set as 1. Red colored triangles represent relative PTGS2 mRNA from individual tumor. Asterisk indicates p-value < 0.05 as compared to Veh group. P-values were calculated using Mann-Whitney U Test.
Prostaglandin-endoperoxide synthase 2 (PTGS2) is a reliable ferroptosis biomarker.(20) To evaluate whether the treatment groups were undergoing ferroptosis, we performed quantitative PCR (QPCR) to measure PTGS2 expression and found that it was significantly elevated in the combination group, while its expression in Cyst(e)inase only and rapamycin only groups remained unchanged (Figure 4F). Thus, the expression of PTGS2 suggests that the combination treatment induces ferroptosis in vivo.
Discussion
HLRCC associated renal cell carcinoma is an aggressive kidney cancer with poor prognosis. To date, there is yet a standardized, safe and effective treatment strategy for this subtype of kidney cancer.(2,3,21,22) In this study, we showed that it is possible to induce ferroptosis in an HLRCC tumor model in vivo using a combination of rapamycin and Cyst(e)inase; two therapeutics with favorable clinical and pharmacological characteristics. The combination treatment was well tolerated and resulted in tumor growth suppression. In agreement with our results, a recent study that used Cyst(e)inase to induce ferroptotic cell death in a pancreatic mouse model system also reported a similar degree of tumor growth suppression with minimal toxicity to the host.(23) Mechanistically, our approach exploits several fundamental biological aspects of HLRCC and of ferroptosis regulations: 1. HLRCC cells harbor high intracellular ferritin levels, 2. Intracellular ferritin promotes HLRCC cell proliferation, 3. HLRCC cells are susceptible to ferroptosis, and 4. Ferritin suppresses ferroptosis.(6,7,15) Therefore, the net effect is that Cyst(e)inase depletes circulatory cystine and cysteine, depriving the tumors of exogenous cysteine supply and impairing GPX4 function. Simultaneously, rapamycin promotes the release of ferritin-stored iron through autophagy induction, increasing cellular labile iron pool, suppressing HLRCC cell proliferation, and enhancing ferroptosis. Thus, the tested treatment modality represents a translation of basic HLRCC biology into in vivo preclinical study.
Although in vitro experiments showed that Cyst(e)inase could induce ferroptosis in HLRCC cell lines when used as a single agent, our in vivo experiment only detected significant tumor reduction and ferroptosis induction when combined with rapamycin. This apparent difference between the in vitro and the in vivo models is likely due to the presence of the tumor microenvironment in the in vivo system. The protective effects of tumor microenvironment against ferroptosis have been reported. For example: The expression of Fatty Acid Biding Protein-4 (FABP4) by the tumor endothelial cells has been shown to protect the cancer cells from ferroptotic cell death.(24) Other evidence of tumor microenvironment conferred protection against ferroptosis involves miR-522 containing exosomes that are secreted by cancer associated fibroblast. Once taken in by the cancer cells, miR-522 can inhibit Arachidonate Lipoxygenase 15 (ALOX15), which in turn protects the cells from undergoing ferroptosis.(25) In addition to encouraging ferritinophagy, the difference in outcomes observed in vitro vs in vivo suggests that rapamycin could circumvent the effects of the tumor microenvironment. The precise mechanism by which the tumor microenvironment contributed to our observed outcome remains unknown. Identifying the mechanism may prove to be beneficial, as circumventing it can enhance the utility of Cyst(e)inase as a cancer therapeutic for HLRCC.
Ferroptosis is an immunogenic cell death mechanism.(26) Recent studies showed that ferroptosis induces secondary immune responses, potentially leading to more robust tumor regression. Unfortunately, the xenograft model precludes the possibility of examining any potential secondary immune responses elicited by the ferroptosis induction. However, early case reports have indicated that certain cases of HLRCC are responsive to immune checkpoint inhibitor-based therapy, indicating that HLRCC tumors may be responsive to immunotherapy.(21,27) Induction of immunogenic cell death such as ferroptosis coupled with immune checkpoint therapy might give rise to more robust responses. A recent finding supports that checkpoint therapy promotes tumor ferroptosis through CD8 positive T-cell released interferon gamma, which suppresses tumor expression of system Xc components, leading to ferroptotic cell death.(28) Further advancement of research in HLRCC immunotherapy is hinged upon developing a syngeneic mouse model system to study immune mediated responses in HLRCC.
Apart from rapamycin, other approaches to pharmacologically release ferritin stored iron are through the use of iron chelators. Rapamycin induces general autophagy, which is not specific towards the release of ferritin stored iron. Unlike rapamycin, iron chelators deplete the labile iron pool, leading to the induction of ferritinophagy, a specialized form of autophagy that releases ferritin-stored iron. However, conventional iron chelators such as deferoxamine and clioquinol are not suitable for such combinations as iron chelated by these compounds cannot catalyze Fenton reactions, leading to a decrease in intracellular lipid peroxidation and ferroptosis suppression. One potential way to circumvent this is with redox-active iron chelators. Iron chelates by these chelators remains redox-active, allowing for the continued generation of radical oxygen species and lipid peroxide that drive ferroptosis. Examples of redox-active iron chelators are Dp44MT and nitrilotriacetate.(29,30) However, most of these chelators do not have favorable clinical and pharmacological characteristics. The development of a redox active iron chelator with favorable pharmacological characteristics could prove to be another important avenue for future investigation.
Overall, our results demonstrate an HLRCC treatment modality that is translatable into clinical trials. Although it remains unclear whether human HLRCC will show similar responses, past studies have shown that rapamycin treatment alone could lead to a partial response in certain cases of HLRCC. The addition of Cyst(e)inase may improve the outcomes of certain cases of HLRCC, providing additional treatment options to HLRCC patients.
Supplementary Material
Acknowledgement:
This project is funded by the funding from the National Cancer Institute 5R01-CA226920 awarded to AO. AO is a member of the University of Arizona Cancer Center (UACC) and all the animal studies were performed by the EMSR funded by Cancer Center Support grant P30-CA023074. AO is partly supported by P42-ES004940. This paper is dedicated to the memory of Hazar Kerimoglu and his grandfather Kazim Kerimoglu
Financial support:
This project is funded by the National Cancer Institute 5R01-CA226920 awarded to AO. AO is a member of the University of Arizona Cancer Center (UACC) and all the animal studies were performed by the EMSR funded by Cancer Center Support grant P30-CA023074. AO is partly supported by P42-ES004940.
Footnotes
Conflict of Interest disclosure statement:
E. Stone is an inventor of intellectual property related to Cyst(e)inase and has equity interest in Aeglea Biotherapeutics, a company pursuing the commercial development of this technology. All other authors declare no potential conflicts of interest.
References:
- 1.Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, et al. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc Natl Acad Sci U S A 2001;98(6):3387–92 doi 10.1073/pnas.051633798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Menko FH, Maher ER, Schmidt LS, Middelton LA, Aittomaki K, Tomlinson I, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer 2014;13(4):637–44 doi 10.1007/s10689-014-9735-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ooi A Advances in hereditary leiomyomatosis and renal cell carcinoma (HLRCC) research. Semin Cancer Biol 2020;61:158–66 doi 10.1016/j.semcancer.2019.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002;30(4):406–10 doi 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 5.Ooi A, Wong JC, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, et al. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 2011;20(4):511–23 doi 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 6.Kerins MJ, Milligan J, Wohlschlegel JA, Ooi A. Fumarate hydratase inactivation in hereditary leiomyomatosis and renal cell cancer is synthetic lethal with ferroptosis induction. Cancer Sci 2018;109(9):2757–66 doi 10.1111/cas.13701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kerins MJ, Vashisht AA, Liang BX, Duckworth SJ, Praslicka BJ, Wohlschlegel JA, et al. Fumarate Mediates a Chronic Proliferative Signal in Fumarate Hydratase-Inactivated Cancer Cells by Increasing Transcription and Translation of Ferritin Genes. Mol Cell Biol 2017;37(11) doi 10.1128/MCB.00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol 2016;26(3):165–76 doi 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cramer SL, Saha A, Liu J, Tadi S, Tiziani S, Yan W, et al. Systemic depletion of L-cyst(e)ine with cyst(e)inase increases reactive oxygen species and suppresses tumor growth. Nat Med 2017;23(1):120–7 doi 10.1038/nm.4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Choueiri TK, Escudier B, Powles T, Mainwaring PN, Rini BI, Donskov F, et al. Cabozantinib versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373(19):1814–23 doi 10.1056/NEJMoa1510016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, et al. Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. Lancet 2008;372(9637):449–56 doi 10.1016/S0140-6736(08)61039-9. [DOI] [PubMed] [Google Scholar]
- 12.Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet 2011;378(9807):1931–9 doi 10.1016/S0140-6736(11)61613-9. [DOI] [PubMed] [Google Scholar]
- 13.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet 2010;196(1):45–55 doi 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perrier-Trudova V, Huimin BW, Kongpetch S, Huang D, Ong P, Le Formal A, et al. Fumarate Hydratase-deficient Cell Line NCCFH1 as a New In Vitro Model of Hereditary Papillary Renal Cell Carcinoma Type 2. Anticancer Res 2015;35(12):6639–53. [PubMed] [Google Scholar]
- 15.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149(5):1060–72 doi 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016;12(8):1425–8 doi 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Salahudeen AA, Thompson JW, Ruiz JC, Ma HW, Kinch LN, Li Q, et al. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009;326(5953):722–6 doi 10.1126/science.1176326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vashisht AA, Zumbrennen KB, Huang X, Powers DN, Durazo A, Sun D, et al. Control of iron homeostasis by an iron-regulated ubiquitin ligase. Science 2009;326(5953):718–21 doi 10.1126/science.1176333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buelow B, Cohen J, Nagymanyoki Z, Frizzell N, Joseph NM, McCalmont T, et al. Immunohistochemistry for 2-Succinocysteine (2SC) and Fumarate Hydratase (FH) in Cutaneous Leiomyomas May Aid in Identification of Patients With HLRCC (Hereditary Leiomyomatosis and Renal Cell Carcinoma Syndrome). Am J Surg Pathol 2016;40(7):982–8 doi 10.1097/PAS.0000000000000626. [DOI] [PubMed] [Google Scholar]
- 20.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156(1–2):317–31 doi 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Iribe Y, Furuya M, Shibata Y, Yasui M, Funahashi M, Ota J, et al. Complete response of hereditary leiomyomatosis and renal cell cancer (HLRCC)-associated renal cell carcinoma to nivolumab and ipilimumab combination immunotherapy by: a case report. Fam Cancer 2020. doi 10.1007/s10689-020-00195-0. [DOI] [PubMed] [Google Scholar]
- 22.Park I, Shim YS, Go H, Hong BS, Lee JL. Long-term response of metastatic hereditary leiomyomatosis and renal cell carcinoma syndrome associated renal cell carcinoma to bevacizumab plus erlotinib after temsirolimus and axitinib treatment failures. BMC Urol 2019;19(1):51 doi 10.1186/s12894-019-0484-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 2020;368(6486):85–9 doi 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luis G, Godfroid A, Nishiumi S, Cimino J, Blacher S, Maquoi E, et al. Tumor resistance to ferroptosis driven by Stearoyl-CoA Desaturase-1 (SCD1) in cancer cells and Fatty Acid Biding Protein-4 (FABP4) in tumor microenvironment promote tumor recurrence. Redox Biol 2021;43:102006 doi 10.1016/j.redox.2021.102006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang H, Deng T, Liu R, Ning T, Yang H, Liu D, et al. CAF secreted miR-522 suppresses ferroptosis and promotes acquired chemo-resistance in gastric cancer. Mol Cancer 2020;19(1):43 doi 10.1186/s12943-020-01168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang D, Kepp O, Kroemer G. Ferroptosis becomes immunogenic: implications for anticancer treatments. Oncoimmunology 2020;10(1):1862949 doi 10.1080/2162402X.2020.1862949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yonese I, Ito M, Takemura K, Kamai T, Koga F. A Case of Metastatic Hereditary Leiomyomatosis and Renal Cell Cancer Syndrome-Associated Renal Cell Carcinoma Treated with a Sequence of Axitinib and Nivolumab Following Cytoreductive Nephrectomy. J Kidney Cancer VHL 2020;7(2):6–10 doi 10.15586/jkcvhl.2020.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature 2019;569(7755):270–4 doi 10.1038/s41586-019-1170-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jansson PJ, Hawkins CL, Lovejoy DB, Richardson DR. The iron complex of Dp44mT is redox-active and induces hydroxyl radical formation: an EPR study. J Inorg Biochem 2010;104(11):1224–8 doi 10.1016/j.jinorgbio.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 30.Jiang D, Li X, Williams R, Patel S, Men L, Wang Y, et al. Ternary complexes of iron, amyloid-beta, and nitrilotriacetic acid: binding affinities, redox properties, and relevance to iron-induced oxidative stress in Alzheimer’s disease. Biochemistry 2009;48(33):7939–47 doi 10.1021/bi900907a. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available within the article and its supplementary data files.