

Abstract
The relationship between the checkpoint kinase Chk1 and the STAT3 pathway was examined in multiple myeloma (MM) cells. Gene expression profiling of U266 cells exposed to low (nM) Chk1 inhibitor (PF-477736) concentrations revealed STAT3 pathway-related gene down-regulation (e.g., BCL-XL, MCL-1, c-Myc), findings confirmed by RT-PCR. This was associated with marked inhibition of STAT3 Tyr705 (but not Ser727) phosphorylation, dimerization, nuclear localization, DNA binding, STAT3 promoter activity by ChIP assay, and down-regulation of STAT-3-dependent proteins. Similar findings were obtained in other MM cells and with alternative Chk1 inhibitors (e.g., prexasertib, CEP3891). While PF did not reduce GP130 expression or modify SOCS or PRL-3 phosphorylation, the phosphatase inhibitor pervanadate antagonized PF-mediated Tyr705 dephosphorylation. Significantly, PF attenuated Chk1-mediated STAT3 phosphorylation in in vitro assays. SPR analysis suggested Chk1/STAT3 interactions and PF reduced Chk1/STAT3 co-immunoprecipitation. Chk1 CRISPR knockout or shRNA knockdown cells also displayed STAT3 inactivation and STAT-3-dependent protein down-regulation. Constitutively active STAT3 diminished PF-mediated STAT3 inactivation and down-regulated STAT3-dependent proteins while significantly reducing PF-induced DNA damage (γH2A.X formation) and apoptosis. Exposure of cells with low basal phospho-STAT3 expression to IL-6 or human stromal cell conditioned medium activated STAT3, an event attenuated by Chk1 inhibitors. PF also inactivated STAT3 in primary human CD138+ MM cells and tumors extracted from an NSG MM xenograft model while inhibiting tumor growth.
Introduction
Signal transducer and activator of transcription 3 (STAT3) is a transcription factor that regulates the expression of diverse genes implicated in tumorigenesis, proliferation, and survival, particularly in the case of hematologic malignancies such as multiple myeloma (MM) 1,2. STAT3 activation has been identified in up to 50% of patients with MM and is associated with a poor prognosis 3. STAT3 responds to diverse growth factors and cytokines (e.g., IL-6) which activate receptor and non-receptor tyrosine kinases such as Janus-activating kinases1/2 (JAK1/2). This leads to phosphorylation of the STAT3 transactivation domain (TAD) on Tyr705, necessary for dimerization and nuclear translocation, culminating in DNA binding 4. STAT3 is also activated by G-protein coupled receptors (GPCRs) and Toll-like receptors (TLRs) 4. Other pathways e.g., MEK1/2/ERK1/2 have been linked to STAT3 Ser727 phosphorylation, which increases transcriptional activity 5,6.
Activation of STAT3 triggers expression of multiple pro-survival and –proliferation genes including MCL-1, BCL-XL, and c-Myc, among others 7. Notably, STAT3 hyper-activation has been implicated in drug resistance 8, particularly micro-environmental forms of resistance 9. While the development of inhibitors of STAT3, like other transcription factors, has been challenging, JAK inhibitors are currently approved for the treatment of patients with myelofibrosis and JAK2 mutations. Although pre-clinical evidence suggests a possible role for such agents in MM 10, the existence of JAK-independent pathways of STAT3 activation may limit the effectiveness of this approach. Other strategies to disrupt STAT3 function include the use of peptidomimetics, anti-sense oligonucleotides, decoy oligonucleotides 4, or small molecule inhibitors/degraders e.g., Stattic, atovaquone 11, SD-36 (PROTAC) 12 However, drug delivery and pharmacokinetic issues continue to represent barriers to successful STAT3 targeting 13.
The Chk1 kinase represents a critical component of the DNA damage response (DDR) 14. Upon genotoxic injury and development of single-strand breaks, Chk1 is activated by ATR (ataxia telangiectasia-mutated- and Rad3-related kinase) and phosphorylates the Cdc25C and Cdc25A phosphatases, leading to their ubiquitination and degradation 15. This promotes inhibitory phosphorylation of cdc2 (CDK1) on Tyr15 and Thr14 sites, allowing cells to arrest in G2 and repair DNA damage 16. Chk1 inhibition spares CDC25A/C, induces “inappropriate” dephosphosphorylation/activation of CDC2, loss of the intra-S-phase checkpoint, increased replication stress, and progression of cells through G2 into mitosis, leading to mitotic catastrophe and cell death 17. In extensive preclinical studies, Chk1 inhibitors synergistically increase the anti-tumor activity of diverse genotoxic agents in vitro and in vivo 18, including in MM 19, prompting the development of multiple clinical Chk1 inhibitor candidates 20. To date, essentially all Chk1 inhibitor strategies have involved combination with genotoxic drugs 21. However, results of such studies have been equivocal, perhaps reflecting the inability of this strategy to improve the therapeutic index of DNA-damaging agents 22. On the other hand, recent attention has focused on novel functions of Chk1 in neoplastic cell survival, proliferation, and DNA repair 17, raising the possibility of alternative Chk1 inhibitor-based approaches in various malignancies.
To date, little information is available concerning the functional relationship between Chk1 and STAT3, although in non-transformed cells, Chk1 has been implicated in STAT3 activation following ischemic injury 23. However, the relationship between Chk1 and STAT3 activation in neoplastic cells in general, and in MM cells in particular, remains largely unexplored. To address this question, we performed gene expression profiling of MM cells exposed to established Chk1 inhibitors. These studies revealed that Chk1 inhibitors, administered at very low (e.g., nM) concentrations, sharply attenuated the STAT3 gene expression profile. Moreover, using both pharmacologic and genetic approaches, we demonstrate that Chk1 inhibition markedly diminishes STAT3 Tyr705 phosphorylation and DNA binding activity accompanied by down-regulation of STAT3-dependent genes (e.g., MCL-1, BCL-XL, c-Myc), events shown to contribute functionally to Chk1 inhibitor lethality. Significantly, STAT3 inactivation by Chk1 inhibition was observed in the presence of IL-6 or stromal cell factors. Collectively, these findings highlight a heretofore unrecognized link between the Chk1 and STAT3 pathways in MM, and identify Chk1 inhibitors as novel and potent candidate STAT3 antagonists.
Materials and Methods
Cell lines and reagents
Human MM cell lines U266, RPMI8226, OPM2, MM1.S, NCI-H929, KMS28BM, KMS28PE, ANBL-6 (1 ng/ml IL-6), and KAS-6/1 (1 ng/ml IL-6) cells were maintained in RPMI-1640 supplemented with 10% fetal bovine serum and penicillin-streptomycin. Bortezomib-resistant cells, U266/PS-R was established and maintained as described previously 24. Human marrow stroma cells (HS-5) were cultured in 10% fetal bovine serum and penicillin-streptomycin.
For information regarding PF-477736 (PF), CEP-3891 (CEP), prexasertib (LY2606368, LY), Cryptotanshinone (Cry), BBI-608 (BBI), TTI-101 (TTI), Stattic, Atovaquone (AQ), Pervanadate (PV), Ruxolitinib (Rux), Recombinant Human Interleukin 6 (IL-6), and plasmids, see Supplemental Table 1.
Human Signal Transduction PathwayFinder RT2 Profiler PCR Array
Total RNA was separated by RNeasy Mini Kit then converted to cDNA using an RT2 First Strand Kit (Qiagen) according to the manufacturer’s instructions. Gene expression profiling was conducted using the Human Signal Transduction PathwayFinder RT2 Profiler PCR Array (Cat. No. PAHS-014Z, Qiagen, SABiosciences, Valencia, CA, USA). Quantitative reverse transcriptase PCR (qRT-PCR) was conducted using an ABI 7900HT Real-time PCR Instrument (Applied Biosystems, Foster City, CA, USA) following the array manufacturer’s instructions. Relative gene expression was determined using the ΔΔCT method. Data was generated using the Qiagen data analysis website.
Surface Plasmon Resonance (SPR)
SPR binding experiments were carried out at 25 °C on a Reichert Technologies (Depew, NY) SR7500DC two-channel system equipped with CM5 chip using D-PBS with 0.05% Tween-20 and 5 μM ATP as the running buffer. Chk1 protein (MW~59000, SignalChem catalog No.C47-10H-100) was injected onto the surface on the left channel and captured to a level of 1727 μRIU, whereas the right channel was to serve as a reference for nonspecific binding of STAT3. Under a flow rate of 25 μl/min, STAT3 protein (MW~95000, SignalChem catalog No. S54-54BH) was injected over both channels at concentrations of 0.1, 0.2, 0.45, 0.9 and 2 μM, respectively, for 6 min followed by a 6 min dissociation in running buffer. The kinetic data were analyzed using TraceDrawer (Ridgeview Instruments) with a one to two model, while the equilibrium data were fitted using Scrubber (Biologic Software) to a one to two binding model.
Immunofluorescence and Flow Cytometry using ImageStream
Cells were fixed in 4% paraformaldehyde for 15 min at room temperature, and permeabilized by incubation in 90% methanol on ice for 30 minutes. Cells were incubated with primary antibodies for 1 hour at room temperature: 1:200 anti-phospho-STAT3(Tyr705) (Cell Signaling Technology Cat# 9145, RRID:AB_2491009), 1:400 anti-phospho-STAT3 (Ser727) (Novus Cat# IC4934U), 1:400 anti-STAT3 (Santa Cruz Biotechnology Cat# sc-8019, RRID:AB_628293), 1:400 anti-Chk1 (GeneTex Cat# GTX83385, RRID:AB_11175647), 1:10 CD-138-PE (Miltenyi Biotec Cat# 130-117-395); and secondary antibodies at room temperature, in the dark for 30 minutes: 1:1000 Alexa Fluor® 488 (Life Technology Cat# A11070) and Alexa Fluor® 647 (Life Technologies Cat# A21235); 1: 500 APC (Invitrogen Cat# 31984). Cells were also stained with DAPI 1:100 for 5 minutes before imaging. The cells were washed in 1000 μl of PBS-2%FBS and recovered by centrifugation at 300 g for 5 min; and the incubation buffer for the antibodies was made from 5% BSA and 0.3% TritonX dissolved in PBS (phosphate-buffered saline). Cells were resuspended in 60 μl of PBS-2%FBS and analyzed with an ImageStreamX (Amnis) image flow cytometer.
Chk1 Kinase Assay (radioactive and non-radioactive)
Kinase assays were performed using recombinant full-length human GST-STAT3 (SignalChem C47-10H) together with recombinant full-length human His-Chk1 (SignalChem S54-54G), in kinase buffer (20 mM Hepes pH 7.4, 10 mM MgCl2, 10 mM MgCl2, 1 mM EGTA, 0.1 mM Na3VO4, 0.5 mM NaF, 50 mM β-glycerophosphatase, 50 mM DTT) in the presence of 50 μM ATP and 5 μCi [32P]-ATP (EasyTide). The kinase reaction proceeded for 45 min at 30 °C, and was stopped by the addition of NuPAGE™ LDS Sample Buffer (4X) (Invitrogen NP0007), boiled for 10 min at 85°C, and analyzed by electrophoresis on 4-12% SDS-polyacrylamide gel. Gels were stained with Coomassie Brilliant Blue (CBB), then fixed, dried and subjected to autoradiography. A non-radioactive Kinase Assay was performed without cold ATP. Kinase products were analyzed by electrophoresis on 4-12% SDS-polyacrylamide gel and transferred to polyvinylidene difluoride membranes, which were further stained Ponceau S. Immunoblotting was analyzed with rabbit anti-phospho-STAT3 (Tyr705, Cell Signaling Technology Cat# 2577), rabbit anti-phospho-STAT3 (Ser727, Cell Signaling Technology Cat# 9134) and rabbit anti-phospho-Chk1 (Ser296, Cell Signaling Technology Cat# 2349). Images were quantified and analyzed using ImageJ software (ImageJ, RRID:SCR_003070).
Isolation of primary MM cells
All studies were obtained with written informed consent from MM patients undergoing routine diagnostic aspirations. These studies were conducted in accordance with recognized ethical guidelines (e.g., the Declaration of Helsinki), and were approved by the Virginia Commonwealth Institutional Review Board (IRB #MCC-8712-3A; MCC-02447; MCC-03340).
For MM primary cell analysis, mononuclear cells were isolated from MM patient bone marrows using the Ficoll Histopaque method (#10771, Sigma-Aldrich, USA). CD138+ and CD138− cells were separated using an MS+/LS+ column and a magnetic separator according to the manufacturer’s instructions (Miltenyi Biotecafter).
Statistical analysis
Values represent the means ±SD for three separate experiments. The significance of differences between experimental variables was determined using the Student’s t-test. Values were considered statistically significant at *, P < 0.05; **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Animal studies
All animal studies were IACUC approved and performed in accordance with AAALAC, USDA, and PHS guidelines. NOD-SCID IL2Rgammanull mice (Jackson Laboratories, Bar Harbor, ME) were subcutaneously injected with 5 × 106 U266 cells into the flank. The Chk1 inhibitor PF-477736 (Abmole, M1764) was prepared in a 1:1 (v/v) mixture of PBS and dimethyl sulfoxide at a concentration of 10.0 mg/ml. When tumors grew to 300 mm3, PF (15 mg/kg per day) was administrated (i.p.) for 3 days. Control animals received equal volumes of vehicle. Tumors were collected to perform immunoblotting and immunohistochemistry staining.
For additional information and methods, see Supplementary Materials and Methods, and Supplemental Table 1.
Results
In view of emerging evidence of non-canonical roles for Chk1 25, gene expression profiling was performed on U266 MM cells exposed (16 hr) to a low (100 nM) concentration of the Chk1 inhibitor PF-477736 26 (Fig 1A). These cells were selected because of their relatively high basal STAT3 activation and pronounced sensitivity to the pro-apoptotic effects of PF (Suppl. Fig S1A, upper panel, and B). Lower basal STAT3 activation was observed in MM1.S, H929, 8226, and OPM2 cells, which was sharply increased by HS-5 conditioned medium (Suppl. Fig S1A, lower panel). Marked reductions (e.g., > 2-fold) in gene expression profiling of multiple STAT3 pathway genes (e.g., MCL-1, BCL-XL, c-Myc) were observed in U266 cells exposed to PF (Fig 1A), results confirmed by quantitative PCR array at both short e.g., 4 hr and longer e.g., 16 hr intervals. (Fig 1B). Parallel reductions in mRNA expression were also observed in IL-6-dependent KAS/6-1 cells exposed (2 hr) to PF or the Chk1 inhibitor CEP3891 27 (Suppl. Fig S1C–E). These results indicate that pharmacologic Chk1 inhibitors repress the transcription of STAT3-dependent genes in MM cells.
Figure 1. Chk1 inhibitors block tyrosine phosphorylation of STAT3 (p-Y705) in MM cells.
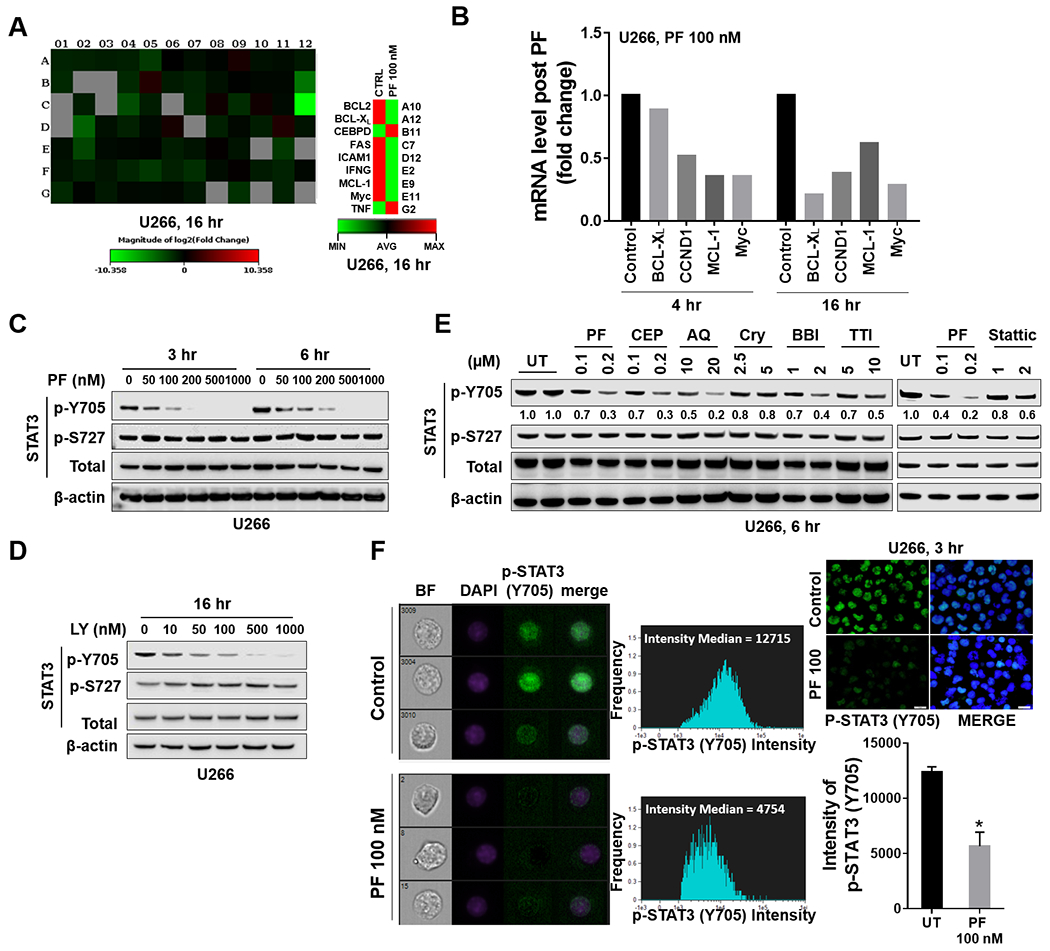
A, U266 cells were treated with 100 nM PF-477736 (PF) for 4 and 16 hr. Gene expression profiling was performed using Human Signal Transduction PathwayFinder R2 PCR Array (84 genes). The heat map shows absolute mRNA copy numbers, which were calculated from PCR cycle thresholds (Ct) and generated from Qiagen RT² Profiler PCR Data Analysis Web page. B, Fold-change of STAT3 pathway-related genes upon PF stimulation in U266 cells as compared to untreated controls. C-E, Western blot analysis of p-Y705 STAT3, p-S727 STAT3, and total STAT3 in U266 cells treated with indicated doses of PF, LY, or other STAT3 inhibitors. CEP = CEP-3891; AQ = Atovaquone; Cry = Cryptotanshinone; BBI = BBI-608; TTI = TTI-101. β-actin controls were assayed to ensure equivalent loading and transfer. Images were quantified and analyzed by using ImageJ software. Data was normalized by the ratio of indicated protein and β-actin vs control. F, Untreated and treated (PF 100 nM, 3 hr) cells were stained with p-Y705 STAT3 and DAPI, and then visualized by fluorescence microscope and by ImageStream and representative cells are shown (BF = brightfield; Scale bar = 20 μM). Histogram of p-Y705 STAT3 intensity and fold change were shown. Representative data of at least three replicates. *, P < 0.05.
Western blot analysis revealed dose-dependent down-regulation of STAT3 Y705 (but not S727) phosphorylation in U266 cells exposed to PF (Fig 1C) or the Chk1 inhibitor LY2603618 (Fig 1D). More detailed time course studies demonstrated prolonged Y705 dephosphorylation in U266 cells by PF e.g., up to 48 hr (Suppl Fig S1F). Notably, comparison studies demonstrated that low (e.g.,100-200 nM) concentrations of PF or CEP were as or more potent than 1-log (Cry, BBI, TTI, Stattic) or 2-log (AQ; atovaquone) higher concentrations of other known STAT3 inhibitors (Fig 1E). Finally, ImageStream flow cytometry demonstrated a significant reduction of DAPI and p-Y705 nuclear co-staining in PF-treated U266 cells (Fig 1F). Similar results were obtained in U266 cells treated with CEP (Suppl. Fig S1G). These findings indicate that Chk1 inhibitors block STAT3-Y705 nuclear translocation in MM cells.
Attempts were then made to replicate these results in other cell types, including those displaying relatively low basal p-Y705 STAT3 activation stimulated by IL-6 or HS-5 (human stroma cell)-conditioned medium (CM). Exposure of IL-6-dependent KAS/6-1 cells to very low concentrations of PF or CEP (50-300 nM) robustly reduced STAT3 Y705 phosphorylation by western blot analysis but had minimal effects on STAT3 S727 (Fig 2A, B). As in the case of U266 cells, PF and CEP were 1-2 logs more potent than other STAT3 inhibitors e.g., Cry 2.5 μM 28 or AQ in diminishing Y705 phosphorylation (Fig 2B) in KAS/6-1 as well as in bortezomib-resistant U266 cells (PS-R; Fig 2C) 24. Analogously, culture of OPM2 cells with CM or IL-6 sharply increased STAT3 Y705 phosphorylation but had lesser effects on S727 (Fig 2D, E). Co-administration of PF diminished both CM- and IL-6-mediated STAT3 Y705 phosphorylation in these cells. Finally, ImageStream analysis confirmed the ability of PF and LY to block Y705 nuclear expression in CM- or IL-6-stimulated OPM2 cells (Fig 2F).
Figure 2. Chk1 inhibitors block tyrosine phosphorylation of STAT3 (p-Y705) in multiple MM cell lines.
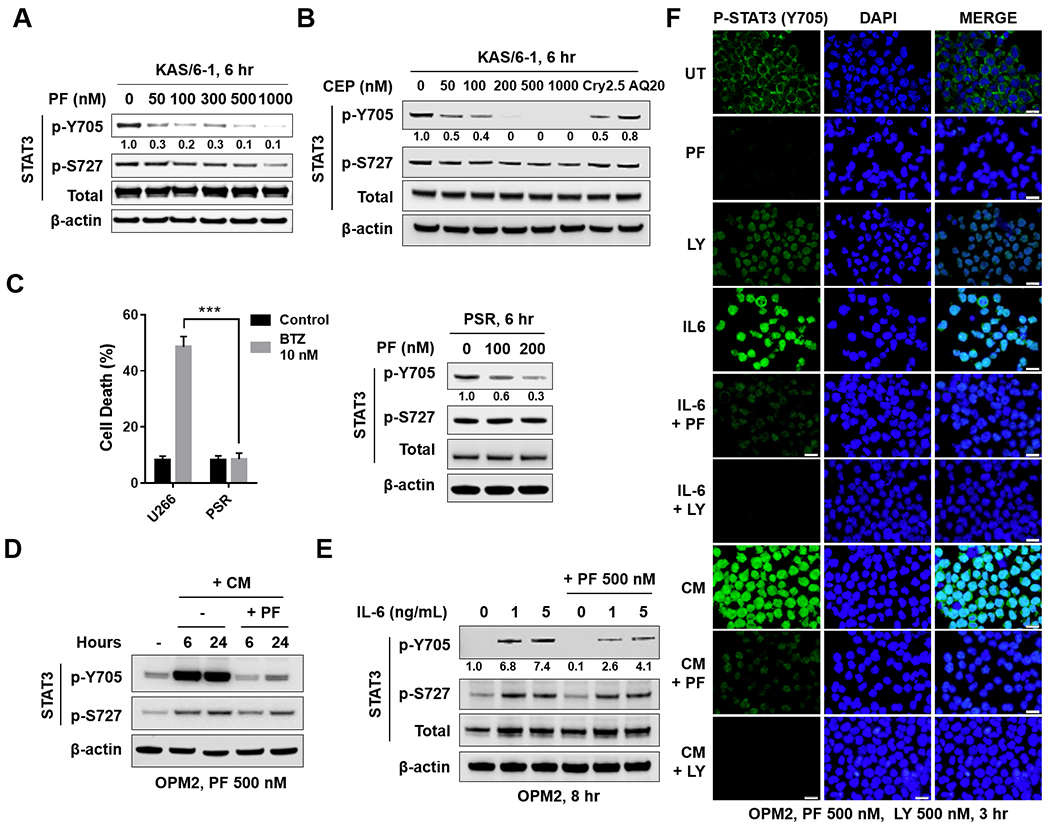
A, B and C (right panel), KAS/6–1, or PSR cells were treated with the indicated concentrations of PF, CEP, Cry 2.5 μM, or AQ 20 μM for 6 hr. Western blot analysis of p-Y705 STAT3, p-S727 STAT3, and total STAT3 were performed. β-actin controls were assayed to ensure equivalent loading and transfer. C (left panel), Bortezomib-resistant PS-R cells were exposed (24 hr) to 10 nM bortezomib (BTZ), followed by flow cytometry to monitor the percentage of apoptotic (7-AAD+) cells. Values represent the means ± S.D. for three experiments performed in triplicate. *** = P < 0.001. D, OPM2 cells were pretreated with HS-5 conditioned medium for 16 hr, and then treated with 500 nM PF for 6 and 24 hr. Western blot analysis was carried out to as mentioned in A-C. E, OPM2 cells were pretreated with IL-6 (1 ng/ml or 5 ng/ml) for 15 min, and then treated with 500 nM PF for 8 hr. Western blot analysis was carried out as in A-C. Images were quantified and analyzed using ImageJ software. Data was normalized by the ratio of indicated protein/β-actin vs control. F, OMP2 cells were pretreated with CM or IL-6 as in D and E, Immunofluorescence staining was performed to monitor p-Y705 STAT3.
Subsequent studies were undertaken to characterize further events involved in Chk1 inhibitor-mediated inactivation of STAT3. Consistent with its capacity to antagonize STAT3 Y705 phosphorylation, PF clearly diminished STAT3 dimerization in U266 cells at both 3 and 6 hr in a concentration-dependent manner (Fig 3A). Electrophoretic mobility shift assays (EMSA) assays demonstrated that PF reduced STAT3-DNA binding in these cells at 6 and 16 hr (Fig 3B). In addition, PF, LY, and CEP significantly reduced STAT3 binding activity at 6 or 16 hr (Fig 3C). In parallel, both PF and CEP significantly diminished STAT3 binding activity in CM-treated OPM2 cells (Suppl. Fig S2A, B).
Figure 3. Chk1 inhibitors disrupt STAT3 activation in MM cells.
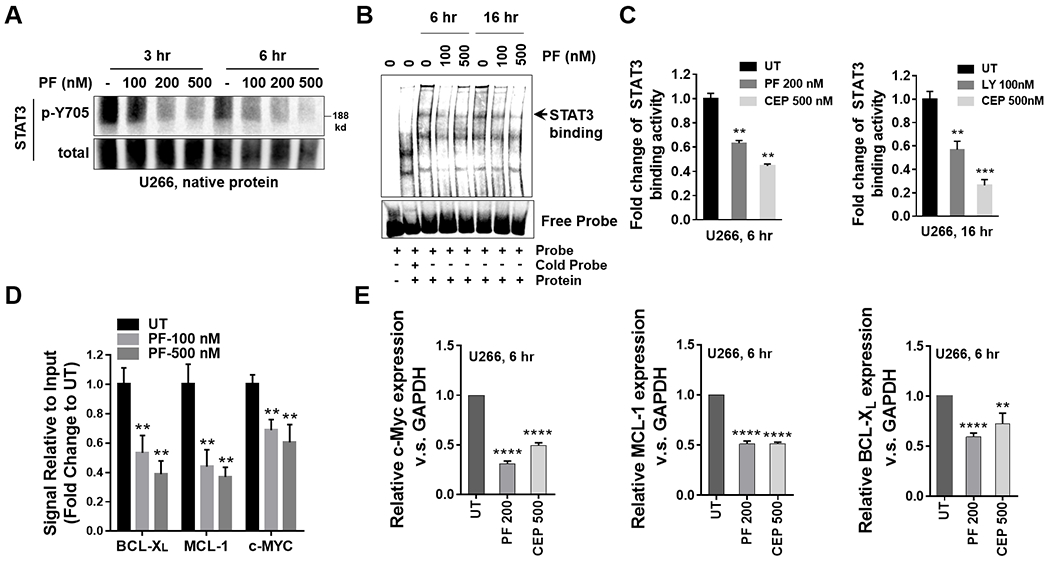
A, U266 cells were treated with indicated concentrations of PF for 3 and 6 hr. Native nuclear protein extracts were prepared and were separated on a 6% native PAGE gel. Electrophoresis was performed in the absence of SDS. Proteins were immunoblotted with p-Y705 STAT3 and total STAT3. Total STAT3 served as a loading control. B, Nuclear extracts from U266 cells treated with the indicated concentrations of PF for 6 and 16 hr were subjected to EMSA analysis. C, U266 cells were treated with indicated concentrations of PF, CEP or LY for 6 and 16 hr. A STAT3 DNA-binding ELISA assay was employed to evaluate the ability of STAT3 in cellular nuclear extracts to bind to its corresponding consensus sequence that had been immobilized on a 96-well plate. D, U266 cells were treated with the indicated concentrations of PF for 16 hr. ChIP experiments were performed to detect the presence of STAT3 at the promotors of the BCL-XL, MCL-1, and c-Myc genes after treatment with PF. Data was normalized with non-immune IgG (negative control) and STAT3 antibody, and the relative fold- change (vs untreated controls; UT) is presented. E, Relative mRNA expressions of c-Myc, MCL-1 and BCL-XL were determined by real-time reverse transcription-PCR analysis. GAPDH served as an internal control. **, P < 0.01; ***, P < 0.001; ****, P < 0.0001.
Based upon these findings, ChIP assays were performed to assess the effects of PF on binding of STAT3 to the promoters of STAT3 downstream target genes. Treatment of cells with PF (100 or 500 nM) significantly reduced STAT3 binding to c-Myc, BCL-XL, and MCL-1 promoters (Fig 3D). Consistently, both PF (200 nM) and CEP (500 nM) significantly diminished c-Myc, BCL-XL, and MCL-1 mRNA levels in U266 cells (Fig 3E), as well as in IL-6-dependent KAS/6.1 cells (Suppl. Fig S2C).
Lastly, STAT3 promoter-reporter transcriptional activity was investigated in a 293t cell reporter system. Exposure of cells to IL-6 sharply increased STAT3 transcriptional activity, and this was dramatically blocked by very low concentrations of PF (e.g., 50 nM), which was more effective than 2-log higher concentrations of AQ (Suppl. Fig S2D). Similar results were seen with CEP and LY (Suppl. Fig S2E, F).
Attempts were then made to investigate the mechanism(s) by which Chk1 inhibition disrupted STAT3 signaling. To assess possible physical interactions between Chk1 and STAT3, surface plasmon resonance (SPR) utilizing recombinant STAT3 and Chk1 was employed. As shown in Fig 4A, a concentration-dependent increase in binding of STAT3 to Chk1 was observed. Furthermore, immunoprecipitation analysis revealed that administration of PF (200 nM; 16 hr) to U266 cells reduced the amount of total STAT3 co-immunoprecipitating with Chk1 in both nuclear and cytoplasmic fractions, and of p-Y705 STAT3 associating with Chk1 in the nuclear fraction (Fig 4B), raising the possibility that direct interactions may be involved in the Chk1/STAT3 nexus.
Figure 4. Mechanisms of STAT3 inhibition by Chk1 inhibitors.
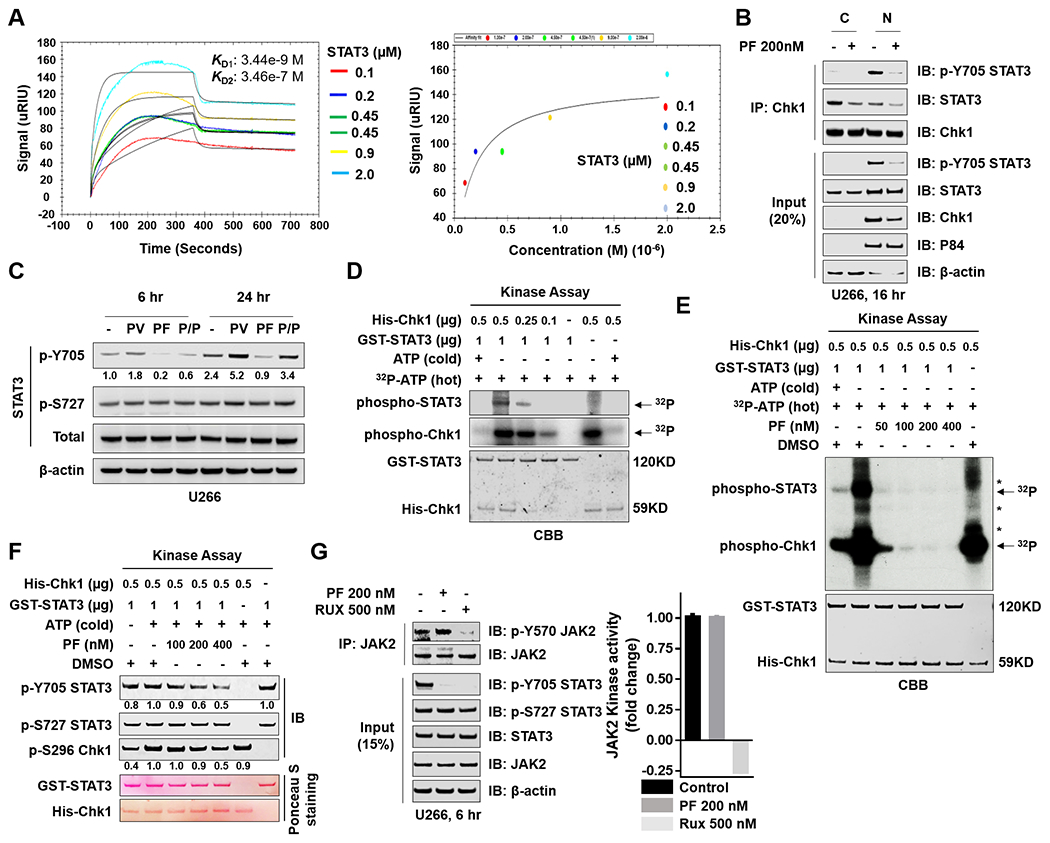
A, Surface plasmon resonance (SPR) analyses is shown to monitor Chk1 and STAT3 interactions. SPR characterizes the interaction between Chk1 and STAT3 by detecting the soluble STAT3 (analyte) binding to immobilized Chk1 on a carboxymethyl dextran sensor chip. The STAT3 was injected over the Chk1 surface in a series of concentrations (0.1, 0.2, 0.45, 0.45, 0.9 and 2 μM that are labeled with different colors). (Left panel) The data were fit to a 1:2 binding model in a kinetic analysis evaluation graph; the black lines show the global 1:2 model fit). The binding constants are reported in the insets. (Right panel) Equilibrium fit plot displays the STAT3 concentration-response curve between Chk1 and STAT3 obtained from affinity SPR analysis. B, U266 cells were treated with 200 nM PF for 16 hr. Nuclear and cytoplasmic extracts were collected for performance of co-immunoprecipitation assays. Chk1 pull-down proteins were immunoblotted with p-Y705 STAT3, total STAT3, and Chk1. Total lysates were immunoblotted for p-Y705 STAT3, total STAT3, and Chk1. P84 and β-actin were used as controls for nuclear and cytoplasmic protein levels, respectively. C, U266 cells were treated with 100 μM pervanadate (PV) and 50 nM PF for 6 hr and 24 hr. Western blot analysis of p-Y705 STAT3, p-S727 STAT3, and total STAT3 were then performed. β-actin controls were assayed to ensure equivalent loading and transfer. D and E, Purified His-Chk1 was used in an in vitro kinase assay using purified GST-STAT3 as substrate. Kinase assay products were separated by SDS-PAGE (6–12%) and visualized by autoradiography (P32) and Coomassie-brilliant blue (CBB). F, A kinase assay was performed as in E in the presence or absence of cold ATP, and kinase assay products were transferred to a nitrocellulose membrane and immunoblotted with p-Y705 STAT3, p-S727 STAT3, and p-S296 Chk1. Ponceau S stained blots served as loading controls. G, (left panel) U266 cells were treated with 200 nM PF or 500 nM Rux (ruxolitinib) for 6 hr, followed by lysis and immunoprecipitation with JAK2. The blots were probed for p-Y570 JAK2 and total JAK2. Total lysates were immunoblotted for p-Y705 STAT3, p-S727 STAT3, total STAT3, and JAK2. β-actin was used as a loading control. (right panel) A non-radioactive JAK2 kinase assay was performed. The graph was generated by expressing fold-phosphorylation changes (normalized to control). Images were quantified and analyzed using ImageJ software. Data were normalized by the ratio of the indicated protein/β-actin or Ponceau S staining vs control.
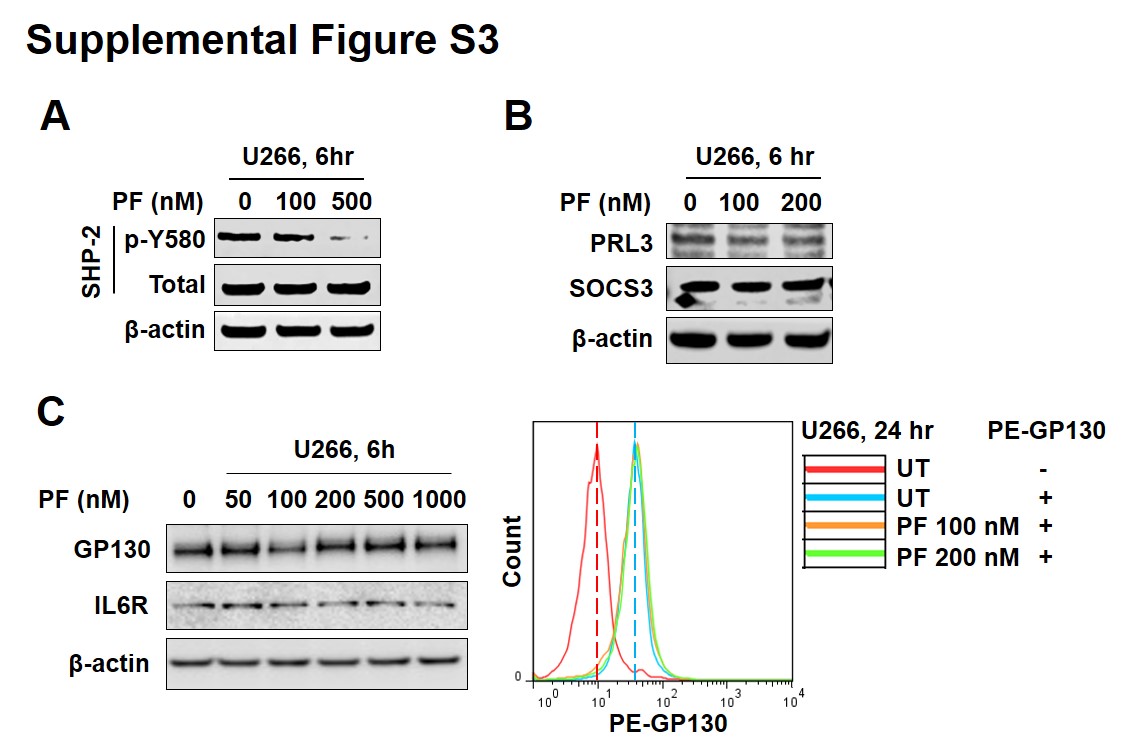
To explore the possible role of phosphatases in inhibition of STAT3 Y705 phosphorylation by Chk1 inhibitors, U266 cells were exposed (6 or 24 hr) to PF in the presence or absence of the specific phosphatase inhibitor pervanadate (PV). Co-exposure to PV antagonized PF-induced p-Y705 down-regulation, particularly at 24 hr, but had little effect on p-S727 expression (Fig 4C), raising the possibility that phosphatase induction may contribute to this interaction. However, PF diminished rather than induced Y580 phosphorylation of the tyrosine phosphatase SHP2 (Suppl. Fig S3A). In contrast to a recent report involving a putative STAT3 inhibitor 29, PF reduced rather than increased expression of the PRL3 phosphatase, and had little effect on the SOCS3 phosphatase (Suppl. Fig S3B). Furthermore, unlike atovaquone 11, PF failed to reduce GP130 or IL6R expression (Suppl. Fig S3C).
An in vitro kinase assay was performed to determine if PF might directly inhibit STAT3 Y705 phosphorylation. Interestingly, addition of His-Chk1 (0.1-0.5 μg) to GST-STAT3 in the presence of 32P increased labeled phospho-STAT3 in a concentration-dependent manner (Fig 4D). This event was attenuated, also in a concentration-dependent fashion, by co-administration of PF, which diminished p-Chk1 signal (Fig 4E). Western blot analysis confirmed reductions in STAT3 Y705 (but not S727) phosphorylation in the presence of increasing concentrations of PF, along with down-regulation of p-Chk1 S296 (Fig 4F), consistent with effects on Chk1 downstream targets, 30 e.g., down-regulation of phospho-CDC25A and upregulation of Cyclin E (Suppl. Fig S4A, B). These findings raise the possibility that Chk1 participates in STAT3 Y705 phosphorylation, and that this process may be directly disrupted by Chk1 inhibition.
Finally, in immunoprecipitation studies, the JAK kinase inhibitor ruxolitinib robustly blocked JAK2 Y570 phosphorylation, whereas PF had no effect (Fig 4G, left panel). Both agents blocked STAT3 Y705 but not S727 phosphorylation. Consistent with these results, ruxolitinib but not PF abrogated JAK2 kinase activity in a cell-free assay (Fig 4G, right panel). Similarly, JAK1, JAK3 and TYK2 were not dephosphorylated by PF (Suppl. Fig S4C, D). These findings argue that PF inhibits STAT3 Y705 activation through a JAK/TYK-independent process.
To confirm the functional contribution of Chk1 to STAT3 activity, U266 cells inducibly expressing Chk1 shRNA were generated (Fig 5A). Addition of doxycycline-inducible shChk1 partially diminished Chk1 expression accompanied by down-regulation of STAT3 p-Y705. In parallel, three stable Chk1 shRNA knock-down clones were generated (KD1-3) which displayed marked down-regulation of STAT3 p-Y705 (Fig 5B). Compatible results were obtained in Chk1 CRISPR knock-down cells (CP1 and CP2; Suppl. Fig S5A). Notably, stable Chk1 KD cells exhibited highly significant reductions in STAT3 binding activity (Fig 5C), a finding confirmed by EMSA assay (Fig 5D). Significantly diminished STAT3 binding activity was also observed in CRISPR knock-down cells (Suppl. Fig S5B). In addition, KD1, KD2, and KD3 cells displayed significantly reduced STAT3 binding to the c-Myc, MCL-1, and BCL-XL promoters (Fig 5E–G) or mRNA expression by RT-PCR (Fig 5H) compared to controls. Finally, Chk1 shRNA or CRISPR KD cells exhibited significantly reduced STAT3 p705 nuclear intensity compared to EV controls (Fig 5I and Suppl. Fig S5C). Together, these findings support a functional contribution of Chk1 in regulating STAT3 nuclear disposition and DNA binding activity in MM cells.
Figure 5. Chk1 plays a critical role in regulating STAT3 signaling.
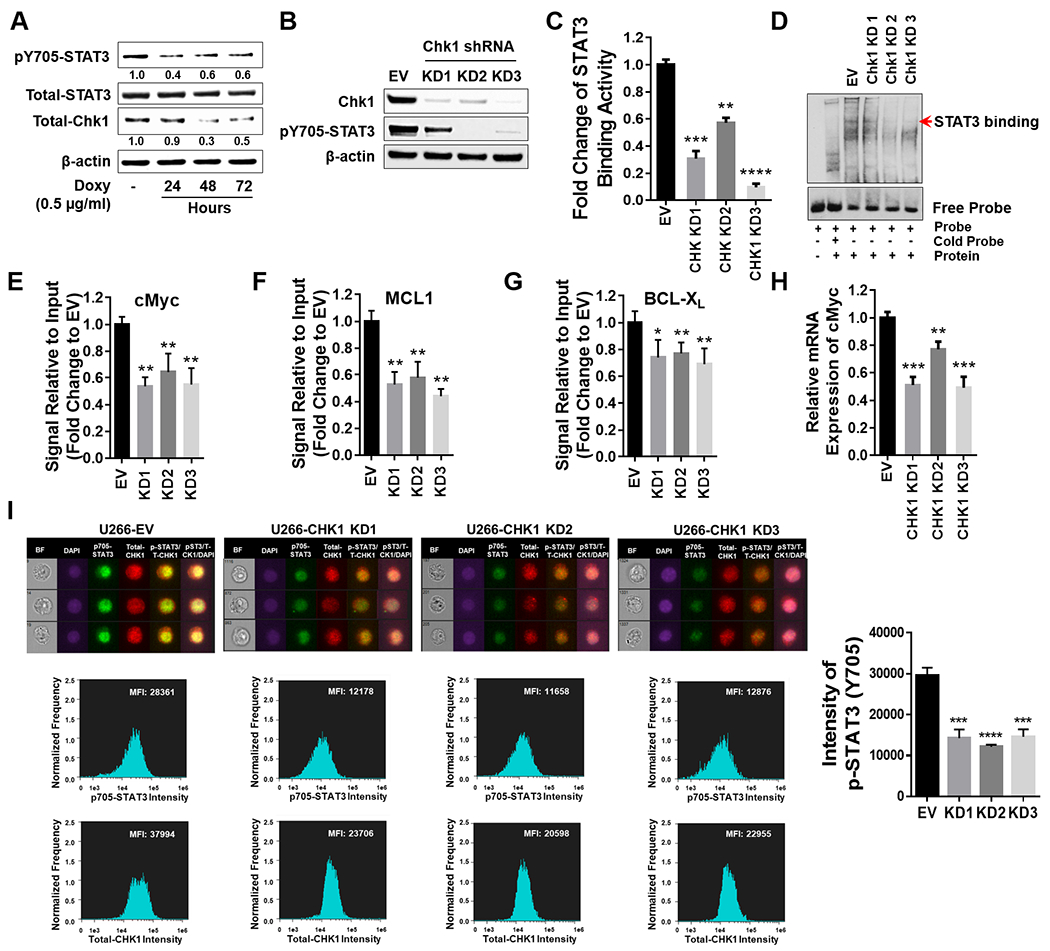
A, U266 cells were transfected with a Tet-on inducible Chk1 shRNA. Doxycycline (0.5 μg/ml) induced cells were collected at 24, 48 and 72 hr. Total lysates were immunoblotted for p-Y705 STAT3, total STAT3, and Chk1. β-actin was used as a loading control. Images were quantified and analyzed using ImageJ software. Data was normalized by the ratio of indicated protein/β-actin vs control. B-I, U266 cells were transfected with a lentivirus harboring Chk1 shRNA. B, Western blot analyses of Chk1 and p-Y705 STAT3 were performed. β-actin controls were assayed to ensure equivalent loading and transfer. C, The STAT3 DNA-binding ELISA was used to evaluate the ability of STAT3 in cellular nuclear extracts to bind to its corresponding consensus sequence immobilized on a 96-well plate. D, EMSA assay of DNA-binding activity of STAT3 in Chk1 knock-down cells. E-G, ChIP experiments were performed to detect the presence of STAT3 at the promotor regions of BCL-XL, MCL-1, and c-Myc in Chk1 knockdown cells. Data was normalized with non-immune IgG (negative control) and STAT3 antibody, and relative fold change (vs UT) is presented. H, Relative mRNA expression of c-Myc was determined by real-time reverse transcription-PCR analysis. GAPDH served as an internal control. Relative fold change (vs EV) is presented. I, Cells were stained with p-Y705 STAT3, Chk1 and DAPI, and then visualized by fluorescence microscope and by ImageStream and representative cells are shown (BF = brightfield). Histograms of p-Y705 STAT3 intensity and fold change are shown. Representative data for at least three replicates. *, P < 0.05; ** = P < 0.01; ***, P < 0.001; **** = P < 0.0001.
To assess the functional significance of diminished STAT3 activation on the survival of cells exposed to PF, U266 cells expressing constitutively active STAT3 (mutation of A662C and N664C) 31 were generated. Such cells, designated CA01 and CA02, displayed significantly increased basal STAT3 binding activity compared to empty-vector controls (Fig 6A). Notably, these cells were significantly less sensitive to PF-induced lethality compared to controls (Fig 6B), and displayed markedly reduced PF-mediated PARP and caspase cleavage as well as DNA damage (γH2A.X; Fig 6C). Parallel studies were performed utilizing cells transfected with dominant-negative STAT3 ( Y705F mutation 32), designated DN01 and DN02, which exhibited significantly reduced STAT3 binding activity compared to controls (Fig 6D). In contrast to CA cells, DN cells were significantly more sensitive to PF-induced cell death (Fig 6E), and displayed diminished STAT3 p-Y705 expression and PARP cleavage (Fig 6F) compared to empty-vector controls. These findings indicate that STAT3 inactivation plays a significant functional role in the lethal consequences of Chk1 inhibition in MM cells.
Figure 6. STAT3 inhibition by Chk1 inhibitor through a STAT3-dependent manner.
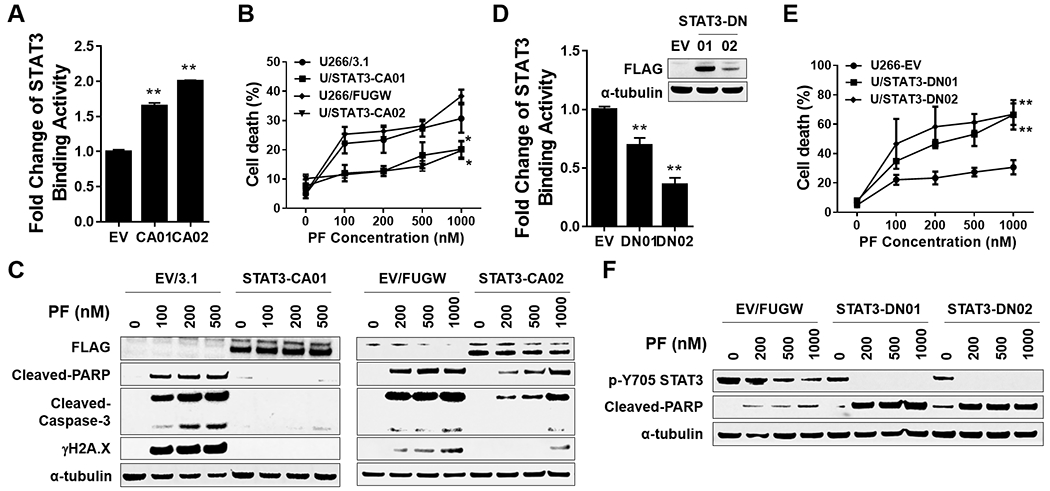
A-C, U266 were transfected with pcDNA3.1 (empty vector) or pcDNA-STAT3 CA (FLAG fusion), and STAT3-CA01 was selected. STAT3-CA02 was selected in U266 cells infected with a lentivirus harboring STAT3 CA (FLAG fusion). A, The STAT3 DNA-binding ELISA was used to evaluate STAT3 activity in STAT3-CA cell. B, cells were exposed (24 hr) to indicated concentrations of PF, followed by flow cytometry to monitor the percentage of apoptotic (7-AAD+) cells. Values represent the means ± S.D. for three experiments performed in triplicate. * = P < 0.05; ** = P < 0.01. C, Western blot analysis of FLAG, cleaved-PARP, cleaved-Caspase-3, and γH2A.X was performed. α-tubulin controls were assayed to ensure equivalent loading and transfer. D-F, U266 cells infected with a lentivirus harboring STAT3 DN (FLAG fusion). STAT3-CA01/02 were selected. Assays were performed as in A-C, ** = P < 0.01.
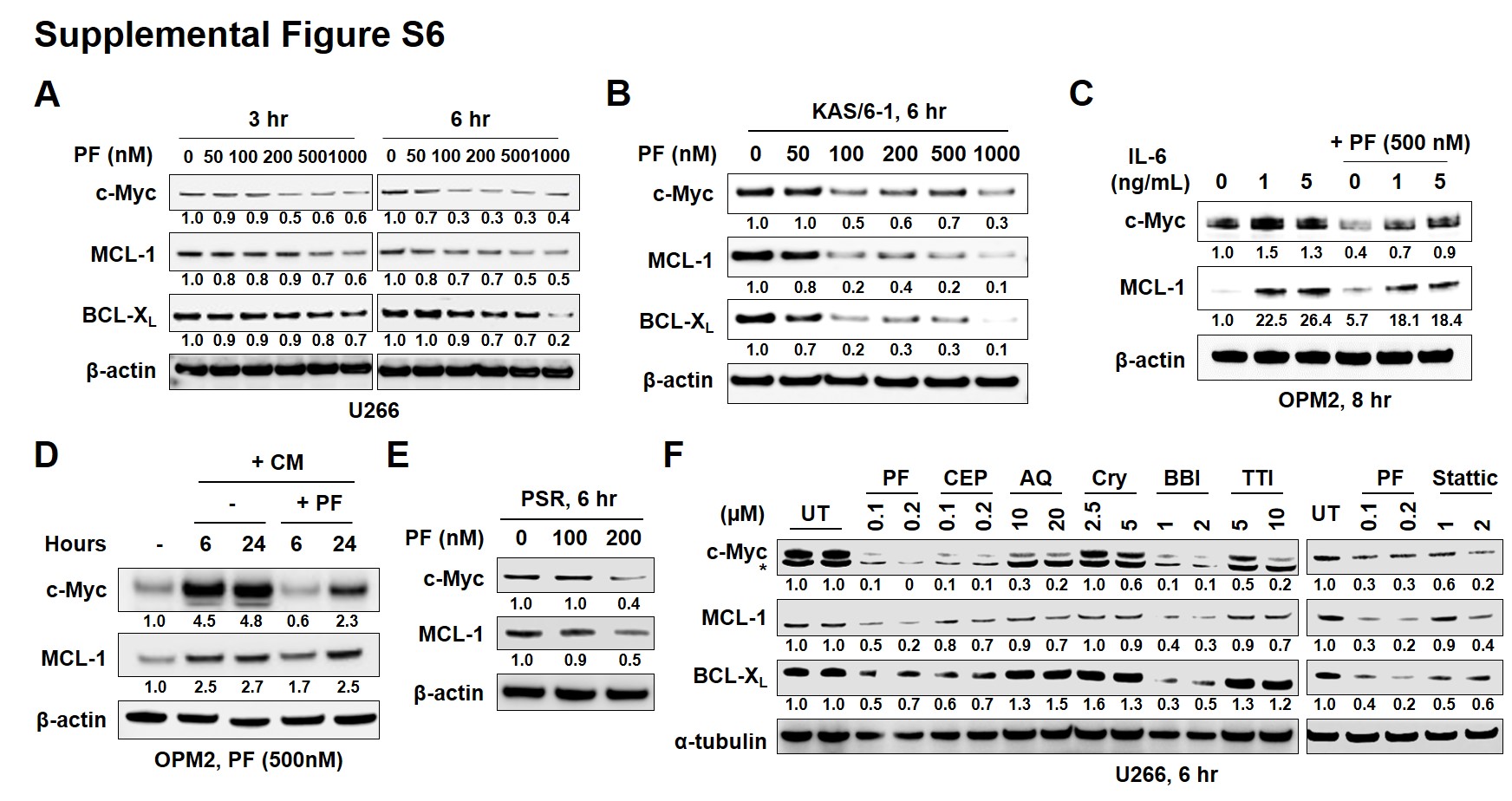
Western blot analysis was performed to assess the impact of Chk1 interruption on expression of STAT3 target proteins. Exposure (3 and particularly 6 hr) of U266 cells to nM concentrations of PF diminished expression of c-Myc, MCL-1, and BCL-XL proteins (Suppl. Fig S6A). Similar results were obtained in IL-6-dependent KAS/6-1 cells (Suppl. Fig S6B), OPM2 cells exposed to IL-6 (Suppl. Fig S6C), OPM2 cells cultured in the presence of conditioned medium (Suppl. Fig S6D), and bortezomib-resistant PS-R cells (Suppl. Fig S6E). In addition, as observed in the case of mRNA levels (Fig 1E), treatment of U266 cells (6 hr) with PF or CEP was as or more effective than 1-2 log higher concentrations of multiple known STAT3 inhibitors in diminishing expression of c-Myc, MCL-1, and BCL-XL protein levels (Suppl. Fig S6F). Of note, Chk1 shRNA knock-down cells induced similar reductions in expression of these proteins (Suppl. Fig S7A). Furthermore, enforced expression of constitutively active STAT3 abrogated PF-mediated protein down-regulation (Suppl. Fig S7B). Finally, enforced expression of MCL-1, c-Myc, or BCL-XL significantly diminished PF-induced cell death (Suppl. Fig S7C–E), arguing that disruption of STAT3 downstream targets plays a functional role in Chk1 inhibitor mediated lethality.
Finally, attempts were made to extend these findings to primary human specimens and in vivo models. Two CD138+ -isolated samples displayed clear p-Y705 and p-S727 activation but not in the CD138− cell population (Fig 7A, top). Interestingly, treatment (24 hr) of a 3rd CD138+-isolated specimen with 500 nM PF diminished p-Y705 expression (Fig 7A, bottom). ImageStream flow cytometry exhibited significantly reduced pSTAT3 705Y nuclear intensity compared to controls (Fig 7B). More detailed analysis of the effects of Chk1 inhibitor treatment was performed on primary samples in which sufficient numbers of CD138+ cells were available for western blot analysis. In three such samples, PF or CEP (24 hr) reduced or abrogated p-Y705 expression. This was accompanied by variable reductions in the expression of c-Myc and MCL-1 protein (Fig 7C).
Figure 7. Chk1 inhibitors decrease p-Y705 STAT3 expression in primary human MM cells ex-vivo and in mice in vivo.
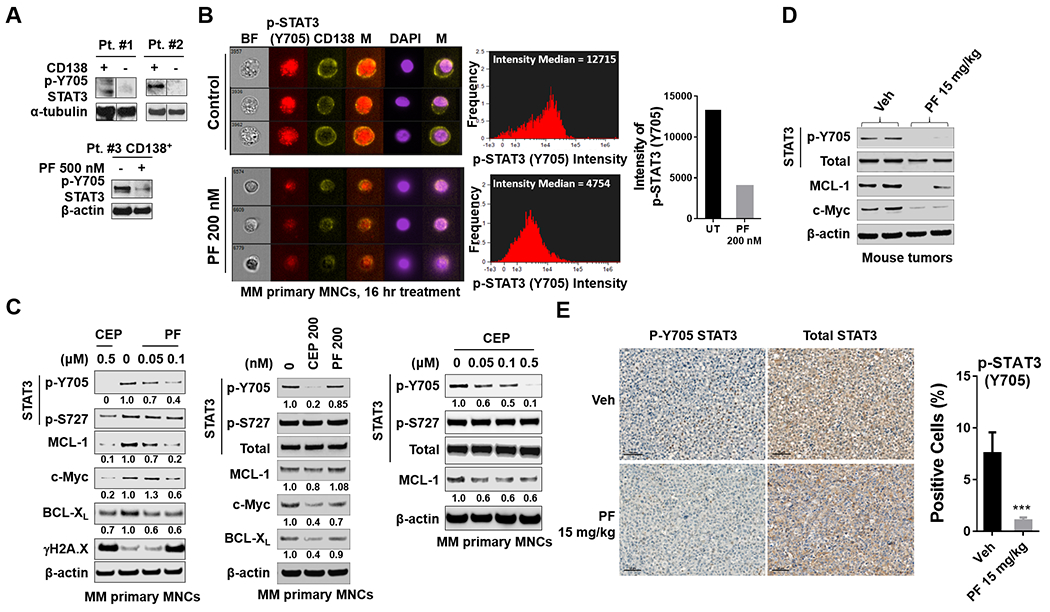
A (upper panel), CD138+ and CD138− MNCs were isolated from MM primary samples. (lower panel) Isolated CD138+ MNCs (mononuclear cells) were treated with 500 nM PF for 16 hr. Western blot analysis of p-Y705 STAT3 and p-S727 STAT3 were then performed. α-tubulin and β-actin controls were assayed to ensure equivalent loading and transfer. A black border indicates that the blots were cut and spliced from the same membrane and after the same exposure interval. B, Isolated MM primary mononuclear cells (MNCs) were stained with p-Y705 STAT3, CD138 and DAPI, and then visualized by fluorescence microscopy and by ImageStream; representative cells are shown (BF = brightfield). Histograms of p-Y705 STAT3 intensity and fold change are shown. C, Isolated MM primary MNCs were treated with indicated doses of PF and CEP for 16 hr. Western blot analysis of p-Y705 STAT3, p-S727 STAT3, STAT3, c-Myc, MCL-1, BCL-XL, and γH2A.X were performed. β-actin controls were assayed to ensure equivalent loading and transfer. D, NOD-SCID IL2Rgammanull mice were subcutaneously injected with 5×106 U266 cells into the flank. When tumors grew to 10 mm (length), PF (15 mg/kg) was administrated (i.p.) for 3 days. Control animals received equal volumes of vehicle. Western blot analysis was performed to monitor the indicated candidate pharmacodynamic markers, identified from in vitro experiments, in tumors excised from representative mice. Images were quantified and analyzed using ImageJ software. Data was normalized by determining the ratio of the indicated protein/β-actin vs control. E, Tumor sections from vehicle and PF-treated mice were stained with p-Y705 STAT3 antibody. Sections were visualized and images captured usingVectra® Polaris Imaging System (Akoya Biosciences) research microscope and quantified using the Polaris software program. Scale bar = 50 μm. ***= P < 0.001.
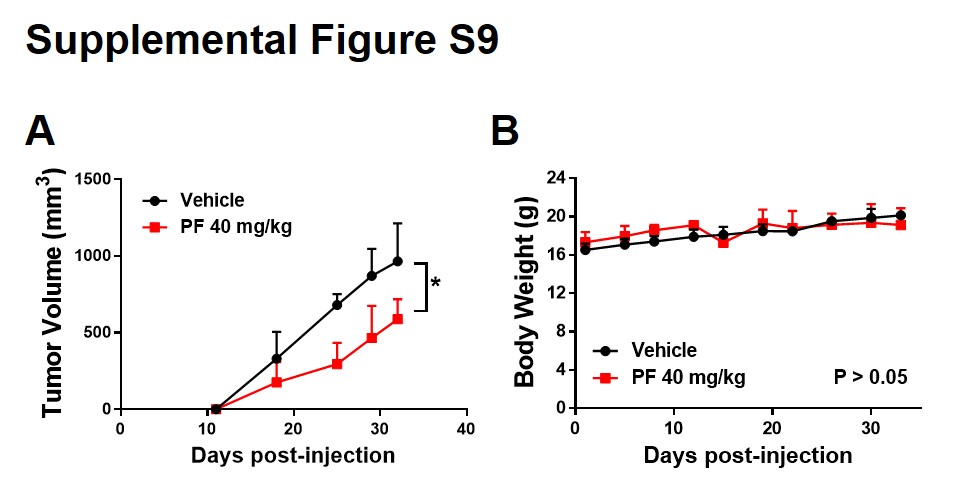
Parallel studies were performed in NSG (NOD-SCID IL2Rgammanull) mice inoculated in the flank with 5×106 U266 cells and treated with PF (15 mg/kg, i.p., daily). Two mice were sacrificed on day 3, after which tumors were excised and proteins subjected to Western blot analysis. As shown in Fig 7D, tumors obtained from PF-treated mice exhibited down-regulation of p-Y705 accompanied by moderate reductions in the expression of c-Myc and MCL-1 proteins. Immunohistochemical analysis confirmed the reduction in p-Y705-staining cells in tumors extracted from PF-treated mice (Fig 7E and Suppl. Fig S8). Furthermore, PF significantly suppressed tumor growth in vivo (P < 0.05; Suppl. Fig S9A), with little evidence of toxicity (Suppl. Fig S9B). Together, these findings suggest that inactivation of STAT3 by Chk1 inhibitors occurs in primary MM cells and can be recapitulated in the in vivo setting.
Discussion
The STAT3 transcription factor plays a key role in oncogenesis, transformation, metastasis, differentiation, and drug resistance, among multiple other activities 33. These characteristics have made it a high-value target in drug development, despite the fact that transcription factors are notoriously difficult to target therapeutically. Nevertheless, several candidate STAT3 inhibitors (e.g., TTI-101, BBI608) have now entered the clinic (NCT03195699, NCT03647839). In addition to the de novo development of STAT3 antagonists, attention has also focused on the ability of other classes of agents to disrupt STAT3 function. For example, CDK7 antagonists have been shown to interfere with STAT3 activity 34, as has the quinone antimicrobial atovaquone 11. The present results identify a heretofore unrecognized link between the Chk1 and STAT3 pathways that offers the potential for therapeutic intervention. Specifically, they demonstrate that a variety of clinically relevant Chk1 inhibitors potently disrupt STAT3 activation at extremely low (e.g., nM) concentrations, both in vitro as well as in vivo. Significantly, the inhibitory effects of Chk1 inhibitors on STAT3 activation were observed at 1-2 logs lower than those reported for other STAT3 antagonists. While previous studies have suggested that STAT3 may regulate Chk1 responses in normal renal cells subjected to oxidative injury 23, this represents the first indication that Chk1 may operate upstream of and play a critical role in STAT3 activation in neoplastic cells.
The mechanism(s) by which Chk1 inhibition disrupts STAT3 function is (are) likely to be multi-factorial. STAT3 activation requires phosphorylation on Tyr705, which promotes dimerization and nuclear localization, necessary for DNA binding and activation 35. In contrast, phosphorylation on Ser727 is primarily responsible for enhancing transactivation 36. In the present studies, Chk1 inhibitors primarily blocked the former action, and had little effect on Ser727 phosphorylation. This resulted in diminished STAT3 dimerization, nuclear translocation, and activation of STAT3-dependent genes. While chemical and genetic approaches may exert disparate activities 37, it is noteworthy that Chk1 knock-down recapitulated the effects of Chk1 inhibitors on STAT3 function, arguing for on-target actions. The question of how Chk1 inhibition opposes STAT3 Tyr705 phosphorylation is clearly important to address. In this regard, activation of STAT3 may be mediated by diverse events, including cytokine-mediated stimulation through JAK2 or diverse alternative upstream kinases (e.g., Src or MEK1/2) 38. However, in contrast to atovaquone, Chk1 inhibition failed to down-regulate expression of the scaffolding protein GP130, implicated in STAT3 activation events 39. Additionally, Chk1 inhibition had no discernible effects on JAK1, JAK2, JAK3, and TYK2 activation, arguing against the participation of these kinases in STAT3 modulation. Significantly, the results of the in vitro kinase assay suggested that PF may act directly to diminish STAT3 Tyr705 phosphorylation, raising the possibility that Chk1 represents a kinase responsible for STAT3 activation at this site. Although such a finding was unanticipated given the canonical view that Chk1 is a serine/threonine kinase 40, several lines of evidence argue strongly for this possibility. These include the findings that a) multiple Chk1 inhibitors or b) Chk1 knock-down selectively blocked STAT3 Tyr705 but not Ser727 phosphorylation; c) results of the in vitro kinase assay demonstrating the capacity of recombinant Chk1 to promote STAT3 Tyr705 phosphorylation; and d) prediction of Tyr sites as potential targets of Chk1 action (Supplemental Table 2, http://www.cbs.dtu.dk/services/NetPhos/; http://sysbio.unl.edu/PhosphoSVM/prediction.php). Finally, the existence of an alternative STAT3-activating tyrosine kinase susceptible to Chk1 inhibitor actions cannot be completely excluded.
In addition to blocking STAT3 phosphorylation, Chk1 inhibitors might potentially act by increasing the expression/activity of phosphatases responsible for dephosphorylation. In this context, up-regulation of the PRL phosphatase has recently been shown to promote IL-6-mediated STAT3 activation in MM cells through a GP130- and SHP2-dependent mechanism 29. However, in the present study, Chk1 inhibition reduced rather than increased PRL and SHP2 expression/activation, arguing against a role for these phosphatases in Chk1 inhibitor action. Additionally, Chk1 inhibitors did not up-regulate the SOCS3 phosphatase, also implicated in STAT3 regulation 29. On the other hand, the specific tyrosine phosphatase inhibitor pervanadate substantially reduced the ability of PF to block STAT3 Tyr705 phosphorylation, raising the possibility that enhanced activity of yet to be defined phosphatases might contribute to STAT3 inactivation in this setting.
Previous reports have shown that STAT3 associates with Chk1 in non-neoplastic cells e.g., normal renal cells subjected to oxidative injury 23. Consistent with these findings, SPR analysis demonstrated a dose- and time-dependent association between Chk1 and STAT3. Moreover, co-administration of PF diminished the amount of STAT3 co-immunoprecipitating with Chk1 in both the cytoplasm and nucleus, although in the case of nuclear proteins, this could be attributed to disruption of STAT3 Y705 phosphorylation and nuclear transport. Consequently, while it is conceivable that direct Chk1/STAT3 interactions may be required for or contribute to STAT3 activation, the functional significance of this association and its disruption remain to be determined. Nevertheless, the possibility exists that in addition to antagonizing STAT3 phosphorylation, dimerization, and nuclear translocation, disruption of direct STAT3/Chk1 interactions by Chk1 inhibitors might contribute to STAT3 antagonism. Further studies will be necessary to confirm or refute this possibility.
Constitutive activation of STAT3 has been reported in 50% or more of MM patients 41. Representative of this population, MM cells with high basal levels of phospho-STAT3 and presumably dependent upon this transcription factor such as U226 cells displayed robust inactivation in the presence of low Chk1 inhibitor concentrations. However, Chk1 inhibitors effectively inactivated STAT3 in cells with low basal STAT3 activation (e.g., OPM2) but stimulated with IL-6 or stromal cell conditioned medium. In this regard, IL-6-mediated STAT3 activation has been implicated in chemoresistance mediated by microenvironmental factors 4. Such findings raise the possibility that interrupting Chk1 may be an effective strategy not only in the subset of MM cells displaying STAT3 addiction, but also in STAT3-independent cells protected from cell death by stromal cell factors. The connection between Chk1 and STAT3 may have specific relevance for MM, a disease which has recently been shown to be particularly vulnerable to replicative stress42. For example, both Chk1 and its immediate upstream effector ATR are prototypical inducers of replicative stress 42. Moreover, STAT3 modulates the DNA damage response pathway, in part by promoting DNA repair 43. Additionally, STAT3 regulates the response to DNA damage through its pro-survival and -proliferation-related downstream targets e.g., MCL-1, BCL-XL, and c-Myc 44. Thus, disruption of STAT3 signaling by Chk1 inhibitors may have dual effects on the DDR e.g., promotion of DNA damage as well as the resulting apoptotic response. In support of this notion, MM cells expressing constitutively active STAT3 displayed diminished DNA damage (γH2A.X formation) and cell death in response to PF. Such a dual mechanism may be particularly relevant to cells such as MM which are intrinsically susceptible to replicative stress.
Collectively, these findings may have implications for MM and potentially other malignances dependent upon STAT3 for survival. Chk1 inhibitors have been evaluated in solid tumors, generally in combination with genotoxic agents, but relatively rarely in hematopoietic malignancies, and to the best of our knowledge, never in MM. Based on the susceptibility of MM to replicative stress, and the ability of Chk1 inhibitors to trigger this phenomenon 45, evaluation of such agents in MM warrants attention. The same could be said of ATR inhibitors, which are also potent inducers of replicative stress 46. Another implication of these findings is that the subset of MM cells that are dependent upon STAT3 for survival might be particularly vulnerable to Chk1 disruption due to a) enhanced DNA damage and b) a lowered apoptotic threshold stemming from STAT3 inactivation. In addition, STAT3 inactivation could promote cell death even in STAT3-independent cells by blocking microenvironmental pro-survival signals mediated by IL-6 and STAT3. Finally, the present findings could provide mechanistic insights supporting possible combination strategies involving Chk1 inhibitors and STAT3. For example, we have previously described synergistic interactions between Chk1 and MEK1/2 inhibitors in MM cells 47. However, whereas Chk1 inhibitors do not inactivate STAT3 Ser727, a process implicated in STAT3 transactivation 48, MEK1/2 is known to phosphorylate STAT3 Ser727 49. It is possible that combined therapy may more profoundly inactivate STAT3 by triggering dephosphorylation at both sites, thereby promoting cell death. Accordingly, studies designed to test this concept, as well as the ability of clinically relevant ATR inhibitors to interact analogously in replication stress- susceptible MM cells are currently underway.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Implications:
These findings identify a heretofore unrecognized link between the Chk1 and STAT3 pathways and suggest that Chk1 pathway inhibitors warrant attention as novel and potent candidate STAT3 antagonists in myeloma.
Acknowledgements
This work was supported by awards 1R01CA205607-01 (S.G.) and an award from the Leukemia and Lymphoma Society of America (R6508-18; S.G.). Services in support of the research project were provided by the Virginia Commonwealth University Massey Cancer Center Tissue and Data Acquisition and Analysis Core, Mouse Models Core Laboratory, and Flow Cytometry Shared Resource, supported, in part, with funding from NIH-NCI Cancer Center Support Grant P30 CA016059.
Footnotes
Conflict-of-interest disclosure: The authors declare no potential conflicts of interest.
References
- 1.Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–154. [DOI] [PubMed] [Google Scholar]
- 2.Chong PSY, Chng WJ, de Mel S. STAT3: A Promising Therapeutic Target in Multiple Myeloma. Cancers (Basel). 2019;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jung SH, Ahn SY, Choi HW, et al. STAT3 expression is associated with poor survival in non-elderly adult patients with newly diagnosed multiple myeloma. Blood Res. 2017;52(4):293–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johnson DE, O’Keefe RA, Grandis JR. Targeting the IL-6/JAK/STAT3 signalling axis in cancer. Nat Rev Clin Oncol. 2018;15(4):234–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aziz MH, Hafeez BB, Sand JM, et al. Protein kinase Cvarepsilon mediates Stat3Ser727 phosphorylation, Stat3-regulated gene expression, and cell invasion in various human cancer cell lines through integration with MAPK cascade (RAF-1, MEK1/2, and ERK1/2). Oncogene. 2010;29(21):3100–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chung J, Uchida E, Grammer TC, Blenis J. STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol Cell Biol. 1997;17(11):6508–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu H, Lee H, Herrmann A, Buettner R, Jove R. Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer. 2014;14(11):736–746. [DOI] [PubMed] [Google Scholar]
- 8.Zhao C, Li H, Lin HJ, Yang S, Lin J, Liang G. Feedback Activation of STAT3 as a Cancer Drug-Resistance Mechanism. Trends Pharmacol Sci. 2016;37(1):47–61. [DOI] [PubMed] [Google Scholar]
- 9.Chatterjee M, Honemann D, Lentzsch S, et al. In the presence of bone marrow stromal cells human multiple myeloma cells become independent of the IL-6/gp130/STAT3 pathway. Blood. 2002;100(9):3311–3318. [DOI] [PubMed] [Google Scholar]
- 10.Scuto A, Krejci P, Popplewell L, et al. The novel JAK inhibitor AZD1480 blocks STAT3 and FGFR3 signaling, resulting in suppression of human myeloma cell growth and survival. Leukemia. 2011;25(3):538–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xiang M, Kim H, Ho VT, et al. Gene expression-based discovery of atovaquone as a STAT3 inhibitor and anticancer agent. Blood. 2016;128(14):1845–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bai L, Zhou H, Xu R, et al. A Potent and Selective Small-Molecule Degrader of STAT3 Achieves Complete Tumor Regression In Vivo. Cancer Cell. 2019;36(5):498–511 e417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang X, Sun Y, Pireddu R, et al. A novel inhibitor of STAT3 homodimerization selectively suppresses STAT3 activity and malignant transformation. Cancer Res. 2013;73(6):1922–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer. 2014;134(5):1013–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanchez Y, Wong C, Thoma RS, et al. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science. 1997;277(5331):1497–1501. [DOI] [PubMed] [Google Scholar]
- 16.Dai Y, Grant S. Targeting Chk1 in the replicative stress response. Cell Cycle. 2010;9(6):1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai Y, Grant S. New insights into checkpoint kinase 1 in the DNA damage response signaling network. Clin Cancer Res. 2010;16(2):376–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zeng L, Beggs RR, Cooper TS, Weaver AN, Yang ES. Combining Chk1/2 Inhibition with Cetuximab and Radiation Enhances In Vitro and In Vivo Cytotoxicity in Head and Neck Squamous Cell Carcinoma. Mol Cancer Ther. 2017;16(4):591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Landau HJ, McNeely SC, Nair JS, et al. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol Cancer Ther. 2012;11(8):1781–1788. [DOI] [PubMed] [Google Scholar]
- 20.McNeely S, Beckmann R, Bence Lin AK. CHEK again: revisiting the development of CHK1 inhibitors for cancer therapy. Pharmacol Ther. 2014;142(1):1–10. [DOI] [PubMed] [Google Scholar]
- 21.Karp JE, Thomas BM, Greer JM, et al. Phase I and pharmacologic trial of cytosine arabinoside with the selective checkpoint 1 inhibitor Sch 900776 in refractory acute leukemias. Clin Cancer Res. 2012;18(24):6723–6731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zenvirt S, Kravchenko-Balasha N, Levitzki A. Status of p53 in human cancer cells does not predict efficacy of CHK1 kinase inhibitors combined with chemotherapeutic agents. Oncogene. 2010;29(46):6149–6159. [DOI] [PubMed] [Google Scholar]
- 23.Ajay AK, Kim TM, Ramirez-Gonzalez V, Park PJ, Frank DA, Vaidya VS. A bioinformatics approach identifies signal transducer and activator of transcription-3 and checkpoint kinase 1 as upstream regulators of kidney injury molecule-1 after kidney injury. J Am Soc Nephrol. 2014;25(1):105–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Zhang Y, Zhou L, et al. A Bim-targeting strategy overcomes adaptive bortezomib resistance in myeloma through a novel link between autophagy and apoptosis. Blood. 2014;124(17):2687–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asano R, Hokari S. Bovine erythrocyte fractionation in Percoll density gradients. Nihon Juigaku Zasshi. 1987;49(3):575–576. [DOI] [PubMed] [Google Scholar]
- 26.Nguyen T, Hawkins E, Kolluri A, et al. Synergism between bosutinib (SKI-606) and the Chk1 inhibitor (PF-00477736) in highly imatinib-resistant BCR/ABL(+) leukemia cells. Leuk Res. 2015;39(1):65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Syljuasen RG, Sorensen CS, Nylandsted J, Lukas C, Lukas J, Bartek J. Inhibition of Chk1 by CEP-3891 accelerates mitotic nuclear fragmentation in response to ionizing Radiation. Cancer Res. 2004;64(24):9035–9040. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Saud SM, Young MR, Colburn NH, Hua B. Cryptotanshinone, a Stat3 inhibitor, suppresses colorectal cancer proliferation and growth in vitro. Mol Cell Biochem. 2015;406(1–2):63–73. [DOI] [PubMed] [Google Scholar]
- 29.Chong PSY, Zhou J, Lim JSL, et al. IL6 Promotes a STAT3-PRL3 Feedforward Loop via SHP2 Repression in Multiple Myeloma. Cancer Res. 2019;79(18):4679–4688. [DOI] [PubMed] [Google Scholar]
- 30.Xiao Z, Chen Z, Gunasekera AH, et al. Chk1 mediates S and G2 arrests through Cdc25A degradation in response to DNA-damaging agents. J Biol Chem. 2003;278(24):21767–21773. [DOI] [PubMed] [Google Scholar]
- 31.Bromberg JF, Wrzeszczynska MH, Devgan G, et al. Stat3 as an oncogene. Cell. 1999;98(3):295–303. [DOI] [PubMed] [Google Scholar]
- 32.Mohr A, Fahrenkamp D, Rinis N, Muller-Newen G. Dominant-negative activity of the STAT3-Y705F mutant depends on the N-terminal domain. Cell Commun Signal. 2013;11:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huynh J, Chand A, Gough D, Ernst M. Therapeutically exploiting STAT3 activity in cancer - using tissue repair as a road map. Nat Rev Cancer. 2019;19(2):82–96. [DOI] [PubMed] [Google Scholar]
- 34.Cayrol F, Praditsuktavorn P, Fernando TM, et al. THZ1 targeting CDK7 suppresses STAT transcriptional activity and sensitizes T-cell lymphomas to BCL2 inhibitors. Nat Commun. 2017;8:14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piao JY, Kim SJ, Kim DH, et al. Helicobacter pylori infection induces STAT3 phosphorylation on Ser727 and autophagy in human gastric epithelial cells and mouse stomach. Sci Rep. 2020;10(1):15711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Decker T, Kovarik P. Serine phosphorylation of STATs. Oncogene. 2000;19(21):2628–2637. [DOI] [PubMed] [Google Scholar]
- 37.Knight ZA, Shokat KM. Chemical genetics: where genetics and pharmacology meet. Cell. 2007;128(3):425–430. [DOI] [PubMed] [Google Scholar]
- 38.Liu AM, Lo RK, Wong CS, Morris C, Wise H, Wong YH. Activation of STAT3 by G alpha(s) distinctively requires protein kinase A, JNK, and phosphatidylinositol 3-kinase. J Biol Chem. 2006;281(47):35812–35825. [DOI] [PubMed] [Google Scholar]
- 39.Garbers C, Aparicio-Siegmund S, Rose-John S. The IL-6/gp130/STAT3 signaling axis: recent advances towards specific inhibition. Curr Opin Immunol. 2015;34:75–82. [DOI] [PubMed] [Google Scholar]
- 40.Lopez-Girona A, Tanaka K, Chen XB, Baber BA, McGowan CH, Russell P. Serine-345 is required for Rad3-dependent phosphorylation and function of checkpoint kinase Chk1 in fission yeast. Proc Natl Acad Sci U S A. 2001;98(20):11289–11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang YH, Molavi O, Alshareef A, et al. Constitutive Activation of STAT3 in Myeloma Cells Cultured in a Three-Dimensional, Reconstructed Bone Marrow Model. Cancers (Basel). 2018;10(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cottini F, Hideshima T, Suzuki R, et al. Synthetic Lethal Approaches Exploiting DNA Damage in Aggressive Myeloma. Cancer Discov. 2015;5(9):972–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Barry SP, Townsend PA, Knight RA, Scarabelli TM, Latchman DS, Stephanou A. STAT3 modulates the DNA damage response pathway. Int J Exp Pathol. 2010;91(6):506–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Banerjee K, Resat H. Constitutive activation of STAT3 in breast cancer cells: A review. Int J Cancer. 2016;138(11):2570–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nazareth D, Jones MJ, Gabrielli B. Everything in Moderation: Lessons Learned by Exploiting Moderate Replication Stress in Cancer. Cancers (Basel). 2019;11(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nagel R, Avelar AT, Aben N, et al. Inhibition of the Replication Stress Response Is a Synthetic Vulnerability in SCLC That Acts Synergistically in Combination with Cisplatin. Mol Cancer Ther. 2019;18(4):762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pei XY, Dai Y, Tenorio S, et al. MEK1/2 inhibitors potentiate UCN-01 lethality in human multiple myeloma cells through a Bim-dependent mechanism. Blood. 2007;110(6):2092–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang R, Cherukuri P, Luo J. Activation of Stat3 sequence-specific DNA binding and transcription by p300/CREB-binding protein-mediated acetylation. J Biol Chem. 2005;280(12):11528–11534. [DOI] [PubMed] [Google Scholar]
- 49.Xuan YT, Guo Y, Zhu Y, et al. Role of the protein kinase C-epsilon-Raf-1-MEK-1/2-p44/42 MAPK signaling cascade in the activation of signal transducers and activators of transcription 1 and 3 and induction of cyclooxygenase-2 after ischemic preconditioning. Circulation. 2005;112(13):1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.