

Abstract
Fine-tuning than complete disruption of 2-arachidonoylglycerol (2-AG) metabolism in the brain represents a promising pharmacological approach to limit potential untoward effects associated with complete blockade of monoacylglycerol lipase (MGL), the primary hydrolase of 2-AG. This could be achieved through α/β-hydrolase domain containing 6 (ABHD6) inhibition, which will provide a smaller and safer contribution to 2-AG regulation in the brain. Pharmacological studies with ABHD6 inhibitors have recently been reported, where modulation of ABHD6 activity either through CB1R-dependent or CB1R-independent processes showed promise in preclinical models of epilepsy, neuropathic pain and inflammation. Furthermore in the periphery, ABHD6 modulates 2-AG and other fatty acid monoacylglycerols (MAGs) and is implicated in Type-2 diabetes, metabolic syndrome and potentially other diseases. Herein, we report the discovery of single-digit nanomolar potent and highly specific ABHD6 inhibitors with >1000-fold selectivity against MGL and FAAH. The new ABHD6 inhibitors provide early leads to develop therapeutics for neuroprotection and the treatment of inflammation and diabetes.
Keywords: α/β-Hydrolase domain containing 6 (ABHD6); α/β-hydrolase domain containing 12 (ABHD12), fatty acid amide hydrolase (FAAH); monoacylglycerol lipase (MGL); N-arachidonoylethanolamine (anandamide, AEA); cannabinoid CB1, CB2 receptors; α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA); endocannabinoid system; diabetes; neuroprotection; neuroinflammation
1. Introduction
The endocannabinoid system (ECS) consists of the cannabinoid receptors CB1 and CB2, the endogenous ligands N-arachidonoylethanolamine (anandamide, AEA) and 2-arachidonoylglycerol (2-AG), and their hydrolytic enzymes fatty acid amide hydrolase (FAAH), monoacylglycerol lipase (MGL), α/β-hydrolase domain containing 6 (ABHD6) and α/β-hydrolase domain containing 12 (ABHD12)[1]. CB1 receptors are widely distributed throughout the brain at presynaptic nerve terminals and account for the majority of the neurobehavioral effects of cannabinoids at the central nervous system (CNS)[2]. In contrast, CB2 receptors are predominantly found in cells of immune origin,[3] including microglia, although low-level expression has been reported in healthy neurons[4]. The endocannabinoids (AEA, 2-AG) are synthesized on-demand and not stored in vesicles. The production of 2-AG is mediated by phospholipase Cβ and diacylglycerol lipase (DAGL),[5] while AEA production is mediated by N-acyl phosphatidylethanolamine phospholipase D (NAPE-PLD)[6]. 2-AG is the most abundant endocannabinoid in the brain, about ~170-fold higher than anandamide[7] and acts as a full agonist at cannabinoid receptors CB1 and CB2, while anandamide functions as a partial agonist[8]. The extent and duration of the endocannabinoids’ signaling is tightly regulated by the balance between their production and hydrolysis. AEA is degraded by FAAH, and 2-AG is hydrolyzed to arachidonic acid (AA) and glycerol by three enzymes MGL, ABHD6 and ABHD12[9]. In the brain, MGL is the primary 2-AG hydrolase accounting for approximately 85% of the total membrane activity[9], however ABHD6 and MGL are each responsible for about half of the total 2-AG hydrolysis in primary neuron homogenates[10]. The three 2-AG hydrolases exhibit unique and distinct cellular and/or subcellular distributions and regulate different pools of 2-AG in intact cells allowing for specific pharmacological modulation[11]. In the brain, several reports indicated that although MGL modulation of 2-AG did not reproduce all untoward effects seen with direct CB1 activation, chronic blockade of MGL caused desensitization and downregulation of CB1 receptors, mirroring the observations seen in MGL knockout mice[12]. Therefore, “fine-tuning” rather than complete disruption of 2-AG metabolism could be a promising pharmacological approach to limit any potential untoward effects with full MGL blockade. This can potentially be achieved through α/β-hydrolase domain containing 6 (ABHD6) inhibition, which will provide a smaller and safer contribution to 2-AG regulation in the brain. Pharmacological studies to delineate the role of ABHD6 inhibition in pathophysiological conditions have been reported, where modulation of ABHD6 activity augments 2-AG signaling and exerts CB1R-mediated anti-inflammatory and neuroprotective effects in Traumatic Brain Injury (TBI)[13]. Conversely, ABHD6 also functions in a CB1-independent manner to affect other pathophysiological conditions showing promise in preclinical models of epilepsy[14], neuropathic pain[15], Type-2 diabetes [16], metabolic syndrome[17] and lung inflammation[18]. ABHD6 inhibitors have also been reported in neurodegenerative and inflammatory diseases such as multiple sclerosis, and a nice review has recently been proposed regarding the endocannabinoid system modulation by ABHD6[19].
ABHD6 enzyme:
ABHD6 is a transmembrane serine hydrolase that hydrolyzes 2-AG and other fatty acid monoacylglycerols (MAGs) substrates. ABHD6 is highly expressed in the immune system, predominantly the spleen and the small intestine, followed by the brain, particularly the hippocampus and cerebral cortex[20]. ABHD6 is characterized by a unique Ser148-Asp278-His306 catalytic triad that is responsible for 2-AG hydrolysis[21]. The infancy of the ABHD6 field when compared to MGL and FAAH, is mostly related to limited direct information of the ABHD6 structure. In this paper, we report the design and structure-activity relationship studies of novel “signature templates” that resulted in highly potent and specific ABHD6 inhibitors.
1.2. Known ABHD6 inhibitors
To date, a small number of carbamates and triazole-ureas ABHD6 inhibitors have been reported, mostly by the Cravatt Lab[22] (Figure 1). In 2007, Cravatt and co-workers reported the identification of WWL70 the first carbamate ABHD6 inhibitor. Its potency (IC50 = 70 nM) and selectivity against other serine hydrolases were determined by application of the activity-based protein profiling (ABPP) assay[22b]. Very recently, JZP-430 (Fig. 1), a 1,2,5-thiadiazole carbamate inhibitor has been reported[23]. Also, the Cravatt Lab has disclosed potent and selective triazole-ureas ABHD6 inhibitors KT-182, KT185 and KT-203 (Fig. 1). Their activity for ABHD6 was determined by using recombinant mouse ABHD6 protein overexpressed in HEK293T cells with inhibitory potency in the range of 3.9–15.1 nM[22a]. The selectivity profile of the triazole-urea inhibitors was determined by the ABPP assay[22a]. All reported ABHD6 inhibitors interacted irreversibly with the enzyme forming carbamylating adducts with the nucleophilic catalytic residue Ser148 of ABHD6[22a, 24].
Figure 1.

Known ABHD6 inhibitors
2. Chemistry
Screening in-house sample collection in ABHD6 fluorescence assays resulted in the identification of carbamate ABHD6 inhibitor 1 (Fig. 2), which showed moderate potency for hABHD6 (IC50 = 198 nM). The discovery of initial lead 1 prompted us to initiate target-based lead optimization to improve potency and selectivity for ABHD6. Toward that end, we have conducted structure-activity relationship (SAR) studies to: (a) probe the pharmacophoric features of the carbamate head group of the molecule; (b) modify the cycloamine-linker moiety; and (c) alter the hydrophobic aromatic region by introducing polar groups to lower ClogP and increase tPSA to improve water solubility and achieve good oral bioavailability.
Figure 2.
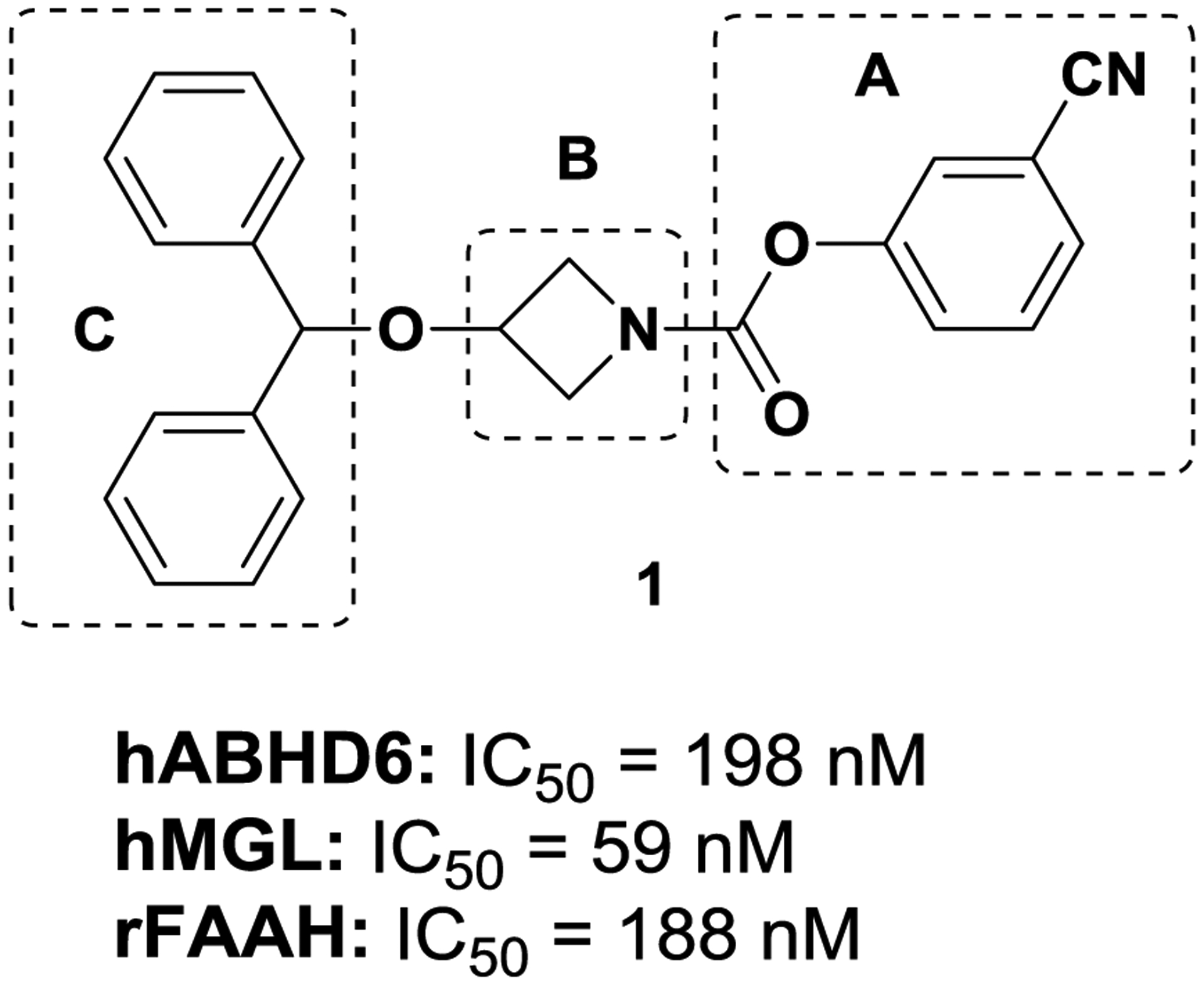
ABHD6 initial screening hit
Region A:
We have incorporated a variety of carbamate groups commonly used in serine hydrolases[25]. The pharmacophoric region of the molecule (A; Fig. 2) is predicted to occupy the hydrophilic region of the enzyme adjacent to the catalytic site where the polar acyl-glycerol group of the substrate 2-AG is presumably positioned. Even though this part of the ligand departs upon formation of a covalent adduct between ligand and catalytic residue Ser148 of ABHD6 (carbamylation step), still, it plays a critical role in forming a favorable adaptive “precovalent” interaction with the protein to facilitate the formation of the covalent adduct.
Region B:
We have modified the cycloamine-linker (B) between the distal aromatic region (C) and the carbamate pharmacophoric region (A) (Fig. 2) within individual analogs to alter the structural geometry and conformation of the molecule to interrogate ligand potency and selectivity for the target.
Region C:
We have introduced structural modifications of region C (Fig. 2) including aromatic and heteroaromatic moieties bearing polar groups to: (1) explore van der Waals hydrophobic contacts with the protein to increase affinity; and (2) exploit a polar region present at the ABHD6’s binding pocket[26] to improve water solubility and bioavailability.
2.1. Synthesis
The compounds needed to delineate the SAR for this study were prepared according to schemes 1–6.
Scheme 1. General strategy for the synthesis of carbamates (1, 5a-6c).
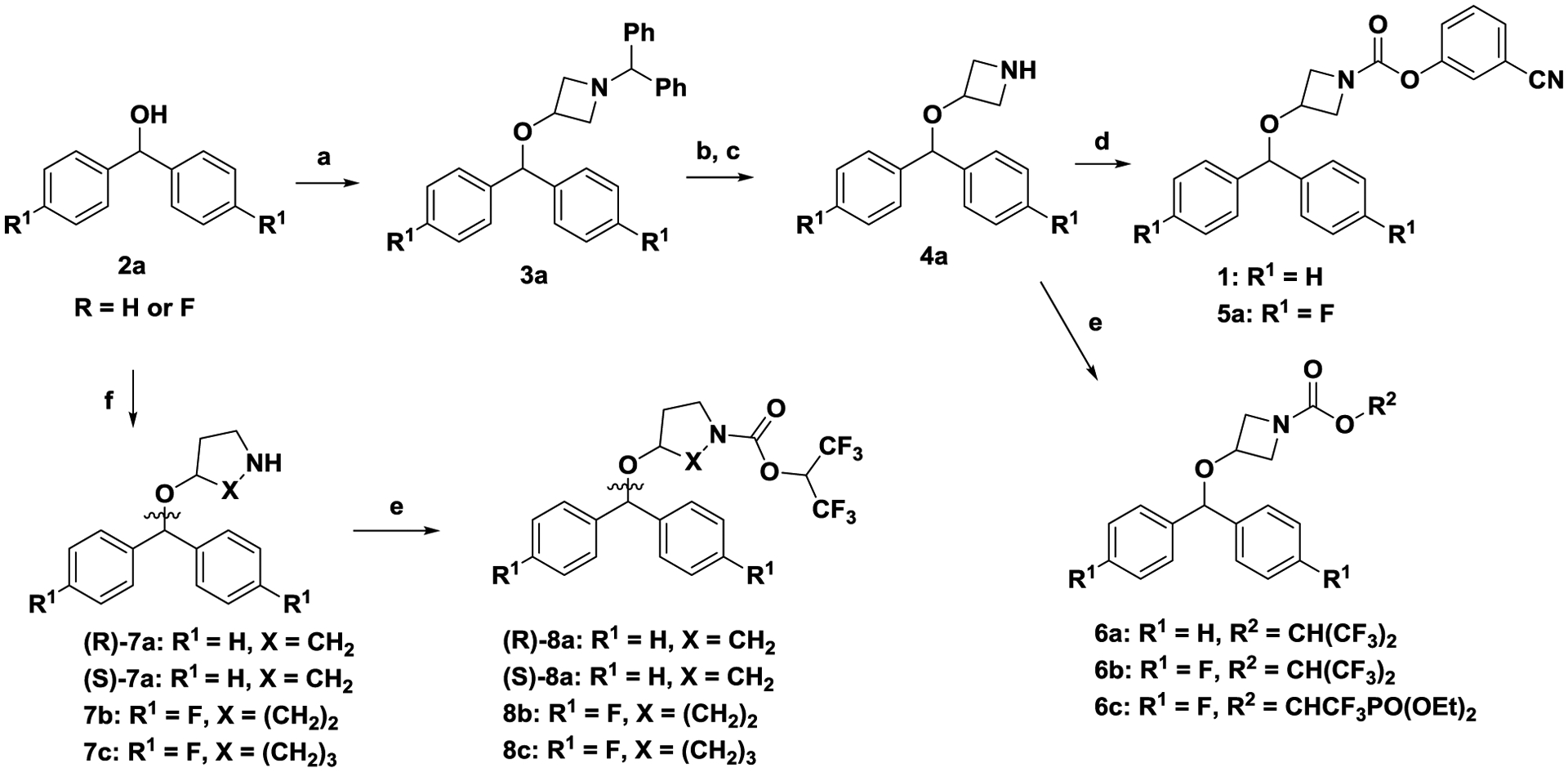
Reagents and conditions. (a) 1-benzhydrylazetidin-3-ol, para-toluenesulfonic acid, toluene, reflux, 3h; (b) a-chloroethyl chloroformate, CH2Cl2, 24 h, RT; (c) NaOH, MeOH, 3h, RT; (d) triphosgene, 3-hydroxybenzonitrile, Et3N, CH2Cl2. 0 °C to RT, 1 h; (e) triphosgene, 1,1,1,3,3,3-hexafluoropropan-2-ol, N,N-diisopropylethylamine, CH2Cl2, 0 °C to RT, 1 h; (f) (R)-pyrrolidin-3-ol, or (S)-pyrrolidin-3-ol, 4-hydroxypiperidine or azepan-4-ol, para-toluenesulfonic acid, toluene, reflux, 3 h.
Scheme 6. General strategy for the synthesis of carbamates (25a-25b, 25c, 26a-16b).
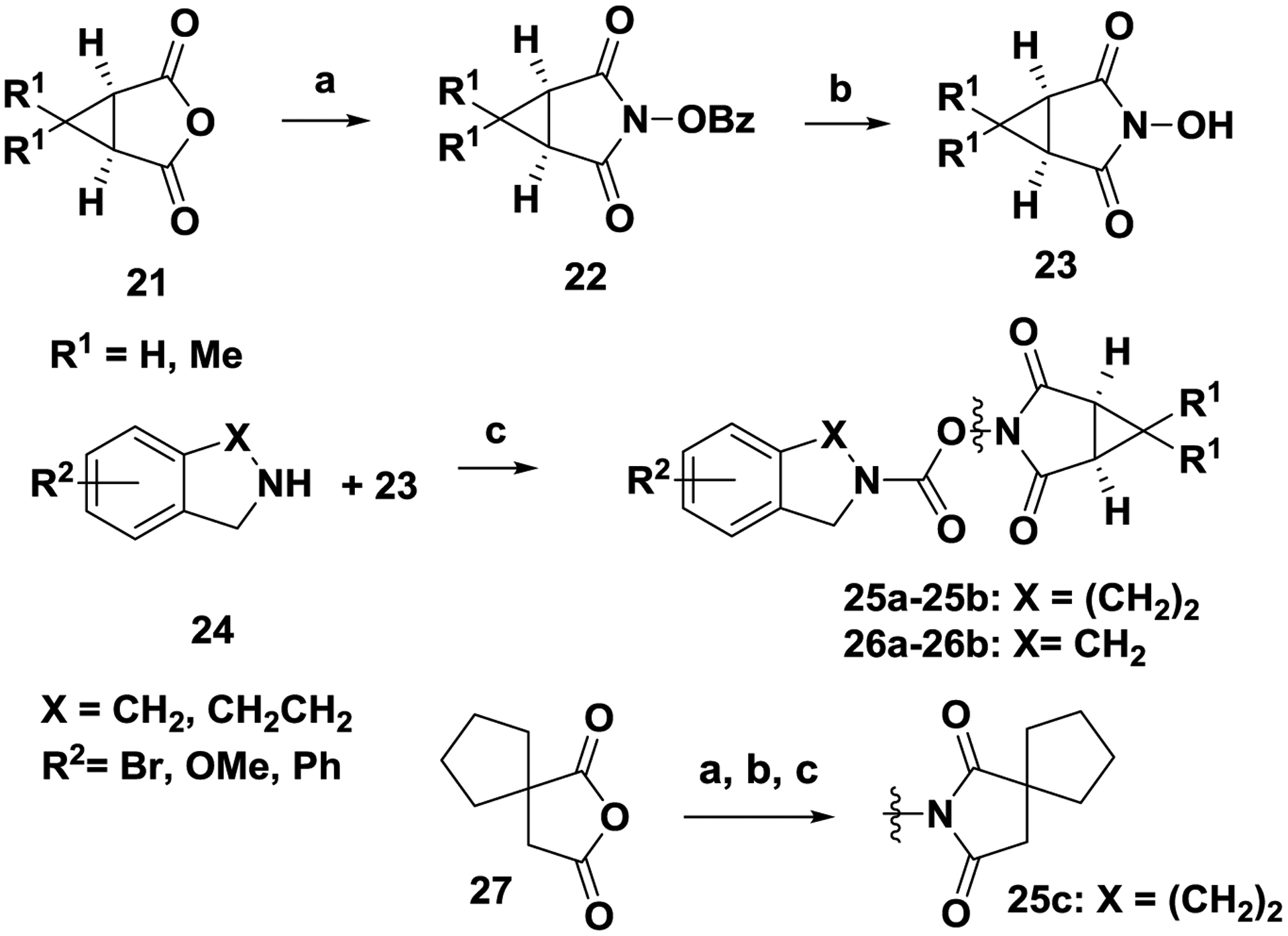
Reagents and conditions. (a) O-benzylhydroxylamine.HCl, N-methylmorpholine, AcOH, toluene, 110 °C, 2 h; (b) H2, Pd/C, 1:1 MeOH/EtOAc, 24 h, RT; (c) 23, triphosgene, N,N-diisopropylethylamine, 0 °C to RT, 1 h.
2.1.1. Synthesis of benzhydryl carbamates hABHD6 inhibitors (Tables 1).
Table 1.
Benzhydryl carbamates hABHD6 inhibitors
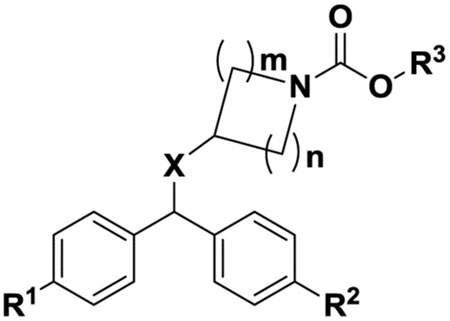
| Compd | R1 | R2 | R3 | X | m | n | hABHD6 | hMGL | hFAAH | ClogPd |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | H | H | 3-CNPh | O | 1 | 1 | 198±2.8 | 59±2.6 | 32% @1 uM | 4.65 |
| 5a | F | F | 3-CNPh | O | 1 | 1 | 581±5.6 | 66.1±2.4 | 33% @ 0.1 uM | 4.93 |
| 6a | H | H | CH(CF3)2 | O | 1 | 1 | 32.4±2.9 | 12.1±2.2 | inactivec | 5.34 |
| 6b | F | F | CH(CF3)2 | O | 1 | 1 | 60.1±3.0 | 6.4±1.5 | inactive | 5.63 |
| 6c | H | H | CF3CHPO(OEt)2 | O | 1 | 1 | 368±9.8 | inactive | inactive | 4.43 |
| (R)-8a | F | F | CH(CF3)2 | O | 2 | 1 | 74%@10uMb | 1337±103 | inactive | 5.42 |
| (S)-8a | F | F | CH(CF3)2 | O | 2 | 1 | 47%@10 uM | 101±11.1 | inactive | 5.42 |
| 8b | F | F | CH(CF3)2 | O | 2 | 2 | 1342±15.6 | 22.8±5.2 | inactive | 5.21 |
| 8c | F | F | CH(CF3)2 | O | 2 | 3 | 21%@10 uM | inactive | inactive | 5.77 |
| 9d | F | F | CH(CF3)2 | NMe | 1 | 1 | 283±5.7 | 83.5±1.8 | inactive | 6.07 |
| 10d |
|
37%@10 uM | 348±7.5 | inactive | 5.35 | |||||
IC50±SEM performed in triplicate and determined from eight concentrations.
% inhibition was conducted in three points assay with inhibitor at concentration 1, 10 and 100 μM.
inactive tested at 1uM.
ClogP values were calculated using ChemDraw, version 18.2. IC50 values were calculated using Prism software (GraphPad).
Scheme 1:
Coupling of bis(4-fluorophenyl) methanol 2a (R = F) with 1-benzhydrylazetidin-3-ol upon treatment with para-toluenesulfonic acid in toluene under reflux with continuous removal of water (Dean-Stark trap) afforded 3a. Debenzylation of 3a in a two-step process[27], first with α-chloroethyl chloroformate in dichloromethane to produce the corresponding α-chloroethyl carbamate (not shown) and second with NaOH/MeOH afforded amine 4a. Amine 4a was treated either with triphosgene/triethylamine and 3-hydroxybenzonitrile to produce carbamate 5a; or with triphosgene/N,N-diisopropylethylamine and 1,1,1,3,3,3-hexafluoropropan-2-ol to produce carbamate 6b. Modifications of the azetidine nucleus of 6a and 6b with introduction of 5–7 ring-sized cyclic produced amines 7a—7c which were converted to carbamates 8a-8c according to according to scheme 1.
Scheme 2:
Scheme 2. Synthesis of carbamate 9d.
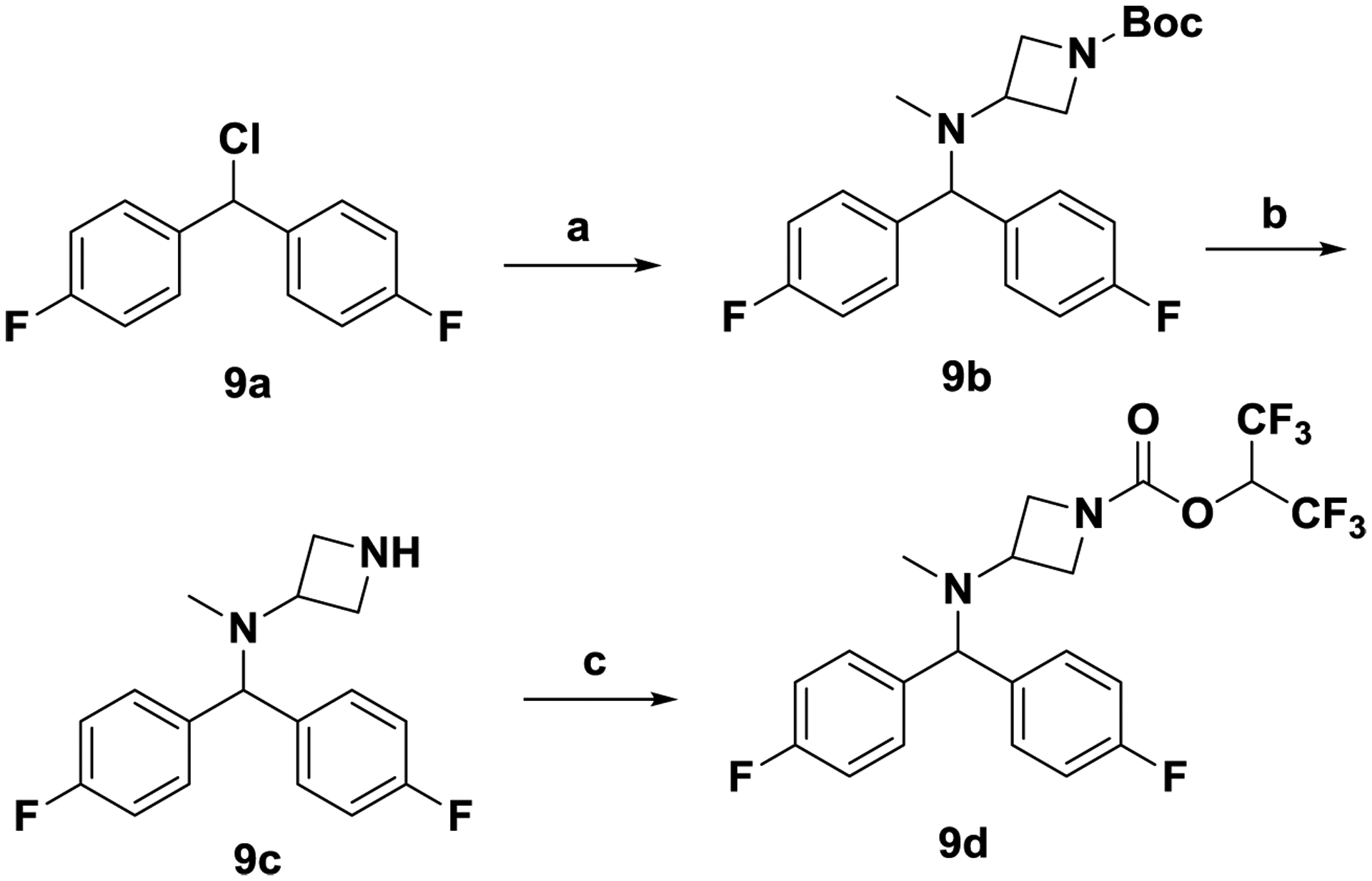
Reagents and conditions. (a) tert-butyl 3-(methylamino)azetidine-1-carboxylate, K2CO3, CH3CN, 24 h, RT; (b) TFA, CH2Cl2, 24 h, RT; (c) triphosgene, 1,1,1,3,3,3-hexafluoropropan-2-ol, N,N-diisopropylethylamine, CH2Cl2, 0 °C to RT, 1 h.
4,4’-(Chloromethylene)bis(fluorobenzene) 9a was treated with tert-butyl 3-(methylamino)azetidine-1-carboxylate and potassium carbonate in acetonitrile to afford 9b. Unmasking the Boc group of 9b with trifluoroacetic acid in dichloromethane afforded amine 9c, which was converted to carbamate 9d as described in scheme 1.
Scheme 3:
Scheme 3. Synthesis of carbamate 10d.
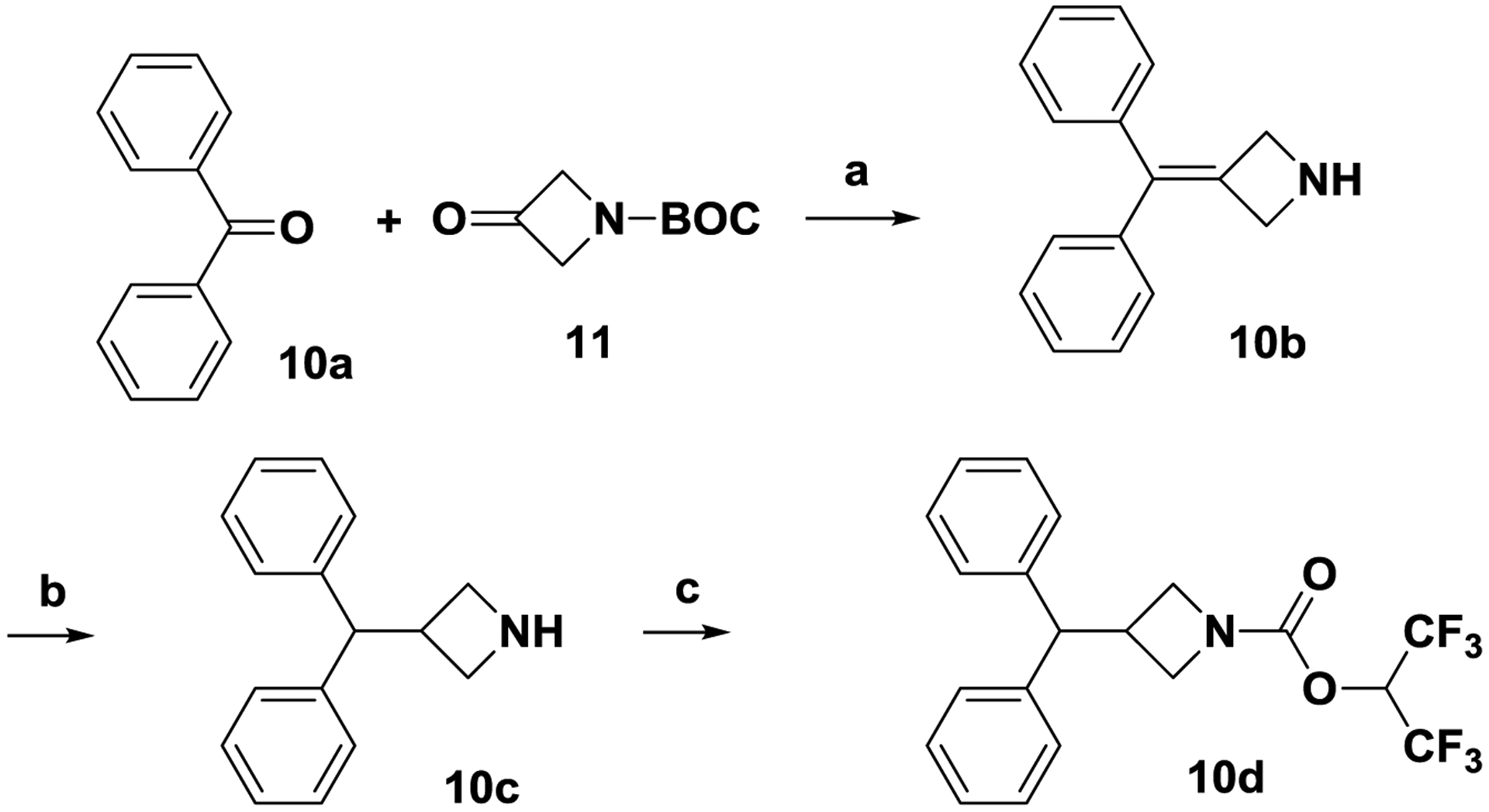
Reagents and conditions. (a) Zn, TiCl4, THF, 60 °C, 8 h; (b) Pd/C, H2, 40 psi, 24h, RT; (c) triphosgene, 1,1,1,3,3,3-hexafluoropropan-2-ol, Et3N, CH2Cl2, 0 °C to RT, 1 h.
The deo-oxygenated benzhydryl carbamate 10d was prepared as follows. Reductive coupling[28] of ketones 10a and 11 in the presence of Zn and TiCl4 afforded olefin 10b. Hydrogenation (H2, Pd/C) of olefin 10b produced amine 10c, which was converted to carbamate 10d as described in scheme 1.
2.1.2. Synthesis of tetrahydroisoquinoline and isoindoline carbamates hABHD6 inhibitors (Tables 2 and 3):
Table 2.
Tetrahydroisoquinoline carbamates hABHD6 inhibitors
| Compd | R1 | R2 | R3 | n | hABHD6 | hMGL | hFAAH | ClogPd |
|---|---|---|---|---|---|---|---|---|
| 14a | H | H | CH(CF3)2 | 2 | 84.2±1.3 | inactive | inactivec | 3.96 |
| (RS)-14b | H | CH3 | CH(CF3)2 | 2 | inactive | inactive | inactive | 4.47 |
| (R)-14c | H | Ph | CH(CF3)2 | 2 | 40%@10 uMb | 40%@ 1uM | inactive | 5.51 |
| 14d | 8-Br | H | CH(CF3)2 | 2 | 58±3.5 | inactive | inactive | 4.82 |
| 14e | 7-Br | H | CH(CF3)2 | 2 | 394±4.3 | inactive | inactive | 4.82 |
| 14f | 6-Br | H | CH(CF3)2 | 2 | 72.7±1.5 | inactive | inactive | 4.82 |
| 14g | 5-Br | H | CH(CF3)2 | 2 | 10±0.5 | inactive | inactive | 4.82 |
| 18a | 5-OMe | H | CH(CF3)2 | 2 | 8±2.3 | inactive | inactive | 3.87 |
| 18b | 5-O(CH2)3F | H | CH(CF3)2 | 2 | 30.6±1.2 | 28%@1 uM | inactive | 4.36 |
| 17a | 5-peperidine | H | CH(CF3)2 | 2 | 66%@10 uM | inactive | inactive | 4.79 |
| 17b | 5-morpholine | H | CH(CF3)2 | 2 | 81%@10 uM | inactive | inactive | 3.41 |
| 14h | 5-Ph | H | CH(CF3)2 | 2 | 49±2.6 | 83%@1 uM | inactive | 3.87 |
| 14i | H | H | CH(CF3)2 | 3 | 29%@10 uM | inactive | inactive | 4.37 |
| 14j | 5-Br | H | CF3CHCH2OH | 2 | 35%@10 uM | inactive | inactive | 3.70 |
| 14k | 5-Br | H | CH(CHF2)2 | 2 | 55%@10 uM | inactive | inactive | 3.64 |
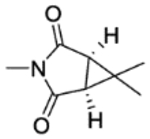
| 25a | 5-Br | H |
|
2 | 252±5.8 | inactive | inactived | 3.03 |
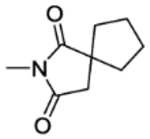
| 25b | 5-Br | H |
|
2 | 22±1.9 | inactive | inactive | 4.33 |
| 25c | 5-Br | H |
|
2 | 128±7.6 | inactive | inactive | 4.25 |
IC50±SEM performed in triplicate and determined from eight concentrations.
% inhibition was conducted in three points assay with inhibitor at concentration 1, 10 and 100 μM.
inactive tested at 1uM.
ClogP values were calculated using ChemDraw, version 18.2. IC50 values were calculated using Prism software (GraphPad).
Table 3.
Isoindoline carbamates hABHD6 inhibitors
| Compd | R1 | R2 | X | hABHD6 | hMGL | hFAAH | ClogPd |
|---|---|---|---|---|---|---|---|
| 15a | 4-Br | CH(CF3)2 | C | 114.7±1.9 | inactive | inactivee | 4.36 |
| 15b | 5-Br | CH(CF3)2 | C | 13.4±3.6 | 29%@1uMb | inactive | 4.36 |
| 15c | 5-Br | CH(CF3)2 | N | 191.2±5.8 | inactive | inactive | 4.36 |
| 22a | 5-OMe | CH(CF3)2 | C | 8.3±0.9 | inactive | inactive | 3.42 |
| 22b | 5-OCH2CHF2 | CH(CF3)2 | C | 6.0±0.3 | 48%@1 uM | inactive | 3.89 |
| 22c | 5-O(CH2)3F | CH(CF3)2 | C | 4.0±0.8 | inactive | inactive | 3.99 |
| 22d | 5-O(CH2cycloprane) | CH(CF3)2 | C | 4.1±0.8 | 45%@1 uM | inactive | 4.39 |
| 18a | pyrrolidine | CH(CF3)2 | C | 51±3.9 | inactive | inactive | 3.77 |
| 18b | morpholine | CH(CF3)2 | C | 18±2.1 | inactive | inactive | 2.95 |
| 22e | dioxothiomorpholine | CH(CF3)2 | C | 42±2.9 | inactive | inactive | 1.99 |
| 22f |
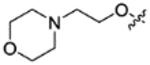
|
CH(CF3)2 | C | 8±0.6 | inactive | inactivef | 3.53 |
| 22g |
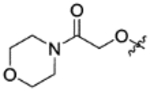
|
CH(CF3)2 | C | 9.9±1.5 | inactive | inactive | 2.73 |
| 22h |
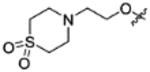
|
CH(CF3)2 | C | 4.3±1.8 | inactive | inactive | 2.57 |
| 22i |
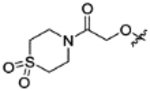
|
CH(CF3)2 | C | 19.7±2.1 | inactive | inactive | 1.76 |
| 22j |
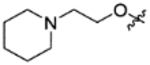
|
CH(CF3)2 | C | 11.9±1.1 | inactive | 71% @ 1uM | 4.75 |
| 22k |
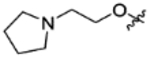
|
CH(CF3)2 | C | 18.6±1.4 | inactive | inactive | 4.19 |
| 22l |
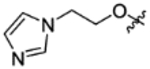
|
CH(CF3)2 | C | 2.5±0.5 | inactive | inactive | 3.11 |
| 23 |
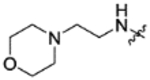
|
CH(CF3)2 | C | 17.9±3.1 | inactive | inactive | 3.21 |
| 26a | 5-OMe |

|
C | 24.4±1.1 | inactive | inactive | 3.03 |
| 26b | 5-morpholine |
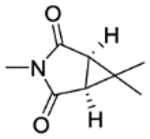
|
C | 44±2.8 | inactive | inactive | 2.59 |
IC50±SEM performed in triplicate and determined from eight concentrations.
% inhibition was conducted in three points assay with inhibitor at concentration 1, 10 and 100 μM.
inactive tested at 1uM.
ClogP values were calculated using ChemDraw, version 18.2. IC50 values were calculated using Prism software (GraphPad).
Synthetic protocols of prepared analogs of Tables 2 and 3 are shown in schemes 4, 5 and 6.
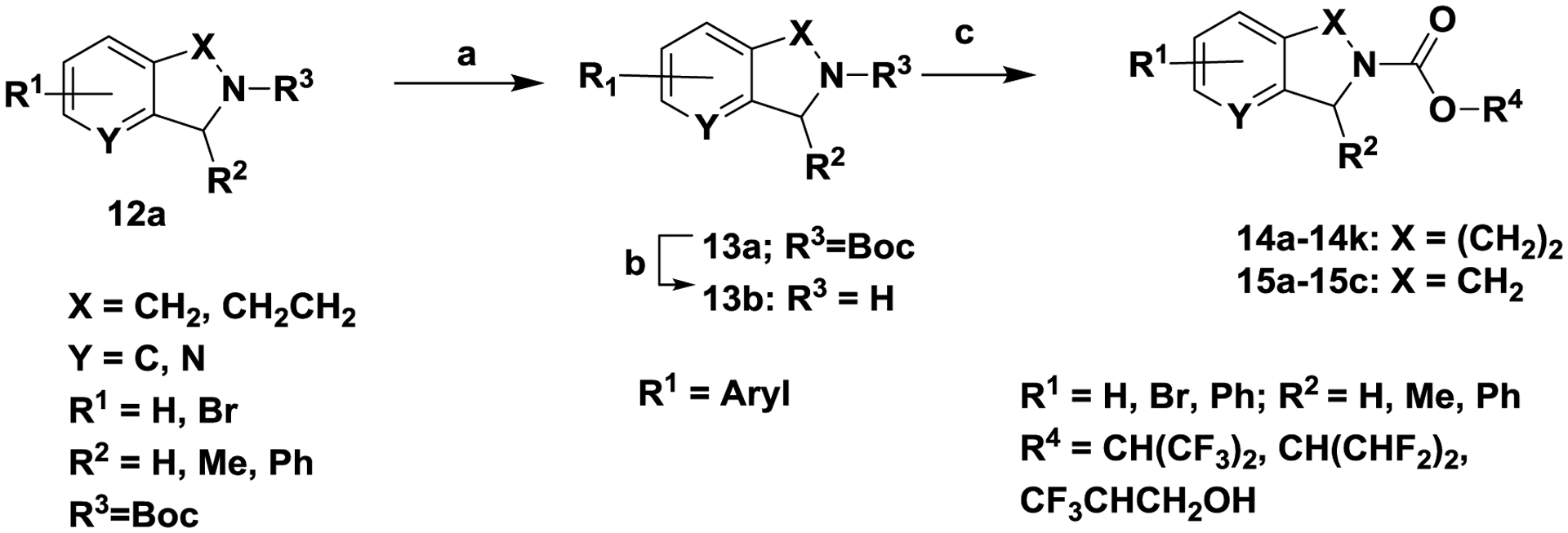
Scheme 4. General strategy for the synthesis of carbamates (14a-14k, 15-a-15c).
Reagents and conditions. (a) Ar-B(OH)2, Pd(PPh3)4, K2CO3, 80 °C, 4 h; (b) trifluoroacetic acid, CH2Cl2; 24 h , RT; (c) 1,1,1,3,3,3-hexafluoropropan-2-ol or 1,1,3,3-tetrafluoropropan-2-ol, triphosgene, N,N-diisopropylethylamine, 0 °C to RT, 1 h
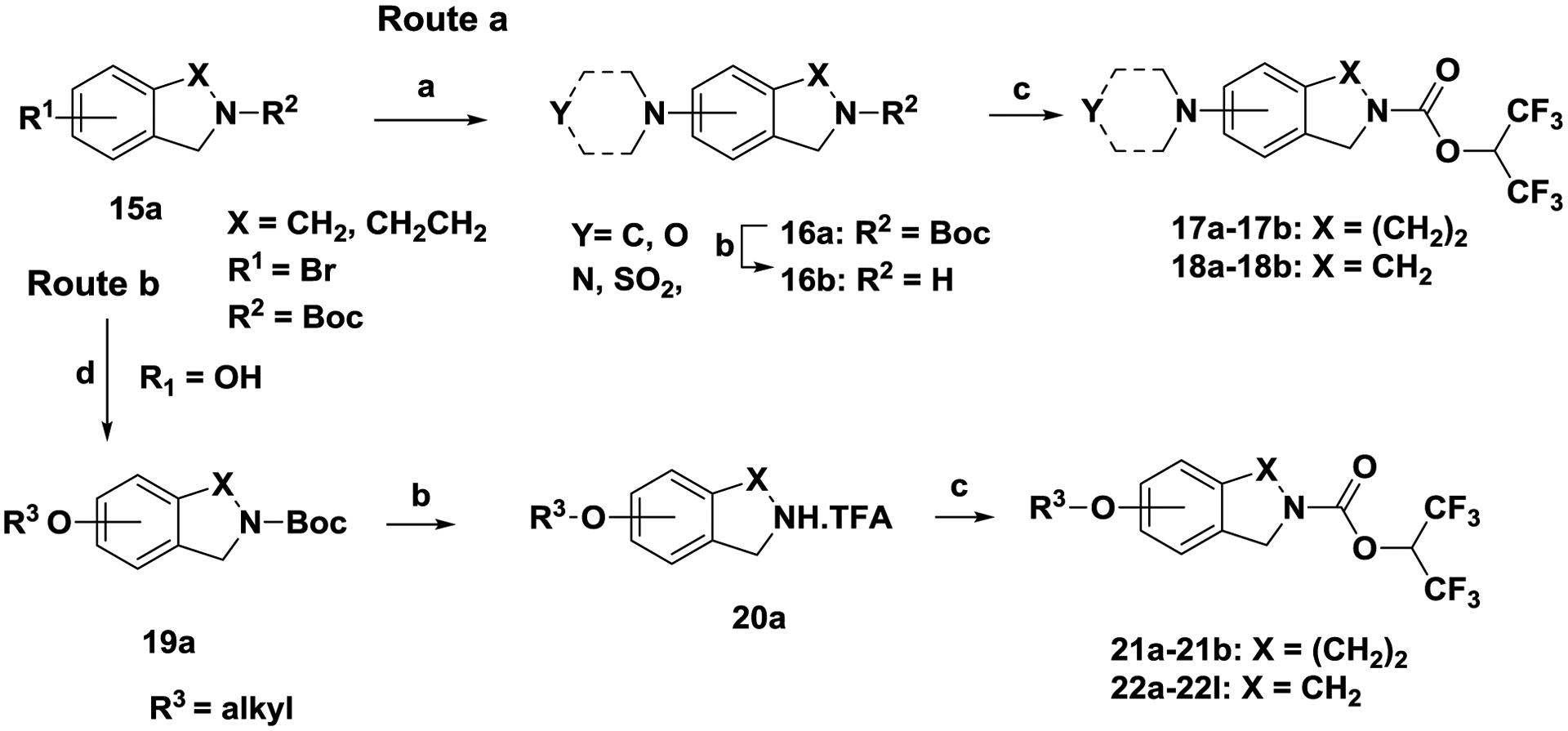
Scheme 5. General strategy for the synthesis of carbamates (17a-17-b, 18a-18b, 21a-21-b, 22a-22l).
Reagents and conditions. (a) Pd2(dba)3, 2-(di-tert-butylphosphino)biphenyl, sodium tert-butoxide, N-methylpiperazine, toluene, 80 °C, 4 h; (b) TFA, CH2Cl2j 24 h, RT; (c) 1,1,1,3,3,3-hexafluoropropan-2-ol, triphosgene, N,N-diisopropylethylamine, 0 °C to RT, 1 h; (d) R3-halide, NaH, DMF, °C to RT, 24 h.
Biaryl analogs
Scheme 4.
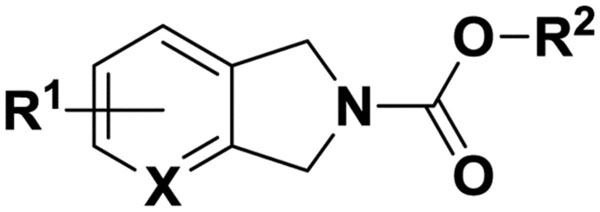
Palladium mediated cross-coupling reaction between aryl-bromide 12a (R1 = Br) and an appropriately substituted aryl boronic acid in the presence of tetrakis (triphenylphosphine) palladium (0), K2CO3, dioxane and water produced 13a (R1 = aryl, heteroaryl). Unmasking the Boc group of 13a with trifluoroacetic acid in dichloromethane afforded amine 13b, which was converted to tetrahydroisoquinoline carbamates 14a-14k and isoindoline carbamates 15a-15c as described in scheme 1.
Nitrogen- and oxygen-linked analogs
Scheme 5
Route a:
Palladium mediated cross-coupling reaction between aryl-bromide 15a (R1 = Br) and an appropriate amine (i.e. morpholine, pyrrolidine) in the presence of Pd2(dba)3 / 2-(di-tert-butylphosphino) biphenyl, sodium tert-butoxide and toluene afforded 16a. Unmasking the Boc group of 16a with trifluoroacetic acid in dichloromethane afforded the amine 16b, which was converted to converted to tetrahydroisoquinoline carbamates 17a-17b and isoindoline carbamates 18a-18b as described in scheme 1.
Route b:
Alkylation of phenol 15a (R1 = OH) with an appropriately substituted alkyl halide (i.e. CHF2CH2Br) in the presence of sodium hydride and N,N-dimethylformamide afforded alkoxy analog 19a. Unmasking the Boc group of 19a with trifluoroacetic acid in dichloromethane afforded amine 20a, which was converted to converted to tetrahydroisoquinoline carbamates 21a-21b and isoindoline carbamates 22a-22l as described in scheme 1.
2.1.3. Synthesis of imide-carbamates hABHD6 inhibitors (Table 2; analogs 25a-25c, and Table 3, analogs 26a,26b).
Scheme 6:
Treatment of anhydride 21 with O-benzylhydroxylamine in the presence of N-methylmorpholine and acetic acid produced imide 22. Debenzylation of 22 with catalytic hydrogenation (H2, Pd/C) afforded hydroxylamine 23. Coupling of 23 with amine 24 in the presence of triphosgene and N-diisopropylethylamine produced tetrahydroisoquinoline imide-carbamates 25a-25b and isoindoline imide-carbamates 26a-26b. The tetrahydroisoquinoline imide-carbamate 25c was similarly prepared from anhydride 27.
3. Results and discussion
All synthesized compounds were assessed in fluorescence-based assays using solubilized membrane fractions containing of full-length hABHD6 with the fluorogenic substrate arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE)[29] and the hΔ29–4-hABHD6 variant and fluorogenic substrate valeroyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (VMMCE)[29]. Both assays were reported[29] to produce similar findings. Compounds that inhibited hABHD6 with an IC50 value of < 30 nM were evaluated in selectivity counterscreens for their ability to inhibit rat FAAH (rFAAH) and human-recombinant FAAH (hFAAH)[29] using the fluorogenic substrate arachidonoyl 7-amino-4-methylcoumarin amide (AAMCA)[30], and recombinant human MGL (hMGL) and purified rat MGL (rMGL) using the fluorogenic substrate arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE)[31]. Furthermore, selected compounds were evaluated for their ability to bind to CB1 and CB2 receptors using rat brain[32] or HEK293 cell membranes expressing mouse CB2 (mCB2) or human CB2 (hCB2),[33] respectively, via competition-equilibrium binding using [3H]CP-55,940[33b, 33c, 34] to minimize/eliminate any potential cross-reactivity due to potential common pharmacophoric features. Below we describe our structure-activity relationship (SAR) studies for a selection of compounds shown in Tables 1–3.
3.1. Benzhydryl-carbamates ABHD6 inhibitors (Table 1).
Our SAR objectives were to design new “signature templates” for ABHD6 bearing the hexafluoroisopropylcarbamate-carbamate (HFIPC), commonly used in serine hydrolase inhibitors[25, 35]. The starting point compound 1 exhibited weak activity for ABHD6 and modest selective against FAAH and MGL. Introduction of the HFIPC pharmacophore[36](entry 6a) resulted in 5-fold potency jump for both ABHD6 and MGL and it was found to be inactive against FAAH. The more polar phosphonate pharmacophore 6c was about 2-fold less active for ABHD6, but it was selective against both MGL and FAAH. Introduction of fluorine atoms at the phenyl moieties (entries 5a, 6b) to protect of potential cytochrome P450 metabolism resulted in about 2-fold loss in potency for ABDH6. Next, we prepared 5–7 ring-sized cycloamine linkers (entries 8a-8c) to further interrogate potency and selectivity against ABHD6. All analogs exhibited marked loss in potency for ABHD6.
Next, we investigated spacers between the azetidine nucleus and the benzhydryl group of 6b. The nitrogen analog 9d was weaker against both ABHD6 and MGL, while the truncated analog 10d was practically inactive against ABHD6 (10d vs 6a). At this stage, we have confirmed that the HFIPC carbamate has shown preference for MGL inhibition, as previously reported[36b, 37], with a few exceptions (entries 6a, 6b), where they also showed good potency for ABHD6. In order to eliminate the MGL activity of the molecule, we have pursued new “signature templates” specific to ABHD6.
3.2. Tetrahydroisoquinoline carbamates ABHD6 inhibitors.
We have limited the rotatable bonds of the benzhydryl group of 9d to explore constrained tetrahydroisoquinoline analog 9e (Fig. 3; ligand design), which showed good potency for both ABHD6 and MGL. The tetrahydroisoquinoline moiety represented a good replacement of the benzhydryl group by improving ABHD6 potency 5-fold (9e vs 9d), but still it lacked good selectivity for ABHD6. Then, we crafted an even smaller constrained ligand tetrahydroisoquinoline 14a by eliminating the azetidine nucleus present at 9e. Gratifyingly, we found that analog 14a exhibited good affinity for ABHD6 (IC50 = 84 nM), while lacked activity against MGL and FAAH at 1 uM concentration.. Armed with this new finding, we decided to expand our SAR studies (Table 2) and further improve ligand potency and selectivity. Introduction of either a methyl group (RS)-14b or a phenyl group (R)-14c at position-2 of the tetrahydroisoquinoline ring of 14a resulted in marked losses in potency. We have postulated that substitutions at the vicinity of the carbamate pharmacophore impacted the interaction between ligand and catalytic residue Ser148 of ABHD6 to form the anticipated carbamylation adduct. Next, we probed the aromatic region of the molecule to explore which substitution position could improve ligand affinity. Introduction of a bromine group at the various aromatic positions (entries 14d-14g) proved that position-5 of the tetrahydroisoquinoline nucleus was the best substitution position to achieve the highest potency (IC50 = 10 nM). Similarly, the 5-methoxy analog 18a was highly potent (IC50 = 8 nM). The longer 5-propyloxy analog 18b resulted in 4-fold loss in potency (18b vs 18a), while the more polar cyclic amines piperidine 17a and morpholine 17b exhibited marked losses in potency. The phenyl analog 14h also lost 5-fold in potency.
Figure 3.
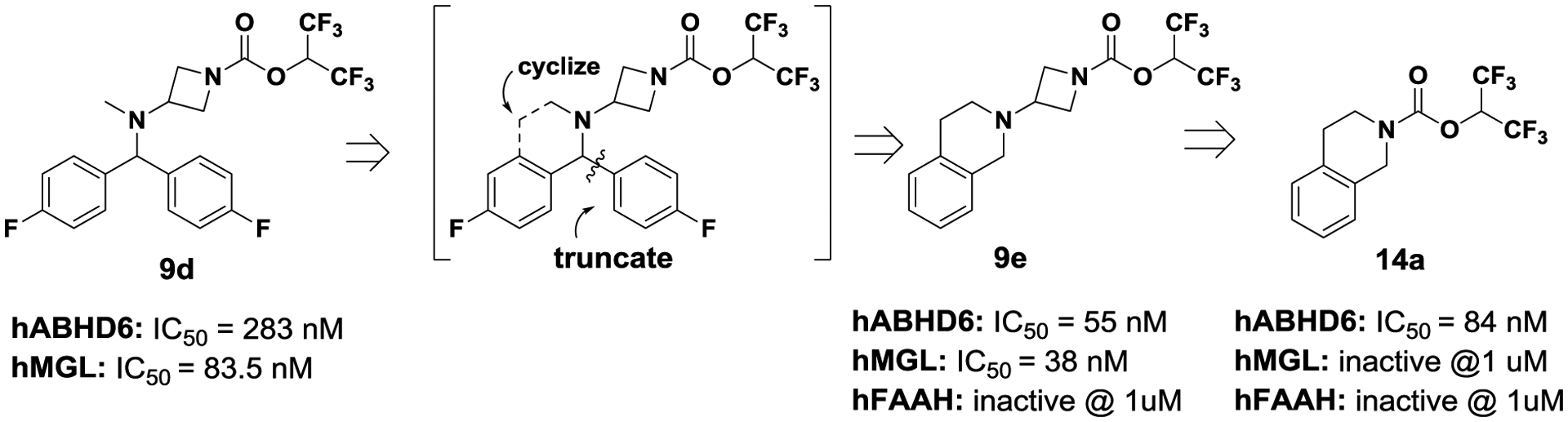
Design of tetrahydroisoquinoline ABHD6 inhibitors
Enlarging the tetrahydroisoquinoline ring of 14a to benzo-azepine 14i was detrimental to the affinity of the ligand. Examining the selectivity profile of the tetrahydroisoquinoline analogs (Table 2), all compounds were practically inactive against both MGL and FAAH at 1 uM concentration. At this stage, we have confirmed that the tetrahydroisoquinoline moiety represented a powerful recognition template for ABHD6. Next, we modified the pharmacophoric region of the molecule to lower hydrophobicity of the molecule by reducing the number of fluorine atoms of the HFIPC pharmacophore group. The trifluoromethyl glycol group (entry 14j), which was previously used in MGL inhibition[38], was found to be practically inactive against ABHD6, as well as the bis(difluoro) analog 14k. We have postulated that the loss of potency of 14j and 14k was due to the decreased electrophilicity of the pharmacophore group impacting its ability to form the carbamylation adduct with the catalytic residue Ser148 of ABHD6.
3.3. Design imide-type ABHD6 inhibitors:
Even though the imide-type pharmacophores had been studied on MGL inhibition[36b], we have decided to use bulkier bicyclic fused- and spiro-imide moieties (entries 25a-25c; Table 2) to introduce steric hindrance at the pharmacophoric region of the ligand as a rational optimization approach to further interrogate potency and good selectivity for the target. The fused cyclopropane-succinimide carbamate 25a was weakly active for ABHD6, while the gem-dimethyl cyclopropane analog 25b exhibited high potency (IC50 = 22 nM) and selectivity for the target. The bulkier spiro-cyclopentane analog 25c lost about 6-fold in potency (25c vs 25b). Noteworthy to mention that several potent tetrahydroisoquinoline ABHD6 inhibitors were evaluated and found to be inactive against cannabinoid receptors CB1 and CB2 at 1 uM concentration (data not shown).
3.4. Isoindoline carbamates hABHD6 inhibitors.
Confident that the tetrahydroisoquinoline template represented a recognition template for ABHD6, next we investigated the smaller isoindoline scaffold (Table 3).
Introduction of a bromine group at position-4 of the isoindoline nucleus (entry 15a) inhibited ABHD6 with about 10-fold loss in potency when compared to the analogous tetrahydroisoquinoline 14g. In contrast, when we moved the bromine at position-5 (entry 15b) we regained the potency for the target with an IC50 value of 13 nM. The more polar pyrrolo-pyridine analog 15c was about 15-fold less potent for ABHD6 (15c vs 15b). We have postulated that the nitrogen containing aromatic moiety forms unfavorable electrostatic repulsions with residue(s) of the binding pocket of ABHD6, which have caused loss in ligand affinity. The 5-methoxy analog 22a exhibited single-digit nanomolar potency for ABHD6 (IC50 = 8 nM). Introduction of longer aliphatic tails (entries 22b, 22c), by masking the terminal carbon of the aliphatic tail with fluorine groups to prevent cytochrome P450 metabolic ω-oxidation, resulted in enhancement of affinity by about 2-fold. Similar high potency (IC50 = 4 nM) was also determined for the cyclopropane analog 22d.
Next, we explored the polar region of the binding pocket of ABHD6[26] with the introduction of hydrophilic groups to improve water solubility and oral bioavailability. To that end, we introduced cyclic amines (entries 18a, 18b), which were found to be potent ABHD6 inhibitors. Then, we introduced polar groups deeper into the binding pocket with the addition of a short alkyl chain attached at position-5 of the aromatic ring (entries 22f-22l, 23). All analogs showed high potency for ABHD6 with IC50 values in the range of 2 to 20 nM. Also, these analogs represented a diverse group of inhibitors with ClogP values ranging within three log units 1.76–4.75. We anticipate the more polar analogs to exhibit reduced blood-brain barrier permeability, representing potential periphery restricted ABHD6 inhibitors. Lastly, we have prepared the dimethyl cyclopropane imide-pharmacophore, which we have identified in the tetrahydroisoquinoline series (entry 22b; Table 2) as a potent pharmacophore for ABHD6 inhibition.. The analogous isoindoline imide-carbamate 26a was found to be potent for ABHD6 with an IC50 value of 24 nM, while the more polar morpholine analog 25b was 2-fold weaker.
Similarly to the tetrahydroisoquinoline analogs (Table 2), all isoindoline ABHD6 inhibitors (Table 3) were also found to be highly selective for ABHD6 when tested against serine hydrolases FAAH and MGL (Table 3), as well as against cannabinoid receptors CB1 and CB2 (not shown).
3.5. Evaluation of ABHD6 inhibitors using in vitro competitive activity-based protein profiling (ABPP) assay.
We have assessed a representative ABHD6 inhibitor in the activity-based protein profiling (ABPP) assay [39] to assess its selectivity in a larger panel of serine hydrolases. Gel-based ABPP analysis was performed using membrane homogenates prepared from rat brain tissue with the rhodamine-tagged fluorophosphonate (FP-Rh) probe that was typically used to profile the serine hydrolase superfamily[11, 40]. The previously reported selective ABHD6 inhibitor WWL70[22b] was used as the positive control[41]. Protein homogenates (10 mg/mL) were incubated with representative ABHD6 inhibitor 22h (0.1, 1 and 10 μM), reference compound ABHD6 inhibitor WWL70[22b] at 10 μM or DMSO for 30 min at room temperature. The tissues were then treated with 10 μM FP-Rh probe for 45 min at room temperature. The reaction was quenched using 2x SDS-PAGE loading buffer and separated with SDS-PAGE (12% acrylamide). Fluorescence was detected using Amersham® Imager 600 with the green epi light (520 nm) as the light source. Selective ABHD6 inhibitor 22h inhibited only ABHD6 without significant inhibition of other serine hydrolases. In our experiments, 22h appeared to share similar selectivity profile, comparable to reference compound WWL70 (Fig. 4). These ABPP studies confirmed that inhibitor 22h was selective for ABHD6 in the brain, since none of the other FP-reactive serine hydrolases in this tissue were inhibited by this agent. In summary, we have identified and validated that the tetrahydroisoquinoline and isoindoline scaffolds represent potent and selective “signature templates” for ABHD6.
Figure 4.
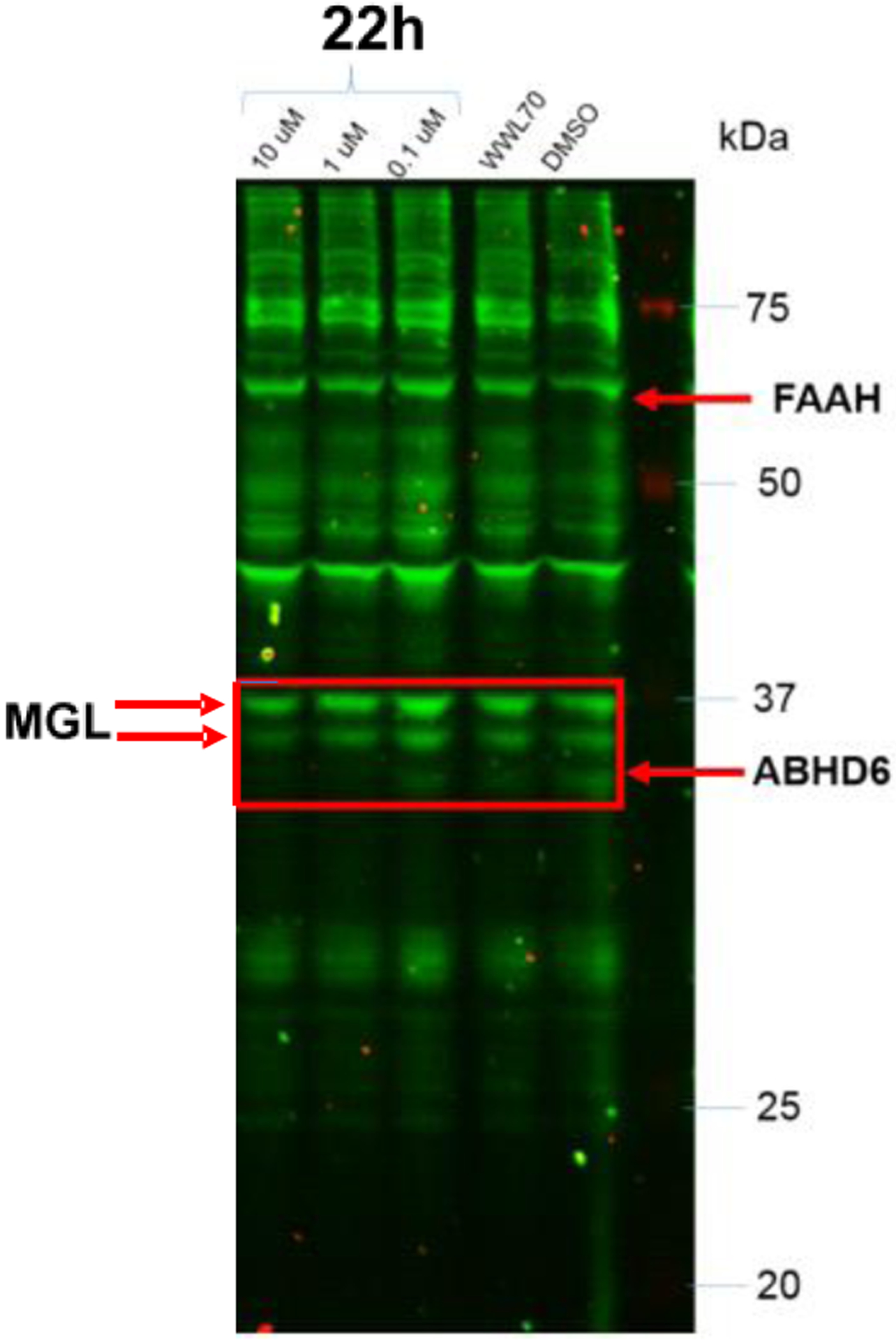
Gel-based ABPP analysis was performed using membrane homogenates prepared from rat brain tissue and the rhodamine-tagged fluorophosphonate (FP-Rh) probe. Specific ABHD6 inhibitor 22h completely inhibited ABHD6 at 0.1, 1 and 10 μM without significant inhibition of other serine hydrolases
4. ADME Properties
We have evaluated early ADME properties of few potent ABHD6 inhibitors. They were found to be stable in human, rat, and mouse plasma (purchased from BioVIT) with t1/2 > 2 hours.
The microsomal stability was assessed in human, rat, and mouse liver microsomal preparations (purchased from BioVIT), where the tested inhibitors found to exhibit stability with t1/2 in the range between 4 to 40 min (Table 4). Inhibitor 22f was the most stable molecule in human microsomes. We have also evaluated inhibitors 15b and 22f (Table 5) in cassette pharmacokinetics experiments in male CD-1 mice (purchased form Charles River) at dose: 2 mg/kg, iv and 8 mg/kg po. Based on the findings (Table 5), both compounds were brain permeable by comparing brain and plasma levels after iv administration at 15 min. Inhibitor 22f exhibited the highest plasma and brain levels drug levels and estimated oral bioavailability.
Table 4.
Stability of selected inhibitors
| Microsomes t1/2 mina | Plasma t1/2 minb | |||
|---|---|---|---|---|
| Compound | Mouse | Rat | Human | Mouse/rat/human |
| 14a | 4 | 12 | 21 | >120 |
| 22a | 5 | 6 | 11 | >120 |
| 22f | 7 | 17 | 40 | >120 |
Inhibitor was pre-incubated with liver microsomes before the reaction was initiated with NADPH or buffer (control). Following protein precipitation, the samples were analyzed using LC-MS/MS.
Inhibitor solution was made in plasma, buffer containing 0.1% BSA. The samples were analyzed by HPLC to predict in vitro plasma half-lives
Table 5.
Cassette pharmacokineticsa
| Compound | 15b | 22f |
|---|---|---|
| 15 min IV plasma (μg/mL) | 0.114 ± 0.015 | 0.633 ± 0.046 |
| 15 min IV brain (μg/g) | 0.192 ± 0.032 | 0.735 ± 0.056 |
| 30 min oral plasma (μg/mL) | 0.006 ± 0.001 | 0.180 ± 0.072 |
| 30 min oral brain (μg/g) | 0.008 ± 0.002 | 0.248 ± 0.089 |
| 60 min oral plasma (μg/mL) | 0.002 ± 0.001 | 0.140 ± 0.031 |
| 60 min oral brain (μg/g) | 0.004 ± 0.002 | 0.227 ± 0.095 |
| IV brain penetration | 1.7 | 1.2 |
| Bioavailability (Estimated) | 8% | 28% |
Male CD-1 mice (Charles River) following a single intravenous dose of ABHD6 inhibitor 2 mg/kg and an oral dose of 8 mg/kg at 15, 30 and 60 minutes timepoints (n = 3 animals per time point). The compounds were brain permeable by comparing brain and plasma levels after iv administration at 15 min.
5. Pharmacology
Retina disease:
The endocannabinoid system (ECS) has been studied in retinal neurodegenerative diseases[42]. Endocannabinoid (2-AG and AEA) levels have been investigated in normal human eyes and ocular tissues from patients with glaucoma, diabetic retinopathy or age-related macular degeneration (AMD)[43]. A recent study showed that 2-AG provided neuroprotection to bNOS-expressing retinal amacrine cells in a dose-dependent manner via the activation of both CB1R and CB2R and the neuroprotective effects of 2-AG in the retina were mediated by CB1 receptor-dependent activation of the PI3K/Akt pathway[44]. Based on the favorable pharmacokinetics properties, inhibitor 22f was assessed in in vivo studies to protect rat retina against AMPA excitotoxicity in neurodegeneration and neuroinflammation retina diseases. We have recently reported that ABHD6 inhibitor 22f attenuated the AMPA-induced glia activation and produced a dose-dependent retina neuroprotection in rats[44]. These early pharmacological findings with ABHD6 inhibitor 22f are promising as a potential treatment of retina disease.
6. Conclusions
In this paper, we report the discovery of tetrahydroisoquinoline and isoindoline “signature templates” for ABHD6 with single-digit nanomolar inhibitory potency and specificity for the target. These new series of ABHD6 inhibitors exhibited >1000-fold selectivity against FAAH and MGL and lacked affinity against the cannabinoid receptors CB1 and CB2. The selectivity of the new series of ABHD6 inhibitors was also confirmed using the in vitro competitive activity-based protein profiling (ABPP) assay. Our target-based ligand design followed traditional structure-activity relationship (SAR) studies and chemoinformatics to introduce polar characteristics in order to improve water solubility and oral bioavailability. Key analogs were evaluated in stability assays and demonstrated good plasma stability (t1/2 > 2 hours; human and rodents) and microsomal stability (t1/2 ~ 4–40 min, rodent and humans liver microsomal preparations). Early pharmacokinetics studies suggested that ABHD6 inhibitor 22f represented a suitable tool compound to study in retina disease, where it was found to attenuate AMPA-induced glia activation and produce a dose-dependent retina neuroprotection in rats.
Accordingly, the new series of potent and specific ABHD6 inhibitors are providing early leads for developing therapeutics against neuroprotection, inflammation, ocular diseases, and diabetes, where ABHD6 has been reported to play a critical role.
9. Experimental Section
9.1. Chemistry
Proton nuclear magnetic resonance spectra were obtained on a VARIAN 400 spectrometer at 500 MHz. Spectra are given in ppm (δ) and coupling constants, J values, are reported in hertz. Splitting patterns are designated as follows: s, singlet; brs, broad singlet; d, doublet; t, triplet; q, quartet; m, multiplet. Tetramethylsilane was used as an internal reference standard. Mass spectra were obtained on a Waters Micromass ZQ spectrometer. Not all compounds ionized under experimental conditions. 1HNMR, LC-MS techniques and high-resolution mass spectrometry were used to determine compound purity. All reagents and solvents were obtained from commercial suppliers and used without further purification. All non-aqueous reactions were carried out in oven-dried glassware under an atmosphere of dried argon or nitrogen. The reactions were monitored by thin layer chromatography (TLC plates F254, Merck) or LC-MS analysis. All products, unless otherwise noted, were purified by flash chromatography using a Biotage Isolera purification system with pre-packed silica cartridges. Purity of all final products was > 96% as determined by 1HNMR and LC-MS using the following protocol. Mobile Phase A = water, B = acetonitrile solvent gradient 95/5 to 5/95 A:B in 11 min; flow rate 1.5 mL/min; Waters XTerra MS C8 column (4.6 × 50 mm) with UV detection at 190–400 nm wavelength.
The following abbreviations were used: CDCl3, deuterated chloroform; EtOAc, ethyl acetate; CH2Cl2, dichlorometane; MgSO4, magnesium sulfate; THF, tetrahydrofuran; NH4Cl, ammonium chloride; MeOH, methanol; NaHCO3, sodium bicarbonate; TFA, trifluoroacetic acid; LC-MS, liquid chromatography with mass spectrometry; MS, mass spectrometry; NMR, nuclear magnetic resonance spectrometry; TLC, thin layer chromatography.
Method A (Scheme 1)
8.1.1 1,1,1,3,3,3-Hexafluoropropan-2-yl 3-(bis(4-fluorophenyl)methoxy)azetidine-1-carboxylate (6b)
Step a). Bis(4-fluorophenyl)methanol (5.0 g, 22.7 mmol) and p-toluenesulfonic acid monohydrate (8.63 g, 45.4 mmol) were added to a suspension of 1-benzhydrylazetidin-3-ol (5.45 g, 22.7 mmol) in toluene (100 mL). The mixture was refluxed for 3 hours with continuous removal of water (Dean-Stark Trap). After completion of the reaction (TLC monitoring), the mixture was cooled to room temperature and the insoluble matter was removed by filtration. The organics were washed three times with sodium hydroxide (2N) solution and dried over anhydrous MgSO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 5/1 ratio) to afford 1-benzhydryl-3-(bis(4-fluorophenyl)methoxy)-azetidine (4 g, 40% yield). 1H NMR (400 MHz, CDCl3) δ 7.37–7.34 (m, 4H), 7.25–7.16 (m, 10H), 6.99–6.95 (m, 4H), 5.24 (s, 1H), 4.33 (s, 1H), 4.33–4.15 (m, 1H), 3.41–3.37 (m, 2H), 2.92–2.88 (m, 2H); MS (ES) m/z 442.1911 [M+H]+.
Step b). To a cooled (0 °C) solution of 1-benzhydryl-3-(bis(4-fluorophenyl)methoxy)azetidine (3.5 g, 7.9 mmol) and dichloromethane (25 mL) was added dropwise 1-chloroethyl 1-chloroformate (1.6 g, 11 mmol) in dichloromethane (5 mL). The mixture was stirred at room temperature overnight. The volatiles were removed under vacuum and the residue was treated with NaOH (188 mg, 15.8 mmol) in methanol (25 mL). The mixture was refluxed for 2 hours, cooled to room temperature and extracted with ethyl acetate. The organics were dried over anhydrous MgSO4. The solvents were removed under vacuum and the product 3-(bis(4-fluorophenyl)methoxy) azetidine (1.16 g) was carried to the next step without any purification. 1H NMR (400 MHz, CDCl3) δ 7.26–7.21 (m, 4H), 7.01–6.97(m, 4H), 5.25 (s, 1H), 4.13–4.11 (m, 1H), 3.40–3.37 (m, 1H), 3.31–3.27 (m, 1H), 2.97–2.92 (m, 2H).
Step c). Hexafluoropropan-2-yl chloroformate solution was prepared in situ by treating triphosgene (60 mg, 0.225 mmol) in dichloromethane (3 mL) with hexafluoroisopropanol (0.115 mL, 0.69 mmol) and N,N-diisopropylethylamine (0.144 mL, 1.035 mmol) at 0 ° C. The mixture was stirred for 30 minutes while allowing the temperature to rise to 15 °C. Then, the mixture was added dropwise to a cold (0 °C) mixture of 3-(bis(4-fluorophenyl)methoxy) azetidine (50 mg, 0.18 mmol) and N,N-diisopropylethylamine (0.41 mL, 101 mmol) in dichloromethane (3 mL). The reaction mixture was stirred at room temperature for 1 hour and then the mixture was diluted in dichloromethane (25 mL) and washed twice with water (15 mL). The organic layer was dried over anhydrous Na2SO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 2/1 ratio) to afford 1,1,1,3,3,3-hexafluoropropan-2-yl 3-(bis(4-fluorophenyl)methoxy)azetidine-1-carboxylate (60 mg, 69% yield). 1H NMR (400 MHz, CDCl3) δ 7.28–7.24 (m, 4H), 7.05–7.01 (m, 4H), 5.64–5.61 (m, 1H), 5.31 (s, 1H), 4.41–4.38 (m, 1H), 4.20–3.98 (m, 4H); MS (ES) m/z 470.1123 [M+H]+.
8.1.2 3-Cyanophenyl 3-(bis(4-fluorophenyl)methoxy)azetidine-1-carboxylate (5a)
To a cooled (0 °C) solution 3-(bis(4-fluorophenyl)methoxy)azetidine (50 mg, 0.18 mmol), Et3N (0.141 mL, 101 mmol) in dichloromethane (3 mL) was added 3-cyanophenyl carbonochloridate (66 mg, 0.36 mmol). The reaction mixture was stirred at room temperature for 1 hour and then diluted with dichloromethane (20 mL) and washed twice with water (15 mL). The organic layer was dried over anhydrous Na2SO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 2/1 ratio) to afford 3-cyanophenyl 3-(bis(4-fluorophenyl)methoxy)azetidine-1-carboxylate (110 mg, 65% yield). 1H NMR (400 MHz, CDCl3) δ 7.47–7.41 (m, 3H), 7.38–7.35 (m, 1H), 7.30–7.25 (m, 4H), 7.06–7.02 (m, 4H), 5.34 (s, 1H), 4.48–4.43 (m, 1H), 4.14–4.00 (m, 4H). HRMS (ESI) m/z calculated for C24H18F2N2O3 [M + H]+ 421.1340. Found 421.1346.
Method B (Scheme 2)
1,1,1,3,3,3-Hexafluoropropan-2-yl 3-((bis(4-fluorophenyl)methyl) (methyl)amino) azetidine-1-carboxylate (9d)
Step a) 4,4’-(Cloromethylene)bis(fluorobenzene) (1.9 g, 8.06 mmol.) was added dropwise into a stirred solution of tert-butyl 3-(methylamino)azetidine-1-carboxylate (501 mg, 2.68 mmol) and potassium carbonate (1.47 g, 10.72 mmol) in acetonitrile (20 mL). The solution was stirred at room temperature overnight. Then, the mixture was concentrated under reduced pressure. The resulting residue was partitioned between EtOAc and brine and the aqueous phase was extracted with EtOAc (3×20 mL). The combined organics were washed with brine and dried over Na2SO4. The organics were concentrated to afford tert-butyl 3-((bis(4-fluorophenyl)methyl)(methyl)amino)azetidine-1-carboxylate (775 mg, 75%), which was used without any further purification. 1H NMR (400 MHz, Chloroform-d) δ 7.34 – 7.30 (m, 1H), 7.27 – 7.23 (m, 3H), 7.11 – 6.79 (m, 4H), 4.52 (s, 1H), 3.78 (dd, J = 8.8, 6.0 Hz, 2H), 3.69 (dd, J = 8.8, 7.3 Hz, 3H), 3.44 – 3.40 (m, 1H), 2.12 (s, 3H), 1.41 (s, 9H).
Step b). To a solution of tert-butyl 3-((bis(4-fluorophenyl)methyl)(methyl)amino)azetidine-1-carboxylate (600 mg, 1.54 mmol) in CH2Cl2 (10 mL) was added TFA (0.88 mL). The mixture was stirred at room temperature for 8 h. The mixture was concentrated to get the hydrochloride salt of the product ( mg), and sue to the next step without purification.
-
Step c). This step was performed according to Method A, step e).
1H NMR (400 MHz, CDCl3) δ 7.26–7.24 (m, 4H), 7.03–7.01 (m, 4H), 5.63–5.60 (m, 1H), 4.53 (s, 1H), 4.033.99 (m, 1H), 3.95–3.91 (m, 2H), 3.83–3.78 (m, 1H), 3.65–3.63 (m, 1H), 2.17 (s, 3H); MS (ES) m/z 483.14 [M+1]+.
Method C (Scheme 3)
8.1.3 1,1,1,3,3,3-Hexafluoropropan-2-yl 3-benzhydrylazetidine-1-carboxylate (10d)
Step a). To a cooled (10 °C) mixture of benzophenone (1.82 g, 10 mmol) and 1-((l1-methyl)(l1-oxidaneyl)boraneyl)azetidin-3-one (1.71 g, 15.8 mmol) in anhydrous THF (50 mL) was added zinc powder (307.4 mg, 4.7 mmol) and stirred for 15 min. To the mixture was added TiCl4, (474 mg. 2.5 mmol) and stirred at 60 °C for 8 hours. Then, the reaction mixture cooled to room temperature, treated with HCl (1N, 20 mL), and extracted with ethyl acetate. The organic layer was dried over anhydrous Na2SO4. The solvents were removed under vacuum and the solids were washed with hexanes to afford product 3-(diphenylmethylene)azetidine (1.5 g, 67% yield), which was carried to the next step without any further purification. 1H NMR (400 MHz, CDCl3) δ 7.37–7.35 (m, 10H), 4.60–4.55 (m, 4H).
Step b). To a mixture of 3-(diphenylmethylene)azetidine (1.5 g, 6.78 mmol) in EtOH (10 mL) at room temperature was added Pd/C (1 g) and the contents were hydrogenated under a hydrogen atmosphere (40 psi) for 24 h. The reaction mixture was filtered through a celite pad and the celite washed with EtOH. The organic layer was dried over anhydrous Na2SO4. The solvents were removed under vacuum to afford 3-benzhydrylazetidine (1.4 g, 99% yield), which was carried to the next step without any further purification. 1H NMR (400 MHz, CDCl3) δ 1H NMR (400MHz, CDCl3) δ 7.36–7.31 (m, 10H), 4.56–4.51 (m, 4H), 4.11 (m, 1H), 3.91 (m, 1H).
Step c). This step was performed according to Method A, steps e). 1H NMR (400 MHz, CDCl3) δ 7.37–7.35 (m, 10H), 5.59–5.56 (m, 1H), 4.60–4.55 (m, 4H), 4.03 (d, J = 9.6 Hz, 1H), 3.96 (d, J = 9.6 Hz 1H); HRMS (ESI) m/z calculated for C25H22F3NO3 [M + H]+ 432.1034. Found 432.1031.
Method D (Scheme 4)
8.1.4 1,1,1,3,3,3-Hexafluoropropan-2-yl 3,4-dihydroisoquinoline-2(1H)-carboxylate (14a)
Step a). To a cold (0 °C) solution of 1,1,1,3,3,3-hexafluoropropan-2-ol (378 mg, 2.25 mmol), and N, N-diisopropylethylamine (0.58 mL, 4.50 mmol) in dichloromethane (5 mL) was added triphosgene (233 mg, 0.78 mmol). The reaction mixture was gradually allowed to come to room temperature and stirred for 30 minutes. Next, the solution was added dropwise to cold (0 °C) solution of 1,2,3,4-tetrahydroisoquinoline (100 mg, 0.75 mmol) and N, N-diisopropylethylamine (0.29 mL, 1.50 mmol) in dichloromethane (5 mL). The reaction mixture was gradually allowed to come to room temperature and stirred for 1 hour. Then, the reaction was diluted in dichloromethane (25 mL) and washed twice with water (15 mL) and brine. The organic extracts were dried over anhydrous Na2SO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 5/1 ratio) to afford 1,1,1,3,3,3-hexafluoropropan-2-yl 3,4-dihydroisoquinoline-2(1H)-carboxylate as colorless oil (220 mg, 90% yield): 1H NMR (400 MHz, CDCl3) δ ppm 7.23–7.15 (m, 4H), 5.80 (sept, 1H), 4.68 (m, 2H), 3.76 (t, J = 6 Hz, 2H), 2.93–2.88 (m, 2H); purity 99%, retention time 5.3 min.
Method E (scheme 5).
8.1.5 1,1,1,3,3,3-hexafluoropropan-2-yl 5-(pyrrolidin-1-yl)isoindoline-2-carboxylate (18a)
Step a) Tris(dibenzylideneacetone)dipalladium(0) (114 mg, 0.12 mmol), 2-(di-tert-butylphosphino) biphenyl (74 mg, 0.05 mmol) and sodium tert-butoxide (6.72 g, 7 mmol) were added into a solution of 5-bromo-1,3-dihydro-isoindole-2-carboxylic acid tert-butyl ester (1.49 g, 5 mmol) in toluene (15 mL). The reaction mixture was purged with anhydrous argon for 5 minutes and then pyrrolidine (426 mg, 6 mmol) was added and the mixture was stirred at 80 °C for 4 hours. After the completion of the reaction (monitored by TLC) the solvent was removed under vacuum, water (3 mL) was added and extracted three times with dichloromethane (30 mL). The combined organic extracts were dried over Na2SO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents EtOAc/hexanes 4/1) to afford tert-butyl 5-(pyrrolidin-1-yl)isoindoline-2-carboxylate as brown solid (850 g, 60 % yield); 1H NMR (400 MHz, DMSO-CDCl3) δ ppm 7.16–7.12 (m, 1 H), 6.92–6.88 (m, 2H), 4.76–4.71 (m, 4H), 3.28–3.26 (m, 4H), 2.63–2.60 (m, 4H), 1.51 (9H, s).
Step b). Trifluoroacetic acid (1.0 mL, 8.68 mmol) was slowly added into a cold (0 °C) solution of tert-butyl 5-(pyrrolidin-1-yl)isoindoline-2-carboxylate (500 mg, 1.74 mmol) and dichloromethane (10 mL). The resulting reaction mixture was stirred at room temperature for 2 hours. Then, the mixture was concentrated under vacuum and CHCl3 (2 × 15mL) was added and evaporated to ensure removal of the trifluoroacetic acid. The product 5-(pyrrolidin-1-yl)isoindoline • TFA was used in the next step without any further purification.
Step c). This step was performed according to Method A, steps e). 1H NMR (400 MHz, CDCl3) δ ppm 7.12–7.06 (m, 1H), 6.53–6.50 (m, 1H), 6.43–6.40 (m, 1H), 5.80 (sept, 1H), 4.76–4.71 (m, 4H), 3.28–3.26 (m, 4H), 2.03–2.00 (m, 4H); MS (ES) m/z 383.1223 [M+H]+.
8.1.6 1,1,1,3,3,3-Hexafluoropropan-2-yl 5-(3-fluoropropoxy)-3,4 dihydro -isoquinolin-2(1H)-carboxylate (22c)
Step a) Sodium hydride (28.9 mg, 1.2 mmol) was added into a cold (°C) solution of tert-butyl 3,4-dihydroisoquinoline-2(1H)-carboxylate (200 mg, 0.8 mmol) and N,N-dimethylformamide (3 mL). The reaction mixture was stirred for 1 hour and then, bromo-fluoropropane (226.2 mg, 1.6 mmol) was added. The reaction mixture was allowed to come to room temperature and stirred overnight. Then, the mixture was quenched with aqueous ammonium chloride and extracted with ethyl acetate. The organic extracts were dried over anhydrous MgSO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 5/1 ratio) to afford tert-butyl 5-(3-fluoropropoxy)-3,4-dihydroisoquinoline-2(1H)-carboxylate as colorless oil (161.2 mg, 65% yield). 1H NMR (500 MHz, CDCl3) δ ppm 7.15 (t, J = 8 Hz, 1H), 6.72 (m, 2H), 4.72 (t, J = 6 Hz, 1H), 4.62 (t, J = 6 Hz, 1H), 4.56 (s, 2H), 4.11 (t, J = 6.5 Hz, 2H), 3.64 (t, J = 6 Hz, 2H), 2.76 (t, J = 6 Hz, 2H), 2.23–2.20 (m, 1H), 2.17–2.15 (m, 1H), 1.48 (s, 9H); purity 98%, retention time 5.35 min.
Step b) This step was performed according to Method D, steps c, d). 1H NMR (500 MHz, CDCl3) δ ppm 7.21–7.16 (m, 1H), 6.76 (d, J = 7.5 Hz, 2H), 5.82–5.79 (sept, 1H), 4.72 (t, J = 6 Hz, 1H), 4.67 (s, 2H), 4.63 (t, J = 6 Hz, 1H), 4.13 (dt, J = 6, 2 Hz, 2H), 3.78 (dt, J = 6, 4 Hz, 2H), 2.86–2.81 (m, 2H), 2.24–2.18 (m, 1H), 2.17–2.15 (m, 1H).
The following compounds were prepared according to Method E.
8.1.18 1,1,1,3,3,3-Hexafluoropropan-2-yl 5-(2-morpholinoethoxy)isoindoline-2-carboxylate (22f).
1H NMR (500 MHz, CDCl3) δ ppm 7.19 (q, J = 8.5 Hz, 1H), 6.88 (d, J = 8.5 Hz, 1H), 6.83 (d, J = 15 Hz, 1H), 5.81–5.78 (sept, 1H), 4.79–4.75 (m, 4H), 4.13–4.09 (m, 2H), 3.75–3.73 (m, 4H), 2.82 (t, J = 5.5 Hz, 2H), 2.59 (d, J = 4 Hz, 4H); MS (ES) m/z 443.56 [M+1]+; ]+; purity 98%, retention time 4.9 min.
8.1.7 1,1,1,3,3,3-Hexafluoropropan-2-yl 5-(2-(piperidin-1-yl)ethoxy)isoindoline-2-carboxylate (22j)
1H NMR (500 MHz, CDCl3) δ ppm 7.14 (dd, J = 16.8, 8.4 Hz, 1H), 6.85 (d, J = 8.4 Hz, 1H), 6.79 (d, J = 16.0 Hz, 1H), 5.78 (dt, J = 12.3, 6.1 Hz, 1H), 4.74 (dd, J = 12.2, 7.6 Hz, 4H), 4.08 (td, J = 6.0, 2.7 Hz, 2H), 2.76 (t, J = 6.0 Hz, 2H), 2.49 (s, 4H), 1.61 – 1.58 (m, 4H), 1.44 (d, J = 5.0 Hz, 2H). MS (ES) m/z 441.60 [M+1]+.
8.1.8 (R)-1,1,1-trifluoro-3-hydroxypropan-2-yl 5-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (14j)
Step a). (R)-perfluorophenyl (1,1,1-trifluoro-3-((4-methoxybenzyl)oxy)propan-2-yl) carbonate (60 mg, 0.13 mmol; Patent US 2017/0029390 Al) in acetonitrile (2 mL) was added into cold 0 °C solution of 5-bromo,1,2,3,4-tetrahydroisoquinoline (30 mg, 0.14 mmol) and N,N-diisopropylethylamine (0.05 mL, 0.39 mmol) in acetonitrile (4 mL). The resulting reaction mixture was allowed to come to room temperature and stirred for 1 hour. Then, the reaction was diluted in dichloromethane (25 mL) and washed with water (2 × 15 mL). The organic extracts were dried over anhydrous NaSO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 4/1 ratio) to afford (R)-1,1,1-trifluoro-3-((4-methoxybenzyl)oxy)propan-2-yl 5-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate as colorless oil. 1H NMR (400 MHz, CDCl3) δ ppm 7.46–7.45 (m, 1H), 7.25–7.16 (m, 2H), 7.08–7.06 (m, 2H), 6.84–6.82 (m, 2H), 5.52–5.51 (sept, 1H), 4.64–4.61 (m, 2H), 4.52–4.44 (m, 2H), 3.78 (s, 3H), 3.76–3.71 (m, 3H), 2.90–2.87 (m, 3H).
Step b). 10% Pd/C (30 mg) was added into a stirred solution of (R)-1,1,1-trifluoro-3-((4-methoxybenzyl)oxy)propan-2-yl 5-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate (50 mg, 0.1 mmol) in ethyl acetate (10 mL). The resulting reaction mixture was stirred at room temperature in under a hydrogen atmosphere for 24 hours. Then, the mixture was filtered through celite pad. The filtrate was concentrated under reduced pressure and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 3/1 ratio) to afford 3-hydroxy-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2,4-dione as colorless oil (30 mg, 81% yield). 1H NMR (400 MHz, CDCl3) δ ppm 7.21–7.12 (m, 3H), 5.32–5.29 (sept, 1H), 4.66–4.65 (m, 2H), 4.04–4.00 (m, 1H), 3.92–3.87 (m, 1H), 3.75–3.72 (m, 2H), 2.89 (brs, 2H); MS (ES) m/z 330.11 [M+H]+.
Method F (scheme 6)
8.1.9 6,6-Dimethyl-2,4-dioxo-3-azabicyclo[3.1.0]hexan-3-yl 5-bromo-3,4-dihydro isoquinoline-2(1H)-carboxylate (25b)
Step a). O-benzylhydroxylamine hydrochloride (0.79 g, 5 mmol) was added into a solution of 6,6-dimethyl-3-oxabicyclo[3.1.0]hexane-2,4-dione (0.7 g, 5 mmol), N-methylmorpholine (1.01 mL, 10 mmol,) and anhydrous toluene (30 mL). The reaction mixture was stirred at room temperature for 30 minutes and then heated to reflux with azeotropic removal of water using Dean-Stark apparatus. Glacial acetic acid (1.0 mL) was added after 2 hours into the mixture and the reaction refluxed for 6 hours. Then, the reaction mixture was allowed to come to room temperature, and the product was extracted with ethyl acetate (3 × 50 mL) and washed with a saturated solution of NaHCO3 and brine. The combined organic layers were dried over anhydrous Na2SO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents dichloromethane/MeOH 10/1 ratio) to afford 3-(benzyloxy)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2,4-dione as a white solid (720 mg, 59% yield). 1H NMR (400 MHz, CDCl3) δ ppm 7.50–7.48 (m, 2H), 7.38–7.36 (m, 3H), 5.08 (s, 2H), 1.24 (s, 3H), 1.20 (s, 3H); MS (ES) m/z 246.12 [M+H]+.
Step b). 10% Pd/C (70 mg) was added into a solution of 3-(benzyloxy)-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2,4-dione (700 mg, 2.85 mmol) in 1:1 ethyl acetate/methanol (20 mL). The reaction mixture was stirred at room temperature under a hydrogen atmosphere for 24 hours. The reaction mixture was filtered through a celite pad. The filtrate was concentrated under reduced pressure to afford 3-hydroxy-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2,4-dione as colorless solid (430 mg, 97% yield), which was used to the next step without any further purification.
Step c). Triphosgene (37 mg, 0.12 mmol) was added into a cold (0 °C) solution of 3-hydroxy-6,6-dimethyl-3-azabicyclo[3.1.0]hexane-2,4-dione (55 mg, 0.36 mmol), N,N-diisopropylethylamine (0.06 mL, 0.47 mmol) and dichloromethane (3 mL). The reaction mixture was allowed to come to room temperature and stirred for 30 minutes. The resulting solution was added dropwise into a cold (0 °C) solution of 5-bromo-1,2,3,4-tetrahydroisoquinoline (50 mg, 0.24 mmol), N,N-diisopropylethylamine (0.06 mL, 0.47 mmol) and dichloromethane (3 mL). The reaction mixture was allowed to come to room temperature and stirred for 1 hour. Then, the reaction was diluted in dichloromethane (25 mL) and washed with water (2 × 15 mL) and brine. The organic extracts were dried over anhydrous NaSO4. The solvents were removed under vacuum and the residue was purified on silica gel (Biotage; eluting solvents hexanes: EtOAc 3/1 ratio) to afford 6,6-dimethyl-2,4-dioxo-3-azabicyclo[3.1.0]hexan-3-yl 5-bromo-3,4-dihydroisoquinoline-2(1H)-carboxylate as colorless solid (56 mg, 65% yield). 1H NMR (400 MHz, CDCl3) δ ppm 7.48–7.46 (m, 1H), 7.10–7.04 (m, 2H), 4.75–4.63 (m, 2H), 3.83–3.74 (m, 2H), 2.99–2.95 (m, 2H).
9.2. Biochemical assays
-
9.2.1
Fluorescence Assays were performed for evaluating the inhibitory activity of the compounds in the endocannabinoid enzymes. In brief, recombinant hABHD6 variants[29], rat ΔTM FAAH,[30] human FAAH (hFAAH) and hMGL[31, 45] were expressed in E. coli and purified. Compounds were screened in 3 points and characterized in 8 points high-throughput as described earlier using substrates arachidonoyl 7-amino-4-methylcoumarin amide (AAMCA) for FAAH[45b] and arachidonoyl, 7-hydroxy-6-methoxy-4-methylcoumarin ester (AHMMCE) for hABHD6[29] and MGL[31]. A high-throughput fluorometric inhibition screening assay using a novel fluorogenic substrate valeroyl-7-hydroxy-6-methoxy-4-methylcoumarin ester (VHMMCE) in combination with the hΔ29–4-ABHD6 variant was also employed[29]. IC50 values were calculated using Prism software (GraphPad).
-
9.2.2
Binding assays against receptors rCB1, hCB2, and mCB2 were performed as follow. HEK293 cells expressing the mCB2 or hCB2 receptor[32] or frozen rat brains[33a] containing rCB1 were adapted in our laboratory[33b, 33c]. Compound affinity was tested by using competition-equilibrium binding with [3H]CP-55,940[33b, 33c, 34]. Nonlinear regression was used to determine the actual IC50 and the Ki values (Prizm by GraphPad Software, Inc.)[46].
-
9.2.3
The selectivity profile of ABHD6 inhibitor was also assessed in the competitive activity-based protein profiling (ABPP) assay. Brain membrane proteomes, made from rat brain homogenate, were incubated with various concentrations of inhibitors for 30 min at 37°C prior to the addition of FP-rhodamine[47]. After quenching, the reactions were visualized via fluorescence from SDS-page gel. IC50 values were calculated using Prism (GraphPad). Animals were sacrificed by cervical dislocation followed by decapitation. This method was consistent with the June 2007 AVMA Guidelines on Euthanasia and our IACUC-approved animal protocol (#16–0402R). Northeastern University’s animal assurance number was #A-3155.
9.3. ADME assays
-
9.3.1
Evaluation of stability in plasma and buffer. Inhibitor solution (200 μM) was made in mouse, rat or human plasma (purchased from BioVIT), buffer containing 0.1% BSA. After quenching with acetonitrile, the samples were analyzed by HPLC to predict in vitro plasma half-lives.[48]
-
9.3.2
Evaluation of metabolic stability. Inhibitors were pre-incubated with mouse, rat or human liver microsomal protein (purchased from BioVIT) at 37°C before the reaction is initiated with NADPH or buffer (control)[49]. Following protein precipitation, the samples were analyzed using a LC-MS/MS in SRM mode. For prospective metabolite identification, the samples were analyzed by LC-MS/MS in data dependent mode.
-
9.3.3
Assessment of the compound pharmacokinetics in brain and plasma. Mice (CD-1, 25–30 g; purchased form Charles River) were injected iv or orally with 0.1–2 mg/kg each compound in a cassette dosing paradigm. Tissue samples (plasma and brain were taken fifteen minutes post-IV injection, or 30 and 60 minutes post-oral administration and flash frozen tissues were extracted[50] and analyzed using LC-MS/MS in SRM mode with internal standards used for quantification. All studies were performed according to protocols consistent with the June 2007 AVMA Guidelines on Euthanasia and our IACUC-approved animal protocol (#16–0402R) and Northeastern University’s animal assurance number was #A-3155.
9.4. Computational Studies
Computational software (Schrodinger Suite 2017-2) were used to assess in silico ligand ADME/pharmacokinetic properties of the proposed compounds in areas as Lipinski rules, ligand efficiency, topological polar surface area, and lipophilicity.
Supplementary Material
8. Acknowledgments.
This work was supported by grant number R01 DA03801.
Footnotes
References
- [1].Mechoulam R, Ben-Shabat S, Hanus L, Ligumsky M, Kaminski NE, Schatz AR, Gopher A, Almog S, Martin BR, Compton DR, et al. , Biochem Pharmacol 1995, 50, 83–90. [DOI] [PubMed] [Google Scholar]
- [2].Herkenham M, Lynn AB, Johnson MR, Melvin LS, de Costa BR, Rice KC, J Neurosci 1991, 11, 563–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Klein TW, Newton C, Friedman H, Immunol Today 1998, 19, 373–381. [DOI] [PubMed] [Google Scholar]
- [4].Fernandez-Ruiz J, Romero J, Velasco G, Tolon RM, Ramos JA, Guzman M, Trends Pharmacol Sci 2007, 28, 39–45. [DOI] [PubMed] [Google Scholar]
- [5].Reisenberg M, Singh PK, Williams G, Doherty P, Philos Trans R Soc Lond B Biol Sci 2012, 367, 3264–3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Zhang H, Hilton DA, Hanemann CO, Zajicek J, Brain Pathol 2011, 21, 544–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stella N, Schweitzer P, Piomelli D, Nature 1997, 388, 773–778. [DOI] [PubMed] [Google Scholar]
- [8].a Savinainen JR, Jarvinen T, Laine K, Laitinen JT, Br J Pharmacol 2001, 134, 664–672; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sugiura T, Waku K, World Rev Nutr Diet 2001, 88, 200–206. [DOI] [PubMed] [Google Scholar]
- [9].Savinainen JR, Saario SM, Laitinen JT, Acta Physiol (Oxf) 2012, 204, 267–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Marrs WR, Blankman JL, Horne EA, Thomazeau A, Lin YH, Coy J, Bodor AL, Muccioli GG, Hu SS, Woodruff G, Fung S, Lafourcade M, Alexander JP, Long JZ, Li W, Xu C, Moller T, Mackie K, Manzoni OJ, Cravatt BF, Stella N, Nat Neurosci 2010, 13, 951–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Blankman JL, Simon GM, Cravatt BF, Chem Biol 2007, 14, 1347–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu QS, Lichtman AH, Cravatt BF, Nat Neurosci 2010, 13, 1113–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tchantchou F, Zhang Y, J Neurotrauma 2013, 30, 565–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Naydenov AV, Horne EA, Cheah CS, Swinney K, Hsu KL, Cao JK, Marrs W, Blankman JL, Tu S, Cherry AE, Fung S, Wen A, Li W, Saporito MS, Selley DE, Cravatt BF, Oakley JC, Stella N, Neuron 2014, 83, 361–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Wen J, Jones M, Tanaka M, Selvaraj P, Symes AJ, Cox B, Zhang Y, J Neuroinflammation 2018, 15, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhao S, Mugabo Y, Ballentine G, Attane C, Iglesias J, Poursharifi P, Zhang D, Nguyen TA, Erb H, Prentki R, Peyot ML, Joly E, Tobin S, Fulton S, Brown JM, Madiraju SR, Prentki M, Cell Rep 2016, 14, 2872–2888. [DOI] [PubMed] [Google Scholar]
- [17].Thomas G, Betters JL, Lord CC, Brown AL, Marshall S, Ferguson D, Sawyer J, Davis MA, Melchior JT, Blume LC, Howlett AC, Ivanova PT, Milne SB, Myers DS, Mrak I, Leber V, Heier C, Taschler U, Blankman JL, Cravatt BF, Lee RG, Crooke RM, Graham MJ, Zimmermann R, Brown HA, Brown JM, Cell Rep 2013, 5, 508–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bottemanne P, Paquot A, Ameraoui H, Alhouayek M, Muccioli GG, FASEB J 2019, 33, 7635–7646. [DOI] [PubMed] [Google Scholar]
- [19].Maramai S, Brindisi M, ChemMedChem 2020, 15, 1985–2003. [DOI] [PubMed] [Google Scholar]
- [20].Baggelaar MP, van Esbroeck AC, van Rooden EJ, Florea BI, Overkleeft HS, Marsicano G, Chaouloff F, van der Stelt M, ACS Chem Biol 2017, 12, 852–861. [DOI] [PubMed] [Google Scholar]
- [21].Navia-Paldanius D, Savinainen JR, Laitinen JT, J Lipid Res 2012, 53, 2413–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a Hsu KL, Tsuboi K, Chang JW, Whitby LR, Speers AE, Pugh H, Cravatt BF, J Med Chem 2013, 56, 8270–8279; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Li W, Blankman JL, Cravatt BF, J Am Chem Soc 2007, 129, 9594–9595. [DOI] [PubMed] [Google Scholar]
- [23].Patel JZ, Nevalainen TJ, Savinainen JR, Adams Y, Laitinen T, Runyon RS, Vaara M, Ahenkorah S, Kaczor AA, Navia-Paldanius D, Gynther M, Aaltonen N, Joharapurkar AA, Jain MR, Haka AS, Maxfield FR, Laitinen JT, Parkkari T, ChemMedChem 2015, 10, 253–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Deng H, Li W, Eur J Med Chem 2020, 198, 112353. [DOI] [PubMed] [Google Scholar]
- [25].Tuo W, Leleu-Chavain N, Spencer J, Sansook S, Millet R, Chavatte P, J Med Chem 2017, 60, 4–46. [DOI] [PubMed] [Google Scholar]
- [26].Bowman AL, Makriyannis A, Chem Biol Drug Des 2013, 81, 382–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Yang B, Li J, O’Rourke D, Synlet 1993, 3, 195–196. [Google Scholar]
- [28].Mukaiyama T, Sato T, Hanna J, Chem Let 1973, 2, 1041–1044. [Google Scholar]
- [29].Shields CM, Zvonok N, Zvonok A, Makriyannis A, Sci Rep 2019, 9, 890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Patricelli MP, Lashuel HA, Giang DK, Kelly JW, Cravatt BF, Biochemistry 1998, 37, 15177–15187. [DOI] [PubMed] [Google Scholar]
- [31].Zvonok N, Pandarinathan L, Williams J, Johnston M, Karageorgos I, Janero DR, Krishnan SC, Makriyannis A, Chem Biol 2008, 15, 854–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Abadji V, Lucas-Lenard JM, Chin C, Kendall DA, J Neurochem 1999, 72, 2032–2038. [DOI] [PubMed] [Google Scholar]
- [33].a Dodd PR, Hardy JA, Oakley AE, Edwardson JA, Perry EK, Delaunoy JP, Brain Res 1981, 226, 107–118; [DOI] [PubMed] [Google Scholar]; b Guo Y, Abadji V, Morse KL, Fournier DJ, Li X, Makriyannis A, J Med Chem 1994, 37, 3867–3870; [DOI] [PubMed] [Google Scholar]; c Morse KL, Fournier DJ, Li X, Grzybowska J, Makriyannis A, Life Sci 1995, 56, 1957–1962. [DOI] [PubMed] [Google Scholar]
- [34].Lan R, Liu Q, Fan P, Lin S, Fernando SR, McCallion D, Pertwee R, Makriyannis A, J. Med. Chem 1999, 42, 769–776. [DOI] [PubMed] [Google Scholar]
- [35].Chicca A, Arena C, Manera C, Recent Pat CNS Drug Discov 2016, 10, 122–141. [DOI] [PubMed] [Google Scholar]
- [36].a Cisar JS, Weber OD, Clapper JR, Blankman JL, Henry CL, Simon GM, Alexander JP, Jones TK, Ezekowitz RAB, O’Neill GP, Grice CA, J Med Chem 2018, 61, 9062–9084; [DOI] [PubMed] [Google Scholar]; b Granchi C, Caligiuri I, Minutolo F, Rizzolio F, Tuccinardi T, Expert Opin Ther Pat 2017, 27, 1341–1351. [DOI] [PubMed] [Google Scholar]
- [37].Bononi G, Poli G, Rizzolio F, Tuccinardi T, Macchia M, Minutolo F, Granchi C, Expert Opin Ther Pat 2021, 31, 153–168. [DOI] [PubMed] [Google Scholar]
- [38].McAllister LA, Butler CR, Mente S, O’Neil SV, Fonseca KR, Piro JR, Cianfrogna JA, Foley TL, Gilbert AM, Harris AR, Helal CJ, Johnson DS, Montgomery JI, Nason DM, Noell S, Pandit J, Rogers BN, Samad TA, Shaffer CL, da Silva RG, Uccello DP, Webb D, Brodney MA, J Med Chem 2018, 61, 3008–3026. [DOI] [PubMed] [Google Scholar]
- [39].Cravatt BF, Wright AT, Kozarich JW, Annu Rev Biochem 2008, 77, 383–414. [DOI] [PubMed] [Google Scholar]
- [40].a Ahn K, Johnson DS, Mileni, Beidler D, Long JZ, McKinney MK, Weerapana E, Sadagopan N, Liimatta M, Smith SE, Lazerwith S, Stiff C, Kamtekar S, Bhattacharya K, Zhang Y, Swaney S, Van Becelaere K, Stevens RC, Cravatt BF, Chemistry & biology 2009, 16, 411–420; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Clapper JR, Henry CL, Niphakis MJ, Knize AM, Coppola AR, Simon GM, Ngo N, Herbst RA, Herbst DM, Reed AW, Cisar JS, Weber OD, Viader A, Alexander JP, Cunningham ML, Jones TK, Fraser IP, Grice CA, Ezekowitz RAB, O’Neill GP, Blankman JL, J Pharmacol Exp Ther 2018, 367, 494–508. [DOI] [PubMed] [Google Scholar]
- [41].Niphakis MJ, Cognetta AB 3rd, Chang JW, Buczynski MW, Parsons LH, Byrne F, Burston JJ, Chapman V, Cravatt BF, ACS Chem Neurosci 2013, 4, 1322–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Rapino C, Tortolani D, Scipioni L, Maccarrone M, Curr Neuropharmacol 2018, 16, 959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Matias I, Wang JW, Moriello AS, Nieves A, Woodward DF, Di Marzo V, Prostaglandins Leukot Essent Fatty Acids 2006, 75, 413–418. [DOI] [PubMed] [Google Scholar]
- [44].Kokona D, Spyridakos D, Tzatzarakis M, Papadogkonaki S, Filidou E, Arvanitidis KI, Kolios G, Lamani M, Makriyannis A, Malamas MS, Thermos K, Neuropharmacology 2021, 185, 108450. [DOI] [PubMed] [Google Scholar]
- [45].a Zvonok N, Williams J, Johnston M, Pandarinathan L, Janero DR, Li J, Krishnan SC, Makriyannis A, J Proteome Res 2008, 7, 2158–2164; [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ramarao MK, Murphy EA, Shen MW, Wang Y, Bushell KN, Huang N, Pan N, Williams C, Clark JD, Anal Biochem 2005, 343, 143–151. [DOI] [PubMed] [Google Scholar]
- [46].Cheng Y, Prusoff WH, Biochem Pharmacol 1973, 22, 3099–3108. [DOI] [PubMed] [Google Scholar]
- [47].Long JZ, Jin X, Adibekian A, Li W, Cravatt BF, J Med Chem 2010, 53, 1830–1842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a Hale JT, Bigelow JC, Mathews LA, McCormack JJ, Biochem Pharmacol 2002, 64, 1493–1502; [DOI] [PubMed] [Google Scholar]; b Wood JT, Smith DM, Janero DR, Zvonok AM, Makriyannis A, Life Sci 2013, 92, 482–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].a Mohutsky MA, Chien JY, Ring BJ, Wrighton SA, Pharm Res 2006, 23, 654–662; [DOI] [PubMed] [Google Scholar]; b Obach RS, Drug Metab Dispos 1999, 27, 1350–1359. [PubMed] [Google Scholar]
- [50].Folch J, Lees M, Sloane Stanley GH, J Biol Chem 1957, 226, 497–509. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.