

Abstract
Xentuzumab is an insulin‐like growth factor (IGF) ligand‐neutralizing antibody. This phase 1 trial assessed xentuzumab in Japanese patients with solid tumors. Patients aged ≥20 y old with solid tumors that were refractory or not amenable to standard therapy were enrolled. Patients received xentuzumab intravenously at a starting dose of 750 mg/wk. Dose escalation used a 3 + 3 design with dose de‐escalation. The primary endpoint was to determine the maximum tolerated dose (MTD) of xentuzumab. Safety, pharmacokinetics, pharmacodynamics, and anti‐tumor activity were also assessed. Fifteen patients received xentuzumab in the dose escalation part (750 mg/wk [n = 6]; 1000 mg/wk [n = 3]; 1400 mg/wk [n = 6]). There were no dose‐limiting toxicities at any dose; the MTD of xentuzumab was not reached. Xentuzumab 1000 mg/wk was recommended as the relevant biological dose. Six further patients received xentuzumab 1000 mg/wk in an expansion cohort. Of 21 patients, 13 (61.9%) experienced a drug‐related adverse event, most commonly fatigue (23.8%), neutropenia (19.0%), diarrhea, nausea, white blood cell count decrease, and muscle spasms (14.3% each). No relevant deviations from dose linearity of xentuzumab exposure were observed during dose escalation. Total IGF‐1 and IGF‐2 levels increased and bioactive IGF levels decreased from baseline to 24 h after the first infusion in cycle 1. Partial response was observed in 2 (9.5%) patients with desmoid‐type fibromatosis. Disease control was achieved in 6 (28.6%) patients (median duration 42.4 mo). Xentuzumab monotherapy was well tolerated in Japanese patients and showed evidence of anti‐tumor activity. This study was registered with www.clinicaltrials.gov (NCT02145741).
Keywords: advanced tumors, insulin‐like growth factor, Japanese, monoclonal antibody, xentuzumab
This phase 1 study assessed the safety and anti‐tumor activity of xentuzumab in 21 Japanese patients with solid tumors. Treatment was generally well tolerated with no significant changes in blood glucose levels and no drug‐related grade ≥3 adverse events. Two patients (9.5%) had a partial response and disease control was achieved in a total of 6 patients (28.6%); both patients with partial responses and 2 patients with stable disease had sarcomas.

1. INTRODUCTION
Insulin‐like growth factor (IGF) signaling plays a role in cancer progression and is associated with the development of resistance to anti‐cancer therapies targeting other signaling pathways. 1 Targeting the IGF axis as a therapeutic strategy has been assessed in a range of cancers. Initial approaches used monoclonal antibodies or tyrosine kinase inhibitors (TKIs) to target the IGF type 1 receptor (IGF‐1R). Unfortunately, late‐stage trials for these agents were largely disappointing, with limited clinical benefit and potential tolerability concerns related to interference with glucose metabolism. 2 , 3 , 4 An alternative therapeutic strategy targets the IGF‐1 and IGF‐2 ligands. This approach may offer advantages vs IGF‐1R‐targeted approaches, as it also inhibits the growth‐promoting activity of IGF‐2 acting via insulin receptor (IR) isoform‐A. Moreover, signaling via IR‐B is not affected, therefore reducing the potential for hyperglycemia. 4
Xentuzumab is a humanized immunoglobulin G1 monoclonal antibody that binds to IGF‐1 and IGF‐2 ligands with high affinity, neutralizing their activity. 5 Xentuzumab given either weekly or every 3 wk has been assessed in patients with advanced or metastatic solid tumors in 2 first‐in‐human phase 1 trials, conducted in Taiwan and the UK, respectively. 6 The relevant biological dose was determined as 1000 mg weekly and was generally well tolerated with evidence of preliminary anti‐tumor activity. 6
Due to the favorable safety profile of xentuzumab and the role of IGF signaling in the development of resistance to other anti‐cancer therapies, early phase trials have also assessed xentuzumab in combination with other therapies. A phase 1b/2 trial evaluated xentuzumab in combination with everolimus and exemestane in patients with advanced hormone receptor‐positive breast cancer. 7 In the overall population, addition of xentuzumab to everolimus and exemestane did not show progression‐free survival benefit vs everolimus and exemestane alone (hazard ratio [HR] 0.97, 95% confidence interval [CI] 0.57‐1.65; P = .91). Nevertheless, a potential benefit was observed in a pre‐specified subgroup analysis of patients who had only non‐visceral metastases at baseline (HR 0.21, 95% CI 0.05‐0.98; P = .029). 7
Xentuzumab has also been assessed in combination with afatinib in patients with advanced epidermal growth factor receptor mutation‐positive non–small‐cell lung cancer (NSCLC). Although treatment was well tolerated, the combination did not show significant clinical activity. 8 The combination of xentuzumab and enzalutamide vs enzalutamide alone was also evaluated in a phase 2 trial in patients with metastatic castration‐resistant prostate cancer that had progressed after docetaxel and abiraterone. Although the safety profile was generally similar between treatment arms, addition of xentuzumab to enzalutamide did not prolong progression‐free survival vs enzalutamide alone. 9
To determine the maximum tolerated dose (MTD) of xentuzumab and evaluate its safety, pharmacokinetics (PK), pharmacodynamics (PD), and anti‐tumor activity also in Japanese patients, we conducted a dedicated phase 1 trial in patients with advanced solid tumors.
2. MATERIALS AND METHODS
2.1. Study design and patients
This phase 1, open‐label, dose escalation, and expansion trial (NCT02145741) was conducted at 1 site (National Cancer Center Hospital East, Chiba) in Japan. Patients aged ≥20 y old with cytologically or histologically confirmed solid tumors that were refractory to standard therapy, for whom no standard therapy of proven efficacy existed, or who were not amenable to established treatment options, were enrolled. Patients were required to have an Eastern Cooperative Oncology Group performance status (ECOG PS) of 0, 1, or 2. Key exclusion criteria were: active infectious disease; inadequate organ function; serious illness or concomitant disease considered by the investigator to be incompatible with study treatment; untreated or symptomatic brain metastases; uncontrolled diabetes mellitus; and treatment within 4 wk before starting trial medication with chemotherapy, immunotherapy, radiotherapy (within 2 wk for local palliative therapy), biologic therapy, or other investigational drugs. Patients receiving hormone therapy within 2 wk before trial treatment (except luteinizing hormone releasing hormone agonists in prostate cancer or bisphosphonates) were also excluded.
The study was conducted in accordance with Japanese Good Clinical Practice regulations and the Declaration of Helsinki and Good Clinical Practice guidelines as defined by the International Conference on Harmonization. All patients provided written informed consent for trial participation.
2.2. Study treatment
Patients received xentuzumab by 1‐h intravenous infusion weekly. The starting dose was 750 mg/wk (based on results from the earlier phase 1 trials). 6 Subsequent dose escalation cohorts were 1000 and 1400 mg/wk. The study used a traditional 3 + 3 dose escalation design with dose de‐escalation; if the MTD could not be identified following escalation to the highest planned dose, a relevant biological dose was to be recommended and an additional 6 patients were to be added in an expansion cohort at this dose. Treatment continued until disease progression, unacceptable toxicity, or consent withdrawal.
2.3. Endpoints
The primary endpoint was the MTD of xentuzumab, which was defined as the highest dose of xentuzumab at which no more than 1 of 6 evaluable patients experienced a dose‐limiting toxicity (DLT) during cycle 1. Other safety endpoints included adverse events (AEs). Efficacy endpoints included objective response, disease control, and duration of disease control.
2.4. Assessments
Intensity of AEs was assessed per National Cancer Institute Common Terminology Criteria for Adverse Events (version 4.03); relationship to study treatment was assessed by the investigator. DLTs were defined as any drug‐related AEs that met the criteria listed in Method S1.
For assessment of PK parameters of xentuzumab and biomarkers, blood samples were collected at pre‐defined intervals. Xentuzumab concentrations were measured using a validated ELISA (Enzyme‐linked immunosorbent assay), with concentrations determined on a standard curve plotting optical density vs concentration. The lower limit of quantification was 200 ng/mL. Total IGF‐1 and IGF‐2 levels were analyzed using 2 different validated sandwich ELISAs that had been developed at Boehringer Ingelheim using selective anti‐IGF‐1 or anti‐IGF‐2 antibodies for capturing the analyte to the surface of 96‐well microtiter plates, as well as for detecting the formed immune complexes. Antibodies were purchased from BioTechne, Peprotech, or generated in‐house. Recombinant standards were purchased from BioTechne. The assays quantified total amounts of IGF using a sample preparation procedure that splits the antibody‐IGF complexes and the IGF and IGF‐binding protein complexes. Concentrations were determined relative to the calibration standard and the antibodies used and reported as mass equivalents per volume (µgEq/L). The lower limit of quantification was validated as 10 ngEq/mL for IGF‐1 and 80 ngEq/L for IGF‐2. The between‐run precision during sample measurements was 4%‐16% coefficient of variation (CV) for IGF‐1 and 3%‐6% CV for IGF‐2 (n = 20). IGF bioactivity was assessed in heparinized plasma samples based on quantification of phosphorylated IGF‐1R. The IGF‐1R phosphorylation assay is a cell‐based immunoassay detecting activated (phosphorylated) IGF‐1R by fluorometric detection. Mouse fibroblasts expressing human IGF‐1R (Boehringer Ingelheim) were cultured in 96‐well plates. Plasma samples were added and could induce phosphorylation. Cells were then fixed to preserve activation‐specific protein modifications, such as phosphorylation, and subsequently permeabilized and incubated with a primary antibody (Cell Signaling; #3024) that recognized intracellular phosphorylation sites of human IGF‐1R. Incubation with a secondary alkaline phosphatase‐conjugated antibody (Dako; #D0487) and a substrate solution (Roche; #11 681 982 001) resulted in a fluorometric signal. IGF bioactivity in post‐treatment plasma samples was compared with the activity in pre‐dose samples.
Tumor assessments were performed after every 2 cycles until the end of cycle 6, and every 3 cycles thereafter. Tumor response, assessed by the investigator, was evaluated per Response Evaluation Criteria in Solid Tumors, version 1.1.
2.5. Statistical analysis
Safety and anti‐tumor activity were assessed in all patients who received at least 1 dose of xentuzumab. All analyses were descriptive and exploratory.
3. RESULTS
3.1. Patients and treatment
In total, 21 patients were treated; 15 patients received xentuzumab in the dose escalation part (6 in the 750 mg/wk group [3 were replaced due to involuntary discontinuation of study treatment before the end of cycle 1]; 3 in the 1000 mg/wk group; and 6 in the 1400 mg/wk group). Six patients received xentuzumab 1000 mg/wk in the expansion part.
Baseline demographics are shown in Table 1. Thirteen (61.9%) patients were male, median age was 66 y and most patients had an ECOG PS of 0 (71.4%). Patients were heavily pre‐treated; 9 (42.9%) patients had received ≥5 previous systemic chemotherapies.
TABLE 1.
Patient demographics and baseline characteristics
| Characteristics | Dose escalation | Expansion | Total (N = 21) | ||
|---|---|---|---|---|---|
| 750 mg (n = 6) | 1000 mg (n = 3) | 1400 mg (n = 6) | 1000 mg (n = 6) | ||
| Gender, n (%) | |||||
| Male | 2 (33.3) | 2 (66.7) | 5 (83.3) | 4 (66.7) | 13 (61.9) |
| Female | 4 (66.7) | 1 (33.3) | 1 (16.7) | 2 (33.3) | 8 (38.1) |
| Median age, years (range) | 66 (46‐70) | 68 (56‐69) | 68 (48‐74) | 38 (32‐71) | 66 (32‐74) |
| ECOG PS, n (%) | |||||
| 0 | 4 (66.7) | 2 (66.7) | 4 (66.7) | 5 (83.3) | 15 (71.4) |
| 1 | 2 (33.3) | 1 (33.3) | 2 (33.3) | 1 (16.7) | 6 (28.6) |
| Primary tumor type, n (%) | |||||
| Adrenal | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (9.5) |
| Colorectal | 1 (16.7) | 0 | 1 (16.7) | 0 | 2 (9.5) |
| Esophagus | 1 (16.7) | 0 | 2 (33.3) | 0 | 3 (14.3) |
| Gastrointestinal tract | 0 | 1 (33.3) | 0 | 0 | 1 (4.8) |
| Non–small‐cell lung cancer | 0 | 1 (33.3) | 1 (16.7) | 0 | 2 (9.5) |
| Soft tissue/ossifying sarcoma | 0 | 0 | 0 | 2 (33.3) | 2 (9.5) |
| Stomach | 1 (16.7) | 1 (33.3) | 1 (16.7) | 1 (16.7) | 4 (19.0) |
| Other | 2 (33.3) | 0 | 0 | 3 (50.0) | 5 (23.8) a |
| Number of metastatic sites at screening, n (%) | |||||
| 1 | 3 (50.0) | 1 (33.3) | 0 | 3 (50.0) | 7 (33.3) |
| 2 | 1 (16.7) | 1 (33.3) | 2 (33.3) | 2 (33.3) | 6 (28.6) |
| ≥3 | 2 (33.3) | 1 (33.3) | 4 (66.7) | 1 (16.7) | 8 (38.1) |
| Prior surgery, n (%) | 4 (66.7) | 2 (66.7) | 5 (83.3) | 4 (66.7) | 15 (71.4) |
| Prior radiotherapy, n (%) | 1 (16.7) | 1 (33.3) | 2 (33.3) | 2 (33.3) | 6 (28.6) |
| Prior hormone therapy, n (%) | 1 (16.7) | 0 | 0 | 2 (33.3) | 3 (14.3) |
| Number of previous systemic chemotherapies, n (%) | |||||
| 0 | 0 | 0 | 0 | 1 (16.7) | 1 (4.8) |
| 1 | 0 | 0 | 1 (16.7) | 2 (33.3) | 3 (14.3) |
| 2 | 0 | 0 | 0 | 1 (16.7) | 1 (4.8) |
| 3 | 2 (33.3) | 0 | 1 (16.7) | 2 (33.3) | 5 (23.8) |
| 4 | 0 | 2 (66.7) | 0 | 0 | 2 (9.5) |
| ≥5 | 4 (66.7) | 1 (33.3) | 4 (66.7) | 0 | 9 (42.9) |
Abbreviation: ECOG PS, Eastern Cooperative Oncology Group performance status.
Other tumor types; 750 mg cohort: neuroendocrine carcinoma and breast cancer; 1000 mg cohort: retroperitoneal solitary fibrous tumor and desmoid‐type fibromatosis (n = 2).
At data cut‐off (20 September 2019), 18 (85.7%) patients had discontinued treatment, primarily due to progressive disease (15 [71.4%] patients). Three patients remained on treatment (all in the 1000 mg dose expansion cohort). Median duration of xentuzumab treatment was 1.2 mo in the dose escalation cohorts (0.6, 1.2, and 0.9 mo in the 750, 1000 and 1400 mg dose cohorts, respectively). In the dose expansion cohort, median duration of xentuzumab treatment was 42.2 mo; of note, 4 patients in the 1000 mg dose expansion cohort received xentuzumab treatment for over 3 y (all 4 patients had a confirmed partial response or confirmed stable disease).
3.2. Identification of MTD
There were no DLTs in the trial and the MTD of xentuzumab was not reached for doses up to 1400 mg weekly. Xentuzumab 1000 mg/wk was recommended as the relevant biological dose for investigation in the expansion part.
3.3. Safety and tolerability
Of 21 patients, 18 (85.7%) had had an AE during the on‐treatment period. The most common AEs were fatigue (28.6%), diarrhea (23.8%), and nausea (23.8%; Table 2). Most AEs were grade 1 or 2. Four patients had grade 3 AEs (reported preferred terms were: gastrointestinal hemorrhage; urinary tract infection; pathological fracture; hypokalemia and general physical health deterioration). One grade 4 AE (tumor hemorrhage) was reported and there were no grade 5 AEs. There were no AEs leading to dose reduction or discontinuation of xentuzumab.
TABLE 2.
Most common any‐cause adverse events with xentuzumab during the on‐treatment period (occurring in ≥2 patients; treated set)
| N (%) | All patients (N = 21) | ||||
|---|---|---|---|---|---|
| All grades | Grade 1 | Grade 2 | Grade 3 | Grade 4 | |
| Patients with any AE a | 18 (85.7) | 5 (23.8) | 9 (42.9) | 3 (14.3) | 1 (4.8) |
| Fatigue | 6 (28.6) | 5 (23.8) | 1 (4.8) | 0 | 0 |
| Diarrhea | 5 (23.8) | 4 (19.0) | 1 (4.8) | 0 | 0 |
| Nausea | 5 (23.8) | 4 (19.0) | 1 (4.8) | 0 | 0 |
| Neutropenia | 4 (19.0) | 1 (4.8) | 3 (14.3) | 0 | 0 |
| Pyrexia | 4 (19.0) | 4 (19.0) | 0 | 0 | 0 |
| Decreased appetite | 4 (19.0) | 4 (19.0) | 0 | 0 | 0 |
| Influenza‐like illness | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| ALT increased | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| AST increased | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| White blood cell count decreased | 3 (14.3) | 0 | 3 (14.3) | 0 | 0 |
| Hyperglycemia | 3 (14.3) | 2 (9.5) | 1 (4.8) | 0 | 0 |
| Muscle spasms | 3 (14.3) | 2 (9.5) | 1 (4.8) | 0 | 0 |
| Cough | 3 (14.3) | 3 (14.3) | 0 | 0 | 0 |
| Anemia | 2 (9.5) | 0 | 2 (9.5) | 0 | 0 |
| Dental caries | 2 (9.5) | 0 | 2 (9.5) | 0 | 0 |
| Hemorrhoids | 2 (9.5) | 1 (4.8) | 1 (4.8) | 0 | 0 |
| Toothache | 2 (9.5) | 2 (9.5) | 0 | 0 | 0 |
| Abnormal hepatic function | 2 (9.5) | 1 (4.8) | 1 (4.8) | 0 | 0 |
| Seasonal allergy | 2 (9.5) | 2 (9.5) | 0 | 0 | 0 |
| Nasopharyngitis | 2 (9.5) | 1 (4.8) | 1 (4.8) | 0 | 0 |
| Blood creatine phosphokinase increased | 2 (9.5) | 0 | 2 (9.5) | 0 | 0 |
| Hypokalemia | 2 (9.5) | 0 | 1 (4.8) | 1 (4.8) | 0 |
| Tumor pain | 2 (9.5) | 1 (4.8) | 1 (4.8) | 0 | 0 |
| Dizziness | 2 (9.5) | 2 (9.5) | 0 | 0 | 0 |
| Dermatitis | 2 (9.5) | 1 (4.8) | 1 (4.8) | 0 | 0 |
| Rash | 2 (9.5) | 2 (9.5) | 0 | 0 | 0 |
Abbreviations: AE, adverse event; ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Table shows maximum Common Terminology Criteria for Adverse Events grade.
Thirteen (61.9%) patients had a drug‐related AE. The most common drug‐related AEs were fatigue (23.8%), neutropenia (19.0%), diarrhea, nausea, white blood cell count decreased, and muscle spasms (14.3% each). All drug‐related AEs were grade ≤2; there was no dose dependency with regard to frequency of xentuzumab‐related AEs. Drug‐related hyperglycemia was reported in 2 patients (both grade 1) and there were no infusion‐related reactions.
3.4. Pharmacokinetics/pharmacodynamics
Exposure of xentuzumab increased with increasing dose; no relevant deviations from dose linearity were observed during the dose escalation part of the study (Figure 1). Mean total serum IGF‐1 and IGF‐2 levels increased from baseline until the end of cycle 1 by approximately 1500%‐2000% and 150%‐180%, respectively, across treatment groups. The increases in both IGF‐1 and IGF‐2 were broadly sustained until the end of treatment. Mean total bioactive IGF (based on phosphorylated IGF‐1R) decreased rapidly from baseline to the end of the first infusion in cycle 1 by approximately 20%‐30% in each treatment group and was sustained until the end of treatment. Changes in total IGF‐1 and IGF‐2 levels, and bioactive IGF, with the xentuzumab 1000 mg dose (n = 9) during cycle 1 and at end of treatment are shown in Table 3.
FIGURE 1.
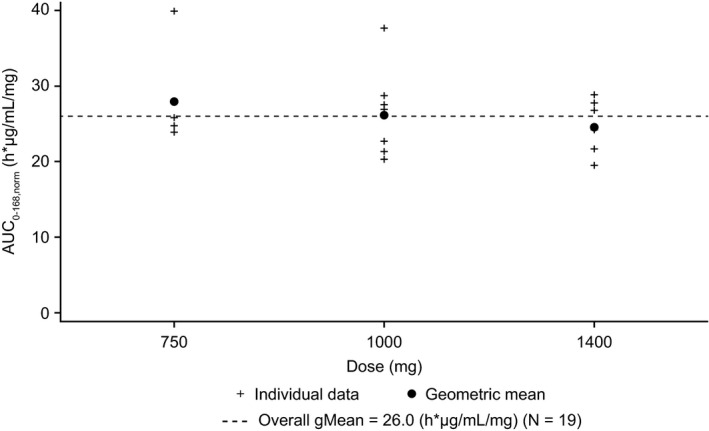
Comparison of individual and geometric mean AUC0‐168,norm values for xentuzumab after weekly intravenous infusions (n = 4/9/6). AUC0‐168,norm, dose normalized area under the curve from time 0 to 168 h post‐dose; gMean, geometric mean
TABLE 3.
Total IGF‐1, total IGF‐2, and bioactive IGF after administration of xentuzumab 1000 mg/wk infusion
| Baseline | Cycle 1 (24 h) | Cycle 1 (168 h) | Cycle 1 (336 h) | End of treatment | |
|---|---|---|---|---|---|
| Total IGF‐1 concentrations, (µgEq/L) | |||||
| N | 9 | 9 | 9 | 8 | 5 |
| gMean | 137 | 416 | 1630 | 2170 | 1420 |
| gCV (%) | 71.4 | 33.7 | 22.9 | 31.9 | 81.6 |
| Total IGF‐2 concentrations, (µgEq/L) | |||||
| N | 9 | 9 | 9 | 8 | 5 |
| gMean | 440 | 618 | 706 | 681 | 495 |
| gCV (%) | 24.8 | 19.2 | 22.9 | 23.7 | 42.2 |
| Bioactive IGF concentrations, (ratio) | |||||
| N | 9 | 9 | 9 | 8 | 5 |
| gMean | 1.00 | 0.215 | 0.220 | 0.186 | 0.204 |
| gCV (%) | 0 | 84.6 | 84.6 | 54.4 | 43.8 |
Table shows cycle 1 and end of treatment timepoints only; additional samples were taken during cycles 2 and 3.
Abbreviations: gCV, geometric coefficient of variation; gMean, geometric mean; IGF, insulin‐like growth factor.
3.5. Anti‐tumor activity
Partial response was observed in 2 patients (both in the expansion cohort of the 1000 mg group), therefore the objective response rate was 9.5% (Table 4). Both patients had desmoid‐type fibromatosis with tumor shrinkage of 36.2% and 32.7% from baseline. Both remained on treatment at data cut‐off (partial response was ongoing for both patients with a duration of 1359 and 1156 d, respectively).
TABLE 4.
Summary of best confirmed overall response (treated set)
| N (%) | Dose escalation | Expansion | Total (N = 21) | ||
|---|---|---|---|---|---|
| 750 mg (n = 6) | 1000 mg (n = 3) | 1400 mg (n = 6) | 1000 mg (n = 6) | ||
| Disease control | 1 (16.7) | 0 | 1 (16.7) | 4 (66.7) | 6 (28.6) |
| Objective response | 0 | 0 | 0 | 2 (33.3) | 2 (9.5) |
| Complete response | 0 | 0 | 0 | 0 | 0 |
| Partial response | 0 | 0 | 0 | 2 (33.3) | 2 (9.5) |
| Stable disease | 1 (16.7) | 0 | 1 (16.7) | 2 (33.3) | 4 (19.0) |
| Progressive disease | 4 (66.7) | 3 (100) | 5 (83.3) | 2 (33.3) | 14 (66.7) |
| Not evaluable | 1 (16.7) | 0 | 0 | 0 | 1 (4.8) |
Stable disease was observed in 4 (19.0%) patients (Table 4). Disease control was therefore achieved in 6 (28.6%) patients, with median duration of 42.4 mo (range 2.1‐48.1).
4. DISCUSSION
This phase 1 trial evaluated weekly infusions of xentuzumab in Japanese patients with advanced solid tumors. There were no DLTs during dose escalation and the MTD of xentuzumab was not reached for doses up to 1400 mg/wk. Xentuzumab 1000 mg/wk was recommended as the relevant biological dose and was investigated in the expansion part of the study. Similarly, in the 2 first‐in‐human phase 1 trials with xentuzumab, MTD was not reached for either weekly (up to 1800 mg) or every 3 wk (up to 3600 mg) dosing. 6 Xentuzumab 1000 mg/wk was selected as the relevant biological dose in these studies based on an integrated analysis of safety, PK, biomarker data, and anti‐tumor activity. Bayesian Logistic Regression modeling—integrating biomarker and response data—confirmed the selection. 6
Xentuzumab was generally well tolerated in Japanese patients. The most commonly reported treatment‐related AEs were fatigue, neutropenia, gastrointestinal disorders (diarrhea and nausea), decreased white blood cell counts, and muscle spasms. This is consistent with data from the earlier phase 1 trials, in which the most common treatment‐related AE across both studies was nausea. 6 The low occurrence of hyperglycemia in this study is of interest, as it is considered a class effect of IGF‐1R‐targeted monoclonal antibodies and TKIs due to interaction with the IR and/or disruption of growth hormone homeostasis. 1 There were 2 treatment‐related events of hyperglycemia (both grade 1). This is in line with results from the earlier phase 1 trials, in which there were only 2 cases of treatment‐related hyperglycemia (grades 1 and 3). 6 Similarly, another IGF‐ligand blocking antibody, dusigitumab, also showed a low incidence of treatment‐related hyperglycemia in phase 1 studies conducted in the United States and Japan (1 event in each trial), 10 , 11 suggesting that targeting IGF ligands had minimal impact on glucose homeostasis.
PK parameters were largely consistent with those reported in the previous phase 1 trials with xentuzumab. 6 Total IGF‐1, total IGF‐2, and bioactive IGF in serum were measured as markers of target engagement. Xentuzumab treatment resulted in a sustained accumulation of total serum IGF‐1 and IGF‐2. This is likely to reflect a reduced elimination rate following their binding to xentuzumab. Furthermore, IGF‐1 secretion may initially be increased following xentuzumab treatment due to a growth‐hormone‐dependent negative feedback mechanism. 6 , 12 Our findings are consistent with a mechanistic PK‐PD modeling study that suggested high neutralization of free IGF‐1 and IGF‐2 by at least 90% and 64%, respectively, for the xentuzumab 1000 mg/wk dose (at steady state vs baseline). 13 However, in the previous phase 1 trials, xentuzumab was associated with an increase in total IGF‐1 (reaching a plateau at doses of ≥1050 mg/wk), but had no clear effect on total IGF‐2 levels. 6 It was suggested that this could be due to a lower binding affinity of xentuzumab to IGF‐2 vs IGF‐1 and differences in regulation of synthesis of IGF‐1 vs IGF‐2 (IGF‐2 is not affected by the negative feedback mechanism). Both the current study and other phase 1 studies also showed a reduction in bioactive IGF (a surrogate for IGF‐1 and IGF‐2 activity), consistent with the mode of action of xentuzumab.
Xentuzumab showed preliminary anti‐tumor activity in patients with advanced solid tumors, with 2 patients achieving partial response and 4 experiencing stable disease. Responses and stable disease were durable, with 5 patients receiving xentuzumab for more than 1 y. Interestingly, both partial responses and 2 of the patients with stable disease were in the expansion cohort and all of these patients had sarcomas. Sarcomas are strongly associated with IGF signaling 14 and IGF‐1R‐targeted antibodies have shown activity in some patients with Ewing sarcoma and soft‐tissue sarcoma. 15 , 16
Specifically, both patients with partial response had desmoid tumors. These are rare tumors (incidence of approximately 2‐4 per million people per year) that arise from connective tissues. 17 The incidence is not well documented in Japan but, according to the Bone and Soft Tissue Tumor Registry database, 530 patients were registered with desmoid‐type fibromatosis between 2006 and 2012. 18 There are 2 distinct tumors: sporadic and familial adenomatous polyposis (FAP)‐related desmoids. Although most sporadic desmoid tumors have somatic mutations in β‐catenin (CTNNB1), desmoid tumors in patients affected by FAP usually contain germline mutations in the APC gene. These mutations lead to stabilization of the β‐catenin protein, a key component of the Wnt signaling pathway, which translocates to the nucleus and binds to the TCF/Lef family of transcription factors, resulting in activation of target genes. 19 Interestingly, IGFBP‐6 (a member of the IGF‐binding protein family), which binds to IGF‐1 and IGF‐2, has been shown to be directly downregulated by the β‐catenin/TCF complex in desmoid tumors. 20
In the earlier phase 1 trials of xentuzumab, 2 partial responses were reported during dose escalation: 1 in a patient with poorly differentiated nasopharyngeal carcinoma and another in a patient with peripheral primitive neuroectodermal tumor (pPNET). Durable stable disease (≥24 wk) was also reported in 8 patients with a range of tumor types. 6 Although the expansion parts of these trials each had a cohort enrolling patients with Ewing sarcoma or pPNET, no objective responses were observed in these patients. Furthermore, patients with desmoid‐type tumors were not included in either of these phase 1 trials. 6
Based on the favorable safety profile observed with xentuzumab and mechanisms of resistance to existing anti‐cancer therapies, ongoing trials are assessing xentuzumab as a combination therapy. Following the identification of a potential benefit for the combination of xentuzumab with everolimus and exemestane among patients with advanced hormone receptor‐positive breast cancer with non‐visceral disease, 7 the phase 2 XENERA™‐1 trial (NCT03659136) was initiated to further assess the combination in this patient population. Furthermore, the combination of xentuzumab with the cyclin‐dependent kinase 4/6 inhibitor, abemaciclib, with or without hormone therapy, is also being assessed in a phase 1 trial in patients with advanced solid tumors, including NSCLC and hormone receptor‐positive breast cancer (NCT03099174).
In conclusion, there were no DLTs in the trial and the MTD of xentuzumab was not reached for doses up to 1400 mg weekly. Xentuzumab 1000 mg/wk was determined as the relevant biological dose in Japanese patients with advanced tumors. Xentuzumab was well tolerated and showed evidence of preliminary anti‐tumor activity. The safety and anti‐tumor activity of xentuzumab will be further assessed in combination with other anti‐cancer therapies in breast cancer.
DISCLOSURE
TD declares research funding from Boehringer Ingelheim. YK declares research funding from Taiho Pharmaceutical, Takeda, Ono, AbbVie, AstraZeneca, Boehringer Ingelheim, Incyte, Amgen, Chugai, GSK, Genmab, Astellas, Daiichi Sankyo. YN reports receipt of lecture fees, honoraria, or other fees from Chugai, Pfizer, Eli Lilly and Novartis; and research funding from Daiichi Sankyo, Taiho Pharmaceutical, Pfizer, and Boehringer Ingelheim. MI and YT are employees of Boehringer Ingelheim. TT is an employee of EPS Corporation.
Supporting information
Methods S1
ACKNOWLEDGMENTS
This study was funded by Boehringer Ingelheim. The sponsor played a role in the design of the study, the collection, management, analysis, and interpretation of the data; preparation, review, and approval of the manuscript; and the decision to submit the manuscript for publication, and as such are included in the author list. The authors received no direct compensation related to the development of the manuscript. Medical writing support for the development of this manuscript, under the direction of the authors, was provided by Caroline Allinson of Ashfield MedComms, an Ashfield Health Company, and was funded by Boehringer Ingelheim.
Doi T, Kuboki Y, Naito Y, Ishida M, Tanaka T, Takeuchi Y. A phase 1 trial of xentuzumab, an IGF‐neutralizing antibody, in Japanese patients with advanced solid tumors. Cancer Sci. 2022;113:1010–1017. doi: 10.1111/cas.15231
DATA AVAILABILITY STATEMENT
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant‐level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer‐reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see https://www.mystudywindow.com/msw/datasharing). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data‐access system for a limited period of 1 y, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de‐identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information.
REFERENCES
- 1. Pollak M. The insulin and insulin‐like growth factor receptor family in neoplasia: an update. Nat Rev Cancer. 2012;12:159‐169. [DOI] [PubMed] [Google Scholar]
- 2. Sclafani F, Kim TY, Cunningham D, et al. A randomized phase II/III study of dalotuzumab in combination with cetuximab and irinotecan in chemorefractory, KRAS wild‐type, metastatic colorectal cancer. J Natl Cancer Inst. 2015;107:djv258. [DOI] [PubMed] [Google Scholar]
- 3. Scagliotti GV, Bondarenko I, Blackhall F, et al. Randomized, phase III trial of figitumumab in combination with erlotinib versus erlotinib alone in patients with nonadenocarcinoma nonsmall‐cell lung cancer. Ann Oncol. 2015;26:497‐504. [DOI] [PubMed] [Google Scholar]
- 4. Simpson A, Petnga W, Macaulay VM, Weyer‐Czernilofsky U, Bogenrieder T. Insulin‐like growth factor (IGF) pathway targeting in cancer: role of the IGF axis and opportunities for future combination studies. Target Oncol. 2017;12:571‐597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Friedbichler K, Hofmann MH, Kroez M, et al. Pharmacodynamic and antineoplastic activity of BI 836845, a fully human IGF ligand‐neutralizing antibody, and mechanistic rationale for combination with rapamycin. Mol Cancer Ther. 2014;13:399‐409. [DOI] [PubMed] [Google Scholar]
- 6. de Bono J, Lin C‐C, Chen L‐T, et al. Two first‐in‐human studies of xentuzumab, a humanised insulin‐like growth factor (IGF)‐neutralising antibody, in patients with advanced solid tumours. Br J Cancer. 2020;122:1324‐1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schmid P, Sablin M‐P, Bergh J, et al. A phase Ib/II study of xentuzumab, an IGF‐neutralising antibody, combined with exemestane and everolimus in hormone receptor‐positive, HER2‐negative locally advanced/metastatic breast cancer. Breast Cancer Res. 2021;23:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Park K, Shao Weng Tan D, Su W‐C, et al. Phase Ib open‐label trial of afatinib plus xentuzumab (BI 836845) in patients with EGFR mutation‐positive NSCLC after progression on EGFR tyrosine kinase inhibitors. JTO Clin Res Rep. 2021;2:100206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hussain SA, Maroto P, Climent MÁ, et al. Targeting IGF‐1/2 with xentuzumab (Xe) plus enzalutamide (En) in metastatic castration‐resistant prostate cancer (mCRPC) after progression on docetaxel chemotherapy (DCt) and abiraterone (Abi): randomized phase II trial results. J Clin Oncol. 2019;37(15_suppl):5030. [Google Scholar]
- 10. Haluska P, Menefee M, Plimack ER, et al. Phase I dose‐escalation study of MEDI‐573, a bispecific, antiligand monoclonal antibody against IGFI and IGFII, in patients with advanced solid tumors. Clin Cancer Res. 2014;20:4747‐4757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Iguchi H, Nishina T, Nogami N, Kozuki T, Yamagiwa Y, Yagawa K. Phase I dose‐escalation study evaluating safety, tolerability and pharmacokinetics of MEDI‐573, a dual IGF‐I/II neutralizing antibody, in Japanese patients with advanced solid tumours. Invest New Drugs. 2015;33:194‐200. [DOI] [PubMed] [Google Scholar]
- 12. Mireuta M, Birman E, Barmash M, Pollak M. Quantification of binding of IGF‐1 to BI 836845, a candidate therapeutic antibody against IGF‐1 and IGF‐2, and effects of this antibody on IGF‐1:IGFBP‐3 complexes in vitro and in male C57BL/6 mice. Endocrinology. 2014;155:703‐715. [DOI] [PubMed] [Google Scholar]
- 13. Parra‐Guillen ZP, Schmid U, Janda A, Freiwald M, Troconiz IF. Model‐informed dose selection for xentuzumab, a dual insulin‐like growth factor‐I/II‐neutralizing antibody. Clin Pharmacol Ther. 2020;107:597‐606. [DOI] [PubMed] [Google Scholar]
- 14. Rikhof B, de Jong S, Suurmeijer AJ, Meijer C, van der Graaf WT. The insulin‐like growth factor system and sarcomas. J Pathol. 2009;217:469‐482. [DOI] [PubMed] [Google Scholar]
- 15. Olmos D, Postel‐Vinay S, Molife LR, et al. Safety, pharmacokinetics, and preliminary activity of the anti‐IGF‐1R antibody figitumumab (CP‐751,871) in patients with sarcoma and Ewing's sarcoma: a phase 1 expansion cohort study. Lancet Oncol. 2010;11:129‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Schöffski P, Adkins D, Blay J‐Y, et al. An open‐label, phase 2 study evaluating the efficacy and safety of the anti‐IGF‐1R antibody cixutumumab in patients with previously treated advanced or metastatic soft‐tissue sarcoma or Ewing family of tumours. Eur J Cancer. 2013;49:3219‐3228. [DOI] [PubMed] [Google Scholar]
- 17. Devata S, Chugh R. Desmoid tumors: a comprehensive review of the evolving biology, unpredictable behavior, and myriad of management options. Hematol Oncol Clin North Am. 2013;27:989‐1005. [DOI] [PubMed] [Google Scholar]
- 18. Nishida Y, Kawai A, Toguchida J, et al. Clinical features and treatment outcome of desmoid‐type fibromatosis: based on a bone and soft tissue tumor registry in Japan. Int J Clin Oncol. 2019;24:1498‐1505. [DOI] [PubMed] [Google Scholar]
- 19. Kotiligam D, Lazar AJ, Pollock RE, Lev D. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol. 2008;23:117‐126. [DOI] [PubMed] [Google Scholar]
- 20. Denys H, Jadidizadeh A, Amini Nik S, et al. Identification of IGFBP‐6 as a significantly downregulated gene by β‐catenin in desmoid tumors. Oncogene. 2004;23:654‐664. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Methods S1
Data Availability Statement
To ensure independent interpretation of clinical study results, Boehringer Ingelheim grants all external authors access to relevant material, including participant‐level clinical study data, as needed by them to fulfill their role and obligations as authors under the ICMJE criteria. Clinical study documents and participant clinical study data are available to be shared on request after publication of the primary manuscript in a peer‐reviewed journal, and if regulatory activities are complete and other criteria met as per the BI Policy on Transparency and Publication of Clinical Study Data (see https://www.mystudywindow.com/msw/datasharing). Bona fide, qualified scientific and medical researchers are eligible to request access to the clinical study data with corresponding documentation describing the structure and content of the datasets. Upon approval, and governed by a Legal Agreement, data are shared in a secured data‐access system for a limited period of 1 y, which may be extended upon request. Prior to providing access, clinical study documents and data will be examined, and, if necessary, redacted and de‐identified, to protect the personal data of study participants and personnel, and to respect the boundaries of the informed consent of the study participants. Researchers should use the https://vivli.org/ link to request access to study data and visit https://www.mystudywindow.com/msw/datasharing for further information.