

Abstract
The challenge to improve the clinical efficacy and enlarge the population that benefits from immune checkpoint inhibitors (ICIs) for non‐small‐cell lung cancer (NSCLC) is significant. Based on whole‐exosome sequencing analysis of biopsies from NSCLC patients before anti‐programmed cell death protein‐2 (PD‐1) treatment, we identified NLRP4 mutations in the responders with a longer progression‐free survival (PFS). Knockdown of NLRP4 in mouse Lewis lung cancer cell line enhanced interferon (IFN)‐α/β production through the cGAS‐STING‐IRF3/IRF7 axis and promoted the accumulation of intratumoral CD8+ T cells, leading to tumor growth retardation in vivo and a synergistic effect with anti‐PD‐ligand 1 therapy. This was consistent with clinical observations that more tumor‐infiltrating CD8+ T cells and elevated peripheral IFN‐α before receiving nivolumab treatment were associated with a longer PFS in NSCLC patients. Our study highlights the roles of tumor‐intrinsic NLRP4 in remodeling the immune contextures in the tumor microenvironment, making regional type I IFN beneficial for ICI treatment.
Keywords: immune checkpoint inhibitor therapy, lung cancer, NLRP4, tumor microenvironment, type I interferon
Our study addressed novel mechanisms for overcoming failures of immune checkpoint inhibitor (ICI) therapy for non‐small‐cell lung cancer (NSCLC) through endogenous regulation of NLRP4 on type I interferon. This is realized mostly through reshaping the tumor microenvironment to a “hot” tumor status, which is beneficial to the treatment of anti‐programmed cell death protein‐1. Furthermore, we provided new evidence on type I interferon (regulated by NLRP4 in our study) as a potential immune adjuvant to improve the efficacy of NSCLC patients ineffective to ICI treatment.
Abbreviations
- Arg1
arginase 1
- GSVA
Gene Set Variation Analysis
- ICI
immune checkpoint inhibitor
- IFA
immunofluorescence assay
- IFN
interferon
- IFNI
type I interferon
- IHC
immunohistochemistry
- IRF
interferon regulatory factor
- KD
knockdown
- LLC
Lewis lung cancer cells
- MDSC
myeloid‐derived suppressive cell
- NC
negative control
- NLR
nod‐like receptor
- NLRP4
NLR family pyrin domain containing 4
- NR
nonresponder
- NSCLC
non‐small‐cell lung cancer
- OE
overexpression
- PD
progressive disease
- PD‐1
programmed cell death protein 1
- PD‐L1
programmed death‐ligand 1
- PFS
progression‐free survival
- PR
responder
- R
partial response
- SD
stable disease
- SJTUSM
Shanghai Jiao Tong University School of Medicine
- STING
stimulator of IFN genes
- TBK1
TANK‐binding kinase 1
- TCGA
The Cancer Genome Atlas
- TME
tumor microenvironment
- WES
whole‐exome sequencing
1. INTRODUCTION
The application of ICIs in the treatment of advanced NSCLC has achieved remarkable accomplishments in the last several years. 1 Despite the clinical progress, only 20% of patients benefit from ICI monotherapy with durable responses. 2 , 3 , 4 Therefore, the development of new therapies to overcome the resistance and expand the beneficial population remains a big challenge.
Immune checkpoint inhibitors are reported to be able to reinvigorate exhausted immune cells, 5 mainly targeting infiltrating immune cells in the TME. 6 The composition, function, density, and compartmentalization of infiltrating immune cells are presumably relevant to the prognosis of ICI treatment. However, the properties of the tumors themselves are also likely to affect the efficacy of ICIs, including expression levels of PD‐L1 on tumor cells 7 or tumor mutation burden. 8 In addition, cytokines including IL‐10 and transforming growth factor‐β secreted by tumor cells affect the outcome of ICI treatment as well. 9 They construct a unique immune‐suppressive TME together with local immune suppressive cells such as regulatory T cells and MDSCs. 10 Antitumor immunity is thus impeded locally, resulting in immune escape. Therefore, to define the inherently tumor‐intrinsic immune‐like properties in the TME to some extent becomes an alternative opportunity to find new strategies to overcome ICI resistance.
In this study, based on the results of WES of tumor biopsies and paired peripheral blood from patients with advanced NSCLC with different responses to nivolumab (anti‐PD‐1 Ab) before treatment, 11 , 12 we identified NLRP4, an NLR family member previously reported as a negative regulator of IFNI signaling, 13 to be a novel tumor‐intrinsic factor modulating the efficacy of ICI therapy largely through reshaping the immunological properties of the TME.
2. MATERIALS AND METHODS
2.1. Human subjects and cell lines
Non‐small‐cell lung cancer patients in this study were those enrolled in CheckMate 078 (NCT02613507), CheckMate 870 (NCT03195491), and NCT03594747 studies from Shanghai Chest Hospital affiliated to Shanghai Jiao Tong University. All patients provided signed informed consent. The study followed the principles of the Declaration of Helsinki and was approved by the Ethics Committee of Shanghai Chest Hospital (No. 2926). Detailed information is provided in Document S1.
Mouse lung cancer cell lines, including LLC and KLN cell lines, mouse colon cancer cell line MC‐38, and macrophage cell line Raw 264.7 were purchased from the Biological Sciences Chinese Academy of Sciences, and maintained routinely in the laboratory.
2.2. Whole‐exome sequencing and identification of putative somatic mutations
Thirty‐one tumor biopsies and matched peripheral blood neutrophils as normal counterparts were subjected to WES by Shanghai Shenyou Biotech Company. Pathway analysis was undertaken by using GSVA. 14
2.3. Cytokine detection
Plasma was collected from whole blood (10 mL) of 59 NSCLC patients before nivolumab treatment. Cytokines were detected using the Inflammation 20‐Plex Human ProcartaPlex Panel (Cat. No. EPX200‐12185‐901; Invitrogen) according to the manufacturer’s instructions.
2.4. Multiplex IFA
Formaldehyde‐fixed and paraffin‐embedded sections from the biopsies before nivolumab treatment were subjected to multiplex IFA by using a tyramide signal amplification multiplex IHC kit (Cat. No. 0004100100; Panovue) according to the manufacturer’s instructions. The slides were scanned using the Mantra multispectral imaging system (PerkinElmer). Different cell types were quantified in each field of the images automatically with inForm software (PerkinElmer) according to the immunofluorescence signals. Cells within a 30‐μm radius around CD8+ T cells were counted and weighd. 15
2.5. Knockdown and overexpression of NLRP4 in mouse tumor cell lines
Lentiviruses containing mouse NLRP4 shRNA (listed in Table S1) or full‐length NLRP4 sequences were purchased from Hanbio. The efficacy of NLRP4 KD and OE together with NC was determined by RT‐PCR and western blot analysis.
2.6. Quantitative real‐time PCR
Total RNA was extracted from cells using TRIzol reagent (Invitrogen) according to the manufacturer’s guideline and quantitative real‐time PCR was used to determine gene expression levels. The primers of the genes are listed in Table S1.
2.7. Cell proliferation assay
Cell Counting Kit‐8 detection reagent (Cat. No. CK04; Dojindo) was used according to the manufacturer’s guideline to determine cell proliferation in vitro. The absorbance at 450 nm was measured using a PowerWaveXS2 microplate spectrophotometer (BioTek Instruments).
2.8. Tumor growth and Ab treatment in C57BL/6 mice
Female C57BL/6 mice (6‐8 weeks old) were purchased from the Shanghai Laboratory Animal Center and maintained under specific pathogen‐free conditions in the animal facility of SJTUSM. For ICI treatment, 200 μg anti‐PD‐L1 mAb (Clone10F.9G2, Cat. No. BE0101; BioXcell was given intraperitoneally from day 12 with a 2‐day interval. In some experiments, 5 × 104 units IFN‐β (R&D Systems) were injected intratumorally twice from day 11 with a 3‐day interval. For the CD8 depletion experiment, 250 μg anti‐mouse CD8 Ab (Clone YTS169.4; BioXcell) was injected intraperitoneally at days 1, 5, and 11. The protocols of animal experiments were approved by the Animal Ethics Committee of SJTUSM, and carried out under the Guide for the Care and Use of Laboratory Animals.
2.9. Western blot analysis
Cells were collected and lysed by RIPA lysis buffer (Cat. No. 89900; Thermo Fisher Scientific) containing cocktail proteinase inhibitors (Cat. No. C0001; TargetMol). Western blotting was carried out routinely. Antibodies used in western blotting are listed in Table S1.
2.10. Flow cytometry
Implanted tumors were harvested and digested into single‐cell suspension using a mouse tumor dissociation kit (Cat. No. 130‐096‐730; Miltenyi Biotec). Cell types and function were assayed by using flow cytometric analysis. Cells were acquired with a Fortessa flow cytometer (BD Biosciences) after staining (Abs used in the assays are listed in Table S1). Data were analyzed using FlowJo software 7.5 (Treestar). The doublet events were excluded by FSC‐A/FSC‐H gating.
2.11. Transwell migration assay
Migration assays were carried out by seeding the splenocytes in the upper chamber, and NC‐, NLRP4 KD‐, or NLRP4 OE‐LLC cells in the bottom chambers. In some experiments, IFN‐α or IFN‐β (200 μg/mL, listed in Table S1) were added in the bottom chambers. CD8+ T cells migrating to the bottom chambers were enumerated by flow cytometry.
2.12. RNA sequencing analysis
Total RNA was extracted using an AllPrep DNA/RNA/miRNA Universal Kit (Cat No 80224; Qiagen) and subjected to the transcriptome assay (Shanghai Shenyou Biotech). Gene ontology enrichment analysis was undertaken using online DAVID (https://david.ncifcrf.gov/). The abundances of cell types were estimated using the CIBERSORT analysis (http://cibersort.stanford.edu/) based on the gene expression data (RNA transcripts). 16
2.13. Prediction of binding capacity of mutant NLRP4 proteins
Meta‐PPISP analysis was used to predict the capacity of protein‐protein interactions with mutant NLRP4 proteins. Binding scores were generated based on secondary structure, atomic distribution, amino acid pairs, and sequence conservation calculated by machine learning.
2.14. Statistical analysis
Data are presented as mean ± SEM or SD. Statistical analysis was undertaken by using SPSS 17.0 (IBM) or GraphPad Prism 5.0 (GraphPad Software). All statistical tests were two‐sided. P values less than .05 were considered statistically significant.
3. RESULTS
3.1. Presence of NLRP4 mutations in tumor biopsies associated with favorable responses to anti‐PD‐1 treatment in advanced NSCLC
Tumor biopsies were obtained from 31 patients with advanced NSCLC before treatment with either anti‐PD‐1 therapy (n = 22) or combined therapy (n = 9). The WES analysis was carried out to determine somatic mutations existing in the tumors from two groups. Patients were defined as R (with PR or SD) or NR (with PD) according to the RECIST 1.1 evaluation at 8 weeks after treatment. A total of 29 genes harboring somatic mutations were identified; of them, IFNI signaling‐related genes, including IRF7, IKBKB, RIPK3, and NLRP4, were enriched in the R group. Among them, NLRP4 was the gene with somatic mutations (Figure S1) existing only in the R group (2/11) whereas no mutations were detectable in the NR group (0/11) (Figure 1A). Two patients harboring NLRP4 mutations had a long PFS of 208 and 565 days, respectively (Figure 1B, arrows). In nine patients receiving combination therapy, three patients harboring NLRP4 mutations (Figure 1C) (including P721Q, R179L, and D80Y) also had a relatively long PFS of 720, 462, and 238 days, respectively (Figure 1D). Moreover, by using the TCGA database, it was also revealed that patients with NLRP4 mutations had a prolonged PFS (median, 30 months) as well when compared to those without the mutations (median, 7.9 months) (Figure 1E). In addition, serum IFN‐α (Figure 1F, left) and IFN‐β (Figure 1F, right) in NSCLC patients with NLRP4 mutations before receiving nivolumab treatment were relatively higher than those without NLRP4 mutations. Therefore, tumor‐intrinsic mutations in NLRP4 are inclined to facilitate anti‐PD‐1 therapy with durable responses in advanced NSCLC, which is probably associated with the elevation of IFNIs.
FIGURE 1.
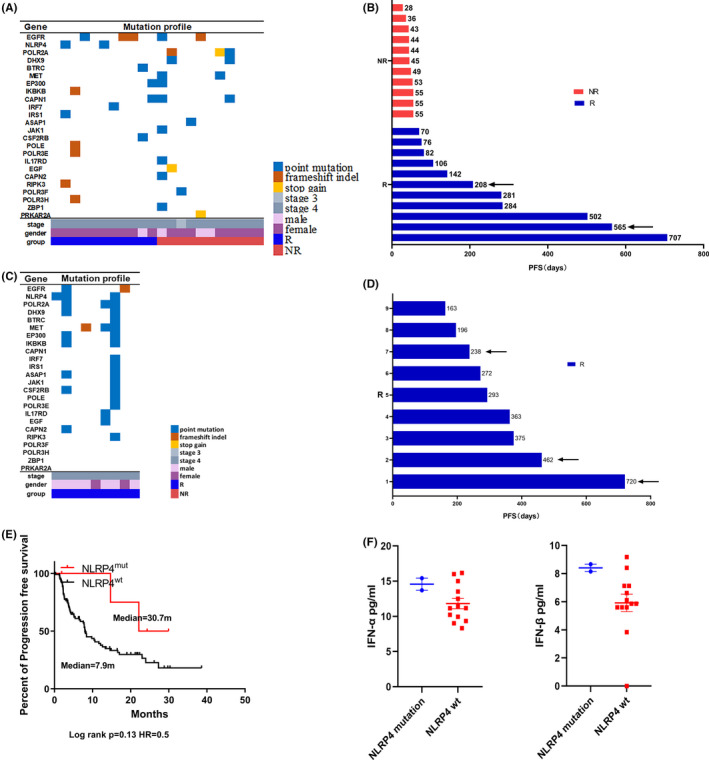
Identification of NLRP4 somatic mutations in patients with advanced non‐small‐cell lung cancer (NSCLC) with a longer progression‐free survival (PFS) following anti‐programmed cell death protein‐1 (PD‐1) treatment. A‐D, Mutation profiles and the PFS of NSCLC patients receiving nivolumab treatment (n = 22) (A,B) or combination therapy (C,D) (n = 9). Arrows indicate patients with NLRP4 mutations. E, Kaplan‐Meier overall survival curves for NSCLC patients (n = 109) with immune checkpoint inhibitor treatment in The Cancer Genome Atlas database. HR, hazard ratio. F, Comparison of serum interferon (IFN)‐α (left) and IFN‐β (right) between responder (R) and nonresponder (NR) groups
3.2. NLRP4 KD shows increased IFNI expression through the STING‐TBK1‐IRF3 axis and retards tumor growth in vivo
The observations on the clinical relevance of NLRP4 mutations to a longer PFS following nivolumab treatment drove us to explore the underlying mechanisms. NLRP4 KD‐LLC and NLRP4 OE‐LLC were constructed to validate the effect of NLRP4 on IFNI production (Figure 2A). It was found that the suppression of NLRP4 in LLC cells (Figure 2B, left; P < .05) significantly increased the mRNA levels of IFN‐α (Figure 2C, left; P < .01) and IFN‐β (Figure 2D, left; P < .001), which was consistent with protein levels in the supernatants of cell cultures (Figure S2). In contrast, overexpression of NLRP4 in LLC cells (Figure 2B, right; P < .001) led to a significant decrease in IFN‐β mRNA levels (Figure 2D, right; P < .05) and no significant alteration in IFN‐α (Figure 2C, right). When detecting the downstream signaling, it was found that the level of phospho‐STING (p‐STING) increased together with the phosphorylations of TBK1 and IRF3 when NLRP4 expression was suppressed in LLC cells (Figure 2E, left), but decreased in NLRP4 OE‐LLC cells compared to those in control LLC cells (Figure 2E, right).
FIGURE 2.
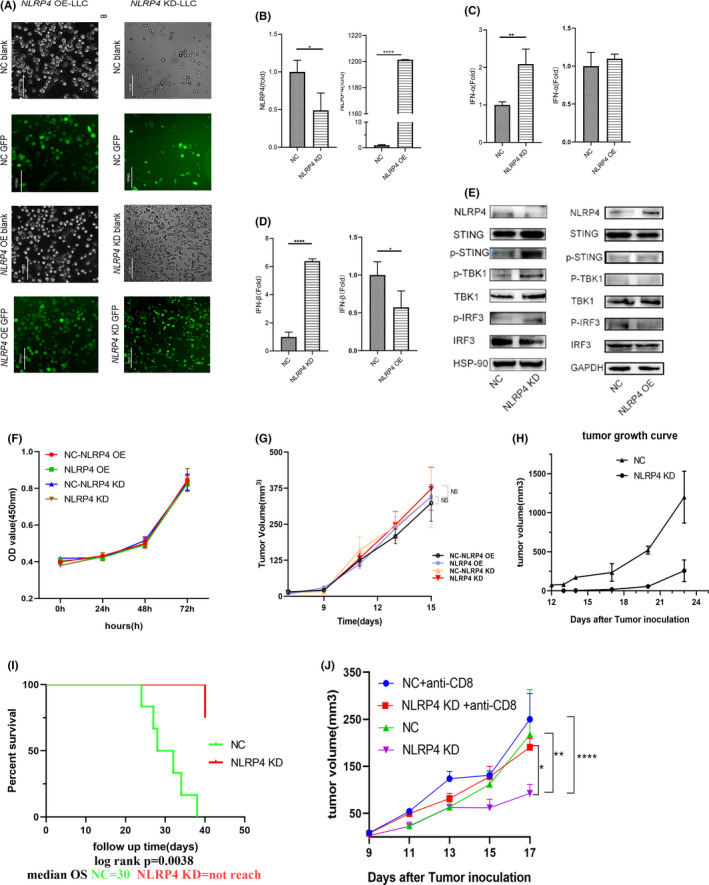
NLRP4 regulates type I interferon (IFN) production through the stimulator of IFN genes (STING)‐TANK‐binding kinase 1 (TBK1)‐IFN regulatory factor 3 (IRF3) axis in Lewis lung cancer cells (LLC) cells and retards tumor growth in vivo. A, GFP signals in LLC cells infected with lentivirus containing negative control (NC) or NLRP4 shRNA (NLRP4 KD) (right), or overexpressed NLRP4 gene (NLRP4 OE) (left). Magnification, ×200. Scale bar, 100 μm. B‐D, NLRP4 (B), IFN‐a (C), and IFN‐β (D) mRNA levels were compared between NC and NLRP4 KD‐LLC or NLRP4 OE‐LLC cells. E, Signaling molecules including phospho (p)‐STING, p‐IRF3, and p‐TBK1 between LLC‐NC cells were analyzed among NC‐, NLRP4 KD‐, or NLRP4 OE‐LLC cells. F, G, Proliferative curves of NLRP4 KD‐ and NLRP4 OE‐LLC cells in vitro (F) and in nude mice (G). H, Tumor growth curves in C57BL/6 mice with LLC cell inoculation. The result is the representative of two independent experiments. I, Survival curves of C57BL/6 mice with 0.5 × 106 LLC cells in‐situ implantation. J, Tumor growth curves in C57BL/6 mice with NLRP4 KD‐ and NC‐LLC cell inoculations following anti‐CD8 Ab treatment. P values were calculated using Student’s t test or ANOVA. *P < .05; **P < .01; ****P < .0001. HSP‐90, heat shock protein‐90; OD, optical density; OS, overall survival
We further determined the impacts of NLRP4 on tumor growth both in vitro and in vivo. Neither NLRP4 KD nor OE influenced the proliferation of LLC cells in vitro (Figure 2F) and in nude mice (Figure 2G). This was also observed in KLN cells (another lung cancer cell line with low expression of NLRP4) with NLRP4 OE (Figure S3). However, when inoculated in C57BL/6 mice, there was a dramatic retardation of tumor growth in NLRP4 KD‐LLC cell‐implanted mice (Figure 2H). The survival rate of the mice with in situ implantation of NLRP4 KD‐LLC cells was significantly prolonged when compared to NC‐LLC cell implanted mice (Figure 2I; P = .0038). Similar phenomena were observed in MC‐38 mouse colon cancer cells with NLRP4 either downregulated or OE (Figure S4). Moreover, depletion of CD8+ T cells in C57BL/6 mice led to the acceleration of tumor growth of NLRP4 KD‐LLC cells in vivo (Figure 2J). These results indicate that tumor‐intrinsic NLRP4 negatively regulates IFNI production through the STING‐TBK1‐IRF3 axis and inhibits tumor growth only in vivo, partially in a CD8+ T cell‐dependent manner.
3.3. Regional CD8+ T cell responses in NLRP4 KD‐LLC tumors are enhanced and associated with the efficacy of anti‐PD‐L1 therapy in mice
To investigate the mechanisms underlying NLRP4 KD inhibition of tumor growth in vivo (Figure 2H), but not in vitro (Figure 2F), we used RNA sequencing analysis of tumor samples derived from NLRP4 KD‐LLC and NC‐LLC tumors. Volcano plots derived from the differential expression gene analysis revealed that CD8+ T cell‐related genes, such as CD8a, CD3e, lat, and Cd8b1 expressions, were enriched in NLRP4 KD‐LLC tumors (Figure 3A,B) with dramatic difference when compared to NC‐LLC tumors (Figure 3C). This was also confirmed by IHC staining of CD8 molecules in grafted tumors (Figure 3D; P < .0001). As high percentages of CD8+ T cells were present in NLRP4 KD‐LLC grafted tumors (Figure S5A), we further determined whether NLRP4 KD in LLC cells triggered the migration of CD8+ T cells into tumor regions. 17 The Transwell migrating culture assay showed that more CD8+ T cells migrated to the lower chamber when incubated with NLRP4 KD‐LLC cells, whereas fewer CD8+ T cells migrated with NLRP4 OE‐LLC cells (P = .031), when compared to control LLC cells (Figure 3E). When IFN‐α and IFN‐β were added directly to the Transwell culture systems directly, it was evident that both IFNs were able to trigger the migration of CD8+ T cells (Figure 3F).
FIGURE 3.
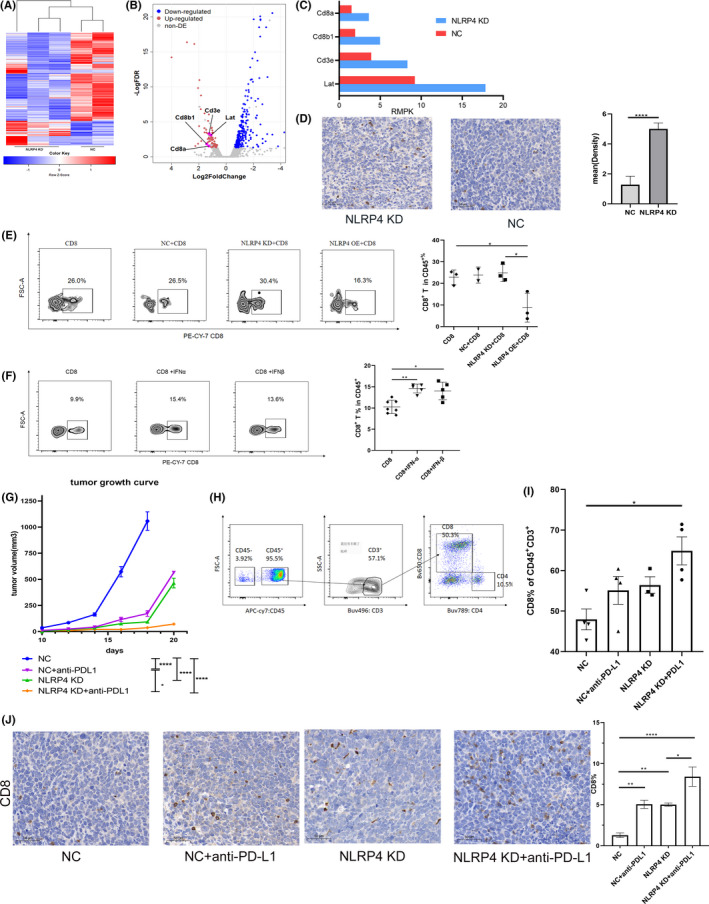
Knockdown (KD) of NLRP4 leads to high infiltration of CD8+ T cells and orchestrates anti‐programmed cell death‐ligand 1 (PD‐L1) treatment. A, Heatmap and hierarchical clustering analysis of NLRP4‐KD and negative control (NC) Lewis lung cancer (LLC) cell tumors in mice. B, Volcano plots showing upregulated genes (red) and downregulated genes (blue) in NLRP4 KD‐LLC‐derived grafted tumors. C, Comparisons of gene expression levels (reads per kilobase per million map reads [RMPK]) related to CD8+ T cell activation based on RNA sequencing data. D, Representative images of CD8 expression in paraffin‐embedded tumors samples (left) (magnification, ×200) and statistical results (right). E, F, Percentages of CD8+ T cells (in CD45+ cells) migrating to the bottom chambers in the Transwell assays when incubating with LLC cells (E) or with the addition of interferon (IFN)‐α (200 unit/mL) or IFN‐β (200 unit/mL) (F). G, Growth curves of inoculated tumors with NC‐LLC and NLRP4 KD‐LLC cells with anti‐PD‐L1 treatment. H, CD8+ T cells gating of flowcytometric analysis. I, Flow cytometry analysis of CD8+ T cells in inoculated tumors among NC‐LLC and NLRP4 KD‐LLC cell‐derived tumors with or without anti‐PD‐L1 treatment. J, Comparison of CD8+ T cell percentages among four groups. Results are representative of two independent experiments. P values were calculated using Student’s t test. *P < .05; **P < .01; ****P < .0001
As higher infiltration of CD8+ T cells, together with the upregulation of PD‐L1 expression on CD45− nonimmune cells, were present in NLRP4 KD‐LLC tumors (Figure S5B‐D), we further treated C57BL/6 mice bearing NC‐LLC or NLRP4 KD‐LLC tumors with a murine anti‐PD‐L1 mAb. It was obvious that anti‐PD‐L1 mAb largely improved tumor control both in NC‐LLC and NLRP4 KD‐LLC tumors (Figures 3G and S6) (WT+PD‐L1 vs WT; NLRP4 KD vs NLRP4 KD+PD‐L1; both P <.001) while anti‐PD‐L1 mAb exhibited the best tumor inhibitory effect in NLRP4 KD‐LLC tumors (Figure 3G, orange line). We also observed significantly higher percentages of CD8+ T cells in NLRP4 KD‐LLC grafted tumors following anti‐PD‐L1 mAb treatment when compared to the tumors from other three groups (Figure 3H,I), which was consistent with the results of CD8 positivity from the IHC staining assay (Figure 3J). These results indicate that NLRP4 KD in LLC cells can improve the therapeutic effects of anti‐PD‐L1 Ab, largely through promoting more infiltration of CD8+ T cells in the TME.
3.4. Anti‐PD‐L1 treatment leads to increase in M1 macrophage infiltration in LLC tumors with NLRP4 KD
With CD8+ T cells, macrophages are the main components of immune cells in the TME. Based on RNA sequencing data and the CIBERSORT 16 analysis on grafted LLC tumors, it was shown that NLRP4 KD led to a significant increase in M2 macrophages in tumors (Figure 4A). When coculturing Raw264.7 cells with NLRP4 KD‐ or NC‐LLC cells in vitro, it was shown that NLRP4 KD‐LLC cells promoted the induction of CD206+F4/80+ M2 macrophages (Figure 4B; P < .01 in 1:1; P < .05 in 1:4) 18 as well. In vivo treatment of anti‐PD‐L1 mAb led to a dramatic decrease in CD11b+F4/80+CD206+CD86− M2 19 macrophages (Figure 4C‐E), but an increase in CD11b+F4/80+CD206−CD86+ M1 macrophages (Figure 4F) in both NC‐LLC and NLRP4 KD‐LLC tumors. In addition, there was a dramatic downregulation of PD‐L1 on CD11b+F4/80+CD206+ M2 macrophages following anti‐PD‐L1 treatment, whether NLRP4 was knocked down or not (Figure 4E). The IHC results of CD206 (Figure 4I) expression in NLRP4 KD‐LLC and NC‐LLC tumors were consistent with those by flow cytometry assay, although the expressions of Arg1 (Figure 4H) and inducible nitric oxide synthase (iNOS) (Figure 4J) maintained in NLRP4 KD‐LLC and NC‐LLC tumors with or without anti‐PD‐L1 treatment. These results indicate that although NLRP4 KD in LLC cells triggers more induction of M2 macrophages both in vitro and in the grafted tumors, anti‐PD‐L1 mAb treatment in vivo leads to an increase in M1 macrophages locally.
FIGURE 4.
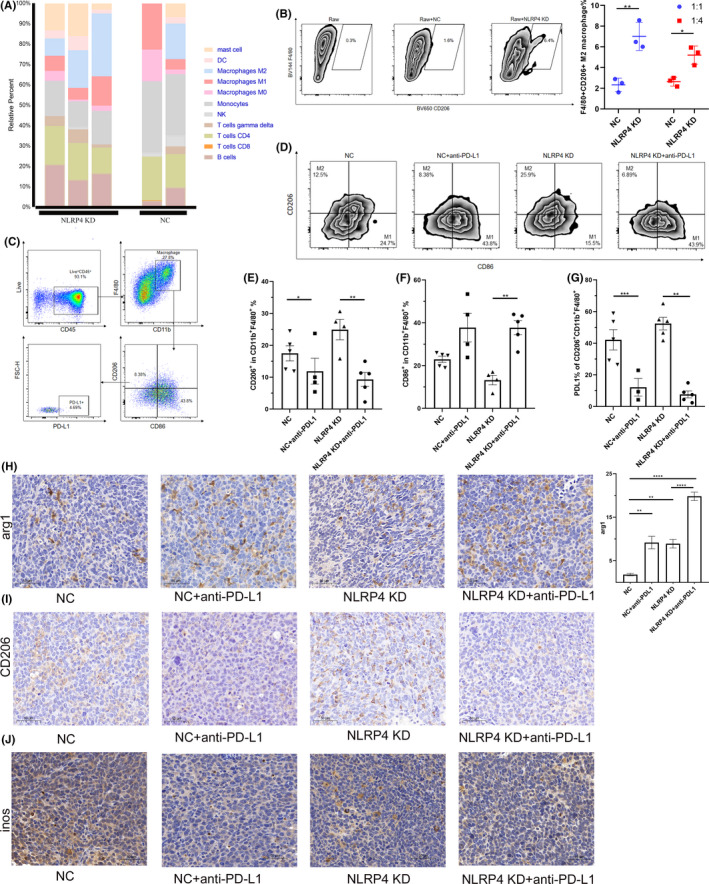
Anti‐programmed cell death‐ligand 1 (PD‐L1) treatment leads to increase in M1 macrophage infiltration in Lewis lung cancer cells (LLC) tumors following NLRP4 knockdown (KD). A, Cell composition analysis in LLC‐derived grafted tumors by CIBERSORT method. B, Phenotypic analysis of Raw 264.7 cells cocultured with NC‐LLC and NLRP4 KD‐LLC cells. Data are the representative of at least two independent experiments. C, D, Flow cytometric analysis of CD11b+F4/80+CD86+CD206− (M1) and CD11b+F4/80+CD206+CD86− (M2) macrophages in LLC inoculated tumors. E‐G, Comparisons of M2 (E) and M1 (F) proportions in CD11b+F4/80+ macrophages and PD‐L1 expression on M2 macrophages (G) in LLC inoculated tumors. H‐J, Immunohistochemical results of Arginase 1 (Arg1) (H), CD206 (I), and inducible nitric oxide synthase (iNOS) (J) in LLC inoculated tumors. Data are shown as mean ± SEM; n = 4‐8 mice per group. P values were calculated using ANOVA and Student’s t test. *P < .05; **P < .01; ****P < .0001. DC, dendritic cell; NC, negative control; NK, natural killer
3.5. Infiltration of CD8+ T cells and higher percentages of M2‐like macrophages in TME associated with better response to nivolumab in NSCLC patients
Due to the enhanced CD8+ T cell and M2 macrophage distributions in NLRP4 KD‐LLC tumors with better responses to experimental anti‐PD‐L1 treatment, we next validated the observations in tumor biopsies from 24 advanced NSCLC patients (NR = 12 and R = 12). There was no significant difference in the age, gender, pathology, or stages between the two groups, except smoking (Table 1). 20 Based on multiplex IFA staining (Figure 5A), it was apparent that NSCLC patients with more CD8+ T cells had a significantly longer PFS (high 94 days vs low 35.5 days; P < .001) (Figure 5B and Table S2). Similar to CD8+ T cell infiltration, more tumor cells surrounding CD8+ T cells were associated with prolonged PFS 17 (P = .036) (Figure 5C). We also analyzed the spatial distributions of CD8+ T cells and CD33+CD11b+ myeloid cells to explore interactions between the two types of immune cells in the TME by counting space distance with Euclidean distance of the distribution (represented by within mean values, calculated by inForm software) (Figure 5D). The average of within mean values was lower in the R group than in the NR group (Figure 5E), 21 implying more interactions between CD8+ T cells and CD33+CD11b+ myeloid cells.
TABLE 1.
Clinical manifestations of patients with non‐small‐cell lung cancer (NSCLC)
| Characteristic | Total (n = 24) | NR (n = 12) | R (n = 12) | P value |
|---|---|---|---|---|
| Age, y | ||||
| Median | 61.64 | 60.67 | 62.33 | >.05 |
| Sex | ||||
| Male | 21 | 9 (75.00) | 12 (100.00) | .064 |
| Female | 3 | 3 (25.00) | 0 (0.00) | .217 |
| History | ||||
| Squamous | 12 | 6 (50.00) | 6 (50.00) | >.05 |
| Nonsquamous | 12 | 6 (50.00) | 6 (50.00) | >.05 |
| Smoking status | ||||
| Smoker | 17 | 6 (50.00) | 11 (91.67) | .025 |
| Nonsmoker | 7 | 6 (50.00) | 1 (8.33) | .069 |
| Disease stage | ||||
| III | 4 | 3 (25.00) | 1 (8.33) | .273 |
| IV | 20 | 9 (75.00) | 11 (91.67) | .590 |
| EGFR mutation | ||||
| Yes | 3 | 1 (8.33) | 2 (16.67) | .534 |
| No | 19 | 10 (83.33) | 9 (75.00) | 1.000 |
| ALK mutation | ||||
| Yes | 0 | 0 (0.00) | 0 (0.00) | |
| No | 20 | 11 (91.67) | 9 (75.00) | |
| T stage | ||||
| 1 | 3 | 2 | 1 | .068 |
| 2 | 8 | 5 | 3 | .081 |
| 3 | 5 | 4 | 1 | |
| 4 | 8 | 1 | 7 | |
| N stage | ||||
| 0 | 4 | 3 | 1 | .392 |
| 1 | 1 | 1 | 0 | .413 |
| 2 | 10 | 5 | 5 | |
| 3 | 9 | 3 | 6 | |
| M stage | ||||
| 0 | 4 | 3 | 1 | .273 |
| 1 | 20 | 9 | 11 | .590 |
Formalin‐embedded sections from 24 patients with advanced NSCLC were subjected to multiplex immunofluorescence assay to determine the infiltrating lymphocyte profiles. Patients were subgrouped into nonresponder (NR; n = 12) and responder (R; n = 12) groups according to disease progression at 8 wk after treatment and were followed up until the end of the treatment for calculating progression‐free survival values. Bold value shows the significant difference (P = .025 < .05).
FIGURE 5.
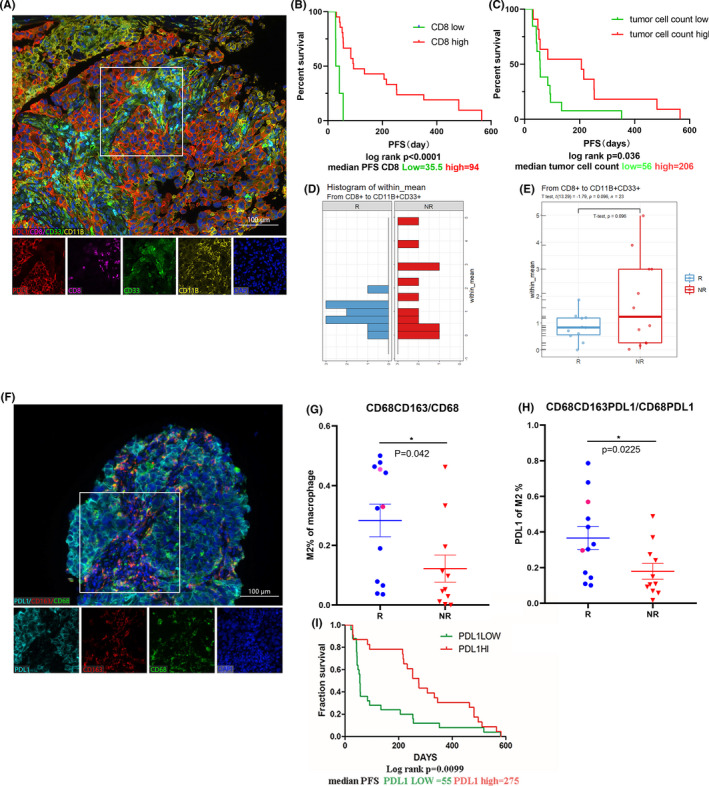
Infiltrating lymphocyte features in the tumor microenvironment of patients with advanced non‐small‐cell lung cancer (NSCLC) before receiving anti‐programmed cell death protein 1 (PD‐1)/PD‐ligand 1 (PD‐L1) treatment. A, Representative images of multiplex immunofluorescence assays (IFAs) with CD8 (pink), CD11B (yellow), PD‐L1 (red), and CD33 (green). DAPI dye (purple) was used to indicate the nuclei. Magnification, ×200; scale bar, 100 μm. B, C, Kaplan‐Meier curves for NSCLC patients (n = 24) with (B) CD8+ T cell percentages and (C) tumor cells around CD8+ T cells. D, E, Euclidean distance of CD8+ T cells and CD33+CD11b+ myeloid cells (D), and comparison between responder (R; n = 12) and nonresponder (NR; n = 12) groups (E). F, Multiplex IFA staining of CD68 (green), CD163 (red), and PD‐L1 (blue). G, H, Percentages of CD68+CD163+ macrophages in CD68+ macrophages (G) and PD‐L1 expression in CD68+CD163+ macrophages (H) between R and NR NSCLC patients. Plots with red circles were two patients with NLRP4 mutations. I, Kaplan‐Meier curves for NSCLC patients (n = 14) with the percentages of PD‐L1 expression plotted. P values were calculated using the Mann‐Whitney U test. Kaplan‐Meier curves were plotted according to the cut‐off values determined by maximum Youden index of the receiver operating characteristic curve. *P < .05. PFS, progression‐free survival
We also determined the proportion of infiltrating macrophages in tumor biopsies collected before nivolumab treatment by using CD68, CD163, and PD‐L1 expression in parallel (Figure 5F). The percentages of CD163+CD68+ macrophages in total CD68+ cells were higher in the biopsies from the R group than the NR group (P = .042; Figure 5G). In addition, PD‐L1 expression was also significantly enhanced in CD163+CD68+ macrophages in the R group (Figure 5H, P = .0225). Higher PD‐L1 expression in tumor biopsies was associated with a longer PFS (P = .0099) (Figure 5I). Two NSCLC patients harboring NLRP4 mutations showed either a high percentage of infiltrating CD8+ T cells (Table S2) or CD163+CD68+ macrophages (Figure 5G, red circle).
Therefore, our results indicate that, consistent with the results from the mouse models, high CD8 expression and infiltration of M2 macrophages are correlated with good response to ICI therapy.
3.6. Type I IFN response influences responses to nivolumab therapy for NSCLC
Because NLRP4 could regulate the production of IFNIs in LLC cell lines and affect ICI treatment, we evaluated the association between IFNIs and responses to ICI immunotherapy. It was found that patients with a high level of IFN‐α (197 pg/mL) had a significantly longer PFS than those with a low level of IFN‐α (56 pg/mL) (high, 197 days vs low, 56 days, P =.0031) (Figure 6A). Clinical characteristics of 59 NSCLC patients are listed in Table S3. The GSVA analysis of the WES data from 33 patients with advanced NSCLC before pembrolizumab therapy revealed that the IFN‐α/β signaling pathway was also enriched in the R group (P = .02364) (Figure 6B). Interferon‐α and IFN‐β mRNA levels were also elevated with NLRP4 mutations (Figure S1C). Furthermore, following intratumoral injections of IFN‐β in LLC mouse models, it was found that IFN‐β alone showed similar suppression on tumor growth following anti‐PD‐L1 treatment. The combination of IFN‐β and anti‐PD‐L1 exerted stronger inhibitory effects on tumor growth (Figure 6C). These results indicate that NLRP4‐regulated IFNIs might become a synergistic cytokine to promote the efficacy of ICI therapy.
FIGURE 6.
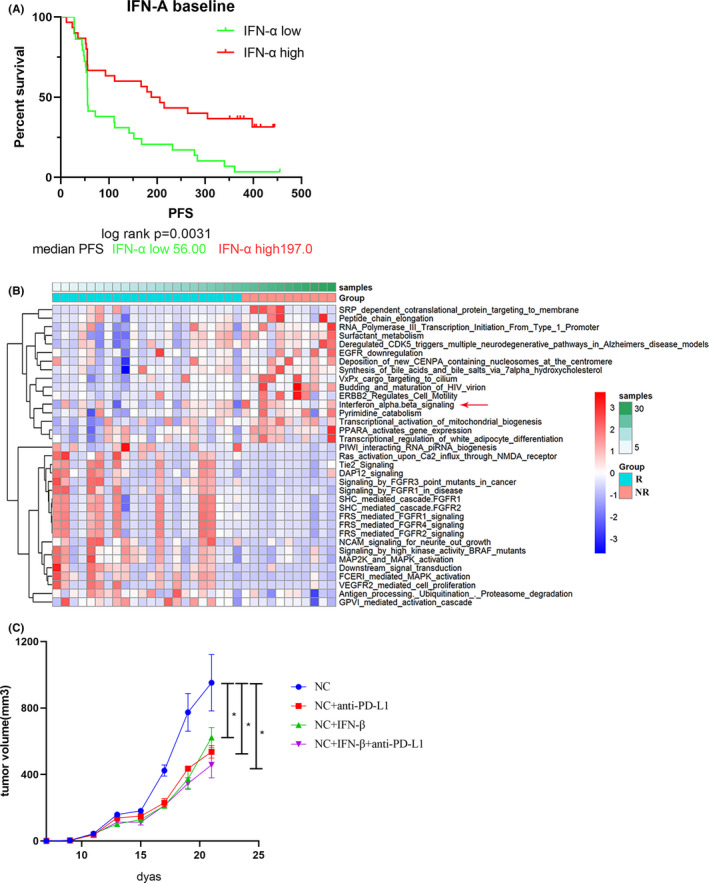
Associations between type I interferon (IFN) and clinical efficacy of nivolumab treatment for patients with advanced non‐small‐cell lung cancer (NSCLC). A, Kaplan‐Meier progression‐free survival (PFS) curves of 59 patients with advanced NSCLC with serum IFN‐α levels before nivolumab treatment. B, Heatmap analysis of signaling pathway enrichment in NSCLC patients classified as responders (R; n = 11) or nonresponders (NR; n = 22) before receiving anti‐programmed death‐ligand 1 (PD‐L1) treatment using Gene Set Variation Analysis of the whole‐exome sequencing data from cBioPortal database. C, Tumor growth curves of Lewis lung cancer tumors in C57BL/6 mice receiving intratumoral IFN‐β injection. The correlation coefficient r and P value were calculated using Pearson’s test. *P < .05
4. DISCUSSION
Although PD‐1/PD‐L1 blockades have revolutionized clinical oncology in the last 10 years, one of the great remaining challenges is to improve the efficacy and enlarge the populations that benefit from ICI therapy. 22 Along with accumulating evidence supporting the critical roles of innate immunity in tumorigenesis and tumor progression, our study identified the roles of tumor‐intrinsic NLRP4 in modulating the inflammatory TME and affecting anti‐PD‐L1 treatment in NSCLC, probably through modulating IFNI responses. Belonging to the NLR family, 23 , 24 NLRP4 reportedly exerts regulatory roles in IFNI production through interacting with the TBK1‐USP38‐DTX axis, 13 which in turn renders resistance to viral infection. In our study, we have addressed an alternative role of NLRP4 in modulating IFNI responses through the cGAS‐STING pathway. This might be triggered by increased cytosolic TERT genomic DNA and D‐loop mtDNA, which we detected in LLC and MC‐38 cells with NLRP4 KD (Figure S7). We have also tried to predict the binding alteration of mutant NLRP4 proteins by using a meta‐PPISP method. 25 It was revealed that the alteration of amino acids derived from point mutations in the NLRP4 gene we detected in the clinical samples putatively led to the reduction of binding values when compared to WT NLRP4 protein (Figure S1D), which might affect their interactions with TBK1 or other molecules. Although the number of NSCLC patients with NLPR4 mutations was limited in our study, five NSCLC patients with NLRP4 mutations in tumor biopsies from our study and the data from the TCGA database showed similar longer PFS values, further supporting the potential involvement of NLRP4 in modulating cancer immunotherapy.
More interestingly, NLRP4 KD‐LLC‐derived grafted tumors exert “hot tumor” contextures, such as more infiltration of CD8+ T cells and upregulation of PD‐L1 (Figure S5) in tumor regions. This is consistent with the synergistic outcome following anti‐PD‐L1 treatment in vivo (Figure 3G). NLRP4 KD also changed the composition and proportion of other immune cells in the tumor microenvironment (Figure 4 and Figure S8), such as promotion M2 polarization both in vivo (Figure 4E) and in vitro (Figure 4B), as well as downregulation of PD‐L1 expression on macrophages (Figure S8D). No effects on infiltrating MDSCs were observed with or without anti‐PD‐L1 treatment (Figure S8C). As CD68+CD163+ M2‐type macrophages were reported as the major PD‐L1‐expressing immune cells, in addition to tumor cells, 19 , 26 the increased intratumoral PD‐L1+ M2 macrophages are reported to be associated with benefit from ICIs. 27 Similar to the mechanisms proposed by Arlauckas et al, 28 the alteration of M1 and M2 distribution in tumor regions following anti‐PD‐L1 mAb treatment in our study further supported the restore of protumor immunity through remodeling macrophage profiles. However, we observed the maintenance of Arg1 expression in LLC‐NLRP4 KO grafted tumors following anti‐PD‐L1 treatment. This might be due to the increased production of IFN‐α with NLRP4 KD. A previous study reported that systemic poly(I:C)/IFN treatment could undesirably trigger M2 type macrophage commitment 29 and high expression of Arg1. 30 We would like to propose that anti‐PD‐L1 treatment probably maintains or increases IFN‐α in grafted tumors, which subsequently leads to Arg1 expression as well.
The contact between immune cells and tumor cells in the TME to some extent determines the outcome of tumorigenesis and tumor progression, 31 , 32 based on immune editing theory. 33 , 34 Cytokines are believed to be crucial factors mediating the cross‐talk between tumor cells and immune cells, and shaping the TEM. 35 , 36 , 37 , 38 Type I IFNs have been regarded as playing a central role in antitumor immunity for a long time. 39 In our study, tumor cell‐derived IFNIs should be one of the key participants dedicated to altered TME due to the mutations (human samples) or depletion (mouse cell lines) of the NLRP4 gene. Although we did not determine the local concentration of IFNIs in grafted tumors, elevated IFN‐α and IFN‐β in NLRP4‐KD‐LLC cells probably affected the higher infiltration of CD8+ T cells in tumor regions, the upregulation of PD‐L1 (Figures 2F and S5D) and IFN‐α/β receptor 1 on CD45− cells (Figure S8A,B), as previously reported. 40 The association of higher IFN‐α in NSCLC patients with a longer PFS in our study also supports this deduction (Figure 6A). Furthermore, our results showing that direct intratumoral injection of IFN‐β with anti‐PD‐L1 Ab had synergistic effects in suppressing tumor growth with few side‐effects in mice provided the preliminary evidence for IFN‐α in future clinical applications. 41 , 42 Interferon‐β and anti‐PD‐L1 alone or in combination induce high infiltration of CD8+ T cells in tumors (Figure S9A) as well as increased percentages of CD8+ T cells in the spleen (Figure S9B), implying a tendency to endow the TME with further “hot” tumor characteristics. This would help to overcome the obstacles in current ICI immunotherapy, making IFNIs a potential immune‐adjuvant to improve the clinical efficacy of ICIs. 43
DISCLOSURE
Lei Jiao is from Panovue Biological Technology Co. The other authors declare no conflict of interests.
Supporting information
Fig. S1‐S9
Table S1‐S3
Doc. S1
ACKNOWLEDGMENTS
We appreciate the technical support of Professor Guoping Zhao and Dr Chen Yang from the Chinese Academy of Science and Professor Bing Su and Dr Liufu Deng from the Shanghai Institute of Immunology for fruitful discussions and advice on the manuscript. This work was supported by a grant from the National Key R&D Program of China (2016YFC1303300) (SL and YW), Shanghai Municipal Science & Technology Commission Research Project (20JC1417500 to YW, 17431906103 to SL), Science and Technology Innovation Program of Shanghai Municipal Government (No.19411950500) (SL and YW), Shanghai Chest Hospital Project of Collaborative Innovation (YJXT20190105) (SL), Natural Science Foundation of China (Key Program 82030045) (SL).
Wang H, Xia L, Yao C‐C, et al. NLRP4 negatively regulates type I interferon response and influences the outcome in anti‐programmed cell death protein (PD)‐1/PD‐ligand 1 therapy. Cancer Sci. 2022;113:838–851. doi: 10.1111/cas.15243
Hui Wang and Liliang Xia contributed equally to this work.
Funding information
National Key R&D Program of China,Grant/Award Number: 2016YFC1303300; Shanghai Municipal Science and Technology Commission Research Project, Grant/Award Number: 20JC1417500 and17431906103; Science and Technology Innovation Program of Shanghai Municipal Government, Grant/Award Number:19411950500; Shanghai Chest Hospital Project of Collaborative Innovation, Grant/Award Number: YJXT20190105; Natural Science Foundation of China, Grant/Award Number: 82030045.
Contributor Information
Ying Wang, Email: ywang@sibs.ac.cn.
Shun Lu, Email: shunlu@sjtu.edu.cn.
REFERENCES
- 1. Gettinger S, Horn L, Jackman D, et al. Five‐year follow‐up of nivolumab in previously treated advanced non‐small‐cell lung cancer: results from the CA209‐003 study. J Clin Oncol. 2018;36:1675‐1684. [DOI] [PubMed] [Google Scholar]
- 2. Herbst RS, Baas P, Kim DW, et al. Pembrolizumab versus docetaxel for previously treated, PD‐L1‐positive, advanced non‐small‐cell lung cancer (KEYNOTE‐010): a randomised controlled trial. Lancet. 2016;387:1540‐1550. [DOI] [PubMed] [Google Scholar]
- 3. Rittmeyer A, Barlesi F, Waterkamp D, et al. Atezolizumab versus docetaxel in patients with previously treated non‐small‐cell lung cancer (OAK): a phase 3, open‐label, multicentre randomised controlled trial. Lancet. 2017;389:255‐265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Sharma P, Hu‐Lieskovan S, Wargo JA, Ribas A. Primary, adaptive, and acquired resistance to cancer immunotherapy. Cell. 2017;168:707‐723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zou W, Wolchok JD, Chen L. PD‐L1 (B7‐H1) and PD‐1 pathway blockade for cancer therapy: mechanisms, response biomarkers, and combinations. Sci Transl Med. 2016;8:328rv324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Haanen J. Converting cold into hot tumors by combining immunotherapies. Cell. 2017;170:1055‐1056. [DOI] [PubMed] [Google Scholar]
- 7. Reck M, Rodríguez‐Abreu D, Robinson AG, et al. Pembrolizumab versus chemotherapy for PD‐L1‐positive non‐small‐cell lung cancer. N Engl J Med. 2016;375:1823‐1833. [DOI] [PubMed] [Google Scholar]
- 8. Goodman AM, Kato S, Bazhenova L, et al. Tumor mutational burden as an independent predictor of response to immunotherapy in diverse cancers. Mol Cancer Ther. 2017;16:2598‐2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dorta‐Estremera S, Hegde VL, Slay RB, et al. Targeting interferon signaling and CTLA‐4 enhance the therapeutic efficacy of anti‐PD‐1 immunotherapy in preclinical model of HPV(+) oral cancer. J Immunother Cancer. 2019;7:252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gangaplara A, Martens C, Dahlstrom E, et al. Type I interferon signaling attenuates regulatory T cell function in viral infection and in the tumor microenvironment. PLoS Pathog. 2018;14:e1006985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Brahmer J, Reckamp KL, Baas P, et al. Nivolumab versus docetaxel in advanced squamous‐cell non‐small‐cell lung cancer. N Engl J Med. 2015;373:123‐135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wu YL, Lu S, Cheng Y, et al. Nivolumab versus docetaxel in a predominantly Chinese patient population with previously treated advanced NSCLC: CheckMate 078 randomized phase III clinical trial. J Thorac Oncol. 2019;14:867‐875. [DOI] [PubMed] [Google Scholar]
- 13. Cui J, Li Y, Zhu L, et al. NLRP4 negatively regulates type I interferon signaling by targeting the kinase TBK1 for degradation via the ubiquitin ligase DTX4. Nat Immunol. 2012;13:387‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA‐seq data. BMC Bioinformatics. 2013;14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsujikawa T, Kumar S, Borkar RN, et al. Quantitative multiplex immunohistochemistry reveals myeloid‐inflamed tumor‐immune complexity associated with poor prognosis. Cell Rep. 2017;19:203‐217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453‐457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen PL, Roh W, Reuben A, et al. Analysis of immune signatures in longitudinal tumor samples yields insight into biomarkers of response and mechanisms of resistance to immune checkpoint blockade. Cancer Discov. 2016;6:827‐837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang Q, Ni H, Lan L, Wei X, Xiang R, Wang Y. Fra‐1 protooncogene regulates IL‐6 expression in macrophages and promotes the generation of M2d macrophages. Cell Res. 2010;20:701‐712. [DOI] [PubMed] [Google Scholar]
- 19. Wang F, Li B, Wei Y, et al. Tumor‐derived exosomes induce PD1(+) macrophage population in human gastric cancer that promotes disease progression. Oncogenesis. 2018;7:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jin Y, Dong H, Xia L, et al. The diversity of gut microbiome is associated with favorable responses to anti‐programmed death 1 immunotherapy in Chinese patients with NSCLC. J Thorac Oncol. 2019;14:1378‐1389. [DOI] [PubMed] [Google Scholar]
- 21. Halse H, Colebatch AJ, Petrone P, et al. Multiplex immunohistochemistry accurately defines the immune context of metastatic melanoma. Sci Rep. 2018;8:11158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pauken KE, Wherry EJ. Overcoming T cell exhaustion in infection and cancer. Trends Immunol. 2015;36:265‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Ng KW, Marshall EA, Bell JC, Lam WL. cGAS‐STING and cancer: dichotomous roles in tumor immunity and development. Trends Immunol. 2018;39:44‐54. [DOI] [PubMed] [Google Scholar]
- 24. Ma X, Qiu Y, Zhu L, et al. NOD1 inhibits proliferation and enhances response to chemotherapy via suppressing SRC‐MAPK pathway in hepatocellular carcinoma. J Mol Med (Berl). 2020;98:221‐232. [DOI] [PubMed] [Google Scholar]
- 25. Qin S, Zhou HX. Meta‐PPISP: a meta web server for protein‐protein interaction site prediction. Bioinformatics. 2007;23:3386‐3387. [DOI] [PubMed] [Google Scholar]
- 26. Liu Y, Zugazagoitia J, Ahmed FS, et al. Immune cell PD‐L1 colocalizes with macrophages and is associated with outcome in PD‐1 pathway blockade therapy. Clin Cancer Res. 2020;26:970‐977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lo Russo G, Moro M, Sommariva M, et al. Antibody‐Fc/FcR interaction on macrophages as a mechanism for hyperprogressive disease in non‐small cell lung cancer subsequent to PD‐1/PD‐L1 blockade. Clin Cancer Res. 2019;25:989‐999. [DOI] [PubMed] [Google Scholar]
- 28. Arlauckas SP, Garris CS, Kohler RH, et al. In vivo imaging reveals a tumor‐associated macrophage‐mediated resistance pathway in anti‐PD‐1 therapy. Sci Transl Med. 2017;9:eaal3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Guo P, Yang L, Zhang M, et al. A monocyte‐orchestrated IFN‐I–to–IL‐4 cytokine axis instigates protumoral macrophages and Thwarts Poly(I:C) therapy. J Immunol. 2021;207:408‐420. [DOI] [PubMed] [Google Scholar]
- 30. Tong Y, Zhou L, Yang L, et al. Concomitant type I IFN and M‐CSF signaling reprograms monocyte differentiation and drives pro‐tumoral arginase production. EBioMedicine. 2019;39:132‐144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dranoff G. Immunotherapy at large: balancing tumor immunity and inflammatory pathology. Nat Med. 2013;19:1100‐1101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Quezada SA, Peggs KS, Simpson TR, Allison JP. Shifting the equilibrium in cancer immunoediting: from tumor tolerance to eradication. Immunol Rev. 2011;241:104‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dunn GP, Bruce AT, Ikeda H, Old LJ, Schreiber RD. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991‐998. [DOI] [PubMed] [Google Scholar]
- 34. Yang X, Zhang X, Fu ML, et al. Targeting the tumor microenvironment with interferon‐β bridges innate and adaptive immune responses. Cancer Cell. 2014;25:37‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunn GP, Bruce AT, Sheehan KC, et al. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6:722‐729. [DOI] [PubMed] [Google Scholar]
- 36. Tétreault MP, Weinblatt D, Ciolino JD, et al. Esophageal expression of active IκB kinase‐β in mice up‐regulates tumor necrosis factor and granulocyte‐macrophage colony‐stimulating factor, promoting inflammation and angiogenesis. Gastroenterology. 2016;150:1609‐1619.e1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka T, Narazaki M, Kishimoto T. IL‐6 in inflammation, immunity, and disease. Cold Spring Harb Perspect Biol. 2014;6:a016295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Arienti F, Gambacorti‐Passerini C, Borin L, et al. Increased susceptibility to lymphokine activated killer (LAK) lysis of relapsing vs. newly diagnosed acute leukemic cells without changes in drug resistance or in the expression of adhesion molecules. Ann Oncol. 1992;3:155‐162. [DOI] [PubMed] [Google Scholar]
- 39. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405‐414. [DOI] [PubMed] [Google Scholar]
- 40. Liang Y, Tang H, Guo J, et al. Targeting IFNα to tumor by anti‐PD‐L1 creates feedforward antitumor responses to overcome checkpoint blockade resistance. Nat Commun. 2018;9:4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Atkins MB, Hodi FS, Thompson JA, et al. Pembrolizumab plus pegylated interferon alfa‐2b or ipilimumab for advanced melanoma or renal cell carcinoma: dose‐finding results from the Phase Ib KEYNOTE‐029 study. Clin Cancer Res. 2018;24:1805‐1815. [DOI] [PubMed] [Google Scholar]
- 42. Davar D, Wang H, Chauvin J‐M, et al. Phase Ib/II study of pembrolizumab and pegylated‐interferon Alfa‐2b in advanced melanoma. J Clin Oncol. 2018;36(35):3450‐3458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Spaapen RM, Leung MY, Fuertes MB, et al. Therapeutic activity of high‐dose intratumoral IFN‐β requires direct effect on the tumor vasculature. J Immunol. 2014;193:4254‐4260. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1‐S9
Table S1‐S3
Doc. S1