

Abstract
Serine/threonine kinase 16 (STK16) is crucial in on regulating tumor cell proliferation, apoptosis, and prognosis. Activated M1 macrophages regulate lung adenocarcinoma (LUAD) growth by releasing exosomes. This study aims to investigate the role of STK16 and then focus on the possible mechanisms through which exosomes derived from M1 macrophages play their roles in LUAD cells by targeting STK16. Clinical LUAD samples were used to evaluate the expression of STK16 and its association with prognosis. Exosomes were isolated from M0 and M1 macrophages by ultracentrifugation and were then identified by electron microscopy and western blotting. In vitro gain‐ and loss‐of‐function experiments with LUAD cells were performed to elucidate the functions of miR‐181a‐5p, ETS1, and STK16, and mouse xenograft models were used to verify the function of STK16 in vivo. Western blotting, quantitative real‐time PCR, CCK‐8 assay, cell apoptosis, immunohistochemistry staining, luciferase assay, ChIP assay, and bioinformatics analysis were performed to reveal the underlying mechanisms. High expression of STK16 was observed in LUAD tissues and cells, and higher expression of STK16 was associated with worse prognosis. Silencing STK16 expression inhibited cell proliferation and promoted apoptosis via the AKT1 pathway. Exosomes from M1 macrophages inhibited viability and promoted apoptosis by inhibiting STK16. Moreover, miR‐181a‐5p is the functional molecule in M1 macrophage‐derived exosomes and plays a vital role in inhibiting cell proliferation and promoting apoptosis by targeting ETS1 and STK16. Hence, exosomes derived from M1 macrophages were capable of inhibiting viability and promoting apoptosis in LUAD via the miR‐181a‐5p/ETS1/STK16 axis.
Keywords: apoptosis, exosome, lung adenocarcinoma, macrophage, STK16
The finding that MiR‐181a‐5p in exosomes from M1‐polarized macrophages inhibits STK16 by targeting ETS1 to regulate cell viability and apoptosis in LUAD provides guidance for the treatment of LUAD and deserves attention in the clinical practice of precision medicine.

1. INTRODUCTION
Lung cancer is the predominant cause of cancer‐related mortality worldwide, with lung adenocarcinoma (LUAD) being one of the most commonly diagnosed histological varieties. 1 , 2 Admittedly, therapeutic strategies for LUAD patients are evolving from the application of surgery, radiation, and cytotoxic chemotherapy as well as from the promotion of targeted therapies to personalized treatment, which has improved a subset of patients’ survival outcomes. However, most patients with metastatic LUAD have an unfavorable prognosis. 3 , 4 A better understanding of the mechanisms of LUAD will facilitate the development of novel medications and improve awareness of the curative nature of treatments.
Serine/threonine kinases (STKs), a group of approximately 125 protein kinases that phosphorylate serines or threonines, are capable of maintaining cellular homeostasis, regulating the cell signaling pathway, and participating in cell proliferation, apoptosis, and metabolism by phosphorylating nuclear effectors, transcription factors, and cell cycle regulators. 5 , 6 , 7 , 8 STK16 is a myristoylated and palmitoylated STK that participates in the regulation of a wide array of cellular processes. 9 , 10 Previous studies have indicated that STK16 knockdown or kinase inhibition leads to cytokinesis arrest, thus affecting cell growth and apoptosis. 11 , 12
Macrophages, one of the most abundant immune cell types, account for approximately 5% to 40% of the tumor environment (TME) of malignant solid tumors. 13 Macrophages exhibit considerable functional plasticity in mediating local microenvironment stimulation and the immune process. 14 , 15 In lung cancer, macrophages have been reported to be able to stimulate tumor angiogenesis and promote tumor cell invasion, migration, and intravasation. 16 Tumor‐associated macrophages (TAM), a heterogeneous class of ubiquitously innate immune cells with high plasticity present in the microenvironment, play indispensable functions of promoting tumor progression and inhibiting antitumor immune processes mediated by T cell regulation. 17 , 18 TAM can contribute to carcinogenesis, neoangiogenesis, immune‐suppressive TME remodeling, cancer chemoresistance, recurrence, and metastasis. 19 Exosomes are small vesicles released by live cells that contain various constituents, including proteins, DNA, and RNAs. 20 As vital messengers, exosomes derived from macrophages can serve as conveyors to transfer multiple types of bioactive substances from macrophages to distant target cells and regulate the biological function of the targeted cells. 21 Previous studies have shown that exosomes isolated from macrophage cell media can deliver proinflammatory signals and, thus, play a significant role in the immune stimulatory microenvironment. 22 , 23 MicroRNA, a small noncoding RNA, suppresses its target gene through RNA silencing or posttranscriptional regulation. 24 MiR‐181a‐5p, as a functional miRNA, has been studied in a multitude of different cancer types, including lung cancer. 25 , 26 , 27 A previous study analyzed the relationship between miR‐181a‐5p and non–small cell lung cancer (NSCLC), and the results implied that miR‐181a‐5p is a potential noninvasive biomarker in terms of diagnosis and prognosis. 28
The role of STK16 and its mechanisms of promoting lung cancer progression have not been explored. We investigated the biological function and prognostic value of STK16. Subsequently, we examined how exosomes derived from M1 macrophages play roles in lung cancer cells, focusing on the mediating role of miR‐181a‐5p and its downstream regulatory gene, STK16.
2. MATERIALS AND METHODS
2.1. Clinical specimens
From October 2012 to March 2015, two independent LUAD cohorts from Huashan Hospital, Fudan University, were recruited in the current study. Patients who underwent neoadjuvant treatment were excluded from the study. In cohort 1, 20 fresh tumor tissues and paired normal nontumor tissues were ultimately selected, while in cohort 2, a total of 113 formalin‐fixed, paraffin‐embedded tumors and 20 histologically adjacent tumor tissues were selected, as illustrated in Table 1. All enrolled patients signed an informed consent form, and all experimental procedures followed the research protocols that were approved by the Ethics Committee of Huashan Hospital, Fudan University.
TABLE 1.
Clinicopathological features of lung adenocarcinoma (LUAD) patients and serine/threonine kinase 16 (STK16) expression in hospital cohort 2
| Clinicopathological parameters | STK16 expression | P value | |
|---|---|---|---|
| Low (No. 48) | High (No. 65) | ||
| Age | |||
| ≤55 | 20 | 35 | .2004 |
| >55 | 28 | 30 | |
| Sex | |||
| Female | 22 | 38 | .1836 |
| Male | 26 | 27 | |
| Size (cm) | |||
| ≤4 | 36 | 32 | .0057 |
| >4 | 12 | 33 | |
| Smoking status | |||
| Never | 21 | 29 | .9271 |
| Former and current smokers | 27 | 36 | |
| Tumor stage | |||
| I | 23 | 11 | .0027 |
| II | 16 | 26 | |
| III | 6 | 20 | |
| IV | 3 | 8 | |
| Spinal metastases | |||
| No | 27 | 23 | .0273 |
| Yes | 21 | 42 | |
P < .05 represents statistical significance (χ2‐test).
2.2. Immunohistochemistry and scoring
Tissues were embedded in paraffin and sliced into 4‐μm‐thick sections. The paraffin sections were incubated with primary antibodies against STK16 (ab228608, Abcam; 1:100 dilution) overnight at 4°C. Subsequently, the sections were incubated with secondary antibodies at room temperature for 1 hour, incubated in HRP‐labeled streptavidin solution for approximately 10 minutes, and then stained with diaminoaniline solution. The intensity of staining defining immunoreactivity was scored for each specimen by two investigators using the H‐score system based on the percentage of positively stained tumor cells (with a range from 0 to 4) and the intensity of staining (with a range from 0 to 3). All patients with a score ≥3 were defined as having high expression, while those with a score of <3 were defined as having low expression.
2.3. Cell culture
The human LUAD cell lines and 16HBE cells were obtained from the ATCC, while the human THP‐1 monocytic cell line was purchased from the cell bank of Shanghai Biology Institute, Chinese Academy of Science. All the cells used were cultured in Roswell Park Memorial Institute 1640 medium (RPMI‐1640) (HyClone) supplemented with 10% FBS (Gibco, Thermo Fisher Scientific) at 37°C and 5% CO2.
2.4. Cell transfection
The coding sequences of STK16 and ETS1 were cloned into pLVX‐Puro plasmids (Clontech) to upregulate STK16 and ETS1 expression, while the RNA interference sequence of STK16 was cloned into a linearized pLKO.1 plasmid (Addgene) to downregulate STK16 expression. The interference sites and corresponding primer sequences were as follows: STK16 shRNA#1,5′‐GCGTGCTATATGCCATGATGT‐3′; shRNA#2,5′‐GTGCTATATGCCATGATGTTT‐3′; shRNA#3,5′‐GCCAACATACTACCCAAATCT‐3′. Recombinant plasmids together with the packaging plasmids psPAX2 and pMD2G were cotransfected into 293T cells. After 48 hours of transfection, recombined lentiviral vectors were collected and enriched for transducing targeted cells. Cells with pLKO.1‐scramble shRNA or blank pLVX‐Puro transduction were employed as the negative controls.
2.5. CCK‐8 assay
Cell viability was assessed by CCK‐8 assay (Dojin Laboratories). Cells were seeded and routinely cultured in 96‐well plates at a density of 3 × 103 cells/well and supplemented with 100 µL of RPMI‐1640 containing 1% FBS for 12 hours. H1299, H1975, and A549 cells were transduced with the indicated lentiviral vectors. After 12, 24, 48, and 72 hours of incubation, 10 µL of CCK‐8 reagent was added to each well for 1 hour of incubation. The absorbance at 450 nm wavelength of cell in each well was determined and analyzed.
2.6. Cell apoptosis assay
Cells were seeded in a six‐well plate at a density of 5 × 105 cells/well and grown to 50% confluence. The cells were stained with 5 μL of propidium iodide and 5 μL of FITC‐labeled recombinant annexin V (annexin V–FITC) in the dark for 15 minutes at 4°C. Cellular apoptosis was profiled using the Beckman CytoFLEX Flow Cytometer (Beckman Coulter).
2.7. Polarization of macrophages
THP‐1 cells were differentiated with phorbol‐12‐myristate‐13‐acetate (PMA, 100 nM; EMD Calbiochem). The medium was changed the next day and subsequently every 2 days for 6 days. Polarization of resting differentiated macrophages (M0 cells) was performed by 48 hours of treatment with 50 U/mL IFN‐γ and 10 ng/mL TNF‐α for M1‐like polarization. The cells were washed and incubated with fresh medium.
2.8. Isolation, experimental analysis, and in vitro supplementation of exosomes
Cells were washed with PBS and then cultured in RPMI 1640 medium with exosome‐free FBS for 2 days. The cultured medium was centrifuged at 2200 g for 15 minutes and 11 000 g for 35 minutes, followed by filtration with a 0.22‐µm filter. The medium was then centrifuged at 110 000 g for 100 minutes. The pelleted exosomes were resuspended and centrifuged at 110 000 g for 100 minutes and resuspended in 50 μL of PBS. Exosomes were placed on a copper grid for examination under an electron microscope. Protein markers of purified exosomes were determined using western blotting with anti–ALIX (ab225555; Abcam), anti–CD63 (ab216130; Abcam), and anti–TSG101 (ab125011; Abcam) antibodies. Exosomes were labeled with PKH67 (Sigma) and cocultured with H1975 cells for 48 hours. The uptake of exosomes by H1975 cells was analyzed with an Olympus FV1200 microscope. H1975 cells were provided with fresh medium plus 100 μg/mL exosomes isolated from M0 or M1 macrophages every 48 hours.
2.9. Coculture assay
To investigate the potential efficacy of exosomes from M1‐polarized macrophages in vitro, an H1975 cell monolayer and M1 macrophage cell Transwell coculture model were established. M1 macrophages were pretreated with or without 20 μM GW4869 for 24 hours before the cell seeding procedure. H1975 cells at a density of 5 × 104 cells/well were seeded in the lower chambers, and M1 macrophages at the same density were seeded in the upper chambers. The coculture Transwell chambers were incubated for 6 days in an incubator at 37°C in a 5% CO2 atmosphere. The RNA and protein of H1975 cells were extracted for further experimental analysis.
2.10. miRNA transfection
miR‐125a‐5p mimic (5′‐UCCCUGAGACCCUUUAACCUGUGA‐3′), miR‐193a‐5p mimic (5′‐UGGGUCUUUGCGGGCGAGAUGA‐3′), miR‐181a‐5p mimic (5′‐AACAUUCAACGCUGUCGGUGAGU‐3′), miR‐181a‐5p inhibitor (5′‐ACUCACCGACAGCGUUGAAUGUU‐3′), and negative control miR‐NC mimics (5′‐CAGUACUUUUGUGUAGUACAA‐3′) were synthesized by Beyotime (Beijing). Cells were transfected using Lipofectamine 2000 reagent (Invitrogen). The transfected cells were digested for subsequent analysis after 48 hours of transfection.
2.11. Luciferase reporter assay
293T cells were seeded into a 24‐well plate at a density of 2 × 104 cells/well and grown to 50% confluence. The ETS1 3′‐UTR was introduced to pGL3 plasmids to construct pGL3‐ETS1‐WT or pGL3‐ETS1‐Mut plasmids. Cells were then transfected with the miR‐181a‐5p and pGL3‐ETS1‐WT plasmids or pGL3‐ETS1‐Mut. Luciferase activity was detected by the Dual‐Luciferase Reporter assay system (Promega) after 2 days of transfection.
2.12. Quantitative real‐time PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen) and reverse‐transcribed with a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher). Quantitative real‐time PCR (qRT‐PCR) was performed using SYBR Green PCR Master Mix (Thermo Fisher) on an ABI 7300 System. The sequences of primers applied in the study are shown in Table S1. The relative abundance of genes was quantified using the comparative 2−ΔΔCt method with GAPDH or U6 as an internal control.
2.13. Western blotting
Proteins were separated by SDS‐PAGE and then transferred onto a nitrocellulose membrane (Millipore). Membranes were further blocked with 5% skim milk at 4°C for 1 hours and incubated with antibody against STK16 (Abcam, ab228608), ETS1 (Abcam, ab220361), p‐AKT1 (Abcam, ab82283), AKT1 (Abcam, 233755), and GAPDH (CST, #5174) at 4°C overnight. The membranes were incubated with the secondary antibody solution linked to HRP (Beyotime) at room temperature for 1 hour. Signals were captured by a chemiluminescence system according to the manufacturer’s instructions.
2.14. ChIP
Cells were fixed with 1% formaldehyde, harvested, sonicated, and incubated with anti–ETS1 (Abcam; ab220361) or control antibody (Proteintech; 30000‐0‐AP) for 12 hours. ETS1 binding was measured using PCR with the following primers: 5′‐AGGACCTCTTTCCTGGAGA‐3′ and 5′‐GACACATGCCTTCAGTAC‐3′.
2.15. In vivo tumor model in mice
A total of 2 × 106 H1975 cells transduced with STK16 shRNA or shNC vector were subcutaneously injected into 6‐week‐old male nude mice. For the exosome treatment model, 2 × 106 H1975 cells transduced with STK16 expression or blank vector per mouse were used to establish xenografts. Twelve days after injection, mice were treated with exosomes (100 μg of total protein in 100 μL of volume) derived from M1 macrophages and injected every 3 days in the vicinity of the subcutaneous tumors. On the 33rd day postinoculation, murine tumors (n = 6 per group) were collected, weighed, photographed, and analyzed by TUNEL staining, qPCR, and western blotting. In vivo surviving H1975 cells were also detected using bioluminescence. All in vivo experiments were performed according to our institution’s guidelines for the use of laboratory animals and were approved by the Committee on the Ethics of Animal Experiments of Shanghai Medical College, Fudan University.
2.16. Bioinformatics analysis
RNA‐seq data related to STK16 expression in various cancer patients were acquired from The Cancer Genome Atlas (TCGA) dataset, which included 526 cases of LUAD tumor tissues and 59 cases of normal lung tissues. Survival data were downloaded from the Kaplan‐Meier plotter database. The gene set enrichment analysis (GSEA) algorithm was used to identify the pathways of significant enrichment between high and low STK16 expression.
2.17. Statistical analysis
Data analysis was performed using GraphPad Prism 8.0.2. The results are presented as the mean ± SD from at least three independent experiments. Comparisons between different experimental groups were achieved with ANOVA or Student’s t‐test. P values <.05 were considered indicative of statistical significance.
3. RESULTS
3.1. Serine/threonine kinase 16 is closely associated with the progression and prognosis of lung adenocarcinoma
To investigate the expression of STK16 in LUAD, we first analyzed the expression of STK16 in the TCGA database (Figure S1A). By analyzing the transcription profiles of 526 LUAD tumors and 59 corresponding nontumorous lung tissues, the results indicated that compared to adjacent normal tissues, STK16 was prominently upregulated in tumors (Figure 1A). In addition, higher expression of STK16 was notably associated with poor overall survival (OS) (P < .001; Figure 1B). Subsequently, qRT‐PCR confirmed that compared to the nontumorous tissues, the level of STK16 was dramatically enhanced in LUAD tissues (Figure 1C). By immunohistochemistry (IHC), we found that the STK16 expression level was higher in 133 LUAD tumor tissues than in 20 nontumorous lung tissues (Figure 1D). Consistently, high expression of STK16 was related to poor OS in hospital cohort 2 (P = .017; Figure 1E).
FIGURE 1.
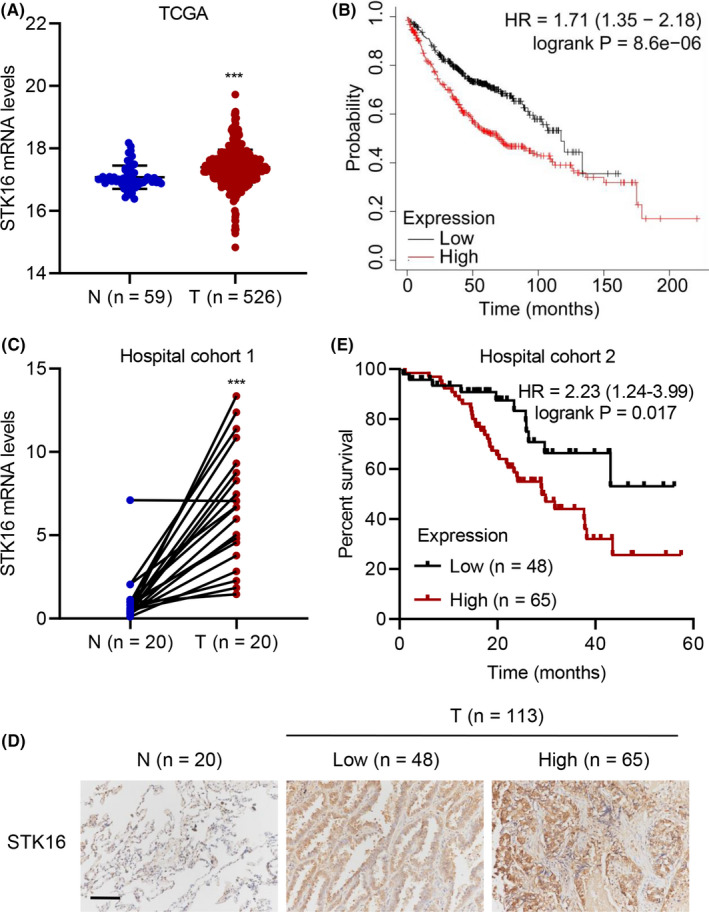
Serine/threonine kinase 16 (STK16) expression is associated with lung adenocarcinoma (LUAD) progression and survival rate. STK16 mRNA expression in (A) TCGA database and (C) tumor and paired normal tissues from 20 LUAD patients in hospital cohort 1. (D) immunohistochemistry (IHC) analysis for STK16 protein expression in tumor and normal tissues from LUAD patients in hospital cohort 2. Scale bars: 100 μm. The overall survival of LUAD patients in (B) Kaplan‐Meier plotter database and (E) hospital cohort 2. ***P < .001 compared with N
3.2. Serine/threonine kinase 16 promotes tumor cell growth both in vitro and in vivo
STK16 was significantly upregulated in lung cancer cell lines compared with nonlung cancer cell lines by qRT‐PCR detection. A similar result was also observed by western blot. Moreover, the STK16 mRNA and protein levels were prominently high in H1299 and H1975 cells (Figure 2A‐B). Given that the expression of STK16 is increased in LUAD, we hypothesize that STK16 might lead to modifications in LUAD cells and play a pivotal role in mediating cancer progression. We established lentiviral‐mediated stable STK16‐silenced H1299 and H1975 cell lines with three different shRNA sequences (shSTK16#1, shSTK16#2, and shSTK16#3) (Figure S2A‐D). The shSTK16#2 and shSTK16#3 cell lines were the most efficient cell lines in terms of specific knockdown of STK16 expression. Then, in vitro experiments evaluating proliferation and apoptosis were designed. The cell viability results showed that knockdown of STK16 dramatically suppressed the viability of the H1299 (Figure 2C) and H1975 (Figure 2D) cell lines. Further study confirmed that STK16 silencing could also promote apoptosis in H1299 and H1975 cell lines (Figure 2E,F). GSEA indicated that high STK16 gene expression was enriched in apoptosis signaling (Figure S1B). Knockdown of STK16 could decrease the expression of p‐AKT1 but did not have an effect on AKT1 expression in STK16‐silenced cell lines H1299 and H1975 (Figure 2G). GSEA also inferred that the expression of STK16 was enriched in the ATK1 pathway (Figure S1C). Therefore, compared with the negative control group, knockdown of STK16 was capable of downregulating the AKT1 signaling pathway.
FIGURE 2.
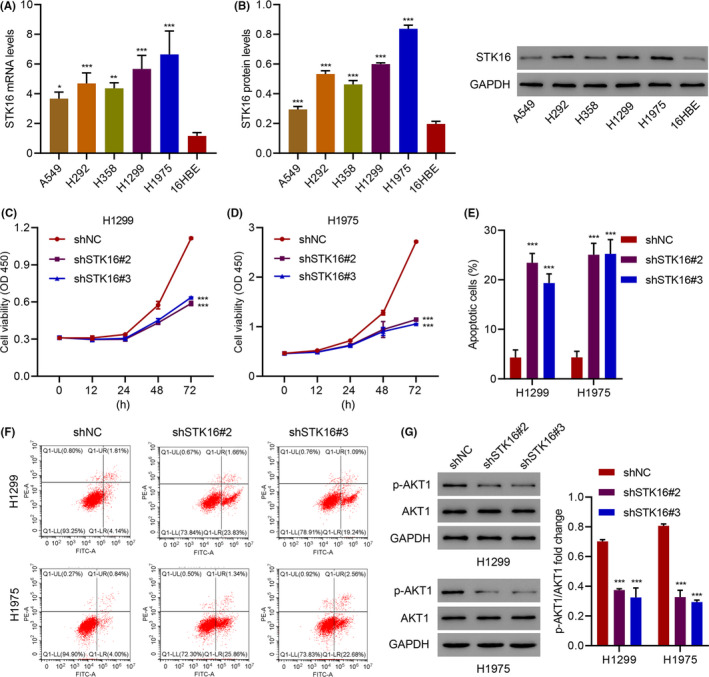
Serine/threonine kinase 16 (STK16) silencing inhibits cell viability and promotes cell apoptosis. (A, B) STK16 expression in lung adenocarcinoma (LUAD) cell lines and 16HBE cells. (C, D) Cell viability, (E, F) apoptosis, and (G) expression of p‐AKT1 and AKT1 in H1299 and H1975 cells transduced with STK16 shRNA vector. *P < .05, **P < .01, ***P < .001 compared with 16HBE or shNC
To determine the effects of STK16 on tumor growth in vivo, H1975 cells were transduced with sh‐NC or sh‐STK16 vector and then subcutaneously injected into nude mice. The tumors derived from the STK16‐silencing group were smaller and lighter than those of the negative control group, which demonstrated that knockdown of STK16 suppressed tumor growth (Figure 3A–C). We also performed TUNEL staining of STK16 knockdown LUAD cells in nude mice (Figure 3D). We also evaluated the expression of p‐AKT1 and AKT1. Similar to the in vitro results, we found that STK16 silencing was able to downregulate the expression of p‐AKT1 rather than the expression of AKT in tumorous tissues (Figure 3E).
FIGURE 3.

Serine/threonine kinase 16 (STK16) silencing inhibits tumor cell growth in vivo. After H1975 cells transduced with STK16 shRNA or shNC vector were injected into the nude mice, the (A) tumor volume, (B) formation, (C) weight, (D) TUNEL staining, and (E) expression of STK16, p‐AKT1, and AKT1 was measured. Scale bars: 50 μm. **P < .01, ***P < .001 compared with shNC
3.3. Serine/threonine kinase 16 promotes cell viability and inhibits cell apoptosis through the AKT1 signaling pathway
To confirm our conclusion, we then overexpressed STK16 in A549 cells and verified by qRT‐PCR (Figure S2E) and western blotting (Figure S2F) that it could specifically increase STK16 expression. In A549 cells, overexpression of STK16 significantly increased LUAD cell proliferation, and when using the AKT inhibitor, A‐674563 reversed the proliferation induced by STK16 overexpression (Figure 4A). STK16 overexpression also inhibited apoptosis in A549 cells, which was reversed by A‐674563 (Figure 4B,C). Finally, we evaluated the expression of p‐AKT1 and AKT1 in A549 cells transduced with the STK16 expression vector and treated with A‐674563 by western blotting (Figure 4D). Unsurprisingly, we found that overexpression of STK16 could increase the expression of p‐AKT1 and that p‐AKT1 expression was decreased when A‐674563 was used (Figure 4E).
FIGURE 4.
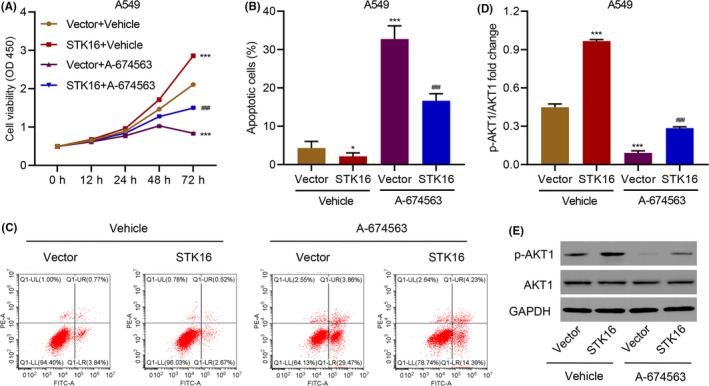
Serine/threonine kinase 16 (STK16) overexpression promotes cell viability and inhibits cell apoptosis through the AKT1 signaling pathway. (A) Cell viability, (B, C) apoptosis, and (D, E) expression of p‐AKT1 and AKT1 in A549 cells transduced with STK16 expression vector and treated with 10 nM A‐674563. ***P < .001 compared with Vector+Vehicle. ### P < .001 compared with STK16+Vehicle
3.4. Exosomes from M1‐polarized macrophages inhibit viability and promote apoptosis by inhibiting serine/threonine kinase 16
To trace the function of STK16 and identify its mechanism in LUAD, we explored the TME, which might be associated with the regulation of STK16. To investigate the relationship between macrophages and STK16, we examined STK16 expression in H1975 cells treated with exosomes derived from M0 macrophages (M0‐exo) or M1 macrophages (M1‐exo) with or without the exosome inhibitor GW4869 by qRT‐PCR and western blotting. M0 macrophages can be induced toward M1 polarization by various environmental signals. Electron microscopy images of exosomes isolated from M0 and M1 macrophages reveal small round nanometer‐sized particles with bilayer membranes (Figure S4A). In addition, western blotting confirmed the presence of the exosomal marker proteins TSG101, CD63, and ALIX (Figure S4B). Confocal imaging of PKH67 (green) dye stained was also used to label the exosomes (Figure S4C). The expression levels of IL‐1β and inducible nitric oxide synthase (iNOS) were analyzed in Mo‐exos and M1‐exos using qRT‐PCR and western blotting analysis. IL‐1β and iNOS were more highly expressed in M1‐exos than in M0‐exos (Figure S3A,B). We found that STK16 expression was significantly decreased when H1975 cells were treated with exosomes derived from M1 macrophages, while there were no differences when cells were treated with exosomes from M0 macrophages (Figure 5A,B). We also found that the gene and protein expression levels of STK16 decreased gradually over time when H1975 cells were treated with exosomes derived from M1 macrophages (Figure S3C,D). However, qRT‐PCR and western blotting analysis indicated that STK16 expression in the M1‐exo group could be reinstated with the application of GW4869, an inhibitor of exosome biogenesis (Figure 5C,D). To further validate the mechanism, we evaluated the viability, apoptosis, and expression of p‐AKT1 and AKT1 in H1975 cells treated with M1‐exos and transduced them with the STK16 expression vector. The cell viability decreased in the M1‐exo group, and the M1‐exo group with the STK16 expression vector had increased cell viability compared with the M1‐exo group (Figure 5E). The percentage of apoptotic cells increased in the M1‐exo group, and the M1‐exo group with the STK16 expression vector had a decreased percentage compared with the M1‐exo group (Figure 5F,G). Finally, we found that M1‐exos with the STK16 expression vector increased p‐AKT1 expression, and M1‐exos with the vector decreased p‐AKT1 expression (Figure 5H,I).
FIGURE 5.
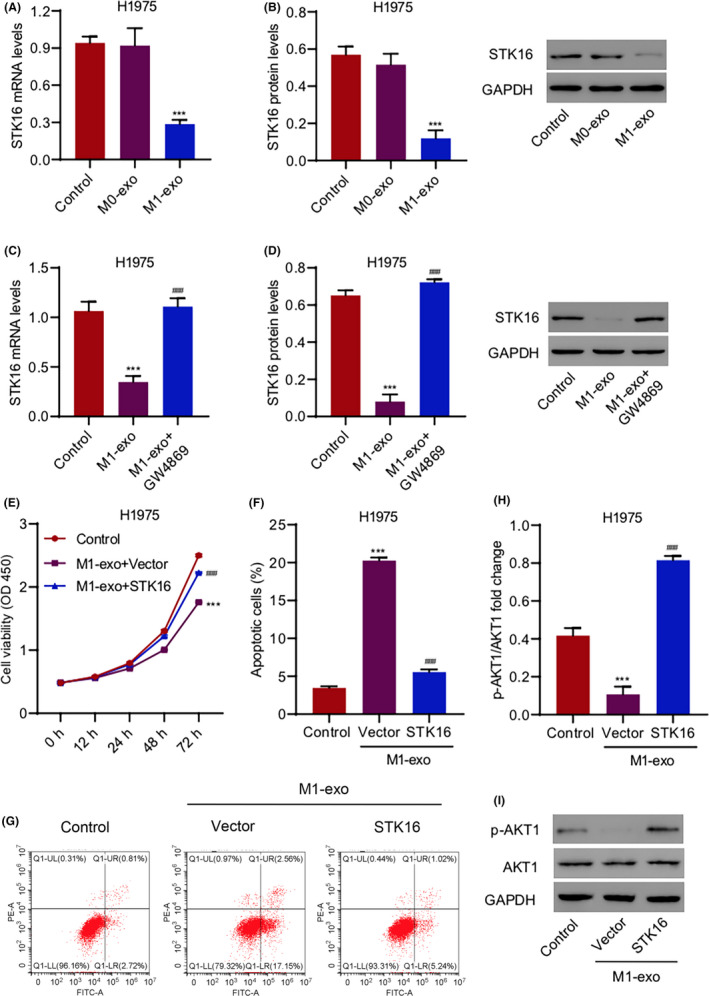
Exosome derived from M1 macrophages inhibits cell viability and promotes cell apoptosis by inhibiting serine/threonine kinase 16 (STK16). (A‐D) STK16 expression in H1975 cells treated with exosomes derived from M0 macrophages (M0‐exo) or M1 macrophages (M1‐exo) with or without 20 μM GW4869. (E) Cell viability, (F, G) apoptosis, and (H, I) expression of p‐AKT1 and AKT1 in H1975 cells treated with M1‐exo and transduced with STK16 expression vector. ***P < .001 compared with control. ### P < .001 compared with M1‐exo or M1‐exo+Vector
To evaluate the effects of M1‐polarized macrophages and STK16 in vivo, H1975 cells transduced with the STK16 expression vector and exosomes derived from M1 macrophages (M1‐exos) were subcutaneously implanted into nude mice, and tumor size was monitored every 3 days. M1‐exos suppressed tumor growth and reduced tumor size and weight compared to the control group (Figure 6A–D). Overexpression of STK16 could promote tumor growth to some extent. We also performed TUNEL staining of tumors in nude mice and found that the percentage of TUNEL‐positive cells in the M1‐exo group was much higher than in the control group, while STK16 overexpression could reduce the percentage (Figure 6E,F). In addition, we evaluated the expression of p‐AKT1 and AKT1. We found that M1‐exo exerted a strong influence in decreasing STK16 expression, that overexpression of STK16 could increase the expression of p‐AKT1, and that AKT1 had no change. The fold change of p‐AKT1/AKT1 was significantly different from that of the control (Figure 6G,H).
FIGURE 6.
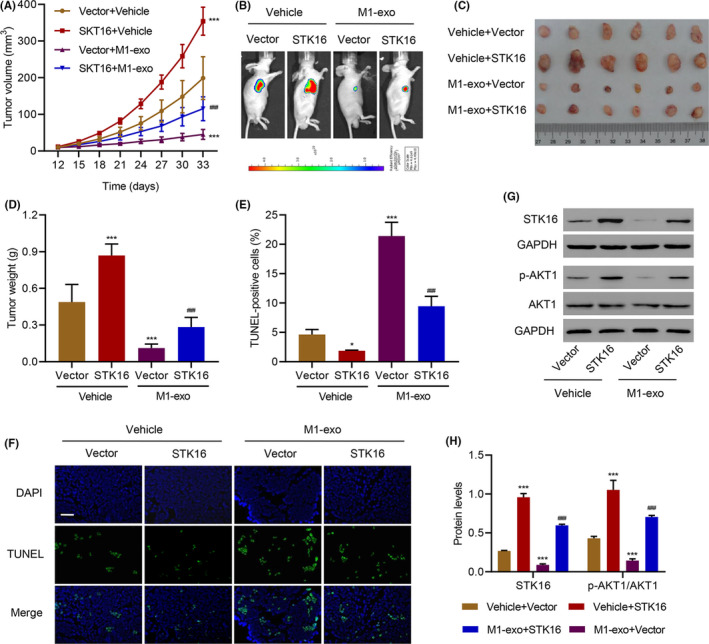
Exosome derived from M1 macrophages inhibits tumor cell growth in vivo. After mice were injected with H1975 cells transduced with serine/threonine kinase 16 (STK16) expression vector and exosome derived from M1 macrophages (M1‐exo), the (A) tumor volume, (B) formation, (C, D) weight, (E, F) TUNEL staining, and (G, H) expression of STK16, p‐AKT1 and AKT1 was measured. Scale bars: 50 μm. *P < .05, ***P < .001 compared with Vector+Vehicle. ### P < .001 compared with Vector+M1‐exo
3.5. miR‐181a‐5p regulates cell viability and apoptosis by inhibiting serine/threonine kinase 16
To investigate the mechanism by which M1‐exos regulate the expression of STK16, we detected the expression levels of several miRNAs in Mo‐exos and M1‐exos, and the results showed that miR‐125a‐5p, miR‐181a‐5p and miR‐193a‐5p were more highly expressed in M1‐exos (Figure S5A,B). The mRNA and protein levels of STK16 were decreased when H1975 cells were treated with miR‐181a‐5p mimics rather than miR‐125a‐5p mimics and miR‐193a‐5p mimics compared with those treated with miR‐NC (Figure S5C,D). To further verify miR‐181a‐5p in exosomes derived from M1‐exos and its relationship with STK16, we evaluated the cell viability, apoptosis, and expression of p‐AKT1 and AKT1 in H1975 cells treated with miR‐181a‐5p mimic and transduced with the STK16 expression vector. The cell viability decreased in the miR‐181a‐5p mimic group, and the miR‐181a‐5p mimic group with the STK16 expression vector recovered and increased cell viability (Figure 7A). The percentage of apoptotic cells increased in the miR‐181a‐5p mimic group, and the miR‐181a‐5p mimic group with the STK16 expression vector had a decreased percentage compared with the miR‐181a‐5p mimic group (Figure 7B,C). We found that the miR‐181a‐5p mimic with the STK16 expression vector increased the expression of p‐AKT1, and the miR‐181a‐5p mimic with the vector decreased STK16 and p‐AKT1 expression. While AKT1 had no change, the fold change of p‐AKT1/AKT1 was significantly different compared with that of the control (Figure 7D,E). In contrast, cell viability increased in the miR‐181a‐5p inhibitor group, and the miR‐181a‐5p inhibitor group with the STK16 shRNA vector exhibited reduced and decreased cell viability (Figure 7F). The percentage of apoptotic cells decreased in the miR‐181a‐5p inhibitor group, and the miR‐181a‐5p inhibitor group with the STK16 shRNA vector increased in percentage compared with the miR‐181a‐5p inhibitor group (Figure 7G,H). We found that the miR‐181a‐5p inhibitor with the STK16 shRNA vector decreased the expression of p‐AKT1, and the miR‐181a‐5p inhibitor with the vector increased STK16 and p‐AKT1 expression. While the expression of AKT1 did not change, the fold change of p‐AKT1/AKT1 was significantly different compared with that of the control (Figure 7I,J).
FIGURE 7.
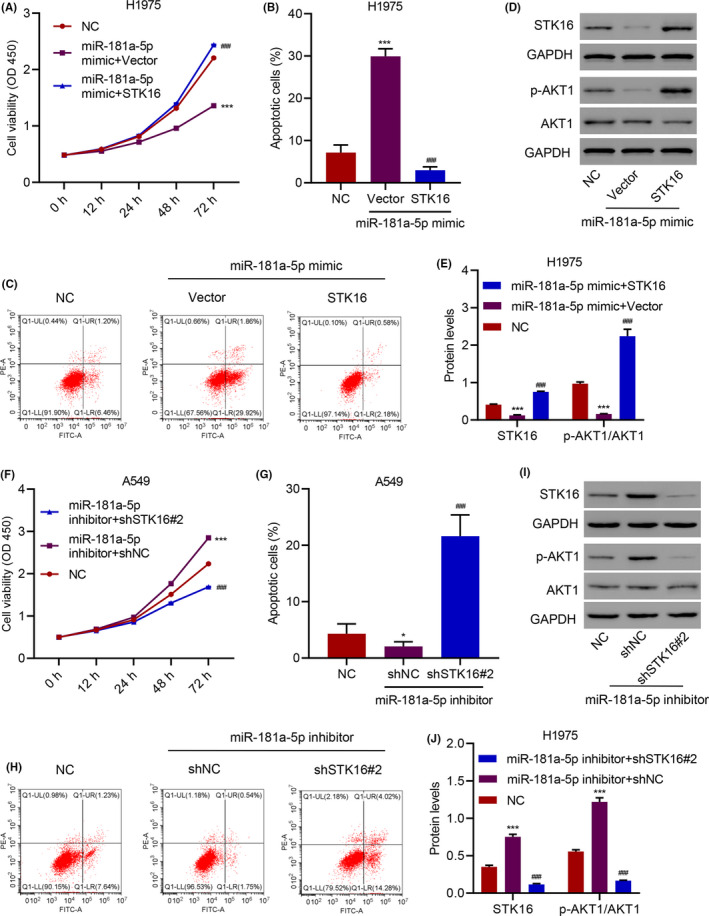
miR‐181a‐5p regulates cell viability and apoptosis through serine/threonine kinase 16 (STK16). (A) Cell viability, (B, C) apoptosis, and (D, E) expression of p‐AKT1 and AKT1 in H1975 cells transfected with miR‐181a‐5p mimic and transduced with STK16 expression vector. (F) Cell viability, (G, H) apoptosis, and (I, J) expression of p‐AKT1 and AKT1 in A549 cells transfected with miR‐181a‐5p inhibitor and transduced with STK16 shRNA vector. ***P < .001 compared with NC. ### P < .001 compared with miR‐181a‐5p mimic+Vector or miR‐181a‐5p inhibitor+shNC
3.6. miR‐181a‐5p inhibits serine/threonine kinase 16 by directly targeting ETS1
To uncover the molecular mechanism underlying the inhibitory effect of miR‐181a‐5p on LUAD viability, we combined bioinformatics approaches to identify the targets of miR‐181a‐5p. Although miR‐181a‐5p could inhibit STK16 expression, miR‐181a‐5p could not directly bind to the 3′UTR region of STK16 (Figure S5E‐G), suggesting that STK16 is not a direct target of miR‐181a‐5p. Therefore, other target candidates were selected based on the predictive binding of miR‐181a‐5p, among which ETS proto‐oncogene 1 transcription factor (ETS1), which has been shown to be significantly associated with LUAD progression, was the selected candidate (Figure 8A). To verify whether ETS1 is the direct target of miR‐181a‐5p, a 3′UTR element of ETS1 with wild‐type or mutated sequences was designed, constructed, cloned into a dual‐luciferase reporter, and then cotransfected with miR‐181a‐5p inhibitor and mimics into HEK‐293T cells. The luciferase activities were significantly increased and decreased in the reporter with wild‐type binding sites along with miR‐181a‐5p inhibitor and mimics but not with mutant (P < .001, Figure 8B), suggesting that miR‐181a‐5p regulated ETS1 expression in a site‐specific manner. Furthermore, the mRNA and protein levels of ETS1 in miR‐181a‐5p inhibitor‐transfected H1975 cells were markedly elevated but were reduced in miR‐181a‐5p mimic‐transfected H1975 cells compared with their control counterparts (Figure 8C,D). We also found that the miR‐181a‐5p mimic‐induced decreases in cell viability and apoptosis could be reversed by ETS1 overexpression (Figure S6A–E). Moreover, STK16 had an identical variation in H1975 cells transfected with miR‐181a‐5p mimic and transduced with an ETS1 expression vector (Figure 8E).
FIGURE 8.
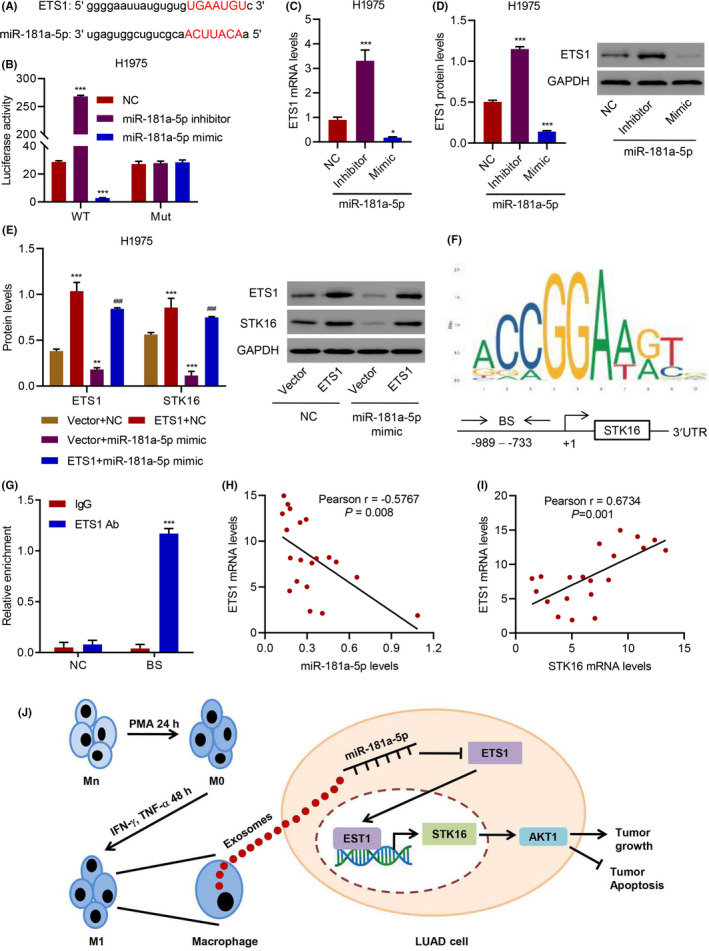
miR‐181a‐5p inhibits serine/threonine kinase 16 (STK16) expression through directly targeting ETS1. (A) The predictive binding of miR‐181a‐5p and ETS1. (B) The luciferase activity of mutant or wild‐type ETS1 mRNA 3′UTR in H1975 cells transfected with miR‐181a‐5p mimic or inhibitor. (C, D) Expression of ETS1 in H1975 cells transfected with miR‐181a‐5p mimic or inhibitor. (E) Expression of ETS1 and STK16 in H1975 cells transfected with miR‐181a‐5p mimic and transduced with ETS1 expression vector. (F) ETS1 binding site (BS) in STK16 promoter schematic diagram. (G) Chromatin immunoprecipitation assay of ETS1 binding with STK16. (H, I) Pearson correlation scatter plots in lung adenocarcinoma (LUAD) tissues (n = 20). *P < .05, **P < .01, ***P < .001 compared with NC or IgG. ### P < .001 compared with miR‐181a‐5p mimic+Vector. (J) Schematic representation showing the mechanism that exosomes from M1‐polarized macrophages inhibit viability and promote apoptosis in LUAD via the miR‐181a‐5p/ETS1/STK16 axis
Bioinformatics analysis revealed the ETS1 binding site in the STK16 promoter schematic diagram (Figure 8F). To further validate STK16 as a bona fide ETS1‐mediated target, a chromatin immunoprecipitation assay was performed and analyzed with qRT‐PCR. The results showed that the ETS1‐specific antibody significantly enriched the ETS1 promoter compared with the immunoglobulin G pull‐down control (Figure 8G). Notably, we observed significant inverse correlations between EST1 expression and miR‐181a‐5p levels and positive correlations between EST1 expression and STK16 expression in LUAD tissues (Figure 8H–I). Taken together, exosomes derived from M1 macrophages could inhibit viability and promote apoptosis in LUAD via the miR‐181a‐5p/ETS1/STK16 axis (Figure 8J).
4. DISCUSSION
Over the past few years, the emerging landscape of immunotherapy in the treatment of lung cancer has become of increasing interest, and the impact of TME has been appreciated progressively. 29 , 30 Crosstalk between lung cancer cells and the TME plays a vital role in contributing to the development of cancer; thus, investigating the possible mechanisms of the TME that regulate lung cancer initiation, progression, and evolution appears to be particularly significant. 31 , 32 , 33 TAM are immune cells in the TME with high heterogeneity and complicated functions for regulating tumor immunity and mediating immunotherapy. 34 Initially, we focused on the role of STK16 in LUAD and found that STK16 expression was higher in cancer tissues than in normal tissues, and higher expression of STK16 was intimately associated with worse prognosis. Then, we explored the possible upstream mechanisms of STK16 and found the breakthrough point; namely, that STK16 expression is closely related to exosomes derived from M1 macrophages. In the present study, the data demonstrated that exosomes derived from M1 macrophages inhibit the progression and promote the apoptosis of LUAD via the miR‐181a‐5p/ETS1/STK16 axis. To the best of our knowledge, our study is the first to investigate the mechanism of STK16 and reveal how STK16 is mediated by exosomes derived from M1 macrophages in LUAD.
Emerging evidence and experimental studies have reported that TAM play crucial roles in the proliferation, invasion, and angiogenesis of solid tumors. 35 Normally, TAM can be polarized to either M1 or M2 macrophages. 36 A considerable number of studies have indicated that M1 macrophages have a proinflammatory phenotype, acting as a secretor of multiple proinflammatory cytokines, such as IL‐1β, IL‐6, IL‐23, TNF‐α, and iNOS, and, thus, participate in immune activity as an immune monitor. 35 However, M2 macrophages mainly secrete anti–inflammatory cytokines, such as IL‐10, IL‐4, and TGF‐β, which have suppressive effects on inflammation and tumor growth promotion. 37 A wide array of studies have reported that M1 macrophages can also secrete exosomes in addition to cytokines, and exosomes exert essential roles in regulating inflammatory processes, mediating cell metabolism, and promoting cell differentiation and progression. 38 , 39 , 40 Exosomes are a subset of membrane‐encased vesicles secreted by many cell types, including immune cells. 41 Multiple components, including proteins, mRNAs, microRNAs, circRNAs, and cholesterol, in the vesicles were delivered and selectively taken up by the targeted cells. 42 Exosomes derived from M1 macrophages are involved in multiple pathophysiological processes, including intercellular communication and signal transduction. Therefore, they play a seminal role in the growth, angiogenesis, metastasis, invasion, and drug resistance of cancer cells. 39 A previous study revealed that M1 macrophage‐derived exosomes delivered the chemotherapeutic agent paclitaxel, serving as a transporter and antitumor effector by activating the NF‐κB pathway. 43 A strong relationship between macrophage‐derived exosomes and neointimal hyperplasia has been reported in the literature. For example, exosomes derived from M1 macrophages can promote the progression of neointimal hyperplasia through the miR‐222/CDKN1B/CDKN1C pathway. 44 Prior studies have noted the importance of M1 macrophage‐derived exosomes in lung cancer and stated that M1 macrophage‐derived exosomes not only possess the potential to suppress tumor growth but also facilitate cisplatin to enhance its antitumor effect in lung cancer, 45 which is consistent with our current results that exosomes from M1‐polarized macrophages inhibit viability and promote apoptosis. Therefore, M1 macrophage‐derived exosomes might represent a promising target for the treatment of lung cancer.
MicroRNAs (miRNAs) are noncoding RNA molecules that have been reported to mediate cellular signals or interactions between cancer cells and macrophages. 46 The effect of exosomal miRNAs from TAM on cancer cell invasion, growth, and anticancer drug resistance reminds us that miRNAs are expected to be biotargets for the development of clinical drugs. 47 Cobos Jiménez et al 48 analyzed the expression signatures of miRNAs in Mo and polarized macrophages, and the results indicated that miR‐125a‐5p, miR‐181a‐5p, miR‐145‐5p, miR‐146a‐5p, miR‐193a‐5p, miR‐29b‐3p, and miR‐99b‐5p are expressed more in M1‐polarized macrophages. Our results showed that miR‐125a‐5p, miR‐181a‐5p, and miR‐193a‐5p were more highly expressed in M1‐exos. The expression of STK16 was decreased when H1975 cells were treated with miR‐181a‐5p mimics rather than miR‐125a‐5p mimics and miR‐193a‐5p mimics. Thus, miR‐181a‐5p, the functional molecule in M1 macrophage‐derived exosomes, was selected as a candidate to function STK16. Previous work has highlighted the biological function of miR‐181‐5p in certain types of tumors. Serum miR‐181a‐5p is a potential noninvasive biomarker for the diagnosis and prognosis of patients with NSCLC. 28 Moreover, overexpression of miR‐181a‐5p possesses an antitumor effect on the progression of NSCLC cells. 49 The expression of miR‐181a‐5p was dramatically decreased in lung cancer tissues and cell lines, and LUAD cells overexpressing miR‐181a‐5p could significantly inhibit the proliferation and migration of the LUAD cell line A549. 50 In our present study, we demonstrated the tumor‐suppressor role of miR‐138‐5p delivered from exosomes in LUAD tumorigenesis in a gain‐of‐function experiment. Exosomes can be employed as an effective vehicle to transport miR‐138‐5p, regulating downstream gene expression.
STK16 is a member of the numb‐associated family of protein kinases and a membrane‐associated kinase that is involved in the regulation of cell proliferation, apoptosis, signaling pathways, and metabolism. 9 To date, the biological functions of STK16 have not been well elucidated. Thus, we first analyzed the expression of STK16 from the TCGA database and verified our prediction using two cohorts from the hospital. We then confirmed that STK16 is more highly expressed in LUAD tissues and cancer cells. Furthermore, higher expression of STK16 is closely associated with worse outcomes of survival in patients with LUAD, suggesting that STK16 is a potential biomarker for predicting survival in LUAD. Previous studies have indicated that STK16 can directly bind and regulate actin dynamics to regulate the cell cycle. 11 STK16 is also involved in the regulation of many different cellular processes, such as the transcription of TGF‐β and VEGF. 51 In this study, it was found that knockdown of STK16 significantly inhibited cell proliferation and promoted cell apoptosis in vitro, and silencing of the STK16 gene markedly reduced the weight of nodules in a subcutaneously implanted tumor model. We further explored the underlying mechanism of STK16 in LUAD and found that STK16 promotes cell viability and inhibits cell apoptosis through the AKT1 signaling pathway. Thus, STK16 plays an indispensable role in the progression of LUAD.
Subsequently, we also demonstrated that miR‐138‐5p inhibits STK16 by targeting ETS1. ETS1 is a transcription factor that plays a crucial role in directly regulating the expression of cytokine and chemokine genes. 52 ETS1 is demonstrated to be involved in cell development, differentiation, and proliferation. 53 , 54 Previously, studies have identified multiple miRNAs that downregulate ETS1 expression by directly targeting the 3′UTR of ETS1. 55 Despite the lack of comprehensive investigations of the miR‐181a‐5p‐associated downstream mechanism in LUAD, the current study showed that miR‐181a‐5p inhibits STK16 expression by targeting ETS1 and might regulate the progression of LUAD through the AKT1 signaling pathway.
In conclusion, the finding that miR‐181a‐5p in exosomes from M1‐polarized macrophages inhibits STK16 by targeting ETS1 to regulate cell viability and apoptosis in LUAD provides guidance for the treatment of LUAD and deserves attention in the clinical practice of precision medicine.
DISCLOSURE
The authors declare that there are no conflicts of interest.
Supporting information
Fig S1‐S6
Table S1
ACKNOWLEDGMENT
Special thanks to Prof. Feng Tang and Zunguo Du, Department of Pathology, Huashan Hospital, Fudan University, who kindly helped us to evaluate the results of IHC score analysis.
Wang X, Huang R, Lu Z, Wang Z, Chen X, Huang D. Exosomes from M1‐polarized macrophages promote apoptosis in lung adenocarcinoma via the miR‐181a‐5p/ETS1/STK16 axis. Cancer Sci. 2022;113:986–1001. doi: 10.1111/cas.15268
Xuan Wang and Renhong Huang contributed equally to this work.
Funding information
This study was supported by the special project of clinical research of Shanghai Health Commission (No.202040454) and the National Natural Science Foundation of China (82002773)
Contributor Information
Zheng Wang, Email: zhengwang@shsmu.edu.cn.
Xiaofeng Chen, Email: chenxiaofengmd@126.com.
Dayu Huang, Email: davidhuang809@126.com.
REFERENCES
- 1. Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer statistics, 2021. CA Cancer J Clin. 2021;71:7‐33. [DOI] [PubMed] [Google Scholar]
- 2. Relli V, Trerotola M, Guerra E, Alberti S. Abandoning the notion of non‐small cell lung cancer. Trends Mol Med. 2019;25:585‐594. [DOI] [PubMed] [Google Scholar]
- 3. Yuan M, Huang L‐L, Chen J‐H, Wu J, Xu Q. The emerging treatment landscape of targeted therapy in non‐small‐cell lung cancer. Signal Transduct Target Ther. 2019;4:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Elmore L, Greer S, Daniels E, et al. Blueprint for cancer research: Critical gaps and opportunities. CA Cancer J Clin. 2021;71(2):107‐139. [DOI] [PubMed] [Google Scholar]
- 5. Edelman A, Blumenthal D, Krebs E. Protein serine/threonine kinases. Annu Rev Biochem. 1987;56:567‐613. [DOI] [PubMed] [Google Scholar]
- 6. Capra M, Nuciforo P, Confalonieri S, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Can Res. 2006;66:8147‐8154. [DOI] [PubMed] [Google Scholar]
- 7. Zhang C, Sun W, Tan M, et al. The eukaryote‐like serine/threonine kinase STK regulates the growth and metabolism of zoonotic streptococcus suis. Front Cell Infect Microbiol. 2017;7:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang Q, Fan Z, Zhang L, You Q, Wang L. Strategies for targeting serine/threonine protein phosphatases with small molecules in cancer. J Med Chem. 2021;64:8916‐8938. [DOI] [PubMed] [Google Scholar]
- 9. Wang J, Ji X, Liu J, Zhang X. Serine/threonine protein kinase STK16. Int J Mol Sci. 2019;20:1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wang J, Liu J, Ji X, Zhang X. Tyr198 is the essential autophosphorylation site for STK16 localization and kinase activity. Int J Mol Sci. 2019;20:4852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu J, Yang X, Li B, et al. STK16 regulates actin dynamics to control Golgi organization and cell cycle. Sci Rep. 2017;7:44607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu F, Wang J, Yang X, et al. Discovery of a highly selective STK16 kinase inhibitor. ACS Chem Biol. 2016;11:1537‐1543. [DOI] [PubMed] [Google Scholar]
- 13. Vinogradov S, Warren G, Wei X. Macrophages associated with tumors as potential targets and therapeutic intermediates. Nanomedicine. 2014;9:695‐707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang S, Song X, Li Y, Ye L, Zhou Q, Yang W. Tumor‐associated macrophages: A promising target for a cancer immunotherapeutic strategy. Pharmacol Res. 2020;161:105111. [DOI] [PubMed] [Google Scholar]
- 15. Stout R, Suttles J. Functional plasticity of macrophages: reversible adaptation to changing microenvironments. J Leukoc Biol. 2004;76:509‐513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010;141:39‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pan Y, Yu Y, Wang X, Zhang T. Tumor‐associated macrophages in tumor immunity. Front Immunol. 2020;11:583084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gun SY, Lee SWL, Sieow JL, Wong SC. Targeting immune cells for cancer therapy. Redox Biol. 2019;25:101174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang N, Wang S, Wang X, et al. Research trends in pharmacological modulation of tumor‐associated macrophages. Clin Transl Med. 2021;11:e288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kalluri R, LeBleu V. The biology function and biomedical applications of exosomes, vol. 367. Science (New York, NY); 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Nam G, Choi Y, Kim G, Kim S, Kim S, Kim I. Emerging prospects of exosomes for cancer treatment: from conventional therapy to immunotherapy. Adv Mater. 2020;32:e2002440. [DOI] [PubMed] [Google Scholar]
- 22. Othman N, Jamal R, Abu N. Cancer‐derived exosomes as effectors of key inflammation‐related players. Front Immunol. 2019;10:2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhou X, Xie F, Wang L, et al. The function and clinical application of extracellular vesicles in innate immune regulation. Cell Mol Immunol. 2020;17:323‐334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ling H, Fabbri M, Calin GA. MicroRNAs and other non‐coding RNAs as targets for anticancer drug development. Nat Rev Drug Discov. 2013;12:847‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ma Z, Qiu X, Wang D, et al. MiR‐181a‐5p inhibits cell proliferation and migration by targeting Kras in non‐small cell lung cancer A549 cells. Acta Biochim Biophys Sin (Shanghai). 2015;47:630‐638. [DOI] [PubMed] [Google Scholar]
- 26. Li Y, Kuscu C, Banach A, et al. miR‐181a‐5p inhibits cancer cell migration and angiogenesis via downregulation of matrix metalloproteinase‐14. Can Res. 2015;75:2674‐2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wen X, Li S, Guo M, et al. miR‐181a‐5p inhibits the proliferation and invasion of drug‐resistant glioblastoma cells by targeting F‐box protein 11 expression. Oncol Lett. 2020;20:235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xue W, Zhang M, Li R, Liu X, Yin Y, Qu Y. Serum miR‐1228‐3p and miR‐181a‐5p as noninvasive biomarkers for non‐small cell lung cancer diagnosis and prognosis. Biomed Res Int. 2020;2020:9601876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iams W, Porter J, Horn L. Immunotherapeutic approaches for small‐cell lung cancer. Nat Rev Clin Oncol. 2020;17:300‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Casanova‐Acebes M, Dalla E, Leader A, et al. Tissue‐resident macrophages provide a pro‐tumorigenic niche to early NSCLC cells. Nature. 2021;595:578‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Quail DF, Joyce JA. Microenvironmental regulation of tumor progression and metastasis. Nat Med. 2013;19:1423‐1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Izumi M, Sawa K, Oyanagi J, et al. Tumor microenvironment disparity in multiple primary lung cancers: impact of non‐intrinsic factors, histological subtypes, and genetic aberrations. Transl Oncol. 2021;14:101102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Altorki NK, Markowitz GJ, Gao D, et al. The lung microenvironment: an important regulator of tumour growth and metastasis. Nat Rev Cancer. 2019;19:9‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369‐382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chen Y, Song Y, Du W, Gong L, Chang H, Zou Z. Tumor‐associated macrophages: an accomplice in solid tumor progression. J Biomed Sci. 2019;26:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Li C, Xu X, Wei S, Jiang P, Xue L, Wang J. Tumor‐associated macrophages: potential therapeutic strategies and future prospects in cancer. J Immunother Cancer. 2021;9:e001341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang L, Zhang S, Wu H, Rong X, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106:345‐358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Du T, Yang CL, Ge MR, et al. M1 macrophage derived exosomes aggravate experimental autoimmune neuritis via modulating Th1 response. Front Immunol. 2020;11:1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Han C, Zhang C, Wang H, Zhao L. Exosome‐mediated communication between tumor cells and tumor‐associated macrophages: implications for tumor microenvironment. Oncoimmunology. 2021;10:1887552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xia Y, He X, Xu X, Tian B, An Y, Chen F. Exosomes derived from M0, M1 and M2 macrophages exert distinct influences on the proliferation and differentiation of mesenchymal stem cells. PeerJ. 2020;8:e8970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Doyle LM, Wang MZ. Overview of extracellular vesicles, their origin, composition, purpose, and methods for exosome isolation and analysis. Cells. 2019;8:727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wang Y, Liu J, Ma J, et al. Exosomal circRNAs: biogenesis, effect and application in human diseases. Mol Cancer. 2019;18:116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang P, Wang H, Huang Q, et al. Exosomes from M1‐polarized macrophages enhance paclitaxel antitumor activity by activating macrophages‐mediated inflammation. Theranostics. 2019;9:1714‐1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang Z, Zhu H, Shi H, et al. Exosomes derived from M1 macrophages aggravate neointimal hyperplasia following carotid artery injuries in mice through miR‐222/CDKN1B/CDKN1C pathway. Cell Death Dis. 2019;10:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li J, Li N, Wang J. M1 macrophage‐derived exosome‐encapsulated cisplatin can enhance its anti‐lung cancer effect. Minerva Medica. 2020; 10.23736/s0026-4806.20.06564-7 [DOI] [PubMed] [Google Scholar]
- 46. Mohapatra S, Pioppini C, Ozpolat B, Calin GA. Non‐coding RNAs regulation of macrophage polarization in cancer. Mol Cancer. 2021;20:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sun Z, Shi K, Yang S, et al. Effect of exosomal miRNA on cancer biology and clinical applications. Mol Cancer. 2018;17:147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cobos Jiménez V, Bradley EJ, Willemsen AM, van Kampen AH, Baas F, Kootstra NA. Next‐generation sequencing of microRNAs uncovers expression signatures in polarized macrophages. Physiol Genomics. 2014;46:91‐103. [DOI] [PubMed] [Google Scholar]
- 49. Wang L, Zhang L, Wang L. SNHG7 contributes to the progression of non‐small‐cell lung cancer via the SNHG7/miR‐181a‐5p/E2F7 axis. Cancer Manag Res. 2020;12:3211‐3222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ma Z, Qiu X, Wang D, et al. MiR‐181a‐5p inhibits cell proliferation and migration by targeting Kras in non‐small cell lung cancer A549 cells. Acta Biochim Biophys Sin. 2015;47:630‐638. [DOI] [PubMed] [Google Scholar]
- 51. Zhang YE. Non‐smad signaling pathways of the TGF‐β family. Cold Spring Harb Perspect Biol. 2017;9:a022129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Garrett‐Sinha L. Review of Ets1 structure, function, and roles in immunity. Cell Mol Life Sci. 2013;70:3375‐3390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dittmer J. The biology of the Ets1 proto‐oncogene. Mol Cancer. 2003;2:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Taveirne S, Wahlen S, Van Loocke W, et al. The transcription factor ETS1 is an important regulator of human NK cell development and terminal differentiation. Blood. 2020;136:288‐298. [DOI] [PubMed] [Google Scholar]
- 55. Taylor MA, Wappett M, Delpuech O, Brown H, Chresta CM. Enhanced MAPK signaling drives ETS1‐mediated induction of miR‐29b leading to downregulation of TET1 and changes in epigenetic modifications in a subset of lung SCC. Oncogene. 2016;35:4345‐4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S6
Table S1