

Summary
Drug resistance and metastasis—the major complications in cancer—both entail adaptation of cancer cells to stress, whether a drug or a lethal new environment. Intriguingly, these adaptive processes share similar features that cannot be explained by a pure Darwinian scheme, including dormancy, increased heterogeneity, and stress-induced plasticity. Here, we propose that learning theory offers a framework to explain these features and may shed light on these two intricate processes. In this framework, learning is performed at the single-cell level, by stress-driven exploratory trial-and-error. Such a process is not contingent on pre-existing pathways but on a random search for a state that diminishes the stress. We review underlying mechanisms that may support this search, and show by using a learning model that such exploratory learning is feasible in a high-dimensional system as the cell. At the population level, we view the tissue as a network of exploring agents that communicate, restraining cancer formation in health. In this view, disease results from the breakdown of homeostasis between cellular exploratory drive and tissue homeostasis.
Subject areas: Evolutionary theories, Cancer systems biology
Graphical Abstract
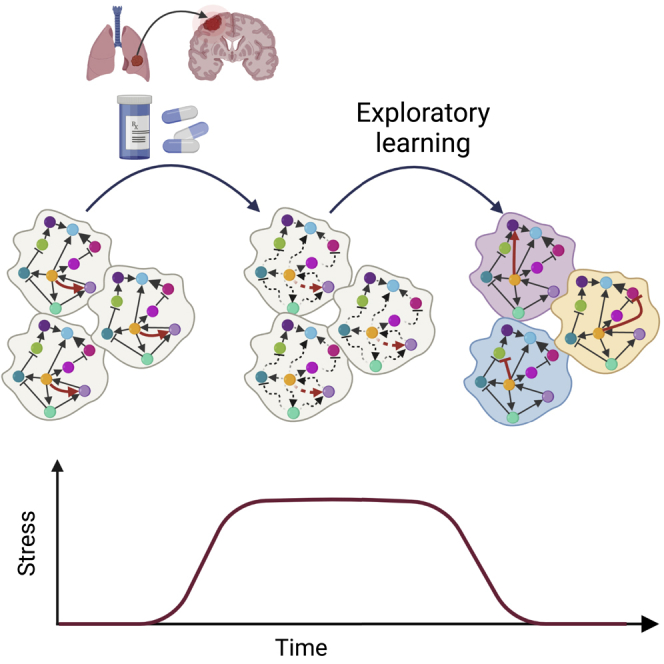
Evolutionary theories; Cancer systems biology
Introduction
Cancer progression is traditionally viewed as the outcome of the accumulation of random genetic mutations and the selection of cells harboring mutations that confer them a growth advantage under certain conditions (Garraway and Lander, 2013; Nowel, 1976; Vogelstein et al., 2013). This Darwinian view is intuitively appealing and provided a powerful framework for cancer research because it reduced cancer to the molecular level of the DNA. Indeed, studies that identified genes in which mutations are causally related to cancer, emerged at a brisk tempo, especially after the Cancer Genome Atlas and the International Cancer Genome Consortium were launched by the end of the 2000s (Martincorena and Campbell, 2015; The International Cancer Genome Consortium, 2010; Weinstein et al., 2013; Zhang et al., 2011).
This reductionist approach motivated developing drugs that target single molecular abnormalities or cancer pathways (Zugazagoitia et al., 2016). Although these drugs have achieved good clinical results, they only increased the overall survival by an average of nearly 3 months (Salas-Vega et al., 2017). A major reason for this is that malignant cells manage to adapt to the drug by bypassing its target, rendering the tumor resistant. In fact, resistance is the norm for all drugs that target specific molecules (Vasan et al., 2019).
Another factor contributing to poor clinical outcome is metastasis—the process of migration and colonization of distant organs by cells from a primary tumor. Metastasis is the major culprit of cancer-associated mortality accounting for almost 90% of deaths (Cheung and Ewald, 2016; Lambert et al., 2017). These two avenues of disease progression and mortality—drug resistance and metastasis—are distinct biological processes. However, they seem to be remarkably analogous in several fundamental properties. In particular, accumulating evidence suggests that both drug resistance and colonization of cancer cells in secondary organs exhibit aspects that do not exclusively follow a Darwinian scheme of mutation and selection (Welch and Hurst, 2019). Despite many years of research, a clear genetic signature of mutations associated with metastasis has not been found (Lambert et al., 2017; Vogelstein et al., 2013). Such a signature was found in some cancers at the initiation stage and was therefore expected and sought also in the context of metastasis. Resistance, in parallel, can arise from purely non-genetic mechanisms (Bell and Gilan, 2020; Marine et al., 2020; Pisco and Huang, 2015).
A cytotoxic treatment or a hostile environment in a secondary organ are both sources of stress to which cancer cells should adapt in order to survive. Plethora of findings showed that such a stress can actively induce an adaptive response and lead to drug resistance and the formation of metastasis (Pisco and Huang, 2015; Welch and Hurst, 2019). Such adaptation is supported by global epigenetic changes that enhance the plasticity of the cells. Various terms have appeared in the literature to describe these phenomena: “alternative pathways”, “acquired”, “adaptive”, or “induced” resistance (Kim et al., 2018; Maynard et al., 2020; Pisco and Huang, 2015; Shaffer et al., 2017; Stewart et al., 2020). Nevertheless, a comprehensive theory and a deeper understanding of these terms are still lacking and are crucial to pave the way to new therapeutic avenues.
In this perspective, we propose that learning theory offers a framework that may shed some light on the two complex processes of drug resistance and metastasis. Learning is the fundamental ability of a system to modify itself with relation to its environment and thus acquire novel functionality. Although most commonly attributed to the brain, we support the view that learning is not an exclusive function of neural networks, but rather a general property of plastic high-dimensional systems coupled to their environment (Baluška and Levin, 2016; Watson and Szathmáry, 2016; Watson et al., 2014). Experiments on yeast cells have revealed that cellular networks can provide a substrate for learning and adaptation in the face of unforeseen challenges to which no pre-programmed response is available (Braun, 2015). This experimental paradigm was based on artificially rewiring the genetic regulation network of yeast cells (Stolovicki et al., 2006). Exposing these cells to an environment where expression of the rewired gene is essential while its natural regulation is compromised, confronts them with regulatory challenging and stressful conditions for which no pre-programmed response is available. Without dedicated sensory information or pre-existing regulatory pathways, the yeast cells adapt through a trial-and-error process (Stern et al., 2007) that can be described as a primitive form of learning by the gene regulatory network (Schreier et al., 2017). In a related experimental setup, developing fly larvae that were exposed to artificial patterns of toxic challenge managed to develop into normal flies through epigenetic modifications (Stern et al., 2012). Such exploratory adaptation was proposed to have relevance to general biological contexts including evolution, development, and cancer (Braun, 2015).
Here, we propose that a similar type of adaptation is implicated in metastasis and resistance, where cancer cells aim to achieve stress reduction in a manner that is not contingent on pre-programmed pathways. Stressful situations, such as drugs and lethal environments, induce plasticity that can initiate an exploratory process by which new states of the cellular networks—regulatory genetic network, metabolic, and signaling networks—can be sampled. As a more adaptive and less stressful state is reached, the drive for additional sampling of new states decreases. The novelty of our approach is to highlight recent evidence for the use of this exploratory mode of adaptation by cancer cells, and to tie these observations to basic tenets of learning theory and thus establish a deep connection between the two.
We combine these insights from learning theory with the tissue population level of organization. While each individual cell can be viewed as a learning system in itself and might explore to find an adaptive state, it is also part of a coupled cell population in a tissue environment and interacts with neighboring cells. Thus, its propensity to explore may be constrained at the population level by a collective force that drives the cells to a local synchronous state.
To make our framework more concrete, we address two corollary questions: First, can one identify empirical signatures of exploratory dynamics in the processes of resistance and metastasis? We will delve into recent corroborative experimental findings that highlight prominent features of exploration and tissue constraints in cancer progression. Second, how is exploration feasible as an effective converging mechanism in a complex high-dimensional system as the cell? We will discuss the feasibility of such a mechanism based on recent modeling work done in our lab.
The fingerprints of exploratory adaptation in resistance and metastasis
Long and variable timescales in dormancy
Dormancy is a prominent feature of both resistance and metastasis, uncharacteristic of a pure Darwinian scheme. Several types of non-proliferative cell states have been characterized in the literature, for example dormant, quiescent, senescent; distinction between different states is often made by identification of specific markers. From a physiological point of view, the crucial phenomenon is the appearance of long timescales of very slow, or complete absence, of cell growth and division. In our context these can be grouped into a non-proliferative phenotype. Cancer cells that arrive at distant organs can remain in a quiescent non-proliferative state for a highly variable time period that extends up to decades (Figure 1A) (Chaffer and Weinberg, 2011; Gupta and Massagué, 2006; Lambert et al., 2017). The clinical implication of this phenomenon is that dormant cells can cause a metastatic relapse even years after radical dissection of the primary tumor.
Figure 1.

The fingerprints of exploratory adaptation in drug resistance and metastasis
(A) Dormancy: stressed cancer cells often enter a non-proliferative state that might be crucial for them to contrive adaptive states.
(B) Induced heterogeneity: despite the selective pressure of the treatment or the secondary organ, resistant cells and metastases exhibit high heterogeneity. This is concordant with exploratory adaptation as it yields multiple solutions to the same problem.
(C) Stress-induced stemness: the transition to a stem cell state is enhanced when cancer cells are exposed to stress such as a new environment. This transition provides cells with transient high plasticity that enables them to search for a new adaptive state. Created with BioRender.com
Similarly, in response to cytotoxic treatments, small sub-populations of cells can survive by initially entering a drug-tolerant state that displays little to no proliferation (Bell and Gilan, 2020; De Angelis et al., 2019; Marine et al., 2020; Ramirez et al., 2016; Sharma et al., 2010). These cells are genetically indistinguishable from the bulk tumor population, and they regain sensitivity to the drug after its withdrawal. Crucially, a fraction of these cells can gain the ability to proliferate in the drug after a long-term treatment (weeks to months). Thus, the quiescent subpopulation provides an adaptive reservoir for the acquisition of resistance mechanisms (Bell and Gilan, 2020).
In the absence of overt proliferation, genetic changes are less likely to occur and to be selected for (Giancotti, 2013). This adds to the arguments suggesting that adaptation to drugs or new environments may not occur through the selection of resistant genotypes. What exactly happens during the dormancy period is not well understood; it has been suggested that stress signaling is involved, as well as the emergence of stem-cell-like properties, which will be discussed in more detail below (Giancotti, 2013). One possibility is that dormancy time is utilized for an active search process in a large space of possible phenotypic configurations toward an epigenetic adaptive state. Once the cells manage to contrive an adaptive phenotype, they exit this state and reactivate growth. The yeast rewiring experiments provide an opportunity to glimpse into the single-cell dynamics during this latent phase. Woronoff et al. (2020) utilized micro droplet techniques to reveal that different yeast cells in the latent phase consume sugar at varying rates. This rate was correlated with their adaptation success, suggesting that the energy invested in metabolic activity might be the cost of contriving adaptation mechanisms during this period.
In cancer cells, no such measurement is available. An indirect clue might be obtained by observing the state of cells at the end of the latent phase. Specifically, does the passage through a dormant state force cancer cells into a single genetic/epigenetic state, or can multiple resistant phenotypes eventually arise? Exploratory adaptation supports the latter scenario. The nature and extent of heterogeneity will be considered next.
Single cell heterogeneity
Both drug resistant and metastatic cells are highly heterogeneous (Figure 1B) (Gupta and Massagué, 2006; Klein, 2013; Lawson et al., 2018; Scott et al., 2012; Vogelstein et al., 2013). The heterogeneity of metastatic lesions is especially intriguing in light of the fact that metastasis is an extremely inefficient process. The bottleneck of metastasis is colonization in distant organs where cancer cells need to adapt to a new deadly environment—even the most congenial environment is still very hostile for them. While 80% of tumor cells that are injected into the circulation manage to survive and extravasate, less than 0.02% form macro-metastases (Brabletz, 2012; Giancotti, 2013; Luzzi et al., 1998; Massagué and Obenauf, 2016). In this process, the miniature fraction of cells that manages to survive the new pernicious environment produces high intra-metastatic heterogeneity. This is somewhat surprising when compared to the effect of bottlenecks in ecology, where the signature of species migration is a dramatic temporary decrease in population heterogeneity (Amos and Harwood, 1998). Such a decrease is expected in any scenario that is based on selection at the population level, regardless of the mechanisms underlying the generation of variability. Thus, the heterogeneity of cancer cells following colonization of a new tissue, consistently with the temporal features discussed above, suggests that the process does not rely solely on the selection of fit cells but contains an element of intracellular dynamics.
Concordantly, recent studies showed a globally increased intra-tumoral heterogeneity following treatment resistance (Jr et al., 2016; Stewart et al., 2020). Careful experimental controls indicate that this increased heterogeneity is drug-induced. In contrast to the induction of pre-existing pathways dedicated to respond to a specific signal, here cells exposed to the same signals activated different pathways.
Taken together, these observations suggest that both metastasis and drug resistance include a crucial element of intracellular dynamics that results in highly variable outcomes. While both dormancy and heterogeneity can be considered indirect evidence of a highly plastic state of cancer cells, the next section highlights a more direct observation of plasticity.
Stress-induced stem cell state
The concept of a stem cell represents an extremely high degree of cellular plasticity, with very little constraints from epigenetic barriers. Although once thought to be a starting point of a unidirectional path in development leading to cell differentiation, it is now known that this path is reversible (Sánchez Alvarado and Yamanaka, 2014). Non-specific induced resistance can be implemented by a stem-like cell state, with a larger potential to acquire new phenotypes that are not accessible to fully differentiated cells; indeed, stemness has been tied to resistance-enhancing plasticity in many studies (Adorno-Cruz et al., 2015; Chang, 2016; De Angelis et al., 2019; Doherty et al., 2016; Lytle et al., 2018; Pisco and Huang, 2015). Induction of stemness can occur through a non-proliferative state (De Angelis et al., 2019). For example, chemotherapy can initiate a sequence of events that result in senescence-associated stemness (Milanovic et al., 2018).
Cancer stem cells are also pivotal players in metastasis, and were suggested to possess a tumor-initiating potential and exhibit a higher degree of plasticity that enables them to adapt to the challenges posed by the new environment (Adorno-Cruz et al., 2015; Chang, 2016; Doherty et al., 2016; Lytle et al., 2018). In particular, stem-like plasticity properties can be induced by external signals that are linked to the epithelial-mesenchymal transition (EMT), which is considered an early step in metastasis formation (Doherty et al., 2016). It is intriguing that the same noxious environment, which is supposed to kill the cells, enables them to acquire essential traits for survival. This dual role of the environment in metastasis is analogous to the role of the drug in resistance; in fact, metastasis can be viewed as resistance to the new environment. However, in contrast to drugs which target one pathway, survival in a new environment requires myriad general and tissue-specific adaptations.
For instance, cancer cells face a higher oxidative stress in the target organs; thus, cells that manage to produce antioxidants are more likely to survive (Piskounova et al., 2015). In addition, many organ-specific adaptations in the lung, bones, brain, and liver were identified (Massagué and Obenauf, 2016; Obenauf and Massagué, 2015). In the brain, for example, astrocytes produce plasminogen activator which induces the production of plasmin that leads to cancer cell death. Thus, to survive under such conditions, cancer cells need to produce serpins that are typically produced by neurons to shield them from plasminogen activator-mediated cell death (Valiente et al., 2014). In the liver, the survival of cancer cells is highly associated with their ability to consume creatinine and ATP to produce phosphocreatine (Loo et al., 2015). Such fundamental phenotypic changes require an extremely plastic cell state, such as that provided by stem cells (see Figure 1C).
Is exploratory adaptation feasible?
Taken together, the three features described in the preceding sections combine to suggest that single cells, in the context of metastasis and resistance, can enter a highly plastic state in order to explore intracellular configurations that may lead to adaptive behavior. Motivated by these observations, we proceed to examine the feasibility of exploratory adaptation.
To this end, two questions should be addressed—pertaining to the parts and to the whole: First, what are the molecular building blocks that could lead to trial-and-error learning? Second, does it all add up? Namely, can the process of exploratory adaptation converge within the context of single cell networks?
What molecular mechanisms support trial-and-error learning?
When drug resistance develops, even if selection of pre-adapted sub-populations is involved, cells still need to undergo epigenetic reprogramming to acquire resistance (Hong et al., 2019; Kim et al., 2018; Maynard et al., 2020; Shaffer et al., 2017; Sharma et al., 2010). Interestingly, the acquired resistance is neither drug-specific nor pathway-specific (Sharma et al., 2010), but rather entails global broad epigenetic changes. The coupling between cellular stress and mechanisms that can promote such changes is a central building block in our suggested framework (Braun, 2015; Soen et al., 2015). Stress can drive temporary plasticity, in turn driving global epigenetic reprogramming, and eventually allowing the acquisition of novel adaptive phenotypes. A recent study has shown that tumors consistently contain a fraction of cells in a stress-like state (Baron et al., 2020). Intriguingly, these cells are more efficient in seeding new tumors and hold drug-resistant properties that can be induced by heat shock.
An important mechanism that can increase cellular plasticity and enable trial-and-error exploration is chromatin remodeling. Disruption of chromatin homeostasis can lower epigenetic landscape barriers, making large regions of the genome accessible to transcription factor (TF) binding, and expanding the attainable space of gene expression patterns (Figure 2A) (Flavahan et al., 2017; Guo et al., 2019). Importantly, chromatin remodeling can be induced by non-genetic factors and provide transient and reversible coupling between stress and epigenetic plasticity. For instance, overexpression of the transcription factor Nfib led to increased accessibility to distal regulatory elements that promote pro-metastatic neural gene expression programs (Figure 2A) (Denny et al., 2016). Chromatin remodeling was also found to dynamically mediate resistance in a subpopulation, in response to drug application (Sharma et al., 2010). Thus, chromatin remodeling emerges as a candidate mechanism for modulating the level of cellular plasticity in a transient and stress-sensitive manner. In support of this picture, yeast cell experiments showing exploratory adaptation following a rewiring perturbation, exhibited modulation in the efficiency of adaptation in strains with mutations associated with chromatin remodeling (Freddolino et al., 2018).
Figure 2.

Molecular mechanisms supporting trial-and-error learning
(A) Chromatin remodeling: (top) Stress induced by the secondary organ or the drug can reshape the chromatin landscape, making it more permissive. (bottom) This endows the cells with higher plasticity to explore alternative states by lowering the barriers of transition between them.
(B) Regulatory network plasticity: (left) Regulatory networks are dynamic and transcription factors can even take contradictory roles depending on context. Arrows represent activating interactions and caps represent inhibitory interactions. (right) Intrinsically disordered proteins can confer plasticity by alternating between conformations that enable them to interact with different proteins. Created with BioRender.com.
Both cellular stress and chromatin remodeling have a global effect on the cellular network. In addition, the local elements of the network are themselves plastic. Many proteins have alternative binding partners that in turn give rise to multiple binding patterns depending on context, each resulting in markedly different network configuration (Figure 2B). For TFs, which mediate regulatory connections, this can induce a flexible network structure. Genome-wide binding assays in yeast have shown that TF binding patterns are context-dependent (Holland et al., 2019; Lee et al., 2002). In mammalian cells, it was demonstrated by computational analysis of time-varying single-cell data that edges in the regulatory networks are modulated during induced EMT (Krishnaswamy et al., 2018).
Alternative binding of transcription factors could be supported by the coexistence of multiple conformations (James and Tawfik, 2003). Intrinsically disordered protein regions, once thought to be a curious feature of a small number of proteins, are now acknowledged as a general property of many proteins, and most notably of TFs (Peng et al., 2015). Such disordered regions can give rise to a large number of folds and binding affinities (Figure 2B). While the role of random protein domains is still under intense study, recent results from plants implicate their connection with adaptation to stressful conditions (Liu et al., 2017). The existence of multiple alternative functional folds of TFs can potentially provide a powerful mechanism to confer plasticity to the genetic network and enable to explore different configurations due to rapid fluctuations between different conformations (Wright and Dyson, 2015). Other molecular mechanisms, such as post-transcriptional modifications, have also been suggested as an important characteristic of TFs that induces flexibility on gene regulation (Niklas et al., 2015).
From a systems-level perspective, such plasticity represents the ability of cellular networks to modify themselves, in analogy to neural networks in learning. The main mechanism thought to support learning in neural networks is synaptic plasticity, the ability of connections between neurons to remodel following signals and experience. Connection remodeling also underlies learning in practically all artificial network algorithms. The analogy with the ability of gene regulatory networks to modify their interactions, as supported by the above mechanisms, is straightforward and would thus endow the network with the ability to learn. This analogy motivated us to address the feasibility of convergence by borrowing concepts from learning models traditionally used to study neural networks.
Can a high-dimensional system converge under exploratory adaptation?
In contrast to evolutionary trial-and-error dynamics, which takes place at the population level, exploratory adaptation involves trial-and-error at the level of the individual. Variation is created in a single system along time and selected by feedback from the environment. Such dynamics have been considered in several biological contexts, such as spindle assembly and bacterial chemotaxis (Figures 3A and 3B) (Gerhart and Kirschner, 1997). However, these instances of exploration take place in three-dimensional space, and therefore, are feasible in terms of convergence success at finite time. For cancer cells to adapt by exploring gene expression, they face a different situation of a high-dimensional system (order of thousands of degrees of freedom) undergoing a random search in its vast configuration space (Figure 3C). With no sophisticated means to compute an error from some target function, the cell has only its global stress level at its disposal to provide feedback on the exploration. Under what conditions can such a learning scheme converge successfully? Because this is a quantitative question, one naturally turns to mathematical modeling: a model that captures the essential features of exploratory adaptation in high-dimensional space can shed light on the conditions for its possibility of convergence.
Figure 3.

Modeling exploratory adaptation
(A) A low-dimensional example of exploratory adaptation is chemotaxis, where bacteria move toward a higher gradient of an attractant (blue gradient) by a biased random walk.
(B) When exposed to stress, a system will search for a new state that relaxes the stress through a trial-and-error exploration. When the system reaches such a state, exploration stops. The time that takes to reach this state is the convergence time (Tconv).
(C) Convergence time increases dramatically with the number of dimensions. In the case of a genetic network, each gene is a dimension. This makes it hard for large networks performing exploratory adaptation to converge. Nevertheless, networks that harbor outgoing hubs—nodes with disproportionately high number of outgoing connections—can converge in a plausible time.
(D) Cells exposed to stress perform exploratory adaptation in which the connections between the genes are modified by a random walk. The strength of the random walk is dictated by the stress , where is the distance between the low dimensional state of the cell, y, and the constraint . Outgoing hubs (big purple nodes) enable exploratory adaptation to converge. Created with BioRender.com
We constructed such a model based on large random networks of interacting elements (Schreier et al., 2017). It draws from classic work on learning in neuroscience (Jaeger and Haas, 2004; Maass et al., 2007; Sussillo and Abbott, 2009). At the same time, random network models have been a popular modeling approach for gene regulation when general properties are of interest, such as number of fixed points, evolvability, or canalization (Drossel, 2008; Kauffman, 1993; Li et al., 2013).
The model (Figure 3D) describes a system of N interacting genes (N on the order of 1,000), whose state (expression level) is represented by the N-dimensional vector . The system's time evolution is determined by a nonlinear dynamic rule governed by a random interaction matrix W , in which the element Wij represents the strength of influence of gene j on gene i. Regulatory plasticity is represented by the ability of the interactions Wij to change their strengths over time; in exploratory dynamics, these changes will be essentially random, and their amplitude is controlled by feedback. The critical ingredient of the model is to define the feedback and close the loop.
A given cellular phenotype can be realized by different expression patterns. Mathematically, this corresponds to a low-dimensional projection, which we take to be linear, with weights given by N arbitrary numbers b:
| (Equation 1) |
The challenge is presented to the system by the constraint of maintaining the phenotype in a range . This allows for multiple gene expression patterns to comply with the constraint in different microscopic implementations. If, however, the projection deviates outside an allowed range, a global cellular stress will emerge, , causing the system to initiate exploratory dynamics in the form of random changes in the interaction matrix W. The amplitude of these changes is dictated by cellular stress S: it relaxes if and when it reaches a stable state that relieves stress, (Figure 3B). In this way, a simple feedback loop couples internal exploratory dynamics with the suitability of the current configuration to relieve stress and to match the environmental demand. This algorithm draws from a classic paradigm of learning in neuroscience based on the concept of drive reduction (Shahaf and Marom, 2001).
One might imagine that such a simple algorithm will not allow convergence to an appropriate phenotype in a reasonable time for large networks, and indeed, this is the case for homogeneous random matrices W, corresponding to identical and independent probability of connections between any two genes. Intriguingly, the main conclusion from the modeling work was that exploratory adaptation is sensitive to network structure; in particular, it is likely to converge for networks with outgoing hubs—a small number of nodes with disproportionately high connectivity (Figure 3C). For example, in a scale-free network topology, there are hubs in the tail of the distribution that are connected to a large fraction of the network (Schreier et al., 2017). However, scale-free networks are merely a mathematical tool to classify topologies; we have shown that a handful of outgoing genes in an arbitrary network are sufficient to induce the effect (Schreier et al., 2017; Rivkind et al., 2020). In real gene networks, master regulators are well known genes that control up to hundreds of other genes (Cai et al., 2020). Such hubs are usually considered in the context of specific gene programs that they regulate, but our model proposes that they can also coordinate network plasticity and drive it more easily through an exploratory process to discover novel stable states. This new role is similar to the stabilizing effect of an external feedback that suppresses irregular network activity (Rivkind et al., 2020). Importantly, it is in line with recent discoveries on master regulators in the context of cancer reprogramming.
For instance, ZEB1 is a key player in EMT, where its most well-known function is the suppression of epithelial genes. Interestingly, under some conditions, the flexible nature of this regulator is revealed and ZEB1 can turn into a transcriptional activator in aggressive cancer types (Lehmann et al., 2016). In fact, this is not a special property of ZEB1, but also exists for other transcription factors (Stemmler et al., 2019). Thus, master regulators can direct alternative expression patterns in different situations. In the absence of an external agent that directs the hubs which program to choose, our framework of exploratory adaptation suggests that the appropriate configuration emerges through stress-mediated feedback.
In conclusion, the feasibility of exploratory adaptation in heterogeneous random networks, and the fact that actual gene regulatory networks fulfill the key requirements of the model, suggest that this mechanism can be implemented in cells adapting to stressful conditions. In particular, it can help explain the behavior of cancer progression in drug resistance and metastasis. Intriguingly, applying this model to experimental data, Celiku et al. (2019) have shown that the adaptation of glioblastoma cells as they spread to diverse tumor microenvironments can be at least partly attributed to exploratory dynamics. Future work may use this framework and model to analyze experimental data in other systems.
The constraining force of the population
The capability of cells to explore for new configurations makes them vulnerable to fall by chance into cancerous configurations when they need to adapt to a stress. Without any constraining force, cancer would be the norm rather than the exception. We will argue below that this is not the case because the tissue constrains exploration and cancer progression. There has been an increasing surge of interest in the effect of cellular communication in cancer and many excellent reviews gather previous findings in this area (Bissell and Hines, 2011; Capp, 2005; Soto and Sonnenschein, 2011). Here, we tie these findings to our learning framework and emphasize the significance of cellular communication as a constraint on exploratory adaptation.
Mutations and growth control in normal tissues
Cancer is traditionally associated with the occurrence of specific patterns of mutations that are thought to drive transformation. It turns out that these mutations are not sufficient for transformation. A recent comprehensive RNA-sequencing analysis detected thousands of somatic mutations across all human tissues and in almost all tested individuals, including mutations at cancer hotspots and other cancer genes (Yizhak et al., 2019). Greater numbers of mutations were observed in tissues that are more exposed to carcinogenic environmental factors, such as sun-exposed skin, esophagus mucosa, and lung (Yizhak et al., 2019). Congruently, a recent analysis of sun-exposed skin tissues revealed a remarkably high burden of somatic mutations of averaged two to six mutations per megabase per cell, similar to that found in many cancers (Martincorena et al., 2015). The frequency of mutations that were previously identified as “driver mutations” in these tissues was surprisingly high. For instance, there were more NOTCH1 mutations in a sun-exposed skin biopsy than have been identified in more than 5,000 cancers sequenced by The Cancer Genome Atlas (Martincorena et al., 2015). Remarkably, clones carrying driver genes expand in normal skin tissues; however, this growth stops early in the expansion, giving rise to limited-sized clones (Martincorena et al., 2015). Some of the analyzed clones carried two to three driver mutations while having a completely normal skin phenotype. Similarly, sequencing studies of normal blood cells revealed signatures of somatic mutations broadly similar to blood cancer (Genovese et al., 2014; Jaiswal et al., 2014). These findings raise the question: how is homeostasis maintained in normal tissues despite the high burden of cancer driver mutations? What prevents the appearance of cancer phenotype in these tissues?
One widely accepted explanation is that driver mutations trigger oncogene-induced senescence (OIS), arresting the proliferation of cells before the accumulation of additional mutations (Bennett, 2003; Huang et al., 2017; Kaplon et al., 2014; Michaloglou et al., 2005; Serrano et al., 1997; Wajapeyee et al., 2008). OIS is usually depicted as a cell-autonomous stress response, namely, expressing an oncogene in a cell leads to stress which induces a growth arrest in that cell. However, a recent study by Ruiz-Vega et al. (Ruiz-Vega et al., 2020), using a mouse model of BRAF-driven nevus formation, showed that this is not necessarily the case. BRAF mutation is the most common driver mutation in melanoma (Davies et al., 2002), yet it is present in 89% of benign nevi (pigmented “moles”) (Pollock et al., 2003). In this work, no evidence supported the senescence of nevus cells, either compared with other skin cells or other melanocytes. Moreover, the nevus size distribution could not be fit by any simple cell-autonomous model of growth arrest, yet was easily fit by models of collective feedback between the cells. This emphasizes the significance of the tissue level of organization, in particular, cell-cell interactions, in maintaining homeostasis and constraining cancer development.
For better or worse: micro-environment controls cell fate
Intriguingly, the effect of cellular communication is so dominant that it not only stops the development of a cancerous phenotype but also can actually turn around cell fate. The classic work of Beatrice Mintz and Karl Illmenesse (Mintz and Illmensee, 1975) showed that placing teratocarcinoma (undifferentiated embryonic carcinoma cells) in a blastocyst gave rise to perfectly normal and tumor-free offspring that displayed many traits of the parental tumor cells. Similarly, transplantation of nuclei from malignant cells in enucleated oocytes gave rise to stem cells that were able to produce mice (Hochedlinger, 2004).
Numerous works provided additional evidence for the ability of the micro-environment to suppress tumor growth and induce differentiation to a variety of functional tissues. In their seminal work, the laboratories of Ole Petersen and Mina Bissel showed that breast cancer cells revert to nearly normal phenotype in three-dimensional culture that mimics the normal breast tissue (Howlett et al., 1994). The genome of the reverted cells was shown to be similar to mutated and malignant cells grown in two-dimensional cultures (Rizki et al., 2008; Weaver et al., 1995). Similarly, highly malignant melanoma cells injected into zebrafish embryos (Kasemeier-Kulesa et al., 2008), mammary carcinoma cells recombined with normal mammary gland stroma (Maffini et al., 2005), and liver cancer cells injected into normal liver (McCullough et al., 1997) are all additional examples for the ability of the micro-environment to normalize cancer cells. More recently, it was shown that converting invasive breast cancer cells into adipocytes by treating them with appropriate cues inhibits cancer metastasis (Ishay-Ronen et al., 2019). Unequivocally, all these findings point to the fact that the environment can play a crucial role in redirecting the phenotype of cancer cells and determining whether cancer is contained or spreads. They also demonstrate the high plasticity of cancer cells which can be exploited to adapt to dynamic changes, triggered by external signals.
Unfortunately, the stroma (fibroblasts, vasculature, immune cells, and interstitial ECM) that suppresses the growth of tumors can induce tumorigenesis when it undergoes detrimental changes. Maffini et al. (2004) showed that combining a carcinogen-exposed mammary stroma with vehicle-exposed mammary epithelium resulted in neoplasm. The reverse combination did not. Similar results were obtained from a normal mammary cell line with an irradiated stroma (Mh and Sa, 2000) and a normal prostate cell line and fibroblasts derived from prostate cancer (Barclay et al., 2005). These findings indicate that carcinogenesis can be the result of an abnormal interaction between the stroma and epithelial cells. Moreover, such interaction can support tumor progression and induce resistance (Chan et al., 2019; Shaked, 2019).
Feedback control from the tissue plays also a crucial role in development and size control (Balázsi et al., 2011; Lander, 2011; Levin, 2021). Such feedback occurs through secreted molecules, mechanical, and electrical feedback. Interestingly, a disruption of resting potential states can induce metastasis in embryos without carcinogen exposure or mutations (Levin, 2021). Conversely, modulation of bioelectric states can prevent or normalize tumors even in the presence of oncogenic mutations (Levin, 2021).
These data suggest a balance between the exploratory drive of individual cells under stress, and the constraining effect of interaction with the environment in a healthy tissue. The emerging picture supports a view of cancer which goes beyond the single cell and places the disease at the level of the cell-tissue interface (Braun, 2015; Capp, 2005). In this framework, cellular communication is a double-edged sword (Bissell and Hines, 2011). It can prevent transformation of normal cells harboring mutations and normalize cancer cells despite their mutations, but also induce tumorigenesis when aberrant. As Smithers stated it “Cancer is no more a disease of cells than a traffic jam is a disease of cars. A lifetime study of the internal combustion engine would not help anyone to understand our traffic problems” (Smithers, 1962).
Conclusion
Taken together, we propose that single cells have the capability to learn novel phenotypes by utilizing their internal plasticity under the guidance of global cellular stress. This proposition is based on analogies between features of cellular adaptation to stress and learning in neural networks; it is quantitatively supported by mathematical modeling that demonstrates its feasibility in high dimensions. We view cells in a tissue as a system of coupled explorers or learners. The state of the entire system is determined by interplay between the constraints of the population and the exploratory drive of individual cells. At one end of the spectrum, normal tissues are characterized by high constraints and low exploration. Tissue homeostasis is maintained by various mechanisms that include biochemical, electrical, and mechanical interactions between cells. At the other end of the spectrum, cancer progression is associated with looser constraints that allow a high exploratory behavior. In line with our framework, Gyurkó et al. (2013) described cancer development as a learning process of increasing network plasticity followed by decreasing plasticity. Our view shares with the tissue organization field theory (TOFT) of cancer premise that cells are not quiescent by default, but rather maintain continuous internal drive, while homeostasis is enforced on them by the tissue level of organization (Sonnenschein and Soto, 2020). It is also consistent with the idea that aging affects the propensity for cancer development through the decrease in effective tissue homeostasis (Capp and Thomas, 2021). Our conceptual framework may open the door to novel directions in cancer research and therapeutic development as it shifts the focus from looking for additional molecular targets to treating the learning cancer ecosystem as a whole (Weinberg, 2014). By providing the right environment and preserving the balance between these two forces, perhaps we can teach cancer cells to act like normal cells. Alternatively, a deeper understanding of the learning process can enable us not only to steer it toward desired phenotypes but also to prevent it to avoid resistance.
Acknowledgments
This work was supported in part by the Israel Science Foundation (Grant no. 346/16, O.B.; and Grant No. 155/18, N.B.). We acknowledge the Adams Fellowship Program of the Israel Academy of Science and Humanities (AS).
References
- Adorno-Cruz V., Kibria G., Liu X., Doherty M., Junk D.J., Guan D., Hubert C., Venere M., Mulkearns-Hubert E., Sinyuk M., et al. Cancer stem cells: targeting the roots of cancer, seeds of metastasis, and sources of therapy resistance. Cancer Res. 2015;75:924–929. doi: 10.1158/0008-5472.CAN-14-3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amos W., Harwood J. Factors affecting levels of genetic diversity in natural populations. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1998;353:177–186. doi: 10.1098/rstb.1998.0200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balázsi G., van Oudenaarden A., Collins J.J. Cellular decision making and biological noise: from microbes to mammals. Cell. 2011;144:910–925. doi: 10.1016/j.cell.2011.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baluška F., Levin M. On having no head: cognition throughout biological systems. Front. Psychol. 2016;7:902. doi: 10.3389/fpsyg.2016.00902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay W.W., Woodruff R.D., Hall M.C., Cramer S.D. A system for studying epithelial-stromal interactions reveals distinct inductive abilities of stromal cells from benign prostatic hyperplasia and prostate cancer. Endocrinology. 2005;146:13–18. doi: 10.1210/en.2004-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron M., Tagore M., Hunter M.V., Kim I.S., Moncada R., Yan Y., Campbell N.R., White R.M., Yanai I. The stress-like cancer cell state is a consistent component of tumorigenesis. Cell Syst. 2020;11:536–546.e7. doi: 10.1016/j.cels.2020.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell C.C., Gilan O. Principles and mechanisms of non-genetic resistance in cancer. Br. J. Cancer. 2020;122:465–472. doi: 10.1038/s41416-019-0648-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett D.C. Human melanocyte senescence and melanoma susceptibility genes. Oncogene. 2003;22:3063–3069. doi: 10.1038/sj.onc.1206446. [DOI] [PubMed] [Google Scholar]
- Bissell M.J., Hines W.C. Why don’t we get more cancer? a proposed role of the microenvironment in restraining cancer progression. Nat. Med. 2011;17:320–329. doi: 10.1038/nm.2328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brabletz T. To differentiate or not — routes towards metastasis. Nat. Rev. Cancer. 2012;12:425–436. doi: 10.1038/nrc3265. [DOI] [PubMed] [Google Scholar]
- Braun E. The unforeseen challenge: from genotype-to-phenotype in cell populations. Rep. Prog. Phys. 2015;78:036602. doi: 10.1088/0034-4885/78/3/036602. [DOI] [PubMed] [Google Scholar]
- Cai W., Zhou W., Han Z., Lei J., Zhuang J., Zhu P., Wu X., Yuan W. Master regulator genes and their impact on major diseases. PeerJ. 2020;8:e9952. doi: 10.7717/peerj.9952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capp J.-P. Stochastic gene expression, disruption of tissue averaging effects and cancer as a disease of development. Bioessays. 2005;27:1277–1285. doi: 10.1002/bies.20326. [DOI] [PubMed] [Google Scholar]
- Capp J.-P., Thomas F. Tissue-disruption-induced cellular stochasticity and epigenetic drift: common origins of aging and cancer? Bioessays. 2021;43:e2000140. doi: 10.1002/bies.202000140. [DOI] [PubMed] [Google Scholar]
- Celiku O., Gilbert M.R., Lavi O. Computational modeling demonstrates that glioblastoma cells can survive spatial environmental challenges through exploratory adaptation. Nat. Commun. 2019;10:5704. doi: 10.1038/s41467-019-13726-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaffer C.L., Weinberg R.A. A perspective on cancer cell metastasis. Science. 2011;331:1559–1564. doi: 10.1126/science.1203543. [DOI] [PubMed] [Google Scholar]
- Chan T.-S., Shaked Y., Tsai K.K. Targeting the interplay between cancer fibroblasts, mesenchymal stem cells, and cancer stem cells in desmoplastic cancers. Front. Oncol. 2019;9:688. doi: 10.3389/fonc.2019.00688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J.C. Cancer stem cells. Medicine (Baltimore) 2016;95:e4766. doi: 10.1097/MD.0000000000004559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung K.J., Ewald A.J. A collective route to metastasis: seeding by tumor cell clusters. Science. 2016;352:167–169. doi: 10.1126/science.aaf6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H., Bignell G.R., Cox C., Stephens P., Edkins S., Clegg S., Teague J., Woffendin H., Garnett M.J., Bottomley W., et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- De Angelis M.L., Francescangeli F., La Torre F., Zeuner A. Stem cell plasticity and dormancy in the development of cancer therapy resistance. Front. Oncol. 2019;9:626. doi: 10.3389/fonc.2019.00626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denny S.K., Yang D., Chuang C.-H., Brady J.J., Lim J.S., Grüner B.M., Chiou S.-H., Schep A.N., Baral J., Hamard C., et al. Nfib promotes metastasis through a widespread increase in chromatin accessibility. Cell. 2016;166:328–342. doi: 10.1016/j.cell.2016.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doherty M.R., Smigiel J.M., Junk D.J., Jackson M.W. Cancer stem cell plasticity drives therapeutic resistance. Cancers. 2016;8:8. doi: 10.3390/cancers8010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drossel B. Reviews of Nonlinear Dynamics and Complexity. John Wiley & Sons, Ltd; 2008. Random boolean networks; pp. 69–110. [Google Scholar]
- Flavahan W.A., Gaskell E., Bernstein B.E. Epigenetic plasticity and the hallmarks of cancer. Science. 2017;357:eaal2380. doi: 10.1126/science.aal2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freddolino P.L., Yang J., Momen-Roknabadi A., Tavazoie S. Stochastic tuning of gene expression enables cellular adaptation in the absence of pre-existing regulatory circuitry. Elife. 2018;7:e31867. doi: 10.7554/eLife.31867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway L.A., Lander E.S. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Genovese G., Kähler A.K., Handsaker R.E., Lindberg J., Rose S.A., Bakhoum S.F., Chambert K., Mick E., Neale B.M., Fromer M., et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. N. Engl. J. Med. 2014;371:2477–2487. doi: 10.1056/NEJMoa1409405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerhart J.C., Kirschner M.W. Blackwell; 1997. Cells, embryos and evolution: toward a cellular and developmental understanding of phenotypic variation and evolutionary adaptability (No. 575.21 GER) [Google Scholar]
- Giancotti F.G. Mechanisms governing metastatic dormancy and reactivation. Cell. 2013;155:750–764. doi: 10.1016/j.cell.2013.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo M., Peng Y., Gao A., Du C., Herman J.G. Epigenetic heterogeneity in cancer. Biomark Res. 2019;7:23. doi: 10.1186/s40364-019-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta G.P., Massagué J. Cancer metastasis: building a framework. Cell. 2006;127:679–695. doi: 10.1016/j.cell.2006.11.001. [DOI] [PubMed] [Google Scholar]
- Gyurkó D.M., Veres D.V., Módos D., Lenti K., Korcsmáros T., Csermely P. Adaptation and learning of molecular networks as a description of cancer development at the systems-level: potential use in anti-cancer therapies. Semin. Cancer Biol. 2013;23:262–269. doi: 10.1016/j.semcancer.2013.06.005. [DOI] [PubMed] [Google Scholar]
- Hochedlinger K. Reprogramming of a melanoma genome by nuclear transplantation. Genes Dev. 2004;18:1875–1885. doi: 10.1101/gad.1213504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland P., Bergenholm D., Börlin C.S., Liu G., Nielsen J. Predictive models of eukaryotic transcriptional regulation reveals changes in transcription factor roles and promoter usage between metabolic conditions. Nucleic Acids Res. 2019;47:4986–5000. doi: 10.1093/nar/gkz253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S.P., Chan T.E., Lombardo Y., Corleone G., Rotmensz N., Bravaccini S., Rocca A., Pruneri G., McEwen K.R., Coombes R.C., et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun. 2019;10:3840. doi: 10.1038/s41467-019-11721-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howlett A.R., Petersen O.W., Steeg P.S., Bissell M.J. A novel function for the nm23-H1 gene: overexpression in human breast carcinoma cells leads to the formation of basement membrane and growth arrest. J. Natl. Cancer Inst. 1994;86:1838–1844. doi: 10.1093/jnci/86.24.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.M., Chikeka I., Hornyak T.J. Melanocytic nevi and the genetic and epigenetic control of oncogene-induced senescence. Dermatol. Clin. 2017;35:85–93. doi: 10.1016/j.det.2016.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishay-Ronen D., Diepenbruck M., Kalathur R.K.R., Sugiyama N., Tiede S., Ivanek R., Bantug G., Morini M.F., Wang J., Hess C., et al. Gain fat—lose metastasis: converting invasive breast cancer cells into adipocytes inhibits cancer metastasis. Cancer Cell. 2019;35:17–32.e6. doi: 10.1016/j.ccell.2018.12.002. [DOI] [PubMed] [Google Scholar]
- Jaeger H., Haas H. Harnessing nonlinearity: predicting chaotic systems and saving energy in wireless communication. Science. 2004;304:78–80. doi: 10.1126/science.1091277. [DOI] [PubMed] [Google Scholar]
- Jaiswal S., Fontanillas P., Flannick J., Manning A., Grauman P.V., Mar B.G., Lindsley R.C., Mermel C.H., Burtt N., Chavez A., et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014;371:2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James L.C., Tawfik D.S. Conformational diversity and protein evolution – a 60-year-old hypothesis revisited. Trends Biochem. Sci. 2003;28:361–368. doi: 10.1016/S0968-0004(03)00135-X. [DOI] [PubMed] [Google Scholar]
- Jr P.B.F., Diggins K.E., Polikowsky H.G., Mohan S.R., Seegmiller A.C., Irish J.M. High-dimensional analysis of acute myeloid leukemia reveals phenotypic changes in persistent cells during induction therapy. PLoS One. 2016;11:e0153207. doi: 10.1371/journal.pone.0153207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon J., Hömig-Hölzel C., Gao L., Meissl K., Verdegaal E.M.E., van der Burg S.H., van Doorn R., Peeper D.S. Near-genomewide RNAi screening for regulators of BRAFV600E-induced senescence identifies RASEF, a gene epigenetically silenced in melanoma. Pigment Cell Melanoma Res. 2014;27:640–652. doi: 10.1111/pcmr.12248. [DOI] [PubMed] [Google Scholar]
- Kasemeier-Kulesa J.C., Teddy J.M., Postovit L.-M., Seftor E.A., Seftor R.E.B., Hendrix M.J.C., Kulesa P.M. Reprogramming multipotent tumor cells with the embryonic neural crest microenvironment. Dev. Dyn. 2008;237:2657–2666. doi: 10.1002/dvdy.21613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauffman S.A. Oxford University Press; 1993. The Origins of Order: Self-Organization and Selection in Evolution. [Google Scholar]
- Kim C., Gao R., Sei E., Brandt R., Hartman J., Hatschek T., Crosetto N., Foukakis T., Navin N.E. Chemoresistance evolution in triple-negative breast cancer delineated by single-cell sequencing. Cell. 2018;173:879–893.e13. doi: 10.1016/j.cell.2018.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C.A. Selection and adaptation during metastatic cancer progression. Nature. 2013;501:365–372. doi: 10.1038/nature12628. [DOI] [PubMed] [Google Scholar]
- Krishnaswamy S., Zivanovic N., Sharma R., Pe’er D., Bodenmiller B. Learning time-varying information flow from single-cell epithelial to mesenchymal transition data. PLoS One. 2018;13:e0203389. doi: 10.1371/journal.pone.0203389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert A.W., Pattabiraman D.R., Weinberg R.A. Emerging biological principles of metastasis. Cell. 2017;168:670–691. doi: 10.1016/j.cell.2016.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander A.D. Pattern, growth, and control. Cell. 2011;144:955–969. doi: 10.1016/j.cell.2011.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson D.A., Kessenbrock K., Davis R.T., Pervolarakis N., Werb Z. Tumour heterogeneity and metastasis at single-cell resolution. Nat. Cell Biol. 2018;20:1349–1360. doi: 10.1038/s41556-018-0236-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T.I., Rinaldi N.J., Robert F., Odom D.T., Bar-Joseph Z., Gerber G.K., Hannett N.M., Harbison C.T., Thompson C.M., Simon I., et al. Transcriptional regulatory networks in Saccharomyces cerevisiae. Science. 2002;298:799–804. doi: 10.1126/science.1075090. [DOI] [PubMed] [Google Scholar]
- Lehmann W., Mossmann D., Kleemann J., Mock K., Meisinger C., Brummer T., Herr R., Brabletz S., Stemmler M.P., Brabletz T. ZEB1 turns into a transcriptional activator by interacting with YAP1 in aggressive cancer types. Nat. Commun. 2016;7:10498. doi: 10.1038/ncomms10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin M. Bioelectrical approaches to cancer as a problem of the scaling of the cellular self. Prog. Biophys. Mol. Biol. 2021;165:102–113. doi: 10.1016/j.pbiomolbio.2021.04.007. [DOI] [PubMed] [Google Scholar]
- Li Z., Bianco S., Zhang Z., Tang C. Generic properties of random gene regulatory networks. Quant Biol. 2013;1:253–260. doi: 10.1007/s40484-014-0026-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y., Wu J., Sun N., Tu C., Shi X., Cheng H., Liu S., Li S., Wang Y., Zheng Y., et al. Intrinsically disordered proteins as important players during desiccation stress of soybean radicles. J. Proteome Res. 2017;16:2393–2409. doi: 10.1021/acs.jproteome.6b01045. [DOI] [PubMed] [Google Scholar]
- Loo J.M., Scherl A., Nguyen A., Man F.Y., Weinberg E., Zeng Z., Saltz L., Paty P.B., Tavazoie S.F. Extracellular metabolic energetics can promote cancer progression. Cell. 2015;160:393–406. doi: 10.1016/j.cell.2014.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzi K.J., MacDonald I.C., Schmidt E.E., Kerkvliet N., Morris V.L., Chambers A.F., Groom A.C. Multistep nature of metastatic inefficiency. Am. J. Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lytle N.K., Barber A.G., Reya T. Stem cell fate in cancer growth, progression and therapy resistance. Nat. Rev. Cancer. 2018;18:669–680. doi: 10.1038/s41568-018-0056-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maass W., Joshi P., Sontag E.D. Computational aspects of feedback in neural circuits. PLoS Comput. Biol. 2007;3:e165. doi: 10.1371/journal.pcbi.0020165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffini M.V., Soto A.M., Calabro J.M., Ucci A.A., Sonnenschein C. The stroma as a crucial target in rat mammary gland carcinogenesis. J. Cell Sci. 2004;117:1495–1502. doi: 10.1242/jcs.01000. [DOI] [PubMed] [Google Scholar]
- Maffini M.V., Calabro J.M., Soto A.M., Sonnenschein C. Stromal regulation of neoplastic development. Am. J. Pathol. 2005;167:1405–1410. doi: 10.1016/S0002-9440(10)61227-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marine J.-C., Dawson S.-J., Dawson M.A. Non-genetic mechanisms of therapeutic resistance in cancer. Nat. Rev. Cancer. 2020;20:743–756. doi: 10.1038/s41568-020-00302-4. [DOI] [PubMed] [Google Scholar]
- Martincorena I., Campbell P.J. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- Martincorena I., Roshan A., Gerstung M., Ellis P., Loo P.V., McLaren S., Wedge D.C., Fullam A., Alexandrov L.B., Tubio J.M., et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–886. doi: 10.1126/science.aaa6806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J., Obenauf A.C. Metastatic colonization by circulating tumour cells. Nature. 2016;529:298–306. doi: 10.1038/nature17038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard A., McCoach C.E., Rotow J.K., Harris L., Haderk F., Kerr D.L., Yu E.A., Schenk E.L., Tan W., Zee A., et al. Therapy-Induced evolution of human lung cancer revealed by single-cell RNA sequencing. Cell. 2020;182:1232–1251.e22. doi: 10.1016/j.cell.2020.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough K.D., Coleman W.B., Smith G.J., Grisham J.W. Transformed rat liver epithelial cells into the liver. Cancer Res. 1997;57:1807–1813. [PubMed] [Google Scholar]
- Mh B.-H., Sa R. Irradiated mammary gland stroma promotes the expression of tumorigenic potential by unirradiated epithelial cells. Cancer Res. 2000;60:1254–1260. [PubMed] [Google Scholar]
- Michaloglou C., Vredeveld L.C.W., Soengas M.S., Denoyelle C., Kuilman T., van der Horst C.M.A.M., Majoor D.M., Shay J.W., Mooi W.J., Peeper D.S. BRAF E600 -associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720–724. doi: 10.1038/nature03890. [DOI] [PubMed] [Google Scholar]
- Milanovic M., Fan D.N.Y., Belenki D., Däbritz J.H.M., Zhao Z., Yu Y., Dörr J.R., Dimitrova L., Lenze D., Monteiro Barbosa I.A., et al. Senescence-associated reprogramming promotes cancer stemness. Nature. 2018;553:96–100. doi: 10.1038/nature25167. [DOI] [PubMed] [Google Scholar]
- Mintz B., Illmensee K. Normal genetically mosaic mice produced from malignant teratocarcinoma cells. Proc. Natl. Acad. Sci. U S A. 1975;72:3585–3589. doi: 10.1073/pnas.72.9.3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niklas K.J., Bondos S.E., Dunker A.K., Newman S.A. Rethinking gene regulatory networks in light of alternative splicing, intrinsically disordered protein domains, and post-translational modifications. Front. Cell Dev. Biol. 2015;3:8. doi: 10.3389/fcell.2015.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowel P.C. The clonal evolution of tumor cell populations. Acquired genetic lability permits stepwise selection of variant sublines and underlies tumor progression. Science. 1976;194:223–228. [Google Scholar]
- Obenauf A.C., Massagué J. Surviving at a distance: organ-specific metastasis. Trends Cancer. 2015;1:76–91. doi: 10.1016/j.trecan.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Z., Yan J., Fan X., Mizianty M.J., Xue B., Wang K., Hu G., Uversky V.N., Kurgan L. Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell. Mol. Life Sci. 2015;72:137–151. doi: 10.1007/s00018-014-1661-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisco A.O., Huang S. Non-genetic cancer cell plasticity and therapy-induced stemness in tumour relapse: ‘what does not kill me strengthens me. ’ Br. J. Cancer. 2015;112:1725–1732. doi: 10.1038/bjc.2015.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E., Agathocleous M., Murphy M.M., Hu Z., Huddlestun S.E., Zhao Z., Leitch A.M., Johnson T.M., DeBerardinis R.J., Morrison S.J. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature. 2015;527:186–191. doi: 10.1038/nature15726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock P.M., Harper U.L., Hansen K.S., Yudt L.M., Stark M., Robbins C.M., Moses T.Y., Hostetter G., Wagner U., Kakareka J., et al. High frequency of BRAF mutations in nevi. Nat. Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Ramirez M., Rajaram S., Steininger R.J., Osipchuk D., Roth M.A., Morinishi L.S., Evans L., Ji W., Hsu C.-H., Thurley K., et al. Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat. Commun. 2016;7:10690. doi: 10.1038/ncomms10690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivkind A., Schreier H., Brenner N., Barak O. Scale free topology as an effective feedback system. PLoS Comput. Biol. 2020;16:e1007825. doi: 10.1371/journal.pcbi.1007825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizki A., Weaver V.M., Lee S.-Y., Rozenberg G.I., Chin K., Myers C.A., Bascom J.L., Mott J.D., Semeiks J.R., Grate L.R., et al. A human breast cell model of preinvasive to invasive transition. Cancer Res. 2008;68:1378–1387. doi: 10.1158/0008-5472.CAN-07-2225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Vega R., Chen C.-F., Razzak E., Vasudeva P., Krasieva T.B., Shiu J., Caldwell M.G., Yan H., Lowengrub J., Ganesan A.K., et al. Dynamics of nevus development implicate cell cooperation in the growth arrest of transformed melanocytes. Elife. 2020;9:e61026. doi: 10.7554/eLife.61026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salas-Vega S., Iliopoulos O., Mossialos E. Assessment of overall survival, quality of life, and safety benefits associated with new cancer medicines. JAMA Oncol. 2017;3:382–390. doi: 10.1001/jamaoncol.2016.4166. [DOI] [PubMed] [Google Scholar]
- Sánchez Alvarado A., Yamanaka S. Rethinking differentiation: stem cells, regeneration, and plasticity. Cell. 2014;157:110–119. doi: 10.1016/j.cell.2014.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier H.I., Soen Y., Brenner N. Exploratory adaptation in large random networks. Nat. Commun. 2017;8:14826. doi: 10.1038/ncomms14826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott J., Kuhn P., Anderson A.R.A. Unifying metastasis — integrating intravasation, circulation and end-organ colonization. Nat. Rev. Cancer. 2012;12:445–446. doi: 10.1038/nrc3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serrano M., Lin A.W., McCurrach M.E., Beach D., Lowe S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. doi: 10.1016/s0092-8674(00)81902-9. [DOI] [PubMed] [Google Scholar]
- Shaffer S.M., Dunagin M.C., Torborg S.R., Torre E.A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P.A., Xiao M., et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahaf G., Marom S. Learning in networks of cortical neurons. J. Neurosci. 2001;21:8782–8788. doi: 10.1523/JNEUROSCI.21-22-08782.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaked Y. The pro-tumorigenic host response to cancer therapies. Nat. Rev. Cancer. 2019;19:667–685. doi: 10.1038/s41568-019-0209-6. [DOI] [PubMed] [Google Scholar]
- Sharma S.V., Lee D.Y., Li B., Quinlan M.P., Takahashi F., Maheswaran S., McDermott U., Azizian N., Zou L., Fischbach M.A., et al. A chromatin-mediated reversible drug tolerant state in cancer cell subpopulations. Cell. 2010;141:69–80. doi: 10.1016/j.cell.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smithers D.W. Cancer an attack on cytologism. Lancet. 1962;279:493–499. doi: 10.1016/s0140-6736(62)91475-7. [DOI] [PubMed] [Google Scholar]
- Soen Y., Knafo M., Elgart M. A principle of organization which facilitates broad Lamarckian-like adaptations by improvisation. Biol. Direct. 2015;10:68. doi: 10.1186/s13062-015-0097-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenschein C., Soto A.M. Over a century of cancer research: inconvenient truths and promising leads. PLoS Biol. 2020;18:e3000670. doi: 10.1371/journal.pbio.3000670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto A.M., Sonnenschein C. The tissue organization field theory of cancer: a testable replacement for the somatic mutation theory. Bioessays. 2011;33:332–340. doi: 10.1002/bies.201100025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stemmler M.P., Eccles R.L., Brabletz S., Brabletz T. Non-redundant functions of EMT transcription factors. Nat. Cell Biol. 2019;21:102–112. doi: 10.1038/s41556-018-0196-y. [DOI] [PubMed] [Google Scholar]
- Stern S., Dror T., Stolovicki E., Brenner N., Braun E. Genome-wide transcriptional plasticity underlies cellular adaptation to novel challenge. Mol. Syst. Biol. 2007;3:106. doi: 10.1038/msb4100147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S., Fridmann-Sirkis Y., Braun E., Soen Y. Epigenetically heritable alteration of fly development in response to toxic challenge. Cell Rep. 2012;1:528–542. doi: 10.1016/j.celrep.2012.03.012. [DOI] [PubMed] [Google Scholar]
- Stewart C.A., Gay C.M., Xi Y., Sivajothi S., Sivakamasundari V., Fujimoto J., Bolisetty M., Hartsfield P.M., Balasubramaniyan V., Chalishazar M.D., et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer. 2020;1:423–436. doi: 10.1038/s43018-019-0020-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolovicki E., Dror T., Brenner N., Braun E. Synthetic gene recruitment reveals adaptive reprogramming of gene regulation in yeast. Genetics. 2006;173:75–85. doi: 10.1534/genetics.106.055442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussillo D., Abbott L.F. Generating coherent patterns of activity from chaotic neural networks. Neuron. 2009;63:544–557. doi: 10.1016/j.neuron.2009.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The International Cancer Genome Consortium International network of cancer genome projects. Nature. 2010;464:993–998. doi: 10.1038/nature08987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valiente M., Obenauf A.C., Jin X., Chen Q., Zhang X.H.-F., Lee D.J., Chaft J.E., Kris M.G., Huse J.T., Brogi E., et al. Serpins promote cancer cell survival and vascular co-option in brain metastasis. Cell. 2014;156:1002–1016. doi: 10.1016/j.cell.2014.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasan N., Baselga J., Hyman D.M. A view on drug resistance in cancer. Nature. 2019;575:299–309. doi: 10.1038/s41586-019-1730-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogelstein B., Papadopoulos N., Velculescu V.E., Zhou S., Diaz L.A., Kinzler K.W. Cancer genome landscapes. Science. 2013;339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajapeyee N., Serra R.W., Zhu X., Mahalingam M., Green M.R. Oncogenic BRAF induces senescence and apoptosis through pathways mediated by the secreted protein IGFBP7. Cell. 2008;132:363–374. doi: 10.1016/j.cell.2007.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson R.A., Szathmáry E. How can evolution learn? Trends Ecol. Evol. 2016;31:147–157. doi: 10.1016/j.tree.2015.11.009. [DOI] [PubMed] [Google Scholar]
- Watson R.A., Wagner G.P., Pavlicev M., Weinreich D.M., Mills R. The evolution of phenotypic correlations and “developmental memory”: the evolution of developmental memory. Evolution. 2014;68:1124–1138. doi: 10.1111/evo.12337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver V.M., Howlett A.R., Langton-Webster B., Petersen O.W., Bissell M.J. The development of a functionally relevant cell culture model of progressive human breast cancer. Semin. Cancer Biol. 1995;6:175–184. doi: 10.1006/scbi.1995.0021. [DOI] [PubMed] [Google Scholar]
- Weinberg R.A. Coming full circle-from endless complexity to simplicity and back again. Cell. 2014;157:267–271. doi: 10.1016/j.cell.2014.03.004. [DOI] [PubMed] [Google Scholar]
- Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013;45:1113–1120. doi: 10.1038/ng.2764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch D.R., Hurst D.R. Defining the hallmarks of metastasis. Cancer Res. 2019;79:3011–3027. doi: 10.1158/0008-5472.CAN-19-0458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woronoff G., Nghe P., Baudry J., Boitard L., Braun E., Griffiths A.D., Bibette J. Metabolic cost of rapid adaptation of single yeast cells. Proc. Natl. Acad. Sci. U S A. 2020;117:10660–10666. doi: 10.1073/pnas.1913767117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P.E., Dyson H.J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015;16:18–29. doi: 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yizhak K., Aguet F., Kim J., Hess J.M., Kübler K., Grimsby J., Frazer R., Zhang H., Haradhvala N.J., Rosebrock D., et al. RNA sequence analysis reveals macroscopic somatic clonal expansion across normal tissues. Science. 2019;364:eaaw0726. doi: 10.1126/science.aaw0726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J., Baran J., Cros A., Guberman J.M., Haider S., Hsu J., Liang Y., Rivkin E., Wang J., Whitty B., et al. International cancer genome consortium data portal—a one-stop shop for cancer genomics data. Database. 2011;2011:bar026. doi: 10.1093/database/bar026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zugazagoitia J., Guedes C., Ponce S., Ferrer I., Molina-Pinelo S., Paz-Ares L. Current challenges in cancer treatment. Clin. Ther. 2016;38:1551–1566. doi: 10.1016/j.clinthera.2016.03.026. [DOI] [PubMed] [Google Scholar]