

Abstract
Mitochondrial quality control (MQC) consists of multiple processes: the prevention of mitochondrial oxidative damage, the elimination of damaged mitochondria via mitophagy and mitochondrial fusion and fission. Several studies proved that MQC impairment causes a plethora of pathological conditions including cardiovascular diseases. However, the precise molecular mechanism by which MQC reverses mitochondrial dysfunction, especially in the heart, is unclear. The mitochondria-specific peroxidase Peroxiredoxin 3 (Prdx3) plays a protective role against mitochondrial dysfunction by removing mitochondrial reactive oxygen species. Therefore, we investigated whether Prdx3-deficiency directly leads to heart failure via mitochondrial dysfunction. Fifty-two-week-old Prdx3-deficient mice exhibited cardiac hypertrophy and dysfunction with giant and damaged mitochondria. Mitophagy was markedly suppressed in the hearts of Prdx3-deficient mice compared to the findings in wild-type and Pink1-deficient mice despite the increased mitochondrial damage induced by Prdx3 deficiency. Under conditions inducing mitophagy, we identified that the damaged mitochondrial accumulation of PINK1 was completely inhibited by the ablation of Prdx3. We propose that Prdx3 interacts with the N-terminus of PINK1, thereby protecting PINK1 from proteolytic cleavage in damaged mitochondria undergoing mitophagy. Our results provide evidence of a direct association between MQC dysfunction and cardiac function. The dual function of Prdx3 in mitophagy regulation and mitochondrial oxidative stress elimination further clarifies the mechanism of MQC in vivo and thereby provides new insights into developing a therapeutic strategy for mitochondria-related cardiovascular diseases such as heart failure.
Keywords: Peroxiredoxin 3, Heart failure, Mitochondrial quality control, Damaged mitochondria, Mitophagy, PINK1
Graphical abstract
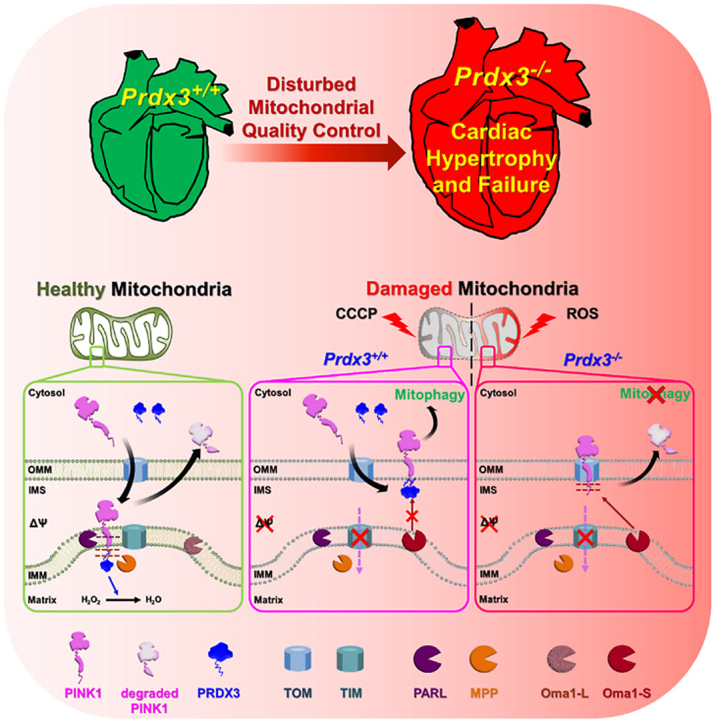
Highlights
-
•
Prdx3 is a master regulator of mitochondrial quality control (MQC).
-
•
Prdx3 deficiency aggravates cardiac hypertrophy by dysfunction in the MQC.
-
•
Prdx3 deficiency markedly decreases in vivo mitophagy.
-
•
Prdx3 protecting PINK1 against Oma1-dependent undergoing mitophagy.
-
•
Investigation of Prdx3 will facilitate further understanding of MQC in vivo.
1. Introduction
The heart has a high number and density of mitochondria. Adult cardiac mitochondria occupy approximately 30% of total cardiomyocytes volume, and primarily supply energy to compensate for the high adenosine triphosphate (ATP) consumption of the beating heart [1]. Mitochondria are the major sites of energy transfer and reactive oxygen species (ROS) production in the heart, and MQC dysfunction is associated with heart failure [2]. Heart failure includes heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction in a 1:1 ratio. Patients with HFpEF commonly have mitochondrial dysfunction. In particular, MQC dysfunction in the heart causes diastolic dysfunction, which decreases stroke volume [3,4]. However, few studies have examined the molecular mechanisms directly linking MQC dysfunction to cardiac function in vivo.
MQC is essential for cellular physiology and homeostasis [5]. Mitochondrial quality is tightly controlled to maintain a healthy mitochondrial network via multiple processes: the prevention of mitochondrial damage, elimination of damaged mitochondria and mitochondrial fission and fusion [6,7]. The prevention of mitochondrial damage primarily requires regulators of ROS, which represent the main cause of mitochondrial damage [8]. These regulators include peroxiredoxin 3 (Prdx3), a mitochondria-specific peroxidase that protects mitochondria against damage by removing H2O2 [9]. The elimination of damaged mitochondria largely relies on mitophagy, a mitochondria-specific type of autophagy [10]. As a key mediator of mitophagy, phosphatase and tensin homolog-induced kinase 1 (PINK1) regulates the selective removal of depolarized (damaged) mitochondria. The MQC also involves changes in mitochondrial shape, number and size through fission and fusion processes, which are regulated by Drp1 and Mfn, respectively [11]. However, it remains unclear whether these processes are linked to coordinate the entire MQC process.
Mammals carry six Prdx isoforms (Prdx1–6) that act as antioxidants and remove cellular ROS. Among them, Prdx3 localizes to the mitochondrial matrix to perform its mitochondrion-specific functions. In addition to their well-characterized role as antioxidants, Prdxs also function as molecular chaperones in a peroxidase-independent manner under stress [12,13]. In this regard, Prdx3 has an additional role as a chaperone in mitochondria. For example, human PRDX3 was reported to form filaments with putative chaperone activity under stress [14]. Taken together, it is likely that Prdx3 contributes to MQC via its dual antioxidant and chaperone functions.
Regarding the elimination of damaged mitochondria, the most well-characterized pathway is the PINK1–Parkin pathway regulating ubiquitin-dependent mitophagy. In healthy mitochondria, PINK1 is first transported to the inner mitochondrial membrane (IMM) and matrix, after which it is proteolytically destabilized by the presenilin-associated rhomboid-like protease (PARL) and mitochondrial processing peptidase (MPP) and constitutively degraded by the ubiquitin–proteasome system. In damaged mitochondria, PINK1 accumulates in the outer mitochondrial membrane (OMM), followed by the activation of its kinase activity and recruitment of Parkin, an E3 ubiquitin ligase, to damaged mitochondria to induce mitophagy through the ubiquitination of mitochondrial proteins, such as mitofusin and VDAC [15]. Of note, mutations in PINK1 or Parkin, which result in the accumulation of damaged mitochondria, are linked to familial Parkinsonism [16]. Despite the crucial roles of PINK1 in mitophagy, previous studies revealed that Pink1-deficient mice display mild, late-onset phenotypes without lethality [17]. Interestingly, recent reports illustrated that in vivo basal mitophagy is not altered in mice or Drosophila lacking PINK1 or Parkin [18,19], whereas stress-induced mitophagy is reduced in these models [20,21]. Thus, PINK1-mediated mitophagy may be associated with the clearance of stress-induced damaged mitochondria. Notably, a recent study reported that the stress-activated metalloprotease the m-AAA protease 1 homolog (OMA1) degrades PINK1 in mitochondria [22]. OMA1 in the IMM is activated by mitochondrial stressors, and it participates in MQC [23]. However, the mechanism protecting PINK1 against activated OMA1, permitting the induction of mitophagy under mitochondrial stress, remains unclear.
In this study, we investigated the regulatory mechanism that PRDX3 coordinates the entire process of MQC to protect heart failure and demonstrated the solid evidence that PRDX3, a master regulator of MQC, is essential for protection of heart failure via PRDX3-dependent regulation of mitophagy.
2. Methods
2.1. Mice
To generate Prdx3-deficient mitochondria-targeted Keima (mt-Keima) mice, Prdx3-deficient mice were crossed with mt-Keima mice [24]. To generate Pink1-deficient mt-Keima mice, Pink1-deficient mice (these mice were provided by Prof. Han-Woong Lee, Yonsei University) were crossed with mt-Keima mice. Mice were backcrossed more than seven times in the C57BL/6J background (Jackson Laboratory). All animal care and experimental procedures were performed in compliance with protocols approved by the Institutional Animal Care and Use Committee of Ewha Womans University.
2.2. Drosophila strains
Mt-Keima transgenic Drosophila (UAS-mt-Keima) was generated previously [21]. The white RNAi (whiteGD14981) lines were purchased from the Vienna Drosophila Resource Center. The da-GAL4 and Prx3 RNAi (Prx3HMJ22845) lines were purchased from the Bloomington Stock Center (Indiana University, Bloomington, IN, USA).
2.3. Mammalian cell culture
Primary mouse embryonic fibroblasts (MEFs) were isolated from wild-type and Prdx3-deficient embryos on embryonic day 13.5. Primary MEFs, HeLa cells, and 293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM, Invitrogen) supplemented with 10% FBS (Invitrogen), 100 U/ml penicillin, and 100 μg/ml streptomycin (Invitrogen). Cardiomyocytes were isolated from wild-type and Prdx3-deficient mice on postnatal day 3 using a Pierce™ Primary Cardiomyocyte Isolation Kit per the manufacturer's instructions. Isolated cells were cultured for 7 days prior to analysis to confirm the proper cell morphology. To restore the expression of Prdx3, Prdx3-deficient cardiomyocytes were infected with Prdx3 adenovirus or dominant-negative Prdx3 (Prdx-DN) adenovirus (Sirion).
2.4. Plasmids and RNAi oligonucleotides
Prdx3-Myc plasmids were generated via PCR amplification of full-length PRDX3 (NCBI accession number NM_006793.5) and cloning of the product into the EcoRI–XhoI sites of pcDNA.3.1/Myc-His (−) A plasmids (Invitrogen). The constructs encoding PINK1-GFP were generated via PCR amplification of full-length PINK1 (NCBI accession number NM_032409.3) and subcloning of the product into the EcoRI–BamHI sites of pEGFP-N3 plasmids (Clontech). The construct encoding Mid49-GFP was generated via PCR amplification of full-length Mid49 (isoform 1; NCBI accession number NM_139162.3) and cloning of the product into the EcoRI–BamHI sites of pEGFP-N3 plasmids (Clontech). The construct encoding the N-terminal sequence of Mid49 (amino acids 1–126, N-Mid49) or amino acids 111–581 of PINK1 (PINK1111−581) was generated via PCR amplification of full-length Mid49 or PINK1, respectively. To generate the fusion construct of N-Mid49 and PINK1111−581, Mid491−126 was subcloned into the XhoI–EcoRI sites of pEGFP-N3 plasmids (Clontech), followed by subcloning of PINK1111−581 into the EcoRI–BamHI sites of N-Mid49 pEGFP-N3 plasmids. Point mutations in PRDX3 (to express PRDX3L53V and PRDX3DN) and PINK1 (to express PINK1A93V) were generated using a Phusion Site-directed Mutagenesis Kit according to the manufacturer's protocol (Thermo Fisher Scientific). The construct encoding PRKN-GFP was generated via PCR amplification of full-length PRKN (isoform 1; NCBI accession number NM_004562.3) and subcloning of the product into the EcoRI–BamHI sites of pERFP-C1 plasmids (Clontech). pDsRed2-Mito (Clontech) was used for mitochondrial matrix staining. The construct encoding GST-Δ1–40PRDX3 was created by cloning the product into the EcoRI-XhoI sites of pGEM-4T-1 plasmids (GE Healthcare). siRNAs were synthesized by GenePharma. Human siPRDX3 targeted the sequence 5′-AAG CCA AGT CCA GCT GCT TCC-3′, human siOMA1 targeted the sequence 5′-GAA GTG CTT TGT CAT CTA ATT-3′ and mouse siOma1 targeted the sequence 5′-GGA TAC AGT CAA AGT TGC AGG-3′. As a control, Silencer Negative Control siRNA (GenePharma) was used.
2.5. Transfection and drug treatment
To overexpress mito-catalase, Prdx3-deficient MEFs or PRDX3-depleted HeLa cells were infected with a mito-catalase adenovirus and cultured for 24 h in DMEM supplemented with 10% FBS. Cells (2 × 105 cells/well) were seeded in six-well plates for 18 h prior to transfection. Plasmid transfection was performed in OPTI-MEM medium (Invitrogen) containing 3 μl of Lipofectamine 2000 for 4 h or the NEON™ transfection system (Invitrogen). The cells were then seeded onto microscope dishes with cover-glass bottoms (SPL) at a density of approximately 0.5 × 105 cells/ml. For RNAi transfection, cells were seeded onto six-well plates at 30–40% confluency (or an equivalent density). Then, 4 μl of Lipofectamine 2000 (Invitrogen) or 5 μl of RNAiMAX (Invitrogen) and 40 nM RNAi oligonucleotides were added to each well. Cells were re-transfected 24 h later with 3 μl of Lipofectamine 2000 and the appropriate plasmid DNA. For mitochondrial staining, cells were treated with 100 nM MitoTracker Red CMXRos (Invitrogen), a live mitochondria-labeling fluorescent dye exhibiting membrane potential-dependent accumulation, in DMEM for 20 min prior to fixation. For live imaging of mitochondria, cells were treated with 150 nM MitoTracker Green (Invitrogen) and 5 μM MitoSOX (Invitrogen), a mitochondrial superoxide indicator in live cells, in DMEM for 20 min. For carbonyl cyanide m-chlorophenylhydrazone (CCCP Sigma), oligomycin (Sigma), antimycin A (Sigma), or MG132 (Sigma) treatment, cells were seeded on poly-l-lysine–coated coverslips (0.1 mg/ml, Sigma) and incubated with 10 μM CCCP for 4–24 h, 2.5 μM oligomycin plus 250 nM antimycin A (OA) for 4 h, or 20 μM MG132 for 4 h in serum-containing medium. DMSO was used as the vehicle control.
2.6. Electron microscopy
To prepare samples for cellular transmission electron microscopy (TEM), heart, liver, skeletal muscle, and brain tissues were isolated from 10-week–old wild–type and Prdx3–deficient mice. The samples were then fixed with 2% glutaraldehyde paraformaldehyde in 0.1 M PBS (pH 7.4) for 2 h and washed three times for 30 min in 0.1 M PBS (pH 7.4, 1 mM). The tissues were then post-fixed with 1% OsO4 dissolved in 0.1 M PBS (pH 7.4) for 2 h, dehydrated in a graded ethanol series (50, 60, 70, 80, 90, 95, and 100%), and incubated with propylene oxide. Specimens were embedded using a Poly/Bed 812 kit (Polysciences, USA). After being embedded in pure fresh resin at 60 °C in an electron microscope oven (TD-700, DOSAKA, Japan) for 24 h, 300-nm-thick sections were initially cut and stained with toluidine blue for observation under a light microscope (Olympus BX40, Japan). Then, 80-nm-thick sections were double-stained with 7% uranyl acetate and lead citrate for contrast staining (20 min). The sections were then cut using a LEICA Ultracut UCT Ultramicrotome (Leica Microsystems, Austria). All of the samples were observed using TEM (JEM-1011, JEOL, Japan) at an acceleration voltage of 80 kV.
2.7. Measurement of mitophagy levels
Mitophagy levels were examined using the pH-dependent fluorescent probe mt-Keima via confocal microscopy as described previously [21,24]. To analyze the mt-Keima fluorescence signals, mt-Keima mouse and Drosophila tissue samples were examined using a Zeiss LSM 800 confocal microscope (Carl Zeiss) equipped with Plan-Apochromat 10 × /0.45 M27, Plan-Apochromat 20 × /0.8 M27, and c-Apochromat 40 × /1.20W Korr lenses. Mt-Keima fluorescence was imaged using two sequential excitation lasers (488 and 555 nm) and a 595–700 nm emission bandwidth. Quantitation of mitophagy based on the mt-Keima confocal images was performed using Zeiss Zen software on a pixel-by-pixel basis, as described previously [21,24]. The mitophagy level (% of mitophagy) was defined as the number of pixels that had a high red/green ratio divided by the total number of pixels. To quantify the mitophagy level in heart, at least five tissue samples were used for quantification, and the average values were calculated. In all confocal microscopy analyses, all imaging parameters remained constant, and only the gain level was adjusted to avoid saturation of any pixel. The results are presented as the mean ± SD.
2.8. Mitochondrial isolation
Mitochondria were isolated from MEF, cardiomyocytes, 293T cell, and HeLa cell lysates using a mitochondria isolation kit for cultured cells according to the manufacturer's protocol (Thermo Fisher Scientific, Inc.).
2.9. Measurement of the ATP level and respiration measurements
Measurement of the ATP level in cardiomyocytes for the ATP assay, mitochondria were isolated from cardiomyocytes lysates using a mitochondria isolation kit (Thermo Fisher Scientific, Inc.). The relative ATP level was calculated by dividing the measured ATP concentration by the 20 μg mitochondria. The ATP concentration was measured by ENLITEN® ATP Assay System Bioluminescence Detection Kit for ATP Measurement (Promega, USA) as previously described (21). Oxygen consumption rate (OCR) of cardiomyocytes was measured by XFp Analyzer (Agilent, USA) as described previously [25].
2.10. Immunoprecipitation and Western blotting
For immunoprecipitation experiments, non-transfected and transfected cells were lysed in whole-cell extraction buffer (10 mM HEPES [pH 7.9], 400 mM NaCl, 0.1 mM EDTA, 5% glycerol, 1 mM DTT, and protease inhibitors). Non-treated cell lysate or anti-PRDX3 (1 μg) was added to the lysate followed by the addition of anti-Myc–tagged agarose (MBL) or protein A–agarose (Upstate Biotech) in TEG reaction buffer (20 mM Tris–HCl at pH 7.4, 1 mM EDTA, 10% glycerol, 1 mM DTT, and 150 mM NaCl), and the mixture was stirred for 3 h or overnight at 4 °C. Immunoprecipitates were washed in TEG washing buffer (TEG reaction buffer containing 0.1% Triton X-100). For Western blotting, cells were lysed in whole-cell extract buffer or homogenized using a MICCRA D-8 homogenizer (ART Moderne Labortechnik) in protein extraction buffer (20 mM HEPES [pH 7.9], 300 mM NaCl, 10 mM EDTA, 0.1% NP40, 100 mM KCl, and protease inhibitors). Total protein was fractionated on a sodium dodecyl sulfate-polyacrylamide gel and transferred to nitrocellulose membranes (Amersham Biosciences). Primary antibodies against the following proteins were used: PRDX3 (1:300, AbFrontier, LF PA0044), catalase (1:100, Labfrontier, BC 100494), PRKN (1:100, Santa Cruz Biotechnology, sc-32282), GFP (1:100, Santa Cruz Biotechnology, sc-9976), TOM20 (1:100, Santa Cruz Biotechnology, sc-11415), tubulin (1:300, Sigma, T6199), OMA1 (1:100, Proteintech, 17116-1-AP), PRKN (1:100, Abcam, ab77924), Myc (1:200, Abcam, ab32), COX4 (1:100, Abcam, ab33985), myomesin (1:100, DSHB, B4–S), and PINK1 (1:50, NOVUS, BC 100494). The secondary antibodies were fluorescein-conjugated anti-rabbit IgG, anti-mouse IgG, anti-goat IgG (Invitrogen) and anti-rabbit, anti-mouse, and anti-goat HRP-conjugated antibodies (Zymed Laboratories). Protein–antibody complexes were detected using the ECL Plus system (Amersham Biosciences).
2.11. Transthoracic echocardiography and MI surgery
Transthoracic echocardiography was performed using the Vevo2100 system (VisualSonics) with a 25–55-MHz linear array transducer in wild-type and Prdx3-deficient mice (10 and 49–55 weeks old). After the induction of anesthesia with 1.5–2% isoflurane, mice were subjected to echocardiography with the heart rate maintained in the physiological range (>450 bpm) by anesthesia, and body temperature was maintained at 37 °C using a homeothermic pad during echocardiography. Left ventricular (LV) variables such as the ejection fraction (EF), fractional shortening (FS), SV, cardiac output (CO), end-diastolic volume, and end-systolic volume were analyzed via the long-axis view using Simpson's monoplane method of disks approach and VevoStrain software provided by the manufacturer. Wild-type and Prdx3-deficient mice (10–12 weeks old) were subjected to cardiac surgery under anesthesia with isoflurane. The left anterior descending coronary artery was permanently occluded using 7-0 silk suture. Transthoracic echocardiography was performed 15 days after surgery using the Vevo2100 system (VisualSonics) with a 25–55-MHz linear array transducer. Anesthesia was conducted and maintained via the inhalation of 1.5–2% isoflurane, and body temperature was monitored during echocardiography. LV variables such as EF, SV, end-diastolic volume, and end-systolic volume were analyzed via the long-axis view Simpson's monoplane method of disks approach using VevoStrain software provided by the manufacturer.
2.12. Cardiac fibrosis analysis
Serial sections of Masson's trichrome-stained hearts were used to assess left ventricular (LV) fibrosis. Paraffin-embedded LV samples were sectioned into 6-μm-thick slices and stained using a Masson's Trichrome Stain Kit (BBC Biochemical) according to the manufacturer's instructions. Tissue sections were scanned using the Vectra Polaris automated quantitative pathology imaging system (PerkinElmer), and the percentage of fibrosis relative to the LV area was measured using inForm Advanced Imaging Analysis Software (PerkinElmer).
2.13. Live imaging and confocal microscopy
The live imaging of cells was performed using a spinning disc confocal system (A1C; Nikon). For live cells, imaging was performed at 37 °C and 5% CO2 in an LCI chamber (Chamlide TC; LCI), and images were taken using a × 60 numerical aperture (NA) 1.4 oil objective lens. For fixed cells, imaging was performed using a laser-scanning confocal microscope (LSM880; Carl Zeiss) and structured illumination microscope (ELYRA S.1; Carl Zeiss). Images were taken with a × 63 oil objective lens (NA 1.4). Images were analyzed using Carl Zeiss Elements, Photoshop (Adobe), IMARIS (Bitplane AG), or ImageJ (US National Institutes of Health) software. Scale bars were generated using Carl Zeiss Elements and ImageJ.
2.14. Damaged mitochondria and mitochondrial ROS analysis
To measure depolarized (damaged) mitochondria, only mitochondria-associated MitoTracker Red CMXRos (Invitrogen) images on consecutive z-planes were quantified using ImageJ software. To analyze mitochondrial ROS, only mitochondria-associated MitoSOX (Invitrogen) images on consecutive z-planes were quantified using ImageJ software. Statistical analyses were performed using SigmaPlot (Systat Software, Inc). Data are presented as the mean ± SEM of at least three experiments.
2.15. Immunofluorescence and histology
Cultured cells were fixed in 4% formaldehyde in PBS for 20 min at room temperature (RT), washed with PBS, and permeabilized with 0.1% Triton X-100 for 15 min. Cells were washed with PBS, blocked with 3% BSA in PBS for 1 h, and incubated with primary antibodies in PBS for approximately –1–3 h at RT. After washing with PBS, the cells were incubated with secondary antibodies for 1 h at RT. The cells were then counterstained with 10 μM 4, 6-diamidino-2-phenylindole and mounted with Vectashield (VECTOR). Paraffin-embedded sections (6 μm thick) of the heart were fixed in 10% buffered formaldehyde and used for TUNEL assays and immunofluorescence analyses.
2.16. Mass spectrometric analysis
The gel PRDX3 band was destained and digested with trypsin, and the resulting peptides were extracted and the peptides were analyzed using nanoAcquity™ UPLC/ESI/q-TOF MS/MS (SYNAPT™ G2Si HDMS™, Waters Co., UK).
2.17. TUNEL assays
Apoptosis in myocardial infarcted heart 24 h after MI was assessed using a TACS 2 TdT fluorescein in situ apoptosis detection kit (Trevigen) according to the manufacturer's protocols.
2.18. Statistical analysis
Differences between two experimental groups were analyzed using Student's t-test or Mann–Whitney's U test. One-way ANOVA with Šidák's correction was used to compare three or more groups. P < 0.05 was considered statistically significant.
3. Results
3.1. Prdx3 deficiency induces cardiac hypertrophy and dysfunction
Mitochondria contribute substantially to cardiovascular homeostasis, and their fine-tuning via MQC is crucial for cardiovascular cell survival and the maintenance of physiological cardiac function [26]. Disruption of MQC is closely linked to cardiac defects. To determine the role of Prdx3 in myocardial MQC, cardiac phenotypes were examined in 10- and 52-week-old wild-type and Prdx3-deficient mice. Prdx3-deficient mice exhibited cardiac hypertrophy, including significant increases of the heart weight to body weight ratio, decreased CO at 52 weeks of age, and decreased SV at both 10 and 52 weeks of age without changes of EF (Fig. 1A–D, Supplementary Fig. 1A). Thus, our results are consistent with previous reports that mitochondrial dysfunction is involved in heart failure caused by diastolic dysfunction, which decreases SV and CO [[2], [3], [4]]. Therefore, the induction of LV remodeling and cardiac hypertrophy by Prdx3 deficiency suggests that mitochondrial dysfunction is a critical risk factor of heart failure, reminiscent of human HFpEF. In addition, Prdx3-deficient mice had a similar heart size as wild-type mice at 10 weeks old but exhibited cardiac hypertrophy with significant increases cardiomyocyte size and LV fibrosis at 52 weeks old (Fig. 1E–I, Supplementary Fig. 1B). Damaged mitochondria in Prdx3-deficient cardiomyocytes were observed in 10-week-old mice, and their numbers were increased markedly at 52 weeks old, with the mice displaying giant and damaged mitochondria with morphologically peculiar shapes and an extremely large size (Fig. 1J and K). These results suggest that MQC dysfunction induced by Prdx3 deficiency leads to the accumulation of dysfunctional and giant mitochondria and causes cardiac dysfunction.
Fig. 1.

Peroxiredoxin 3 (Prdx3) deficiency causes cardiac hypertrophy with giant and damaged mitochondria.
(A) Heart weight/body weight ratio between Prdx3 wild-type (Prdx3 WT) and Prdx3-deficient (Prdx3 KO) mice at 10 (n = 5/group) and 52 weeks of age (n = 6/group). (B–D) Analysis of cardiac function by echocardiography in 10-week-old Prdx3 WT (n = 6), 52-week-old Prdx3 WT (n = 10), 10-week-old Prdx3 KO (n = 6), and 52-week-old Prdx3 KO mice (n = 6). Stroke volume (SV) (B), cardiac output (CO) (C), and ejection fraction (EF) (D) were measured in the left ventricular (LV) long-axis view using Simpson's monoplane method of disks approach. (E) Hematoxylin and eosin staining of Prdx3 WT and Prdx3 KO hearts. Scale bar, 1 mm. (F and G) Wheat germ agglutinin staining of LV muscle sections to visualize cell boundaries and quantify LV fibrosis. Scale bars, 20 μm. (H and I) Masson's trichrome staining of LV muscle sections and quantification of cardiac fibrosis. Scale bars, 50 μm. (J and K) Representative electron micrographs and quantification of damaged mitochondria (red arrows) in the hearts of Prdx3 WT and Prdx3 KO mice. Scale bars in 10-week-old Prdx WT and Prdx3 KO mice, 0.5 (boxed areas) or 1 μm. Scale bars in 52-week-old Prdx WT and Prdx KO mice, 1 (boxed areas) or 2 μm *P < 0.05, **P < 0.01 and ***P < 0.001. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.2. Prdx3 deficiency aggravates cardiac dysfunction associated with myocardial infarction (MI)
Mitochondrial injury caused by oxidative stress, a hallmark of infarcted cardiomyopathy, promotes excessive cardiomyocyte loss and LV remodeling [27]. To assess whether Prdx3 deficiency exacerbates heart failure after MI, cardiac function was evaluated using echocardiography 15 days after the induction of MI (Supplementary Fig. 2A). EF and SV were significantly lower in Prdx3-deficient mice than in wild-type mice. LV end-systolic volume was significantly increased in Prdx3-deficient mice, but LV end-diastolic volume was not significantly different between wild-type and Prdx3-deficient mice (Fig. 2A–E). These results indicate that Prdx3 deficiency aggravates cardiac dysfunction after MI. Moreover, Masson's trichrome staining illustrated that Prdx3 deficiency exacerbated cardiac fibrosis and LV remodeling (Fig. 2F). In addition, we observed that the number of damaged mitochondria was higher in the infarcted hearts of Prdx3-deficient mice than in those of wild-type mice (Fig. 2G). Disruption of the MQC-driven elimination of damaged mitochondria leads to pernicious cell death, which is characterized by the activation of apoptotic cascades. We performed in situ apoptosis analysis using the infarcted hearts of wild-type and Prdx3-deficient mice following MI. We observed that apoptotic cell death was significantly increased in the infarcted hearts of Prdx3-deficient mice (Fig. 2H). Although 10-week-old Prdx3-deficient mice did not exhibit obvious cardiac dysfunction under physiological conditions (Supplementary Fig. 2B), an increased number of damaged mitochondria attributable to impaired MQC might explain the prevalence of heart failure after MI. Thus, under MI, Prdx3 deficiency results in increased mitochondrial damage and consequent apoptosis in infarcted hearts.
Fig. 2.

Peroxiredoxin 3 (Prdx3) deficiency accelerates left ventricular (LV) remodeling and cardiac failure after myocardial infarction (MI).
(A–E) Analysis of cardiac function using echocardiography (A) in Prdx3 wild-type (Prdx3 WT; n = 7) and Prdx3 deficient (Prdx KO, n = 8) mice 15 days after MI. LV end-diastolic volume (EDV) (B) and end-systolic volume (ESV) (C) were measured in the LV long-axis view using Simpson's monoplane method of disks approach for stroke volume (SV) (D) and ejection fraction (EF) (E). (F) Serial sections of Masson's trichrome-stained hearts from Prdx3 WT and Prdx KO mice 15 days after MI. The percentage of fibrosis was calculated relative to that in the total LV area. (G) Representative electron micrographs of the hearts of wild-type and Prdx KO mice 1 day after MI. Red arrows indicate damaged mitochondria. Scale bar, 0.5 (boxed areas) or 2 μm. (H) TUNEL assay of the infarcted areas of Prdx3 WT (n = 5) and Prdx KO mice (n = 5) 1 day after MI. Scale bar, 10 (boxed areas) or 50 μm *P < 0.05 and **P < 0.01. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.3. Prdx3 deficiency induces mitochondrial damage but suppresses in vivo mitophagy in the heart
To determine further whether mitochondrial damage caused by Prdx3 deficiency results from mitochondrial ROS accumulation, we examined mitochondrial phenotypes in Prdx3-deficient mice. Mitochondrial membrane potential was analyzed in MEFs isolated from Prdx3-deficient mice, revealing increased numbers of damaged or depolarized mitochondria (Fig. 3A and C). Next, an analysis of mitochondrial ROS in Prdx3-deficient MEFs revealed that Prdx3 deficiency led to increased mitochondrial ROS levels (Fig. 3B and D). Given the primary molecular function of Prdx3 in preventing ROS accumulation, we next examined whether overexpression of mito-catalase, a mitochondrial ROS scavenger, could ameliorate the mitochondrial damage associated with Prdx3 deficiency. Mito-catalase overexpression appeared to reduce mitochondrial damage caused by Prdx3 deficiency (Fig. 3A–E). These findings indicate that Prdx3 deficiency-induced mitochondrial damage is closely associated with aberrant ROS accumulation. Furthermore, Prdx3 deficiency in vivo causes mitochondrial damage in the heart and other tissues of Prdx3-deficient mice (Supplementary Figs. 3A and B). The ATP level and oxygen consumption rate (OCR) were also significantly reduced in Prdx3-deficient cardiomyocytes, demonstrating that Prdx3 deficiency causes cardiac mitochondrial dysfunction (Supplementary Figs. 4A–G). The numbers of damaged mitochondria were increased in the hearts, livers, skeletal muscles, and brains of 52-week-old Prdx3-deficient mice (Fig. 1J and K, Supplementary Figs. 3A and B), suggesting that Prdx3-deficient mice represent a suitable animal model for studying MQC.
Fig. 3.

Increases mitochondrial damage is attributed to reactive oxygen species (ROS) accumulation by peroxiredoxin (Prdx3) deficiency.
(A and B) Representative low- and high-magnification images of the boxed areas of Prdx3 wild-type (Prdx3 WT) and Prdx3-deficient (Prdx3 KO) mouse embryonic fibroblasts (MEFs). MEFs were infected with mito-catalase adenovirus for 24 h and immunostained with an antibody against Tom20 (green) plus 100 nM MitoTracker (red) (A) or 150 nM MitoTracker (green) plus 5 μM MitoSOX (red) (B) for 25 min. Scale bar, 2 (boxed areas) or 10 μm. (C and D) Quantification of depolarized (damaged) mitochondria (C) or mitochondrial ROS levels (D) in Prdx3 WT and Prdx3 KO MEFs (n = 25–33 cells). (E) Immunoblotting of whole-cell lysates of Prdx3 WT and Prdx3 KO MEFs infected with mito-catalase adenovirus for 24 h using catalase, Prdx3, and Tom20 antibodies. **P < 0.01. Data (A–E) are representative of three independent experiments. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Next, to address the possible coordination between the two MQC processes in the prevention of mitochondrial damage through the regulation of ROS by mitochondria-specific Prdx3 and the elimination of damaged mitochondria via PINK1-mediated mitophagy, we compared mitochondrial phenotypes in the hearts of Prdx3-and Pink1-deficient mice. Whereas mitochondrial damage was increased in the hearts and other tissues of Prdx3-deficient mice (Fig. 1J and K, Supplementary Figs. 3A and B), mitophagy was markedly lower in the hearts and other tissues of Prdx3-deficient mice than in wild-type and Pink1-deficient mice (Fig. 4A and B, Supplementary Figs. 5A–C). We next determined the pathophysiological outcome after MI, specifically whether the modality of mitophagy is influenced by Prdx3. We characterized the cardiac function of Prdx3 in mitophagy using Prdx3-deficient mt-Keima mice with MI, mitophagy was significantly increased in the hearts of wild-type mt-Keima mice 4 h after MI, while it was markedly reduced in the hearts of Prdx3-deficient mt-Keima mice regardless of MI (Supplementary Fig. 5D). We also found that Prdx3 deficiency significantly attenuated mitophagy in cardiac tissue 24 h following MI (Fig. 4C and D). Consistently, depletion of Prx3, the Drosophila ortholog of Prdx3, led to decreased mitophagy in wing disks from Drosophila larvae and the Drosophila adult fat body (Supplementary Fig. 5E). These results suggest a potential role of Prdx3 in the regulation of mitophagy in addition to its intrinsic role in preventing mitochondrial damage by regulating ROS levels.
Fig. 4.

Peroxiredoxin 3 (Prdx3) deficiency reduces mitophagy in the heart.
(A and B) Representative images (A) and quantification (B) of mitophagy in the hearts of 10-week-old Prdx3 wild-type (Prdx3 WT; n = 9), 52-week-old Prdx3 WT (n = 6), 10-week-old Pink1-deficient (Pink1 KO; n = 7), 52-week-old Pink KO (n = 6), 10-week-old Prdx3-deficient (Prdx3 KO; n = 8), and 52-week-old Prdx3 KO (n = 6) mitochondria-targeted Keima (mt)-Keima mice. Scale bar, 10 μm. (C and D) Representative images (C) and quantification (D) of mitophagy in the infarcted hearts of Prdx3 WT (n = 5) and Prdx3 KO mt-Keima mice (n = 7) 1 day after the induction of myocardial infarction (MI). Scale bar, 10 μm ***P < 0.001. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.4. Prdx3 regulates mitophagy via the Prdx3-dependent localization and degradation of PINK1 in mitochondria
The reduction of mitophagy in the absence of Prdx3 prompted us to investigate its possible direct involvement in the regulation of mitophagy. In healthy mitochondria, PINK1 localizes to the mitochondrial matrix, in which its N-terminus is cleaved by PARL and MPP via sequential proteolysis. In response to mitochondrial damage, PINK1 accumulates on the OMM of damaged mitochondria, in which it mediates mitophagy [15]. To understand the contribution of Prdx3 to the regulation of mitophagy, we first examined whether PINK1 protein levels are altered by Prdx3 deficiency. To this end, we compared PINK1 protein levels between wild-type and Prdx3-deficient MEFs. PINK1 protein levels were higher in Prdx3-deficient and proteasome inhibitor MG132-treated Prdx3-deficient MEFs than in wild-type MEFs (Fig. 5A and B, Supplementary Fig. 6A). Because Prdx3 deficiency leads to reduced mitophagy despite the upregulation of PINK1, we next examined whether the submitochondrial localization of PINK1 is altered by Prdx3 deficiency. We overexpressed GFP-tagged PINK1 in control and Prdx3-deficient MEFs and compared its localization in mitochondria labeled with the OMM marker Tom20 using Airyscan super-resolution microscopy. In Prdx3-deficient MEFs or PRDX3-depleted HeLa cells, PINK1-GFP co-localized with Tom20 in the OMM, whereas PINK1-GFP preferentially localized to the matrix in wild-type MEFs and HeLa cells (Fig. 5C, Supplementary Fig. 6B). These results suggest that Prdx3 affects the submitochondrial localization of PINK1 and thereby modulates its degradation.
Fig. 5.

Peroxiredoxin 3 (Prdx3) is required for PINK1–Parkin-mediated mitophagy.
(A and B) Western blot analysis (A) and quantification (B) of PINK1 expression in the mitochondrial fractions of Prdx3 wild-type (Prdx3 WT) and Prdx3-deficient (Prdx3 KO) mouse embryonic fibroblasts (MEFs) treated with DMSO, 20 μM MG132, or 10 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 4 h. The mitochondrial fractions were immunoblotted with PINK1 and Tom20 antibodies. (C) Immunostaining of Prdx3 WT and Prdx3 KO MEFs transfected with PINK1-GFP using Tom20 antibody (red). The graphs present the fluorescent intensities of Tom20 and PINK1-GFP measured at the dotted line. Scale bar, 1 (boxed areas) or 10 μm. (D and E) Western blot analysis (D) and quantification (E) of Pink1 expression in the mitochondrial fractions of Prdx3 WT and Prdx3 KO MEFs treated with 10 μM CCCP or 2.5 μM oligomycin and 250 nM antimycin A (OA) for 4 h. The mitochondrial fractions were immunoblotted with PINK1 and Tom20 antibodies. Hashes indicate non-specific bands. (F–I) Immunostaining and quantification of PINK1-GFP (F and G) and Parkin-GFP levels (H and I) in Prdx3 WT and Prdx3 KO MEFs. MEFs were infected with mito-catalase adenovirus for 24 h, transfected with PINK1-GFP or Parkin-GFP, and then treated with DMSO or 10 μM CCCP for 4 h, followed by immunostaining with Tom20 antibody (red). Scale bar, 10 μm *P < 0.05 and **P < 0.01. Data (A–I) are representative of three independent experiments. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
We next assessed whether Prdx3 affected PINK1 expression under pro-mitophagy conditions. We treated wild-type and Prdx3-deficient MEFs with the mitochondrial uncoupling agent CCCP or the mitochondrial respiration inhibitors oligomycin and antimycin A (OA) and examined PINK1 protein levels. Previous studies indicated that CCCP or OA treatment strongly increases PINK1 accumulation in mitochondria, thereby inducing mitophagy. Interestingly, the induction of PINK1 accumulation in mitochondria by CCCP or OA treatment was almost completely abolished by Prdx3 deficiency (Fig. 5A, B, D and E). Consistently, when we overexpressed PINK1-GFP or Parkin-GFP in MEFs, the CCCP-dependent mitochondrial accumulation of PINK1-GFP and Parkin-GFP was strongly inhibited in Prdx3-deficient MEFs even though ROS-induced mitochondrial damage linked to Prdx3 deficiency was ameliorated by mito-catalase overexpression (Fig. 5F–I). In addition, when we overexpressed PINK1-GFP or Parkin-GFP in MEFs and HeLa cells, the CCCP- or OA-dependent mitochondrial accumulation of PINK1-GFP and Parkin-GFP was inhibited in Prdx3-deficient MEFs or PRDX3-depleted HeLa cells (Supplementary Figs. 6C and D). These results suggest that Prdx3 acts as a regulator of PINK1–Parkin-mediated mitophagy by modulating the submitochondrial localization and degradation of PINK1.
3.5. Prdx3 interacts with the mitochondrial targeting sequence of PINK1
We examined whether PRDX3 interacts with PINK1 using co-immunoprecipitation experiments, finding that endogenous PRDX3 binds with endogenous PINK1 (Fig. 6A). To identify the critical binding sites between PRDX3 and PINK1, we created truncated and point-mutated constructs of PRDX3 and PINK1 and found that N-terminal domain of PINK1 is required for binding with PRDX3 (Supplementary Figs. 7A and B). To determine further the N-terminal domain of PRDX3 critical for binding with PINK11−110, we performed IP experiments using PRDX363−256, PRDX337−256, PRDX31−256, and PINK11−110. We found that PINK11−110 binds with PRDX337−256 and PRDX31−256, but not with PRDX363−256, indicating that N-terminal sequence (amino acids 37–62) of PRDX3 is required for binding with PINK1 (Supplementary Fig. 7C). Utilizing PRDX3Leu53Val and PINK1Ala93Val mutants, we observed that Leu53 in PRDX3 and Ala93 in PINK1 are critical for the binding between PRDX3 and PINK1 (Fig. 6B and C). Dominant-negative PRDX3Cys108Ser/Cys229Ser, which blocks ROS removal by PRDX3, could still bind to PINK1, indicating that ROS-counteracting activity remained functionally intact in dominant-negative PRDX3 (Fig. 6B). GST pull-down experiments confirmed that Leu53 in PRDX3 and Ala93 in PINK1 are critical for the direct binding between these proteins (Fig. 6D and E). It has been reported that the N-terminal sequence (amino acids 1–62) of PRDX3 serves as a mitochondrial targeting signal [28]. Mass spectrometric analysis to further clarify the mitochondrial targeting signal sequences of PRDX3 predicted that the N-terminal sequence (amino acids 1–36) of PRDX3 is essential for mitochondrial targeting and revealed that mature PRDX3 contains Leu53, ensuring its binding to PINK1 (Fig. 6F and G). Crystallography of mitochondrial Prdx, the Leishmania infantum ortholog of Prdx3, illustrated that the N-terminal sequence outside the mitochondrial targeting sequence is critical for the formation of structurally stable mitochondrial Prdx [12]. Thus, we demonstrated that the N-terminal sequence (amino acids 37–62) of PRDX3 binds to the mitochondrial targeting sequence of PINK1.
Fig. 6.

Peroxiredoxin 3 (Prdx3) interacts with PINK1.
(A) Interaction between endogenous PINK1 and PRDX3. Whole-cell lysates (WCLs) of 293T
cells were immunoprecipitated with control immunoglobulin G (IgG) and anti
-Prdx3 antibody and then blotted with anti-PINK1 antibody. (B) WCLs of 293T
cells co-transfected with various combinations of constructs expressing PINK1-GFP, PRDX3-Myc, L53V PRDX3-Myc, and PRDX3-Myc dominant-negative (DN; Cys108Ser, Cys229Ser) were used for immunoprecipitation with anti
-Myc beads and subsequent immunoblotting with GFP-specific antibody. Hashes indicate IgG. (C) WCLs of 293T
cells co-transfected with various combinations of constructs expressing PINK1-GFP, A93V PINK1-GFP, and PRDX3-Myc were used for immunoprecipitation with anti
-Myc beads and subsequent immunoblotting with GFP-specific antibody. Hashes indicate IgG. (D) WCLs of 293T
cells transfected with constructs expressing PINK1-GFP were used for immunoprecipitation with GST-Δ1–40PRDX3, GST-Δ1–40PRDX3 L53V, or GST-Δ1–40PRDX3 DN-conjugated beads and subsequent immunoblotting with PINK1- or GFP-specific antibody. Hashes indicate non-specific bands. (E) WCLs of 293T cells transfected with constructs expressing PINK1-GFP or A93V PINK1-GFP were used for immunoprecipitation with GST-Δ1–40PRDX3 and subsequent immunoblotting with a PINK1- or GFP-specific antibody. Hashes denote non-specific bands. (F) WCLs of 293T cells transfected with Myc-His, 63–256 PRDX3-Myc-His, or 1–256 PRDX3-Myc-His expression vectors were used for immunoblotting with PRDX3 and tubulin antibodies. (G) Mass spectrometric analysis of 1–256 PRDX3-Myc-His (red box in f) and predicted MPP cleavage site of premature sequences (residues 1–36 of the PRDX3 mitochondrial targeting sequence) and mature PRDX3. Data (A–E) are representative of three independent experiments. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.6. Prdx3 regulates PINK1 stability via its inhibition of Oma1 during mitophagy
We examined whether Prdx3 protein expression changes as PINK1 levels increase in damaged mitochondria. We found that Prdx3 protein levels also increased in damaged mitochondria, mimicking the upregulation of PINK1 (Fig. 7A and B). We also investigated whether the submitochondrial localization of Prdx3 switches from the mitochondrial matrix to the OMM upon binding with PINK1 under CCCP-induced mitochondrial damage. Co-immunostaining of Prdx3 with Tom20 revealed that Prdx3 localized to the mitochondrial matrix in healthy mitochondria, whereas it translocated to the OMM in damaged mitochondria, as confirmed by its co-localization with either Tom20 or PINK1 (Fig. 7C and D).
Fig. 7.

Peroxiredoxin 3 (Prdx3) regulates the stability of PINK1.
(A and B) Western blot analysis (A) and quantification (B) of Pink1 and Prdx3 expression in the mitochondrial fractions of mouse embryonic fibroblasts (MEFs) treated with DMSO or 10 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 4 h. The mitochondrial fractions were immunoblotted with PINK1, Prdx3, and Cox4 antibodies. (C) Immunostaining using Tom20 (green) and/or PRDX3 antibodies (red) in HeLa cells transfected with pDsRed2-Mito (MTX; matrix) and treated with DMSO or 10 μM CCCP for 4 h. Scale bar, 2 (boxed areas) or 10 μm. (D) Immunostaining using Tom20 (green) and/or PRDX3 antibodies (red) in HeLa cells transfected with PINK1-GFP and treated with 10 μM CCCP for 4 h. Scale bar, 2 (boxed areas) or 10 μm. (E and F) Western blot analysis (E) and quantification (F) of Pink1 in the mitochondrial fractions of Prdx3 wild-type (Prdx3 WT) and Prdx3-deficient (Prdx3 KO) MEFs transfected with Oma1 siRNA and treated with 10 μM CCCP for 4 h. The mitochondrial fractions were immunoblotted with PINK1, Oma1, Prdx3, and Cox4 antibodies. (G and H) Immunostaining using Tom20 antibody (G) and quantification (H) of PINK1-GFP in Prdx3 WT and Prdx3 KO MEFs transfected with Oma1 siRNA and PINK1-GFP. Scale bar, 10 μm *P < 0.05 and **P < 0.01. Data (A–H) are representative of three independent experiments. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
Diverse cellular stresses can drive mitochondrial dysfunction, which can itself promote the activation of the IMM-resident stress-induced protease OMA1 [23]. Upon activation in damaged mitochondria, Oma1 cleaves Dele1 and initiates a stress-induced proteolytic cascade [29]. A recent study reported that OMA1 degrades Parkinson's disease-related PINK1 mutants that are imported into damaged mitochondria [22]. Prdxs, including Prdx3, serve dual functions as peroxidases under normal conditions and as chaperones upon exposure to diverse stresses [12]. Human PRDX3 acts as a self-assembling chaperone that is catalytically active under stress conditions [14]. To determine whether PINK1 is degraded by Oma1 regardless of the presence of Prdx3, we examined the stability of PINK1 in damaged mitochondria in wild-type and Prdx3-deficient MEFs following Oma1 depletion and CCCP treatment. In damaged mitochondria undergoing mitophagy, PINK1 was not degraded by Oma1 in the presence of Prdx3 (Fig. 7E and F). Interestingly, PINK1 was degraded by Oma1 in the absence of Prdx3, but its levels were restored by Oma1 depletion using siRNA (siOma1). PINK1-GFP was also completely lost in Prdx3-deficient MEFs and PRDX3-depleted HeLa cells after CCCP treatment. However, Oma1 depletion using siOma1 dramatically restored PINK1-GFP levels in cells treated with CCCP (Fig. 7G and H, Supplementary Fig. 8). Thus, the binding of Prdx3 to the N-terminus of PINK1 protects the latter protein against degradation by blocking its cleavage by Oma1. This indicates that Prdx3 acts as a chaperone for PINK1.
To confirm that Prdx3 regulates PINK1–Parkin-mediated mitophagy by modulating the stability of PINK1 in damaged mitochondria, we performed PINK1 recruitment/rescue experiments. Mid49 regulates the mitochondrial recruitment of Drp1, which controls fission in mitochondrial dynamics, and it is an OMM protein with an N-terminal mitochondrial targeting sequence and OMM domain. Using N-Mid49, which contains the mitochondrial targeting sequence and OMM domain [30], we created a mitochondrial targeting domain-truncated form of PINK1 (PINK1111−581) that lacked both the PRDX3-binding region and OMA1 cleavage site and localized persistently to the OMM (Supplementary Fig. 9A). When N-Mid49–PINK1111−581 was overexpressed in Prdx3-deficient MEFs, the construct localized normally to mitochondria irrespective of CCCP-induced mitochondrial damage (Supplementary Fig. 9B). When N-Mid49–PINK1111−581 was overexpressed in Prdx3-deficient MEFs or PRDX3-depleted HeLa cells, the construct only recruited Parkin to damaged mitochondria following CCCP treatment (Supplementary Figs. 9C and D). N-Mid49–PINK1111−581 was expressed stably in damaged mitochondria regardless of the presence of Prdx3 and CCCP-induced mitochondrial damage (Supplementary Fig. 9E). Moreover, overexpression of N-Mid49–PINK1111−581 in Prdx3-deficient MEFs resulted in decreased mitochondrial depolarization via the occurrence of normal mitophagy compared to the findings in Prdx3-deficient MEFs (Supplementary Fig. 9F). To induce mitochondrial depolarization in most cells, Prdx3-deficient MEFs and N-Mid49–PINK1111−581–overexpressing Prdx3-deficient MEFs were treated with CCCP for 24 h. The number of depolarized mitochondria was increased in Prdx3-deficient MEFs, indicating defective mitophagy. However, mitochondrial depolarization following CCCP treatment was reduced in N-Mid49–PINK1111−581–overexpressing Prdx3-deficient MEFs, demonstrating the clearance of damaged mitochondria via N-Mid49–PINK1111−581–mediated mitophagy (Supplementary Fig. 9G). Thus, Prdx3 maintains PINK1 stability in damaged mitochondria by acting as a chaperone and binding with the protein, thus protecting it from cleavage by activated Oma1.
3.7. Prdx3 regulates MQC processes in cardiomyocytes
Further, to determine whether the suppression of mitophagy in the Prdx3-deficient heart has a functional link with PINK1–Parkin-mediated mitophagy, we examined the localization of Parkin in damaged mitochondria in wild-type and Prdx3-deficient cardiomyocytes after CCCP treatment. We found that endogenous Parkin localized to damaged mitochondria in wild-type cardiomyocytes, but not in Prdx3-deficient cardiomyocytes (Fig. 8A, Supplementary Fig. 10), indicating that Prdx3 regulates PINK1–Parkin-mediated mitophagy in cardiomyocytes. Next, we examined whether either Prdx3 or Prdx3-DN adenovirus can rescue mitophagy under CCCP-induced mitophagy. Prdx3-deficient cardiomyocytes were infected with adenovirus carrying Prdx3 or Prdx3-DN following CCCP treatment. Prdx3-DN is defective for ROS removal but functional for PINK1-mediated mitophagy. Prdx3 or Prdx3-DN expression restored mitophagy in Prdx3-deficient cardiomyocytes to a level similar to that in wild-type cardiomyocytes (Fig. 8B and C). Additionally, the number of damaged mitochondria in Prdx3-deficient cardiomyocytes was decreased by adenoviral Prdx3 or Prdx3-DN transfection, suggesting that increased mitochondrial damage upon Prdx3 deficiency can be reduced by functional mitophagy activated by Prdx3-DN with defective ROS removal (Fig. 8D and E). Taken together, Prdx3 acts as a key regulator via the interplay between the prevention of mitochondrial damage and the elimination of damaged mitochondria, and it is essential for cardiac function.
Fig. 8.

Peroxiredoxin 3 (Prdx3) is a key modulator of the mitochondrial quality control (MQC) process in cardiomyocytes.
(A) Tom20 (red) and Parkin (green) immunostaining of cardiomyocytes from Prdx3 wild-type (Prdx3 WT) and Prdx3-deficient (Prdx3 KO) mice treated with DMSO or 10 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) for 4 h. Scale bar, 2 (boxed areas) or 10 μm. (B and C) Representative images (B) and quantification (C) of mitophagy in cardiomyocytes from Prdx3 WT and Prdx3 KO mitochondria-targeted Keima (mt-Keima) mice. Cardiomyocytes were infected with Prdx3 adenovirus or Prdx3 dominant-negative (DN) adenovirus for 24 h and 10 μM CCCP for 4 h. Scale bar, 10 μm. (D and E) Representative electron micrographs (D) and quantification (E) of damaged mitochondria (red arrows) in cardiomyocytes from Prdx3 WT and Prdx3 KO mt-Keima mice. Cardiomyocytes were infected with Prdx3 or Prdx3 DN adenovirus for 24 h. Arrowheads indicate myofilaments, and open arrowheads indicate the Z-line of the sarcomere. Scale bar, 1 (boxed areas) or 2 μm *P < 0.05, **P < 0.01 and ***P < 0.001. . (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
4. Discussion
Heart failure includes heart failure with preserved ejection fraction (HFpEF) and heart failure with reduced ejection fraction in half-and-half ratio. Patients with HFpEF have commonly exercise intolerance, mainly due to mitochondrial dysfunction [31]. For this reason, mitochondrial abnormalities affect skeletal muscle, respiratory muscle and myocardial function. Patients with HFpEF have typical mitochondrial abnormalities in both skeletal muscle and cardiac muscle [32]. Similarly, Prdx3-deficient mice exhibited mitochondrial abnormalities with giant and damaged mitochondria in cardiac muscle, as well as in skeletal muscle. It was also reported that Prdx3 has a crucial role in the contractile function of skeletal muscle by regulating mitochondrial homeostasis [33]. In this study, we proposed a new conceptual model in which Prdx3 maintains MQC through the integrative and comprehensive regulation of mitochondrial damage prevention and clearance by modulating the localization and stability of PINK1, implying the role of Prdx3 as a master regulator of MQC for proper heart function.
It is well known that mitochondrial dysfunction induces LV diastolic dysfunction in the heart [3,4]. Typically, LV diastolic dysfunction is related to abnormal LV relaxation and elevated filling pressure from increased myocardial stiffness. As LV remodeling and cardiac hypertrophy induced by Prdx3 deficiency are attributable to disturbed MQC, Prdx3 deficiency may be directly involved in the induction of diastolic dysfunction in the heart. However, the higher heart rate of mice relative to other experimental models makes it difficult to assess diastolic dysfunction via echocardiography in mice. Therefore, further studies are needed to examine whether Prdx3 deficiency is associated with LV diastolic dysfunction in the heart.
Mitochondrial diseases induced by MQC dysfunction include acute and chronic cardiovascular and neurodegenerative diseases such as cardiac ischemia/reperfusion injury, Parkinson's disease, Alzheimer's disease, and stroke [5]. However, little is known regarding the precise role of MQC in acute and chronic mitochondrial diseases. To date, mouse models for studying MQC-related mitochondrial diseases in vivo have not been reported, and only a few studies on changes of in vivo mitophagy are available. Because Prdx3-deficient mice have damaged mitochondria and defective mitophagy, they can serve as an excellent in vivo model to study the mechanisms of mitophagy by evaluating MQC. Furthermore, the generation of conditional Prdx3-knockout mice featuring the tissue-specific deletion of Prdx3, in which MQC becomes dysfunctional in a tissue-specific manner, will help elucidate the balanced role of MQC in acute and chronic mitochondrial diseases and thereby provide therapeutic strategies for the treatment of acute and chronic mitochondrial diseases.
Basal in vivo mitophagy is not altered by the presence or absence of the key mitophagy regulator PINK1 [18,19], suggesting that mitochondrial homeostasis is maintained by the tight regulation of MQC in vivo. Under stress, PINK1 deficiency results in reduced mitophagy in vivo [20,21]. However, mitophagy-inducing methods are limited by the fact that stress occurs in all organelles instead of specifically in mitochondria. Contrarily, the reduced in vivo mitophagy observed in Prdx3-deficient mice was caused by ROS-induced damaged mitochondria and defective mitophagy. Therefore, Prdx3-deficient mice will be useful for systematically analyzing the mechanism by which MQC dysfunction affects pathophysiology.
There are several possible explanations for our findings of reduced mitophagy upon Prdx3 ablation. First, Prdx3 is a key regulator of PINK1 in damaged mitochondria undergoing mitophagy, enabling us to detect the reduction of mitophagy caused by Prdx3 deficiency. Second, although there are no tools for inducing mitochondria-specific damage in vivo, Prdx3-deficient mice have mitochondria-specific damage, and the effects of mitophagy can be examined in this context. Third, given that in vivo mitophagy is not altered by PINK1 deficiency but is reduced by Prdx3 deficiency, Prdx3 may play a critical role in regulating mitophagy in vivo via an alternative mitophagy pathway, in addition to standard PINK1–Parkin-mediated mitophagy.
In healthy mitochondria, PINK1 is continuously imported to the IMM and matrix and then destabilized by PARL and MPP in the IMM and mitochondrial matrix, respectively. In damaged mitochondria, PINK1 is not imported to the IMM and mitochondrial matrix, and it stably accumulates in the OMM in the absence of proteolytic cleavage by PARL and MPP [15]. However, a recent report illustrated that a Parkinson's disease-related PINK1 mutant can be cleaved by Oma1 in damaged mitochondria [22]. In addition, OMA1 activity is modulated by prohibitin, which functions as a chaperone and regulates PINK stability. Oma1, an AAA protease that regulates mitochondrial dynamics and function, is activated under stress [23]. This raises the possibility that in damaged mitochondria, activated Oma1 can regulate the stability of PINK1 and that a protector that blocks the action of activated Oma1 exists. In normal conditions, Prdxs form a stable structure that associates into donut-shaped ring-like decamers [14]. The N-terminal sequence of Prdxs is essential for maintaining their stability [12]. Under stress, Prdxs act as chaperones. Among PRDXs, human PRDX3 is known to self-associate to form filaments that have chaperone activity under stress [14]. Therefore, Prdx3 has a dual function, acting as an antioxidant peroxidase under normal conditions and as a chaperone under stress.
Disruption of mitochondrial homeostasis has been implicated in the development of cardiovascular diseases. Prdx3 is a scavenger of hydrogen peroxide that effectively removes mitochondrial ROS, and it is regulated via its deacetylation by the mitochondrial deacetylase SIRT3 [34]. Mitophagy removes damaged mitochondria for the maintenance of mitochondrial homeostasis, and mitophagy induced by the polyamine drug spermidine attenuates cardiovascular conditions such as aortic mitochondrial dysfunction and atherosclerosis [35]. Dysregulation of the prevention of mitochondrial damage through the removal of ROS or the clearance of damaged mitochondria via mitophagy in vivo has been reported to lead to heart failure [36]. Whether dysfunction in the interplay between mitochondrial ROS removal and mitophagy can influence the development of heart failure has not been studied. Our finding that Prdx3 regulates both ROS removal and mitophagy in mitochondria highlights the importance of the maintenance of mitochondrial homeostasis for proper heart function. Further studies on the physiological phenotypes in developing and adult Prdx3-deficient mice and mitochondria-targeted agents to mimic the function of Prdx3 will provide new insights into developing an effective therapeutic strategy to prevent and treat mitochondria-related cardiovascular diseases such as HFpEF.
Author contributions
Conceptualization, S.K.S. and G.T.O.; Methodology, S.K.S., E.J.S., S.S., and G.T.O.; Formal analysis, S.K.S., E.J.S., S.S., J.C., J.Y., E.Y. C., and G.T.O.; Investigation, S.K.S., E.J.S., S.S., Y.Y.K., J.H.U., F.J.Y., D.S.L., S.J., M.N.L., J.J., H.Y.K., T.K.K., S.K., S.H.M., J.C., J.Y., J.H., E.Y. C., and J.Y.; Writing – Original Draft, S.K.S., E.J.S., S.S., and G.T.O.; Writing –Review and Editing, G.T.O., S.G.R., and S.B.L.; Funding Acquisition, G.T.O.; Supervision, G.T.O.
Declaration of competing interest
We declare that there are no conflicts of interest.
Acknowledgments
The authors thank Drs. David C. Chan, Ki-Hwan Han, and Hyun Ae Woo for their critical reading of this manuscript. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (No. 2020RIA3B2079811), (No. 2021M3E5E7023628) and (No. 2021R1I1A1A01049288).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2022.102275.
Contributor Information
Seong Keun Sonn, Email: ssk999@ewha.ac.kr.
Goo Taeg Oh, Email: gootaeg@ewha.ac.kr.
Appendix A. Supplementary data
The following is the Supplementary data to this article:
References
- 1.Vásquez-Trincado C., García-Carvajal I., Pennanen C., Parra V., Hill J.A., Rothermel B.A., Lavandero S. Mitochondrial dynamics, mitophagy and cardiovascular disease. J. Physiol. 2016;594(3):509–525. doi: 10.1113/JP271301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar A.A., Kelly D.P., Chirinos J.A. Mitochondrial dysfunction in heart failure with preserved ejection fraction. Circulation. 2019;139(11):1435–1450. doi: 10.1161/CIRCULATIONAHA.118.036259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heinonen I., Sorop O., van Dalen B.M., Wüst R.C.I., van de Wouw J., de Beer V.J., Octavia Y., van Duin R.W.B., Hoogstrate Y., Blonden L., Alkio M., Anttila K., Stubbs A., van der Velden J., Merkus D., Duncker D.J. Cellular, mitochondrial and molecular alterations associate with early left ventricular diastolic dysfunction in a porcine model of diabetic metabolic derangement. Sci. Rep. 2020;10:13173. doi: 10.1038/s41598-020-68637-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koncsos G., Varga Z.V., Baranyai T., Boengler K., Rohrbach S., Li L., Schlüter K.D., Schreckenberg R., Radovits T., Oláh A., Mátyás C., Lux Á., Al-Khrasani M., Komlódi T., Bukosza N., Máthé D., Deres L., Barteková M., Rajtík T., Adameová A., Szigeti K., Hamar P., Helyes Z., Tretter L., Pacher P., Merkely B., Giricz Z., Schulz R., Ferdinandy P. Diastolic dysfunction in prediabetic male rats: role of mitochondrial oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2016;311(4):H927–H943. doi: 10.1152/ajpheart.00049.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Craven L., Alston C.L., Taylor R.W., Turnbull D.M. Recent advances in mitochondrial disease. Annu. Rev. Genom. Hum. Genet. 2017;18:257–275. doi: 10.1146/annurev-genom-091416-035426. [DOI] [PubMed] [Google Scholar]
- 6.Shadel G.S., Horvath T.L. Mitochondrial ROS signaling in organismal homeostasis. Cell. 2015;163(3):560–569. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chan D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 2020;15:235–259. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- 8.Zorov D.B., Juhaszova M., Sollott S.J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol. Rev. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chang T.S., Cho C.S., Park S., Yu S., Kang S.W., Rhee S.G. Peroxiredoxin III, a mitochondrion-specific peroxidase, regulates apoptotic signaling by mitochondria. J. Biol. Chem. 2004;279(40):41975–41984. doi: 10.1074/jbc.M407707200. [DOI] [PubMed] [Google Scholar]
- 10.Shirihai O.S., Song M., Dorn G.W. How mitochondrial dynamism orchestrates mitophagy. Circ. Res. 2015;116(11):1835–1849. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song M., Franco A., Fleischer J.A., Zhang L., Dorn G.W. Abrogating mitochondrial dynamics in mouse hearts accelerates mitochondrial senescence. Cell Metabol. 2017;26(6):872–883. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Teixeira F., Tse E., Castro H., Makepeace K.A., Meinen B.A., Borchers C.H., Poole L.B., Bardwell J.C., Tomás A.M., Southworth D.R., Jakob U. Chaperone activation and client binding of a 2-cysteine peroxiredoxin. Nat. Commun. 2019;10:659. doi: 10.1038/s41467-019-08565-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang H.H., Lee K.O., Chi Y.H., Jung B.G., Park S.K., Park J.H., Lee J.R., Lee S.S., Moon J.C., Yun J.W., Choi Y.O., Kim W.Y., Kang J.S., Cheong G.W., Yun D.J., Rhee S.G., Cho M.J., Lee S.Y. Two enzymes in one; two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117(5):625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Yewdall N.A., Venugopal H., Desfosses A., Abrishami V., Yosaatmadja Y., Hampton M.B., Gerrard J.A., Goldstone D.C., Mitra A.K., Radjainia M. Structures of human peroxiredoxin 3 suggest self-chaperoning assembly that maintains catalytic state. Structure. 2016;24(7):1120–1129. doi: 10.1016/j.str.2016.04.013. [DOI] [PubMed] [Google Scholar]
- 15.Okatsu K., Kimura M., Oka T., Tanaka K., Matsuda N. Unconventional PINK1 localization to the outer membrane of depolarized mitochondria drives Parkin recruitment. J. Cell Sci. 2015;128(5):964–978. doi: 10.1242/jcs.161000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kitada T., Asakawa S., Hattori N., Matsumine H., Yamamura Y., Minoshima S., Yokochi M., Mizuno Y., Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 17.Kitada T., Tong Y., Gautier C.A., Shen J. Absence of nigral degeneration in aged parkin/DJ-1/PINK1 triple knockout mice. J. Neurochem. 2009;111(3):696–702. doi: 10.1111/j.1471-4159.2009.06350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee J.J., Sanchez-Martinez A., Martinez Zarate A., Benincá C., Mayor U., Clague M.J., Whitworth A.J. Basal mitophagy is widespread in Drosophila but minimally affected by loss of Pink1 or parkin. J. Cell Biol. 2018;217(5):1613–1622. doi: 10.1083/jcb.201801044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.McWilliams T.G., Prescott A.R., Montava-Garriga L., Ball G., Singh F., Barini E., Muqit M.M.K., Brooks S.P., Ganley I.G. Basal mitophagy occurs independently of PINK1 in mouse tissues of high metabolic demand. Cell Metabol. 2018;27(2):439–449. doi: 10.1016/j.cmet.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sliter D.A., Martinez J., Hao L., Chen X., Sun N., Fischer T.D., Burman J.L., Y L.i., Zhang Z., Narendra D.P., Cai H., Borsche M., Klein C., Youle R.J. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. doi: 10.1038/s41586-018-0448-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim Y.Y., Um J.H., Yoon J.H., Kim H., Lee D.Y., Lee Y.J., Jee H.J., Kim Y.M., Jang J.S., Jang Y.G., Chung J., Park H.T., Finkel T., Koh H., Yun J. Assessment of mitophagy in mt-Keima Drosophila revealed an essential role of the PINK1-Parkin pathway in mitophagy induction in vivo. Faseb. J. 2019;33(9):9742–9751. doi: 10.1096/fj.201900073R. [DOI] [PubMed] [Google Scholar]
- 22.Sekine S., Wang C., Sideris D.P., Bunker E., Zhang Z., Youle R.J. Reciprocal roles of Tom7 and OMA1 during mitochondrial import and activation of PINK1. Mol. Cell. 2019;73(5):1028–1043. doi: 10.1016/j.molcel.2019.01.002. [DOI] [PubMed] [Google Scholar]
- 23.Ehses S., Raschke I., Mancuso G., Bernacchia A., Geimer S., Tondera D., Martinou J.C., Westermann B., Rugarli E.I., Langer T. Regulation of OPA1 processing and mitochondrial fusion by m-AAA protease isoenzymes and OMA1. J. Cell Biol. 2009;187(7):1023–1036. doi: 10.1083/jcb.200906084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun N., Yun J., Liu J., Malide D., Liu C., Rovira I.I., Holmström K.M., Fergusson M.M., Yoo Y.H., Combs C.A., Finkel T. Measuring in vivo mitophagy. Mol. Cell. 2015;60(4):685–696. doi: 10.1016/j.molcel.2015.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tachibana S., Chen C., Zhang O.R., Schurr S.V., Hill C., Li R., Manso A.M., Zhang J., Andreyev A., Murphy A.N., Ross R.S., Cho Y. Analyzing oxygen consumption rate in primary cultured mouse neonatal cardiomyocytes using an extracellular flux analyzer. JoVE. 2019;13(144):59052. doi: 10.3791/59052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manevski M., Muthumalage T., Devadoss D., Sundar I.K., Wang Q., Singh K.P., Unwalla H.J., Chand H.S., Rahman I. Cellular stress responses and dysfunctional Mitochondrial–cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020;33:101443. doi: 10.1016/j.redox.2020.101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hori M., Nishida K. Oxidative stress and left ventricular remodelling after myocardial infarction. Cardiovasc. Res. 2009;81(3):457–464. doi: 10.1093/cvr/cvn335. [DOI] [PubMed] [Google Scholar]
- 28.Ismail T., Kim Y., Lee H., Lee D.S., Lee H.S. Interplay between mitochondrial peroxiredoxins and ros in cancer development and progression. Int. J. Mol. Sci. 2019;20:4407. doi: 10.3390/ijms20184407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo X., Aviles G., Liu Y., Tian R., Unger B.A., Lin Y.T., Wiita A.P., Xu K., Correia M.A., Kampmann M. Mitochondrial stress is relayed to the cytosol by an OMA1-DELE1-HRI pathway. Nature. 2020;579:427–432. doi: 10.1038/s41586-020-2078-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kalia R., Wang R.Y., Yusuf A., Thomas P.V., Agard D.A., Shaw J.M., Frost A. Structural basis of mitochondrial receptor binding and constriction by DRP1. Nature. 2018;558:401–405. doi: 10.1038/s41586-018-0211-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haykowsky M.J., Kitzman D.W. Exercise physiology in heart failure and preserved ejection fraction, Heart Fail. Clin. 2014;10(3):445–452. doi: 10.1016/j.hfc.2014.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maurer M.S., Schulze P.C. Exercise intolerance in heart failure with preserved ejection fraction: shifting focus from the heart to peripheral skeletal muscle. J. Am. Coll. Cardiol. 2012;60(2):129–131. doi: 10.1016/j.jacc.2012.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee K.P., Shin Y.J., Cho S.C., Lee S.M., Bahn Y.J., Kim J.Y., Kwon E.S., Jeong D.Y., Park S.C., Rhee S.G., Woo H.A., Kwon K.S. Peroxiredoxin 3 has a crucial role in the contractile function of skeletal muscle by regulating mitochondrial homeostasis. Free Radic. Biol. Med. 2014;77:298–306. doi: 10.1016/j.freeradbiomed.2014.09.010. [DOI] [PubMed] [Google Scholar]
- 34.Wang Z., Sun R., Wang G., Chen Z., Li Y., Zhao Y., Liu D., Zhao H., Zhang F., Yao J., Tian X. Thiol peroxidases ameliorate SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 2020;28:101343. doi: 10.1016/j.redox.2019.101343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Madeo F., Eisenberg T., Pietrocola F., Kroemer G. Spermidine in health and disease. Science. 2018;359 doi: 10.1126/science.aan2788. [DOI] [PubMed] [Google Scholar]
- 36.Shao D., Kolwicz S.C., Wang P., Roe N.D., Villet O., Nishi K., Hsu Y.A., Flint G.V., Caudal A., Wang W., Regnier M., Tian R. Increasing fatty acid oxidation prevents high-fat diet-induced cardiomyopathy through regulating parkin-mediated Mitophagy. Circulation. 2020;142:983–997. doi: 10.1161/CIRCULATIONAHA.119.043319. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.