

Abstract
Chimeric antigen receptor natural killer (CAR-NK) cells have remarkable cytotoxicity against hematologic malignancies; however, they may also attack normal cells sharing the target antigen. Since human leukocyte antigen DR (HLA-DR) is reportedly lost or downregulated in a substantial proportion of hematologic malignancies, presumably a mechanism to escape immune surveillance, we hypothesize that the anti-cancer specificity of CAR-NK cells can be enhanced by activating them against cancer antigens while inhibiting them against HLA-DR. Here, we report the development of an anti-HLA-DR inhibitory CAR (iCAR) that can effectively suppress NK cell activation against HLA-DR-expressing cells. We show that dual CAR-NK cells, which co-express the anti-CD19 or CD33 activating CAR and the anti-HLA-DR iCAR, can preferentially target HLA-DR-negative cells over HLA-DR-positive cells in vitro. We find that the HLA-DR-mediated inhibition is positively correlated with both iCAR and HLA-DR densities. We also find that HLA-DR-expressing surrounding cells do not affect the target selectivity of dual CAR-NK cells. Finally, we confirm that HLA-DR-positive cells are resistant to dual CAR-NK cell-mediated killing in a xenograft mouse model. Our approach holds great promise for enhancing CAR-NK and CAR-T cell specificity against malignancies with HLA-DR loss.
Keywords: chimeric antigen receptor, natural killer cell engineering, on-target off-tumor toxicity, loss of HLA-DR expression, hematologic malignancies, cancer immunotherapy, CD19, CD33
Graphical abstract
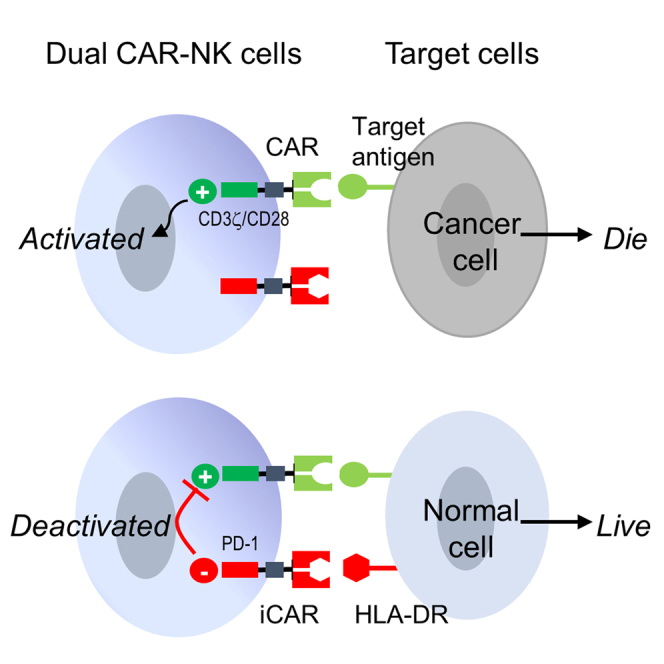
CAR-engineered NK cells have a remarkable ability to kill cancer cells, but they may also destroy normal cells sharing the target antigen. To enhance their anti-cancer specificity, Xie and colleagues developed an inhibitory receptor, which can serve as an immunological brake to stop CAR-NK cells from attacking normal cells.
Introduction
Genetic engineering of T cells and natural killer (NK) cells with chimeric antigen receptors (CAR) has emerged as a powerful new therapeutic approach for cancer and, in particular, hematologic malignancies.1,2 However, most target antigens are not cancer specific, but instead are also expressed on normal cells (albeit sometimes at lower levels). As such, CAR-T and CAR-NK cells can also attack normal cells—a severe adverse effect known as on-target off-tumor toxicity.3,4 For example, anti-CD19 CAR-T cell therapy against B cell leukemia and lymphoma leads to unwanted depletion of normal B cells with prolonged B cell aplasia. Likewise, anti-CD33 CAR-T cells, which have shown potent cytotoxicity to acute myeloid leukemia (AML), can attack healthy myeloid cells in the blood and bone marrow, inducing severe cytopenias.5,6 Clearly, there is a critical need to enhance the specificity of CAR-T and CAR-NK cells against cancer.
While cancer-specific surface antigens are rarely available, cancer cells often lose or downregulate the expression of human leukocyte antigens (HLA) as a mechanism of escape from immune surveillance.7, 8, 9, 10 For example, HLA-DR, the most abundant HLA class II molecule, is reportedly absent or downregulated in up to 33% of all cases of diffuse large B cell lymphoma (DLBCL)7,11,12 and over 50% of those arising in immune-privileged sites, such as the central nervous system and the testis.13 HLA-DR loss is also found in other types of hematologic malignancies, including approximately 15%–17% of AML,14,15 40% of classical Hodgkin lymphoma,12 23% of chronic myelomonocytic leukemia (CMML),16 and some cases of chronic myeloid leukemia (CML).17 As an immune escape mechanism, HLA-DR loss is frequently correlated with lower T cell infiltration and reduced patient survival.7,15,17,18 Importantly, loss of HLA antigen expression is also recognized as a resistance mechanism associated with post-transplant relapses after allogeneic hematopoietic stem cell transplantation, a curative therapy for all types of hematologic malignancies. Two recent studies reported that up to 50% of AML patients with post-transplant relapse are associated with complete or partial loss of HLA-DR and other HLA class II molecules.19,20 Thus, a substantial proportion of hematologic malignancies are HLA-DR deficient at diagnosis and/or relapse.
Based on the above, we speculate that the anti-cancer specificity of CAR-NK cells can be enhanced by activating them in response to cancer antigens while inhibiting them in response to HLA-DR (Figure 1). Previously, Fedorov et al. reported that an inhibitory CAR (iCAR), which contained an intracellular domain derived from immunosuppressive receptors PD-1 or CTLA-4, could inhibit T cell activation in response to the target antigen.21 Inspired by their work, we investigated whether an iCAR could be designed to inhibit unwanted CAR-NK cell activation against HLA-DR+ cells.
Figure 1.

Schematic illustration of an anti-HLA-DR iCAR to reduce on-target off-tumor toxicity
The dual CAR-NK cell expresses an anti-CD19 CAR and an anti-HLA-DR iCAR. The cancer cell, which expresses CD19 but not HLA-DR, would be killed. The normal cell, which expresses both CD19 and HLA-DR, would inhibit CAR-NK cell-mediated cytotoxicity via engaging the anti-HLA-DR iCAR. The scheme is also applicable if CD33 replaces CD19.
Here, we report a PD-1-based anti-HLA-DR iCAR that can effectively inhibit NK cells in response to HLA-DR expression on target cells. We show that dual CAR-NK cells, which co-express the anti-HLA-DR iCAR with a CD28/CD3ζ-based anti-CD19 CAR, can preferentially target CD19+HLA-DRneg cells over CD19+HLA-DR+ cells. We find that the iCAR-mediated inhibition is positively correlated with the densities of both the iCAR and HLA-DR. We also find that HLA-DR-expressing surrounding cells do not affect the target selectivity of dual CAR-NK cells. Furthermore, we confirm that HLA-DR+ cells are resistant to dual CAR-NK cell-mediated killing in vivo using a xenograft mouse model. Finally, we show that the anti-HLA-DR iCAR is also compatible with the anti-CD33 CAR, enabling NK cells to preferentially target HLA-DRneg AML cells. Our study lays a solid foundation for the future development of safer CAR-NK cell therapy against malignancies with HLA-DR loss.
Results
Generation and characterization of single and dual CAR-NK cells
We first constructed an anti-HLA-DR iCAR to pair with a conventional anti-CD19 CAR (Figure 2A). The anti-CD19 CAR consists of an N-terminal HA tag (for detecting CAR expression), a single-chain variable fragment (scFv) derived from the anti-CD19 antibody clone FMC63, a CD8α hinge domain, a CD28 transmembrane domain, and CD28/CD3ζ intracellular domains.22 The anti-HLA-DR iCAR consists of an N-terminal FLAG tag (for detecting iCAR expression), an extracellular scFv derived from the humanized anti-HLA-DR antibody 1D09C323 and PD-1 hinge, transmembrane, and intracellular domains. The CAR and iCAR DNAs were cloned into the pFUW lentiviral vector,24 and lentiviral particles were generated using HEK293T cells.
Figure 2.

Engineering NK-92MI cells to express an anti-CD19 CAR with or without an anti-HLA-DR iCAR
(A) Schematic design of the anti-CD19 CAR and the anti-HLA-DR iCAR. (B) Flow cytometric analysis of untransduced and transduced NK-92MI cells. The CD19 CAR and the HLA-DR iCAR were stained with a PE-labeled anti-HA tag antibody and an APC-labeled anti-FLAG tag antibody, respectively. Data are representative of three independent experiments.
Next, we engineered NK-92MI cells—a human NK cell line—to express the anti-CD19 CAR with or without the anti-HLA-DR iCAR by lentiviral transduction. NK-92MI is an IL-2-expressing derivative of NK-92, both of which have been broadly used in the development of CAR-NK and other cellular therapeutics.25, 26, 27 In our study, NK-92MI cells were first transduced to generate anti-CD19 CAR-NK cells, hereafter referred to as “single CAR-NK cells.” After enrichment by fluorescence-activated cell sorting (FACS), single CAR-NK cells were transduced again to express the anti-HLA-DR iCAR. The cells expressing both CAR and iCAR were purified by FACS, referred to as “dual CAR-NK cells.” Flow cytometric analysis confirmed that both single and dual CAR-NK cells had a purity greater than 97% (Figure 2B).
To ensure that the single and dual CAR-NK cells generated above were directly comparable, we further characterized their expression levels of CARs, antigens, and phenotype markers by surface staining and flow cytometry. Our observations were as follows: (1) single and dual CAR-NK cells expressed the anti-CD19 CAR at the same level, suggesting that they would have a similar activation potential against CD19; (2) neither of them expressed CD19 or HLA-DR, suggesting that they would not undergo anti-CD19 CAR-mediated fratricide or anti-HLA-DR iCAR-mediated self-inhibition; and (3) they had the same expression levels of the activation marker CD56 and the inhibitory receptors PD-1 and LAG-3 as unmodified NK-92MI cells, thus confirming a lack of phenotypic changes induced by CAR or iCAR expression (Figure S1). These results showed that single and dual CAR-NK cells were essentially identical except for the expression of the anti-HLA-DR iCAR.
Verification of CD19 and HLA-DR expression on different target cells
To examine the target specificity of single and dual CAR-NK cells, we selected a panel of six cell lines as target cells. These include K562 and its two derivatives: K562 is a CML cell line that is negative for both CD19 and HLA-DR;28 K562-CD19 is a modified cell line expressing CD19 but not HLA-DR;29 K562-CD19-HLA-DR is a new cell line we generated that expresses both CD19 and HLA-DR. The other three cell lines are KOPN1, Nalm6, and Raji, which are positive for CD19 and HLA-DR at different densities. We verified the expression (or lack thereof) of CD19 and HLA-DR on these cells by staining with PE-conjugated antibodies followed by flow cytometry (Figure 3).
Figure 3.

Verification of the expression of CD19 and HLA-DR on the surface of target cells
Flow cytometric analysis of CD19 (A) and HLA-DR (B) on the cell surface of six cell lines. Cells were stained with PE-conjugated anti-CD19 and anti-HLA-DR antibodies, respectively. Isotype antibodies were used as negative controls. Images are representative of three independent experiments with similar results.
Dual CAR-NK cells exhibit reduced IFN-γ production, CD69 expression, degranulation, and cytotoxicity against HLA-DR+ cells in vitro
Next, we compared and contrasted the activation levels of single and dual CAR-NK cells against different target cells. We first assayed the production of interferon-γ (IFN-γ), a cytokine indicator of NK cell activation and cytotoxicity. To this end, single and dual CAR-NK cells were incubated with each of the six target cells at a 1:1 effector-to-target (E:T) ratio for 4 h, and the concentration of IFN-γ in the cell culture supernatant was measured using an enzyme-linked immunosorbent assay (ELISA). We found that both single and dual CAR-NK cells had enhanced IFN-γ production after incubation with K562-CD19 cells (CD19+HLA-DRneg). Importantly, there was no significant difference between the amounts of IFN-γ they produced. Against K562-CD19-HLA-DR cells, however, dual CAR-NK cells had an approximately 70% reduction in IFN-γ production compared with single CAR-NK cells. A similar reduction was observed when dual CAR-NK cells targeted other HLA-DR+ cells, including KOPN1, Nalm6, and Raji (Figure 4A). These results suggest that dual CAR-NK cells were inhibited by target cells expressing HLA-DR.
Figure 4.

Dual CAR-NK cells preferentially recognize and kill HLA-DRneg cells over HLA-DR+ cells in vitro
(A) Comparison of IFN-γ production by single and dual CAR-NK cells in response to different target cells. Single and dual CAR-NK cells were incubated with each of the six target cells (K562, K562-CD19, K562-CD19-HLA-DR, KOPN1, Nalm6, and Raji) at an E:T ratio of 1:1 for 4 h at 37°C. The cell culture supernatant was collected, and the concentration of IFN-γ was measured by ELISA. Data are shown as mean ± SEM of triplicates. (B) Comparison of CD107a expression by single and dual CAR-NK cells in response to different target cells. Single and dual CAR-NK cells were incubated with each of the six target cells for 1 h at 37°C, in the presence of a PE-conjugated anti-CD107a antibody. Cells were then treated with monensin (GolgiStop), incubated for 4 h, stained with an anti-CD56 antibody, and analyzed by flow cytometry. The percentage of CD107a+ cells was determined, which indicates the level of degranulation. Unmodified NK-92MI cells were used as a negative control. (C) Comparison of the cytotoxicity of single and dual CAR-NK cells against different target cells. The percentage of cytotoxicity was measured by LDH release. Unmodified NK-92MI cells were used as the negative control. Data are shown as mean ± SEM of three independent experiments. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05; n.s., not significant.
We also monitored CD69 upregulation, an early marker of NK cell activation. We observed that single and dual CAR-NK cells had similar percentages of CD69+ population after incubation with K562-CD19 cells (20.7% and 21.3%, respectively). However, dual CAR-NK cells had a significantly lower CD69+ population than single CAR-NK cells after incubation with KOPN1 cells (3.77% versus 14.8%) or Nalm6 cells (4.74% versus 15.5%) (Figure S2). The results were thus consistent with those of the IFN-γ production assay.
To further assess NK cell activation, we performed a CD107a degranulation assay.30 For this purpose, effector cells (NK cells, single CAR-NK cells, and dual CAR-NK cells) were incubated with different target cells at a 1:1 E:T ratio for 4 h. Cells were then stained for CD56 (a marker for NK cells) and CD107a (a marker for degranulation) and subjected to flow cytometric analysis. We demonstrated the following: (1) against K562 cells (negative control), neither single nor dual CAR-NK cells developed a CD107a+ population, suggesting no or little degranulation; (2) against K562-CD19 cells, both single and dual CAR-NK cells developed a CD107a+ population (50.5% and 53.7%, respectively), suggesting a similar level of degranulation; (3) against any of the four HLA-DR+ target cells (K562-CD19-HLA-DR, KOPN1, Nalm6, and Raji), single CAR-NK cells consistently had a 2- to 3-fold higher percentage of CD107a+ population than dual CAR-NK cells had, suggesting that the latter were inhibited by HLA-DR-expressing targets (Figure 4B).
Finally, we examined the cytotoxicity of single and dual CAR-NK cells against HLA-DR+ and HLA-DRneg target cells. NK, single CAR-NK, and dual CAR-NK cells were cocultured with different target cells at three E:T ratios (0.2:1, 1:1, and 5:1). After a 4-h incubation, the cytotoxicity was determined using a lactate dehydrogenase (LDH) release assay. The results showed that single and dual CAR-NK cells had similar killing ability against K562-CD19 cells. However, dual CAR-NK cells were significantly less cytotoxic than single CAR-NK cells in targeting HLA-DR+ cells, including K562-CD19-HLA-DR, KOPN1, Nalm6, and Raji (Figure 4C). We also performed a flow cytometry-based cytotoxicity assay by determining the percentage of viable target cells after coculture.31 The results were consistent with the above, confirming that HLA-DR+ target cells were resistant to dual CAR-NK cell-mediated cytotoxicity (Figure S3).
Collectively, our data strongly suggest that dual CAR-NK cells preferentially recognize and kill HLA-DRneg cells over HLA-DR+ cells.
The inhibition of dual CAR-NK cells is positively correlated with both HLA-DR and iCAR densities
Our results showed that dual CAR-NK cells were inhibited by HLA-DR+ cells but not by HLA-DRneg cells. We hypothesized that the level of inhibition would be positively correlated with the densities of both HLA-DR on target cells and iCAR on NK cells. To test the effect of HLA-DR, we generated recombinant scFv (the same clone as the anti-HLA-DR iCAR) in E. coli followed by in vitro refolding and size-exclusion chromatography (Figure S4). We then used this scFv at different concentrations (0–20 nM) to block HLA-DR on the surface of K562-CD19-HLA-DR cells. This approach ensured that the only changing variable of target cells was the level of exposed HLA-DR, which was not attainable by directly comparing different cell lines as they could differ in other aspects. Next, we incubated single and dual CAR-NK cells with these target cells for 4 h and assessed the activation of CAR-NK cells by IFN-γ production. We found that the activation level of dual CAR-NK cells gradually increased with increasing anti-HLA-DR scFv concentrations and reached that of single CAR-NK cells when scFv was used at 20 nM, the highest concentration tested (Figure 5A). These results suggest that the anti-HLA-DR iCAR-mediated inhibition is positively correlated with the density of HLA-DR on the target cell surface.
Figure 5.

The level of iCAR-mediated inhibition is dependent on the availability of HLA-DR on target cells and iCAR on effector cells
(A) The HLA-DR antigens on K562-CD19-HLA-DR cells were blocked with different concentrations of anti-HLA-DR scFv. These cells were then cocultured with NK, single CAR-NK, and dual CAR-NK cells, respectively. After 4 h, the IFN-γ level in the coculture supernatant was assessed by ELISA. Data are shown as mean ± SEM of two independent experiments. (B) Flow cytometric analysis of single CAR-NK cells and three dual CAR-NK cell populations expressing HLA-DR iCAR at different levels. Cells were stained with a PE-labeled anti-FMC63 scFv antibody (for CD19 CAR) and a PE-labeled anti-FLAG tag antibody (for HLA-DR iCAR) at saturating concentrations, respectively. Untransduced NK-92MI cells were used as the negative control. (C) Comparison of IFN-γ production by single and the three dual CAR-NK cells against K562-CD19, KOPN1, or Nalm6 cells. Cells were incubated at a 1:1 E:T ratio for 4 h. The concentrations of IFN-γ were measured by ELISA. Data are shown as mean ± SEM of two independent experiments. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01, ∗p < 0.05; n.s., not significant.
To assess the effect of the iCAR density, we sorted dual CAR-NK cells into three populations that expressed iCAR at high, intermediate, and low densities, respectively. We verified them by staining with two separate antibodies: (1) a PE-conjugated anti-FMC63 scFv antibody that recognizes the anti-CD19 CAR, and (2) a PE-conjugated anti-FLAG antibody that recognizes the anti-HLA-DR iCAR via its N-terminal FLAG tag. Flow cytometric analysis confirmed that they expressed the iCAR at varying levels, while the CAR was the same level as on single CAR-NK cells (Figure 5B). Since both antibodies were used at saturating concentrations (Figures S5A and S5B), it was reasonable to assume that the mean fluorescence intensities of stained cells were directly proportional to the level of CAR or iCAR. Based on this, we estimated that the iCAR-to-CAR ratios were 0.25:1, 1:1, and 4:1 for the iCARlow, iCARint, and iCARhigh populations, respectively (Figure S5C). Next, we compared the activation levels of these three dual CAR-NK cells as well as the single CAR-NK cells against different target cells. IFN-γ secretion assays showed that they performed equally well against HLA-DRneg target cells. However, against HLA-DR+ target cells, the activation level decreased in the following order: single CAR-NK (i.e., iCARneg) > dual CAR-NK (iCARlow) > dual CAR-NK (iCARint) > dual CAR-NK (iCARhigh) (Figure 5C). These results suggest that the anti-HLA-DR iCAR-mediated inhibition is positively correlated with the level of iCAR (or the iCAR/CAR ratios) on NK cells.
HLA-DR+ surrounding cells do not affect the target selectivity of dual CAR-NK cells
We showed that dual CAR-NK cells were active toward CD19+HLA-DRneg cells but inhibited by CD19+HLA-DR+ cells in vitro. A key question was whether the target selectivity of dual CAR-NK cells would be affected by HLA-DR+ cells in proximity to the CD19+HLA-DRneg target cells (e.g., in the bloodstream or bone marrow microenvironment).
To address this concern, we used the KG-1 cell line as a model of surrounding cells. KG-1 cells expressed HLA-DR but not CD19, as shown by flow cytometry (Figure 6A), and they alone could not activate anti-CD19 CAR-NK cells, as confirmed by cell coculture and IFN-γ production analysis (Figure 6B). We then compared the activation levels of single and dual CAR-NK cells against K562-CD19 cells (CD19+HLA-DRneg) in the presence or absence of KG-1 cells at five different levels, ranging from none to ten times of target cells. With increasing KG-1 cells, we found that both single and dual CAR-NK cells gradually reduced IFN-γ production, probably because they had a decreasing chance of finding K562-CD19 cells in the mixture. Nevertheless, dual CAR-NK cells remained equally reactive as single CAR-NK cells under all conditions, suggesting that they were not inhibited by HLA-DR+ surrounding cells (Figure 6C). Next, we performed the same experiment using K562-CD19-HLA-DR cells (CD19+HLA-DR+) as target cells. We found that dual CAR-NK cells were consistently and considerably less reactive than single CAR-NK cells, suggesting that the iCAR-mediated protective effect was not compromised by surrounding KG-1 cells (Figure 6D).
Figure 6.

The target selectivity of dual CAR-NK cells is not affected by HLA-DR-expressing surrounding cells
(A) Flow cytometric analysis of KG-1 cells stained with PE-conjugated antibodies against CD19 and HLA-DR, separately. Isotype antibodies were used as the negative control. Images are representative of three independent experiments with similar results. (B) ELISA analysis of IFN-γ production by single CAR-NK cells against KG-1 cells or K562-CD19 cells after a 4-h incubation. Data are shown as mean ± SEM of three independent experiments. (C and D) Comparison of the activation levels of single and dual CAR-NK cells against K562-CD19 cells (C) or K562-CD19-HLA-DR cells (D) in the presence or absence of KG-1 cells. CAR-NK cells, target cells, and surrounding KG-1 cells were cocultured at the indicated E:T:S ratios. After a 4-h incubation, cell culture supernatants were collected to assess for IFN-γ secretion by ELISA. Data are shown as mean ± SEM of three independent experiments. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001, ∗∗p < 0.01; n.s., not significant.
To validate the above findings, we also used primary human myeloid cells as surrounding cells. For this purpose, we used immunomagnetic negative selection to isolate human monocytes (CD11b+CD14+), the main type of HLA-DR+ myeloid cells, from human peripheral blood mononuclear cells (PBMCs) (Figure S6A). Like the KG-1 cell line, these primary cells expressed HLA-DR but did not express CD19 or stimulate anti-CD19 CAR-NK cells (Figures S6A and S6B). Next, we used them to substitute for KG-1 cells in the CAR-NK cell activation assays described above. The results again showed that dual CAR-NK cells were equally effective as single CAR-NK cells in targeting CD19+HLA-DRneg cells but significantly less reactive than the latter in targeting CD19+HLA-DR+ cells (Figures S6C and S6D).
Collectively, these results suggest that the target selectivity of dual CAR-NK cells is not affected by HLA-DR+ surrounding cells.
HLA-DR+ cells are resistant to dual CAR-NK cell-mediated killing in vivo
To further evaluate HLA-DR-mediated inhibition of dual CAR-NK cells, we performed an in vivo killing assay using mouse xenograft models. We first compared the ability of single and dual CAR-NK cells to kill K562-CD19-HLA-DR cells (CD19+HLA-DR+, as a model of HLA-DR-expressing cells). Immunocompromised NOD/SCID/gamma (NSG) mice were inoculated with firefly luciferase-expressing K562-CD19-HLA-DR cells. Mice were then treated with either unmodified NK cells (control), single CAR-NK cells, or dual CAR-NK cells (Figure 7A). The growth of K562-CD19-HLA-DR cells in mice was monitored by bioluminescence imaging. On day 3, there was no visible difference among all three groups. On days 10, 17, and 24, we observed a much weaker bioluminescence signal in mice treated with single CAR-NK cells than in those treated with dual CAR-NK cells or NK cells (Figures 7B and 7C). We continued to monitor the survival of mice and found that the group treated with dual CAR-NK cells had a shorter survival time than those treated with single CAR-NK cells (Figure 7D). Similar results were observed in a separate experiment in which we used Raji cells (CD19+HLA-DR+) as target cells (Figure S7). Thus, it is clear that dual CAR-NK cells have diminished cytotoxicity in vivo against target cells expressing both CD19 and HLA-DR.
Figure 7.

HLA-DR+ cells, but not HLA-DRneg cells, are resistant to dual CAR-NK cell-mediated cytotoxicity in vivo
(A) Schematic diagram of the in vivo killing assay. NSG mice were inoculated with 5 × 105 K562-CD19-HLA-DR-Luc or K562-CD19-Luc cells through tail vein injection on day 0 and then treated with 1 × 107 NK cells, single CAR-NK cells, or dual CAR-NK cells through tail vein injection on day 3. Tumor growth was monitored by in vivo bioluminescence imaging on days 3, 10, 17, and 24. (B) Bioluminescence images of K562-CD19-HLA-DR tumor growth in mice treated with NK cells (left), single CAR-NK cells (middle), and dual CAR-NK cells (right). (C) Quantification of bioluminescence in each treatment group (n = 5) on day 24. Data are shown as mean ± SEM. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗∗∗∗p < 0.0001, ∗∗p < 0.01; n.s., not significant. (D) Survival of mice in each treatment group (n = 5) are shown in Kaplan-Meier curves. Statistical significance was calculated by log rank (Mantel-Cox) test. ∗∗p < 0.01; n.s., not significant. (E) Bioluminescence images of K562-CD19 tumor growth in mice treated with NK cells (left), single CAR-NK cells (middle), and dual CAR-NK cells (right). (F) Quantification of bioluminescence in each treatment group (n = 5) on day 24. Data are shown as mean ± SEM. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗p < 0.05; n.s., not significant. (G) Survival of mice in each treatment group (n = 5) are shown in Kaplan-Meier curves. Statistical significance of survival data was calculated by log rank (Mantel-Cox) test. ∗∗p < 0.01; n.s., not significant.
We also compared the in vivo killing ability of single and dual CAR-NK cells against K562-CD19 cells (CD19+HLA-DRneg, as a model of HLA-DR loss in cancer). To this end, we inoculated NSG mice with firefly luciferase-expressing K562-CD19 cells, followed by the same treatment scheme as above (Figure 7A). In this case, we found that dual CAR-NK cells performed similarly to single CAR-NK cells. They both significantly inhibited tumor growth and prolonged mouse survival compared with unmodified NK cells (Figures 7E–7G). Therefore, dual CAR-NK cells are as effective as single CAR-NK cells in killing target cells expressing CD19 but not HLA-DR.
These data were thus consistent with those from in vitro assays (Figure 4), suggesting that CD19+HLA-DR+ double-positive cells have reduced killing by anti-CD19 CAR-NK cells bearing the anti-HLA-DR iCAR.
Anti-CD33 CAR-NK cells bearing the anti-HLA-DR iCAR preferentially target HLA-DRneg AML cells
To examine the generality of the anti-HLA-DR iCAR, we also tested the anti-HLA-DR iCAR in combination with an anti-CD33 CAR. For this purpose, we engineered NK-92MI cells to express a CD28/CD3ζ-based anti-CD33 CAR with or without the anti-HLA-DR iCAR (Figure 8A). We then compared their activation levels against HL-60 (CD33+HLA-DRneg) and KG-1 cells (CD33+HLA-DR+) cells. Both IFN-γ production and CD107a degranulation assays showed that: (1) dual CAR-NK cells were as active as single CAR-NK cells toward HL-60 cells; (2) dual CAR-NK cells were significantly less active than were single CAR-NK cells toward KG-1 cells—the production of IFN-γ was reduced by more than 80%, and the percentage of the CD107a+ population was reduced by approximately 50% (Figures 8B and 8C). Therefore, the anti-HLA-DR iCAR can potentially be used to enhance the anti-leukemia specificity of anti-CD33 CAR-NK cells against HLA-DRneg AML, which has been observed at diagnosis14,15 or relapse after transplantation.19,20
Figure 8.

Anti-CD33 CAR-NK cells bearing the anti-HLA-DR iCAR preferentially target HLA-DRneg AML cells
(A) Flow cytometric analysis of NK cells, and CD33-targeted single and dual CAR-NK cells. Cells were stained for flow cytometry with a PE-labeled anti-HA antibody and an APC-labeled anti-FLAG antibody to assess for CD33 CAR and HLA-DR iCAR expression, respectively. (B) IFN-γ production of CD33-targeted single and dual CAR-NK cells against HL-60 (CD33+HLA-DRneg) and KG-1 (CD33+HLA-DR+) cells. CAR-NK cells were incubated with either HL-60 or KG-1 cells for 4 h. The supernatant was collected to assess for the IFN-γ level by ELISA. (C) CD107a degranulation assays of single and dual CAR-NK cells cocultured with different target cells. CAR-NK cells were incubated with HL-60 or KG-1 for 1 h at 37°C with a PE-conjugated anti-CD107a antibody. Monensin (GolgiStop) was added to the cell culture. After incubation for 4 h, cells were stained with an anti-CD56 antibody. The CD107a+ population in CD56+ cells was determined by flow cytometry. Data are shown as mean ± SEM of two independent experiments. Statistical significance is calculated by unpaired two-tailed Student's t test. ∗∗∗∗p < 0.0001, ∗∗∗p < 0.001; n.s., not significant.
Discussion
On-target off-tumor toxicity presents a significant safety concern for CAR-T and CAR-NK cell therapy because target antigens are rarely cancer specific. Since HLA-DR loss has been well documented in a substantial proportion of hematologic malignancies, we propose to enhance anti-cancer specificity through dual targeting: activating CAR-T and CAR-NK cells in response to cancer antigens while inhibiting them in response to HLA-DR. To this end, we have developed a PD-1-based anti-HLA-DR iCAR that inhibits NK cell activation. We show that anti-CD19 or anti-CD33 CAR-NK cells bearing the anti-HLA-DR iCAR can preferentially target HLA-DRneg cells over HLA-DR+ cells. We also find that the level of CAR-NK cell inhibition is positively correlated with the densities of HLA-DR and iCAR. Moreover, we find that HLA-DR-expressing surrounding cells do not affect the target selectivity of dual CAR-NK cells. Thus, our approach can potentially be used to enhance the specificity of CAR-NK cell therapy against various malignancies with HLA-DR loss.
Fedorov et al. pioneered the development of iCAR as a strategy to enhance the specificity of CAR-T cells.21 They utilized an iCAR with an extracellular domain recognizing the prostate-specific membrane antigen (PSMA) and an intracellular domain derived from immunosuppressive receptors PD-1 or CTLA-4, which they showed could suppress anti-CD19 CAR-T cell activation against CD19+PSMA+ cells. Our study makes two novel contributions to this field: we use iCAR to target HLA-DR, a self-antigen critical to the immune system but frequently lost on malignant cells; and we demonstrate the feasibility of using iCAR to enhance the targeting specificity of CAR-NK cells. In contrast to T cells, allogeneic NK cells have reduced risk of inducing graft-versus-host diseases. There is thus increasing interest in engineering CAR-NK cells as potential off-the-shelf cellular therapeutics.2 The iCAR platform can potentially further enhance the safety of CAR-NK cell therapy. While this study is focused on CAR-NK cell targeting, the anti-HLA-DR iCAR could also be incorporated in CAR-T cells. It should be noted that T cells express HLA-DR upon activation.32,33 Whether that could affect the expression or function of anti-HLA-DR iCAR in T cells warrants further investigation.
In line with our expectation, dual CAR-NK cells preferentially recognize and kill HLA-DRneg cells over HLA-DR+ cells. In comparison with single CAR-NK cells, dual CAR-NK cells were equally reactive against HLA-DRneg cells but were up to 50%–80% less reactive against HLA-DR+ cells in vitro (Figures 4A–4C, 5A, 5C, 6D, 8B, 8C, S2, S3, and S6D). We also confirmed that HLA-DR expression could effectively protect target cells from cytotoxicity mediated by dual CAR-NK cells in vivo (Figures 7 and S7). Nevertheless, there is room for further improvement of both the potency and the selectivity of dual CAR-NK cells. A promising approach is to utilize signaling domains derived from NK cells instead of T cells. NK cell signaling differs significantly from T cell signaling.34 NK cells express a series of germline-encoded activating receptors, such as the NK group 2 member D receptor (NKG2D), the Fc receptor CD16 (FcγRIIIa), and 2B4 (CD244), and these receptors signal through molecules, such as DAP10, FcεRIγ, and CD3ζ.34 Recently, Li et al. reported that NKG2D/2B4/DAP10/CD3ζ-based CARs were more effective than CD28/4-1BB/CD3ζ-based CARs in enabling NK cells to kill ovarian cancer cells.35 NK cells also express strong inhibitory receptors, such as the NK group 2 member A receptor (NKG2A) and killer cell immunoglobulin-like receptors (KIRs).34 Future research should focus on optimizing CAR and iCAR using NK cell-derived signaling domains. Another approach to enhance dual CAR-NK cells is to elevate the expression levels of anti-HLA-DR iCAR, as we observed that the level of inhibition was positively correlated with the iCAR density (Figures 5B and 5C). Currently, the expression of the iCAR used in our construct is controlled by the transcriptional promotor UBC1. The expression density of the iCAR could potentially be increased by using a stronger transcriptional promoter, such as EF1α or MSCV.36,37
A potential limitation of our approach is that cancer cells might reverse HLA-DR downregulation to escape dual CAR-NK cell attack. The loss of HLA-DR expression can be reversible or irreversible depending on the mechanism of loss. HLA-DR loss via genetic deletion is irreversible. For example, HLA-DR loss in DLBCL in immune-privileged sites is mainly due to homozygous deletion of the HLA II region on chromosome 6.13 HLA-DR can also be lost by a combination of hemizygous deletions and stop-codon mutations in the other allele.38 On the other hand, HLA-DR downregulation through transcriptional regulation is reversible. For example, a common mechanism of HLA-DR downregulation is decreased expression of the MHC class II transactivator CIITA,17,19,20,39,40 and it is known that CIITA can be upregulated by IFN-γ.17 However, the expression of HLA-DR would expose cancer cells to host immune surveillance, as demonstrated by increased tumor-infiltrating T cells in HLA-DR+ cases.7 Therefore, it remains to be investigated whether cancer cells can and will upregulate HLA-DR expression to escape from dual CAR-NK cell attack. If that proves to be an issue, a potential solution would be to combine dual CAR-NK cells with another therapy that can target HLA-DR+ cancer cells, e.g., donor lymphocyte infusion41 and TCR-based immunotherapy.42
While this study is focused on targeting HLA-DR loss in hematologic malignancies, the same approach can potentially be used to target HLA class I loss in solid tumors. On-target off-tumor toxicity may prove to be particularly problematic in the treatment of solid tumors because target antigens may be expressed in multiple organs. For example, liver toxicity was observed in a renal carcinoma clinical trial testing CAR-T cells against carbonic anhydrase IX, which is also expressed on the epithelial cells of bile ducts.43,44 Moreover, a colon cancer patient died of respiratory failure after infusion of anti-ERBB2 CAR-T cells, probably due to off-tumor recognition of lung epithelial cells that express ERBB2 at low levels.45 Loss of the expression of HLA class I alleles (including HLA-A, HLA-B, and HLA-C) has been well documented in a variety of solid tumors,46 including up to 40%–50% of breast,47,48 prostate,49 and lung cancers,50, 51, 52 and about 15%–25% of colorectal53 and bladder cancers.54 Notably, three very recent studies reported that iCARs specific for different HLA class I alleles (HLA-A2 and HLA-C1) could inhibit CAR-T cell activation against target cells expressing the respective HLA class I allele.55, 56, 57 These studies and ours are thus complementary, which collectively suggest that CAR-T and CAR-NK cells can be engineered to target malignancies with HLA class I and II loss specifically.
In summary, we have developed novel dual CAR-NK cells that can kill HLA-DRneg cells but spare HLA-DR+ cells. Future studies will investigate how to further enhance the potency and selectivity of dual CAR-NK cells, e.g., by using NK cell-derived activating and inhibitory signaling domains. We also plan to validate the anti-cancer specificity of engineered NK-92 and primary human NK cells using patient-derived xenograft mouse models. Our strategy can potentially enhance the specificity of CAR-NK cell therapy against various hematologic malignancies with HLA-DR loss. It may also be adopted to enhance the targeting of CAR-NK cells and CAR-T cells against solid tumors with HLA class I loss.
Materials and methods
Cell lines and cell culture
NK-92MI cells (ATCC) were maintained in RPMI-1640 medium supplemented with 20% fetal bovine serum (FBS). K562-CD19 cells were a gift from Dr. Pin Wang (USC Viterbi School of Engineering). K562-CD19-HLA-DR cells were generated by lentiviral transduction of K562-CD19 cells to express full-length HLA-DR molecules. K562, K562-CD19, K562-CD19-HLA-DR, Nalm6, KOPN1, and Raji cells were cultured in complete RPMI-1640 medium supplemented with 10% FBS. 293T cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS.
Plasmid construction
The lentiviral vector expressing the anti-CD19 CAR (pFUW-HA-anti-CD19-CD28-CD3ζ) was a gift from Dr. Pin Wang (USC Viterbi School of Engineering). The lentiviral vector expressing the anti-HLA-DR iCAR (pFUW-FLAG-anti-HLA-DR-PD1) was constructed as follows. The anti-HLA-DR scFv in the format of VL-(GGGGS)3-VH was designed based on a full human HLA-DR antibody (1D09C3). The anti-HLA-DR iCAR consists of an N-terminal FLAG tag, an anti-HLA-DR scFv, and PD-1 hinge, transmembrane, and intracellular domains. The whole gene of iCAR was synthesized as a gBlocks gene fragment (IDT Technologies). The plasmid pFUW-FLAG-anti-HLA-DR-PD1 was constructed by inserting the iCAR gene into the pFUW linear vector (BamHI, EcoRI).
Lentiviral production and transduction
293T cells were seeded 1 day before transduction to achieve 90% confluence in two 100-mm dishes (VWR). The plasmid encoding the anti-CD19 CAR or the anti-HLA-DR iCAR was mixed thoroughly with the three packaging plasmids (encoding RRE, REV, and VSVG) in 2× HEPES-buffered saline (HBS). The plasmid-containing HBS buffer was slowly added to 0.25 M CaCl2 solution. The whole mixture was then added to the 293T cell culture from the side of the dish. After a 4-h incubation, the medium was replaced with fresh DMEM supplemented with 10% FBS. Three days after infection, lentiviruses were harvested and filtered through a 0.45-μm filter (Pall), concentrated using 100-kDa ultrafilters (Amicon), and loaded into 1 × 106 NK-92MI cells in a 24-well untreated plate. NK cells were supplemented with 8 μg/mL protamine sulfate (Sigma-Aldrich) and 6 μM BX795 (Invivogen) to enhance transduction efficiency, as reported.58 Cells were then centrifuged for 90 min at 2,400 rpm at room temperature, followed by overnight incubation at 37°C. On the next day, cells were washed twice and supplemented with human recombinant IL-2 (BioLegend, San Diego, CA) to enhance viability.
Generation of the K562-CD19-HLA-DR cell line
The parent lentiviral vector pCDH-EF1-Nluc-P2A-copGFP-T2A-Puro was a gift from Kazuhiro Oka (Addgene, plasmid no. 73022; http://n2t.net/addgene:73022; RRID: Addgene_73022). The HLA-DR alpha chain (UniProtKB-P01903) and beta chain (UniProtKB-P04229) were linked by a furin recognition sequence and a P2A peptide.59,60 The gene of HLA-DRα-Furin-P2A-HLA-DRβ was synthesized as a gBlocks gene fragment (IDT Technologies) and inserted into the pCDH vector (BamHI, EcoRI). Lentiviral particles were generated by co-transfecting HEK293T cells with pCDH-EF1-HLA-DRα-Furin-P2A-HLA-DRβ and three packaging plasmids (encoding RRE, REV, and VSVG). K562-CD19 cells were then modified to express HLA-DR by lentiviral transduction. The transduced cells were stained with a PE-conjugated anti-HLA-DR antibody (BioLegend) and purified using the FACSAria Fusion Cell Sorter.
Analysis of CD19 and HLA-DR expression on the target cell surfaces
Cells (1 × 106) in FACS buffer (PBS with 2% FBS, 2 mM EDTA, 0.05% sodium azide) were stained with PE-conjugated anti-CD19 or anti-HLA-DR antibodies (BioLegend) at saturation on ice for 30 min. PE-conjugated mouse IgG-κ (BioLegend) was used as an isotype control. Cells were washed three times, resuspended in FACS buffer, and analyzed by flow cytometry. Data were analyzed with FlowJo software (TreeStar).
Comparison of CAR and iCAR expression on single and dual CAR-NK cells
Different CAR-NK cells were stained with PE-conjugated anti-FMC63 scFv antibody (ACRO Biosystems, DE, USA) and anti-FLAG-tag antibody (BioLegend) at saturating concentrations, respectively. Cells were analyzed by flow cytometry, and data were analyzed using FlowJo software. The saturating concentration of each PE-conjugated antibody was determined by increasing the antibody concentration until there was no further increment of mean fluorescence intensity of stained cells.
Flow cytometry analysis
All antibodies were purchased from Thermo Fisher Scientific (MA, USA), BioLegend, and ACRO Biosystems. Cells were harvested from in vitro culture, washed twice with FACS buffer (PBS with 2% FBS and 2 mM EDTA), and stained with fluorophore-conjugated antibodies for 30 min on ice before analysis. Flow cytometry was performed on the BD LSR II analyzer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo.
Cytokine production assays
Single or dual CAR-NK cells (1 × 105) were cocultured with target cells (1 × 105) in a 96-well U-bottom plate. For testing the effect of surrounding cells, the indicated amounts of KG1 or myeloid cells were added into the coculture at the beginning of the experiment. After a 4-h incubation, the cell culture supernatant was harvested, and the concentration of IFN-γ in each sample was determined using a human IFN-γ ELISA kit (Invitrogen, Carlsbad, CA) in accordance with the manufacturer's instructions. The chemiluminescence was measured using a Synergy H1 Hybrid Multiplate reader (BioTek). Data are presented as mean ± SEM of triplicates.
CD107a degranulation assays
NK cells, single CAR-NK cells, or dual CAR-NK cells (1 × 105) were cocultured with each of the six target cells at a 1:1 ratio in 200 μL medium with a PE-conjugated anti-CD107a antibody (BioLegend). After a 1-h incubation, 100 μg/mL monensin (GolgiStop, BD Bioscience) was added to the coculture medium. After a 4-h incubation, cells were collected and washed twice using FACS buffer before staining on ice with an APC-conjugated anti-CD56 antibody (BD Bioscience) to differentiate NK cells from target cells. After washing three times, cells were resuspended in FACS buffer for flow cytometry analysis. The degranulated NK cells were identified as the CD107a+ population in CD56+ cells. Non-transduced NK cells were used as the negative control. Data were analyzed using FlowJo.
LDH cytotoxicity assays
Totals of 5 × 104 NK cells, single CAR-NK cells, or dual CAR-NK cells were incubated with targeted cells at three different E:T ratios (0.2:1, 1:1, and 5:1) in a total volume of 100 μL for 4 h. The supernatants were collected, and the released LDH was measured by a colorimetric reaction using a Pierce LDH Cytotoxicity Assay Kit (Thermo Fisher, Rockford, IL). The chemiluminescence was measured using a Synergy H1 Hybrid Multiplate reader (BioTek). Spontaneous LDH release controls of effector and target cells were included by incubating the effector and target cells alone. The maximum LDH release control of each target cell was calculated by adding the lysis buffer 45 min before supernatant collection. The percentage of cytotoxicity (% Cytotoxicity) was calculated by: (Experimental value – Effector Cells Spontaneous Control – Target Cells Spontaneous Control) × 100/(Target Cell Maximum Control – Target Cells Spontaneous Control).
Myeloid cell isolation
Human PBMCs were isolated from the buffy coat (ZenBio, NC) using Ficoll-Paque (GE Healthcares, IL) density gradient centrifugation at 800 × g for 30 min at room temperature. Myeloid cells were further enriched by negative selection using a Human Monocyte Enrichment Kit (STEMCELL Technologies).
Animal studies
All animal protocols and experiments were approved by the USC Institutional Animal Care and Use Committee. Eight- to 10-week-old NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from The Jackson Laboratory (ME, USA). The K562-CD19-HLA-DR-Luc and K562-CD19-Luc cells were generated by transducing K562-CD19-HLA-DR and K562-CD19 cells with pCDH-EF1-Luc2-P2A-copGFP (a gift from Kazuhiro Oka; Addgene. plasmid no. 72,485; http://n2t.net/addgene:72485; RRID: Addgene_72485), followed by purification using the FACSAria Fusion Cell Sorter. On day 0, NSG mice were intravenously inoculated with 5 × 105 K562-CD19-HLA-DR-Luc cells or K562-CD19-Luc cells. On day 3, the bioluminescence of the engrafted tumor cells was monitored by IVIS imaging (PerkinElmer, Waltham, MA). Mice were then randomly divided into three groups (n = 5). Each group was treated with 1 × 107 NK cells, single CAR-NK cells, or dual CAR-NK cells by tail vein injection on day 3. Tumor growth was monitored by bioluminescence imaging once a week. Survival data were reported in Kaplan-Meier plots and analyzed by log rank test. Images were analyzed using Caliper Life Sciences software (PerkinElmer).
Construction, expression, refolding, and purification of the anti-HLA-DR scFv
The gene of the anti-HLA-DR scFv (clone 1D09C3) in the format of VL-(GGGGS)3-VH was inserted into a linear vector pET22b (NdeI, XhoI) to construct pET22b-anti-HLA-DR-scFv. The plasmid was transformed into E. coli BL21 (DE3) competent cells. Cells harboring the plasmid were grown in the LB medium containing 100 μg/mL ampicillin. When OD600 reached 0.5, protein expression was induced by adding 1 mM isopropyl-β-D-thiogalactopyranoside. After 4 h, cells were harvested by centrifugation and lysed by sonication. The inclusion body of the scFv was isolated by centrifugation and solubilized in a denaturation buffer (100 mM Tris-HCl [pH 8.0], 200 mM NaCl, 1 mM EDTA, and 6 M GuHCl). The native scFv protein was obtained by in vitro refolding using a published method.61 The refolded scFv was further purified by gel filtration chromatography using a HiLoad Superdex 200 16/600 column by FPLC (GE Healthcare).
Statistical analysis
Data are presented as means ± SEM. The survival statistics were calculated using the log rank (Mantel-Cox) test. The other differences between groups were analyzed by unpaired two-tailed Student's t test. The statistical significance was defined at p < 0.05 (ns, p > 0.05, ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001). All statistical analyses were performed using GraphPad Prism 8.0.
Acknowledgments
We thank Prof. Pin Wang (University of Southern California) for the gift of the parental anti-CD19 CAR lentiviral vector and the K562-CD19 cell line. We also thank Zipeng Zeng for his experimental assistance. This work was partially supported by funding from the Ming Hsieh Institute for Research of Engineering-Medicine for Cancer (to J.X. and A.S.W.), NCI award P30CA014089 (to A.S.W.), the Margaret E. Early Medical Research Trust (to J.X.), the STOP CANCER Chang & Dodd Family Memorial Seed Grant (to J.X.), and the Theodore and Wen-Hui Chen Endowed Graduate Fellowship (to F.F.).
Author contributions
J.X., F.F., L.R., and A.S.W. designed the project. F.F. and L.R. performed the experiments. F.F., L.R., and J.X. analyzed data. N.J. provided experimental assistance. A.S.W. reviewed the data. J.X., F.F., L.R., and A.S.W. wrote the manuscript. All authors read and approved the manuscript.
Declaration of interests
A.S.W. receives research funding from Kite Pharma and Institut de Recherches Internationales Servier. F.F., L.R., and J.X. have filed a provisional patent application on the use of anti-HLA-DR iCAR to target HLA-DR loss in hematologic malignancies.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.11.013.
Supplemental information
References
- 1.June C.H., Sadelain M. Chimeric antigen receptor therapy. N. Engl. J. Med. 2018;379:64–73. doi: 10.1056/NEJMra1706169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daher M., Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov. 2021;11:45–58. doi: 10.1158/2159-8290.CD-20-0556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fesnak A.D., June C.H., Levine B.L. Engineered T cells: the promise and challenges of cancer immunotherapy. Nat. Rev. Cancer. 2016;16:566–581. doi: 10.1038/nrc.2016.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rafiq S., Hackett C.S., Brentjens R.J. Engineering strategies to overcome the current roadblocks in CAR T cell therapy. Nat. Rev. Clin. Oncol. 2020;17:147–167. doi: 10.1038/s41571-019-0297-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kenderian S.S., Ruella M., Shestova O., Klichinsky M., Aikawa V., Morrissette J.J., Scholler J., Song D., Porter D.L., Carroll M., et al. CD33-specific chimeric antigen receptor T cells exhibit potent preclinical activity against human acute myeloid leukemia. Leukemia. 2015;29:1637–1647. doi: 10.1038/leu.2015.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Q.S., Wang Y., Lv H.Y., Han Q.W., Fan H., Guo B., Wang L.L., Han W.D. Treatment of CD33-directed chimeric antigen receptor-modified T cells in one patient with relapsed and refractory acute myeloid leukemia. Mol. Ther. 2015;23:184–191. doi: 10.1038/mt.2014.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rimsza L.M., Roberts R.A., Miller T.P., Unger J.M., LeBlanc M., Braziel R.M., Weisenberger D.D., Chan W.C., Muller-Hermelink H.K., Jaffe E.S., et al. Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood. 2004;103:4251–4258. doi: 10.1182/blood-2003-07-2365. [DOI] [PubMed] [Google Scholar]
- 8.Aptsiauri N., Ruiz-Cabello F., Garrido F. The transition from HLA-I positive to HLA-I negative primary tumors: the road to escape from T-cell responses. Curr. Opin. Immunol. 2018;51:123–132. doi: 10.1016/j.coi.2018.03.006. [DOI] [PubMed] [Google Scholar]
- 9.Garrido F., Ruiz-Cabello F., Cabrera T., Pérez-Villar J.J., López-Botet M., Duggan-Keen M., Stern P.L. Implications for immunosurveillance of altered HLA class I phenotypes in human tumours. Immunol. Today. 1997;18:89–95. doi: 10.1016/s0167-5699(96)10075-x. [DOI] [PubMed] [Google Scholar]
- 10.Khong H.T., Restifo N.P. Natural selection of tumor variants in the generation of “tumor escape” phenotypes. Nat. Immunol. 2002;3:999–1005. doi: 10.1038/ni1102-999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Higashi M., Tokuhira M., Fujino S., Yamashita T., Abe K., Arai E., Kizaki M., Tamaru J. Loss of HLA-DR expression is related to tumor microenvironment and predicts adverse outcome in diffuse large B-cell lymphoma. Leuk. Lymphoma. 2016;57:161–166. doi: 10.3109/10428194.2015.1038708. [DOI] [PubMed] [Google Scholar]
- 12.Nijland M., Veenstra R.N., Visser L., Xu C., Kushekhar K., van Imhoff G.W., Kluin P.M., van den Berg A., Diepstra A. HLA dependent immune escape mechanisms in B-cell lymphomas: implications for immune checkpoint inhibitor therapy? Oncoimmunology. 2017;6 doi: 10.1080/2162402X.2017.1295202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Riemersma S.A., Jordanova E.S., Schop R.F., Philippo K., Looijenga L.H., Schuuring E., Kluin P.M. Extensive genetic alterations of the HLA region, including homozygous deletions of HLA class II genes in B-cell lymphomas arising in immuneprivileged sites. Blood. 2000;96:3569–3577. [PubMed] [Google Scholar]
- 14.Oelschlaegel U., Mohr B., Schaich M., Schakel U., Kroschinsky F., Illmer T., Ehninger G., Thiede C. HLA-DRneg patients without acute promyelocytic leukemia show distinct immunophenotypic, genetic, molecular, and cytomorphologic characteristics compared to acute promyelocytic leukemia. Cytometry B Clin. Cytometry. 2009;76:321–327. doi: 10.1002/cyto.b.20475. [DOI] [PubMed] [Google Scholar]
- 15.Wetzler M., McElwain B.K., Stewart C.C., Blumenson L., Mortazavi A., Ford L.A., Slack J.L., Barcos M., Ferrone S., Baer M.R. HLA-DR antigen-negative acute myeloid leukemia. Leukemia. 2003;17:707–715. doi: 10.1038/sj.leu.2402865. [DOI] [PubMed] [Google Scholar]
- 16.Gorczyca W., Sun Z.-Y., Cronin W., Li X., Mau S., Tugulea S. In: Darzynkiewicz Z., Holden E., Orfao A., Telford W., Wlodkowic D., editors. Vol. 103. Academic Press; 2011. Chapter 10—immunophenotypic pattern of myeloid populations by flow cytometry analysis; pp. 221–266. (Methods in Cell Biology). [DOI] [PubMed] [Google Scholar]
- 17.Tarafdar A., Hopcroft L.E., Gallipoli P., Pellicano F., Cassels J., Hair A., Korfi K., Jorgensen H.G., Vetrie D., Holyoake T.L., Michie A.M. CML cells actively evade host immune surveillance through cytokine-mediated downregulation of MHC-II expression. Blood. 2017;129:199–208. doi: 10.1182/blood-2016-09-742049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts R.A., Wright G., Rosenwald A.R., Jaramillo M.A., Grogan T.M., Miller T.P., Frutiger Y., Chan W.C., Gascoyne R.D., Ott G., et al. Loss of major histocompatibility class II gene and protein expression in primary mediastinal large B-cell lymphoma is highly coordinated and related to poor patient survival. Blood. 2006;108:311–318. doi: 10.1182/blood-2005-11-4742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Christopher M.J., Petti A.A., Rettig M.P., Miller C.A., Chendamarai E., Duncavage E.J., Klco J.M., Helton N.M., O'Laughlin M., Fronick C.C., et al. Immune escape of relapsed AML cells after allogeneic transplantation. N. Engl. J. Med. 2018;379:2330–2341. doi: 10.1056/NEJMoa1808777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Toffalori C., Zito L., Gambacorta V., Riba M., Oliveira G., Bucci G., Barcella M., Spinelli O., Greco R., Crucitti L., et al. Immune signature drives leukemia escape and relapse after hematopoietic cell transplantation. Nat. Med. 2019;25:603–611. doi: 10.1038/s41591-019-0400-z. [DOI] [PubMed] [Google Scholar]
- 21.Fedorov V.D., Themeli M., Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci. Transl. Med. 2013;5:215ra172. doi: 10.1126/scitranslmed.3006597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cheng Z., Wei R., Ma Q., Shi L., He F., Shi Z., Jin T., Xie R., Wei B., Chen J., et al. In vivo expansion and antitumor activity of coinfused CD28- and 4-1BB-engineered CAR-T cells in patients with B cell leukemia. Mol. Ther. 2018;26:976–985. doi: 10.1016/j.ymthe.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nagy Z.A., Hubner B., Lohning C., Rauchenberger R., Reiffert S., Thomassen-Wolf E., Zahn S., Leyer S., Schier E.M., Zahradnik A., et al. Fully human, HLA-DR-specific monoclonal antibodies efficiently induce programmed death of malignant lymphoid cells. Nat.Med. 2002;8:801–807. doi: 10.1038/nm736. [DOI] [PubMed] [Google Scholar]
- 24.Lois C., Hong E.J., Pease S., Brown E.J., Baltimore D. Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science. 2002;295:868–872. doi: 10.1126/science.1067081. [DOI] [PubMed] [Google Scholar]
- 25.Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W., et al. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 26.Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 27.Zhang C., Oberoi P., Oelsner S., Waldmann A., Lindner A., Tonn T., Wels W.S. Chimeric antigen receptor-engineered NK-92 cells: an off-the-shelf cellular therapeutic for targeted elimination of cancer cells and induction of protective antitumor immunity. Front. Immunol. 2017;8:533. doi: 10.3389/fimmu.2017.00533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papadimitriou L., Morianos I., Michailidou V., Dionyssopoulou E., Vassiliadis S., Athanassakis I. Characterization of intracellular HLA-DR, DM and DO profile in K562 and HL-60 leukemic cells. Mol. Immunol. 2008;45:3965–3973. doi: 10.1016/j.molimm.2008.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Siriwon N., Kim Y.J., Siegler E., Chen X., Rohrs J.A., Liu Y., Wang P. CAR-T cells surface-engineered with drug-encapsulated nanoparticles can ameliorate intratumoral T-cell hypofunction. Cancer Immunol. Res. 2018;6:812–824. doi: 10.1158/2326-6066.CIR-17-0502. [DOI] [PubMed] [Google Scholar]
- 30.Alter G., Malenfant J.M., Altfeld M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods. 2004;294:15–22. doi: 10.1016/j.jim.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Jedema I., van der Werff N.M., Barge R.M., Willemze R., Falkenburg J.H. New CFSE-based assay to determine susceptibility to lysis by cytotoxic T cells of leukemic precursor cells within a heterogeneous target cell population. Blood. 2004;103:2677–2682. doi: 10.1182/blood-2003-06-2070. [DOI] [PubMed] [Google Scholar]
- 32.Viallard J.-F., Blanco P., André M., Etienne G., Liferman F., Neau D., Vidal E., Moreau J.-F., Pellegrin J.-L. CD8+HLA-DR+ T lymphocytes are increased in common variable immunodeficiency patients with impaired memory B-cell differentiation. Clin. Immunol. 2006;119:51–58. doi: 10.1016/j.clim.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 33.Baecher-Allan C., Wolf E., Hafler D.A. MHC class II expression identifies functionally distinct human regulatory T cells. J. Immunol. 2006;176:4622–4631. doi: 10.4049/jimmunol.176.8.4622. [DOI] [PubMed] [Google Scholar]
- 34.Long E.O., Kim H.S., Liu D., Peterson M.E., Rajagopalan S. Controlling natural killer cell responses: integration of signals for activation and inhibition. Annu. Rev. Immunol. 2013;31:227–258. doi: 10.1146/annurev-immunol-020711-075005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li Y., Hermanson D.L., Moriarity B.S., Kaufman D.S. Human iPSC derived natural killer cells engineered with chimeric antigen receptors enhance anti-tumor activity. Cell Stem Cell. 2018;23:181–192. doi: 10.1016/j.stem.2018.06.002. e185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Varma N., Janic B., Ali M., Iskander A., Arbab A. Lentiviral based gene transduction and promoter studies in human hematopoietic stem cells (hHSCs) J. Stem Cells Regen. Med. 2011;7:41–53. doi: 10.46582/jsrm.0701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jones S., Peng P.D., Yang S., Hsu C., Cohen C.J., Zhao Y., Abad J., Zheng Z., Rosenberg S.A., Morgan R.A. Lentiviral vector design for optimal T cell receptor gene expression in the transduction of peripheral blood lymphocytes and tumor-infiltrating lymphocytes. Hum. Gene Ther. 2009;20:630–640. doi: 10.1089/hum.2008.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jordanova E.S., Philippo K., Giphart M.J., Schuuring E., Kluin P.M. Mutations in the HLA class II genes leading to loss of expression of HLA-DR and HLA-DQ in diffuse large B-cell lymphoma. Immunogenetics. 2003;55:203–209. doi: 10.1007/s00251-003-0563-z. [DOI] [PubMed] [Google Scholar]
- 39.Steidl C., Shah S.P., Woolcock B.W., Rui L., Kawahara M., Farinha P., Johnson N.A., Zhao Y., Telenius A., Neriah S.B., et al. MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature. 2011;471:377–381. doi: 10.1038/nature09754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cycon K.A., Rimsza L.M., Murphy S.P. Alterations in CIITA constitute a common mechanism accounting for downregulation of MHC class II expression in diffuse large B-cell lymphoma (DLBCL) Exp. Hematol. 2009;37:184–194. doi: 10.1016/j.exphem.2008.10.001. [DOI] [PubMed] [Google Scholar]
- 41.Dietz A.C., Wayne A.S. Cells to prevent/treat relapse following allogeneic stem cell transplantation. Hematology. 2017;2017:708–715. doi: 10.1182/asheducation-2017.1.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Biernacki M.A., Brault M., Bleakley M. T-Cell receptor-based immunotherapy for hematologic malignancies. Cancer J. 2019;25:179–190. doi: 10.1097/PPO.0000000000000378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lamers C.H.J., Sleijfer S., Vulto A.G., Kruit W.H.J., Kliffen M., Debets R., Gratama J.W., Stoter G., Oosterwijk E. Treatment of metastatic renal cell carcinoma with autologous T-lymphocytes genetically retargeted against carbonic anhydrase IX: first clinical experience. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2006;24:e20–e22. doi: 10.1200/JCO.2006.05.9964. [DOI] [PubMed] [Google Scholar]
- 44.Lamers C.H.J., Sleijfer S., Van Steenbergen S., Van Elzakker P., Van Krimpen B., Groot C., Vulto A., Den Bakker M., Oosterwijk E., Debets R., Gratama J.W. Treatment of metastatic renal cell carcinoma with CAIX CAR-engineered T cells: clinical evaluation and management of on-target toxicity. Mol. Ther. 2013;21:904–912. doi: 10.1038/mt.2013.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morgan R.A., Yang J.C., Kitano M., Dudley M.E., Laurencot C.M., Rosenberg S.A. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 2010;18:843–851. doi: 10.1038/mt.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garrido F., Aptsiauri N. Cancer immune escape: MHC expression in primary tumours versus metastases. Immunology. 2019;158:255–266. doi: 10.1111/imm.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kaneko K., Ishigami S., Kijima Y., Funasako Y., Hirata M., Okumura H., Shinchi H., Koriyama C., Ueno S., Yoshinaka H., Natsugoe S. Clinical implication of HLA class I expression in breast cancer. BMC Cancer. 2011;11:454. doi: 10.1186/1471-2407-11-454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Madjd Z., Spendlove I., Pinder S.E., Ellis I.O., Durrant L.G. Total loss of MHC class I is an independent indicator of good prognosis in breast cancer. Int. J. Cancer. 2005;117:248–255. doi: 10.1002/ijc.21163. [DOI] [PubMed] [Google Scholar]
- 49.Carretero F.J., del Campo A.B., Flores-Martín J.F., Mendez R., García-Lopez C., Cozar J.M., Adams V., Ward S., Cabrera T., Ruiz-Cabello F., et al. Frequent HLA class I alterations in human prostate cancer: molecular mechanisms and clinical relevance. Cancer Immunol. Immunother. 2016;65:47–59. doi: 10.1007/s00262-015-1774-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baba T., Shiota H., Kuroda K., Shigematsu Y., Ichiki Y., Uramoto H., Hanagiri T., Tanaka F. Clinical significance of human leukocyte antigen loss and melanoma-associated antigen 4 expression in smokers of non-small cell lung cancer patients. Int. J. Clin. Oncol. 2013;18:997–1004. doi: 10.1007/s10147-012-0491-8. [DOI] [PubMed] [Google Scholar]
- 51.McGranahan N., Rosenthal R., Hiley C.T., Rowan A.J., Watkins T.B.K., Wilson G.A., Birkbak N.J., Veeriah S., Van Loo P., Herrero J., Swanton C. Allele-specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171:1259–1271. doi: 10.1016/j.cell.2017.10.001. e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perea F., Bernal M., Sanchez-Palencia A., Carretero J., Torres C., Bayarri C., Gomez-Morales M., Garrido F., Ruiz-Cabello F. The absence of HLA class I expression in non-small cell lung cancer correlates with the tumor tissue structure and the pattern of T cell infiltration. Int. J. Cancer. 2017;140:888–899. doi: 10.1002/ijc.30489. [DOI] [PubMed] [Google Scholar]
- 53.Cabrera T., Collado A., Fernandez M.A., Ferron A., Sancho J., Ruiz-Cabello F., Garrido F. High frequency of altered HLA class I phenotypes in invasive colorectal carcinomas. Tissue Antigens. 1998;52:114–123. doi: 10.1111/j.1399-0039.1998.tb02274.x. [DOI] [PubMed] [Google Scholar]
- 54.Maleno I., Romero J.M., Cabrera T., Paco L., Aptsiauri N., Cozar J.M., Tallada M., López-Nevot M.A., Garrido F. LOH at 6p21.3 region and HLA class altered phenotypes in bladder carcinomas. Immunogenetics. 2006;58:503–510. doi: 10.1007/s00251-006-0111-8. [DOI] [PubMed] [Google Scholar]
- 55.Tao L., Farooq M.A., Gao Y., Zhang L., Niu C., Ajmal I., Zhou Y., He C., Zhao G., Yao J., et al. CD19-CART cells bearing a KIR/PD-1-based inhibitory CAR eradicate CD19(+)HLA-C1(–) malignant B cells while sparing CD19(+)HLA-C1(+) healthy B cells. Cancers (Basel) 2020;12:2612. doi: 10.3390/cancers12092612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hamburger A.E., DiAndreth B., Cui J., Daris M.E., Munguia M.L., Deshmukh K., Mock J.-Y., Asuelime G.E., Lim E.D., Kreke M.R., et al. Engineered T cells directed at tumors with defined allelic loss. Mol. Immunol. 2020;128:298–310. doi: 10.1016/j.molimm.2020.09.012. [DOI] [PubMed] [Google Scholar]
- 57.Hwang M.S., Mog B.J., Douglass J., Pearlman A.H., Hsiue E.H.-C., Paul S., DiNapoli S.R., Konig M.F., Pardoll D.M., Gabelli S.B., et al. Targeting loss of heterozygosity for cancer-specific immunotherapy. Proc. Natl. Acad. Sci. 2021;118 doi: 10.1073/pnas.2022410118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sutlu T., Nyström S., Gilljam M., Stellan B., Applequist S.E., Alici E. Inhibition of intracellular antiviral defense mechanisms augments lentiviral transduction of human natural killer cells: implications for gene therapy. Hum. Gene Ther. 2012;23:1090–1100. doi: 10.1089/hum.2012.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chng J., Wang T., Nian R., Lau A., Hoi K.M., Ho S.C.L., Gagnon P., Bi X., Yang Y. Cleavage efficient 2A peptides for high level monoclonal antibody expression in CHO cells. MAbs. 2015;7:403–412. doi: 10.1080/19420862.2015.1008351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu Z., Chen O., Wall J.B.J., Zheng M., Zhou Y., Wang L., Ruth Vaseghi H., Qian L., Liu J. Systematic comparison of 2A peptides for cloning multi-genes in a polycistronic vector. Sci. Rep. 2017;7:2193. doi: 10.1038/s41598-017-02460-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsumoto K., Shinoki K., Kondo H., Uchikawa M., Juji T., Kumagai I. Highly efficient recovery of functional single-chain Fv fragments from inclusion bodies overexpressed in Escherichia coli by controlled introduction of oxidizing reagent—application to a human single-chain Fv fragment. J. Immunol. Methods. 1998;219:119–129. doi: 10.1016/s0022-1759(98)00127-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.