

Abstract
Aims
Traditional atherosclerotic cardiovascular disease (ASCVD) risk factors fail to address the full spectrum of the complex interplay of atherosclerotic and atherothrombotic factors integral to ASCVD events. This study sought to examine the association between atherothrombotic biomarkers and ASCVD events.
Methods and results
The association between atherothrombotic biomarkers and 877 ASCVD events with and without adjustment for traditional risk factors was evaluated via Cox proportional hazards models and factor analysis in 5789 Multi-Ethnic Study of Atherosclerosis participants over a median follow-up of 14.7 years. Factor analysis accounted for multidimensional relationship and shared variance among study biomarkers, which identified two new variables: a thrombotic factor (Factor 1), principally defined by shared variance in fibrinogen, plasmin–antiplasmin complex, factor VIII, D-dimer, and lipoprotein(a), and a fibrinolytic factor (Factor 2), principally defined by shared variance of plasminogen and oxidized phospholipids on plasminogen. In a model including both factors, the thrombotic factor was associated with the higher risk of ASCVD events [hazard ratio (HR) 1.57, 95% confidence interval (CI) 1.45, 1.70], while the fibrinolytic factor was associated with the lower risk of ASCVD events (HR 0.76, 95% CI 0.70, 0.82), with estimated ASCVD free survival highest for low atherothrombotic Factor 1 and high atherothrombotic Factor 2.
Conclusion
Two atherothrombotic factors, one representative of thrombotic propensity and the other representative of fibrinolytic propensity, were significantly and complementarily associated with incident ASCVD events, remained significantly associated with incident ASCVD after controlling for traditional risk factors, and have promise for identifying patients at high ASCVD event risk specifically due to their atherothrombotic profile.
Keywords: Atherosclerotic cardiovascular disease, Atherothrombosis, Fibrinolysis, Risk prediction, Factor analysis
Graphical Abstract
See the editorial comment for this article ‘Thrombosis and fibrinolysis in atherosclerotic cardiovascular disease: it takes two to tango’, by A. Fedorowski et al., https://doi.org/10.1093/eurheartj/ehab710.
Introduction
Epidemiological studies have led to the identification and association of risk factors for the development and progression of atherosclerosis. This knowledge has resulted in preventive strategies that have contributed to the significant decline in deaths due to atherosclerotic cardiovascular disease (ASCVD) in the USA and Europe over the last four decades.1,2 However, traditional risk factors incompletely capture all cardiovascular disease risk, and ASCVD continues to be the leading cause of deaths worldwide.2 ASCVD events result from a combination of atherosclerosis, which develops over years, and a thrombotic response to an acute disruption of atherosclerotic plaque—atherothrombosis.
The balance between fibrin formation and degradation is fundamental to the atherothrombotic process, and is therefore key in determining the clinical consequence of plaque disruption, i.e. thrombus formation and the ensuing ASCVD events.3–8 While associations between individual thrombotic and fibrinolytic biomarkers and ASCVD events have been demonstrated, an assessment of the balance between the opposing processes of thrombosis and fibrinolysis, assessed via multiple biomarkers, and ASCVD events has not been adequately explored. Quantification of an individual’s propensity to form or resolve an arterial thrombus may have significant implications for ASCVD risk prediction scores (currently dominated by atherosclerosis risk factors); more efficacious use of antithrombotic medication (e.g. oral anticoagulation only for patients with high thrombotic but low fibrinolytic capacity) and the development of new therapeutics targeted to an individual’s atherothrombotic risk.
The Multi-Ethnic Study of Atherosclerosis (MESA) is an NIH-funded, sex-balanced, multi-ethnic, contemporary prospective cohort of approximately 7000 participants at baseline with adjudicated ASCVD events over a median Follow-up of 14.7 years.9 MESA participants have undergone extensive phenotyping, with measurement of multiple key atherothrombotic biomarkers, including a newly studied atherothrombotic biomarker—oxidized phospholipids (OxPLs) bound to plasminogen (OxPL-PLG). Elevated levels of OxPL-PLG have been associated with enhanced fibrinolysis in vitro and therefore higher levels would be expected to lower risk of acute ASCVD events.10 For example, plasma OxPL-PLG levels are lower among patients at the time of an acute myocardial infarction (MI) and remain lower than in patients with stable coronary artery disease (CAD) ∼3 months later.11
To more fully characterize atherothrombotic risk, we sought to examine the association of key atherothrombotic biomarkers (including the novel biomarker OxPL-PLG) with each other, and with adjudicated ASCVD events in MESA.
Methods
Study participants and design
MESA began enrolling participants in the year 2000, and as of 2017, has adjudicated all cardiovascular events over a median of 14.7 years of follow-up. The design and methods of the MESA study have been previously published.9 Briefly, 6814 participants aged 45–84 years and representing four different ethnic backgrounds (White, Chinese, Black, Hispanic) were recruited from six communities in the United States (Forsyth County, NC; Northern Manhattan and the Bronx, NY; Baltimore, MD; St. Paul, MN; Chicago, IL; and Los Angeles County, CA) between 2000 and 2002. All participants were free of clinical ASCVD at study enrolment. An approximately equal number of men and women were recruited according to pre-specified age and race/ethnicity strata. MESA-1000 is a random sample of 1000 participants that underwent additional laboratory tests using baseline blood samples. For these participants, the baseline blood repository volumes are smaller than for the other 5814 participants. To minimize depletion of these specimens, ancillary studies, without specific justification, exclude MESA-1000 participants, and those with missing data leaving 5789 available participants. Medical history, anthropometric measurements, and laboratory data were assessed as previously described.9
Median follow-up of the MESA cohort as of 2017 is 14.7 years, and ascertainment of new events occurs according to a planned schedule. At intervals of 9–12 months, an interviewer contacts each participant or a family member by telephone to inquire about interim hospital admissions, outpatient diagnoses of ASCVD, and deaths. To verify self-reported diagnoses, all death certificates and medical records for all hospitalizations and outpatient cardiovascular diagnoses are requested. Next-of-kin interviews for out-of-hospital cardiovascular deaths may be obtained. Trained personnel abstract data from medical records reporting possible cardiovascular events. Two physician members of the MESA mortality and morbidity review committee independently classify events, and in the event of disagreement, the full committee makes the final classification. ASCVD events consist of MI; definite/probable angina, resuscitated cardiac arrest; stroke (not transient ischaemic attack); and death from atherosclerotic coronary heart disease, stroke, or ‘atherosclerotic disease other than coronary disease, stroke’. A more detailed description of the MESA follow-up methods is available at www.mesa-nhlbi.org.
Laboratory variables
Factors evaluated in this study included all biomarkers with known association with thrombosis and fibrinolysis and which were available among non-MESA-1000 subjects at baseline. Measurements of plasminogen and OxPL-PLG and were performed at the University of California San Diego laboratory of Dr Sotirios Tsimikas. Samples were deidentified prior to measurement. In brief, to measure plasminogen levels, microtiter well plates (Dynex Technologies, Chantilly, VA, USA) were incubated with a mouse monoclonal anti-human plasminogen antibody (Meridian Life Science, Saco, ME, USA) at 5 µg/mL overnight at 4°C; the plates were then washed, and human plasma was added (1:32 000 dilution), and plasminogen detected with biotinylated guinea pig anti-human plasminogen antibody using chemiluminescence enzyme-linked immunosorbent assay (ELISA).10 A standard curve of purified human plasminogen was used to assign values. OxPL-PLG was detected with biotinylated murine monoclonal antibody E06, which recognizes the phosphocholine group on oxidized but not native phospholipids in a chemiluminescence ELISA at a plasma dilution of 1:400.10–12 The assay’s lower and uppers limit of quantification are 0.01 and 500 nM/L of phosphocholine equivalents, with a coefficient of variance of 5–10%. This assay was normalized in all wells to the same amount of plasminogen from each sample, so that the measure essentially reflects the carrying capacity of OxPL by a fixed and similar amount of plasminogen from each subject.13 All measurements were performed in triplicate. Lipoprotein(a) [Lp(a)] was measured in the laboratory under the direction of Dr Michael Y. Tsai at the University of Minnesota, with a latex-enhanced turbidimetric immunoassay (Denka Seiken, Tokyo, Japan). This assay has an upper and lower limit of quantification (LOQ) of 80 and 2 mg/dL with a mean coefficient of variance of <3%. Values above LOQ were diluted and re-assayed until within the LOQ. Given the very low lower LOQ no adjustment was needed for values below the LOQ, with the exception of zero which was offset by 0.1 to allow for log-transformation.
All additional measures of atherothrombotic risk available in MESA were included in this study: D-dimer, fibrinogen antigen, factor VIIIc (FVIII), plasmin–antiplasmin; all were measured in a central laboratory (University of Vermont, Burlington, VT, USA). D-dimer was measured by immunoturbidometry (Liatest D-DI; Diagnostica Stago, NJ, USA) on the Sta-R analyzer (Diagnostica Stago), with analytical coefficients of variation (CVs) of 8%. Fibrinogen antigen was measured by immunonephelometry with the BNII nephelometer (N Antiserum to Human Fibrinogen, Dade Behring, IL, USA), with CVs of 2.6%. FVIII coagulant activity was measured as the clotting time of a sample in FVIII-deficient plasma in the presence of activators with the Sta-R analyzer (STA-Deficient VIII), with a CV of 10%. Plasmin–antiplasmin was measured using a two-site ELISA that utilizes two monoclonal antibodies; the inter-assay CV was 6.7–11.1%.
Statistical analysis
Baseline cohort characteristics, including atherothrombotic factors and ASCVD event rates were determined. Traditional ASCVD risk factors included those variables included in the American College of Cardiology/American Heart Association (ACC/AHA) ASCVD pooled cohort equations.14 Atherothrombotic biomarkers included fibrinogen, plasmin–antiplasmin complex, FVIII, D-dimer, and Lp(a), plasminogen and OxPL-PLG. Pairwise associations between study atherothrombotic biomarkers (log-transformed) were evaluated by determining pairwise Pearson correlation coefficients. We used Bartlett’s test of sphericity to determine if the shared variance between the biomarkers could be represented in a lower dimension using factor analysis.15 Factor analysis was utilized to determine two new atherothrombotic factors, Factor 1 and Factor 2, that quantify the multidimensional relationship and shared variance among study atherothrombotic biomarkers. Two factors (as opposed to one or three factors) were determined as eigenvalue analysis revealed this to be the optimal number for capturing a significant amount of the variance in a lower dimension. This factor analysis was conducted using seven plasma biomarkers at baseline from all non-MESA-1000 participants and did not include other attributes of the subject (e.g. race, sex, age, eventual ASCVD status). Thus, the atherothrombotic components, called ‘Factor 1’ and ‘Factor 2’, describe only variance in the plasma atherothrombotic biomarkers. Prior to factor analysis, atherothrombotic biomarkers were transformed: log(analyte + 1), which resulted in less departure from a normal distribution, and missing values were imputed using a multivariate imputations by chained equations approach.16–18 One or more values were imputed for 3.76% of the study participants included in the analysis. The factor analysis was conducted using the minimum residual method19 followed by the application of an ‘oblimin’ rotation20 to satisfy Thurstone’s criteria for ensuring a simple structure.21
The distribution of ASCVD risk factors, cohort characteristics, and incident ASCVD events across quartiles of Factors 1 and 2 were determined. To evaluate how Factors 1 and 2, representing the multidimensional relationship between atherothrombotic biomarkers, were associated with ASCVD events, a Cox proportional hazards model was estimated (unadjusted model). A model with Factors 1 and 2 was compared to a model with all the atherothrombotic biomarkers included. This comparison was made with and without adjustment for traditional risk factors. These models were compared utilizing the Bayesian information criterion which measures goodness-of-fit, penalizes overfitting, and is appropriate for comparing non-nested models.22 Treating Factors 1 and 2 as continuous variates was the primary analysis. An a priori planed secondary analysis compared ASCVD event-free survival when factors were balanced and unbalanced by utilizing extreme (1st and 4th) quartiles of Factors 1 and 2 in a Kaplan–Meier ASCVD event-free survival analysis. To determine the contributions of Factors 1 and 2 in explaining risk beyond traditional risk factors, multivariable Cox proportional hazards models were fit with traditional risk factors included and Factors 1 and 2 left out or included. A likelihood ratio test was conducted in order to determine whether Factors 1 and 2 explained variability in ASCVD risk that was not explained by traditional risk factors. To determine whether inclusion of Factors 1 and 2 improved prediction, Harrel’s C-index was estimated via 10-fold cross-validation.23 The relationship between Factors 1 and 2 and individual components of the ASCVD outcome variable was evaluated via risk factor-adjusted multivariable Cox proportional hazards models. We performed an exploratory analysis to examine the relationship between Factors 1 and 2 and incident cancer and venous thromboembolic events using the same modelling design. To explore whether the association between the factors and ASCVD events differs by baseline coronary atherosclerotic burden, interaction terms between the factors and coronary artery calcification scoring were added to the original adjusted model. Coronary artery calcification (Agatston score) was evaluated as a log-transformed continuous variable. Likelihood ratio tests were conducted to determine if the addition improved model fit. Statistical analyses were conducted using the R statistical language (version 4.0.2) and the following packages: survival, mice, emmeans, dplyr, and ggplot2.
The MESA study protocol was approved by the institutional review board of each field centre; all participants provided written informed consent. Secondary analysis of deidentified MESA data does not qualify as ‘human subject’ research per §46.102 US Department of Health and Human Services.
Results
Baseline demographics of the 5789 participants with available plasminogen, OxPL-PLG and ASCVD risk factor data, including, mean and median values of traditional cardiovascular risk factors, atherothrombotic biomarkers, and number of adjudicated ASCVD events are reported in Table 1.
Table 1.
Cohort characteristics for the 5789 MESA participants included in the analysis
| Variable | |
|---|---|
| Age, years | 62.6 (10.2) |
| Male sex | 2777 (48.0) |
| Ethnicity | |
| White | 2156 (37.2) |
| Chinese | 705 (12.2) |
| Black | 1666 (28.8) |
| Hispanic | 1262 (21.8) |
| Diabetes | 742 (12.8) |
| Hypertension | 2635 (45.5) |
| Anti-hypertensive medication | 2186 (37.8) |
| Total cholesterol, mg/dL | 194.0 (35.8) |
| HDL cholesterol, mg/dL | 50.9 (14.9) |
| Systolic BP, mmHg | 127.0 (21.5) |
| Diastolic BP, mmHg | 72.0 (10.3) |
| Smoking history | |
| Never | 2903 (50.1) |
| Former | 2139 (36.9) |
| Current | 727 (12.6) |
| BMI, kg/m2 | 28.3 (5.5) |
| Family history of MI | 2313 (40.0) |
| hs-CRP, mg/L | 1.90 [0.84, 4.20] |
| Calibrated factor VIII | 94 [73, 120] |
| Fibrinogen antigen, mg/dL | 338 [295, 389] |
| Plasmin–antiplasmin complex, nM | 4.44 [3.45, 5.70] |
| D-dimer, μg/mL | 0.23 [0.13, 0.37] |
| PLG, mg/dL | 11.8 [8.1, 15.3] |
| OxPL-PLG, nM | 82.6 [60.5, 110.8] |
| Lp(a), mg/dL | 17.8 [7.75, 41.0] |
| Atherosclerotic cardiovascular disease eventsa | 877 (15.1) |
| Myocardial Infarction | 282 (4.9) |
| Definite/probable angina | 306 (5.3) |
| Resuscitated cardiac arrest | 34 (0.6) |
| Stroke (not TIA) | 275 (4.8) |
| Death from ASCVD | 211 (3.6) |
Data are shown as mean (standard deviation), n (%), or median [Q1, Q3].
ASCVD, atherosclerotic cardiovascular disease; BMI, body mass index; HDL, high-density lipoprotein; BP, blood pressure; hs-CRP, high-sensitivity C-reactive protein; LDL, low-density lipoprotein; Lp(a), lipoprotein(a); MI, myocardial infarction; TIA, transient ischemic attack; OxPL, oxidized phospholipids; PLG, plasminogen; Q1, first quartile (25th percentile); Q3, third quartile (75th percentile).
Total represents first events.
Four of the seven individual atherothrombotic biomarkers were associated with ASCVD event risk in a Cox proportional hazards model of all seven atherothrombotic biomarkers, with each biomarker adjusted for all others (Table 2), and three remained so after controlling for traditional ASCVD risk factors (Table 3). Significant, but modest, pairwise correlations were observed between several atherothrombotic biomarkers (Figure 1). Bartlett’s test of sphericity furnished evidence that significant correlation exists between some or all of the seven atherothrombotic biomarkers (P < 0.0001). Factor analysis produced two new variables (Factors) based on the multidimensional relationships between the seven atherothrombotic biomarkers but uninformed about any other study variables or outcomes. These two new factors explained approximately half of the total variance (48.0%) in atherothrombotic biomarkers and aligned well with known atherothrombotic pathology. Factor 1 was largely determined by shared variance in biomarkers related to thrombotic propensity, namely fibrinogen, plasmin–antiplasmin complex, FVIII, and D-dimer, Lp(a) and is therefore referred to as the ‘thrombotic factor’. Factor 2 was largely determined by shared variance in biomolecules related to fibrinolytic propensity, namely plasminogen and OxPL-PLG and is therefore referred to as the ‘fibrinolytic factor’. The contribution of each atherothrombotic biomarker measurement to the factors and the mathematical definitions of the factor analysis components (factor loadings) is provided in Supplementary material online, Figure S1 and Supplementary material online, Table S1. Thrombotic factor and fibrinolytic factor scores are moderately correlated (Pearson r = 0.456). This indicates that while the factor analysis recognizes the two components as being representative of distinct processes, a higher value of one factor is associated with a higher value in the other. In other words, participants with high thrombotic factor (thrombotic propensity) are more likely to have high fibrinolytic factor (fibrinolytic propensity). The positive association (correlation) between the factors is important to consider when interpreting the observed association between increasing quartiles of thrombotic and fibrinolytic factors and multiple ASCVD risk factors and outcomes (Supplementary material online, Table S2).
Table 2.
Cox proportional hazards model of all seven atherothrombotic biomarkers for the prediction of atherosclerotic cardiovascular disease risk
| Variable | Hazard ratio (95% CI) | P-value |
|---|---|---|
| Plasminogena | 0.98 (0.82, 1.18) | 0.86 |
| OxPL-plasminogenb | 0.63 (0.53, 0.74) | <0.0001 |
| Fibrinogena | 2.88 (1.99, 4.17) | <0.0001 |
| Plasmin–antiplasmin complexb | 1.20 (0.95, 1.52) | 0.12 |
| D-dimerc | 1.63 (1.32, 2.00) | <0.0001 |
| Factor VIIId | 1.45 (1.20, 1.76) | <0.0001 |
| Lp(a)a | 1.03 (0.97, 1.10) | 0.36 |
Prior to estimating models, each biomarker was log-transformed. Cross-validation estimated C-index = 0.603. Table presents individual biomarker associations following adjustment for all other variables included in the model.
CI, confidence interval; Lp(a), lipoprotein(a); OxPL, oxidized phospholipids.
Log(mg/dL).
Log(nM).
Log(μg/mL).
Log(calibrated %).
Table 3.
Cox proportional hazards model for the prediction of atherosclerotic cardiovascular disease risk from individual atherothrombotic biomarkers in combination with traditional atherosclerotic cardiovascular disease risk factors
| Variable | Hazard ratio (95% CI) | P-value |
|---|---|---|
| Male sex | 1.66 (1.42, 1.94) | <0.0001 |
| Race | ||
| White | 1.00 | |
| Chinese | 0.72 (0.56, 0.91) | 0.007 |
| Black | 0.74 (0.62, 0.89) | 0.002 |
| Hispanic | 0.95 (0.80, 1.14) | 0.57 |
| Age, years | 1.04 (1.04, 1.05) | <0.0001 |
| Total cholesterol, mg/dL | 1.002 (1.000, 1.004) | 0.02 |
| HDL, mg/dL | 0.990 (0.984, 0.996) | 0.0005 |
| Systolic BP, mmHg | 1.011 (1.007, 1.014) | <0.0001 |
| Diabetes | 1.72 (1.46, 2.04) | <0.0001 |
| Smoking status | ||
| Never | 1.00 | |
| Former | 1.17 (1.01, 1.36) | 0.03 |
| Current | 1.54 (1.24, 1.92) | <0.0001 |
| Hypertension medications | 1.34 (1.16, 1.55) | <0.0001 |
| Plasminogena | 1.03 (0.86, 1.24) | 0.76 |
| OxPL-plasminogenb | 0.78 (0.65, 0.92) | 0.005 |
| Fibrinogena | 2.24 (1.52, 3.30) | <0.0001 |
| Plasmin–antiplasmin complexb | 1.07 (0.83, 1.38) | 0.62 |
| D-dimerc | 1.16 (0.91, 1.46) | 0.23 |
| Factor VIIId | 1.24 (1.02, 1.50) | 0.03 |
| Lp(a)a | 1.08 (1.01, 1.15) | 0.03 |
The table presents individual associations following adjustment for all other variables included in the model. Prior to estimating model, each biomarker [plasminogen, OxPL-plasminogen, fibrinogen, plasmin–antiplasmin complex, D-dimer, factor VIII, and Lp(a)] was log-transformed.
CI, confidence interval; HDL, high-density lipoprotein; BP, blood pressure; Lp(a), lipoprotein(a); OxPL, oxidized phospholipids.
Log(mg/dL).
Log(nM).
Log(μg/mL).
Log(calibrated %).
Figure 1.
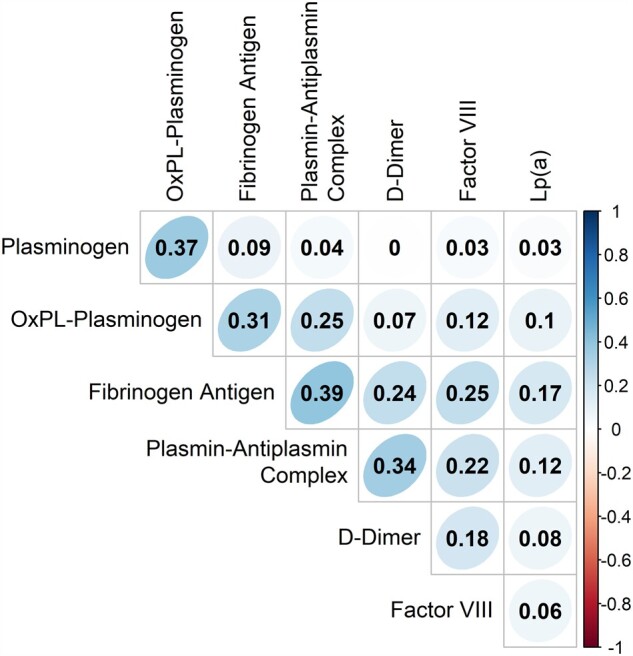
Continuous variable associations of study atherothrombotic biomarkers. Values represent the Pearson correlation coefficient between log-transformed values of each biomarker. Graded colours are utilized to depict strength of linear association (red represents negative association; blue represents positive association). Bartlett’s test of sphericity provided evidence that the covariance matrix for the biomarkers differed from the identity matrix (P < 0.0001).
In a multivariable Cox proportional hazards model, thrombotic factor (thrombotic propensity) was positively associated with incident ASCVD events before (Table 4) and after adjustment for traditional ASCVD risk factors (Table 5 and Supplementary material online, Table S3). In contrast, fibrinolytic factor (fibrinolytic propensity) was negatively associated with incident ASCVD events before (Table 4) and after adjustment for traditional ASCVD risk factors (Table 5 and Supplementary material online, Table S3). Without adjustment for traditional ASCVD risk factors, thrombotic factor was associated with an increased risk of a future ASCVD event with a hazard ratio (HR) and 95% confidence interval (CI) of 1.57 (95% CI 1.45, 1.70) for each unit increase (Table 4). Conversely, without adjustment for traditional ASCVD risk factors, higher fibrinolytic factor values were significantly (P < 0.0001) associated with a lower risk of ASCVD events with a HR of 0.76 (95% CI 0.70, 0.82) for each unit increase (Table 4). Considering the factors individually, rather than together, resulted in a substantial attenuation of the association of each factor with ASCVD risk (Supplementary material online, Table S4) indicating that the balance between thrombotic factor and fibrinolytic factor (i.e. thrombotic and fibrinolytic potential) contributes to the association with ASCVD beyond what can be observed from either factor alone. In the secondary analysis comparing estimated survival by quartiles of thrombotic factor and fibrinolytic factor, survival was greatest in combination (thrombotic factor: lowest quartile, fibrinolytic factor: highest quartile) with a Kaplan-Meier-estimated survival of 92.0% (95% CI 87.2%, 97.2%) at 15.25 years (Figure 2). Estimated survival was lowest in the combination (thrombotic factor: highest quartile, fibrinolytic factor: lowest quartile) with a Kaplan–Meier-estimated survival of 67.0% (95% CI 57.4%, 78.25%) (Figure 2). Estimated survival was similar and in-between the discordant factor quartiles combinations when thrombotic factor and fibrinolytic factor were most balanced (both low or both high) (Figure 2).
Table 4.
Cox proportional hazards model for atherosclerotic cardiovascular disease events utilizing thrombotic factor and fibrinolytic factor with both factors in the model
| Variable | Coefficient | Hazard ratio (95% CI) | P-value |
|---|---|---|---|
| Thrombotic factor (Factor 1) | 0.453 | 1.57 (1.45, 1.70) | <0.00001 |
| Fibrinolytic factor (Factor 2) | −0.275 | 0.76 (0.70, 0.82) | <0.00001 |
Concordance C-index = 0.600. Univariate Cox proportional hazards model analysis of atherothrombotic Factors 1 and 2 presented in Supplementary material online, Table S4.
Table 5.
Multivariable Cox proportional hazards model for atherosclerotic cardiovascular disease events with main effects for each covariate that is included in the pooled cohort equations model as well as thrombotic factor and fibrinolytic factor
| Variable | Hazard ratio (95% CI) | P-value |
|---|---|---|
| Male sex | 1.60 (1.37, 1.87) | <0.0001 |
| Race | ||
| White | ||
| Chinese | 0.72 (0.57, 0.92) | 0.009 |
| Black | 0.79 (0.66, 0.94) | 0.01 |
| Hispanic | 0.96 (0.81, 1.15) | 0.66 |
| Age, years | 1.04 (1.03, 1.05) | <0.0001 |
| Total cholesterol, mg/dL | 1.003 (1.001, 1.005) | 0.005 |
| HDL, mg/dL | 0.998 (0.983, 0.994) | <0.0001 |
| Systolic BP, mmHg | 1.011 (1.008, 1.014) | <0.0001 |
| Diabetes | 1.77 (1.50, 2.09) | <0.0001 |
| Smoking status | ||
| Never | ||
| Former | 1.51 (1.22, 1.88) | 0.05 |
| Current | 1.34 (1.16, 1.55) | 0.0002 |
| Hypertension medications | 1.36 (1.18, 1.57) | <0.0001 |
| Thrombotic factora | 1.28 (1.17, 1.40) | <0.0001 |
| Fibrinolytic factora | 0.87 (0.81, 0.95) | 0.001 |
Cross-validation estimated C-index = 0.721. The table presents individual associations following adjustment for all other variables included in the model.
CI, confidence interval; HDL, high-density lipoprotein; BP, blood pressure.
Standardized unit.
Figure 2.

Kaplan–Meier plot for participants with discordant levels of the factors (low thrombotic factor and high fibrinolytic factor; high thrombotic factor and low fibrinolytic factor), and two concordant levels (low thrombotic factor and low fibrinolytic factor; high thrombotic factor and high fibrinolytic factor).
The likelihood ratio test demonstrates that there is significant evidence that the thrombotic and fibrinolytic factors explain variability in ASCVD event risk that is not explained by traditional risk factors. The addition of thrombotic factor and fibrinolytic factor to a multivariable Cox proportional hazards model that includes the variables of the ACC/AHA ASCVD risk prediction calculators (AKA: pool cohort equation) resulted in significant improvement in model fit (LR test χ2 P-value <0.0001) and discrimination—moving the C-index from 0.718 to 0.721 as estimated by cross-validation (Supplementary material online, Table S5). Evidence of an interaction between the factors and coronary artery calcification was not observed (P = 0.37). The model that includes both factors also demonstrates the lowest Bayesian information criterion value (best model) in comparison with Cox proportional hazards models with the individual atherothrombotic biomarkers, with and without traditional ASCVD risk factors (Table 6).
Table 6.
Bayesian information criteria
| Model | Bayesian information criteria | Comparison | ΔBIC | Bayes factor conclusion |
|---|---|---|---|---|
| M1: All atherothrombotic biomarkers | 14 695.38 | |||
| M2: Thrombotic factor and fibrinolytic factor | 14 683.56 | M2 vs. M1 | −11.82 | Very strong evidence M2 is better than M1 |
| M3: Traditional risk factors | 14 342.06 | |||
| M4: Traditional risk factors + all atherothrombotic biomarkers | 14 345.84 | M4 vs. M3 | 3.78 | No evidence M4 is better than M3 |
| M5: Traditional risk factors + thrombotic factor and fibrinolytic factor | 14 327.85 | M5 vs. M3 | −14.21 | Very strong evidence M5 is better than M3; very strong evidence M5 is better than M4 |
Analyses of the association of the individual components of the ASCVD outcome via multivariable Cox proportional hazards model were consistent with the main findings of the study (Supplementary material online, Table S6–S9). In the exploratory multivariable Cox proportional hazards analysis of incident venous thromboembolic events as the outcome, we observed a HR of 1.195 (95% CI 0.993, 1.436, P = 0.06) for thrombotic factor and HR of 0.987 (95% CI 0.839, 1.161, P = 0.87) for fibrinolytic factor (Supplementary material online, Table S10). A significant association was not observed between thrombotic factor [HR 1.055 (95% CI 0.956, 1.165), P = 0.29], or fibrinolytic factor [HR 0.928 (95% CI 0.854, 1.008), P = 0.08] and incident cancer (Supplementary material online, Table S11).
Discussion
This study demonstrates a strong association between atherothrombotic biomarkers and ASCVD events over >14 years of follow-up. These biomarkers explain variability in ASCVD event risk that is not accounted for by traditional risk factors. Atherothrombotic propensity was best represented by two new variables (factors) that represent the complex interplay between multiple components of the atherothrombotic process. Factor analysis produced two factors that represented the multidimensional relationship between all seven atherothrombotic biomarkers. Factor 1 represents a new ‘axis’ along which the propensity to form thrombus varies (i.e. thrombotic factor) while Factor 2 represents a new ‘axis’ along which fibrinolytic propensity varies (i.e. fibrinolytic factor) (Graphical abstract). These factors were moderately positively correlated, which provides evidence that when thrombus (fibrin) formation is high, fibrin dissolution is also high. This suggests that increased thrombotic propensity is most often balanced by increased fibrinolytic propensity. However, ASCVD risk was highest and lowest when thrombotic propensity and fibrinolytic propensity were most unbalanced. For example, the highest ASCVD risk combination by factor quartiles was the combination of a high thrombotic factor score (high thrombotic propensity) and a low fibrinolytic factor score (low fibrinolytic propensity). Consistent with this finding, the lowest ASCVD event risk combination was found in participants with a low thrombotic factor score (low thrombotic propensity) and a high fibrinolytic factor score (high fibrinolytic propensity). Participants with high fibrinolytic and high thrombotic, or low fibrinolytic and low thrombotic potential/propensity, as determined by thrombotic and fibrinolytic factor scores, were at relative intermediate risk. The finding that the association of the factors with ASCVD event risk is stronger when both factors are evaluated together and attenuated when evaluated individually provides further evidence that the balance (or imbalance) between thrombotic propensity and fibrinolytic propensity, represented by these factors, contributes to ASCVD event risk. These data suggest that thrombotic factor and fibrinolytic factor scores are reflective of an individual’s thrombotic and fibrinolytic propensity and together are a measure of propensity for clinically meaningful atherothrombotic ASCVD events.
Graphical Abstract.

While atherosclerotic plaque is a prerequisite for ASCVD events, it alone is not sufficient for ASCVD events. The lack of evidence for an interaction between the thrombotic factor, fibrinolytic factor, coronary artery calcium score and ASCVD events suggests that the association between these novel thrombotic and fibrinolytic factors and ASCVD events are likely independent of the association of stable atherosclerosis, as assessed by coronary artery calcification, and ASCVD events. These factors were developed specifically for atherothrombotic events and no statistically significant associations were observed between these factors and venous thromboembolic events and incident cancer. However, these analyses were exploratory, and the findings do not exclude the possibility that one of both factors may be related to thrombotic events more broadly, including venous thromboembolic events and thrombotic propensity associated with cancer—hypotheses which warrant further investigation.
The study of atherothrombotic factors is complicated by the multiple biological interactions dictating their activity in vivo—a process which cannot be reproduced and therefore cannot be studied in vitro or duplicated in animal models.24 The factor analysis performed in this study allowed us to represent the multidimensional relationships between multiple in vivo measures of atherothrombotic biomarkers. Factor analysis is a multivariate statistical technique that seeks to identify latent (or unobservable) structure present in multivariate data.25,26 Latent factors represent unobservable variables in a biological process that are determined by accounting for how multiple biomarkers within a specific domain, such as thrombogenicity, interact with each other. These factors then can be evaluated for impact on an outcome (e.g. ASCVD events). For example, in the case of atherothrombosis we cannot directly measure biological processes such as in vivo fibrinolytic or thrombotic potential—these are latent processes. However, we do measure the concentrations of biomarkers, as well as the interrelatedness of biomarkers, which drive or are driven by the biological (latent) process (e.g. thrombosis and fibrinolysis). The application of factor analysis in this study determined two new factors that capture the shared communality in sets of biomarkers to allow for greater understanding of the relationship between processes (thrombosis and fibrinolysis) and the outcomes (ASCVD events). These approaches address limitations of incorporating multiple (dependent) biomarkers in linear models, including issues of collinearity and misspecification of biomarker interactions. The two factors (thrombotic factor and fibrinolytic factor) determined by this factor analysis explained ∼50% of the variability in these factors among the individuals in the MESA cohort.
Consistent with our findings that the thrombotic factor score is higher with higher fibrinogen, D-dimer, plasmin–antiplasmin, FVIII, and Lp(a), higher levels of these analyses have independently been associated with higher risk of ASCVD events.27–40 Plasminogen is the precursor to plasmin, which digests fibrin.41 In this study, higher fibrinolytic factor scores were associated with higher plasminogen levels and lower ASCVD event rates.
Our analysis also included examining the impact of a newly studied atherothrombotic factor—OxPL-PLG. OxPL are carried by apoB-containing lipoproteins, primarily by Lp(a) among lipoproteins13,42 and by plasminogen in circulation.10,43 When OxPL are bound to plasminogen, it was demonstrated that the OxPL component on plasminogen facilitates fibrinolysis in vitro.10 Extension of these findings to in vivo data from our laboratory linked OxPL-PLG to atherothrombosis by demonstrating that OxPL-PLG levels are lower among subjects with an acute MI vs. stable CAD.11 More specifically, OxPL-PLG levels were lower among thrombotic MI vs. non-thrombotic (type II) MI subjects, suggesting such patients have a lower propensity for fibrinolysis.11 In this study, higher levels of OxPL-PLG were associated with higher fibrinolytic factor scores, consistent with greater fibrinolytic propensity and with our observation that higher fibrinolytic factor scores were associated with fewer ASCVD events.
These data underscore the importance of an individual’s propensity for thrombus formation/degradation in future ASCVD events. While the use of antithrombotic agents has been shown to significantly reduce ASCVD events, the efficacy of these agents is limited by iatrogenic bleeding, particularly anticoagulant therapy.44–46 The thrombotic and fibrinolytic factors, developed with multivariate statistical methodology in this study, allow for a more complete representation of the balance of thrombosis and fibrinolysis which determines atherothrombosis. These factors hold promise for predicting an individual’s propensity to form a thrombus resulting in an ASCVD event. Such information may allow for precision medicine when selecting individuals for antithrombotic therapy to more effectively balance risk of iatrogenic bleeding with the benefits of ASCVD event risk reduction, resulting in safer treatment and cost savings for an increasingly resource-strained health care system.47
Limitations and future directions
The dynamic nature of the atherothrombotic biomarkers measured in this study was not analysed secondary to measurements at baseline alone. How these factors change over time and how such change is related to ASCVD events is of interest for a future study. Platelet activation resulting in platelet aggregation is fundamental to the atherothrombotic process,7 and measures of platelet activation and aggregation may provide additional information but were not available in this project. Although the association of the factors studied with fibrinolysis and thrombosis is well established, it is possible that these factors are associated with ASCVD event by mechanisms in addition to fibrinolysis and thrombosis. While only a slight improvement in the cross-validation estimated C-index was observed (from 0.718 to 0.721), likelihood ratio test shows significant evidence of a better model fit with the incorporation of the thrombotic and fibrinolytic factors over a model without these factors. This finding provides evidence that these factors provide information about ASCVD event prediction beyond what is ascertained from traditional risk factors alone. Nevertheless, in current risk prediction models, this degree of C-index change is not likely to provide a meaningful clinical impact on ASCVD risk prediction for the average patient. However, our multivariable Cox proportional hazards model illustrates that substantial imbalance of thrombotic and fibrinolytic factors may more clearly identify patients which are significantly protected or prone to ASCVD events; identification of such patients will allow for testing of predicted atherothrombotic risk specific treatment strategies. Finally, aetiological subtyping of ASCVD events (e.g. atherothrombotic MI vs. non-atherothrombotic MI) was not available in MESA at the time of this study.48 Work is currently underway to provide this aetiological differentiation of ASCVD event types in MESA. We hypothesize that thrombotic and fibrinolytic factor balance will provide clinical actionable risk prediction data specific to atherothrombotic ASCVD events (e.g. type 1 vs. type 2 MI).
Conclusions
Atherothrombotic biomarkers explain variability in ASCVD event risk that is not accounted for by traditional risk factors. Factor analysis was successful in developing two new variables (factors) that appear to be representative of an individual’s propensity to form thrombus (thrombotic factor) and propensity to degrade fibrin (fibrinolytic factor). The balance or imbalance between a patient’s thrombotic and fibrinolytic propensity, as represented by thrombotic factor and fibrinolytic factor scores, is associated with improved ASCVD event risk prediction. The improvement in risk prediction is likely secondary to accounting for atherothrombotic risk not addressed by traditional ASCVD risk factors. Accurate characterization of an individual patient’s atherothrombotic event risk holds great promise for more efficacious and judicious use of antithrombotic therapy for the prevention of ASCVD events.
Supplementary material
Supplementary material is available at European Heart Journal online.
Supplementary Material
Acknowledgements
We acknowledge Allison E. Smith at the University of Louisville for her editing assistance.
Contributor Information
Andrew P DeFilippis, Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University Medical Center, 1215 21st Avenue South, MCE 5th Floor, North Tower, Nashville, TN 37232, USA.
Patrick J Trainor, Department of Chemistry and Biochemistry, New Mexico State University, 1175 N Horseshoe Dr., Las Cruces, NM 88003, USA.
George Thanassoulis, Department of Medicine, Division of Experimental Medicine, McGill University Health Center, 1001 Decarie Boulevard, Montreal, QC H4A 3J1, Canada.
Lyndia C Brumback, Department of Biostatistics, University of Washington, 1959 NE Pacific Street Seattle, WA 98105, USA.
Wendy S Post, Department of Medicine, Division of Cardiology, Johns Hopkins University, School of Medicine, 600 N. Wolfe Street, Baltimore, MD 21287, USA.
Michael Y Tsai, Department of Laboratory Medicine and Pathology, University of Minnesota, 420 Delaware ST SE, Minneapolis, Minnesota 55455, USA.
Sotirios Tsimikas, Division of Cardiology, Department of Medicine, University of California San Diego, 9500 Gilman Dr., La Jolla, CA 92093, USA.
Funding
This research was supported by a Heart to Heart Grant awarded by the Alpha Phi Foundation and by an Institutional Development Award (IDeA) from the National Institute of General Medical Sciences of the National Institutes of Health (NIH) under grant number P20GM103451, NIH grant GM139730 (P.T.), and from NIH grant number HL128550 (G.T., S.T.). S.T. has received research support from the Fondation Leducq and partial support from NIH grants HL136275, HL106579, HL108735, HL136098, and HL135737.
This MESA study used in this analysis was supported by contracts HHSN268201500003I, N01-HC-95159, N01-HC-95160, N01-HC-95161, N01-HC-95162, N01-HC-95163, N01-HC-95164, N01-HC-95165, N01-HC-95166, N01-HC-95167, N01-HC-95168, and N01-HC-95169 from the National Heart, Lung, and Blood Institute and by grants UL1-TR-000040, UL1-TR-001079, and UL1-TR-001420 from the National Center for Advancing Translational Sciences (NCATS). The authors thank the other investigators, the staff, and the participants of the MESA study for their valuable contributions. A full list of participating MESA investigators and institutions can be found at http://www.mesa-nhlbi.org.
Conflict of interest: S.T. is a co-inventor and receives royalties from patents owned by UCSD on oxidation-specific antibodies and of biomarkers related to oxidized lipoproteins and is a co-founder and has an equity interest in Oxitope, Inc. and its affiliates (‘Oxitope’) as well as in Kleanthi Diagnostics, LLC (‘Kleanthi’). Although these relationships have been identified for conflict of interest management based on the overall scope of the project and its potential benefit to Oxitope and Kleanthi, the research findings included in this particular publication may not necessarily relate to the interests of Oxitope and Kleanthi. The terms of this arrangement have been reviewed and approved by the University of California, San Diego, in accordance with its conflict of interest policies. S.T. is an employee of Ionis Pharmaceuticals and of the University of California San Diego. The other authors have no relationships with industry pertinent to this work.
Data availability
Interested investigators may request access to the data by contacting the MESA data coordinating center (chsccweb@u.washington.edu).
References
- 1. Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics-2021 update: a report from the American Heart Association. Circulation 2021;143:e254–e743. [DOI] [PubMed] [Google Scholar]
- 2. Fifth Joint Task Force of the European Society of Cardiology; European Association of Echocardiography; European Association of Percutaneous Cardiovascular Interventions; European Heart Rhythm Association; Heart Failure Association; European Association for Cardiovascular Prevention and Rehabilitation; European Atherosclerosis Society; International Society of Behavioural Medicine; European Stroke Organisation; European Society of Hypertension; European Association for the Study of Diabetes; European Society of General Practice/Family Medine; International Diabetes Federation Europe; European Heart Network. European Guidelines on cardiovascular disease prevention in clinical practice (version 2012): the Fifth Joint Task Force of the European Society of Cardiology and Other Societies on Cardiovascular Disease Prevention in Clinical Practice (constituted by representatives of nine societies and by invited experts). Eur J Prev Cardiol 2012;19:585–667. [DOI] [PubMed] [Google Scholar]
- 3. Maseri A, Fuster V. Is there a vulnerable plaque? Circulation 2003;107:2068–2071. [DOI] [PubMed] [Google Scholar]
- 4. Naghavi M, Libby P, Falk E, Casscells SW, Litovsky S, Rumberger J, Badimon JJ, Stefanadis C, Moreno P, Pasterkamp G, Fayad Z, Stone PH, Waxman S, Raggi P, Madjid M, Zarrabi A, Burke A, Yuan C, Fitzgerald PJ, Siscovick DS, de Korte CL, Aikawa M, Juhani Airaksinen KE, Assmann G, Becker CR, Chesebro JH, Farb A, Galis ZS, Jackson C, Jang IK, Koenig W, Lodder RA, March K, Demirovic J, Navab M, Priori SG, Rekhter MD, Bahr R, Grundy SM, Mehran R, Colombo A, Boerwinkle E, Ballantyne C, Insull W Jr., Schwartz RS, Vogel R, Serruys PW, Hansson GK, Faxon DP, Kaul S, Drexler H, Greenland P, Muller JE, Virmani R, Ridker PM, Zipes DP, Shah PK, Willerson JT. From vulnerable plaque to vulnerable patient: a call for new definitions and risk assessment strategies: part I. Circulation 2003;108:1664–1672. [DOI] [PubMed] [Google Scholar]
- 5. Sambola A, Osende J, Hathcock J, Degen M, Nemerson Y, Fuster V, Crandall J, Badimon JJ. Role of risk factors in the modulation of tissue factor activity and blood thrombogenicity. Circulation 2003;107:973–977. [DOI] [PubMed] [Google Scholar]
- 6. Arbab-Zadeh A, Fuster V. The myth of the "vulnerable plaque": transitioning from a focus on individual lesions to atherosclerotic disease burden for coronary artery disease risk assessment. J Am Coll Cardiol 2015;65:846–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angiolillo DJ, Ueno M, Goto S. Basic principles of platelet biology and clinical implications. Circ J 2010;74:597–607. [DOI] [PubMed] [Google Scholar]
- 8. Ferreira-González I, Marsal JR, Ribera A, Permanyer-Miralda G, García-Del Blanco B, Martí G, Cascant P, Martín-Yuste V, Brugaletta S, Sabaté M, Alfonso F, Capote ML, De La Torre JM, Ruíz-Lera M, Sanmiguel D, Cárdenas M, Pujol B, Baz JA, Iñiguez A, Trillo R, González-Béjar O, Casanova J, Sánchez-Gila J, García-Dorado D. Background, incidence, and predictors of antiplatelet therapy discontinuation during the first year after drug-eluting stent implantation. Circulation 2010;122:1017–1025. [DOI] [PubMed] [Google Scholar]
- 9. Bild DE, Bluemke DA, Burke GL, Detrano R, Diez Roux AV, Folsom AR, Greenland P, Jacob DR Jr., Kronmal R, Liu K, Nelson JC, O'Leary D, Saad MF, Shea S, Szklo M, Tracy RP. Multi-ethnic study of atherosclerosis: objectives and design. Am J Epidemiol 2002;156:871–881. [DOI] [PubMed] [Google Scholar]
- 10. Leibundgut G, Arai K, Orsoni A, Yin H, Scipione C, Miller ER, Koschinsky ML, Chapman MJ, Witztum JL, Tsimikas S. Oxidized phospholipids are present on plasminogen, affect fibrinolysis, and increase following acute myocardial infarction. J Am Coll Cardiol 2012;59:1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. DeFilippis AP, Chernyavskiy I, Amraotkar AR, Trainor PJ, Kothari S, Ismail I, Hargis CW, Korley FK, Leibundgut G, Tsimikas S, Rai SN, Bhatnagar A. Circulating levels of plasminogen and oxidized phospholipids bound to plasminogen distinguish between atherothrombotic and non-atherothrombotic myocardial infarction. J Thromb Thrombolysis 2016;42:61–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Byun YS, Lee JH, Arsenault BJ, Yang X, Bao W, DeMicco D, Laskey R, Witztum JL, Tsimikas S; TNT Trial Investigators. Relationship of oxidized phospholipids on apolipoprotein B-100 to cardiovascular outcomes in patients treated with intensive versus moderate atorvastatin therapy: the TNT trial. J Am Coll Cardiol 2015;65:1286–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsimikas S, Bergmark C, Beyer RW, Patel R, Pattison J, Miller E, Juliano J, Witztum JL. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J Am Coll Cardiol 2003;41:360–370. [DOI] [PubMed] [Google Scholar]
- 14. Goff DC Jr., Lloyd-Jones DM, Bennett G, Coady S, D'Agostino RB, Gibbons R, Greenland P, Lackland DT, Levy D, O'Donnell CJ, Robinson JG, Schwartz JS, Shero ST, Smith SC Jr., Sorlie P, Stone NJ, Wilson PW, Jordan HS, Nevo L, Wnek J, Anderson JL, Halperin JL, Albert NM, Bozkurt B, Brindis RG, Curtis LH, DeMets D, Hochman JS, Kovacs RJ, Ohman EM, Pressler SJ, Sellke FW, Shen WK, Smith SC Jr., Tomaselli GF; American College of Cardiology/American Heart Association Task Force on Practice Guidelines. 2013 ACC/AHA guideline on the assessment of cardiovascular risk: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation 2014;129:S49–73. [DOI] [PubMed] [Google Scholar]
- 15. Bartlett MS. The effect of standardization on a Chi-2 approximation in factor analysis. Biometrika 1951;38:337–344. [Google Scholar]
- 16. Stef van Buuren KG-O. mice: multivariate imputation by chained equations in R. J Stat Softw 2011;45. [Google Scholar]
- 17. van Buuren S, Boshuizen HC, Knook DL. Multiple imputation of missing blood pressure covariates in survival analysis. Stat Med 1999;18:681–694. [DOI] [PubMed] [Google Scholar]
- 18. van Buuren S. Multiple imputation of discrete and continuous data by fully conditional specification. Stat Methods Med Res 2007;16:219–242. [DOI] [PubMed] [Google Scholar]
- 19. Harman HH, Jones WH. Factor analysis by minimizing residuals (minres). Psychometrika 1966;31:351–368. [DOI] [PubMed] [Google Scholar]
- 20. Clarkson DB, Jennrich RI. Quartic rotation criteria and algorithms. Psychometrika 1988;53:251–259. [Google Scholar]
- 21. Thurstone LL. Multiple factor analysis. Psychol Rev 1931;38:406–427. [Google Scholar]
- 22. Kadane JB, Lazar NA. Methods and criteria for model selection. J Am Stat Assoc 2004;99:279–290. [Google Scholar]
- 23. Harrell F, Regression Modeling Strategies: With Applications to Linear Models, Logistic and Ordinal Regression, and Survival Analysis. New York, USA: Springer International Publishing; 2015. [Google Scholar]
- 24. Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, Lopez CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG; Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc Natl Acad Sci U S A 2013;110:3507–3512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. HäRdle WaS Lo, Applied Multivariate Statistical Analysis. 3rd ed. Springer, Heidelberg/New York: 2012. [Google Scholar]
- 26. Johnson RA, Wichern DW, Applied Multivariate Statistical Analysis. 6th ed. Upper Saddle River, NJ: Pearson Prentice Hall; 2007. [Google Scholar]
- 27. Danesh J, Collins R, Appleby P, Peto R. Association of fibrinogen, C-reactive protein, albumin, or leukocyte count with coronary heart disease: meta-analyses of prospective studies. JAMA 1998;279:1477–1482. [DOI] [PubMed] [Google Scholar]
- 28. Ernst E, Resch KL. Fibrinogen as a cardiovascular risk factor: a meta-analysis and review of the literature. Ann Intern Med 1993;118:956–963. [DOI] [PubMed] [Google Scholar]
- 29. Maresca G, Di Blasio A, Marchioli R, Di Minno G. Measuring plasma fibrinogen to predict stroke and myocardial infarction: an update. Arterioscler Thromb Vasc Biol 1999;19:1368–1377. [DOI] [PubMed] [Google Scholar]
- 30. Alehagen U, Dahlstrom U, Lindahl TL. Elevated D-dimer level is an independent risk factor for cardiovascular death in out-patients with symptoms compatible with heart failure. Thromb Haemost 2004;92:1250–1258. [DOI] [PubMed] [Google Scholar]
- 31. Chang SS, Lee SH, Wu JY, Ning HC, Chiu TF, Wang FL, Chen JH, Li CH, Lee CC, Chan RC. Evaluation of the value of rapid D-dimer test in conjunction with cardiac troponin I test for early risk stratification of myocardial infarction. J Thromb Thrombolysis 2010;30:472–478. [DOI] [PubMed] [Google Scholar]
- 32. Cushman M, Lemaitre RN, Kuller LH, Psaty BM, Macy EM, Sharrett AR, Tracy RP. Fibrinolytic activation markers predict myocardial infarction in the elderly. The Cardiovascular Health Study. Arterioscler Thromb Vasc Biol 1999;19:493–498. [DOI] [PubMed] [Google Scholar]
- 33. Lowe GD, Rumley A, McMahon AD, Ford I, O'Reilly DS, Packard CJ, West of Scotland Coronary Prevention Study Group. Interleukin-6, fibrin D-dimer, and coagulation factors VII and XIIa in prediction of coronary heart disease. Arterioscler Thromb Vasc Biol 2004;24:1529–1534. [DOI] [PubMed] [Google Scholar]
- 34. Menown IB, Mathew TP, Gracey HM, Nesbitt GS, Murray P, Young IS, Adgey AA. Prediction of Recurrent Events by D-Dimer and Inflammatory Markers in Patients with Normal Cardiac Troponin I (PREDICT) Study. Am Heart J 2003;145:986–992. [DOI] [PubMed] [Google Scholar]
- 35. Smith A, Patterson C, Yarnell J, Rumley A, Ben-Shlomo Y, Lowe G. Which hemostatic markers add to the predictive value of conventional risk factors for coronary heart disease and ischemic stroke? The Caerphilly Study. Circulation 2005;112:3080–3087. [DOI] [PubMed] [Google Scholar]
- 36. Gorog DA. Prognostic value of plasma fibrinolysis activation markers in cardiovascular disease. J Am Coll Cardiol 2010;55:2701–2709. [DOI] [PubMed] [Google Scholar]
- 37. Folsom AR, Rosamond WD, Shahar E, Cooper LS, Aleksic N, Nieto FJ, Rasmussen ML, Wu KK. Prospective study of markers of hemostatic function with risk of ischemic stroke. The Atherosclerosis Risk in Communities (ARIC) Study Investigators. Circulation 1999;100:736–742. [DOI] [PubMed] [Google Scholar]
- 38. Rossouw JE, Cushman M, Greenland P, Lloyd-Jones DM, Bray P, Kooperberg C, Pettinger M, Robinson J, Hendrix S, Hsia J. Inflammatory, lipid, thrombotic, and genetic markers of coronary heart disease risk in the women's health initiative trials of hormone therapy. Arch Intern Med 2008;168:2245–2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Labudovic D, Kostovska I, Tosheska Trajkovska K, Cekovska S, Brezovska Kavrakova J, Topuzovska S. Lipoprotein(a)—link between atherogenesis and thrombosis. Prague Med Rep 2019;120:39–51. [DOI] [PubMed] [Google Scholar]
- 40. Tsimikas S, Fazio S, Ferdinand KC, Ginsberg HN, Koschinsky ML, Marcovina SM, Moriarty PM, Rader DJ, Remaley AT, Reyes-Soffer G, Santos RD, Thanassoulis G, Witztum JL, Danthi S, Olive M, Liu L. NHLBI Working Group recommendations to reduce lipoprotein(a)-mediated risk of cardiovascular disease and aortic stenosis. J Am Coll Cardiol 2018;71:177–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mosesson MW. Fibrinogen and fibrin structure and functions. J Thromb Haemost 2005;3:1894–1904. [DOI] [PubMed] [Google Scholar]
- 42. Tsimikas S, Lau HK, Han KR, Shortal B, Miller ER, Segev A, Curtiss LK, Witztum JL, Strauss BH. Percutaneous coronary intervention results in acute increases in oxidized phospholipids and lipoprotein(a): short-term and long-term immunologic responses to oxidized low-density lipoprotein. Circulation 2004;109:3164–3170. [DOI] [PubMed] [Google Scholar]
- 43. Edelstein C, Pfaffinger D, Yang M, Hill JS, Scanu AM. Naturally occurring human plasminogen, like genetically related apolipoprotein(a), contains oxidized phosphatidylcholine adducts. Biochim Biophys Acta 2010;1801:738–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005;143:241–250. [DOI] [PubMed] [Google Scholar]
- 45. Andreotti F, Testa L, Biondi-Zoccai GG, Crea F. Aspirin plus warfarin compared to aspirin alone after acute coronary syndromes: an updated and comprehensive meta-analysis of 25,307 patients. Eur Heart J 2006;27:519–526. [DOI] [PubMed] [Google Scholar]
- 46. Mega JL, Braunwald E, Wiviott SD, Bassand JP, Bhatt DL, Bode C, Burton P, Cohen M, Cook-Bruns N, Fox KA, Goto S, Murphy SA, Plotnikov AN, Schneider D, Sun X, Verheugt FW, Gibson CM; ATLAS ACS 2-TIMI 51 Investigators. Rivaroxaban in patients with a recent acute coronary syndrome. N Engl J Med 2012;366:9–19. [DOI] [PubMed] [Google Scholar]
- 47. Joshi PH, Chaudhari S, Blaha MJ, Jones SR, Martin SS, Post WS, Cannon CP, Fonarow GC, Wong ND, Amsterdam E, Hirshfeld JW, Blumenthal RS. A point-by-point response to recent arguments against the use of statins in primary prevention: this statement is endorsed by the American Society for Preventive Cardiology. Clin Cardiol 2012;35:404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. DeFilippis AP, Nasir K, Blaha MJ. Myocardial infarction as a clinical end point in research. Circ Res 2019;124:1701–1703. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Interested investigators may request access to the data by contacting the MESA data coordinating center (chsccweb@u.washington.edu).