

Abstract
The effect of immunotherapy is limited by oncometabolite D-2-hydroxyglutarate (D2HG). D2HGDH is an inducible enzyme that converts D2HG into the endogenous metabolite 2-oxoglutarate. We aimed to evaluate the impairment of CD8 T lymphocyte function in the high-D2HG environment and to explore the phenotypic features and anti-tumor effect of D2HGDH-modified CAR-T cells. D2HG treatment inhibited the expansion of human CD8 T lymphocytes and CAR-T cells, increased their glucose uptake, suppressed effector cytokine production, and decreased the central memory cell proportion. D2HGDH-modified CAR-T cells displayed distinct phenotypes, as D2HGDH knock-out (KO) CAR-T cells exhibited a significant decrease in central memory cell differentiation and intracellular cytokine production, while D2HGDH over-expression (OE) CAR-T cells showed predominant killing efficacy against NALM6 cancer cells in high-D2HG medium. In vivo xenograft experiments confirmed that D2HGDH-OE CAR-T cells decreased serum D2HG and improved the overall survival of mice bearing NALM6 cancer cells with mutation IDH1. Our findings demonstrated that the immunosuppressive effect of D2HG and distinct phenotype of D2HGDH modified CAR-T cells. D2HGDH-OE CAR-T cells can take advantage of the catabolism of D2HG to foster T cell expansion, function, and anti-tumor effectiveness.
Keywords: immunotherapy, oncometabolites, D-2-hydroxyglutarate, chimeric antigen receptor, D2HGDH
Graphical abstract
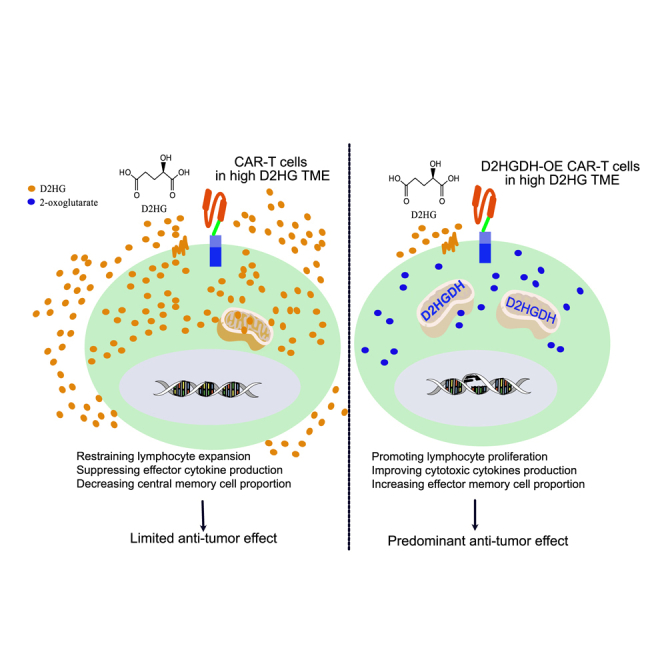
Guo et al. reveal the immunosuppressive effect of D2HG and explore the phenotypic characteristics of modified CAR-T cells through knock-out and over-expression of D2HGDH. D2HGDH over-expression in CAR-T cells improves the killing effect against cancer cells through the potential of metabolic adaptability in an immunosuppressive environment.
Introduction
The treatment of relapsed and refractory hematopoietic malignancies remains challenging despite advancements in immunotherapy and targeted therapy.1 Specifically, chimeric antigen receptor (CAR)-modified T cell therapy targeting CD19 has demonstrated substantial efficacy in highly refractory hematologic malignancies; thus, application of this therapeutic approach is currently the hope for patients when the standard cancer treatment fails.1,2 CAR-T cell therapy works by harnessing the host immune system via importing a functional domain in an antigen-specific manner to recognize and eliminate cancer cells through direct cytolytic function and indirect induction of the destruction of tumor cells by external inflammatory stimuli.3 However, this strategy remains challenging, since immune cells can develop tolerance within the tumor microenvironment (TME).4,5 Active research is now underway to overcome the resistance of the “immunologically cold” TME by uncovering key molecules that impair the cytotoxicity of CAR-T cells to cancer cells.6,7
Metabolic reprogramming in lymphocytes is accomplished by phenotypic shifts in T cell-specific function, differentiation, expansion, and fate.8 D-2-hydroxyglutarate (D2HG) is an oncometabolite in the TME and has emerged as a critical regulator of tumorigenesis, maintenance, genetic instability, and ultimately malignant transformation.9,10 D2HG is produced via somatic gain-of-function mutations in IDH1/2 in cancer cells in the TME and is now recognized as an immunosuppressive mediator.11 Previous studies have revealed that D2HG treatment impaired activated T cell migration, proliferation, and cytokine secretion.12 In vivo experiments showed that mice bearing mutational IDH1 gliomas displayed accumulated D2HG, down-regulated leukocyte chemotaxis, and reduced immune cell infiltration of macrophages, monocytes, and neutrophils.13 Clinical evidence revealed that D2HG concentration could be significantly increased up to the millimole level in IDH1-mutant leukemia and glioma patients.14,15 These patients with mutational IDH1 quickly develop tolerance to immunotherapy and have a poor prognosis.16,17 In this research study, we modified a CAR vector to metabolize high accumulated D2HG and subsequently evaluated its effectiveness against anti-tumor activities in a metabolically immunosuppressive TME.
Results
Phenotypic characteristics of CD8+ T cells treated with D2HG
It was reported that D2HG could accumulate up to millimole levels in cancer cells through gain-of-function of IDH1/2 mutations.14,15 To verify the effects of D2HG on CD8+ T lymphocyte proliferation, activation, cytotoxicity, and differentiation, we isolated peripheral blood mononuclear cells (PBMCs) from healthy donors, purified CD8+ T lymphocytes through positive selection, and treated the purified cells with D2HG (Figure 1A) in accordance with the related protocol.18,19 After incubation with anti-human CD3/CD28 antibody-coated microbeads, CD8+ T lymphocytes were activated, as indicated by positive CD45RO expression (Figure 1B). Activated T cell proliferation is susceptible to glucose metabolism; thus, we measured the proliferation by a carboxyfluorescein succinimidyl ester (CFSE) assay. To reveal the metabolic intervention effect, we added D2HG stock solution to the medium to form a series of D2HG solutions with concentrations ranging from 100 to 10 mM. After purified CD8+ T lymphocytes had been cultured for 5 days, the T cell proliferation was significantly inhibited after D2HG treatment, and there was a concentration-effect relationship of D2HG on the inhibition of T cell proliferation (Figures 1C and 1D). Activation of CD8+ T lymphocytes relies on glycolytic metabolism to sustain effector function, as glucose is the primary source of energy.20 Glucose uptake was then measured by a 2-NBDG assay after the preliminary experiment (Figure 1E). We found that glucose uptake was significantly increased when the final concentration of D2HG reached above 100 μM (Figure 1F). Moreover, 500 μM D2HG treatment resulted in scattered 2-NBDG-positive cells. The D2HG concentration of 500 μM was a physiologically relevant concentration and was thus used for the following experiments. The unique metabolic characteristics of T cell were associated with memory subsets; thus, we investigated the protein expression of CD62L and CD45RO after treatment with 500 μM D2HG for 7 days. A typical FACS plot depicted an increased effector memory cell (Tem, CD45RO + CD62L–) proportion and a decreased central memory cell (Tcm, CD45RO + CD62L+) proportion from 3 different biological duplications (Figure 1G). Previous studies reported that memory cell subsets were related to intracellular metabolism and functional properties.21 Then, we treated activated CD8+ T lymphocytes with D2HG solution and found that 500 μM D2HG suppressed effector cytokine production, as intracellular cytokine IFN-γ+, TNF-α+ and lytic granule protease granzyme B+ cells were significantly decreased when the cells were cultured for more than 7 days (Figure 1H). Furthermore, D2HG treatment resulted in significantly increased proportions of PD-1- and TIM-3-positive cells when they were co-cultured with NALM6 cells for 7 days (Figure 1I).
Figure 1.

Phenotypic characteristics of CD8+ T cells treated with D2HG
(A) Flowchart showing the strategy for experimental D2HG treatment on CD8+ T lymphocytes.
(B) Representative FACS plots showing CD8 and CD45RO with gating selection of activated T lymphocytes.
(C) Representative FACS plots showing CFSE with gating selection of proliferating cells after different concentration of D2HG treatment for 3 days from 3 biological replicates for each group.
(D) Histogram showing the cell proliferating differences after gradient final concentration of D2HG.
(E) Representative FACS plots showing 2-NBDG with gating selection of glucose uptake after different concentration of D2HG treatment from 3 biological replicates for each group.
(F) Histogram showing the glucose uptake differences after gradient final concentration of D2HG.
(G) Representative FACS plots showing CD45RO versus CD62L for the memory subset after D2HG treatment of 500 μM from 3 biological replicates for each group. Differences of memory cell subsets are labeled in the histogram.
(H) Representative FACS plots showing CD8 versus the intracellular cytokines IFN-γ, TNF-α, and lytic granule protease granzyme B after 500 μM D2HG treatment from 3 biological replicates for each group. Differences are labeled in the histogram.
(I) Representative FACS plots showing PD-1 and Tim-3 for CD8 T cells after 500 μM D2HG treatment from 3 biological replicates for each group. Differences are labeled in the histogram. The data and statistical results are in Table S1.
CAR-T cell-mediated cytotoxicity was impaired in high-D2HG medium
To evaluate CAR-T cell-mediated cytotoxicity in high-D2HG medium, we used second-generation anti-CD19 CAR-T cells (Figure 2A). CAR expression was detected with an anti-FLAG-tag antibody. It was found that CAR-T cells gradually decreased from approximately 70% on day 3–to 20% on day 14 (Figure 2B). Interestingly, CAR-positive cells were dispersible before day 10 and scattered beginning on day 14. The scattered CAR-positive cells were sorted and cultured. On day 7, cells were harvested for the detection of CD3ε protein expression using western blotting (Figure 2C). CD8+ T cells expressed only endogenous CD3ε, whereas CAR-T cells expressed monomeric CD3ε and dimeric CD3ε22 (Figure 2C). This indicated that CAR expression was consistent with the FACS results. Moreover, CAR expression in CD8+ T lymphocytes resulted in a slightly increased percentage of apoptotic cells after culturing for 14 days (Figure 2D).
Figure 2.

CAR-T cell-mediated cytotoxicity was impaired in high-D2HG medium
(A) Flowchart showing the strategy for CAR-T cell production, phenotype analysis, and killing assay.
(B) Representative FACS plots of CAR expression in CD8+ T lymphocytes.
(C) Western blot analysis showing endogenous CD3ε, monomeric CD3ε, and dimeric CD3ε in CD8 and CAR-T cells.
(D) Representative FACS plots showing annexin V and 7-AAD with gating selection of apoptotic cells.
(E) Lysis of cancer cells in normal medium and in 500 μM D2HG medium at different effector CAR-T cell-to-target NALM6-GL cell ratios. Each groups included 5 biological replicates.
(F) Representative FACS plots showing CD8 and CAR for surviving cells in the killing assay. Most cancer cells were killed, and the surviving cells were mostly CAR-T cells.
(G) Representative FACS plot based on CD62L and CD45RO for surviving cells in the killing assay and the memory subset from 3 biological replicates for each group.
(H) Histograms of memory cell subsets at different times and the differences are marked. The data and statistical results are in Table S2.
We then evaluated CAR-T cell-mediated cytotoxicity through co-culture of effector anti-CD19 CAR-T cells and target NALM6 cancer cells (with high CD19 expression) at different ratios. To detect the lysis of cancer cells, a lentivirus carrying luciferase-green fluorescent protein (GL) was introduced through lentiviral inoculation, and NALM6-GL monoclonal stable cell lines were then generated and sorted. As predicted, approximately 84% of the target cancer cells were lysed by effector CAR T cells at an effector-to-target ratio of 1:1 when co-cultured for 24 h, and the lysis rate gradually decreased when the effector CAR T cell proportion decreased (Figure 2E). This indicated that our CAR-T cells effectively killed target NALM6 cancer cells. However, when we added D2HG to a final concentration of 500 μM into the medium, the proportion of lysed cancer cells significantly decreased compared with the normal medium at all effector-to-target ratios. Cells co-cultured at an effector-to-target ratio of 1:1 were washed and used to verify the CAR-T cell and cancer cell proportions (Figure 2F). None of the cancer cells expressed CD8 or DYKDDDDK (FLAG tag)-CAR, whereas CAR-T cells exhibited high expression of CAR and CD8. It was noted the CAR-T cell proportion was approximately 87.8% in normal medium and 70.9% in high-D2HG medium. After antigenic stimulation, the formation and development of different memory subsets were measured. D2HG treatment resulted in a significant decrease of Tcm after co-culture of CAR-T cells with NALM6 cells for more than 7 days (Figures 2G and 2H). This was consistent with previous results that high D2HG inhibited the formation of Tcm cells.
Catabolism of D2HG by D2HGDH over-expression enhanced CAR-T cell-mediated cytotoxicity
To overcome the immunosuppressive effect of D2HG on CD8+ T lymphocytes and CAR-T cells, we analyzed the catabolic pathway of D2HG and regulatory proteins. The metabolic pathway analysis points enzyme D2HGDH, since it catalyzes irreversible catabolism of D2HG. Several studies have focused on the value of D2HGDH for the treatment of D-2-hydroxyglutaric aciduria.23 D2HGDH encodes D-2-hydroxyglutarate dehydrogenase and participates in reactions that produce energy for cell activities.24 Specifically, D2HGDH converts D2HG into 2-oxoglutarate, which is an intermediate metabolite of the citrate cycle.25 Across two independent experiments, we produced D2HGDH-over-expression (OE) and D2HGDH-knock-out (KO) CAR-T cells. First, we obtained the cDNA sequence of D2HGDH and added T2A before the sequence. Then, we designed oligos with restriction sites and synthesized the oligos. The T2A-D2HGDH cDNA products were cloned through PCR amplification, and cDNA was inserted into a digested CAR shuttle plasmid. Afterward, we produced D2HGDH-OE CAR-T cells with the previously described CAR-T cell production process. D2HGDH-KO CAR-T cells were produced through CRISPR/CAS9-mediated knock-out and ribonucleoprotein (RNP) delivery methods in CAR-T cells. A schematic illustration of the anti-CD19 CAR tandem gene cDNA construct is included, and the experiment strategy is shown (Figure 3A). There was similar high-CAR expression among the D2HGDH -modified CAR-T cells (Figure 3B). The mRNA expression of D2HGDH in CAR-T cells was similar to that in CD8+ T lymphocytes but was significantly decreased in the D2HGDH-KO group and increased in the D2HGDH-OE group (Figure 3C). The protein expression of D2HGDH showed consistent results of the mRNA expression (Figure 3D). To confirm the KO of D2HGDH in CAR-T cells, we extracted DNA from cells and added the T7E1 enzyme to catalyze the cleavage of DNA mismatches.26 Digestion of the amplicon yielded fragments of ∼200 and ∼400 bp in addition to the parental band of ∼650 bp. The T7E1 mismatch cleavage assay showed a high-DNA mismatch for D2HGDH in CAR-T cells and indicated high KO of D2HGDH (Figure 3E). A CFSE-based proliferation assay showed that D2HGDH-KO resulted in a significant delay in CAR-T cell expansion, whereas OE of D2HGDH resulted in a significant acceleration of CAR-T cell expansion (Figure 3F). A glucose uptake assay utilizing 2-NBDG indicated accelerated CAR-T cell metabolism in the D2HGDH-KO group and similar glucose uptake in the D2HGDH-OE group compared with the CAR-T cell group (Figure 3G). The release of the effector cytokines IFN-γ and TNF-α was decreased in D2HGDH-KO CAR-T cells but significantly increased in D2HGDH-OE CAR-T cells (Figure 3H). This indicated differential cytotoxicity of D2HGDH modified CAR-T cells. As predicted, in normal medium, D2HGDH-OE CAR-T cells showed predominant lysis of cancer cells, whereas D2HGDH-KO CAR-T cells showed limited lysis of cancer cells (Figure 3I). Furthermore, in medium supplemented with 500 μM D2HG, D2HGDH-OE CAR-T cells also showed predominant lysis of cancer cells, whereas D2HGDH-KO CAR-T cells showed poor lysis of cancer cells. To reveal the effects of the metabolite D2HG and enzyme D2HGDH on CAR-T cell-mediated cytotoxicity, we compared the killing effect of D2HGDH-modified CAR-T cells in normal and high-D2HG media. Interestingly, cancer cell lysis by normal CAR-T cells and D2HGDH-KO CAR-T cells was decreased after co-culture in high-D2HG medium; however, D2HGDH-OE CAR-T cells killed almost all cancer cells when they were co-cultured in a high-D2HG medium. The surviving resident cells in high-D2HG medium from the killing assay were then used to analyze the CAR-T cell proportions. Surviving cancer cells were rare when the number of CAR-T effector cells was equal to that of NALM6-GL target cells after co-culturing for 24 h (Figure 3J). However, there was an obvious decrease of CAR-T cells in the groups of CAR-T cells and D2HGDH-KO CAR-T cell groups when the co-cultured effector CAR-T cell-to-target NALM6-GL cell ratio was 0.1:1. Notably, the relative stability of the D2HGDH-OE CAR-T cells in high-D2HG medium indicated that D2HGDH-OE CAR-T cells took advantage of the D2HG catabolism to adapt to the high-D2HG environment.
Figure 3.

CAR-T cell-mediated cytotoxicity was impaired in high-D2HG medium
(A) Flowchart showing the strategy for D2HGDH-OE/KO CAR-T cell production, phenotype analysis, and killing assay.
(B) Representative FACS plots of CAR expression after D2HGDH OE/KO, histogram displaying CAR-T cells percentage.
(C) mRNA expression of D2HGDH after D2HGDH OE/KO.
(D) Western blot analysis showing the expression of D2HGDH after D2HGDH OE/KO.
(E) Surveyor assay showing the DNA mismatches of D2HGDH in CAR-T cells after D2HGDH KO.
(F) Representative FACS plots showing CFSE with gating selection of proliferating cells from 3 biological replicates for each group. Differences are labeled in the histogram.
(G) Representative FACS plots showing 2-NBDG with gating selection of glucose uptake from 3 biological replicates for each group. Differences are labeled in the histogram.
(H) The release of the cytokines IFN-γ and TNF-α into the supernatant was determined using ELISA, and differences are labeled in the histogram. Each group included 3 biological replicates.
(I) Lysis of cancer cells by D2HGDH-KO/OE CAR-T cells in normal medium and in 500 μM D2HG medium at different CAR-T cell effector-to-NALM6-GL target cell ratios. Three biological replicates were used for the normal medium, and 4 biological replicates were used for the high-D2HG medium.
(J) Histogram of the CAR-T cell proportion in the D2HGDH-modified CAR-T cells among the surviving cells in the killing assay. The data and statistical results are in Table S3.
D2HGDH-modified CAR-T cells have distinct phenotypic characteristics
D2HG impaired CD8+ T lymphocyte and CAR-T cell glucose uptake, expansion, and function, whereas D2HGDH-OE CAR-T cells exhibited enhanced cytotoxicity in the high-D2HG medium. It was reasonable to analyze the metabolic function through measuring oxygen consumption rate (OCR). D2HGDH-modified CAR-T cells showed different metabolic capacities due to their different basic and maximal OCR (Figure 4A). Quantitative analysis displayed similar basal OCR of both CAR-T cells and CD8+ T cells while increasing basal OCR in D2HGDH-OE CAR-T cells and decreasing basal OCR in D2HGDH-KO CAR-T cells (Figure 4B). It was noted that CAR-T cells and D2HGDH-KO CAR-T cells showed significantly increased maximal OCR, whereas D2HGDH-OE CAR-T cells showed significantly decreased maximal OCR. These results indicated the excellent metabolic adaptability of D2HGDH-OE CAR-T cells. The metabolic characteristics of CAR-T cells shaped memory subsets, since D2HGDH-KO and D2HGDH-OE resulted in significant decreases in central memory cells, while D2HGDH-OE also resulted in significant increases in effector memory cells (Figures 4C and 4D). Interestingly, D2HGDH-KO in CAR-T cells resulted in significant increases in the expression of PD-1 and TIM-3, whereas D2HGDH-OE resulted in significant decreases in the expression of PD-1 and TIM-3 after being co-cultured with cancer cells for 7 days (Figures 4E and 4F). Intracellular cytokine staining results showed different cytotoxicity of D2HGDH-modified CAR-T cells. D2HGDH-KO CAR-T cells displayed a decreased producing potential of IFN-γ and TNF-ɑ, whereas D2HGDH-OE CAR-T cells displayed an increased producing potential of TNF-ɑ after these CAR-T cells were co-cultured with cancer cells for 7 days (Figure 4G). Intracellular lytic granule protease granzyme B staining results showed similar trends (Figure 4H). Moreover, we measured the intracellular D2HG and 2-oxoglutarate and found that D2HGDH-KO CAR-T cells accumulated about six times the D2HG, while D2HGDH-OE CAR-T cells catabolized 93% of D2HG compared with CAR-T cells (Figure 4I). The production of 1.86 times the 2-oxoglutarate in D2HGDH-OE CAR-T cells indicated the metabolic adaptability of D2HGDH-OE CAR-T cells in the high-D2HG environment. These results indicated that D2HGDH-OE CAR-T cells took advantage of D2HG catabolism to drive the metabolism of 2-oxoglutarate as additional energy sources for their function of increasing intracellularly produced cytokines and outstanding cytotoxicity in high-D2HG medium.
Figure 4.

D2HGDH-modified CAR-T cells have distinct phenotypic characteristics
(A) Representative Seahorse extracellular flux analysis of OCR of D2HGDH-modified CAR-T cells.
(B) Histogram showed the differences of basic OCR and maximal OCR of D2HGDH-modified CAR-T cells.
(C) Representative FACS plots showing CD62L versus CD45RO for the memory subsets from 3 biological replicates for each group after co-culture for 7 days.
(D) Histogram showing the differences of memory cell subsets of D2HGDH-modified CAR-T cells.
(E) Representative FACS plots showing PD-1 and Tim-3 for CAR-T cells from 3 biological replicates for each group after co-culture for 7 days.
(F) Histogram showing the differences of PD-1 and TIM-3 expression of D2HGDH-modified CAR-T cells.
(G) Representative FACS plots showing CD8 versus the intracellular IFN-γ and TNF-α for specific cytokine-producing cells from 3 biological replicates for each group. Histogram showing the differences of IFN-γ- and TNF-α- positive cells of D2HGDH-modified CAR-T cells after co-culture for 7 days.
(H) Representative FACS plots showing CD8 versus the intracellular granzyme B for specific cytokine-producing cells from 3 biological replicates for each group. Histogram showing the differences of granzyme B-positive cells of D2HGDH modified CAR-T cells after co-culture for 7 days.
(I) The intracellular concentration change of D2HG and 2-oxoglutarate from 3 biological replicates for each group. The data and statistical results are in Table S4.
NALM6-mIDH1 cancer cells were not sensitive to CAR-T cell therapy through production of D2HG
To reveal the high-D2HG environment resulting from mutational IDH1 in cancer cells, we cloned mutant IDH1 into a plasmid including luciferase-green fluorescent protein, and produced, sorted, and cultured NALM6-mIDH1 monoclonal stable cells. Then, D2HGDH-modified CAR-T and NALM6-mIDH1 cancer cells were co-cultured for killing assay. We included 500 μM D2HG in a separate group as an experimental control to match the original assays after co-culture for 48 h. With additional D2HG supplements in the medium, D2HGDH-OE CAR-T cells showed increasing killing potential against NALM6 cancer cells, whereas D2HGDH-KO CAR-T cells exhibited inferior killing potential against NALM6 cancer cells (Figure 5A). It was interesting that the import of mutational IDH2 into NALM6 cancer cells also impaired the killing effect of CAR-T cells in normal medium (Figure 5B). Moreover, D2HGDH-OE CAR-T showed an outstanding killing effect on NALM6-mIDH1 cancer cells, whereas D2HGDH-KO CAR-T showed an attenuated killing effect on NALM6-mIDH1 cancer cells. The intracellular D2HG concentration was measured using a LC/MS system. There was more accumulated D2HG in the D2HGDH-KO CAR-T group, but less intracellular D2HG in the D2HGDH-KO CAR-T group after the additional D2HG supplement in the medium when co-cultured with NALM6 cancer cells (Figure 5C). The results were consistent with groups from co-culture of D2HGDH-modified CAR-T and NALM6-mIDH1 cancer cells (Figure 5D). The absence of intracellular D2HG concentration in the D2HGDH-OE CAR-T group indicated the catabolism of D2HG by D2HGDH-OE CAR-T cells.
Figure 5.

NALM6-mIDH1 cancer cells inhibit the killing potential of CAR-T cells
(A) The percentage of lysed cancer cells after co-culture for 48 h of D2HGDH-modified CAR-T cells with NALM6 cancer cells in the additional 500 μM D2HG medium.
(B) The percentage of lysed cancer cells after co-culture for 48 h of D2HGDH-modified CAR-T cells with NALM6-mIDH1 cancer cells in normal medium.
(C) The intracellular fold change of D2HG in the resident cells after co-culture for 48 h of D2HGDH -modified CAR-T cells with NALM6 cancer cells in the additional 500 μM D2HG medium.
(D) The intracellular fold change of D2HG in the resident cells after co-culture for 48 h of D2HGDH-modified CAR-T cells with NALM6-mIDH1 cancer cells in normal medium. The data and statistical results are in Table S5.
D2HGDH-OE CAR-T cell treatment improved the survival of mice bearing NALM6-mIDH1 tumors and extended the survival of CAR-T cells in vivo
To evaluate the in vivo effects of D2HGDH-modified CAR-T cells on mice, we designed animal experiments, as shown in a flowchart (Figure 6A). Mice bearing NALM6-mIDH1 cancer cells showed relatively short survival compared with mice bearing wild-type (WT) NALM6 cancer cells (Figure 6B). Furthermore, three types of CAR-T cells were injected into mice for treatment on day 3 after implantation of NALM6-mIDH1 cancer cells. CAR-T cell treatment could prolong mouse overall survival, since anti-CD19 CAR-T cells could target the CD19 antigen of NALM6-mIDH1 cells. Furthermore, D2HGDH-OE CAR-T cell treatment resulted in a significant improvement in survival, whereas D2HGDH-KO CAR-T cell treatment resulted in significantly shorter survival compared with only CAR-T cell therapy (Figure 6C). The body weight of the D2HGDH-OE CAR-T cell treatment group steadily increased, whereas that of the D2HGDH-KO CAR-T cell treatment group decreased (Figure 6D). As expected, NALM6-mIDH1 cancer cells could produce D2HG, whereas CAR-T cells therapy decreased the serum D2HG level. Furthermore, D2HGDH-KO CAR-T cells therapy did not significantly change serum D2HG levels, whereas D2HGDH-OE CAR-T cells therapy significant decreased serum D2HG levels compared with only CAR-T cell therapy (Figure 6E). These results implied that D2HGDH-OE CAR-T cells enhanced their anti-tumor effect through catabolism of D2HG. Serum cytokine levels were measured using ELISA, and the results showed that CAR-T cell treatment significantly enhanced cytotoxicity, as indicated by high serum IFN-γ, TNF-ɑ, and IL-2 levels (Figure 6F). D2HGDH-OE CAR-T cell treatment resulted in increased serum IFN-γ, TNF-ɑ, and IL-2 concentrations, whereas D2HGDH-KO CAR-T cell treatment resulted in decreased serum IFN-γ, TNF-ɑ, and IL-2 concentrations. On day 14 after injection of CAR-T cells, blood was collected, and circulating CAR-T cells were analyzed using FACS (Figure 6G). The percentage of residual CAR-T cells was approximately 2.39% in the CAR-T-cells-only treatment group. Conversely, the percentage of residual CAR-T cells was decreased to 0.11% in the D2HGDH-KO CAR-T-cell-treated group, while increasing to 4.07% in the D2HGDH-OE CAR-T-cell-treated group (Figure 6H).
Figure 6.

D2HGDH-OE CAR-T cell treatment improved the survival of mice bearing NALM6-mIDH1 tumors and extended the survival of CAR-T cells in vivo
(A) Flowchart showing the experimental strategy for D2HGDH-modified CAR-T cell treatment of mice bearing NALM6-mIDH1 cancer cells.
(B) Survival plot for mice bearing NALM6-mIDH1 cancer cells and wild-type NALM6 cancer cells.
(C) Survival plot for mice bearing NALM6-mIDH1 cancer cells treated with D2HGDH-modified CAR-T cells.
(D) Body weight changes of mice bearing NALM6-mIDH1 cancer cells treated with D2HGDH-modified CAR-T cells.
(E) Serum D2HG changes of mice bearing NALM6-mIDH1 cancer cell after D2HGDH-modified CAR-T cells treatments.
(F) Release of the cytokines IFN-γ, TNF-α, and IL-2 in the sera of NALM6-mIDH1 cancer cell-bearing mice treated with D2HGDH-modified CAR-T cells.
(G) Representative FACS plot showing CD8 and CAR expression for CAR-T cells in the sera of D2HGDH-modified CAR-T cell-treated mice.
(H) The resident CAR-T cells in the blood after treatment of D2HGDH-modified CAR-T cells. Five biological replicates were included in each group, and differences are labeled in the histogram. The data and statistical results are in Table S6.
Discussion
In the present study, we confirmed that the high D2HG concentration impaired CD8+ and CAR-T cell proliferation, glucose metabolism, and cytokines production. D2HGDH-OE CAR-T cells took advantage of their own metabolic adaptability in the high-D2HG environment to improve the anti-tumor effect in vitro and in vivo. D2HGDH-OE in CAR-T cells displayed multiple distinct phenotypic characteristics, including the formation and differentiation of effector memory cells and enhanced cytokine production after antigen stimulation.
The field of immunometabolism has recently attracted growing attention.27 CAR-T cell-based immunotherapy has revolutionized immuno-oncology and highlighted the use of adoptive cell transfer for the treatment of cancer. However, many patients fail to respond to these therapies, and metabolic barriers in the TME mainly contribute to T cell disorder.28 Cancer cells fuel their rapid growth and proliferation mainly via aerobic glycolysis.29 Activated T cells also undergo a metabolic switch from oxidative metabolism to aerobic glycolysis.30 Competition for glucose and amino acids between cancer cells and immune cells impairs the proliferation of CD8 T cells and their development of effector functions. The inadequate metabolism also leads to the accumulation of metabolic waste or intermediate metabolites. Oncometabolites derived from dysfunctional regulation of metabolism result in multiple adverse effects. The oncometabolite D2HG is considered a biomarker of poor prognosis in recognition of its critical regulation of tumorigenesis, maintenance, genetic instability, and ultimately malignant transformation.30 It was reported to impair T cell metabolic adaptation and function.11, 12, 13 Here, in vitro, we elucidated the immunosuppressive characteristics of D2HG, including impaired T lymphocyte proliferation, decreased intracellular production of the cytokines, and inhibited effector memory cell formation and differentiation. The effect of accumulated D2HG defined the metabolic TME as immunologically cold. The catabolism of D2HG is a fascinating strategy for immunotherapy.31
The importation of exogenous genes would induce different metabolic and phenotypic characteristics. It was known that the editing of co-stimulatory factors of 4-1BB and CD28 in the second-generation CAR-T cells reprogram metabolic adaptability as well as differentiation and exhaustion characteristics of CAR-T cells.32 In the present study, we found that D2HGDH-modified CAR-T cells displayed distinct phenotypic characteristics. D2HGDH-KO in CAR-T cells resulted in significant inhibition of the differentiation of central memory cells, decreases in the production of cytokines IFN-γ and TNF-ɑ, and lytic granule protease granzyme B and increases of PD-1 and TIM-3 expression. In contrast, D2HGDH-OE in CAR-T cells improved the CAR-T cell differentiation of effector memory cells and enhanced the production of IFN-γ, TNF-ɑ, and granzyme B. The phenotypic characteristics of D2HGDH-OE CAR-T cells were consistent with the excellent cytotoxicity in high-D2HG medium. These results prompted D2GHDH as an immune-regulated molecule, since the modification of D2HGDH in CAR-T cells resulted in distinct phenotypic characteristics.
Based on previous studies of the effects of D2HG on immunosuppression, D2HG was produced through mutated IDH1/2 and was found in many cancer types, such as leukemia,33 glioblastoma,34 lung cancer,35 and cholangiocarcinoma.36 Patients with IDH1/2 mutation showed worse outcomes and shorter overall survival.37 Moreover, IDH1/2-mutant patients were not sensitive to immunotherapy.38 IDH encodes isocitrate dehydrogenases that catalyze the oxidative decarboxylation of isocitrate to 2-oxoglutarate. IDH mutation resulted in a gain of function to catalyze 2-oxoglutarate into D2HG. D2HG was found only in cancer patients with IDH mutations and gradually accumulated up to the millimole level.14,15 The most frequent mutation sites of IDH1/2 were found to be R132 (IDH1) and R172 (IDH2), which involve the active site and result in neomorphic enzyme activity. We cloned IDH1-mutant cancer cells and found that mice bearing IDH1 (R132H)-mutant cancer cells had shorter overall survival. The improved survival of D2HGDH-OE CAR-T cells and the poor survival of D2HGDH-KO CAR-T cells proved that catabolism of D2HG is beneficial for CAR-T cell therapy in IDH1/2-mutant cancer. The effectiveness of D2HGDH-OE CAR-T cells in the treatment of IDH1-mutant cancer was consistent with the increase in the CAR-T cell percentage and high production of IFN-γ and TNF-ɑ.
On the basis of the fact that the phenotypic characteristics and effector function of CD8+ T lymphocytes were impaired by the oncometabolite D2HG, we stated the hypothesis that catabolism of D2HG would improve CAR-T cell-mediated anti-tumor activity. D2HGDH modification was achieved through synthetic biology, such as introduction of enzymes D2HGDH into CAR-T cells. Then, in vitro and in vivo experiments clarified the phenotypic characteristics and effector functions of CAR-T cells after D2HGDH modification. Furthermore, metabolic reprogramming of D2HGDH-modified CAR-T cells also interpreted the functional adaptability in the high-D2HG metabolic environment. This study illustrates the roles of D2HG in inhibiting tumor immunotherapy and in functioning D2HGDH as a signaling molecule that reprograms the effector characteristics of T lymphocytes.
In conclusion, D2HG accumulation in the body restrained CD8+ T lymphocyte expansion, increased glucose uptake, suppressed effector cytokine production, and decreased the central memory cell proportion. Catabolism of D2HG through D2HGDH-OE in CAR-T cells enhanced anti-tumor activity by improving T lymphocyte expansion, glucose uptake, cytokine production, and memory cell differentiation. Moreover, D2HGDH-OE CAR-T cells took advantage of the catabolism of D2HG to improve anti-tumor effectiveness through an increase in the CAR-T cell proportion in an in vivo experiment. This study not only demonstrated the effects of the oncometabolite D2HG as an immunosuppressive mediator in the TME but also explored the potential of D2HGDH-OE CAR-T cells for immunotherapy. D2HGDH-OE CAR-T cells take advantage of the catabolism of D2HG to support T cell expansion and function, thus generating a distinct phenotype for outstanding anti-tumor effectiveness.
Materials and methods
Isolation and expansion of primary human T lymphocytes
This study was approved by the Human Research Ethics Board of the Shanghai Jiao Tong University Affiliated Sixth People's Hospital and conformed to the Declaration of Helsinki. Informed consent was obtained from participants. PBMCs were isolated from the blood of healthy donors by using Ficoll-Hypaque (Sigma, St. Louis, MO, USA) density gradient according to the manufacturer’s instructions. T cells were positively selected from PBMCs using the MACS CD8+ T cell Isolation Kit (Miltenyi Biotec, Bergisch Gladbach, Germany) and activated using microbeads coated with anti-human CD3 and anti-human CD28 antibodies at a 1:2 bead/cell ratio. The cells were cultured at a density of 2 × 106 cells/mL for 2 days in RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, Grand Island, NY, USA), 30 IU/mL recombinant human IL-2, 10 mM HEPES, 2 mM glutamine, and 1% penicillin-streptomycin.
Recombinant vector constructs
The anti-CD19 CAR vector was composed of an anti-CD19 FMC-63 single-chain variable fragment (scFv), a human IgD hinge, transmembrane domains, a CD137 (4-1BB) costimulatory moiety, and a CD3zeta activation domain.39 The FLAG tag sequence (5′-GATTACAAAGACGATGACGATAAG-3′) was inserted before the CD8a leader peptide fragment and was used for detection of the CAR. CAR-modified sequences were inserted into a second-generation recombinant lentiviral plasmid backbone under the regulation of the EF-1α promoter. The human D2HGDH domain sequence was obtained from the UniProt database (Q8N465), and the cDNA template (CCDS33426.1) was procured by PCR amplification. The T2A linker was used for co-expression of the CAR and D2HGDH.
Cells and culture conditions
Low-passage HEK293T cells were maintained in Dulbecco's modified Eagle's medium (DMEM) (GIBCO). NALM-6 cells were maintained in RPMI 1640 medium. DMEM and RPMI 1640 medium were supplemented with 10% heat-inactivated FBS, 10 mM HEPES, 2 mM glutamine, and 1% penicillin-streptomycin. All cells were cultured at 37°C in an atmosphere of 5% carbon dioxide. NALM6 cells were infected with a lentivirus carrying luciferase-green fluorescent protein (GFP) by spinoculation. After incubation for 2 days, GFP + cells (NALM6-GL) were sorted, and NALM6-GL stable cell lines were generated. IDH1-mutant NALM6 (NALM6-mIDH1) cells were generated using the above method. Tests for mycoplasma on culture fluid confirmed that all the cells were mycoplasma free.
Lentivirus production
Lentiviral particles were produced in HEK293T cells after polyethyleneimine (Polysciences, Warrington, PA, USA)-mediated transfection. The shuttle plasmid and two packaging plasmids, psPAX2 and pMD.2G, were co-transfected into HEK293T cells. Lentivirus-containing supernatants were harvested at 48 and 72 h posttransduction. Cell debris was removed by centrifugation (5 min at 400 rpm). Lentiviruses were concentrated using AmiconUltra 100-kDa ultrafiltration centrifugal tubes (Millipore, Burlington, MA, USA). All virus was aliquoted and stored at −80°C.
Over-expression of the CAR in CD8+ T cells via lentivirus spinfection
For integration of the CAR-modified DNA cassette and D2HGDH-OE modified CAR into CD8+ T cell genomic DNA, spinfection was used to enhance the transduction efficacy of the T cells. Briefly, 3 days before spinfection, T cells were activated with a 1:1 ratio of human anti-CD3/anti-CD28 beads (CD3/CD28 Dynabeads; Thermo Fisher, Danvers, MA, USA) for 16 h, which were later removed with a magnetic separation rack. T cells were incubated for 2 days. Then, CD8 T cells were prepared at a density of 2 × 106 cells/mL in medium and seeded in 0.8 mL in each well of a 24-well plate. For each 0.8 mL of cell suspension, 0.2 mL of concentrated lentivirus and 1.5 μL of polybrene stock (8 mg/mL) were added, and the solution was mixed well by pipetting up and down several times. The plate was then spin-infected at 900 × g for 60 min at 37°C and cultured for 16 h. The next day, all the cells were counted and suspended in RPMI 1640 medium supplemented with 5% FBS and recombinant human IL-2 (30 U/mL). Every 2 or 3 days, fresh medium was added, and T cells were seeded in wells at a density within the range of 0.5–2×106 cells/mL.
Knock-out of the D2HGDH gene in CAR-T cells via CRISPR/CAS9-based ribonucleoprotein delivery
The CRISPR/Cas9 sgRNA sequences for D2HGDH-KO were designed using CRISPick (https://portals.broadinstitute.org/gppx/crispick/public). The optimal sgRNA sequence for D2HGDH was identified as 5′-GAAGCAGCTGTTCATCGGGT-3′, based on a preliminary experiment. tracrRNA and crRNA were synthesized by Genscript Biotech (Nanjing, China). Each gRNA was formed by mixing tracrRNA and the corresponding crRNA at a 1:1 ratio through an annealing process. Stocked Cas9 Protein v2 (Thermo Fisher) was then mixed with diluted Cas9 protein at a molar ratio of 1:1. The RNP complex was formed after incubation at room temperature for 15 min. T cells were prepared at a density of 5 × 106 cells per 50-μL tip reaction in electroporation Buffer R (Neon Transfection System Kits). D2HGDH KO was performed via RNP-based delivery through electroporation at 1,600 V, 10 ms, and three pulses. Immediately, the cells were transferred into prewarmed RPMI 1640 medium supplemented with 5% FBS and recombinant human IL-2 (30 U/mL) for continued incubation.
T7E1 surveyor assay
D2HGDH-KO CAR-T cells were lysed with QuickExtract Buffer according to the manufacturer's recommendation. One microliter of genomic DNA was used for PCR with a 50-μL reaction volume that contained DreamTaq Green 2X PCR Master Mix (Thermo Fisher), primer pairs and nuclease-free water. The PCR conditions were 1 cycle of 3 min at 95°C; 36 cycles of 30 s at 95°C, 30 s at 60°C, and 45 s at 72°C; and, finally, a 4-min incubation at 72°. The PCR products were 550–800 bp. Then, 200 ng of PCR products obtained from WT and KO samples were included in a 19-μL heteroduplex reaction by heating at 95°C for 5 min in a thermocycler following an annealing process. Heteroduplex digestion was processed by adding 1 μL of T7 endonuclease I (T7E1; NEB, Rowley, MA, USA) and incubating at 37°C for 45 min. All 20 μL of digested heteroduplexes was run on a 2% agarose gel stained with ethidium bromide, and the 100-bp DNA ladder was run with the sample for reference.26
RNA extraction, reverse transcription, and quantitative PCR
Total RNA was extracted using TRIzol Reagent (Invitrogen, Carlsbad, CA, USA) following standard RNA extraction protocols. First-strand cDNA was synthesized from the isolated RNA with SuperScript IV Reverse Transcriptase (Invitrogen). After normalizing the concentrations of cDNA with nuclease-free water, quantitative PCR (qPCR) was performed by adding designated primers for genes of interest, and beta-actin was used as an internal positive control.
Protein extraction and western blotting
Protein was extracted from cells with ice-cold 1× RIPA buffer containing protease inhibitors (Beyotime Biotech, Shanghai, China) by following standard RNA extraction protocols. After incubation on ice for 30 min, the protein supernatant was collected after centrifugation at 12,000 × g and at 4°C for 15 min. The protein concentration was measured using a BCA assay kit (Thermo Fisher), and the samples from different groups were adjusted to the same concentration by adding 1× RIPA buffer. Then, 5× loading buffer was added and boiled for 5 min. Protein from each sample (30 μg) was loaded into a 10% SDS-PAGE gel for electrophoresis. Proteins were separated in the gel and transferred to PVDF membranes. The membranes were blocked at room temperature for 1 h using 3% BSA in TBST buffer. The primary antibodies were diluted with 3% BSA in TBST buffer at a ratio of 1:1,000. The membranes were then incubated with the indicated primary antibody at 4°C overnight and washed three times with TBST. Horseradish peroxidase (HRP)-conjugated secondary antibodies were diluted with TBST buffer at a ratio of 1:5,000. The membranes were then incubated with the appropriate diluted secondary antibody at room temperature for 1 h and washed three times with TBST. A chemiluminescent substrate was mixed and added to the membranes for the detection of protein expression. The blots were imaged using a camera-based CCD (Biotanon, Shanghai, China).
Chemical modulation
We used D-α-hydroxyglutaric acid disodium (catalog no. S7873 from Selleckchem) for all the in vitro experiments. For a killing assay performed with a high concentration of D2HG, we prepared prewarmed medium with increasing concentrations of D2HG from 100 to 10 mM.15 To evaluate the effect of D2HG on CD8+ T lymphocytes and CAR-T cells, we gradually added D2HG to the final concentration of 500 μM.
Flow cytometry
Conjugated antibodies for flow cytometry were reacted with human cells and were mainly obtained from BioLegend (San Diego, CA, USA) or BD Biosciences (San Jose, CA, USA). Transduced T cells were stained with APC-conjugated anti-DYKDDDDK (FLAG tag) for detection of the CAR. PE-conjugated anti-CD8, BV510-conjugated anti-CD8, APC/Cyanine7-conjugated anti-CD3, PE/Cyanine7-conjugated anti-CD44 and Alexa Fluor 610-conjugated anti-CD45RO antibodies were used for the detection of activated CD8+ T cells and CAR-T cells. PerCP/Cyanine5.5-conjugated anti-CD62L, APC/Cyanine7-conjugated anti-CD197 (CCR7), PE/Cyanine7-conjugated anti-CD44 and FITC-conjugated anti-CD45RO antibodies were used for the detection of memory T cells. APC/Cyanine7-conjugated anti-CD366 (Tim-3) and PE/Cyanine7-conjugated anti-CD279 (PD-1) antibodies were used for the detection of exhausted T cells. Pacific blue-conjugated anti-granzyme B, APC/Cyanine7-conjugated anti-IFN-γ, and FITC-conjugated anti-TNF-α antibodies were used for the detection of intracellular cytokines. Brilliant violet 605-conjugated anti-mouse IgG was used as a control. Surface protein staining was performed with diluted antibodies (1:200) on ice for 30 min, and the cells were then washed with PBS containing 1% FBS before flow cytometry analysis using a CytoFLEX LX flow cytometer (Beckman Coulter) or Attune NxT flow cytometer (Thermo Fisher). Intracellular staining was achieved with a fixation/permeabilization solution (plus brefeldin A from eBioscience). Peripheral blood from mouse xenografts was treated with red blood cell lysis buffer (BioLegend, San Diego, CA, USA), and cells were stained with the corresponding antibodies. Each group included at least three biological repetitions, and data were analyzed using FlowJo software version 10.3 (TreeStar, Ashland, OR, USA).
CFSE labeling and an ex vivo proliferation assay
CD8+ T cells or CAR-T cells from different groups were incubated with PBS (without Ca2+ and Mg2+) containing 5 μM CFSE (Thermo Fisher) at 37°C for 8 min. Heat-inactivated FBS was then added for 1 min, and the cells were washed three times in RPMI medium +5% FBS. CFSE-labeled cells (2 × 105) were seeded in triplicate in a 96-well flat-bottom plate for 5 days. Proliferation was evaluated by flow cytometry by calculating the CFSE-FITC + cell proportion.
2-NBDG glucose uptake assay
CD8+ T cells or CAR-T cells from different groups were washed with PBS and treated with 400 μL of PBS with 0.5% FBS. 2-NBDG reagent (Thermo Fisher) was added to the cells at a final concentration of 200 μg/mL, and the plate was then incubated at 37°C for 60 min. After the incubation, the cells were collected from the plate and washed with PBS, and glucose uptake was immediately determined by flow cytometry analysis. 2-NBDG glucose uptake was evaluated by calculating the 2-NBDG-FITC + cell proportion.
Analysis of metabolic parameters
Mitochondrial function was assessed with an extracellular flux analyzer of XF24 cell-culture microplates (Seahorse Bioscience). When CD8+ T cells and CAR-T cells were cultured for more than 14 days and displayed a distinct phenotype, the cells were used to assay mitochondrial function. Briefly, the cells were centrifuged at 1,200 × g for 5 min and then suspended in XF assay medium containing 5.5 mM glucose, 2.0 mM L-glutamine, and 1.0 mM sodium pyruvate and seeded at 1 × 106 cells per well. Then the microplate was centrifuged at 1,000 × g for 5 min and incubated in standard culture conditions for 60 min after individual wells were coated with CellTak in accordance with the manufacturer's instructions. Cell adherence and equilibration were processed, and cellular OCRs were measured under basal conditions and, following treatment with 1.5 μM oligomycin, 1.5 μM FCCP, and 40 nM rotenone, with 1 μM antimycin A (XF Cell Mito Stress kit, Seahorse Bioscience).
Metabolite extraction and quantitative determination of D2HG
Two million CAR-T cells were extracted in 1,000 μL of prechilled 80% MS-grade methanol. After centrifugation at 14,000 ×g for 15 min, the supernatants were transferred to polypropylene tubes and evaporated under vacuum. The residues were reconstituted with 50 μL of methanol-water (90%:10%, v/v), and 10 μL was injected into an Agilent 6490 Triple Quadrupole LC/MS System. Liquid chromatography was optimized with the Kinetex 2.6-μm PS C18, LC column 150 × 2.1 mm (Phenomenex, Torrance, CA, USA). Each sample was tested using three replicates. Multiple reaction monitoring was used for the qualitative and quantitative analysis of purified standards. The features of spectra were extracted using Agilent Mass Hunter Qualitative Analysis Software (version B 6.0.633.0). Sera D2HG were measured with the same methods after extraction with MS-grade methanol.
Cytotoxicity assays
A total of 100 μL of 2×105 NALM6-GL cells (target cells) were seeded in each well of flat-bottom 96-well plates. Modified CAR-T cells were then added to each well at the indicated ratio and cultured for 24 or 48 h. Target cell viability was evaluated by adding 10 μL/well 150 μg/mL D-luciferin (potassium salt) (Thermo Fisher). The fluorescence signal was measured within 10 min, and background luminescence was negligible (<1% of the signal from wells containing only target cells). The percent viability (%) was calculated as experimental signal/maximal signal × 100, and the percent lysis was equal to 100 – percent viability. The remaining viable cells were harvested and stained for the CAR and CD8 markers to distinguish different subsets. Viable cells were then quantitatively analyzed through flow cytometry.
Enzyme-linked immunosorbent assay
NALM6-GL cells (target) were co-cultured with modified CAR-T cells (effector) at different effector-to-target ratios for 24 h without exogenous cytokines. Then, the supernatant was collected and analyzed for secretion of the cytokines IFN-γ (DIF50C), granzyme B (DGZB00), and TNF-α (DTA00D) with cytokine-specific ELISA kits (R&D Systems, Minneapolis, MN, USA) according to the kit instructions.
Xenograft models and in vivo assessment
All animal procedures were approved by the Animal Welfare Committee of Shanghai Jiao Tong University Affiliated Sixth People's Hospital, and animal experiments were performed in the Laboratory Animal Center according to the Institutional Animal Care and Use Committee (IACUC). All M-NSG mice (NOD-PrkdcscidIl2rgem1/Smoc) were maintained in specific pathogen-free (SPF)-grade cages and provided autoclaved food and water. To develop the xenograft models, 1 × 106 NALM6 cells in 100 μL of PBS were injected into each mouse via the tail vein as in a previous report.40 Modified CAR-T cells in RPMI 1640 medium were injected via the tail vein 3 days after the injection of NALM6 cells. Peripheral blood was collected from three to five biological replicate mice in each group after the treatment of CAR-T cells for 14 days. Body weight was measured every other day, and the survival/disease status was recorded as per a veterinarian’s instructions.
Statistics
GraphPad Prism 8.0 was applied for statistical analysis. The data were considered parametric and are shown as the means ± SD. For comparing two groups, student's t test was performed. For analyzing more than two groups, one-way ANOVA followed by Sidak’s or Tukey’s multiple comparison test was performed. For analyzing two variables, two-way ANOVA followed by Tukey’s multiple comparison test was performed. Survival analysis was done by the Kaplan-Meier method, and significance was evaluated with the log rank test. Statistical tests were two-sided where data were considered, with a p value < 0.05 where the order of significant defined by ∗∗∗∗p < 1 × 10−4, ∗∗∗p < 2 × 10−4, ∗∗p < 2 × 10−3, ∗p < 0.0332. Statistical details of experiments are provided in the figure panels, figure legends, and supplementary materials.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81872494, 81873042, 82074063, and 81803874).
Author contributions
C.G. and Q.Y. designed the experiments. Q.Y., M.C., Y.W., and J. Li performed the experiments. J. Huang, S. Zheng, S. Zhou, J. Lu, and J.Z. analyzed the data. C.G. supervised the project. Q.Y., J. Hao, C.G., and M.Z. wrote the manuscript. Z. Sheng, T.Y., and J. Hao provided experimental materials.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.007.
Supplemental informationa
References
- 1.Porter D.L., Levine B.L., Kalos M., Bagg A., June C.H. Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N. Engl. J. Med. 2011;365:725–733. doi: 10.1056/NEJMoa1103849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Efficace F., Vignetti M. Quality of life and CAR-T cell therapy in children, adolescents, and young adults with haematological malignancies. Lancet Oncol. 2019;20:1625–1626. doi: 10.1016/S1470-2045(19)30641-2. [DOI] [PubMed] [Google Scholar]
- 3.Cho J.H., Collins J.J., Wong W.W. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell. 2018;173:1426–1438 e11. doi: 10.1016/j.cell.2018.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fraietta J.A., Lacey S.F., Orlando E.J., Pruteanu-Malinici I., Gohil M., Lundh S., Boesteanu A.C., Wang Y., O’Connor R.S., Hwang W.T., et al. Determinants of response and resistance to CD19 chimeric antigen receptor (CAR) T cell therapy of chronic lymphocytic leukemia. Nat. Med. 2018;24:563–571. doi: 10.1038/s41591-018-0010-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murad J.P., Tilakawardane D., Park A.K., Lopez L.S., Young C.A., Gibson J., Yamaguchi Y., Lee H.J., Kennewick K.T., Gittins B.J., et al. Pre-conditioning modifies the TME to enhance solid tumor CAR T cell efficacy and endogenous protective immunity. Mol. Ther. 2021;29:2335–2349. doi: 10.1016/j.ymthe.2021.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vitale I., Shema E., Loi S., Galluzzi L. Intratumoral heterogeneity in cancer progression and response to immunotherapy. Nat. Med. 2021;27:212–224. doi: 10.1038/s41591-021-01233-9. [DOI] [PubMed] [Google Scholar]
- 7.Giuffrida L., Sek K., Henderson M.A., House I.G., Lai J., Chen A.X.Y., Todd K.L., Petley E.V., Mardiana S., Todorovski I., et al. IL-15 preconditioning augments CAR T cell responses to checkpoint blockade for improved treatment of solid tumors. Mol. Ther. 2020;28:2379–2393. doi: 10.1016/j.ymthe.2020.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DePeaux K., Delgoffe G.M. Metabolic barriers to cancer immunotherapy. Nat. Rev. Immunol. 2021;21:785–797. doi: 10.1038/s41577-021-00541-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dang L., White D.W., Gross S., Bennett B.D., Bittinger M.A., Driggers E.M., Fantin V.R., Jang H.G., Jin S., Keenan M.C., et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2009;462:739–744. doi: 10.1038/nature08617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang H., Ye D., Guan K.L., Xiong Y. IDH1 and IDH2 mutations in tumorigenesis: mechanistic insights and clinical perspectives. Clin. Cancer Res. 2012;18:5562–5571. doi: 10.1158/1078-0432.CCR-12-1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bunse L., Pusch S., Bunse T., Sahm F., Sanghvi K., Friedrich M., Alansary D., Sonner J.K., Green E., Deumelandt K., et al. Suppression of antitumor T cell immunity by the oncometabolite (R)-2-hydroxyglutarate. Nat. Med. 2018;24:1192–1203. doi: 10.1038/s41591-018-0095-6. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L., Sorensen M.D., Kristensen B.W., Reifenberger G., McIntyre T.M., Lin F. D-2-hydroxyglutarate is an intercellular mediator in IDH-mutant gliomas inhibiting complement and T cells. Clin. Cancer Res. 2018;24:5381–5391. doi: 10.1158/1078-0432.CCR-17-3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Amankulor N.M., Kim Y., Arora S., Kargl J., Szulzewsky F., Hanke M., Margineantu D.H., Rao A., Bolouri H., Delrow J., et al. Mutant IDH1 regulates the tumor-associated immune system in gliomas. Genes Dev. 2017;31:774–786. doi: 10.1101/gad.294991.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yong C., Stewart G.D., Frezza C. Oncometabolites in renal cancer. Nat. Rev. Nephrol. 2020;16:156–172. doi: 10.1038/s41581-019-0210-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Suh C.H., Kim H.S., Jung S.C., Choi C.G., Kim S.J. 2-Hydroxyglutarate MR spectroscopy for prediction of isocitrate dehydrogenase mutant glioma: a systemic review and meta-analysis using individual patient data. Neuro Oncol. 2018;20:1573–1583. doi: 10.1093/neuonc/noy113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Megias-Vericat J.E., Ballesta-Lopez O., Barragan E., Montesinos P. IDH1-mutated relapsed or refractory AML: current challenges and future prospects. Blood Lymphat Cancer. 2019;9:19–32. doi: 10.2147/BLCTT.S177913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nayak L., Molinaro A.M., Peters K., Clarke J.L., Jordan J.T., de Groot J., Nghiemphu L., Kaley T., Colman H., McCluskey C., et al. Randomized phase II and biomarker study of pembrolizumab plus bevacizumab versus pembrolizumab alone for patients with recurrent glioblastoma. Clin. Cancer Res. 2021;27:1048–1057. doi: 10.1158/1078-0432.CCR-20-2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lugli E., Gattinoni L., Roberto A., Mavilio D., Price D.A., Restifo N.P., Roederer M. Identification, isolation and in vitro expansion of human and nonhuman primate T stem cell memory cells. Nat. Protoc. 2013;8:33–42. doi: 10.1038/nprot.2012.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abdelsamed H.A., Moustaki A., Fan Y., Dogra P., Ghoneim H.E., Zebley C.C., Triplett B.M., Sekaly R.P., Youngblood B. Human memory CD8 T cell effector potential is epigenetically preserved during in vivo homeostasis. J. Exp. Med. 2017;214:1593–1606. doi: 10.1084/jem.20161760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Phan A.T., Doedens A.L., Palazon A., Tyrakis P.A., Cheung K.P., Johnson R.S., Goldrath A.W. Constitutive glycolytic metabolism supports CD8+ T cell effector memory differentiation during viral infection. Immunity. 2016;45:1024–1037. doi: 10.1016/j.immuni.2016.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Klein Geltink R.I., Kyle R.L., Pearce E.L. Unraveling the complex interplay between T cell metabolism and function. Annu. Rev. Immunol. 2018;36:461–488. doi: 10.1146/annurev-immunol-042617-053019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bridgeman J.S., Hawkins R.E., Bagley S., Blaylock M., Holland M., Gilham D.E. The optimal antigen response of chimeric antigen receptors harboring the CD3ζ transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. J. Immunol. 2010;184:6938–6949. doi: 10.4049/jimmunol.0901766. [DOI] [PubMed] [Google Scholar]
- 23.Kranendijk M., Struys E.A., Salomons G.S., Van der Knaap M.S., Jakobs C. Progress in understanding 2-hydroxyglutaric acidurias. J. Inherit. Metab. Dis. 2012;35:571–587. doi: 10.1007/s10545-012-9462-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin A.-P., Abbas S., Kim S.-W., Ortega M., Bouamar H., Escobedo Y., Varadarajan P., Qin Y., Sudderth J., Schulz E., et al. D2HGDH regulates alpha-ketoglutarate levels and dioxygenase function by modulating IDH2. Nat. Commun. 2015;6:1–14. doi: 10.1038/ncomms8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Qiu Z., Lin A.-P., Jiang S., Elkashef S.M., Myers J., Srikantan S., Sasi B., Cao J.Z., Godley L.A., Rakheja D., et al. MYC regulation of D2HGDH and L2HGDH influences the epigenome and epitranscriptome. Cell Chem. Biol. 2020;27:538–550.e7. doi: 10.1016/j.chembiol.2020.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lomov N., Viushkov V., Petrenko A., Syrkina M., Rubtsov M. Methods of evaluating the efficiency of CRISPR/Cas genome editing. Mol. Biol. 2019;53:862–875. doi: 10.1134/S0026898419060119. [DOI] [PubMed] [Google Scholar]
- 27.Lee Y.S., Wollam J., Olefsky J.M. An integrated view of immunometabolism. Cell. 2018;172:22–40. doi: 10.1016/j.cell.2017.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mohammed S., Sukumaran S., Bajgain P., Watanabe N., Heslop H.E., Rooney C.M., Brenner M.K., Fisher W.E., Leen A.M., Vera J.F. Improving chimeric antigen receptor-modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 2017;25:249–258. doi: 10.1016/j.ymthe.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhu J., Thompson C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell Biol. 2019;20:436–450. doi: 10.1038/s41580-019-0123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chang C.-H., Curtis J.D., Maggi L.B., Jr., Faubert B., Villarino A.V., O’Sullivan D., Huang S.C., van der Windt G.J., Blagih J., Qiu J., et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang M., Wang S., Desai J., Trapani J.A., Neeson P.J. Therapeutic strategies to remodel immunologically cold tumors. Clin. Transl. Immunol. 2020;9:e1226. doi: 10.1002/cti2.1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baixauli F., Acín-Pérez R., Villarroya-Beltrí C., Mazzeo C., Nuñez-Andrade N., Gabandé-Rodriguez E., Ledesma M.D., Blázquez A., Martin M.A., Falcón-Pérez J.M., et al. Mitochondrial respiration controls lysosomal function during inflammatory T cell responses. Cell Metab. 2015;22:485–498. doi: 10.1016/j.cmet.2015.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kattih B., Shirvani A., Klement P., Garrido A.M., Gabdoulline R., Liebich A., Brandes M., Chaturvedi A., Seeger T., Thol F., et al. IDH1/2 mutations in acute myeloid leukemia patients and risk of coronary artery disease and cardiac dysfunction—a retrospective propensity score analysis. Leukemia. 2020;35:1–16. doi: 10.1038/s41375-020-01043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yan H., Parsons D.W., Jin G., McLendon R., Rasheed B.A., Yuan W., Kos I., Batinic-Haberle I., Jones S., Riggins G.J., et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toth L.N., de Abreu F.B., Tafe L.J. Non–small cell lung cancers with isocitrate dehydrogenase 1 or 2 (IDH1/2) mutations. Hum. Pathol. 2018;78:138–143. doi: 10.1016/j.humpath.2018.04.014. [DOI] [PubMed] [Google Scholar]
- 36.Abou-Alfa G.K., Macarulla T., Javle M.M., Kelley R.K., Lubner S.J., Adeva J., Cleary J.M., Catenacci D.V., Borad M.J., Bridgewater J., et al. Ivosidenib in IDH1-mutant, chemotherapy-refractory cholangiocarcinoma (ClarIDHy): a multicentre, randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol. 2020;21:796–807. doi: 10.1016/S1470-2045(20)30157-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hartmann C., Hentschel B., Simon M., Westphal M., Schackert G., Tonn J.C., Loeffler M., Reifenberger G., Pietsch T., von Deimling A., et al. Long-term survival in primary glioblastoma with versus without isocitrate dehydrogenase mutations. Clin. Cancer Res. 2013;19:5146–5157. doi: 10.1158/1078-0432.CCR-13-0017. [DOI] [PubMed] [Google Scholar]
- 38.Molenaar R.J., Radivoyevitch T., Nagata Y., Khurshed M., Przychodzen B., Makishima H., Xu M., Bleeker F.E., Wilmink J.W., Carraway H.E., et al. IDH1/2 mutations sensitize acute myeloid leukemia to PARP inhibition and this is reversed by IDH1/2-mutant inhibitors. Clin. Cancer Res. 2018;24:1705–1715. doi: 10.1158/1078-0432.CCR-17-2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cheng Z., Wei R., Ma Q., Shi L., He F., Shi Z., Jin T., Xie R., Wei B., Chen J., et al. In vivo expansion and antitumor activity of coinfused CD28- and 4-1BB-engineered CAR-T cells in patients with B cell leukemia. Mol. Ther. 2018;26:976–985. doi: 10.1016/j.ymthe.2018.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bluhm J., Kieback E., Marino S.F., Oden F., Westermann J., Chmielewski M., Abken H., Uckert W., Höpken U.E., Rehm A. CAR T cells with enhanced sensitivity to B cell maturation antigen for the targeting of B cell non-Hodgkin’s lymphoma and multiple myeloma. Mol. Ther. 2018;26:1906–1920. doi: 10.1016/j.ymthe.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.