

Abstract
Physiological and pathological cardiovascular processes are tightly regulated by several cellular mechanisms. Non-coding RNAs, including long non-coding RNAs (lncRNAs), represent one important class of molecules involved in regulatory processes within the cell. The lncRNA non-coding repressor of NFAT (NRON) was described as a repressor of the nuclear factor of activated T cells (NFAT) in different in vitro studies. Although the calcineurin/NFAT-signaling pathway is one of the most important pathways in pathological cardiac hypertrophy, a potential regulation of hypertrophy by NRON in vivo has remained unclear. Applying subcellular fractionation and RNA fluorescence in situ hybridization (RNA-FISH), we found that, unlike what is known from T cells, in cardiomyocytes, NRON predominantly localizes to the nucleus. Hypertrophic stimulation in neonatal mouse cardiomyocytes led to a downregulation of NRON, while NRON overexpression led to an increase in expression of hypertrophic markers. To functionally investigate NRON in vivo, we used a mouse model of transverse aortic constriction (TAC)-induced hypertrophy and performed NRON gain- and loss-of-function experiments. Cardiomyocyte-specific NRON overexpression in vivo exacerbated TAC-induced hypertrophy, whereas cardiomyocyte-specific NRON deletion attenuated cardiac hypertrophy in mice. Heart weight, cardiomyocyte cell size, hypertrophic marker gene expression, and left ventricular mass showed a NRON-dependent regulation upon TAC-induced hypertrophy. In line with this, transcriptome profiling revealed an enrichment of anti-hypertrophic signaling pathways upon NRON-knockout during TAC-induced hypertrophy. This set of data refutes the hypothesized anti-hypertrophic role of NRON derived from in vitro studies in non-cardiac cells and suggests a novel regulatory function of NRON in the heart in vivo.
Keywords: non-coding RNA, NRON, cardiac hypertrophy, NFAT signaling, heart failure
Graphical abstract
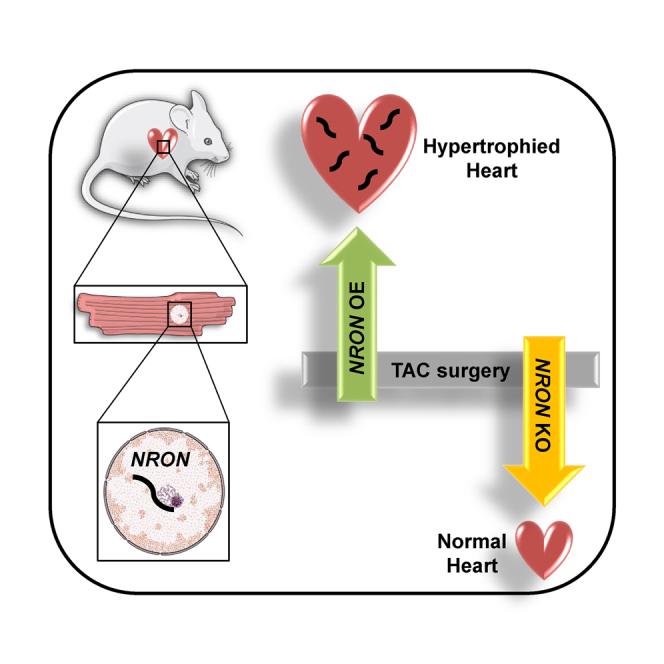
The long non-coding RNA NRON is enriched in cardiomyocytes where it localizes in the nucleus. Unexpectedly, NRON-overexpression in a transverse aortic constriction-induced hypertrophy mouse model exacerbated hypertrophy, whereas cardiomyocyte-specific NRON deletion attenuated cardiac hypertrophy, thereby suggesting a novel regulatory role of NRON in the heart.
Introduction
Cardiovascular diseases display the most common cause of death worldwide. Long non-coding RNAs (lncRNAs) are a subtype of non-coding RNAs with a size of more than 200 nucleotides1 and play important roles in the cardiovascular system on many different levels.2 Hence, lncRNAs might be considered as targets for future treatment strategies. The lncRNA Braveheart was reported to be involved in cardiac lineage development,3 whereas Fendrr is needed for the control of chromatin modification.4 Especially the role of lncRNAs in the development of disease conditions is of high interest. For instance, Meg3 is a cardiac-fibroblast-enriched lncRNA whose expression level decreases during the late cardiac remodeling phase after chronic pressure overload.5 Another lncRNA, called Chast, was discovered to be upregulated and pro-hypertrophic in mouse hearts after transverse aortic constriction (TAC)-induced cardiac hypertrophy,6 whereas the lncRNA H19 was shown to have an anti-hypertrophic effect.7 A therapy using antisense oligonucleotides to silence Chast and a gene therapy approach to overexpress H19, respectively, even prevented and attenuated pathological cardiac remodeling and thereby reinforced the therapeutic potential of ncRNA-based therapies.8,9
The development of pathological cardiac hypertrophy can be triggered through different derailed regulatory signaling pathways. One important pathway is the calcineurin/nuclear factor of activated T cells (NFAT)-signaling pathway. A sustained elevated level of intracellular calcium leads to the activation of calcineurin, which in turn dephosphorylates the NFAT transcription factor family.10,11 The activation of NFAT through dephosphorylation leads to its translocation into the nucleus, where it can induce the expression of several genes, leading to the maladaptive pro-hypertrophic response including the transcriptional switch to the fetal gene program.12 Interestingly, it has been shown that the formation of a RNA-protein scaffold complex inhibits the action of NFAT in vitro. In 2005, Willingham et al. identified a lncRNA that functioned as a key repressor of NFAT in HEK293 cells and named it “noncoding repressor of NFAT (NRON)”.13 Further studies described NRON as a cytoplasmic scaffold for a RNA-protein complex regulating the localization and activity of NFAT in T cells.14 To date, the exact role and mode of action of NRON in vivo still remain unclear. So far, NRON was shown to have a role in the life cycle of HIV-1,15,16 as well as in cancer cells.17,18 In the cardiac system, circulating NRON levels can serve as a biomarker for heart failure19 and can reduce atrial fibrosis by promoting the phosphorylation of NFATc3 in fibroblasts.20
Since NFAT plays a crucial role in the development of cardiac hypertrophy and NRON was suggested as a mediator of nuclear NFAT exclusion, we now hypothesized that NRON acts as anti-hypertrophic lncRNA. We thus characterized the role of NRON in the murine cardiac system by performing loss- and gain-of-function studies in a mouse model of pressure-overload-induced heart failure.
Results
NRON is enriched in cardiomyocytes and regulated during the development of pathological hypertrophy
We first investigated the subcellular distribution of NRON in cardiomyocytes. Using neonatal mouse cardiomyocytes (NMCMs), we found that 85.9% of NRON transcripts associated to chromatin, whereas only 2.19% were detected in the cytoplasmic fraction and 11.91% in the nuclear soluble fraction (Figure 1A). These data were in marked contrast to previous studies showing a cytosolic localization of NRON in blood-derived cells.14 We speculated that NRON may play a distinct role in the cardiac system and thus further investigated its role in vivo. We measured NRON expression levels in murine heart fractions. Whole hearts from adult mice were harvested and fractionated into cardiomyocytes, endothelial cells, and fibroblasts. The expression of NRON was highest in cardiomyocytes and significantly lower in endothelial cells and fibroblasts (Figure 1B; cycle threshold [ct] values are given in Table S1). Since NFAT plays an important role during the development of cardiac hypertrophy, we speculated that the expression of NRON might be also affected by pressure-overload-induced cardiac hypertrophy in vivo. We measured NRON expression in murine heart samples 2–13 weeks after TAC and Sham surgery (Figure 1C). NRON levels showed a biphasic regulation with significant downregulations (early) 2 weeks and (late) 13 weeks after TAC. We next tested the role of NRON in primary NMCMs and exposed the cells to hypertrophic stimuli using isoprenaline and phenylephrine (100 μM) for 48 h followed by measurements of the level of known hypertrophic markers as well as from NRON (Figure 1D). Brain natriuretic peptide (Bnp) and myocyte-enriched calcineurin-interacting protein (Mcip1.4) were significantly upregulated, while the level of NRON RNA was significantly decreased. We performed copy-number qPCR to get quantitative data about NRON expression changes and detected 30 ± 4.7 NRON molecules in 500 ng total RNA in untreated NMCMs versus 24 ± 2.7 molecules in NMCMs after treatment with phenylephrine/isoprenaline (Pe/Iso) for 48 h (Figure 1E).
Figure 1.

NRON level in neonatal mouse cardiomyocytes and heart fractions and after transaortic constriction
(A) Subcellular distribution of NRON in NMCM subfractions. Gapdh, ActB, and Xist mRNA levels were measured as positive controls. Data are calculated as fold change (FC) to the main fraction of the transcript measured by qPCR (mean ± SD; n = 5). (B) Distribution of NRON in murine heart fractions: cardiomyocytes (CMCs), cardiac fibroblasts (CFs), and endothelial cells (ECs). The expression level was measured by qPCR, and ActB was used as a reference gene. Data are calculated as FC to the main fraction (CMCs) of the transcript (mean ± SD; n = 5–7). ∗∗p < 0.01; one-way Anova with Tukey's multiple comparisons test. (C) The expression of NRON in the whole mouse heart after 2, 4, 6, 8, and 13 weeks of TAC surgery. The expression level was measured by qPCR, and ActB was used as a reference gene. Data are calculated relative to Sham samples of each time point (mean ± SD; n = 5–8). ∗∗∗p < 0.001; one-way ANOVA with Sidak multiple comparisons test. (D) RNA levels in neonatal mouse cardiomyocytes (NMCMs) after treatment with 100 μM Pe/Iso for 48 h. The RNA level was measured by qPCR relative to 18S RNA. The FC was calculated using the respective RNA levels in not-treated controls (ntcs). All data are mean ± SD; n = 3. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; Student's t test. (E) Absolute quantification of NRON molecules in NMCMs. A plasmid standard containing murine NRON was used for quantification, and the level of NRON was measured by qPCR. NMCMs were either not treated (ntcs) or treated with 100 μM Pe/Iso for 48 h. n = 6 technical replicates. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; Student's t test. (F) RNA levels in NMCMs after the transduction of AAV6-NRON. The RNA level was measured by qPCR relative to 18S RNA. The FC was calculated using the respective RNA level in AAV6-empty-treated cells as control. All data are mean ± SD; n = 3. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; Student's t test. (G) Subcellular distribution of NRON in NMCM subfractions of cells treated with 5 × 103 AAV6-empty or AAV6-NRON viral particles. Data are calculated by taking the sum of all fractions as 100% (mean ± SD; n = 3). The RNA level was measured by qPCR. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; two-way ANOVA with Tukey's multiple comparisons test. (H) Staining of NRON in NMCMs using RNA-FISH. (Top panel) RNA-FISH of a single nucleus from untreated NMCMs stained with no probe as control and NMCMs infected with AAV6-empty (ntcs) or AAV6-NRON (overexpressed [OE]) viral particles stained with probe for NRON. (Bottom panel) Orthogonal sections from z stack confocal images of NMCMs infected with AAV6-empty (ntcs) or AAV6-NRON (OE) viral particles and stained with RNA-FISH. DAPI was used to stain the nucleus, and NRON was stained using an Alexa Fluor 546 tagged probe set. Representative images were taken using a confocal microscopy. Scale bar, 10 μm.
To further study the effect of NRON in vitro, we infected NMCMs with adeno-associated virus 6 (AAV6)-empty or AAV6-NRON viral particles at an MOI of 5 × 103 for 72 h and measured the expression of hypertrophic markers and NRON. Surprisingly, this led to an increase in hypertrophic markers in NMCMs (Figure 1F). To examine the effects of NRON overexpression in more detail, we fractionated NMCMs after viral infection and measured the distribution of NRON. As is already shown in Figure 1A, the majority of NRON is detectable as a chromatin-associated fraction. After overexpression, the largest proportion of nuclear NRON is still chromatin-associated. In total, the distribution is partially shifted toward the cytoplasmic fraction (Figure 1G). In addition to the assessment of NRON distribution in subcellular fractions of NMCMs using qPCR, we performed RNA fluorescence in situ hybridization (RNA-FISH) to visualize the lncRNA under basal conditions and after AAV6-NRON transduction. To do so, we used a NRON-specific probe set in NMCMs, validating the previously identified subcellular distribution obtained by fractionation and qPCR studies (Figure 1H).
These interesting findings prompted us to perform further NRON loss- and gain-of-function experiments in mice undergoing TAC and Sham surgery.
AAV9-mediated overexpression of NRON exacerbated pressure-overload-induced cardiac hypertrophy
For NRON gain-of-function experiments in murine hearts, we used an AAV9-cardiac troponin T (cTnT) vector to overexpress NRON selectively in cardiomyocytes in comparison to an empty control. After a tail vein injection of 1.5 × 1012 viral particles into adult mice and subsequent TAC surgery, we detected an overexpression of the NRON transcript in the murine heart (Figure 2A and S1). The effect of AAV9-mediated NRON overexpression on cardiac hypertrophy was measured 6 weeks after TAC or Sham surgery. As expected, the TAC surgery led to an increase in the cardiac weight of the animals. When comparing the animals that underwent TAC surgery, NRON overexpression led to a significant increase in heart weight as compared to AAV9-empty-treated mice (Figure 2B). This effect was even more pronounced when comparing the cardiomyocyte cross-sectional area after TAC surgery between AAV9-empty- and AAV9-NRON-treated mice. The cross-sectional area of the cardiomyocytes was highest in the animals that received a TAC surgery in combination with an overexpression of NRON (Figures 2C and 2D). To characterize the different animal groups in more detail, RNA was isolated, and the mRNA level of different cardiac hypertrophy marker genes was measured. The expression levels of Bnp and Mcip1.4 were highest in TAC-operated animals treated with AAV9-NRON (Figures 2E and 2F). When measuring cardiac dimensions after performing echocardiographic analysis of the mice, an increase in left ventricular (LV) mass in AAV9-NRON-treated and TAC-operated mice became apparent in comparison to AAV9-empty TAC mice (Figure 2G). The ejection fraction showed a tendency toward reduced cardiac function upon NRON overexpression but did not reach significance (Table S2). Thereby, these data indicate a pro-hypertrophic effect of NRON in the murine cardiac system.
Figure 2.

TAC surgery of mice after injection of AAV9-NRON
1.5 × 1012 AAV9-NRON or AAV9-empty viral particles were injected following TAC or Sham surgery. Mice were sacrificed after 6 weeks. (A) NRON level was measured by qPCR. (B) Heart weight (HW) was measured relative to the length of the murine tibia (TL). (C and D) The cardiomyocyte cross-sectional area was analyzed by WGA staining, and the fold change was calculated relative to Sham AAV9-empty samples. Scale bar, 100 μm. (E and F) The levels of (E) Bnp and (F) Mcip1.4 mRNA were measured by qPCR relative to 18S RNA. (G) Left ventricular mass corrected (LV mass corr) was analyzed by echocardiography. All data are mean ± SD; n = 5–10; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001; two-way ANOVA with Tukey's multiple comparisons test or Student's t test.
Cardiac hypertrophy is attenuated in NRON KO mice
We next investigated the consequences of a NRON knockout in vivo on cardiac hypertrophy. Therefore, we generated mice with a Myh7-Cre-mediated, cardiomyocyte-specific knockout for NRON and verified this knockout with a phenotyping polymerase chain reaction (PCR) using the primers as depicted in Figure 3A. When comparing wild type (WT) with NRON knockout (KO) mice 6 and 13 weeks after Sham surgery, no differences were visible regarding heart weight and the cardiomyocyte cross-sectional area (Figures 3B–3E), indicating that NRON is dispensable for cardiac function under homeostatic conditions.
Figure 3.

Cardiac hypertrophy in NRON KO mice
Cardiac hypertrophy was analyzed after Sham or TAC surgery in NRON+/+ (WT) and NRON−/− (KO) mice. (A) Cardiomyocyte-specific NRON KO is mediated by cross-breeding Cretg/0 mice with NRON-floxed mice. For genotyping the DNA from the heart and liver tissues of NRON+/+, floxed NRON and NRON−/− mice were extracted, and a PCR was performed using primers 01_F, 02_R, and 03_F for amplification. As a marker (M), 100 bp DNA-Marker (New England Biolabs) was used. (B and C) HW after (B) 6 and (C) 13 weeks of surgery was measured relative to the length of the murine tibia (TL). (D and E) The cardiomyocyte cross-sectional area after 13 weeks of surgery was analyzed by WGA staining and is given in μm2. Scale bar, 100 μm. (F and G) Levels of (F) Bnp and (G) Mcip1.4 mRNA after 6 weeks of surgery were measured by qPCR and normalized to 18S RNA. (H and I) Levels of (H) Bnp and (I) Mcip1.4 after 13 weeks of surgery were measured by qPCR and normalized to 18S RNA. (J and K) LV mass corr was analyzed by echocardiography after (J) 6 and (K) 13 weeks of surgery. All data are mean ± SD; n = 7–12. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Two-way ANOVA with Tukey's multiple comparisons test.
To investigate the effect of NRON KO in vivo on the development of pressure-overload-induced cardiac hypertrophy, we performed TAC or Sham surgeries in WT and NRON KO mice. The mice were sacrificed 2, 6, or 13 weeks after surgery, and different cardiac parameters were measured. NRON KO mice exposed to 6 and 13 weeks of TAC showed a significantly lower heart weight than the respective WT mice (Figures 3B and 3C). This trend was also apparent when comparing the cardiomyocyte cross-sectional area (Figures 3D and 3E). However, no difference was detectable after 2 weeks (data not shown). When comparing the gene expression of hypertrophic markers after 6 weeks of TAC, significantly decreased expression levels of Bnp and Mcip1.4 were detectable upon NRON KO (Figures 3F and 3G). Although less pronounced, these trends were also visible after 13 weeks of TAC (Figures 3H and 3I). Cardiac dimensions after performing echocardiographic analysis of the mice showed a lower LV mass in NRON KO mice 6 weeks after TAC surgery (Figure 3J). This effect was even more pronounced and significant after 13 weeks of TAC (Figure 3K; Table S3). In contrast to the gain-of-function data described above, a NRON loss of function resulted in an anti-hypertrophic effect in the murine cardiac system, thus corroborating a function of NRON in cardiovascular disease in vivo.
Transcriptome profiling of NRON KO hearts reveals anti-hypertrophic pathway enrichment
To gain insight into the molecular processes affected by NRON KO in cardiac hypertrophy, we performed RNA sequencing of heart samples from WT and NRON KO mice 6 weeks after Sham and TAC surgery. Global gene expression was compared among three groups: WT mice exposed to Sham surgery (Sham WT), WT mice exposed to TAC surgery (TAC WT), and NRON KO mice exposed to TAC surgery (TAC KO). Since an anti-hypertrophic effect of NRON KO after TAC surgery was observed in previous experiments (see above), we had a special focus on those transcripts that showed a reverse regulation between Sham WT versus TAC WT and TAC WT versus TAC KO groups. Transcripts with a significantly different abundance in the three animal groups plus the described reverse regulation were displayed in a heat map (Figure 4A). A clear difference in transcript expression was detectable when comparing Sham WT and TAC WT groups. Interestingly, the investigated transcriptome of TAC KO mice was very similar to the one of Sham WT mice, thereby underlining the anti-hypertrophic effect of NRON KO observed before. Further analysis of significantly different regulated transcripts using the Gene Ontology Biological Process (GOBP) analysis revealed a role of NRON in the cellular response to mechanical stimulus as well as in heart-and cardiac-related functions (Figure 4B). Several genes known to associate with cardiac disease (e.g., Nppa, Nppb) were among the top 100 deregulated transcripts (Figure 4C and Table S4) and confirmed the anti-hypertrophic effect of NRON KO. We validated these findings in RNA samples from NRON WT and NRON KO mice exposed to 6 and 13 weeks of TAC or Sham surgery and confirmed the effect by studying Anp, Tgfb2, and Ctgf mRNA levels (Figures 4D–4F and S2).
Figure 4.

Transcriptomic changes upon NRON KO
RNA sequencing was performed in hearts from NRON+/+ (WT) and NRON−/− (KO) mice after 6 weeks of Sham or TAC surgery. (A) A heat map shows significantly different mRNAs with reverse regulation between the three groups: Sham WT, TAC WT, and TAC KO. (B) Gene Ontology Biological Process (GOBP) analysis of significantly different mRNAs with reverse regulation between the three groups and a minimum FC of 1.5. n = 4; one-way ANOVA. (C) Cardiac-related genes among the top 100 deregulated mRNAs. (D–F) Levels of Tgfb2 (D), Anp (E), and Ctgf (F) mRNA after 6 weeks of surgery were measured by qPCR and normalized to 18S RNA. All data are mean ± SD; n = 7–12. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; ∗∗∗∗p < 0.0001. Two-way ANOVA with Tukey's multiple comparisons test.
These data clearly indicate a role of NRON during the induction of or the response to pressure-overload-induced cardiac hypertrophy.
Discussion
Non-coding RNAs play important roles in the regulation of physiological and pathological processes. However, the specific role of the vast majority of ncRNAs is still under investigation. First insights into the function of the lncRNA NRON revealed an association with immunological- and cancer-related processes as well as a biomarker role in heart disease.15, 16, 17, 18, 19 Since NRON was described as a repressor of the hypertrophy-associated transcription factor NFAT, we hypothesized that NRON might interfere with the regulation of hypertrophy in the cardiac system. Hence, the aim of this study was to investigate the role of NRON in a cardiac hypertrophy mouse model.
Indeed, NRON expression was clearly downregulated during pressure-overload-induced cardiac hypertrophy and significantly enriched in cardiomyocytes. In contrast to previous studies demonstrating a cytoplasmic localization of NRON in T cells,13,14 we showed a mainly chromatin-associated localization of NRON in cardiomyocytes. To get the first details into the mechanistic role of NRON in a cardiovascular setting, we studied the lncRNA in primary NMCMs. Again, a clear downregulation was visible after hypertrophic induction, while hypertrophy markers were upregulated. Surprisingly, an overexpression using AAV6 viral particles led to a pro-hypertrophic effect in NMCMs.
These findings are in stark contrast to our own hypothesis that NRON functions as a repressor of NFAT-mediated hypertrophy. Gain-of-function experiments in vivo by cardiomyocyte-specific NRON overexpression also revealed a pro-hypertrophic effect of NRON in the murine TAC model. Accordingly, loss of function by cardiomyocyte-specific NRON KO showed an anti-hypertrophic effect upon TAC surgery in mice. Elevated or reduced heart weight, cardiomyocyte cell size, hypertrophic marker gene expression, and LV mass were clearly apparent for the gain- and loss-of-function models, respectively. Detailed transcriptome analyses strengthened our previous findings by demonstrating similar transcriptomic profiles of hypertrophy-induced NRON KO mice and Sham WT mice. Cardiac-disease-related genes, which are especially associated with hypertrophy, were among the most strongly deregulated transcripts, showing a significant upregulation upon the induction of hypertrophy by TAC surgery along with a significant downregulation upon NRON KO. Thereby, our data suggest a novel yet undescribed role of NRON in cardiomyocytes.
The subcellular localization of lncRNAs and their accompanied interaction with binding partners significantly impact their function.21 Hence, lncRNAs can function differentially in distinct cellular compartments. The lncRNA PYCARD-AS1, for example, was described to function as an epigenetic regulator of its target in the nucleus, whereas it functioned as a translational regulator in the cytoplasm. Both regulatory functions were mediated by an interaction with location-specific binding partners.22 It is postulated that a nuclear localization of lncRNAs is mediated by nuclear retention elements within the lncRNA sequence.23 However, nuclear retention of lncRNAs might also depend on their interaction with binding partners that are involved in the nuclear retention process. Nuclear ribonucleoprotein K is an example of a binding partner that mediates the nuclear enrichment of its target. However, nuclear ribonucleoprotein K showed a cell-type-specific expression,24 and, hence, subcellular localization of its target lncRNAs is cell-type-specific as well.
The lncRNA NRON shows a cell-type-specific localization to different subcellular compartments accompanied with a dual role in the regulation of cellular processes like hypertrophy. Therefore, the unexpected promotion of cardiac hypertrophy by NRON observed in this study might be a result of the differential subcellular localization of NRON in cardiomyocytes compared to the cytoplasmic localization in other, previously investigated cell types. Since this is the first study investigating NRON in cardiomyocytes, further studies need to reveal the molecular mechanisms leading to the observed pro-hypertrophic effect of NRON in cardiomyocytes.
Materials and methods
Animal procedures
Animal procedures were approved by Niedersächsisches Landesamt für Verbraucherschutz und Lebensmittelsicherheit (LAVES; approval number 15/1914). All animal experiments were performed in accordance with regulatory standards of the German animal protection law.
For NRON overexpression experiments, male C57BL/6J mice (8- to 10-weeks-old) were purchased from Charles River. WT mice (B6.129S1-Tg(Myh7-cre)1Jmk; kindly provided by Prof. Heineke, German Center for Cardiovascular Research partner site Mannheim/Heidelberg, Heidelberg, Germany) were crossbred with conditional NRON KO mice (C57BI/6J –Nrontm2Thum; Ozgene, Bentley, Australia) to obtain cardiomyocyte-specific NRON KO mice (B6-Tg(Mhy7)1Jmk-NRONtm2Thum).
TAC, as a model of pressure-overload-induced cardiac hypertrophy, and Sham surgery, a similar procedure to TAC surgery but without occlusion of the aorta, were applied to 8-week-old male mice, as described previously.25 AAV9-cTnT-NRON and AAV9-cTnT-empty viral particles were kindly provided by Prof. Engelhardt, Institute for Pharmacology and Toxicology, Technical University Munich, Munich, Germany. Intravenous injection of 1.5 × 1012 AAV9-NRON or AAV9-empty viral particles was performed at the end of the surgery process. Animals were anesthetized using 5% isoflurane in 100% O2. Anesthesia was maintained by 1.3%–1.5% isoflurane in 100% O2 and monitored via the toe pinch reflex. Butorphanol (2 mg/kg body weight) and Novalgin (62.5 mg/kg body weight) were subcutaneously injected pre-surgery and 6 h post-surgery. In addition, Novalgin (200 mg/kg body weight) was provided in the drinking water for 3 days after the surgery.
Cardiac function was assessed by echocardiography (Vevo2100, Fujifilm/VisualSonics, Canada) at the indicated time points. Animals were anesthetized using 5% isoflurane in 100% O2, and anesthesia was maintained by 0.8%–2% isoflurane in 100% O2. Mice were sacrificed by cervical dislocation under anesthesia at the indicated endpoints, and the heart tissue was used for histological and biochemical analyses.
For TAC and Sham surgeries, animals from the same litter were taken. An AAV9 injection was done in a blinded way by the animal technician. Subsequently, echocardiography was performed in a blinded manner. Only after the analysis the data were unblinded.
Cardiac cell fractionation
Cardiomyocytes, endothelial cells, and fibroblasts were isolated from mouse hearts by retrograde perfusion.26 Mice were anesthetized with 5% isoflurane in 100% O2 followed by 1%–2% isoflurane in 100% O2. Mice were injected intraperitoneally with 0.1 mL heparin (500 IU/mL), and Butorphanol (5 mg/kg body weight) was applied subcutaneously pre-surgery. Hearts were cannulated through the aorta and perfused with a pre-warmed perfusion buffer (113 mM NaCl, 4.7 mM KCl, 0.6 mM KH2PO4, 0.6 mM Na2HPO4, 1.2 mM MgSO4-7H2O, 0.032 mM phenol red, 12 mM NaHCO3, 10 mM KHCO3, 10 mM HEPES, 30 mM taurine, 0.1% glucose, 10 mM 2,3-butanedione monoxime) followed by perfusion with a pre-warmed digestion buffer (perfusion buffer supplemented with 700 U/mL collagenase II (Worthington Biochemical Cooperation, Lakewood, NJ, USA) and 12.5 μM CaCl2 for 10–15 min.
The atria were removed, and ventricles were transferred into a digestion buffer and cut into small pieces. Subsequently, anesthetized animals were sacrificed by cervical dislocation.
Tissue was dissociated using a 1 mL syringe. Stop buffer (perfusion buffer containing 10% FBS and 12.5 μM CaCl2) was added, and shearing was continued. A cell suspension was filtered through a 100 μm cell strainer, and a plating medium (minimum essential medium [MEM] supplemented with 10% FBS, 2 ng/mL vitamin B12, 4.2 mM NaHCO3, 100 U/mL penicillin, 100 μg/mL streptomycin) was added. Cardiomyocytes were allowed to sediment for 10 min at room temperature. A supernatant containing endothelial cells and fibroblasts was transferred to a fresh tube. Cardiomyocytes were washed with PBS, pelleted for 5 min at 3,000 rpm and 4°C, and flash-frozen in liquid nitrogen.
A cell suspension containing endothelial cells and fibroblasts was centrifuged for 5 min at 1,500 rpm and room temperature. Pelleted cells were resuspended in a plating medium, seeded into a tissue culture plate, and incubated at 37°C and 1% CO2 for 60 min. A supernatant containing endothelial cells was transferred into a fresh tube. Endothelial cells were purified by CD146-based MACS separation (Miltenyi, Bergisch Gladbach, Germany) according to the manufacturer's protocol. Briefly, endothelial cells were pelleted for 5 min at 1,500 rpm and 4°C, resuspended in MACS buffer, and mixed with CD146 MicroBeads. After incubation for 15 min at 4°C, cells were washed with MACS buffer and centrifuged for 5 min at 1,500 rpm and 4°C. Cells were resuspended in MACS buffer and applied onto MS column. CD146-positive cells were isolated in the magnetic field, centrifuged for 5 min at 3,000 rpm and 4°C, and flash-frozen in liquid nitrogen.
Fibroblasts were detached from the tissue culture plate using a cell scraper, centrifuged for 5 min at 3,000 rpm and 4°C, and flash-frozen in liquid nitrogen.
Cell culture and subcellular fractionation
NMCMs/neonatal rat cardiomyocytes (NRCMs) were isolated from 1-to-3-day-old mouse babies using the Neonatal Heart Dissociation Kit (Miltenyi). The primary cardiomyocytes were maintained in MEM (Bioconcept) with 5% FBS (Gibco), 1% Penicillin/streptomycin (Life Technologies), 100 nM BrdU (Sigma), and 2 μg/mL vitamin B12 (Sigma).
AAV6-cTnT-NRON and AAV6-cTnT-empty viral particles were kindly provided by Prof. Engelhardt, Institute for Pharmacology and Toxicology, Technical University Munich, Munich, Germany. The infection of NMCMs was done using viral particles at an MOI of 5 × 103 for 72 h.
For hypertrophic treatment, NMCMs were cultured in MEM with 1% FBS (Gibco), 1% Penicillin/streptomycin (Life Technologies), 100 nM BrdU (Sigma), and 2 μg/mL vitamin B12 (Sigma) supplemented with 100 μM phenylephrine hydrochloride (Sigma) and isoprenaline hydrochloride (Sigma) for 48 h. The subcellular fractionation of NMCMs into cytoplasmic, nuclear-soluble, and chromatin-associated fractions was performed as described previously.27 In brief, cells were dissociated by trypsin, washed with PBS, and centrifuged for 5 min at 500 × g. Pelleted cells were lysed in RLN1 buffer (50 mM Tris-HCl pH 8.0, 140 mM NaCl, 1.5 mM MgCl2, 0.5% Igepal, 2 mM Vanadyl Ribonucleosid Complex) and incubated for 5 min on ice. The lysate was centrifuged for 2 min at 300 × g and 4°C. The supernatant, which contains the cytoplasmic fraction, was transferred to a fresh tube and stored on ice. The pellet was lysed with RLN2 buffer (50 mM Tris-HCl pH 8.0, 500 mM NaCl, 1.5 mM MgCl2, 0.5% Igepal, 2 mM Vanadyl Ribonucleoside Complex) for 5 min on ice. The lysate was centrifuged for 2 min at 16,400 × g. The supernatant, which contains the nuclear-soluble fraction, and the pellet, which contains the chromatin-associated fraction, were each transferred into fresh tubes and used for RNA isolation and gene expression analyses.
RNA-FISH
NRON RNA-FISH was performed by using the ViewRNA Cell Plus Assay (Thermo Fisher Scientific, Waltham, MA, USA) in NMCM cells. All the buffers mentioned here were provided by the kit except for the PBS. Cells were cultured in an 8-well chamber slide (Thermo Fisher Scientific, 8 × 104 cells/well) and first fixed and permeabilized with a Fixation/Permeabilization solution for 30 min at room temperature. After being washed three times with 1× PBS/RNase inhibitor, the fixed cells were first incubated with NRON-specific Probe Sets (1:100, type I probe, 546 nm) for 2 h at 40°C. NMCM cells were next incubated one after one with PreAmplifier Mix (1:25), Amplifier Mix (1:25), and Label Probe Mix (1:25, incubated in the dark) for 1 h at 40°C. In between the probe incubation, cells were washed with Wash Buffer Solution five times. After all four Probe Sets/Mix stainings, cells were stained with DAPI for 5–15 min at room temperature in the dark. Images were taken with the Leica SP8 confocal microscope.
Cardiomyocyte cross-sectional area measurement
Cryo sections of mouse heart tissue were fixed with 4% paraformaldehyde and permeabilized with 0.1% Triton X-100 in PBS. The sections were stained with wheat germ agglutinin (WGA) coupled with Alexa Flour 488 (1:100 in PBS; Thermo Fisher Scientific, Waltham, MA, USA) for 40 min in the dark. Nuclei were counterstained with 5 μg/mL Dapi (Sigma-Aldrich, St. Louis, MO, USA). Images were acquired with a Nikon Eclipse Ti-U microscope (Nikon, Melville, NY, USA) and NIS-Elements BR 3.2 software (Nikon Instruments, Tokyo, Japan).
RNA isolation and gene expression analysis
Total RNA was isolated using the TriFast method (VWR, Darmstadt, Germany) according to the manufacturer's protocol. RNA was quantified using Synergy HT Bioreader (Biotek, Winooski, VT, USA). Isolated RNA (100–500 ng, according to sample concentration) was reverse-transcribed using the iScript Select cDNA Synthesis Kit (Bio-Rad, Hercules, CA, USA). For the quantification of gene expression, gene-specific primer sets (Table S5) and the iQ Sybr Green Mastermix were used. Gene expression was measured by ViiA7 (Thermo Fisher Scientific, Waltham, MA, USA) or the CFX384 Touch (BioRad, Hercules, CA, USA) real-time PCR detection system.
Absolute mRNA quantification
The copy number of NRON was quantified by qPCR and a plasmid standard curve, as described previously.28
PCR for genotyping
For the genotyping of mouse samples, genomic DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany) performing RNAse A digest according to the manufacturer's instructions. For the amplification of specific DNA fragments, PCR was performed with the reverse and forward primers for the specific gene product using the HotStarTaq Master Mix Kit (Qiagen) and 200 ng gDNA. The cycling conditions were 94°C for 3 min, 35 cycles of 94°C for 30 s, 65°C for 30 s, 72°C for 50 s, the final elongation at 72°C for 5 min, and hold at 4°C. For visualization, we performed gel electrophoresis using a 2% agarose gel. We used the 100 bp DNA-Marker (New England Biolabs, Ipswich, Massachusetts, USA) as a reference.
Transcriptome profiling
Total RNA was isolated from mouse hearts after 6 weeks of TAC or Sham surgery using the MiRNeasy Kit (Qiagen, Hilden, Germany) in four biological replicates per group. RNA quantity and quality were analyzed by Agilent Bioanalyzer and the RNA 6000 Nano Kit (Agilent Technologies, Santa Clara, CA, USA). Subsequent mRNA sequencing (NextSeq 550, 1 × 75 bp) was performed at the Research Core Unit Transcriptomics of Hannover Medical School (Hannover, Germany). 250 ng of total RNA per sample were utilized as input for the mRNA enrichment procedure with “NEBNext Poly(A) mRNA Magnetic Isolation Module” (E7490L; New England Biolabs) followed by stranded cDNA library generation using “NEBNext Ultra II Directional RNA Library Prep Kit for Illumina” (E7760L; New England Biolabs). Raw reads were aligned to the mouse genome (GRCm38 release M18) using STAR,29 and quantification of the mapped reads was performed with featureCounts.30 Deseq231 was then used for read counts normalization and differential expression analysis. RNA sequencing (RNA-seq) data have been deposited in the GEO database (GEO: GSE188762).
Statistics
Statistical analyses were performed using Graph Pad Prism software. Unpaired two-tailed Student’s t-test, one-way ANOVA with Sidak multiple comparisons test, and two-way ANOVA with Tukey's multiple comparisons test were applied as appropriate and as indicated in the figure legends. Data are shown as mean ± SD of biologically independent experiments. Differences were considered statistically significant at p values below 0.05 and are marked with asterisks (∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001).
Acknowledgments
We acknowledge the support of the Core Facility for Genomics (RCUG) at Hannover Medical School. We acknowledge the support of the Research Core Unit for Laser Microscopy at Hannover Medical School. We thank Jörg Heineke for providing Myh7-cre mice. We thank H. Janssen-Peters for supporting animal experiments. We thank K. Zimmer, K. Scherf, A. Gietz, N. Ernst, A. Korte, I. Riedel, and J. Bode for technical support. This study was funded by the Deutsche Forschungsgemeinschaft (RE 3523/1-1 | TH 903/11-1 given to K.R. and T.T.) and SFB/Transregio (TRR267 to S.E., C.B., and T.T.).
Author contributions
J.H. and J.L. designed research studies, conducted the majority of experiments, acquired and analyzed data, and wrote the manuscript. D.L, K.S., S.B., and H.J.H. acquired data. A.F. conducted echocardiography experiments and acquired and analyzed data. K.X. analyzed RNA-seq data. K.R. designed research studies. D.R. and S.E. provided AAV9 and AAV6 viral particles. C.B. and T.T. designed research studies and revised the manuscript.
Declaration of interests
T.T. and C.B. filed patents in the field of ncRNAs. T.T. is founder and holds shares of Cardior Pharmaceuticals GmbH. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2021.11.018.
Contributor Information
Christian Bär, Email: baer.christian@mh-hannover.de.
Thomas Thum, Email: thum.thomas@mh-hannover.de.
Supplemental information
References
- 1.Mattick J.S., Rinn J.L. Discovery and annotation of long noncoding RNAs. Nat. Struct. Mol. Biol. 2015;22:5–7. doi: 10.1038/nsmb.2942. [DOI] [PubMed] [Google Scholar]
- 2.Hobuß L., Bär C., Thum T. Long non-coding RNAs: at the heart of cardiac dysfunction? Front Physiol. 2019;10:30. doi: 10.3389/fphys.2019.00030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Klattenhoff C.A., Scheuermann J.C., Surface L.E., Bradley R.K., Fields P.A., Steinhauser M.L., Ding H., Butty V.L., Torrey L., Haas S., et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell. 2013;152:570–583. doi: 10.1016/j.cell.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grote P., Wittler L., Hendrix D., Koch F., Währisch S., Beisaw A., Macura K., Bläss G., Kellis M., Werber M., et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell. 2013;24:206–214. doi: 10.1016/j.devcel.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Piccoli M.T., Gupta S.K., Viereck J., Foinquinos A., Samolovac S., Kramer F.L., Garg A., Remke J., Zimmer K., Batkai S., et al. Inhibition of the cardiac fibroblast-enriched lncRNA Meg3 prevents cardiac fibrosis and diastolic dysfunction. Circ. Res. 2017;121:575–583. doi: 10.1161/CIRCRESAHA.117.310624. [DOI] [PubMed] [Google Scholar]
- 6.Viereck J., Kumarswamy R., Foinquinos A., Xiao K., Avramopoulos P., Kunz M., Dittrich M., Maetzig T., Zimmer K., Remke J., et al. Long noncoding RNA Chast promotes cardiac remodeling. Sci. Transl Med. 2016;8:326ra22. doi: 10.1126/scitranslmed.aaf1475. [DOI] [PubMed] [Google Scholar]
- 7.Viereck J., Bührke A., Foinquinos A., Chatterjee S., Kleeberger J.A., Xiao K., Janssen-Peters H., Batkai S., Ramanujam D., Kraft T., et al. Targeting muscle-enriched long non-coding RNA H19 reverses pathological cardiac hypertrophy. Eur. Heart J. 2020;41:3462–3474. doi: 10.1093/eurheartj/ehaa519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chatterjee S., Bär C., Thum T. Linc-ing the noncoding genome to heart function: beating hypertrophy. Trends Mol. Med. 2017;23:577–579. doi: 10.1016/j.molmed.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 9.Bär C., Chatterjee S., Thum T. Long noncoding RNAs in cardiovascular pathology, diagnosis, and therapy. Circulation. 2016;134:1484–1499. doi: 10.1161/CIRCULATIONAHA.116.023686. [DOI] [PubMed] [Google Scholar]
- 10.Wilkins B.J., Dai Y.S., Bueno O.F., Parsons S.A., Xu J., Plank D.M., Jones F., Kimball T.R., Molkentin J.D. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ. Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 11.Wilkins B.J., Molkentin J.D. Calcium-calcineurin signaling in the regulation of cardiac hypertrophy. Biochem. Biophys. Res. Commun. 2004;322:1178–1191. doi: 10.1016/j.bbrc.2004.07.121. [DOI] [PubMed] [Google Scholar]
- 12.Dirkx E., da Costa Martins P.A., De Windt L.J. Regulation of fetal gene expression in heart failure. Biochim. Biophys. Acta. 2013;1832:2414–2424. doi: 10.1016/j.bbadis.2013.07.023. [DOI] [PubMed] [Google Scholar]
- 13.Willingham A.T., Orth A.P., Batalov S., Peters E.C., Wen B.G., Aza-Blanc P., Hogenesch J.B., Schultz P.G. A strategy for probing the function of noncoding RNAs finds a repressor of NFAT. Science. 2005;309:1570–1573. doi: 10.1126/science.1115901. [DOI] [PubMed] [Google Scholar]
- 14.Sharma S., Findlay G.M., Bandukwala H.S., Oberdoerffer S., Baust B., Li Z., Schmidt V., Hogan P.G., Sacks D.B., Rao A. Dephosphorylation of the nuclear factor of activated T cells (NFAT) transcription factor is regulated by an RNA-protein scaffold complex. Proc. Natl. Acad. Sci. U S A. 2011;108:11381–11386. doi: 10.1073/pnas.1019711108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imam H., Bano A.S., Patel P., Holla P., Jameel S. The lncRNA NRON modulates HIV-1 replication in a NFAT-dependent manner and is differentially regulated by early and late viral proteins. Sci. Rep. 2015;5:8639. doi: 10.1038/srep08639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J., Chen C., Ma X., Geng G., Liu B., Zhang Y., Zhang S., Zhong F., Liu C., Yin Y., et al. Long noncoding RNA NRON contributes to HIV-1 latency by specifically inducing tat protein degradation. Nat. Commun. 2016;7:11730. doi: 10.1038/ncomms11730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yao Z., Xiong Z., Li R., Liang H., Jia C., Deng M. Long non-coding RNA NRON is downregulated in HCC and suppresses tumour cell proliferation and metastasis. Biomed. Pharmacother. 2018;104:102–119. doi: 10.1016/j.biopha.2018.05.006. [DOI] [PubMed] [Google Scholar]
- 18.Niu L., Fan Q., Yan M., Wang L. LncRNA NRON down-regulates lncRNA snaR and inhibits cancer cell proliferation in TNBC. Biosci. Rep. 2019;39 doi: 10.1042/BSR20190468. BSR20190468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xuan L., Sun L., Zhang Y., Huang Y., Hou Y., Li Q., Guo Y., Feng B., Cui L., Wang X., et al. Circulating long non-coding RNAs NRON and MHRT as novel predictive biomarkers of heart failure. J. Cell Mol Med. 2017;21:1803–1814. doi: 10.1111/jcmm.13101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang Y., Xu P., Zhang C., Feng J., Gong W., Ge S., Guo Z. LncRNA NRON alleviates atrial fibrosis via promoting NFATc3 phosphorylation. Mol. Cell Biochem. 2019;457:169–177. doi: 10.1007/s11010-019-03521-y. [DOI] [PubMed] [Google Scholar]
- 21.Bridges M.C., Daulagala A.C., Kourtidis A. LNCcation: lncRNA localization and function. J. Cell Biol. 2021;220:e202009045. doi: 10.1083/jcb.202009045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miao H., Wang L., Zhan H., Dai J., Chang Y., Wu F., Liu T., Liu Z., Gao C., Li L., et al. A long noncoding RNA distributed in both nucleus and cytoplasm operates in the PYCARD-regulated apoptosis by coordinating the epigenetic and translational regulation. Plos Genet. 2019;15:e1008144. doi: 10.1371/journal.pgen.1008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Palazzo A.F., Lee E.S. Sequence determinants for nuclear retention and cytoplasmic export of mRNAs and lncRNAs. Front Genet. 2018;9:440. doi: 10.3389/fgene.2018.00440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lubelsky Y., Ulitsky I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature. 2018;555:107–111. doi: 10.1038/nature25757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rockman H.A., Ross R.S., Harris A.N., Knowlton K.U., Steinhelper M.E., Field L.J., Ross J., Jr., Chien K.R. Segregation of atrial-specific and inducible expression of an atrial natriuretic factor transgene in an in vivo murine model of cardiac hypertrophy. Proc. Natl. Acad. Sci. U S A. 1991;88:8277–8281. doi: 10.1073/pnas.88.18.8277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.O'Connell T.D., Rodrigo M.C., Simpson P.C. Isolation and culture of adult mouse cardiac myocytes. Methods Mol. Biol. 2007;357:271–296. doi: 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- 27.Cabianca D.S., Casa V., Bodega B., Xynos A., Ginelli E., Tanaka Y., Gabellini D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in FSHD muscular dystrophy. Cell. 2012;149:819–831. doi: 10.1016/j.cell.2012.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feretzaki M., Nunes P.R., Lingner J. Expression and differential regulation of human TERRA at several chromosome ends. RNA. 2019;25:1470–1480. doi: 10.1261/rna.072322.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liao Y., Smyth G.K., Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014;30:923–930. doi: 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- 31.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.