

Abstract
Prime editor (PE) has tremendous promise for gene therapy. However, it remains a challenge to deliver PE (>6.3 kb) in vivo. Although PE can be split into two fragments and delivered using dual adeno-associated viruses (AAVs), choice of split sites within Cas9—which affects editing efficiency—is limited due to the large size of PE. Furthermore, overexpressing reverse transcriptase in mammalian cells might disrupt translation termination via its RNase H domain. Here, we developed a compact PE without the RNase H domain that showed editing comparable with full-length PE. With compact PE, we used a Cas9 split site (Glu 573) that supported robust editing in cells (up to 93% of full-length PE) and in mouse liver. We then demonstrated that split-cPE573 delivered by dual-AAV8 efficiently mediated a 3-bp TGA insertion in the Pcsk9 gene in mouse liver. Compact PE without the RNase H domain abolished its binding to peptidyl release factor 1 (eRF1) and mitigated the stop codon readthrough effect observed with full-length PE. This study identifies a compact PE with a flexible split design to advance utility of prime editing in vivo.
Keywords: prime editing, reverse transcriptase, split prime editor, mouse liver, AAV
Graphical abstract
Delivering prime editor (PE) in vivo is challenging due to its large size. Here, we developed a compact PE without the RNase H domain. Using compact PE, we developed an optimized split-PE to support robust prime editing in vivo. The compact PE abolished its binding to eRF1 and showed minimal effect on stop codon readthrough.
Introduction
PE is a new gene editing tool consisting of a Cas9 nickase fused to an engineered Moloney murine leukemia virus (M-MLV) reverse transcriptase (RT). In the PE construct, Cas9 nickase is guided by RNA (single guide RNA [sgRNA]) to a target site, where the nickase cleaves only one DNA strand. M-MLV RT then uses an extension of the sgRNA, called prime editing guide RNA (pegRNA), as an RT template to generate complementary DNA (cDNA) for precise repair of the nicked site. By erasing the need to generate double-strand breaks to direct changes in the genome, PE overcomes limitations of previous gene editing tools that have caused bottlenecks in their application as therapeutics. Moreover, PE can flexibly generate all possible base substitutions, small insertions (up to 44 bp), and small deletions (up to 80 bp).1,2 With enhanced precision and versatility, PE promises to accelerate the clinical development of gene editors.
For PE to succeed as a tool for gene therapy, it is important to achieve efficient delivery in vivo. Adeno-associated virus (AAV) is the most widely used vector for in vivo delivery of gene therapies. However, the packaging capacity of AAV vectors is limited to ∼5 kb,3,4 which is insufficient for the large PE construct (>6.3 kb). To address this issue, Cas9 is split into two vectors,5, 6, 7, 8 often using the extensively studied split-intein system, and delivered by dual AAVs. Choice of split site affects the structural stability, and thus the reconstituting activity, of Cas9, resulting in variable editing efficiency. Due to the large size of PE, Cas9 can only be split within a small window to fit in AAV vectors. Unfortunately, split sites within this window do not support efficient Cas9 editing. Reducing PE size would confer flexibility in the choice of split site, and provide an opportunity to achieve more efficient prime editing in vivo.
It may be possible to engineer a compact PE by truncating RT. M-MLV RT is a monomeric polypeptide containing fingers/palm, thumb, connection, and RNase H domains (residues 1–275, 276–361, 362–496, and 497–671, respectively),9, 10, 11 and has two active sites (DNA polymerase and RNase H) that reside in separate, distinct structural domains12 (Figure 1A). Mutating the RNase H domain, which is responsible for degrading RNA in the RNA/DNA hybrid during DNA synthesis, improves efficiency of cDNA synthesis from the protected RNA.13 We hypothesize, therefore, that the RNase H domain is not essential for prime editing.
Figure 1.

The RT RNase H domain in PE3 is dispensable for prime editing
(A) Top: crystal structure of M-MLV RT (PDB: 5dmq.1). Bottom: schematic representations of RT variants with C-terminal truncations. (B) Top: schematic of an mCherry reporter containing a premature stop codon in HEK293T cells. PEs can generate a TAG-to-CAA transition to correct the premature stop codon and trigger mCherry expression. Bottom: editing efficiencies of RT variants (RT362, RT418, RT474, and RT497) and full-length PE3 were measured by flow cytometry. Results were obtained from six independent experiments, shown as mean ± SD. (C) TLR-MCV1 HEK293T cells containing a GFP with a 39-bp insertion, P2A, and out-of-frame mCherry. A precise insertion of 18 bp to replace the 39-bp insertion leads to GFP expression, while indels cause a frameshift that leads to mCherry expression. The frequencies of precise insertion (GFP+) or indel (mCherry+) were measured by flow cytometry. Results were obtained from five independent experiments, shown as mean ± SD. (D) TLR HEK293T cells containing GFP with a 47-bp insertion. Deletion of the 47-bp insertion leads to GFP expression, while indels cause a frameshift that leads to mCherry expression. Frequencies of precise deletion (GFP+) or indel (mCherry+) were measured by flow cytometry. Results were obtained from four independent experiments, shown as mean ± SD. NS, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Removing the RNase H domain may also improve safety of PE in cells. Previous studies show that the RNase H domain of native M-MLV RT interacts with eRF1,14 which is essential for termination of protein translation.15 During translation termination, eRF1 recognizes a stop codon and recruits other termination factors (e.g., eRF3) through its C-terminal domain to form the pre-termination complex. Because eRF1 also interacts with RT through its C-terminal domain, RT (when overexpressed) might occlude binding of regulatory factors to eRF1, promoting stop codon readthrough.14,16 Overexpressing PE in cells shows minimal perturbation of the transcriptome,1 but the effect of PE on protein translation has not been investigated.
In this study, we developed a compact PE without the RNase H domain of RT. The compact PE increased the flexibility of split site choice in Cas9 while maintaining editing efficiency comparable with PE in HEK293T cells. Using the compact PE, we developed an optimized split-PE to support robust prime editing in cells and in vivo. We also found that PE2 interacts with eRF1 through the RNase H domain of RT to promote readthrough in HEK293T cells, whereas compact PE2 without the RNase H domain does not interact with eRF1, abolishing the readthrough effect. Compact PE has the potential to improve the safety and delivery efficiency of prime editing for in vivo applications.
Results
Development of a compact PE lacking the RNase H domain
We constructed four compact PE variants—RT497, RT474, RT418, and RT362—with progressive truncation of the RNase H and connection domains (Figure 1A). After transfection into HEK293T cells, we observed expression of each PE variant by western blot (Figure S1A).
To evaluate the editing efficiency of compact PE3 variants in cells, we delivered each construct to a HEK293T reporter cell line with a premature TAG stop codon in the mCherry coding sequence (Figure 1B). Each compact PE3 variant was programmed to convert the stop codon to CAA to induce mCherry expression, which is measured by flow cytometry.17 We found that RT497, in which the RNase H domain is removed, exhibited prime editing efficiency that was similar to PE3 (11.0% versus 10.2%, respectively; Figures 1B and S1B). By contrast, RT474, RT418, and RT362, which lack not only the RNase H domain but also parts of the connection domain, showed significantly lower editing efficiencies (7.3%, <0.1%, and <0.1%, respectively).
Next, we tested the efficiency of precise genomic insertion by compact PE3 variants in a Traffic Light Reporter Multi-Cas Variant 1 (TLR-MCV1) HEK293T reporter line. Each variant was programmed to insert an 18-bp sequence at the target site to induce GFP expression. Imprecise insertion/deletion (indels) by compact PE3 variants will induce mCherry expression (Figure S2A).17 RT474, RT497, and PE3 exhibited similar editing efficiencies (3.4%, 4.9%, and 4.9%, respectively) and indel rates (0.9%, 1.3%, and 2.0%, respectively; Figure 1C), whereas RT362 and RT418 showed lower on-target editing efficiencies (0.1% for both).
Finally, we tested the compact PE3 variants to precisely delete a 47-bp insertion in a second TLR reporter cell line (Figure S2B).17 RT497 showed higher on-target editing efficiency (4.4% versus 3.5%) and similar indel rate (0.74% versus 0.75%), whereas RT474 and PE3 exhibited similar editing efficiencies (3.2% versus 3.5%) and indel rates (0.83% versus 0.75%). By contrast, editing efficiencies of RT362 and RT418 were significantly lower (0.1% and 0.2%) (Figure 1D).
Collectively, these results demonstrate that the RT RNase H domain is dispensable for prime editing. We found that RT497 exhibits nucleotide substitutions, precise small insertions and deletions, and indel rates at efficiencies comparable with full-length PE3.
Compact PE efficiently introduces mutations at endogenous loci
To show that the observed editing efficiencies of compact PE3 variants are not specific to fluorescent reporters, we programmed each compact PE3 variant to edit at the +1 position (1 bp after the Cas9 nicking site) of four endogenous loci (FANCF, VEGFA, RNF2, and HEK3) previously validated by amplicon sequencing in HEK293T cells.1 Using deep sequencing, we found that RT497 induced on-target editing efficiencies comparable with PE3 at all four loci. Indeed, RT497 induced a 1-bp substitution at a frequency of 51.1%, 50.7%, and 20.7% at FANCF, VEGFA, and RNF2, respectively. PE3 resulted in 1-bp substitutions at a frequency of 46.6%, 46.9%, and 20.3% at FANCF, VEGFA, and RNF2, respectively. RT497 and PE3 also induced 3-bp insertions at a frequency of 40.9% and 37.7%, respectively, in the HEK3 locus (Figures 2A, S3A–S3D). RT474 exhibited lower editing efficiency for 1-bp substitutions at RNF2 (12.6%), but comparable editing efficiencies for 1-bp substitutions at FANCF (46.7%) and VEGFA (48.9%), and 3-bp insertion at HEK3 (37.8%).
Figure 2.

Precise editing by compact PE3 at endogenous loci
(A) Comparison of precise editing and indel efficiency for substitutions and insertions induced by RT362, RT474, RT497, and PE3 at FANCF, VEGFA, RNF2, and HEK3 loci in HEK293T cells. (B) Left: schematic of single nucleotide substitution at positions +1, +12, and +30 of HEK3. Right: comparison of precise editing efficiency for single nucleotide substitution at positions +1, +12, and +30 of HEK3 by RT474, RT497, and PE3. Results were obtained from three independent experiments (RT474) or four independent experiments (RT362, RT497, PE3), shown as mean ± SD. NS, not significant; ∗p < 0.05; ∗∗∗p < 0.001.
Although the RNase H domain of M-MLV RT is not required for DNA polymerase activity, the residues in the C helix may stabilize enzyme/primer-template interactions. Thus, loss of the RNase H domain could affect the processivity of DNA synthesis, leading to a shorter RT product.18 To determine whether the best-performing compact PE3 variants (RT497 and RT474) have a narrower editing window than PE3, we programmed each for 1-bp substitutions at positions +1, +12, and +30 in the HEK3 locus in HEK293T cells (Figure 2B).1 RT474, RT497, and PE3 had similar editing efficiencies at the +1 position (32.3%, 36.5%, and 35.8%, respectively), +12 position (32.3%, 38.0%, and 38.2%, respectively), and +30 position (24.4%, 31.7%, and 31.2%, respectively) (Figure 2B). These data suggest that truncating the RNase H domain only does not affect the processivity of RT for short products (∼30 bp). Based on these results, we moved forward with RT497 to develop an efficient split-PE2.
Development of efficient split-PE2
Due to the packaging limit of AAV (<5 kb) and the large size of SpyCas9 (4.1 kb) and M-MLV RT (2 kb), Cas9 can only be split after residue R691 to ensure the C-terminal fragment of PE2 can be packaged with a U1a promoter, RBG poly(A), and Nostoc punctiforme (Npu) intein into a single AAV vector (Figures 3A and S4A). The smaller size of RT497 allows Cas9 to be split up to residue L551 from the C terminus (Figure S4A), enabling the use of a potentially more efficient Cas9 split site.3,19 To identify an optimal split-RT497 (referred to as split-cPE hereafter), we tested four different split sites that have previously been tested for split-Cas9 or split-base editors: E573 (split-cPE2-573), K637 (split-cPE2-637), Q674 (split-cPE2-674), or Q713 (split-cPE2-713).3,8,19,20 To determine the effect of each split site on RT497 fusion, we co-transfected the N-terminal and C-terminal fragments of each split-cPE2 variant into HEK293T cells and measured protein expression using western blot. We observed spliced full-length RT497 expression after transfection of three out of the four variants: split-cPE2-573, split-cPE2-674, and split-cPE2-713. Among them, split-cPE2-573 showed the highest expression of full-length RT497 (Figure S4B).
Figure 3.

Development of a split-intein PE3 for in vivo delivery
(A) Map of split-cPE2 plasmids. CMV, cytomegalovirus promoter; NLS, SV40 nuclear localization sequence; N-Cas9 nickase, N-terminal Cas9; C-Cas9 nickase, C-terminal Cas9; poly(A), bGH poly(A). (B) Editing frequencies of split-cPE3 at FANCF and HEK3 in HEK293T cells. Results were obtained from three independent experiments, shown as mean ± SD. (C) PE2 or split-cPE2-573, pegRNA, nicking sgRNA targeting Ctnnb1, MYC transposon, and transposase plasmids were hydrodynamically injected into wild-type FVB mice to generate liver tumors. PE2, non-target pegRNA, MYC transposon, and transposase plasmids were injected in the control group. (D) Representative images of liver tumor burden 28 days after plasmid injection. (E) Mean number of visible tumor nodules. Results were obtained from three (control and PE3) or four (split-cPE3-573) mice, shown as mean ± SD. (F) Sanger sequencing of control liver and representative tumors. NS, not significant; ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Next, we tested the prime editing efficiency of split-cPE3 variants at two endogenous loci (FANCF and HEK3) in HEK293T cells. Using deep sequencing, we found that split-cPE3-573 showed the highest precise editing efficiency among split-cPE3 variants. Split-cPE3-573 editing efficiency was 92.6% of full-length RT497 at HEK3 (38.9% versus 42.0%), and 74.2% of RT497 at FANCF (28.2% versus 38.0%) (Figures 3B, S4C, and S4D). Split-cPE3-573 outperformed split-PE3 variants from previous studies with regard to maintaining editing efficiency of full-length counterparts. Indeed, editing efficiencies of PE3 split at 1,005 and 1,024 with Rhodothermus marinus intein21 were ∼72.0% and ∼75.8% of full-length PE3 at HEK3, and ∼65.6% and ∼68.4% of full-length PE3 at VEGFA.21 For comparison, split-cPE3-573 editing efficiency was ∼92.6% of RT497 at HEK3 (Figure 3B) and ∼78.5% of RT497 at VEGFA (Figure S5A). To compare the off-target effects induced by cPE3 versus split-cPE3-573, we analyzed off-target effects at the top four known Cas9 off-target sites for the three pegRNAs at the HEK3, FANCF, and VEGFA loci. Deep sequencing showed no detectable off-target editing at any of the 12 sites (Figure S5A).
Next, we sought to determine whether split-cPE3-573 could induce efficient prime editing in vivo. We designed a pegRNA to generate a C:G to T:A transversion in Ctnnb1 (β-Catenin) that produces the oncogenic S45F mutation frequently observed in liver cancer.22,23 PE2 or split-cPE2-573, pegRNA, and nicking sgRNA were delivered to the liver of Friend leukemia virus B (FVB) mice via hydrodynamic tail-vein injection. Deep sequencing data showed that the average precise substitution of S45F in Ctnnb1 of PE3 and split-cPE3-573 were 0.87% and 0.63% (n = 3), respectively (Figure S5B). Next, we co-injected mice with MYC transposon to provide a second oncogenic driver for liver cancer formation with the Ctnnb1 mutation (Figure 3C).24 Mice were sacrificed after 28 days. Mice injected with PE3 (n = 3) and split-cPE3-573 (n = 4) developed a similar number of liver tumor nodules, with an average of 10.7 tumors and 9.5 tumors per mouse, respectively (Figures 3D and 3E). Sanger sequencing of tumor nodules showed precise substitution of S45F in Ctnnb1 in both groups (Figure 3F). As a control, a non-target pegRNA was injected and no tumor was formed. Together, these data demonstrate that split-cPE3-573 could induce efficient prime editing in vivo. Using split-cPE-573, the coding sequence of PE could be distributed on a dual-vector system and packaged into AAV.
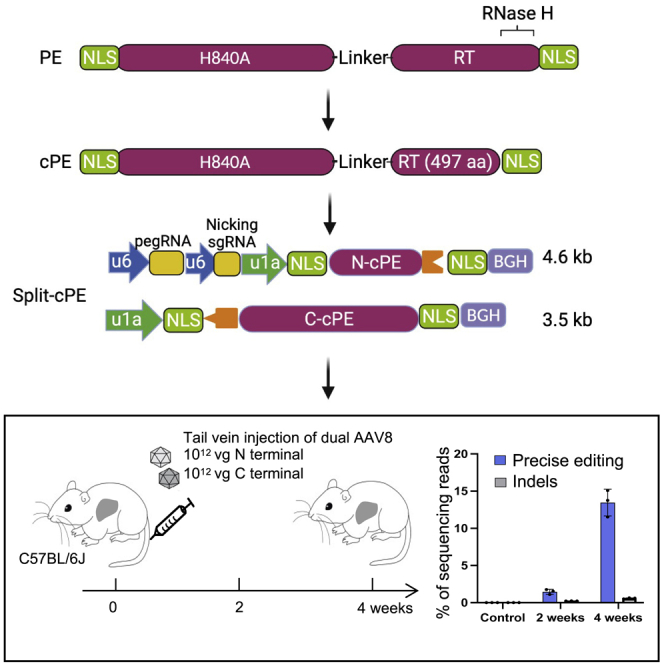
Delivery of split-cPE using AAV for in vivo prime editing
We next investigated whether split-cPE-573 could be delivered in vivo using dual AAVs. We designed a pegRNA and nicking sgRNA targeting the mouse proprotein convertase subtilisin/kexin type 9 (Pcsk9)25,26 gene to insert a TGA stop codon in the first exon, which could abolish Pcsk9 function in familial hypercholesterolemia. We first evaluated the TGA insertion efficiency by co-transfecting PE3 with pegRNA and sgRNA in Hepa 1-6 cells. Sanger sequencing showed that PE3 could efficiently insert TGA at Pcsk9 (Figure 4A).
Figure 4.

Efficient editing of Pcsk9 in mouse liver by dual AAV8 split-intein cPE-573
(A) Introducing a TGA stop codon in the Pcsk9 gene by PE. Sanger sequencing of control and PE3-treated Hepa1-6 cells is shown. Arrows denote TCA insertion (reverse complementary of TGA). (B) Schematic of the split-intein dual-AAV cPE-573 and tail-vein injection experiments. Five-week-old C57BL/6 mice were injected with a total of 2 × 1012 vg AAV8. After 2 and 4 weeks, liver genomic DNA was sequenced. (C) Frequencies of precise editing and indel in AAV-treated mice. Error bars are SD (n = 3). Untreated mice were used as negative controls.
Next, we generated two AAV vectors; the first vector encodes the N-terminal half of cPE2, pegRNA, and nicking sgRNA targeting Pcsk9, and the second vector encodes the C-terminal half of cPE2 (Figure 4B). These two vectors were packaged into AAV8 and a total of 2 × 1012 vector genome (vg) was injected into 5-week-old C57BL/6J mice through the tail vein (Figure 4B). Mouse livers were harvested at 2 weeks and 4 weeks after injection. Deep sequencing showed that the average precise editing efficiencies were 1.5% (n = 3) and 13.5% (n = 3), respectively (Figure 4C). These data suggest that split-cPE2-573 can be delivered in vivo using dual-AAV.
PE2 but not RT497 promotes stop codon readthrough
M-MLV RT interacts with the C-terminal domain of eRF1 via the RNase H domain (Figure 5A).14 Moreover, structures of the eRF1/eRF3 complex and the M-MLV RT/eRF1 complex show that eRF1 interacts with M-MLV RT and eRF3 through overlapping surface regions14; therefore, RT likely competes with eRF3 to bind eRF1. However, it is unclear whether PE2, which contains an engineered variant of M-MLV RT with five mutations (D200N, L603W, T330P, T306K, and W313F), interacts with eRF1 in cells. Using co-immunoprecipitation (coIP) assays, we confirmed that eRF1 interacts with PE2 (Figure 5B).
Figure 5.

PE2 increases stop codon readthrough through the RNase H domain
(A) Structure of the M-MLV RNase H domain/eRF1 complex (PDB: 5DMQ). (B) CoIP of PE2 with eRF1. Plasmid constructs of PE2-FLAG or RT497-FLAG were co-transfected with eRF1-HA into HEK293T cells. Empty vectors expressing each tag (FLAG, HA) were used as negative controls. (C) Schematic of the dual luciferase bicistronic vector used in translational readthrough assays. The red dot represents the stop codon. (D) PE2 but not RT497 overexpression promotes stop codon readthrough. PE2, RT497, or control vector were co-transfected with dual luciferase reporter in HEK293T cells. The ratio of Firefly:Renilla from each experimental group was normalized to a control reporter lacking a stop codon between Firefly and Renilla. (E) Model of PE2 promoting stop codon readthrough. PE2 interacts with eRF1 via the RNase H domain, which inhibits the binding of eRF1 with eRF3, promoting stop codon readthrough.
eRF1 plays an essential role in the termination of protein translation and nonsense-mediated decay (NMD) of mRNA molecules.15 During translational termination, eRF1 recognizes all three stop codons and recruits other termination factors, such as eRF3, to form the pre-termination complex. However, when eRF1 detects a premature stop codon or an irregularly spliced mRNA, it will recruit upframeshift protein 1 (UPF1) to trigger NMD.15 eRF1 interacts with RT and other regulatory factors through its C-terminal domain. RT may compete with eRF3, UPF1, and other regulatory factors' binding to eRF1, thereby promoting stop codon readthrough and bypassing mRNA degradation by NMD. To test whether PE2 outcompetes eRF3 to promote readthrough, we used an established stop codon readthrough reporter containing Renilla luciferase, a recoding window that includes a stop codon, and firefly luciferase (Renilla:TAG:firefly)27 (Figure 5C). Stop codon readthrough is measured by firefly relative to Renilla luciferase activities. A control reporter containing a recoding window lacking a stop codon (Renilla:CAG:firefly) was used for normalization. PE2 overexpression in cells containing the dual luciferase reporter led to a ∼1.6-fold increase in readthrough compared with an empty control (7.6% versus 4.6%, respectively) (Figure 5D). To test whether removing the RNase H domain from PE2 can abolish its interactions with eRF1, we performed coIPs and the stop codon readthrough assay with RT497. RT497 did not interact with eRF1 (Figure 5B), and its overexpression in cells led to readthrough rates comparable with the control (Figure 5D). Taken together, these data suggest PE2 interacts with eRF1 via the RT RNase H domain, thereby promoting stop codon readthrough (Figure 5E).
Discussion
PEs have the potential to correct almost any human pathogenic mutations by generating precise transitions, transversions, small insertions, or small deletions. However, the large size of PEs makes them more difficult to deliver in vivo than Cas9 or base editors, a limitation impeding the development of PE-based gene therapies. In vivo delivery of PEs using dual AAVs has been reported.17,21 However, these studies used a Cas9 split site at 713 with strict limitations in promoter and poly(A) length,17 or used suboptimal Cas9 split sites 1,005 and 1,024.21 In these cases, prime editing efficiency was low in vivo.
In this work, we report that the M-MLV RT RNase H domain is dispensable for prime editing. By removing the RNase H domain, we developed a cPE with editing and indel efficiencies comparable with full-length PE. Using cPE, we screened four split sites combined with Npu intein. Consistent with previous studies using base editors, we found that split-cPE-573 showed the highest on-target editing efficiency among different split sites at endogenous loci.3,8,19 We also developed an optimized dual-AAV split-cPE573 using compact PE, which support robust editing in mouse liver.
It has been reported that expression of PEs perturbs the transcriptome minimally relative to Cas9 nickase and PEs lacking active RT.1 However, the potential effects of PEs on other cellular processes have not been well studied. Here, we uncovered a previously unknown role of PE in promoting stop codon readthrough using a reporter assay, which is mediated by the RT RNase H domain. Further studies are required to determine how PE modulates the stop codon readthrough of endogenous genes. Given that PEs may be used for gene therapy in the future, continued investigations into their potential effects on global protein translation are warranted.
In summary, our study identified a compact PE by removing the RNase H domain of RT. Compact PE can efficiently induce precise editing in vitro and in vivo, with efficiencies similar to full-length PE. This compact PE will enable more flexible vector designs for in vivo delivery.
Material and methods
Plasmid construction
Plasmids expressing sgRNA were generated by ligation of annealed oligos into a BfuA I-digested vector.17 For plasmids, gBlocks gene fragments were inserted into a BfuA I/EcoR I-digested vector by Gibson assembly.17 Compact PE2 variants were generated by PCR using Phusion master mix (Thermo Fisher Scientific). Sequences of sgRNA and pegRNA are listed in Table S1. All primers used for cloning are listed in Table S2. All plasmids used for mammalian cell experiments were purified using plasmid Miniprep kits (Qiagen).
Cell culture
Human embryonic kidney (HEK293T) cells were purchased from ATCC and cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (Gibco) and 1% (v/v) penicillin/streptomycin (Gibco). Cells were cultured at 37°C with 5% CO2 and tested negative for mycoplasma.
HEK293T transfection and genomic DNA extraction
HEK293T and HEK293T reporter cells were seeded on 12-well plates at 100,000 cells per well. Twenty-four hours after seeding, cells were transfected using Lipofectamine 3000 reagent (Invitrogen) following the manufacturer's protocol. Briefly, 1 μg of PE2, 330 ng of pegRNA, and 110 ng of nicking sgRNA were transfected using 3 μL of Lipofectamine 3000 and 3 μL of P3000 (2 μL/μg DNA). For split-cPE2, HEK293T cells were co-transfected with 1 μg of N-terminal fragment, 1 μg of C-terminal fragment, 330 ng of pegRNA, and 110 ng of nicking sgRNA using 3 μL of Lipofectamine 3000 and 5 μL of P3000. For genomic DNA extraction from cells, cells were cultured for 3 days after transfection, washed with PBS, pelleted, lysed with 100 μL of Quick extraction buffer (Epicenter), and incubated at 65°C for 15 min and 98°C for 5 min. To extract genomic DNA from mouse liver tissue, PureLink Genomic DNA Mini Kit (Thermo Fisher) was used following manufacturer's protocol.
CoIP assay
Plasmid construct of PE2-FLAG or RT497-FLAG were co-transfected with eRF1-HA into HEK293T cells. Empty vectors were used as negative controls. Forty-eight hours after transfection, cells were harvested and lysed in radio immunoprecipitation assay (RIPA) buffer with 1:100 Halt phosphatase cocktail inhibitor (Thermo Fisher 78420) and 1:50 Roche Complete protease inhibitor (11836145001) for 30 min on ice, and cleared at 13,000× g for 15 min at 4°C. Cell lysate was incubated with anti-FLAG (Sigma) at 4°C with gentle agitation overnight, then 150 μL of Protein A/G-coated magnetic beads were added, and the mixture incubated for 2 h at 4°C. Forty microliters of 1× loading buffer were added to the beads to extract proteins for SDS-PAGE.
Flow cytometry analysis
Reporter cells were cultured for 5 days after transfection, trypsinized, washed with PBS, and resuspended in PBS for flow cytometry analysis (MACSQuant VYB). For each sample, 50,000 cells were analyzed. All data were analyzed by FlowJo 10.0 software.
Western blotting
Cells were lysed 48 h after transfection using cold RIPA buffer (Boston Bioproducts) supplemented with phosphatase inhibitor cocktail (Thermo Fisher Scientific) and protease inhibitor (Roche). Lysates were quantified using the bicinchoninic acid assay (BCA, Thermo Fisher Scientific). For each sample, 10 μg of protein were loaded onto a 15-well NuPAGE 4%–12% Bis-Tris Protein Gels (Invitrogen), run at 100 V for 2.5 h, then transferred to the nitrocellulose membrane (Thermo Fisher Scientific). The primary antibodies anti-β-actin (#4970, Cell Signaling Technology, dilution: 1:2,000) or anti-Cas9 (A-9000-050, Epigentek Group, dilution: 1:2,000) were used, followed by incubation with fluorophore-conjugated secondary antibodies. Bands were visualized using the Odyssey imaging system (Li-Cor Biosciences).
Deep sequencing and data analysis
Deep sequencing of genomic DNA was performed as previously described.1 For the first round of PCR, genomic sites of interest were amplified from 100 ng of genomic DNA using Phusion Hot Start II PCR Master Mix with the primers containing Illumina forward and reverse adapters (listed in Table S3). Briefly, 20 μL of PCR 1 reaction was performed with forward and reverse primers each at 0.5 μM, 1 μL of genomic DNA extract, and 10 μL of Phusion Flash PCR Master Mix (Thermo Fisher). PCR 1 reactions were carried out as follows: 98°C for 10 s; then 20 cycles of 98°C for 1 s, 55°C for 5 s, and 72°C for 6 s; followed by a final 72°C extension for 2 min. To add a unique Illumina barcode, 30 μL of a given PCR 2 contained 1 μL of unpurified PCR product; unique forward and reverse Illumina barcoding primer pair, each at 0.5 μM; and 15 μL of Phusion Flash PCR Master Mix. Primers are listed in Table S3. PCR 2 reactions were carried out as follows: 98°C for 10 s; then 20 cycles of 98°C for 1 s, 60°C for 30 s, and 72°C for 6 s; followed by a final 72°C extension for 2 min. PCR 2 products were purified by electrophoresis with a 1% agarose gel using a QIAquick Gel Extraction Kit (Qiagen) and eluting with 30 μL of water. DNA concentration was quantified by Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific) or qPCR (KAPA Biosystems). The library was sequenced using an Illumina MiniSeq instrument according to the manufacturer's protocols.
MiniSeq data analysis was done as previously reported.1 Sequencing reads were demultiplexed using bcl2fastq (Illumina). Alignment of amplicon reads to a reference sequence and prime editing efficiency calculations were performed using CRISPResso2.28 To quantify the frequency of precise editing and indels, CRISPResso2 was run in standard mode with “discard_indel_reads” on. Precise editing efficiency was calculated as (number of reads with precise edit)/(number of total reads), while indel efficiency was calculated as 100% − precise editing efficiency − wild-type read rate.
Analysis of off-targets effects
Potential off-targets sites were amplified by PCR using the primer sequences listed in Table S3, then sequenced on the Illumina MiniSeq platform. To determine the on-target and off-target editing efficiency of PE3 or split-cPE3-573, MiniSeq data analysis was performed using CRISPResso2. Untreated cells were used as negative controls to exclude errors originating from PCR amplification and sequencing.
Animal studies
All animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at UMass Medical School. All plasmids used for hydrodynamic tail-vein injection were prepared using EndoFree Plasmid Maxi kit (Qiagen). No randomization or blinding was used. For cancer model generation, 8-week-old FVB/NJ (strain #001800) mice were injected with 2.5 mL of saline containing 30 μg of PE2 or 30 μg each of N-terminal RT497 and C-terminal RT497, 15 μg of pegRNA, 15 μg of nicking sgRNA, 5 μg of pT3 EF1a-MYC (Addgene plasmid # 92046), and 1 μg of CMV-SB10 (Addgene plasmid # 24551) via the tail vein in 5–7 s. Thirty micrograms of PE2, 15 μg of non-target pegRNA, 5 μg of pT3 EF1a-MYC, and 1 μg of CMV-SB10 were injected in the control group. Mice were euthanized 28 days after injection, and livers were isolated.
Production of AAV
HEK293T cells were transfected with AAV genome, pHelper, and Rep/Cap plasmids at about 70% confluency. Three days after transfection, cells were dislodged from the bottom of the plates. Pure chloroform (0.1 vol) was added and the mixture was shaken vigorously at 37°C. NaCl was added to a final concentration of 1 M and then centrifuged at 20,000× g at 4°C for 15 min. The supernatant was collected and processed with PEG8000 (Sigma) for virus precipitation. The pellet was resuspended in DPBS plus MgCl2 and treated with Benzonase (Sigma). The aqueous layer was ultrafiltered through 100-kDa MWCO columns (Millipore). The titer of the virus was quantified via qPCR.29
Readthrough assay
PE2, RT497, or empty vector were transfected with dual luciferase reporter in HEK293T cells. Cells were collected 48 h after transfection. Readthrough assays were performed using the Dual-Glo Luciferase Assay System and following the manufacturer's protocol (Promega, E2920).
Statistical analysis
For all experiments, the sample size and the number of biological replicates are provided in the figure legends. GraphPad Prism 8 was used to analyze the data. All numerical values are presented as mean ± SD. The significance of normally distributed data was tested using a two-tailed unpaired Student's t test and p values of less than 0.05 were considered significant.
Data availability
The raw sequencing data have been deposited to the NCBI Sequence Read Archive under accession PRJNA797253. All raw data are available from the corresponding author upon request.
Acknowledgments
We thank E. Sontheimer for helpful discussions and E. Haberlin for editing the manuscript. We thank Y. Liu in the UMass Morphology Core for support and G. Gao, Q. Su, and J. Xie in the UMass viral vector core for advice on AAV packaging. This work was supported by grants from the National Institutes of Health (DP2HL137167, P01HL131471, and UG3HL147367), American Cancer Society (129056-RSG-16-093), the Lung Cancer Research Foundation, and the Cystic Fibrosis Foundation. P.L. and S.A.W. were supported in part by the National Institutes of Health (R01GM115911 and UG3TR002668) and the Rett Syndrome Research Trust.
Author contributions
W.X., C.Z., and S.A.W. designed the research. C.Z. performed experiments, analyzed the data, and wrote the manuscript. S.L., B.L., and P.L. generated reporter cell lines and performed pegRNA cloning. All authors reviewed, edited, and approved the manuscript.
Declaration of interests
The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.005.
Supplemental information
References
- 1.Anzalone A.V., Randolph P.B., Davis J.R., Sousa A.A., Koblan L.W., Levy J.M., Chen P.J., Wilson C., Newby G.A., Raguram A., Liu D.R. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature. 2019;576:149–157. doi: 10.1038/s41586-019-1711-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lin Q., Zong Y., Xue C., Wang S., Jin S., Zhu Z., Wang Y., Anzalone A.V., Raguram A., Doman J.L., et al. Prime genome editing in rice and wheat. Nat. Biotechnol. 2020;38:582–585. doi: 10.1038/s41587-020-0455-x. [DOI] [PubMed] [Google Scholar]
- 3.Levy J.M., Yeh W.H., Pendse N., Davis J.R., Hennessey E., Butcher R., Koblan L.W., Comander J., Liu Q., Liu D.R. Cytosine and adenine base editing of the brain, liver, retina, heart and skeletal muscle of mice via adeno-associated viruses. Nat. Biomed. Eng. 2020;4:97–110. doi: 10.1038/s41551-019-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schmelas C., Grimm D. Split Cas9, not hairs – advancing the therapeutic index of CRISPR technology. Biotechnol. J. 2018;13 doi: 10.1002/biot.201700432. [DOI] [PubMed] [Google Scholar]
- 5.Wright A.V., Sternberg S.H., Taylor D.W., Staahl B.T., Bardales J.A., Kornfeld J.E., Doudna J.A. Rational design of a split-Cas9 enzyme complex. Proc. Natl. Acad. Sci. U S A. 2015;112:2984–2989. doi: 10.1073/pnas.1501698112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zetsche B., Volz S.E., Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nihongaki Y., Kawano F., Nakajima T., Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 8.Truong D.J., Kuhner K., Kuhn R., Werfel S., Engelhardt S., Wurst W., Ortiz O. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43:6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das D., Georgiadis M.M. The crystal structure of the monomeric reverse transcriptase from Moloney murine leukemia virus. Structure. 2004;12:819–829. doi: 10.1016/j.str.2004.02.032. [DOI] [PubMed] [Google Scholar]
- 10.Cote M.L., Roth M.J. Murine leukemia virus reverse transcriptase: structural comparison with HIV-1 reverse transcriptase. Virus Res. 2008;134:186–202. doi: 10.1016/j.virusres.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Telesnitsky A., Blain S.W., Goff S.P. Defects in Moloney murine leukemia virus replication caused by a reverse transcriptase mutation modeled on the structure of Escherichia coli RNase H. J. Virol. 1992;66:615–622. doi: 10.1128/JVI.66.2.615-622.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kotewicz M.L., Sampson C.M., D'Alessio J.M., Gerard G.F. Isolation of cloned Moloney murine leukemia virus reverse transcriptase lacking ribonuclease H activity. Nucleic Acids Res. 1988;16:265–277. doi: 10.1093/nar/16.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gerard G.F., Potter R.J., Smith M.D., Rosenthal K., Dhariwal G., Lee J., Chatterjee D.K. The role of template-primer in protection of reverse transcriptase from thermal inactivation. Nucleic Acids Res. 2002;30:3118–3129. doi: 10.1093/nar/gkf417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tang X., Zhu Y., Baker S.L., Bowler M.W., Chen B.J., Chen C., Hogg J.R., Goff S.P., Song H. Structural basis of suppression of host translation termination by Moloney murine leukemia virus. Nat. Commun. 2016;7:12070. doi: 10.1038/ncomms12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hellen C.U.T. Translation termination and ribosome recycling in eukaryotes. Cold Spring Harb Perspect. Biol. 2018;10 doi: 10.1101/cshperspect.a032656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Orlova M., Yueh A., Leung J., Goff S.P. Reverse transcriptase of Moloney murine leukemia virus binds to eukaryotic release factor 1 to modulate suppression of translational termination. Cell. 2003;115:319–331. doi: 10.1016/s0092-8674(03)00805-5. [DOI] [PubMed] [Google Scholar]
- 17.Liu P., Liang S.Q., Zheng C., Mintzer E., Zhao Y.G., Ponnienselvan K., Mir A., Sontheimer E.J., Gao G., Flotte T.R., et al. Improved prime editors enable pathogenic allele correction and cancer modelling in adult mice. Nat. Commun. 2021;12:2121. doi: 10.1038/s41467-021-22295-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Telesnitsky A., Goff S.P. RNase H domain mutations affect the interaction between Moloney murine leukemia virus reverse transcriptase and its primer-template. Proc. Natl. Acad. Sci. U S A. 1993;90:1276–1280. doi: 10.1073/pnas.90.4.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y., Zhi S., Liu W., Wen J., Hu S., Cao T., Sun H., Li Y., Huang L., Liu Y., et al. Development of highly efficient dual-AAV split adenosine base editor for in vivo gene therapy. Small Methods. 2020;4 doi: 10.1002/smtd.202000309. [DOI] [Google Scholar]
- 20.Ma D., Peng S., Xie Z. Integration and exchange of split dCas9 domains for transcriptional controls in mammalian cells. Nat. Commun. 2016;7:13056. doi: 10.1038/ncomms13056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhi S., Chen Y., Wu G., Wen J., Wu J., Liu Q., Li Y., Kang R., Hu S., Wang J., et al. Dual-AAV delivering split prime editor system for in vivo genome editing. Mol. Ther. 2021 doi: 10.1016/j.ymthe.2021.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moon R.T., Kohn A.D., De Ferrari G.V., Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat. Rev. Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 23.Zafra M.P., Schatoff E.M., Katti A., Foronda M., Breinig M., Schweitzer A.Y., Simon A., Han T., Goswami S., Montgomery E., et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol. 2018;36:888–893. doi: 10.1038/nbt.4194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu P., Ge M., Hu J., Li X., Che L., Sun K., Cheng L., Huang Y., Pilo M.G., Cigliano A., et al. A functional mammalian target of rapamycin complex 1 signaling is indispensable for c-Myc-driven hepatocarcinogenesis. Hepatology. 2017;66:167–181. doi: 10.1002/hep.29183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Q., Strong A., Patel K.M., Ng S.L., Gosis B.S., Regan S.N., Cowan C.A., Rader D.J., Musunuru K. Permanent alteration of PCSK9 with in vivo CRISPR-Cas9 genome editing. Circ. Res. 2014;115:488–492. doi: 10.1161/CIRCRESAHA.115.304351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yin H., Song C.Q., Suresh S., Wu Q., Walsh S., Rhym L.H., Mintzer E., Bolukbasi M.F., Zhu L.J., Kauffman K., et al. Structure-guided chemical modification of guide RNA enables potent non-viral in vivo genome editing. Nat. Biotechnol. 2017;35:1179–1187. doi: 10.1038/nbt.4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grentzmann G., Ingram J.A., Kelly P.J., Gesteland R.F., Atkins J.F. A dual-luciferase reporter system for studying recoding signals. RNA. 1998;4:479–486. [PMC free article] [PubMed] [Google Scholar]
- 28.Clement K., Rees H., Canver M.C., Gehrke J.M., Farouni R., Hsu J.Y., Cole M.A., Liu D.R., Joung J.K., Bauer D.E., Pinello L. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol. 2019;37:224–226. doi: 10.1038/s41587-019-0032-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su Q., Sena-Esteves M., Gao G. Titration of recombinant adeno-associated virus (rAAV) genome copy number using real-time quantitative polymerase chain reaction (qPCR) Cold Spring Harbor Protoc. 2020;2020 doi: 10.1101/pdb.prot095646. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data have been deposited to the NCBI Sequence Read Archive under accession PRJNA797253. All raw data are available from the corresponding author upon request.