

Abstract
Background and purpose
Signal transducer and activator of transcription 3 (STAT3) may contribute to the proinflammation in the central nervous system diseases by modulating the microglial responses. Thus, this study was intended to investigate the effect of STAT3 on microglia-dependent neuroinflammation and functional outcome after experimental subarachnoid haemorrhage (SAH).
Methods
The SAH model was established by endovascular perforation in the mouse. Real-time PCR (RtPCR) and western blot were used to examine the dynamic STAT3 signalling pathway responses after SAH. To clarify the role of the STAT3 signalling pathway in the microglia-dependent neuroinflammation after SAH, the microglia-specific STAT3 knockout (KO) mice were generated by the Cre-LoxP system. The neurological functions were assessed by Catwalk and Morris water maze tests. Neuronal loss after SAH was determined by immunohistochemistry staining. Microglial polarisation status after STAT3 KO was then examined by RtPCR and immunofluorescence.
Results
The STAT3 and Janus kinase-signal transducer 2 activated immediately with the upregulation and phosphorylation after SAH. Downstream factors and related mediators altered dynamically and accordingly. Microglial STAT3 deletion ameliorated the neurological impairment and alleviated the early neuronal loss after SAH. To investigate the underlying mechanism, we examined the microglial reaction after STAT3 KO. STAT3 deletion reversed the increase of microglia after SAH. Loss of STAT3 triggered the early morphological changes of microglia and primed microglia from M1 to M2 polarisation. Functionally, microglial STAT3 deletion suppressed the SAH-induced proinflammation and promoted the anti-inflammation in the early phase.
Conclusions
STAT3 is closely related to the microglial polarisation transition and modulation of microglia-dependent neuroinflammation. Microglial STAT3 deletion improved neurological function and neuronal survival probably through promoting M2 polarisation and anti-inflammatory responses after SAH. STAT3 may serve as a promising therapeutic target to alleviate early brain injury after SAH.
Keywords: subarachnoid, hemorrhage, inflammation
Introduction
Subarachnoid haemorrhage (SAH) is a life-threatening disease, which accounts for the second mortality among the stroke population. Thirty per cent of victims develop a permanent disability, which severely affects patients’ quality of life.1 2 Recent evidence indicates the critical role of neuroinflammation in the pathogenesis of SAH. Neuroinflammation evolves with glial cell activation and the release of a variety of cytokines.3 4 Microglia, considered as the immune cells of the central nervous system (CNS), have both beneficially and detrimentally in CNS diseases.5 6 Microglia change morphology from ramified to amoeboid shape when activated and obtain bidirectional polarisation in response to the adverse stimulus with the release of distinct profiles of cytokines. The ‘classical’ M1 polarisation is related to the proinflammation and is regarded as the detrimental phenotype. The ‘alternative M2’ polarisation is responsibile for anti-inflammation and contributes to tissue repair and recovery.5 6 The manipulation of microglial polarisation arises as an innovative approach to reverse the unfavourable inflammatory responses, which has been tentatively investigated in experimental SAH. Several mediators including mammalian target of rapamycin and tumor-specific glycoprotein-6 are evidenced contributing to the skewed M2 polarisation.7–9
Signal transducer and activator of transcription 3 (STAT3) is a member of the STAT family and involves the proinflammatory responses in various CNS abnormalities, including traumatic brain injury, multiple sclerosis and glioma.10–12 Suppression of STAT3 signalling pathway activation decreases the infarct volume, number of apoptotic cells and restores the neurological deficits in ischaemic stroke.13 14 Recent studies imply that STAT3/Janus kinase-signal transducer 2 (JAK2) signalling pathway participates in the microglia-dependent neuroinflammatory responses, and this participation may relate to the modulation of the microglial polarisation.8 13 In SAH, activation of the STAT3/JAK2 signalling pathway contributes to early brain injury, cerebral vasculopathy and neurological deficit.15 16 The mediator erythropoietin suppresses the STAT3/JAK2 pathway activation, resulting in the reduction of M1-like inflammatory responses and the amelioration of brain injury after SAH.17 Therefore, we hypothesise that STAT3 may promote the proinflammation-related brain injury after SAH and the inhibition of STAT3 may reverse the unfavourable inflammatory responses and alleviate neurological deficits through the microglia-specific neuroinflammatory modulation.
STAT3 is crucial in early embryogenesis. Lack of STAT3 results in embryo lethality, even in the embryonic stem cells.18 19 The emergence of the Cre-loxP recombination system enables the investigation of STAT3 in adult tissues, which allows the ablation of specific genes in later life. The Cre-loxP recombination system is a site-specific recombinase technology allowing the manipulation of DNA in targeted cell types by deletions, insertions, translocations and inversions at specific sites.20 21 The applications of conditional STAT3 deletion are achieved in the cardiomyocytes, neutrophils, neurons and macrophages.22–24 In this study, we apply the Cre-loxP recombination technique to generate the microglia-specific STAT3 knockout mice under the control of Cx3Cr1. Cx3Cr1 is considered as the microglia-specific marker, which is exclusively expressed in microglia in adult mice.25 The transcription of Cre protein could be navigated by Cx3Cr1 promotor, which then permits the expression of Cre recombinase in the microglia.
In the present study, the temporal STAT3 activation status after SAH was assayed and the downstream factor changes of the STAT3 signalling pathway were investigated first. By generating microglia-specific STAT3 knock-out (KO) mice, we demonstrated the essential role of microglial STAT3 in the development of neurological deficits and neuronal loss after SAH. Finally, we provide a possible treatment strategy for the further treatment of SAH.
Materials and methods
Animals and SAH model
Male, wild-type C57BL/6, average weighing 25–30 g, were obtained from the Laboratory Animal Services Centre of the Chinese University of Hong Kong. Transgenic mice used in this study including B6.129S1-Stat3tm1Xyfu/J (homozygous STAT3flox/flox ) and B6J.B6N(Cg)-Cx3Cr1tm1.1(cre)Jung/J (homozygous Cx3Cr1Cre/Cre ) were purchased from the Jackson Laboratory (JAX Stock #016923 and #25524; JAX, Maine, USA). SAH models were induced by the endovascular perforation as previously described.26 27 STAT3 conditional knockout mice were achieved with Cre-LoxP system (figure 1).
Figure 1.

Characterisation of microglia-specific STAT3 KO mice. (A) The schematic description of the breeding strategy. (B) PCR analysis of genotypes. STAT3flox allele was electrophoretically separated and demonstrated as a 187 bp fragment. Wild-type STAT3 allele was 146 bp. Cx3Cr1cre fragment was 380 bp and Cx3Cr1 − was 302 bp. (C) Biochemical determination of STAT3 KO at the gene and protein levels. RNA and protein were extracted from the CAPS (C), M1 cortex (M) and hippocampus (H) of the SAH brain. RtPCR examination was performed to determine the mRNA expression of STAT3 in STAT3 KO mice compared with the control. WB was performed to determine the STAT3/JAK2 phosphorylation status of STAT3 KO mice. (D) The STAT3 signalling pathway activation status of STAT3 KO mice at 1 and 5 days after SAH. The mRNA expression of the mediators involved in the STAT3 pathway is shown in the fold change in the CAPS, M1 cortex and hippocampus of STAT3 KO mice compared with the control. n=3 per group. Values are mean±SEM. *P≤0.05, **P0.01, ***P≤0.001. CAPS, cortex adjacent to the perforated site; JAK2, Janus kinase-signal transducer 2; KO, knockout; RtPCR, real-time PCR; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3; WB, western blot.
See online supplementary material for full experimental methods (online supplemental material 1).
svn-2021-001028supp001.pdf (19.4MB, pdf)
Results
STAT3 signalling pathway activated after SAH
STAT3 is predominantly expressed and initially activated in microglia rather than astrocytes and neurons.8 15 16 28 29 Based on the pilot study,26 three brain areas, including the cortex adjacent to the perforated site (CAPS), M1 cortex and hippocampus, were chosen for the examination (n=3–4/group/time points). The STAT3 signalling pathway was activated acutely after SAH with the subsequent alteration of the downstream factors and related mediators. The activation of the STAT3 pathway was generally in the same pattern through the brain areas. STAT3 demonstrated significant upregulation in transcriptome within the first 3 days in SAH mice compared with the sham control (p<0.05 for all the three brain areas). The increased expression of upstream regulator JAK2 and downstream factor nuclear factor-kappa B (NF-κB) was appreciated. The pattern of NF-κB expression is consistent with our previous results of microglia accumulation from CAPS to motor area.26 The negative regulator suppressor of cytokine signalling-3 (SOCS3) was reactively increased as well. On day 10, Src showed a significant downregulation in SAH mice, which suggested the STAT3 signalling pathway responded to SAH injury through the negative modulation of Src path at the delayed stage (figure 2A). In protein expression, the levels of phosphorylated STAT3 and JAK2 increased within the first 3 days after SAH and then restored to the unphosphorylated status. The total STAT3 and JAK2 of SAH mice were comparable to the sham control (figure 2B, C). The clarification of the dynamics of STAT3 signalling pathway changes provided the fundamental understanding of how the STAT3 signalling pathway was involved and activated after SAH and the optimal time window for the targeted intervention in the subsequent experiments.
Figure 2.

STAT3 signalling pathway activated after experimental SAH. (A) RNA extract was prepared from CAPS, M1 cortex and hippocampus of SAH brain at 1, 3, 5 and 10 days. The mRNA expression of mediators involved in the STAT3 pathway after SAH was determined by RtPCR and shown in the fold change compared with the sham-operated group. n=3–4/group/time point. Values were mean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. (B) STAT3/JAK2 phosphorylation status was detected by WB at 1, 3, 5 and 10 days. The levels of phosphorylated and total STAT3/JAK2 relative to internal control GAPDH are shown in the cortex adjacent to the perforated site (C), M1 cortex (M) and hippocampus (H), respectively, of SAH and sham-operated mice. n=3/group/time point. (C) The quantitative analysis of the pSTAT3/STAT3 and pJAK2/JAK of (B) to show the trend of pSTAT3/STAT3 and pJAK2/JAK in the SAH and sham groups. CAPS, cortex adjacent to the perforated site; JAK2, Janus kinase-signal transducer 2; NF-κB, nuclear factor-kappa B; RtPCR, real-time PCR; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3; WB, western blot.
Generation of microglia-specific STAT3 knockout (KO) mice
To confirm the efficiency of Cre-mediated deletion, we analysed the expression of STAT3 at the gene and protein levels. The significantly decreased STAT3 expression of mRNA and protein was defined in STAT3 KO mice compared with the control (n=3/group, p<0.005). The phosphorylation level of STAT3 and JAK2 was decreased as well. It was noted that there was still a certain amount of STAT3 expression recognised in PCR products and blotted protein, which might derive from the other CNS cell pools, except microglia, in the mixture of the brain tissue extraction (figure 1C). Other evidence shows that brain inflammation caused by tumour necrosis factor α (TNF-α)-induced interleukin (IL)-6 release in astrocytes was mediated by NF-κB but not by STAT3.29 Several studies found modulators can relieve brain inflammation of SAH that is through the microglia-mediated JAK–STAT3 signal pathway.8 16 So the crosstalk of the JAK–STAT3 pathway in different brain cells during SAH needs further study to clarify. An overall downregulation of STAT3 signalling pathway was appreciated in STAT3 KO mice in the early phase after SAH. On day 5, SOCS3 and NF-κB showed the reversed upregulation, which suggested that microglia-specific STAT3 deletion had a critical effect on the related mediators over time (figure 1D).
Microglial STAT3 deficiency ameliorated the neurological impairment after SAH
Mice of both STAT3 KO and control group presented the SAH-like symptoms after the operation, which occurred mostly within the first 24–48 hours. Mortality was 7.55% in STAT3 KO mice and 14.29% in mice of the control group. The average body weight loss of STAT3 KO mice was 2.4 g, 9.85% of the original body weight, which was comparable to the control ones, which was 2.59 g, accounting for 10.37% of the original body weight. On day 10, body weight returned to the level of original status in the two groups (figure 3A).
Figure 3.
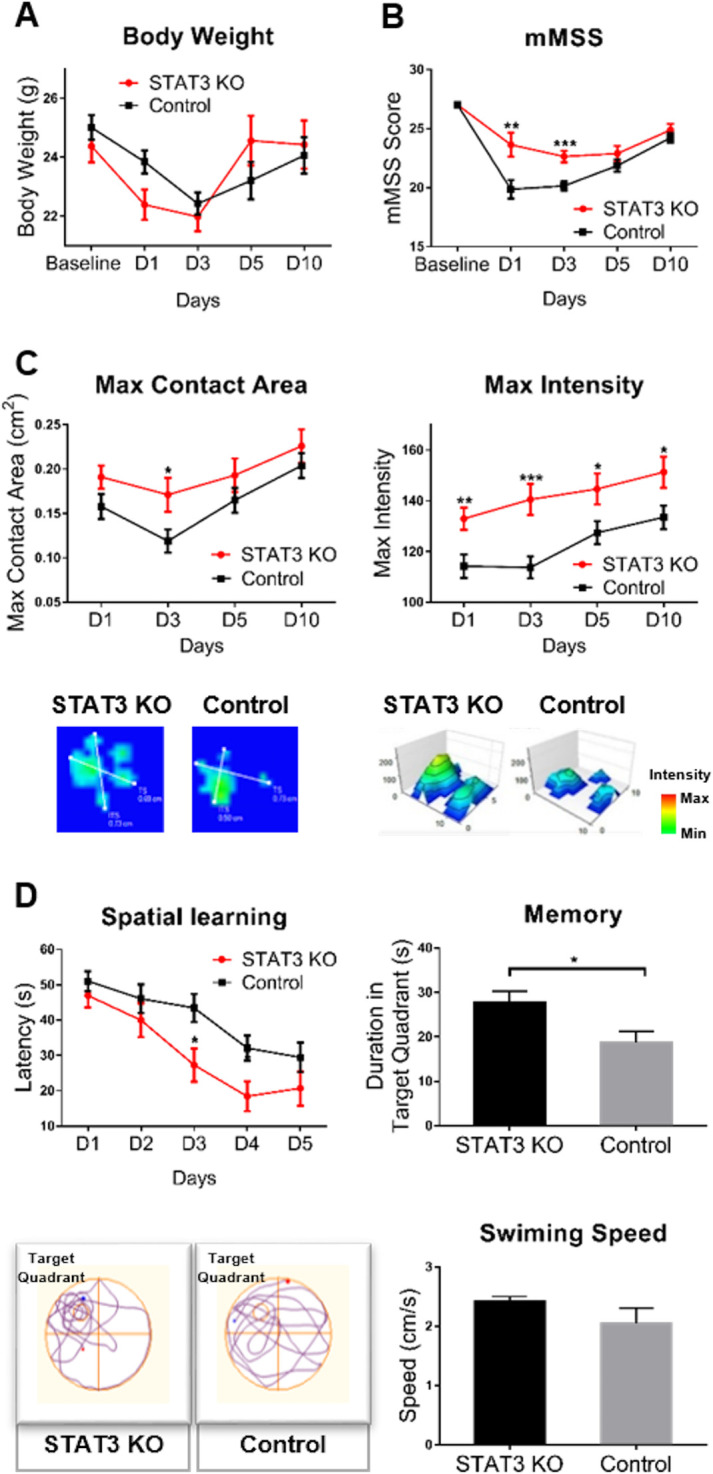
Microglial STAT3 deletion ameliorated the neurological impairment after SAH. (A) The time-course body weight changes were examined in STAT3 KO and control groups of mice. (B)The sensorimotor functions were assessed by the mMSS (5–27 scores). (C) Gait analysis was performed by the Catwalk system. The max contact area (CM2) and max intensity of paw were detected. STAT3 KO mice showed the alleviated paw contraction and increased paw intensity compared with the control. (D) Cognitive functions including learning ability and memory were assessed by the MWM test. Spatial learning ability was defined as the latency to escape the platform. Memory was defined as the time mice spent in the target quadrant where the platform was placed previously. The bottom figure shows the typical swim path and average swimming speed of STAT3 KO and control groups of mice. n=8–16 per group. Values are mean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. KO, knockout; mMSS, Mouse Motor and Sensory Scale; MWM, Morris water maze; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3.
To investigate the effect of microglial STAT3 ablation on the neurological function in SAH, we employed a battery of neurobehavioural tests (n=8–16/group). STAT3 KO mice exhibited an improved sensorimotor function with significantly higher mMSS scores compared with the control ones (group effect: p=0.015). The post hoc statistic tests indicated that the improvement was significant from day 1 to 3 after SAH. The sensorimotor function gradually recovered in both groups of mice in the delayed phase (figure 3B).
Gait analysis was performed by the Catwalk system. The data showed the gait parameters of the right front paws of mice, and the gait analysis of the other three paws presented similar results for the bilateral lesion. STAT3 KO mice showed the alleviated static gait deficits after SAH, compared with the control ones. The max contact area and max intensity of paws significantly increased in STAT3 KO mice (group effect: p<0.005, figure 3C). Other static gait parameters including mean intensity, max contact max intensity, max contact max intensity of paws increased as well in online supplemental figure ⅠA, which indicated microglial STAT3 deletion alleviated the muscle weakness and paw contracture after SAH. The dynamic gait function was not significantly improved in STAT3 KO mice (online supplemental figure ⅠB), which suggested that the microglial STAT3 deletion might not affect the running speed and gait pattern after SAH.
In the training phase of Morris water maze tests, STAT3 KO mice showed remarkably decreased latency to find the escape platform (group effect: p=0.044), which indicated the improved learning ability after SAH. In the probe trial, STAT3 KO mice showed a remarkable amelioration of long-term memory deterioration, presenting significantly increased exploration in the target quadrant compared with the control ones (p=0.024). SAH mice of the control group failed to recall the memory of the position where the platform was previously placed after the interval. In addition, mice showed comparable swim speeds between the two groups, suggesting the equivalent swimming skills in the testing (figure 3D).
Microglial STAT3 deficiency contributed to the neuroprotection in SAH
To investigate if the microglial STAT3 ablation could positively affect neuronal survival in haemorrhage stroke, we subsequently determined the neuronal survival in STAT3 KO mice by using NeuN immunohistochemistry (STAT3 KO n=7, control n=8). In the NeuN immunohistochemistry, SAH mice showed significantly increased neuronal numbers after STAT3 deletion in CAPS and hippocampus on day 5 (p<0.05) (figure 4A, B). The results indicated that microglial STAT3 deficiency had a positive effect on neuroprotection in SAH, which was remarkable in the bleeding area.
Figure 4.
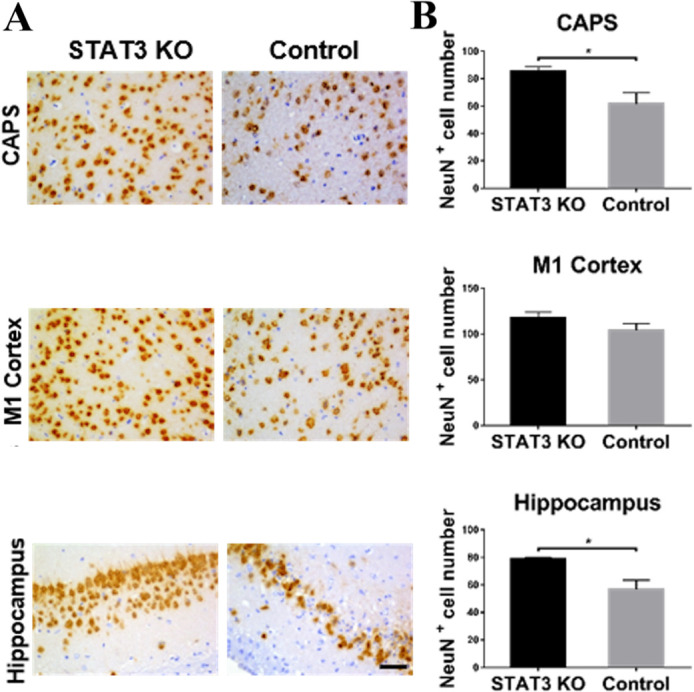
Microglial STAT3 deletion alleviated the neuronal loss after SAH. (A) Neuronal loss was detected by NeuN immunohistochemistry in the CAPS, M1 cortex and hippocampus of the brain. The representative NeuN labelled coronal brain sections were shown on day 5 after SAH. (B) NeuN-positive neurons were quantified in the aforementioned brain areas of STAT3 KO and control groups of mice. Bar=50 µm. STAT3 KO n=7, control n=8. Values are ean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. CAPS, cortex adjacent to the perforated site; KO, knockout; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3.
STAT3 ablation primed microglia to M2 polarisation
To identify key factors alleviating the neurological impairment caused by depletion of microglial STAT3, we first examined the microglial reaction after STAT3 ablation. In iba1 immunohistochemistry (STAT3 KO n=7, control n=8), microglia demonstrated the decreased accumulation after STAT3 deletion in SAH, which was significant in the M1 cortex and hippocampus (figure 5A, B). We used immunofluorescence and confocal microscopy to determine and visualise the polarisation status and morphological changes of microglia (n=6). Microglia showed an obvious reduction of M1 polarisation after STAT3 deletion with the decreased CD16/32 expression in SAH. Strikingly, M1-possessed microglia of STAT3 KO mice showed an early morphological transformation with the condensed processes. The CD206-positive M2 microglia appeared earlier in CAPS after STAT3 deletion (figure 5C and online supplemental figure 2). The quantification of M1 and M2 microglia ratio indicated that microglial STAT3 deletion suppressed M1 polarisation on day 1 after SAH, which was significant in CAPS and M1 cortex (p<0.05). The M2 polarisation was significantly promoted over all the brain areas (p<0.05). The suppression of M1 polarisation was still noted in the delayed phase of SAH on day 5 (p<0.005). The increase of M2 polarisation was significantly in the M1 cortex and hippocampus in this phase (p<0.05, figure 5D). Real-time PCR (RtPCR) measurement demonstrated identical results regarding the mRNA expression of microglial phenotype-specific markers (online supplemental figure 3). Thus, STAT3 deletion reduced the microglial responses toward M1 polarisation after SAH and triggered the early morphological transformation. STAT3 deletion primed microglia to M2 polarisation after SAH.
Figure 5.
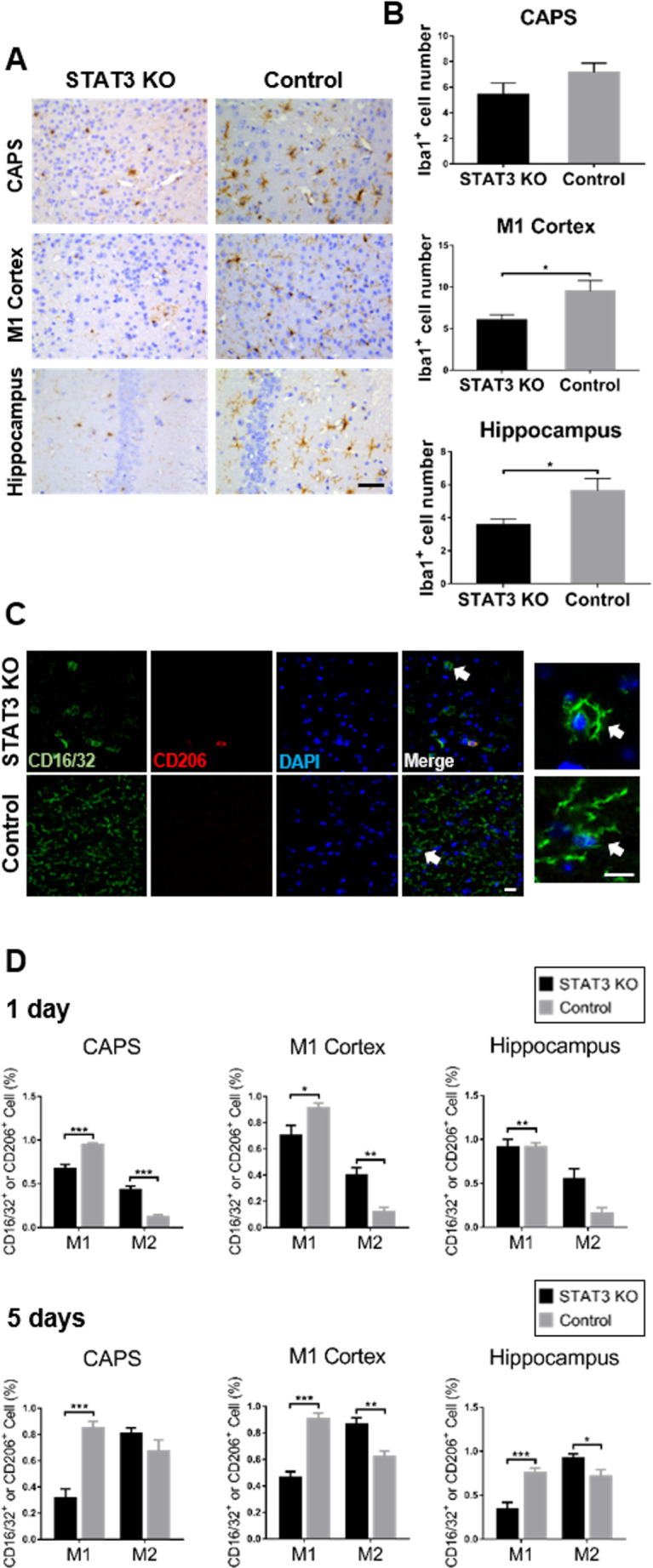
Stat3 deletion primed microglia to M2 polarisation. (A) Microglia was examined by iba1 immunohistochemistry in CAPS, M1 cortex and hippocampus of the brain. The representative iba1 labelled coronal brain sections were shown on day 1 after SAH. Bar=50 µm. (B) Iba1-positive microglia were quantified in the aforementioned brain areas of STAT3 KO and control groups of mice. (C) Microglial polarisation status was determined by immunofluorescence. The representative confocal microscopic images showed the visualisation of CD16/32 (M1, green), CD206 (M2, red) and DAPI (nuclei, blue) coexpression in CAPS on day 1 after SAH. The white arrows indicate the highly magnified view of the typical microglial processes. Bar=20 µm for all magnifications. (D) CD16/32-positive (M1) or CD206-positive (M2) cells were quantified in the aforementioned brain areas of STAT3 KO and control groups of mice at 1 and 5 days after SAH. n=5–8 per group. Values were the mean±SEM. *P≤0.05, **P≤0.01, ***P≤0.001. CAPS, cortex adjacent to the perforated site; KO, knockout; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3.
Microglial STAT3 deficiency promoted the anti-inflammation after SAH
The expression of microglia-related inflammatory cytokines after SAH was detected by RtPCR (n=3). The expression of anti-inflammatory factor-induced IL-4 increased significantly in the condition of microglial STAT3 deletion compared with the control in the early phase after SAH (p<0.05). The change of proinflammatory cytokine expression was not remarkable except in the hippocampus (figure 6A). On day 5 after SAH, IL-4 was continuously maintained at a higher level in STAT3 KO mice, especially in CAPS and hippocampus (p<0.05). The significant upregulating of transforming growth factor-β was also noted in the M1 cortex and hippocampus (p<0.05). The effect of microglial STAT3 deletion on proinflammatory cytokines was heterogeneous in this phase. IL-6 demonstrated the downregulation. However, TNF- α was observed upregulated in CAPS (figure 6B). Thus, the microglial STAT3 deletion suppressed the proinflammation in specific brain areas in addition to promoting the anti-inflammation after SAH. The alteration of cytokine expression after microglial STAT3 deficiency suggested a functional transition of microglia from M1 to M2 polarisation.
Figure 6.
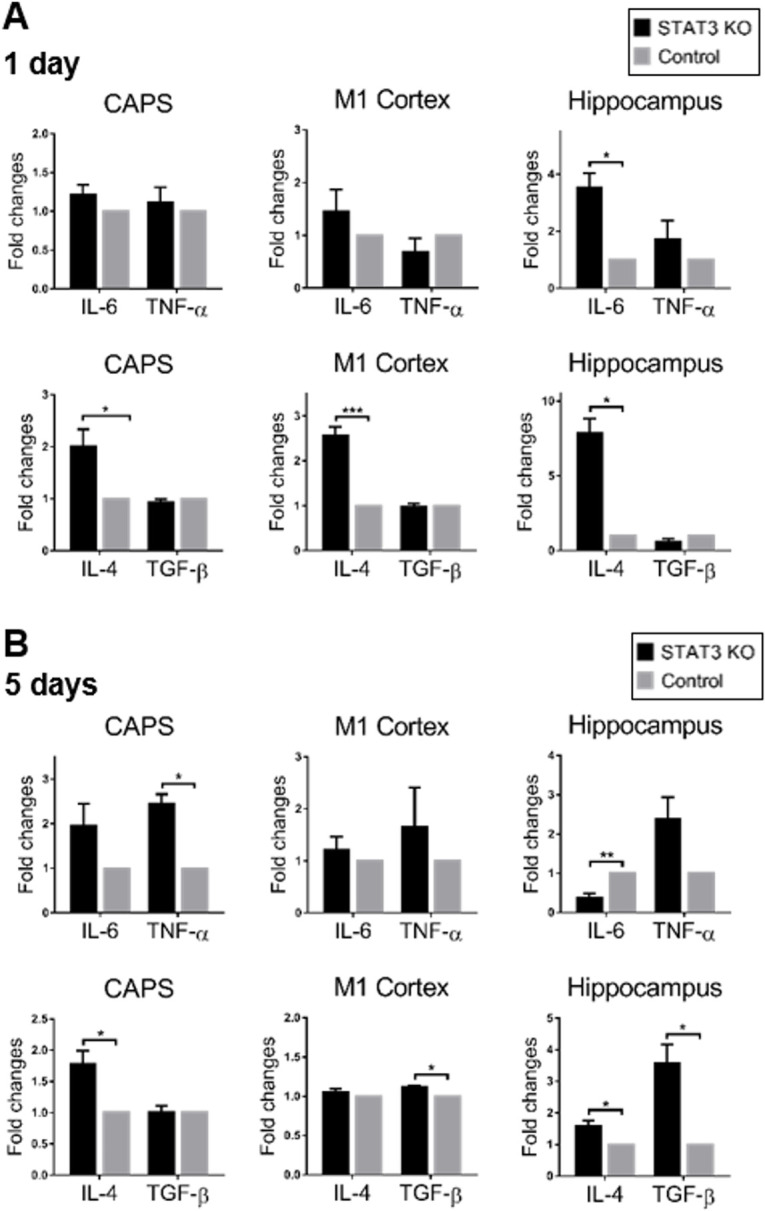
Microglial STAT3 deletion promoted anti-inflammation after SAH. (A, B) The expression of M1/M2 microglia-related cytokines were detected by RtPCR at 1 and 5 days after SAH. The mRNA expression of M1-phenotype related pro-inflammatory factors (IL-6 and TNF-α) and M2-phenotype related anti-inflammatory factors (IL-4 and TGF-β) was shown in fold change in STAT3 KO mice compared with the control. Values were the mean±SEM *p≤0.05, **p≤0.01, ***p≤0.001. CAPS, cortex adjacent to the perforated site; IL, interleukin; KO, knockout; SAH, subarachnoid haemorrhage; STAT3, signal transducer and activator of transcription 3; TGF-β, transforming growth factor-β.
Discussion
The activation of STAT3 was observed in a number of CNS diseases, such as traumatic brain injury, brain tumours and cerebral ischaemia.12 14 30 Especially recent studies found that several modulators can relieve early brain injury of SAH and act as an anti-inflammation medication through suppressing microglia-mediated JAK–STAT3 signal pathway.8 16 17 By using microglia-specific STAT3 KO SAH mice, the activation and biology effect of microglia-mediated JAK–STAT3 signal pathway was studied. In our study, the gene expression of STAT3 was consistent with STAT3/JAK2 protein level, upregulated immediately after SAH. As the disease progressed, STAT3 activation was alleviated at the late stage with the upregulation of the downstream regulators. The negative regulator SOCS3 showed temporal feedback with the upregulated expression to the STAT3 signalling modulation. The delayed downregulation of Src might be a considerable reason for neuronal apoptosis in the delayed phase. Determining the time course STAT3 signalling pathway activation status illustrated the dynamics of how the STAT3 pathway responded to SAH. However, the biology effect of the STAT3 pathway is controversial in some situations,28 31 and it is not fully understood in SAH conditions at present. The present study also investigated the relationship between microglia-specific STAT3 KO and outcomes in SAH, thus giving new insights on the pathological mechanism and possible treatment strategy.
The Cre-loxP recombination system has been particularly useful in neurosciences targeting the various cell types and complex neural circuits of the brain which integrate to form cognition and behaviours.32 33 Studies by Tsien et al have applied the Cre-loxP recombination system in the adult mouse forebrain, in which the various type of neurons are evidenced to be postmitotic.32 33 Analyses of tissue-specific STAT3-deficient mice indicate that STAT3 involves a variety of biological functions including cell growth, suppression and induction of apoptosis, and cell motility.34 In this study, we first demonstrated the microglia-specific STAT3 knockout on mice by using this technique. The dispersion of the genotypes of the F2 hybrids is basically aligned to the expectation according to the principles of Mendel’s genetics. The behaviours and social performance of STAT3 KO are normally through close observation. The presence of Stat3+/+Cx3Cr1−/− , in which the genotype is the same as wild-type mice, is considered derived from the occasional mutation with extremely low incidence. In addition, two STAT3flox/flox Cx3Cr1cre/− mice presented with cataract. Although the abnormal mice were excluded from the subsequent experiments, it was not clear about the underlying mechanism of the appearance of cataracts in STAT3 KO mice. STAT3 KO mice showed pronounced alleviation of the STAT3 pathway activation after SAH. The early downregulation of JAK2 might be due to the lack of a downstream phosphorylated target. The decrease of phosphorylated groups delivery resulted in the negative feedback of JAK2 expression. In the absence of an upstream mediator, the downstream factors and related regulators showed decreased expression after the microglial STAT3 deletion. The modulation of microglia-specific STAT3 deletion in the downstream mediators varied in the delayed phase after SAH. However, that the alteration was from the STAT3 deletion or other signalling input remained to be defined. Other than microglia, the Cx3Cr1 is also expressed in neuron and periphery macrophages, so the STAT3 KO effect could be triggered by these cells; these effects need further studies to be clarified.
The present study found that the microglial STAT3 deletion significantly improved neurological function and neuronal survival in SAH. The ablation of the inflammation-related STAT3 pathway might largely blunt the unfavourable neuroinflammatory responses and thus ameliorate the neuronal damage. For investigating the protective mechanism of microglial in SAH, our previous study found that the reactive immune cells in SAH mice brain were largely from resident microglia pool rather than infiltrating macrophages by using Cx3cr1GFP/GFP Ccr2RFP/RFP transgenic mice.26 A significantly continuous (p<0.001) accumulation of microglia (Iba1+) cells in CAPS was found after SAH. While at the M1 cortex and hippocampus, microglia showed an acute and temporary accumulation within the first 3 days after SAH. Moreover, we also found the microglia reserved as M1 microglia in the early stage of SAH, which acts as a proinflammation function and the morphology, are dendritic. While in the late stage of SAH, microglia transfer to M2 microglia, which were amoeboid/bushy ‘activated’ morphology connected with anti-inflammation effect.26 In the present study, STAT3 deletion reduced the microglial responses with the decreased accumulation in the early phase after SAH. In brief, the CD16/32+ and CD206+ cells in STAT3 KO mice are more reminiscent of the activated microglia/macrophage morphology, which shows that STAT3 KO can reverse or induce polarisation microglia to M2 state. M2 microglia shows more neuroprotective characteristics, which is consistent with the neurobehavioural test and cytokine test that STAT3 KO can suppress the inflammation in SAH. The finding that the inhibition of STAT3 pathway activation ameliorated the M1-like microglial polarisation was also reported in other published data.13 35 36 Microglia after STAT3 deletion demonstrated curved and circled processes rather than the remarkably decreased processes compared with the control. The morphological transformation did not limit the process motility, which was regarded as the dynamic surveillants of brain parenchymal.37
The critical role of the STAT3 signalling pathway in the mediation of the function-related microglial polarisation was also evidenced by the expressed cytokine profile. The microglial STAT3 deletion upregulated the expression of IL-4 dominant anti-inflammatory cytokines after SAH, which implied the protective function in neuroinflammation. Although the increase of TNF-α was observed in D5 after microglial STAT3 deletion, the overall cytokine level tends to be anti-inflammatory. Also, TNF-α has been reported that in brain inflammation, microglia can be activated by IL-6, which is released from TNF-α-sensitive brain pericytes cooperating with IκB-NF-κB and JAK–STAT3 pathways,29 and the changes in TNF- α level in different time and brain area in SAH need further study to detect. STAT3 signalling pathway contributed to the acute M1 related proinflammation, which might contribute to the early brain injury in SAH. The STAT3 deletion primed the microglia-dependent neuroinflammation to M2 directed anti-inflammation. Indeed, we could not exclude the effect of infiltrating peripheral immune cells. However, the proportion of the peripheral immune cells was relatively small compared with the resident microglia, especially at the early stages after SAH.38 The microglial STAT3 ablation-induced anti-inflammatory responses ameliorated neurological impairment and neuronal loss. This neuroprotection might be the result of microglia–neuron interactions. However, the direct effect of neuronal STAT3 on neuronal survival was not investigated in this study. Future studies might make use of the Cre-LoxP system to create the neuronal STAT3 deficiency model to address this issue.
Conclusions
In conclusion, the STAT3 signalling pathway played a critical role in the mediation of the function-related microglial polarisation, which might be prone to an M1 proinflammatory direction. The microglia-specific STAT3 deletion alleviated the unfavourable neuroinflammation and enhanced the anti-inflammatory function in experimental SAH probably by triggering the microglial transition from M1 to M2 polarisation. The results suggested that microglia-specific STAT3 deletion might have a positive effect on the functional outcome after SAH. It might provide us a potential therapeutic approach aiming at the modulation of neuroinflammation for SAH.
Footnotes
ZVZ and JC contributed equally.
Contributors: ZVZ and GKCW designed the experiments; ZVZ, JC, HL and SYEL conducted the experiments; GL and WYC advised on the design and supervised the study together with GKCW; ZVZ, JC and GKCW drafted the manuscript; all authors contributed to the revision of and approved the manuscript.
Funding: The study was partly supported by the Direct Grant for Research, the Chinese University of Hong Kong (grant number 2016.112).
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available upon reasonable request. Experiment data are available upon reasonable request.
Ethics statements
Patient consent for publication
Not required.
Ethics approval
The study was approved by the Animal Experimentation Ethics Committee, the Chinese University of Hong Kong (reference number 19-108-GRF).
References
- 1. Cahill J, Zhang JH. Subarachnoid hemorrhage: is it time for a new direction? Stroke 2009;40:S86–7. 10.1161/STROKEAHA.108.533315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Rooij NK, Linn FHH, van der Plas JA, et al. Incidence of subarachnoid haemorrhage: a systematic review with emphasis on region, age, gender and time trends. J Neurol Neurosurg Psychiatry 2007;78:1365–72. 10.1136/jnnp.2007.117655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zheng VZ, Wong GKC. Neuroinflammation responses after subarachnoid hemorrhage: a review. J Clin Neurosci 2017;42:7–11. 10.1016/j.jocn.2017.02.001 [DOI] [PubMed] [Google Scholar]
- 4. van Dijk BJ, Vergouwen MDI, Kelfkens MM, et al. Glial cell response after aneurysmal subarachnoid hemorrhage - functional consequences and clinical implications. Biochim Biophys Acta 2016;1862:492–505. 10.1016/j.bbadis.2015.10.013 [DOI] [PubMed] [Google Scholar]
- 5. Saijo K, Glass CK. Microglial cell origin and phenotypes in health and disease. Nat Rev Immunol 2011;11:775–87. 10.1038/nri3086 [DOI] [PubMed] [Google Scholar]
- 6. Kettenmann H, Hanisch U-K, Noda M, et al. Physiology of microglia. Physiol Rev 2011;91:461–553. 10.1152/physrev.00011.2010 [DOI] [PubMed] [Google Scholar]
- 7. Zheng ZV, Wong KCG. Microglial activation and polarization after subarachnoid hemorrhage. Neuroimmunol Neuroinflamm 2019;6:1–10. 10.20517/2347-8659.2018.52 [DOI] [Google Scholar]
- 8. Li R, Liu W, Yin J, et al. Tsg-6 attenuates inflammation-induced brain injury via modulation of microglial polarization in SAH rats through the SOCS3/STAT3 pathway. J Neuroinflammation 2018;15:231. 10.1186/s12974-018-1279-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. You W, Wang Z, Li H, et al. Inhibition of mammalian target of rapamycin attenuates early brain injury through modulating microglial polarization after experimental subarachnoid hemorrhage in rats. J Neurol Sci 2016;367:224–31. 10.1016/j.jns.2016.06.021 [DOI] [PubMed] [Google Scholar]
- 10. Abou-Ghazal M, Yang DS, Qiao W, et al. The incidence, correlation with tumor-infiltrating inflammation, and prognosis of phosphorylated STAT3 expression in human gliomas. Clin Cancer Res 2008;14:8228–35. 10.1158/1078-0432.CCR-08-1329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qin H, Yeh W-I, De Sarno P, et al. Signal transducer and activator of transcription-3/suppressor of cytokine signaling-3 (STAT3/SOCS3) axis in myeloid cells regulates neuroinflammation. Proc Natl Acad Sci U S A 2012;109:5004–9. 10.1073/pnas.1117218109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Oliva AA, Kang Y, Sanchez-Molano J, et al. Stat3 signaling after traumatic brain injury. J Neurochem 2012;120:710–20. 10.1111/j.1471-4159.2011.07610.x [DOI] [PubMed] [Google Scholar]
- 13. Ding Y, Qian J, Li H, et al. Effects of SC99 on cerebral ischemia-perfusion injury in rats: selective modulation of microglia polarization to M2 phenotype via inhibiting JAK2-STAT3 pathway. Neurosci Res 2019;142:58–68. 10.1016/j.neures.2018.05.002 [DOI] [PubMed] [Google Scholar]
- 14. Satriotomo I, Bowen KK, Vemuganti R. Jak2 and STAT3 activation contributes to neuronal damage following transient focal cerebral ischemia. J Neurochem 2006;98:1353–68. 10.1111/j.1471-4159.2006.04051.x [DOI] [PubMed] [Google Scholar]
- 15. Parker BL, Larsen MR, Edvinsson LIH, et al. Signal transduction in cerebral arteries after subarachnoid hemorrhage—a phosphoproteomic approach. J Cereb Blood Flow Metab 2013;33:1259–69. 10.1038/jcbfm.2013.78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. An J-Y, Pang H-G, Huang T-Q, et al. Ag490 ameliorates early brain injury via inhibition of JAK2/STAT3-mediated regulation of HMGB1 in subarachnoid hemorrhage. Exp Ther Med 2018;15:1330–8. 10.3892/etm.2017.5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wei S, Luo C, Yu S, et al. Erythropoietin ameliorates early brain injury after subarachnoid haemorrhage by modulating microglia polarization via the EPOR/JAK2-STAT3 pathway. Exp Cell Res 2017;361:342–52. 10.1016/j.yexcr.2017.11.002 [DOI] [PubMed] [Google Scholar]
- 18. Matsuda T, Nakamura T, Nakao K, et al. Stat3 activation is sufficient to maintain an undifferentiated state of mouse embryonic stem cells. Embo J 1999;18:4261–9. 10.1093/emboj/18.15.4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Takeda K, Noguchi K, Shi W, et al. Targeted disruption of the mouse STAT3 gene leads to early embryonic lethality. Proc Natl Acad Sci U S A 1997;94:3801–4. 10.1073/pnas.94.8.3801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A 1988;85:5166–70. 10.1073/pnas.85.14.5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sauer B. Functional expression of the cre-lox site-specific recombination system in the yeast saccharomyces cerevisiae. Mol Cell Biol 1987;7:2087–96. 10.1128/MCB.7.6.2087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schweizer U, Gunnersen J, Karch C, et al. Conditional gene ablation of STAT3 reveals differential signaling requirements for survival of motoneurons during development and after nerve injury in the adult. J Cell Biol 2002;156:287–98. 10.1083/jcb.200107009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Takeda K, Clausen BE, Kaisho T, et al. Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of STAT3 in macrophages and neutrophils. Immunity 1999;10:39–49. 10.1016/S1074-7613(00)80005-9 [DOI] [PubMed] [Google Scholar]
- 24. Jacoby JJ, Kalinowski A, Liu M-G, et al. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A 2003;100:12929–34. 10.1073/pnas.2134694100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wolf Y, Yona S, Kim K-W, et al. Microglia, seen from the CX3CR1 angle. Front Cell Neurosci 2013;7:26. 10.3389/fncel.2013.00026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Zheng ZV, Lyu H, Lam SYE, et al. The dynamics of microglial polarization reveal the resident neuroinflammatory responses after subarachnoid hemorrhage. Transl Stroke Res 2020;11:433–49. 10.1007/s12975-019-00728-5 [DOI] [PubMed] [Google Scholar]
- 27. Du GJ, Lu G, Zheng ZY, et al. Endovascular perforation murine model of subarachnoid hemorrhage. Acta Neurochir Suppl 2016;121:83–8. 10.1007/978-3-319-18497-5_14 [DOI] [PubMed] [Google Scholar]
- 28. Riley JK, Takeda K, Akira S, et al. Interleukin-10 receptor signaling through the JAK-STAT pathway. requirement for two distinct receptor-derived signals for anti-inflammatory action. J Biol Chem 1999;274:16513–21. 10.1074/jbc.274.23.16513 [DOI] [PubMed] [Google Scholar]
- 29. Matsumoto J, Dohgu S, Takata F, et al. TNF-α-sensitive brain pericytes activate microglia by releasing IL-6 through cooperation between IκB-NFκB and JAK-STAT3 pathways. Brain Res 2018;1692:34–44. 10.1016/j.brainres.2018.04.023 [DOI] [PubMed] [Google Scholar]
- 30. Siveen KS, Sikka S, Surana R, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta 2014;1845:136–54. 10.1016/j.bbcan.2013.12.005 [DOI] [PubMed] [Google Scholar]
- 31. Zhong Z, Wen Z, Darnell J. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 1994;264:95–8. 10.1126/science.8140422 [DOI] [PubMed] [Google Scholar]
- 32. Tsien JZ, Chen DF, Gerber D, et al. Subregion- and cell type-restricted gene knockout in mouse brain. Cell 1996;87:1317–26. 10.1016/S0092-8674(00)81826-7 [DOI] [PubMed] [Google Scholar]
- 33. Tsien JZ. Cre-Lox neurogenetics: 20 years of versatile applications in brain research and counting…. Front Genet 2016;7:19. 10.3389/fgene.2016.00019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene 2000;19:2607–11. 10.1038/sj.onc.1203478 [DOI] [PubMed] [Google Scholar]
- 35. Chao J, Zhang Y, Du L, et al. Molecular mechanisms underlying the involvement of the sigma-1 receptor in methamphetamine-mediated microglial polarization. Sci Rep 2017;7:11540. 10.1038/s41598-017-11065-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hao Y, Yang X, Chen C, et al. Stat3 signalling pathway is involved in the activation of microglia induced by 2.45 GHz electromagnetic fields. Int J Radiat Biol 2010;86:27–36. 10.3109/09553000903264507 [DOI] [PubMed] [Google Scholar]
- 37. Nimmerjahn A, Kirchhoff F, Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005;308:1314–8. 10.1126/science.1110647 [DOI] [PubMed] [Google Scholar]
- 38. Xu Z, Shi W-H, Xu L-B, et al. Resident microglia activate before peripheral monocyte infiltration and p75NTR blockade reduces microglial activation and early brain injury after subarachnoid hemorrhage. ACS Chem Neurosci 2019;10:412–23. 10.1021/acschemneuro.8b00298 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
svn-2021-001028supp001.pdf (19.4MB, pdf)
Data Availability Statement
Data are available upon reasonable request. Experiment data are available upon reasonable request.