

Abstract
Autoinflammatory diseases are a heterogenous group of disorders defined by fever and systemic inflammation suggesting involvement of genes regulating innate immune responses. Patients with homozygous loss‐of‐function variants in the OTU‐deubiquitinase OTULIN suffer from neonatal‐onset OTULIN‐related autoinflammatory syndrome (ORAS) characterized by fever, panniculitis, diarrhea, and arthritis. Here, we describe an atypical form of ORAS with distinct clinical manifestation of the disease caused by two new compound heterozygous variants (c.258G>A (p.M86I)/c.500G>C (p.W167S)) in the OTULIN gene in a 7‐year‐old affected by a life‐threatening autoinflammatory episode with sterile abscess formation. On the molecular level, we find binding of OTULIN to linear ubiquitin to be compromised by both variants; however, protein stability and catalytic activity is most affected by OTULIN variant p.W167S. These molecular changes together lead to increased levels of linear ubiquitin linkages in patient‐derived cells triggering the disease. Our data indicate that the spectrum of ORAS patients is more diverse than previously thought and, thus, supposedly asymptomatic individuals might also be affected. Based on our results, we propose to subdivide the ORAS into classical and atypical entities.
Keywords: autoinflammation, linear ubiquitin, LUBAC, ORAS, OTULIN
Subject Categories: Genetics, Gene Therapy & Genetic Disease
The OTULIN‐Related Autoinflammatory Syndrome (ORAS) is known to be caused by homozygous variants in the OTULIN gene. This study discovers that two new compound‐heterozygous variants in OTULIN are associated with an atypical, but potentially fatal, late‐onset form of ORAS.
The paper explained.
Problem
Ubiquitination and its reversal process, deubiquitination, are crucial for the regulation of immune signaling pathways. Thus, it is not surprising that defective (de)ubiquitination has been identified as the underlying cause of a new category of autoinflammatory disorders. One of these disorders, caused by homozygous variants in the deubiquitinase OTULIN, is known as OTULIN‐Related Autoinflammatory Syndrome (ORAS) or Otulipenia. The ORAS is characterized by neonatal‐onset fever, panniculitis, diarrhea, and arthritis. How compound heterozygous mutations in OTULIN manifest clinically is currently unknown.
Results
In this study, we identify two new compound heterozygous variants in OTULIN in a patient without overt signs of autoinflammation until the age of seven who then encountered a severe autoinflammatory episode with multiorgan abscess formation. In‐depth biochemical analysis revealed how both OTULIN variants contribute to diminished OTULIN protein expression and defective hydrolysis of linear ubiquitin linkages observed in patient‐derived cells: whereas binding of OTULIN to linear ubiquitin was compromised by both variants, protein stability and catalytic activity was most affected by the OTULIN variant p.W167S. Although this patient differs clinically from published patients with ORAS, the pathogenesis of the disease seems to be similar and also triggered by perturbed TNF signaling.
Impact
The number of patients with ORAS, first described in 2016, is still very limited. Thus, we can learn a lot from each additional ORAS patient. This study indicates that the clinical spectrum of ORAS patients is more diverse than previously thought and multiorgan sterile abscess formation and potential clinical inapparency should be added to the list of ORAS symptoms. Due to the differences in clinical presentation between this patient with compound heterozygous OTULIN variants and previously identified patients with homozygous OTULIN variants, we suggest to divide the ORAS into classical and atypical entities.
Introduction
Autoinflammation describes a group of inherited, mostly monogenic disorders with recurrent fever and systemic inflammation in the absence of identifiable infectious agents (Manthiram et al, 2017). The post‐translational modification of proteins by ubiquitin plays an essential role in the regulation of immune signaling pathways, in particular in the innate immune response (Zinngrebe et al, 2014). Variants in genes involved in ubiquitination and its reversal process, deubiquitination, have been identified as underlying cause of a new category of autoinflammatory diseases (Aksentijevich & Zhou, 2017; Beck & Aksentijevich, 2019).
Ubiquitination links ubiquitin molecules to substrate proteins, or to one another via the C‐terminal carboxyl group of the donor ubiquitin and one of the seven internal lysine (K) residues or the N‐terminal methionine (M) 1 of the acceptor ubiquitin. This results in a total of eight different inter‐ubiquitin linkage types (Spit et al, 2019). M1‐linkages, also known as linear ubiquitin linkages, are assembled in a head‐to‐tail fashion (Kirisako et al, 2006) by a tripartite protein complex called linear ubiquitin chain assembly complex (LUBAC) consisting of Shank‐Associated RH Domain‐Interacting Protein (SHARPIN), Heme‐Oxidized IRP2 Ubiquitin Ligase 1 (HOIL‐1), and HOIL‐1‐Interacting Protein (HOIP) (Gerlach et al, 2011; Ikeda et al, 2011; Tokunaga et al, 2011).
Ubiquitin linkages are disassembled by so‐called deubiquitinating enzymes (DUBs). In 2013, the OTU‐deubiquitinase with linear linkage specificity (OTULIN; also known as FAM105B or Gumby) was identified to specifically bind to and hydrolyze linear ubiquitin linkages assembled by LUBAC (Keusekotten et al, 2013; Rivkin et al, 2013).
Dysregulation of linear ubiquitin linkages is associated with numerous human diseases, including immune disorders, cancer, and neurodegeneration (Jahan et al, 2021). Variants affecting OTULIN’s catalytic activity, resulting in increased linear ubiquitin linkages, cause embryonic lethality in mice (Rivkin et al, 2013; Heger et al, 2018). Moreover, two independent groups identified a surplus of linear ubiquitin linkages in humans due to homozygous variants in OTULIN to result in an autoinflammatory disease: OTULIN‐Related Autoinflammatory Syndrome (ORAS) or Otulipenia (Damgaard et al, 2016; Zhou et al, 2016). Eight patients with ORAS carrying homozygous missense or premature stop variants in the OTULIN gene have been identified to date (Damgaard et al, 2016, 2019, 2020; Zhou et al, 2016; Nabavi et al, 2019) (Table EV1). All reported patients were born prematurely, showed first signs of disease within weeks after birth, and suffered from fever, nodular panniculitis, failure to thrive, diarrhea, and arthritis accompanied by increased levels of leukocytes, neutrophils, and C‐reactive protein (CrP) (Damgaard et al, 2016, 2019, 2020; Zhou et al, 2016; Nabavi et al, 2019). One ORAS patient carrying a homozygous missense mutation in OTULIN (Damgaard et al, 2016) additionally suffered from steatosis and hepatocyte degeneration with abnormal liver values (Damgaard et al, 2020) suggesting that functioning OTULIN is also essential for liver health. This is further supported by the fact that mice with liver‐specific deletion of OTULIN show a similar disease phenotype with liver inflammation and apoptosis ultimately leading to formation of hepatocellular carcinoma (Damgaard et al, 2020; Verboom et al, 2020).
In the present study, we identified a 7‐year‐old boy with compound heterozygosity in OTULIN carrying two different heterozygous variants with one variant on each allele of the OTULIN gene. He suffered from an atypical form of ORAS with late‐onset manifesting as a fulminant autoinflammatory episode with sterile abscess formation in different organs including skin, lung, and spleen. By performing structural and biochemical analyses, OTULIN gene deletion and reconstitution experiments with different OTULIN variants in a heterologous cell system and by assessing response of patient‐derived fibroblasts and B cells to immune stimuli, we provide characterization of the combined impact of the two different OTULIN variants on OTULIN’s function on both, molecular and functional levels.
Results
Sterile abscess formation in a patient with compound‐heterozygous missense variants in the OTULIN gene
A 7‐year‐old male patient of Greek origin was admitted with abdominal pain and subfebrile temperatures. The boy’s psychomotor development was age‐appropriate, and he was obese (body weight: 36.7 kg, height: 1.29 m, body mass index (BMI): 22.1 kg/m2 (97th age‐specific BMI percentile)). He had previously suffered from a pneumonia at the age of 6 months, an appendicitis at the age of 6 years, and a gluteal abscess which had been difficult to treat. Initially, he presented with leukocytosis (25.5 G/l; normal range: 4.5–13.5 G/l), neutrophilia (14.93 G/l; normal range: 1.8–8 G/l), and highly elevated levels of CrP (241 mg/l; normal range: < 10 mg/l) (Fig 1A). Shortly after admission, he developed spiking fevers with continuously increasing inflammatory parameters (Fig 1A). Treatment with broad‐spectrum antibiotics did not influence the course of systemic inflammatory response syndrome. During further course, the patient developed inflammatory lesions on the left and right wrists and the right ankle (Fig 1B). Total body magnetic resonance imaging (MRI) further revealed abscess formation in the left lower pulmonary lobe, in the left axilla, and in the spleen (Fig 1C). The patient was transferred to intensive care unit (ICU) and underwent the following surgical procedures: debridement of lesions on the wrists, axillary dissection, partial resection of lung and pancreas, and splenectomy. Pus was drained from multiple sites of inflammation; however, biopsies and smears remained sterile (Appendix Table S1). All blood cultures, stool cultures, throat and anal swabs, and tracheal fluids remained sterile (Appendix Table S2). Histopathological analysis of the skin (Fig 1D) revealed massive inflammatory infiltrates of the corium, predominantly consisting of granulocytes, monocytes, and macrophages. In the lung, we found partially necrotizing infiltrates with neutrophils, and also, the spleen showed signs of inflammation and necrosis (Fig 1D). Eosinophils or giant cells were not detected. A monoclonal antibody directed against actin to visualize small blood vessels showed disruption of vessel walls by inflammatory cells in all three organs (Fig 1D). Although the patient’s urine was positive for Pneumococcal antigen (Appendix Table S2), gram‐positive bacteria were not detectable in biopsies of lung, spleen, and skin (Appendix Table S3).
Figure 1. Sterile abscess formation in a patient with compound‐heterozygous missense variants in the OTULIN gene.
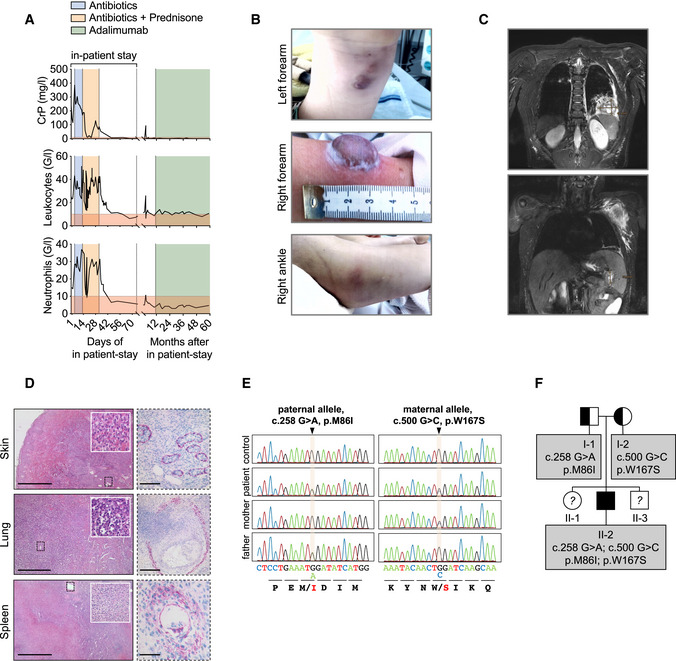
- Blood parameters of patient are depicted.
- Patient’s skin alterations are depicted.
- T2‐weighted MR images in coronal plane show abscess formation in the left lower pulmonary lobe (upper panel) and in spleen and left axilla (lower panel).
- Histological sections of patient biopsies stained with hematoxylin and eosin (left panel) or with an actin antibody (right panel). Scale bars: skin: left 1,000 µm, right 100 µm; lung: left 500 µm, right 250 µm; spleen: left 1,000 µm, right 75 µm.
- Whole Exome Sequencing (WES) and targeted Sanger sequencing identified compound heterozygous variants in OTULIN at position c.258 G > A, p.M86I on the paternal allele and at position c.500 G > C, p.W167S on the maternal allele.
- The pedigree is depicted. The patient (II‐2) is the second child of non‐consanguineous parents.
Since no infectious agent was identified and broad‐spectrum antibiotics had not improved the patient’s condition, an autoinflammatory syndrome was suspected, and additional treatment with corticosteroids was started on day 14 of in‐patient stay (Fig 1A). This resulted in a decline of body temperature and CrP levels and in marked improvement of the patient’s condition. Liver enzymes such as aspartate transaminase (AST), alanine transaminase (ALT), or gamma‐glutamyltransferase (GGT) were elevated at this time and returned to normal in the further course of the disease (Fig EV1A). Alkaline phosphatase (AP) (Fig EV1A), but also total bilirubin, prothrombin, and activated partial thromboplastin were within normal range at all times, and an ultrasound examination of the liver during the autoinflammatory episode showed no abnormalities (Fig EV1B, upper panel). During recovery, the patient developed a pneumothorax and suffered from several bleeding duodenal ulcer requiring application of endoclips. No signs of pathology apart from uncharacteristic inflammation in the gastric antrum (Appendix Table S3) were found in biopsies. Two months following admission, the patient was discharged in good condition.
Figure EV1. Abnormal liver function test and development of steatosis hepatis grade II.
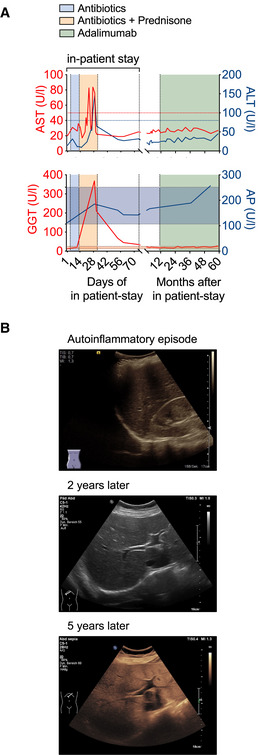
- The course of the patient’s liver enzymes AST (aspartate transaminase) and ALT (alanine transaminase) (upper panel) or GGT (γ‐glutamyltransferase) and AP (alkaline phosphatase) (lower panel) are depicted along the time axis with the upper limit of normal indicated by red and blue lines for AST and ALT and the normal range in shades of red and blue for GGT and AP, respectively.
- Liver ultrasound B‐mode images of the patient during the initial phase of autoinflammation and about 2 and 5 years thereafter display diffuse increase of liver echogenicity and finally slightly impaired appearance of portal vein wall and diaphragm indicative of steatosis hepatis grade II.
To identify the underlying cause of the severe and life‐threatening autoinflammatory episode in this patient, we performed whole exome sequencing (WES) and targeted Sanger sequencing revealing compound heterozygous missense variants in exon 3 (c.258G>A; p.M86I) and exon 5 (c.500G>C; p.W167S) (Fig 1E) of the OTULIN gene, respectively. WES revealed no other homozygous, compound heterozygous or pathogenic variants likely to explain the observed disease phenotype (Appendix Tables S4–S7; for filtering strategy see Appendix Fig S1). The patient inherited the p.M86I variant from his father, whereas his mother is a heterozygous carrier of the p.W167S mutation. The patient is the second child born to non‐consanguineous parents (patient II‐2; Fig 1F). Both, his parents and siblings are clinically well.
The compound‐heterozygous missense variants p.M86I and p.W167S affect OTULIN protein expression and function
Missense or premature stop variants in OTULIN cause ORAS (Damgaard et al, 2016, 2019; Zhou et al, 2016; Nabavi et al, 2019). All published disease‐causing variants in OTULIN are homozygous (Damgaard et al, 2016, 2019; Zhou et al, 2016; Nabavi et al, 2019) (Fig EV2A). As the patient’s phenotype differed considerably from published patients with ORAS (Table EV1), we assessed whether the identified compound heterozygosity in the OTULIN gene affected OTULIN protein expression and/or function. OTULIN protein expression was diminished in patient‐derived fibroblasts and B cells compared to control (Fig 2A). As loss of OTULIN was shown to result in downregulation of LUBAC components in B cells (Damgaard et al, 2016; Heger et al, 2018), we also determined protein expression of SHARPIN, HOIL‐1, and HOIP. Expression of the different LUBAC components remained stable in patient‐derived cells (Fig 2A). To assess whether the diminished OTULIN expression was due to compromised antibody binding to OTULIN variant p.M86I, we applied a second commercially available antibody and confirmed equal detection of wildtype (OTULINWT) and variant (OTULINM86I or OTULINW167S) OTULIN protein overexpressed in A549 OTULIN KO cells (Fig EV2B). OTULIN mRNA expression was unchanged in patient‐derived fibroblasts and B cells (Fig EV2C). The homozygous OTULIN mutation p.G281R was reported to result in diminished protein expression due to increased OTULIN degradation via the proteasome (Damgaard et al, 2019). The half‐life of wildtype and variant OTULIN was similar (Fig EV2D). However, neither co‐incubation with the proteasome inhibitor Bortezomib (BTZ) nor with Bafilomycin A1 (Baf A1), an inhibitor of lysosomal protein degradation, was capable of stabilizing recombinant wildtype and variant OTULIN protein upon incubation with CHX (Fig EV2E). MCL‐1, a protein with high turnover (Wu et al, 2020), and LC3‐II, a marker of autophagosomes (Yoshii & Mizushima, 2017), served as positive controls for BTZ and Baf A1, respectively (Fig EV2E).
Figure EV2. Diminished OTULIN protein expression in patient‐derived cells is not due to reduced mRNA expression or increased degradation via the proteasome or the lysosome.

- OTULIN protein is depicted.
- OTULIN antibodies by Abcam (antigen corresponding to AA 60–158) or by Cell Signaling (recombinant fragment surrounds S76 without spanning M86) were tested in parallel on A549 OTULIN KO cells transfected with different OTULIN constructs as indicated. One representative of two independent experiments is shown.
- Relative mRNA expression of OTULIN in fibroblasts and B cells with two different primer pairs is depicted. Data are presented as mean ± SD of six independent experiments; dots represent individual experiments performed in three technical replicates; ns, non‐significant, unpaired t‐test.
- A549 OTULIN KO cells were transfected with the different OTULIN constructs as indicated. The following day, cells were treated with 50 µg/ml cycloheximide (CHX) for the indicated times, harvested and analyzed by Western blot for OTULIN protein expression. Tubulin served as loading control. One representative of three independent experiments is shown.
- A549 OTULIN KO cells were transfected with the different OTULIN constructs as indicated. The following day, cells were treated with 50 µg/ml CHX alone or in combination with 1 µM Bortezomib (BTZ) or 1 µM Bafilomycin A1 (Baf A1) for 8 h or left untreated (DMSO). Expression of the proteins indicated was analyzed by Western blot. One representative of three independent experiments is shown.
Figure 2. The compound‐heterozygous missense variants p.M86I and p.W167S affect OTULIN protein expression and function.
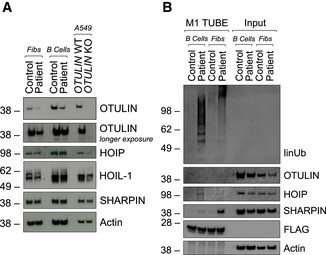
- Expression of indicated proteins was determined by Western blot in patient‐derived and control fibroblasts and B cells and A549 OTULIN WT and KO cells.
- FLAG‐tagged tandem ubiquitin binding entity (TUBE) assay was performed to pull down linear ubiquitin linkages in patient‐derived and control fibroblasts and B cells. One representative (A, B) out of three independent experiments is shown.
Source data are available online for this figure.
One function of OTULIN is the cleavage of linear ubiquitin linkages (Verboom et al, 2021; Weinelt & van Wijk, 2021). Numerous studies demonstrated that OTULIN downregulation or knockout as well as the expression of certain OTULIN variants lead to accumulation of linear ubiquitin linkages in cells (Fiil et al, 2013; Keusekotten et al, 2013; Rivkin et al, 2013; Elliott et al, 2014; Draber et al, 2015; Damgaard et al, 2016; Hrdinka et al, 2016; Zhou et al, 2016; van Wijk et al, 2017; Heger et al, 2018). Thus, we next assessed the level of linear ubiquitin linkages in patient‐derived and control cells. To enrich for linear ubiquitin, a FLAG‐tagged tandem ubiquitin binding entity (TUBE) reagent was used to pull down linear ubiquitin linkages and associated proteins (Fig 2B). Importantly, linear ubiquitin chains and associated LUBAC components SHARPIN and HOIP were increased in both, patient‐derived fibroblasts and B cells (Fig 2B).
Taken together, these results show that the compound heterozygous variants identified by WES in the OTULIN gene compromised OTULIN’s expression and DUB activity in patient‐derived cells.
Pathogenic potential of OTULIN variants p.M86I and p.W167S
The potential pathogenicity of the two novel gene variants identified in this study was further analyzed using Ensembl Variant Effect Predictor (McLaren et al, 2016). Sorting Intolerant From Tolerant (SIFT) (Sim et al, 2012) and PolyPhen‐2 (Adzhubei et al, 2010) algorithms predicted the maternal, rather than the paternal, variant to affect OTULIN protein function (Table 1). Apart from one intronic variant (Nabavi et al, 2019), all published homozygous variants in the OTULIN gene (Damgaard et al, 2016, 2019; Zhou et al, 2016) were located in the OTU domain of OTULIN containing its catalytic activity (Keusekotten et al, 2013). In agreement, the two novel variants identified in our study are also localized in the OTU domain (Fig EV2A). In the OTULIN 3D‐structure (3ZNV, 3ZNZ; Keusekotten et al, 2013), M86 and W167 are, however, located on different ends of the protein (Fig 3A). The catalytic core of OTULIN is composed of N341, H339, and C129 (Fig 3A; pale cyan) (Rivkin et al, 2013). The crystal structure of this catalytic triad was shown to exist in two alternate conformations: “active” and “inhibited” (Keusekotten et al, 2013). The coordination of N341 is a key event in OTULIN activation (Keusekotten et al, 2013). One of the residues coordinating the N341 side chain is Y91 (Keusekotten et al, 2013) (wheat in Fig 3B). Y91, in turn, is involved, through its hydroxyl group, in an intense network of interactions between different OTULIN residues, among them its catalytic residues H339 and N341 (Fig 3B). This network is important for changing OTULIN’s conformation from “inhibitory” to “active” and co‐ordinates the catalytic triad residue N341, aligning it to H339 (Keusekotten et al, 2013). Importantly, the aromatic ring of Y91 approaches the sulfur center of M86 to a distance of 4.1 Å (Fig 3B; sulfur in yellow). The sulfur center of sulfur‐containing amino acids (M, C) and aromatic side chains of Y, W, or F are involved in close (< 5 Å) and frequent contacts in proteins (sulfur‐arene interactions) (Meyer et al, 2003). This interaction is lost in the mutant M86I (Fig 3B; mutant in pale green). We hypothesize that this loss alters the conformation of the catalytic core of OTULIN, enhancing the proportion of the inactive conformation, and thereby reducing the enzyme’s turnover number (kcat ). Consistent with this notion is the observation that replacement of Y91 by F resulted in a 20‐fold reduction of kcat while not affecting KM (Keusekotten et al, 2013). Moreover, when in complex with M1‐diubiquitin (3ZNZ), Y91 is one of OTULIN’s S1' contact sites with the F4 patch of proximal ubiquitin (Fig 3C). The interaction between Y91 and M86, which is lost in the mutant M86I, might, thus, also play a role in binding of OTULIN to linear ubiquitin (Fig 3C). The position of the maternal variant W167 is in close proximity to OTULIN’s helical arm containing the S1 contact site with the I44 patch of distal ubiquitin. This helical arm comprises or adjoins the three residues Y244, L272, and G281, replaced in the known homozygous variants of OTULIN (Damgaard et al, 2016, 2019, 2020; Zhou et al, 2016) (Fig 3D). The mutation of W167 to S results in a replacement of the bulky, apolar indole side chain of W167 by a much smaller, polar hydroxymethyl side chain (Fig 3D; close‐up). It is likely, therefore, that the W167S replacement affects the orientation of the helical arm's S1 site, made up of L259, A262, and R263 toward the I44 patch of distal ubiquitin, thereby indirectly interfering with OTULIN’s binding (KM ) to linear ubiquitin (Fig 3D).
Table 1.
Compound heterozygous variants in the OTULIN gene in a patient with autoinflammation and sterile abscess formation.
| Nucleotide alteration | CDS position | AA alteration | domain | SIFT | PolyPhen | |
|---|---|---|---|---|---|---|
| Paternal allele | Chr5: 14678818G>A | c.258G>A | p.M86I | OTU | 0.11 | 0.31 |
| Maternal allele | Chr5: 14687661G>C | c.500G>C | p.W167S | OTU | 0 | 0.998 |
CDS, coding sequence; AA, amino acid; SIFT, Sorting Intolerant From Tolerant (< 0.05 = deleterious); PolyPhen, Polymorphism Phenotyping (> 0.908 “Probably Damaging”, 0.446–0.908 “Possibly Damaging”, < 0.446 “Benign”).
Figure 3. Pathogenic potential of OTULIN variants p.M86I and p.W167S.

- The overall structure of OTULINWT (3ZNV) is depicted with its catalytic triad (C129, H339, N341; pale cyan) and positions of variants identified in this study (M86, W167).
- Close‐up of M86 (wheat) and M86I (pale green) with the interaction network around OTULIN’s catalytic center.
- Close‐up of OTULINC129A and its interaction with the proximal ubiquitin (Ub). M86, Y91, E95, and R122 of OTULIN and their interaction with each other and with the F4 patch of the proximal Ub are depicted. Interactions (dotted red lines) and water molecules (light blue) < 5 Å are shown. Residues of the catalytic triad (A129, H339, N341) in pale cyan.
- OTULINW167S in complex with di‐Ub. Contact sites of W167 (L237, I241, L273, and R274) in pale yellow. Positions of known homozygous OTULIN substitutions (Y244, L272, G281) in pale green. I44 patch of distal Ub in magenta and its contact sites L259, A262, and R263 in pale yellow. Close‐up of W167 and S167: loss of size and gain of polarity from W to S.
Collectively, this structural analysis suggests that both substitutions, M86I and W167S, may interfere with binding of OTULIN to linear ubiquitin (Fig 3C and D). Moreover, M86I might additionally reduce OTULIN’s catalytic turnover number (kcat ) (Fig 3B).
OTULIN variants compromise binding of OTULIN to linear ubiquitin and differentially affect OTULIN’s intrinsic stability and catalytic activity
To assess how the variants p.M86I and p.W167S affect OTULIN’s intrinsic thermal stability, we purified recombinant wildtype (OTULINWT) and variant OTULIN (OTULINM86I and OTULINW167S) and determined their melting points (T m ) by means of differential scanning fluorimetry (Fig 4A). While OTULINWT and OTULINM86I had a similar T m (OTULINWT: 53.7°C, OTULINM86I: 54°C), OTULINW167S unfolded at a significantly lower temperature (T m 47.4°C) in this assay (Fig 4A). These results are in line with the structural observation that W167 is part of the protein’s hydrophobic core and indicates that replacement with a smaller and more hydrophilic side chain of serine (W167S) influences its integrity. In contrast, the structurally more conservative mutation M86I appears to affect the overall conformation of the protein much less.
Figure 4. OTULIN variants compromise binding of OTULIN to linear ubiquitin and differentially affect OTULIN’s intrinsic stability and catalytic activity.
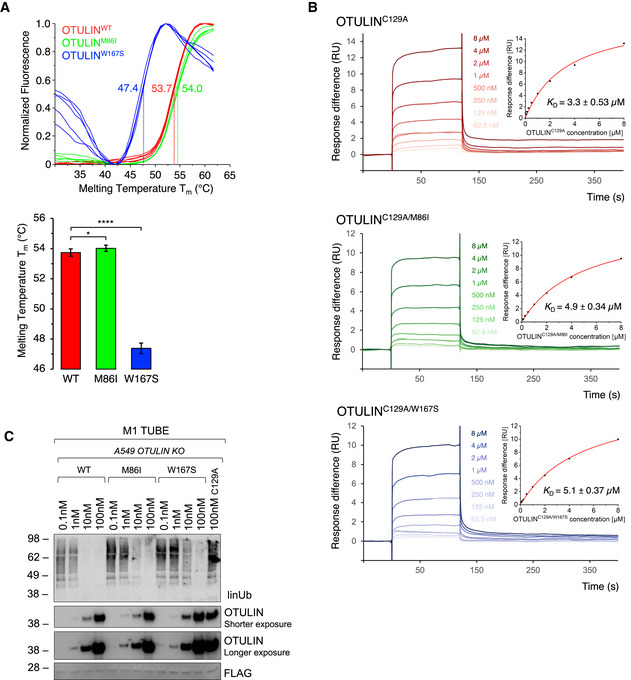
- DSF measurements with OTULINWT, OTULINM86I, or OTULINW167S. Melting temperatures (T m ) are calculated from five independent experiments with standard deviations. *P = 0.011; ****P = 6.64 × 10−18, unpaired t‐test.
- SPR measurements and steady‐state binding curves with calculated dissociation constants (Kd ) after injection of a concentration series of OTULINC129A, OTULINC129A/M86I, or OTULINC129A/W167S to CM5‐immobilized di‐ubiquitin chains.
- Linear ubiquitin linkages isolated from A549 OTULIN KO cells by M1 TUBE assay were incubated with increasing concentrations of recombinant OTULINWT, OTULINM86I, and OTULINW167S or catalytically inactive OTULINC129A as control for 1 h. Afterward, samples were subjected to analysis by Western blot for the indicated proteins. Images are representative of three independent experiments.
To measure the effect of the M86I and W167S mutations on the binding affinity of OTULIN to linear di‐ubiquitin chains, surface Plasmon resonance (SPR) measurements were performed. To prevent ubiquitin cleavage during SPR measurements, a catalytically inactive mutant (OTULINC129A) was used. OTULINC129A bound linear di‐ubiquitin chains with a K d of 3.3 µM in this assay, while lower affinities were determined for both variants OTULINC129A/M86I and OTULINC129A/W167S (4.9 µM and 5.1 µM, respectively) (Fig 4B).
To further evaluate to which extent OTULIN’s catalytic activity was impaired by the two variants, we performed a DUB assay using recombinant OTULINWT, OTULINM86I, and OTULINW167S on endogenous linear ubiquitin linkages isolated from A549 OTULIN KO cells (Fig 4C). Whereas OTULINWT was able to cleave all linear ubiquitin chains, residual amounts of linkages were still present in samples incubated with OTULINW167S. Moreover, also in samples treated with OTULINM86I, we detected remains of linear ubiquitin linkages, although to a lesser extent than in OTULINW167S‐treated samples. In summary, we find binding of OTULIN to linear ubiquitin to be compromised by both variants; however, protein stability and catalytic activity is most affected by the OTULIN variant p.W167S.
Parental variants both contribute to defective hydrolysis of linear ubiquitin linkages in patient‐derived cells, but to different extents
To validate these biochemical findings in a cellular context, we assessed the contribution of both parental variants to defects in hydrolysis of linear ubiquitin linkages in a heterologous cell system. Plasmid vectors encoding wildtype OTULIN (OTULINWT), the paternal p.M86I variant (OTULINM86I) and the maternal p.W167S variant (OTULINW167S) were transiently expressed in A549 OTULIN KO cells. Plasmid vectors encoding both parental variants (OTULINM86I;W167S) and the published variant p.L272P (OTULINL272P) (Damgaard et al, 2016; Zhou et al, 2016) were used for comparison. OTULINWT and OTULINM86I were more efficient in hydrolysis of linear ubiquitin linkages than OTULINW167S or OTULINM86I;W167S when re‐expressed in OTULIN‐deficient cells (Fig 5A). The amount of linear ubiquitin in cells transfected with OTULINL272P was similar to empty vector (EV)‐transfected cells (Fig 5A) indicating that OTULIN’s catalytic activity was most compromised by this variant. The compromised DUB activity of OTULINW167S compared to OTULINWT and OTULINM86I was stable over a range of different DNA concentrations (Fig EV3A). As the double‐mutant OTULINM86I;W167S was less efficient in cleaving linear ubiquitin linkages than a mix of OTULINM86I and OTULINW167S (Fig EV3B), we reasoned that OTULINM86I might be able to substitute for the defect of OTULINW167S. Indeed, defective hydrolysis of linear ubiquitin linkages by OTULINW167S was rescued by co‐expression of OTULINWT and, importantly, also by OTULINM86I (Fig 5B), at least when equal amounts of OTULINM86I and OTULINW167S were present in cells. When we next transfected A549 OTULIN KO cells with the two constructs OTULINM86I and OTULINW167S in different proportions (Fig 5C), we found a surplus of OTULINW167S to result in increased linear ubiquitin linkages suggesting that OTULINM86I can rescue the defect of OTULINW167S only to some extent. To investigate the regulation of linear ubiquitin linkages by the different OTULIN variants in a setting that is close to the natural situation, we assessed linear ubiquitin linkages in EBV‐transformed B cell lines from patient, mother, father and healthy control (Fig 5D). Surprisingly, not only the patient’s B cells but also the mother’s and the father’s B cells had a surplus of linear ubiquitin linkages, although to a lesser extent (Fig 5D). OTULIN protein expression was again diminished in patient‐derived B cells, but, interestingly, also in B cells derived from parents, as compared to control (Figs 5D and EV3C; see Fig 2 for comparison), whereas levels of OTULIN mRNA were similar (Fig EV3D). Together, these results suggest that OTULIN’s catalytic activity is compromised by both parental variants, with the maternal variant p.W167S, however, more severely impairing hydrolysis of linear ubiquitin linkages than the paternal variant p.M86I.
Figure 5. Parental variants both contribute to defective hydrolysis of linear ubiquitin linkages in patient‐derived cells, but to different extents.

-
A–CA549 OTULIN KO cells were transfected with empty vector or different OTULIN constructs as indicated and analyzed by Western blot.
-
DFLAG‐tagged tandem ubiquitin binding entity (TUBE) assay was performed in B cells from control, patient, mother, and father to pull down linear ubiquitin linkages. One representative (A–D) of three independent experiments is shown.
Source data are available online for this figure.
Figure EV3. The maternal OTULIN variant p.W167S more severely impairs hydrolysis of linear ubiquitin linkages than the paternal OTULIN variant p.M86I.
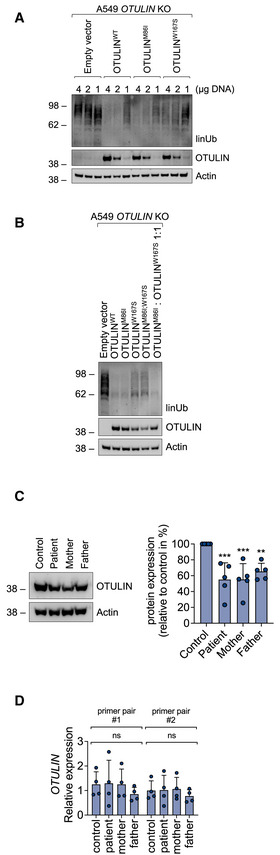
-
A, BA549 OTULIN KO cells were transfected with different OTULIN constructs as indicated. After 24 h, cells were lysed and subjected to analysis by Western blot. One representative of three independent experiments is shown.
-
COTULIN protein expression in B cells derived from control, patient, mother, and father is shown. Blots (left panel) are representative of five independent experiments analyzed by densitometry (right panel). Data are presented as mean ± SD (dots represent individual experiments); ***P = 0.0008, **P = 0.0069, ordinary one‐way ANOVA, Dunnett’s multiple comparisons test.
-
DRelative mRNA expression of OTULIN in B cells derived from control, patient, mother, and father determined with two different primer pairs is depicted. Data are presented as mean ± SD of four independent experiments; dots represent individual experiments performed in three technical replicates; ns, non‐significant, ordinary one‐way ANOVA, Dunnett’s multiple comparisons test.
TNF signaling is altered in patient‐derived cells
Both, mice and humans with defects in OTULIN suffer from autoinflammation mediated by TNF receptor 1 (TNFR1) (Damgaard et al, 2016, 2019; Heger et al, 2018). Importantly, patients with ORAS have successfully been treated with anti‐TNF therapy (Damgaard et al, 2016, 2019; Zhou et al, 2016) (Table EV1). Thus, we investigated whether the compound heterozygosity in OTULIN affected TNF signaling in patient‐derived cells. We found that TNF stimulation resulted in increased formation of linear ubiquitin linkages in patient‐derived fibroblasts as compared to control (Fig 6A). Surprisingly, increased presence of linear ubiquitin linkages and LUBAC components was detectable in the TNFR1‐signaling complex (SC) in patient‐derived cells as compared to control (Fig 6B) although it was recently shown that recruitment of LUBAC to the TNFR1‐SC is compromised when OTULIN’s catalytic activity is perturbed (Heger et al, 2018). OTULIN itself was not recruited to the TNFR1‐SC (Fig 6B) in line with published data (Draber et al, 2015; Elliott et al, 2016; Hrdinka et al, 2016). Of note, OTULIN compound heterozygosity only marginally affected nuclear factor kappa B (NF‐κB) and mitogen‐activated protein kinase (MAPK) signaling activation upon TNF stimulation (Fig 6C). Secretion of interleukin‐6 (IL‐6) upon stimulation with TNF, however, was lower in patient‐derived fibroblasts compared to control (Fig 6D). Lipopolysaccharide (LPS) stimulation resulted in diminished activation of NF‐κB signaling (Fig EV4A) along with reduced secretion of IL‐6 (Fig EV4B) in patient‐derived fibroblasts as compared to control. Moreover, IκBα degradation induced by cluster of differentiation 40 ligand (CD40L) was more pronounced in control than in patient‐derived B cells (Fig EV4C). These data are supported by the fact that IκBα, assessed by cycloheximide (CHX) chase analysis, was more stable in patient‐derived B cells (Fig 6E) and fibroblasts (Fig EV4D) as compared to control suggesting basal NF‐κB activity to be diminished in patient‐derived cells. This is in line with the observation that basal TNF levels were lower in supernatants of patient‐derived B cells as compared to control (Fig 6F).
Figure 6. TNF signaling is altered in patient‐derived cells.
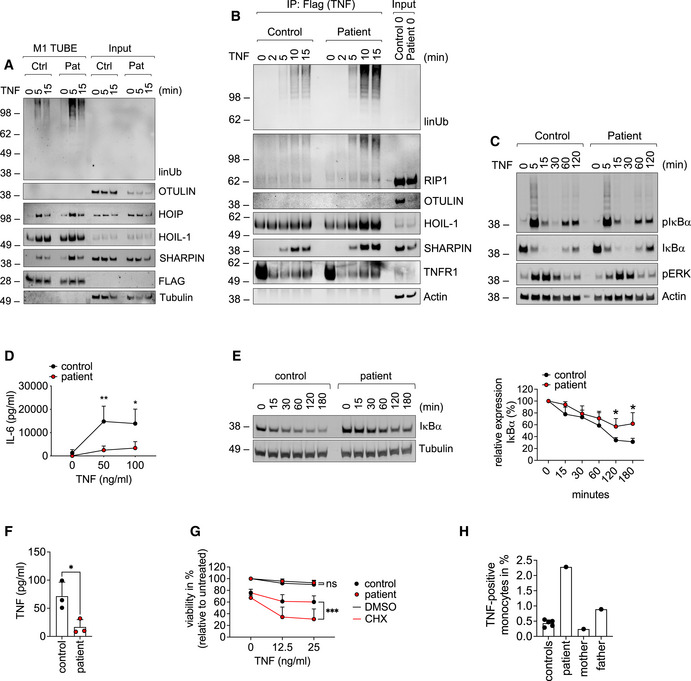
-
A–DPatient‐derived and control fibroblasts were used. (A) FLAG‐tagged tandem ubiquitin binding entity (TUBE) assay was performed to pull down linear ubiquitin linkages upon stimulation with 500 ng/ml TNF. (B) The TNFR1‐SC was isolated using 500 ng/ml TAP‐TNF. (C) Cells were stimulated with 100 ng/ml TNF for the indicated times and analyzed by Western blot. One representative (A–C) of three independent experiments is shown. (D) Cells were stimulated with TNF for 24 h and IL‐6 was determined by ELISA. Data are presented as mean ± SD of three individual experiments performed in two technical replicates; *P = 0.014; **P = 0.007; multiple t‐test corrected for multiple comparisons using the Holm–Sidak method.
-
EB cells were incubated with 50 µg/ml cycloheximide (CHX) for the indicated times and subjected to analysis by Western blot. One representative (left panel) out of 4 individual experiments analyzed by densitometry (right panel) is shown. Data are presented as mean ± SD. *P = 0.02, unpaired t‐test.
-
FPatient‐derived and control B cells were cultivated for 24 h and TNF was determined by ELISA. Data are presented as mean ± SD of 3 individual experiments performed in two technical replicates; *P = 0.028; unpaired t‐test.
-
GFibroblasts were incubated with 50 µg/ml cycloheximide (CHX) or DMSO and stimulated with TNF as indicated. Viability was measured after 24 h. Data are presented as mean ± SD of 5 individual experiments performed in three technical replicates; ***P = 0.0008, mixed‐effects analysis with Tukey’s multiple comparisons test.
-
HIntracellular TNF in CD11b‐positive cells from five healthy controls, patient (before anti‐TNF treatment), mother, and father was determined by FACS.
Source data are available online for this figure.
Figure EV4. Signaling output of different innate immune stimuli in patient‐derived cells.
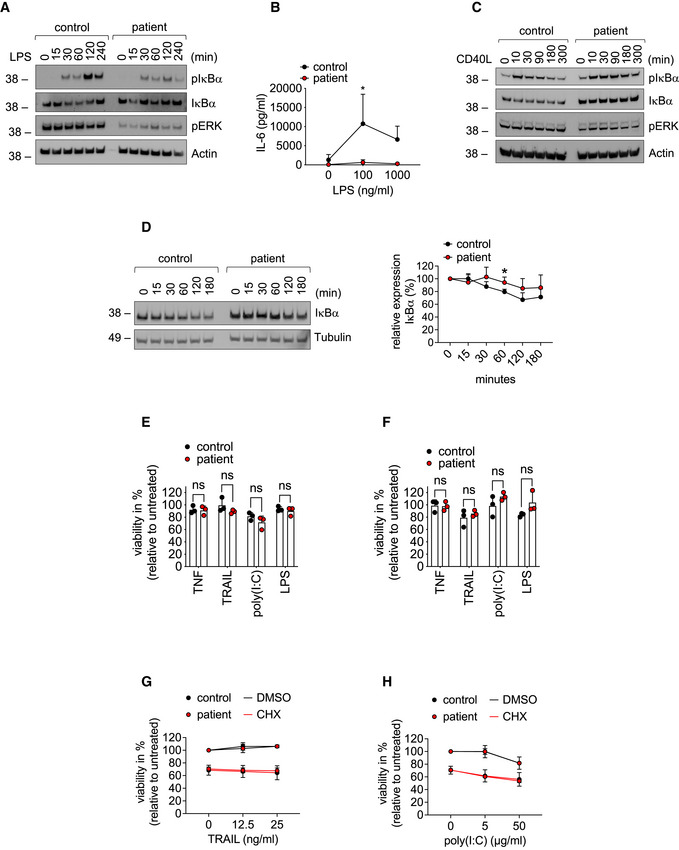
-
AFibroblasts were stimulated with 1 µg/ml LPS for the indicated times and subjected to analysis by Western blot. One representative out of three independent experiments is shown.
-
BFibroblasts were stimulated with LPS for 24 h. Concentration of IL‐6 in the supernatant was determined by ELISA. Data are presented as mean ± SD of three individual experiments performed in two technical replicates; *P = 0.012; multiple t‐tests corrected for multiple comparisons using the Holm–Sidak method.
-
CB cells were treated with 100 ng/ml CD40L for the indicated times and subjected to analysis by Western blot. One representative out of three independent experiments is shown.
-
DFibroblasts were incubated with 50 µg/ml cycloheximide (CHX) for the indicated times and subjected to analysis by western blot. One representative (left panel) out of 4 individual experiments analyzed by densitometry (right panel) is shown. Data are presented as mean ± SD. *P = 0.02, unpaired t‐test.
-
E, FFibroblasts (E) or B cells (F) were treated with 0.1 µg/ml TNF, 0.1 µg/ml TRAIL, 100 µg/ml poly(I:C), 1 µg/ml LPS or left untreated, and viability was determined after 24 h. Data are presented as mean ± SD of three independent experiments; dots represent individual experiments performed in three technical replicates; ns, non‐significant, unpaired t‐test.
-
G, HFibroblasts were incubated with 50 µg/ml cycloheximide (CHX) or DMSO and stimulated with the indicated concentrations of TRAIL (G) or poly(I:C) (H). Viability was measured after 24 h. Data are presented as mean ± SD of three individual experiments performed in three technical replicates.
Source data are available online for this figure.
TNF induces cell death under certain circumstances (Peltzer et al, 2016). However, patient‐derived fibroblasts or B cells and respective control cells were mostly resistant to cell death induction by TNF or by other innate immune stimuli such as TNF‐related apoptosis‐inducing ligand (TRAIL), poly(I:C) or LPS (Fig EV4E and F). It was reported that variants in OTULIN can affect sensitivity of cells to TNF‐induced death in the presence of CHX (Damgaard et al, 2019). Of note, in the presence of CHX, TNF‐induced cell death was significantly higher in patient‐derived fibroblasts than in control cells (Fig 6G). Interestingly, however, patient‐derived fibroblasts were not sensitized to cell death induction by TRAIL or poly(I:C) (Fig EV4G and H).
Myeloid cells of ORAS patients are suggested to be hyperinflammatory and to show increased production of TNF and other inflammatory mediators (Zhou et al, 2016; Damgaard et al, 2019). Indeed, patient‐derived monocytes showed higher intracellular TNF staining as compared to monocytes of healthy controls, mother, or father (Fig 6H). ORAS patients were identified to benefit from TNF blockers (Damgaard et al, 2016, 2019; Zhou et al, 2016) underlining the importance of TNF in the pathogenesis of the disease. Thus, this study’s patient received 30 mg Adalimumab applied subcutaneously every 14 days (Fig 1A) as preventive measure after WES revealed his compound heterozygosity in OTULIN (Fig 1E). The Adalimumab dosage was subsequently adjusted to 40 mg. To assess whether Adalimumab treatment had a beneficial effect on the patient’s inflammatory state, we compared the patient’s plasma protein profile before anti‐TNF treatment and thereafter with plasma from two healthy controls (Fig EV5A). Whereas the patient’s plasma protein profile differed from that of healthy controls before anti‐TNF treatment with upregulation of C‐X‐C motif chemokine ligand (CXCL) 11, C‐C motif chemokine ligand (CCL) 5, IL‐18, and serpin E1, it was reverted to healthy condition after start of anti‐TNF treatment. The Adalimumab concentration in the patient’s serum has always been within therapeutic range (Fig EV5B), and importantly, the patient has not encountered any additional autoinflammatory episodes for 5 years. Laboratory checks and abdominal ultrasound examinations were carried out regularly every 6 months. The liver had initially been unconspicuous on ultrasound; however, the patient has developed signs of liver steatosis over the past 2 years (currently grade II) (Fig EV1B).
Figure EV5. The patient’s inflammatory plasma protein profile is converted to normal after treatment with the anti‐TNF therapeutic Adalimumab.
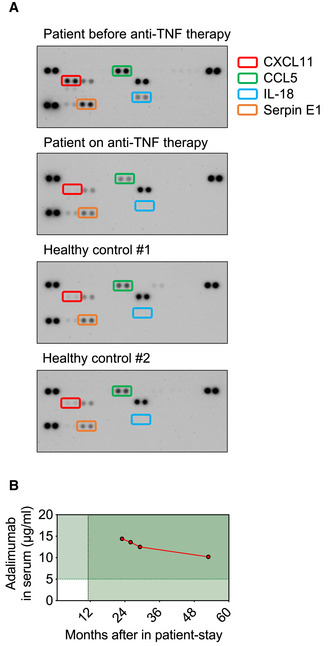
- Plasma was collected from the patient before and after start of therapy with the anti‐TNF therapeutic Adalimumab. Plasma from two healthy controls was used as comparison. The Proteome Profiler Human Cytokine Array was performed according to the manufacturer’s instructions.
- The concentration of Adalimumab in the patient’s serum is shown (target concentration: > 5 µg/ml).
Discussion
Classical OTULIN‐related autoinflammatory syndrome (ORAS) is a rare disease with neonatal‐onset autoinflammation due to homozygous variants in the OTULIN gene (Damgaard et al, 2016, 2019; Zhou et al, 2016; Nabavi et al, 2019). We show here that compound heterozygous variants in OTULIN result in atypical ORAS with distinct clinical manifestation of the disease. The described patient had a previously unremarkable medical history prior to encountering a fulminant autoinflammatory episode at the age of 7 years presenting with fever and severe sterile abscess formation at multiple organ sites but, importantly, without suffering from diarrhea or arthritis (Table EV1).
At the molecular level, we demonstrate that patient‐derived cells express less OTULIN protein than control cells (Fig 2A) and increased amounts of linear ubiquitin linkages under basal conditions (Fig 2B), and, importantly, also upon stimulation with TNF (Fig 6A), the cytokine identified as primary driver of inflammation in mice and humans with defective OTULIN function (Damgaard et al, 2016, 2019; Heger et al, 2018). In addition, we find linear ubiquitin linkages to be more abundant in the TNFR1‐SC along with increased presence of LUBAC components as compared to control (Fig 6B). Two separate pools of cytoplasmic LUBAC complexes were identified as abundant in cells (Draber et al, 2015): (i) LUBAC in complex with OTULIN, and (ii) LUBAC in complex with CYLD, a DUB known to antagonize K63‐ and linear ubiquitin linkages in SCs (Kovalenko et al, 2003), and spermatogenesis‐associated protein (SPATA) 2 (Elliott et al, 2016; Kupka et al, 2016; Schlicher et al, 2016; Wagner et al, 2016). The latter complex can be recruited to SCs, including the TNFR1‐SC (Elliott et al, 2016; Kupka et al, 2016; Schlicher et al, 2016; Wagner et al, 2016). The binding of SPATA2‐CYLD or OTULIN to HOIP was shown to be mutually exclusive (Draber et al, 2015; Schlicher et al, 2016). Thus, a downregulation in OTULIN expression levels, potentially due to reduced thermal stability of OTULINW167S (Fig 4A), as observed in this patient, might result in increased LUBAC‐SPATA2‐CYLD complex formation, and, in turn, increased recruitment of LUBAC to the TNFR1‐SC (Fig 6B). This is an intriguing finding as linear ubiquitin linkages in the TNFR1‐SC were unaffected by a complete absence of OTULIN (Draber et al, 2015), and expression of the catalytically inactive OTULINC129A resulted in even reduced presence of LUBAC and linear ubiquitin linkages in the TNFR1‐SC (Heger et al, 2018). OTULIN deficiency or expression of catalytically inactive OTULINC129A cause increased LUBAC autoubiquitination and degradation (Damgaard et al, 2016, 2019; Heger et al, 2018; Verboom et al, 2020). In patient‐derived cells, LUBAC components were pulled down together with increased amounts of linear ubiquitin linkages in patient‐derived cells (Figs 2B and 6A). Whether LUBAC components are modified by linear ubiquitin linkages in patient‐derived cells, however, requires further investigation. Expression levels of LUBAC components were not altered (Fig 2A) and, in addition, we observed augmented LUBAC recruitment to the TNFR1‐SC (Fig 6B) along with increased ubiquitination of the TNFR1‐SC component RIPK1 (Fig 6B). This is in line with previous reports demonstrating that OTULIN can regulate ubiquitination of substrates other than LUBAC, for example, RIPK1 and TNFR1 (Fiil et al, 2013; Keusekotten et al, 2013).
Thus, even subtle changes in the expression of OTULIN, LUBAC, and probably also CYLD and SPATA2, and their catalytic activities may affect the composition of the TNFR1‐SC and its signaling output.
Analysis of the OTULIN structure suggested the variants in the described patient to interfere with the protein’s catalytic activity and binding to linear di‐ubiquitin (Fig 3). Our in‐depth biochemical analyses indicate that both variant OTULIN proteins, p.M86I and p.W167S, have reduced binding affinities to linear di‐ubiquitin in SPR experiments (Fig 4B). The OTULIN variant p.W167S, however, is the one with lower thermal stability (Fig 4A) and catalytic activity (Figs 4C and 5). Performing reconstitution experiments in a heterologous cell system, we discovered that the paternal mutation p.M86I can compensate the reduced catalytic activity of p.W167S to a certain extent (Fig 5C). Moreover, the published homozygous OTULIN variant p.L272P (Damgaard et al, 2016; Zhou et al, 2016) compromised OTULIN’s DUB activity more than the combined action of the OTULIN variants p.M86I and p.W167S (OTULINM86I;W167S) (Fig 5A). Thus, the degree of impairment of linear chain hydrolysis might correlate with ORAS severity and these data could provide the molecular explanation why the patient of this study was supposedly asymptomatic until the age of 7 years while ORAS patients carrying the homozygous OTULIN variant p.L272P diseased already after birth. This idea is in line with the fact that homozygous loss‐of‐function variants in the LUBAC components HOIL‐1 and HOIP were identified to result in a syndrome encompassing autoinflammation and immunodeficiency with variable manifestation depending on the site of the mutation (Boisson et al, 2012, 2015; Nilsson et al, 2013; Wang et al, 2013; Oda et al, 2019). Interestingly, the analysis of EBV‐transformed B cells clearly showed that both parental variants affect hydrolysis of linear chains, even in the heterozygous setting, as linear ubiquitin linkages were more abundant in B cells from patient, mother, and father (Fig 5D). This data suggests that even heterozygous variants in the OTULIN gene are sufficient to affect the level of linear ubiquitin linkages, at least in vitro.
The trigger of the autoinflammatory exacerbation in our patient is currently unknown. Another ORAS patient who carried the homozygous OTULIN variant p.L272P died of pneumococcal septicemia (Damgaard et al, 2016). As the urine of our patient was tested positive for pneumococcal antigen (Appendix Table S2), it is tempting to hypothesize that ORAS patients might exhibit increased vulnerability to infections with S. pneumoniae. This is of particular interest as TNF signaling was demonstrated to play a protective role in the pathogenesis of S. pneumoniae infections (Takashima et al, 1997; Wellmer et al, 2001). Thus, altered TNF signaling might result in enhanced susceptibility of ORAS patients to infections with S. pneumoniae. It is therefore reasonable to speculate that they may benefit from pneumococcal vaccinations.
Whereas recently identified patients with ORAS presented with failure to thrive (Damgaard et al, 2016, 2019, 2020; Zhou et al, 2016), this study’s patient suffers from obesity. Whether the patient’s obesity is due to his compound heterozygosity in OTULIN is currently unknown. Known obesity‐related genes (Appendix Table S8; Chesi & Grant, 2015; Rouillard et al, 2016; Warner et al, 2021)) were not affected in the patient apart from FAT1 (FAT atypical cadherin 1) and WWOX (WW Domain Containing Oxidoreductase) (Appendix Fig S2). The single nucleotide variants (SNV) described to be associated with obesity are rs9923451 (16: 1677509940) and rs925642 (4: 187915860) for WWOX and FAT1, respectively (Wang et al, 2011). The patient’s WWOX and FAT1 genes, however, showed a heterozygous SNV resulting in a missense mutation of yet unknown clinical impact. Thus, the underlying cause for the patient’s obesity remains currently uncertain.
In addition, one ORAS patient was identified to suffer from progressive steatotic liver disease (Damgaard et al, 2020) suggesting that OTULIN is crucial for liver homeostasis. This assumption was confirmed in mice with liver‐specific OTULIN deficiency which suffered from steatohepatitis, fibrosis, and hepatocellular carcinoma (Damgaard et al, 2020; Verboom et al, 2020). Although our patient showed elevated liver enzymes during his autoinflammatory episode (Fig EV1A), the laboratory values normalized in the further course and always remained within the normal range during follow‐up. Routine abdominal ultrasound examination, however, revealed that the patient had since developed sonographic alterations of the liver indicative of grade II hepatic steatosis (Fig EV1B) while liver function tests remained normal. Whether the steatosis is caused by the patient’s obesity, a risk factor for liver steatosis (Guzzaloni et al, 2000), or directly related to his ORAS, and further fueled by his treatment with Adalimumab (Haas et al, 2017), cannot be clarified with certainty at the moment. That hepatocytes are vulnerable to changes in the amount of linear ubiquitination is further highlighted by the fact that liver tumorigenesis is also promoted in mice with liver‐specific deletion of HOIP (Shimizu et al, 2017). Future studies will be required to assess how exactly the interplay of OTULIN and LUBAC determines liver health to be able to predict whether OTULIN variants identified in this patient, and others, bear the risk of increased liver tumorigenesis. In any case, ORAS patients should be examined regularly for the development of morphological and functional liver abnormalities.
In summary, we identified compound heterozygous variants in the OTULIN gene to be responsible for an atypical late‐onset ORAS phenotype with severe autoinflammation and sterile abscess formation in a hitherto supposedly asymptomatic patient. We identified variants in the OTULIN gene to affect OTULIN protein expression and to result in increased linear ubiquitin linkage formation under basal conditions, and, importantly also upon stimulation with TNF in the TNFR1‐SC. Whereas the symptoms identified in the patient are distinct from symptoms identified in patients with classical ORAS, we found similarities between classical and atypical ORAS on the molecular level: Monocytes showed enhanced TNF production as reported previously, confirming that TNF secreted from myeloid cells is the primary driver of the inflammation in patients with ORAS (Damgaard et al, 2016, 2019). Moreover, TNF‐induced gene activation was downregulated, but TNF‐mediated cell death enhanced in the presence of CHX in patient‐derived fibroblasts, in line with previous reports (Damgaard et al, 2016, 2019; Zhou et al, 2016).
Our study illustrates that the spectrum of patients with ORAS is wider than so far reported. Upon encountering certain triggers, previously asymptomatic individuals—like the patient described here—may develop a severe and life‐threatening ORAS phenotype.
Materials and Methods
Antibodies
Phospho‐IkBα (9246, IgG1, Cell Signaling Technology (CST), dilution 1:1,000), IκBα (9242, rabbit, CST, dilution 1:1,000), phospho‐ERK (9101, rabbit, CST, dilution 1:2,000), MCL‐1 (94296, rabbit, CST, dilution 1:1,000), Tubulin (2148, rabbit, CST, dilution 1:1,000), Tubulin (12004166, Rhodamine‐labelled, Bio‐Rad, dilution 1:5,000), OTULIN (14127, rabbit, CST, dilution 1:2,000), OTULIN (ab151117, rabbit, Abcam, dilution 1:2,000), Actin (A1987, IgG1, Sigma‐Aldrich (SA), dilution 1:5,000), FLAG (F3165, IgG1, SA, dilution 1:1,000), HOIP (SAB2102031, rabbit, SA, dilution 1:1,000), SHARPIN (14626‐1‐AP, rabbit, Proteintech, dilution 1:2,000), TNFR1 (sc‐8436, IgG2b, Santa Cruz, dilution 1:1,000), RIP1 (610459, IgG2a, BD Bioscience, dilution 1:1,000). Antibody against HOIL‐1 (IgG2a) was previously described (dilution 1:2,000) (Haas et al, 2009; Gerlach et al, 2011). Antibody detecting linear ubiquitin (rabbit) was generated using amino acid sequences as described previously (dilution 1:1,000) (Matsumoto et al, 2012). In brief, FreeStyle CHO‐S cells (Thermo Fisher) were transfected with 100 µg of plasmid (pVITRO1‐Rabbit‐Ubiq‐Ab) using Amaxa Nucleofector. 5 days after transfection, the supernatant was collected, purified with a protein A/G column, and eluted with 0.2 M glycine. 40% of glycerol was added to the eluate before storage at −20°C. pVITRO1‐trastuzumab (Addgene, 61883) was used as backbone plasmid, in which the signal peptide of IL‐2 was added and the variable region substituted with the sequence of interest. Plasmid sequence information is available upon request.
Cell lines and materials
The ethics committee of Ulm University provided guidelines for study procedures. The study complies with relevant ethical regulations. Written informed consent was obtained from all subjects. The patient’s parents gave written informed consent to this study and its publication. This study is a case report with an individual attempt at healing. The principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report have been respected where applicable. Epstein–Barr virus (EBV)‐transformed B cell lines were generated at the Institute of Virology, Ulm University Medical Center, Ulm, Germany. B cell lines were cultured in RPMI 1640 containing 20% FCS, 2% l‐Glutamine and 1% Penicillin/Streptomycin (P/S). Normal Human Dermal Fibroblasts (NHDF) were obtained from Cambrex Bio Science and patient‐derived fibroblasts were generated at the Department of Pediatrics and Adolescent Medicine, Ulm University Medical Center, Ulm, Germany. Fibroblasts were cultured in DMEM (ATCC, 30‐2002) containing 20% FCS and 1% P/S. A549 cells were purchased from Caliper Life Science. A549 OTULIN KO and control cells were derived as previously described (Draber et al, 2015) and cultured in Ham’s F‐12K (Kaighn’s)‐Medium containing 10% FCS and 1% P/S. All cell lines used in this study were determined to be free of mycoplasma using MycoAlert Mycoplasma Detection kit (Lonza). Cell lines were not authenticated. Biological material derived from patient and parents is restricted.
Genomic sequencing
Whole exome sequencing of the patient's genomic DNA was performed using an Illumina sequencing platform. Bioinformatics analysis for detection of rare sequence variants following Mendelian inheritance patterns were performed as described previously (Field et al, 2015). Confirmation of the segregation of the OTULIN variants considered to be pathogenic was obtained by Sanger sequencing of genomic DNA (ABI 3130XL; Thermo Fisher) with the use of Big Dye Terminator (v.1.1) chemistry (Thermo Fisher). Primers used for sequencing were designed using ENSG00000154124.5 as reference sequence.
Histology
Tissue was fixed in 5% buffered formalin (pH 7,4) for 24 h. Paraffin sections of about 3 µm were stained with hematoxylin–eosin; immunohistochemistry with a monoclonal antibody specific for smooth muscle actin (Clone HHF35, IR700, Dako Denmark) was used according to standard protocols on a Dako stainer (Wildermann et al, 2021).
3D structural modeling
The 3D structures of OTULIN variants were predicted by protein structure homology‐modeling using SWISS‐MODEL (Waterhouse et al, 2018) using the known structures 3ZNV and 3ZNZ (Keusekotten et al, 2013) as templates. The structures were visualized and analyzed using the PyMOL software (version 2.4.1; Schroedinger).
Cloning
Wildtype OTULIN DNA was amplified from DNA, and the PCR product was cloned into the BamHI and XhoI site of pcDNA3.1+. Point mutations in the OTULIN‐DNA to generate DNA encoding variants OTULINM86I, OTULINW167S, OTULINM86I;W167S, and OTULINL272P were introduced by site‐directed mutagenesis according to the manufacturer’s instructions (QuikChange II Site‐Directed Mutagenesis Kit, Agilent). Nucleotide sequence information of the primers used for site‐directed mutagenesis is available upon request.
For protein expression the DNA encoding wildtype OTULIN, OTULINC129A, OTULINM86I, OTULINW167S, OTULINM86I/C129A, and OTULINW167S/C129A were cloned into the BamHI and XhoI in a modified pGEX‐4T‐2 (kindly provided by Michael Meister) carrying a sequence encoding a HRV3C protease site. The GST‐OTULIN proteins were expressed in E. coli strain BL21 (DE3). Cells were grown at 37°C in LB medium containing 50 μg/ml ampicillin to an OD600 of 0.8. The culture was cooled to 20°C prior to induction with 400 μM IPTG and harvested 20 h post‐induction.
Purification of OTULIN
For purification of GST‐tagged OTULINWT, OTULINC129A, OTULINM86I, OTULINW167S, OTULINM86I/C129A, and OTULINW167S/C129A, cells were resuspended in lysis buffer (500 mM NaCl, 50 mM Tris–HCl pH 8.0, 2 mM DTT) and disrupted using an LM10 microfluidizer (Microfluidics). The clarified lysate (30,000 g, 30 min, 4°C) containing GST‐OTULIN was applied onto equilibrated GSH‐Sepharose 4B beads (Cytiva) and unbound proteins were removed by washing with 2 column volumes of lysis buffer. OTULIN was released by on‐column cleavage with 0.1 mg HRV3C protease overnight, collected and concentrated using Amicon Ultra Centrifugal Filter Units (Merck). A final polishing step was performed by gel filtration (Superdex 200 XK 16/600, Cytiva) in a buffer containing 150 mM NaCl, 20 mM Tris–HCl pH 8.0, and 2 mM DTT. OTULIN containing fractions were pooled, concentrated, frozen in liquid nitrogen, and stored at −80°C.
Binding assay
Binding between OTULINC129A (catalytically inactive background), OTULINM86I/C129A or OTULINW167S/C129A and di‐ubiquitin chains was analyzed using a Biacore S200 instrument (Cytiva). Di‐ubiquitin chains (UbiQ) were amine‐coupled to the CM5 sensor chip (Cytiva) at a concentration of 10 µg/ml in 10 mM sodium acetate buffer pH 4.0. Concentration series of OTULINC129A, OTULINM86I/C129A, or OTULINW167S/C129A (0.065, 0.125, 0.25, 0.5, 1, 2, 4, and 8 µM) were injected in a multi‐cycle approach onto the sensor chip at a flow rate of 30 µl/min in running buffer (150 mM NaCl, 20 mM HEPES/NaOH pH 7.5, 2 mM DTT, 0.005% Tween 20) at 20°C. The surface was regenerated with 10 mM Glycine pH 2.2 between the runs. Acquired binding curves were double‐referenced against buffer runs and a ligand‐free reference channel. The equilibrium dissociation constants (K d) were calculated from steady‐state measurements using the BIAevaluation program (Cytiva).
Differential scanning fluorimetry (DSF)
OTULINWT, OTULINM86I, and OTULINW167S were subjected to thermal denaturation in a real‐time thermal cycler (qTOWER3 G touch, Analytik Jena) using emission and excitation wavelengths of 490 and 580 nm, respectively. Quintuplicates were measured in 2× SYPRO®‐Orange (SA) at a protein concentration of 4 µM in gel filtration buffer. Experiments were performed in white 96‐well qPCR plates (Sarstedt), which were sealed with adhesive qPCR seal (Sarstedt) and centrifuged at 1,000 g for 1 min and 20°C to remove air bubbles. A temperature gradient from 30 to 90°C was applied with a ramping rate of 1°C/s and a ΔT of 1°C while fluorescence was detected after an equilibration time of 30 s. Curves were referenced against buffer controls and T m values calculated and averaged from the derivatives of the melting curves (‐dRn/dT) as implemented in qPCRsoft 4.0 (Jena Analytik).
Intracellular TNF staining in monocytes
4 × 106 Ficoll‐isolated PBMCs were seeded in a 24‐well plate in 2 ml RPMI‐1640 (+ 10% FCS, 2 mM l‐glutamine, 1 mM Sodium‐Pyruvate, 50 μM β‐Mercaptoethanol) for 4 h in presence of 10 µg/ml Brefeldin A (B7651, SA). Cell surface was stained using CD11b‐PB antibody (dilution 1:200; 301315, BioLegend) for 20 min at 4°C in the dark. Intracellular TNF was stained using TNF‐PE (dilution 1:20; 502909, BioLegend) or IgG1‐PE (dilution 1:20, 400140, BioLegend) as isotype control for 30 min at 4°C in the dark.
Western blot
Cells were lysed in lysis buffer (30 mM Tris–HCl, pH 7.4, 150 mM NaCl, 2 mM EDTA, 2 mM KCl, 10% Glycerol) supplemented with 1% Triton X‐100, 1× complete EDTA‐free protease‐inhibitor mix (Roche), and 1× phosphatase‐inhibitor cocktail 2 (SA). Lysates were denatured in reducing sample buffer before separation by SDS–PAGE (BoltTM 4–12% Bis‐Tris). Membranes were incubated with primary antibodies at 4°C overnight or for 1 h at room temperature (RT). Washing of membranes was performed in 1xPBS containing 0.1% Tween‐20 (SA) for 3 × 10 min prior to incubation with the HRP‐conjugated or StarBright Blue 700 fluorescent secondary antibody for 1 h at RT. Membranes were subsequently developed on a Protec Optimax developing machine or imaged on a ChemiDoc Imaging System.
Transfection
A549 OTULIN KO cells were seeded and transfected on the following day with OTULIN constructs using Lipofectamine 2000 according to the manufacturer’s instructions. After 24 h, cells were lysed and subjected to further experimental analysis.
TNFR1‐SC analysis
Cells were seeded and stimulated with FLAG‐TNF (Draber et al, 2015) as indicated in DMEM with 20% FCS. Cells were washed twice with ice‐cold PBS and harvested in 0.5 ml lysis buffer (100 mM NaCl, 40 mM Tris–HCl, pH 7.5, 1 mM CaCl2, 1 mM MgCl2, and 1× complete EDTA‐free protease‐inhibitor mix) supplemented with 0.5% Triton X‐100. Lysates were cleared by centrifugation at 18,000 g for 20 min. The remaining pellet was resuspended in 0.5 ml of lysis buffer supplemented with 1% Triton X‐100 and 0.1% SDS and placed into an ultrasonic bath for 2 × 20 sec prior to centrifugation at 18,000 g for 20 min. Lysates of centrifugation step 1 and 2 were combined. For immunoprecipitation of FLAG‐TNF and associated proteins, 10 µl M2 beads (A2220, SA) were added to each sample and incubated on an overhead wheel at 4°C overnight. Samples were washed at least five times with lysis buffer containing 1% Triton X‐100. After the last centrifugation step, beads were sucked dry and resuspended in 2× reducing sample buffer to elute proteins. Eluted proteins were separated by SDS–PAGE before being examined by Western blotting.
RNA isolation and qRT–PCR
RNA was isolated with the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. cDNA was synthesized using SuperScript II Reverse Transcriptase (Thermo Fisher) with random primers (Thermo Fisher). qRT–PCR was performed using SsoAdvanced Universal SYBR Green Supermix (Bio‐Rad) on a Bio‐Rad CFX Connect Real‐Time PCR Detection System with the following protocol: 95°C for 30 s, then 40 cycles of 95°C for 5 s followed by 60°C for 30 s.
The following primer sequences were used:
OTULIN #1: TATTGACCGCTTCCGTCTTGC (f), GGGTGAGGAGGTGAGACAGAA (r).
OTULIN #2: GGGGAAGATCCTCCAGACCT (f), CACACCAAACCTCTTTGCGG (r).
GAPDH: GAAGGTGAAGGTCGGAGTC (f), GAAGATGGTGATGGGATTTC (r).
ELISA
Cells were stimulated as indicated for 24 h and supernatants were analyzed with DuoSet ELISA for human IL‐6 (DY206, R&D Systems) according to the manufacturer's instructions.
Cell viability assay
Cell viability was determined using CellTiter‐Glo® assay (G7572, Promega) according to the manufacturer’s instructions.
M1 TUBE
Anti‐M1 TUBE (FLAG, UM606, Life Sensors) was performed according to the manufacturer’s instructions.
DUB assay
DUB assay was performed as previously described (Draber et al, 2015). In brief, M1‐linked ubiquitin chains were isolated from A549 OTULIN KO cells with anti‐M1 TUBE (FLAG, UM606, Life Sensors) according to the manufacturer’s instructions. After washing, dried M2‐beads were resuspended in DUB‐buffer (50 mM HEPES (pH 7.6), 150 mM NaCl, 5 mM DTT) and incubated with recombinant OTULINWT, OTULINM86I, or OTULINW167S at 37°C for 1 h. Reducing sample buffer was added to stop the reaction and proteins were eluted by boiling the beads for 10 min at 90°C. Samples were then further subjected to analysis by Western blot.
Human cytokine array
The Proteome Profiler Human Cytokine Array Kit (ARY005B, R&D) was performed according to the manufacturer’s instructions.
Statistical analysis
Data were analyzed using GraphPad Prism software (version 9.1.2) (San Diego, CA). Statistical significance between groups was determined using appropriate statistical tests. For comparison of two groups, unpaired t‐test or multiple t‐tests corrected for multiple comparisons using the Holm–Sidak method were applied. For comparison of more than two groups, ordinary one‐way ANOVA with Dunnett’s multiple comparisons test or mixed‐effects analysis with Tukey’s multiple comparisons test were used. Normal (Gaussian) distribution of data was assumed. As the sample size was small (always n < 7 biological replicates), this assumption was tested using Shapiro–Wilk test. In all quantitative results, standard deviation (SD) is reported to show the variation around the mean. Equal standard deviations were assumed when performing the statistical analysis. Exact p values are stated in the figure legends.
Author contributions
Julia Zinngrebe: Conceptualization; Investigation; Writing—original draft; Writing—review & editing. Barbara Moepps: Investigation. Thomas Monecke: Investigation. Peter Gierschik: Investigation. Ferdinand Schlichtig: Investigation. Thomas Barth: Investigation. Gudrun Strauss: Investigation. Elena Boldrin: Investigation. Carsten Posovszky: Investigation. Ansgar Schulz: Investigation. Ortraud Beringer: Investigation. Eva Rieser: Investigation. Eva‐Maria Jacobsen: Investigation. Myriam Lorenz: Investigation. Klaus Schwarz: Investigation. Ulrich Pannicke: Investigation. Henning Walczak: Investigation. Dierk Niessing: Investigation. Catharina Schuetz: Conceptualization; Investigation. Pamela Fischer‐Posovszky: Conceptualization; Investigation; Writing—original draft; Writing—review & editing. Klaus‐Michael Debatin: Conceptualization; Writing—original draft; Writing—review & editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
JZ, CS, PF‐P, and K‐MD conceived the study; JZ, BM, TM, PG, FS, TFEB, GS, and EB performed research; CP, AS, OB, E‐MJ, MRL, KS, and UP provided patient samples and data; ER and HW provided reagents; JZ, BM, TM, PG, FS, TB, GS, EB, CP, OB, ER, MRL, KS, UP, HW, DN, CS, PF‐P, K‐MD analyzed and interpreted data; JZ, PF‐P, and K‐MD co‐wrote the manuscript. All authors read and approved the manuscript.
For more information
The portal for rare diseases and orphan drugs: https://www.orpha.net/
The human gene database: https://www.genecards.org/
MedlinePlus: https://medlineplus.gov/genetics/condition/otulipenia/
Genomics England PanelApp: https://panelapp.genomicsengland.co.uk/
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Source Data for Expanded View
Source Data for Figure 2
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
JZ received support from Else Kroener‐Forschungskolleg Ulm (Else Kroener‐Fresenius‐Stiftung), Hertha‐Nathorff‐Programm and Bausteinprogramm of Ulm University and is a fellow of the Margarete von Wrangell‐Programm. EB received support from International Graduate School in Molecular Medicine, Ulm. HW is supported by the AvH Foundation, DFG (SFB1399, and SFB1403–414786233), Wellcome Trust (214342/Z/18/Z), MRC (MR/S00811X/1), and Cancer Research UK (A17341). PF‐P holds a Heisenberg Professorship (German Research Association, Fi1700/7‐1). We thank Linda Wolf (Department of Pediatrics and Adolescent Medicine, Ulm University Medical Center) and Sandra Beier (Institute of Pharmacology and Toxicology, Ulm University) for excellent technical support, and Marlies Just (Institute of Virology, Ulm University Medical Center) for generating EBV‐transformed B cell lines, and Michael Meister for providing us with the modified pGEX‐4T‐2 expression vector.
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
EMBO Mol Med (2022) 14: e14901.
Data availability
Explicit consent for disclosure of entire WES data sets in a public data base had not been obtained. Therefore, there are no such data deposited in external repositories.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR (2010) A method and server for predicting damaging missense mutations. Nat Methods 7: 248–249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aksentijevich I, Zhou Q (2017) NF‐kappaB pathway in autoinflammatory diseases: dysregulation of protein modifications by ubiquitin defines a new category of autoinflammatory diseases. Front Immunol 8: 399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck DB, Aksentijevich I (2019) Biochemistry of autoinflammatory diseases: catalyzing monogenic disease. Front Immunol 10: 101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B, Laplantine E, Prando C, Giliani S, Israelsson E, Xu Z, Abhyankar A, Israël L, Trevejo‐Nunez G, Bogunovic D et al (2012) Immunodeficiency, autoinflammation and amylopectinosis in humans with inherited HOIL‐1 and LUBAC deficiency. Nat Immunol 13: 1178–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisson B, Laplantine E, Dobbs K, Cobat A, Tarantino N, Hazen M, Lidov HGW, Hopkins G, Du L, Belkadi A et al (2015) Human HOIP and LUBAC deficiency underlies autoinflammation, immunodeficiency, amylopectinosis, and lymphangiectasia. J Exp Med 212: 939–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chesi A, Grant SFA (2015) The genetics of pediatric obesity. Trends Endocrinol Metab 26: 711–721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Elliott PR, Swatek KN, Maher ER, Stepensky P, Elpeleg O, Komander D, Berkun Y (2019) OTULIN deficiency in ORAS causes cell type‐specific LUBAC degradation, dysregulated TNF signalling and cell death. EMBO Mol Med 11: e9324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Jolin HE, Allison MED, Davies SE, Titheradge HL, McKenzie ANJ, Komander D (2020) OTULIN protects the liver against cell death, inflammation, fibrosis, and cancer. Cell Death Differ 27: 1457–1474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Damgaard RB, Walker JA, Marco‐Casanova P, Morgan NV, Titheradge HL, Elliott PR, McHale D, Maher ER, McKenzie ANJ, Komander D (2016) The deubiquitinase OTULIN is an essential negative regulator of inflammation and autoimmunity. Cell 166: 1215–1230.e20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Draber P, Kupka S, Reichert M, Draberova H, Lafont E, de Miguel D, Spilgies L, Surinova S, Taraborrelli L, Hartwig T et al (2015) LUBAC‐recruited CYLD and A20 regulate gene activation and cell death by exerting opposing effects on linear ubiquitin in signaling complexes. Cell Rep 13: 2258–2272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott P, Leske D, Hrdinka M, Bagola K, Fiil B, McLaughlin S, Wagstaff J, Volkmar N, Christianson J, Kessler B et al (2016) SPATA2 Links CYLD to LUBAC, activates CYLD, and Controls LUBAC signaling. Mol Cell 63: 990–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott PR, Nielsen SV, Marco‐Casanova P, Fiil BK, Keusekotten K, Mailand N, Freund SM, Gyrd‐Hansen M, Komander D (2014) Molecular basis and regulation of OTULIN‐LUBAC interaction. Mol Cell 54: 335–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field MA, Cho V, Andrews TD, Goodnow CC (2015) Reliably detecting clinically important variants requires both combined variant calls and optimized filtering strategies. PLoS One 10: e0143199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiil BK, Damgaard RB, Wagner SA, Keusekotten K, Fritsch M, Bekker‐Jensen S, Mailand N, Choudhary C, Komander D, Gyrd‐Hansen M (2013) OTULIN restricts Met1‐linked ubiquitination to control innate immune signaling. Mol Cell 50: 818–830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong W‐L et al (2011) Linear ubiquitination prevents inflammation and regulates immune signalling. Nature 471: 591–596 [DOI] [PubMed] [Google Scholar]
- Guzzaloni G, Grugni G, Minocci A, Moro D, Morabito F (2000) Liver steatosis in juvenile obesity: correlations with lipid profile, hepatic biochemical parameters and glycemic and insulinemic responses to an oral glucose tolerance test. Int J Obes Relat Metab Disord 24: 772–776 [DOI] [PubMed] [Google Scholar]
- Haas L, Chevalier R, Major BT, Enders F, Kumar S, Tung J (2017) Biologic agents are associated with excessive weight gain in children with inflammatory bowel disease. Dig Dis Sci 62: 3110–3116 [DOI] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T et al (2009) Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF‐R1 signaling complex and is required for TNF‐mediated gene induction. Mol Cell 36: 831–844 [DOI] [PubMed] [Google Scholar]
- Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, Maltzman A, de Sousa e Melo F, Hung J, Zeng YI et al (2018) OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature 559: 120–124 [DOI] [PubMed] [Google Scholar]
- Hrdinka M, Fiil B, Zucca M, Leske D, Bagola K, Yabal M, Elliott P, Damgaard R, Komander D, Jost P et al (2016) CYLD limits Lys63‐ and Met1‐linked ubiquitin at receptor complexes to regulate innate immune signaling. Cell Rep 14: 2846–2858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda F, Deribe YL, Skanland SS, Stieglitz B, Grabbe C, Franz‐Wachtel M, van Wijk SJ, Goswami P, Nagy V, Terzic J et al (2011) SHARPIN forms a linear ubiquitin ligase complex regulating NF‐kappaB activity and apoptosis. Nature 471: 637–641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahan AS, Elbaek CR, Damgaard RB (2021) Met1‐linked ubiquitin signalling in health and disease: inflammation, immunity, cancer, and beyond. Cell Death Differ 28: 473–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keusekotten K, Elliott P, Glockner L, Fiil B, Damgaard R, Kulathu Y, Wauer T, Hospenthal M, Gyrd‐Hansen M, Krappmann D et al (2013) OTULIN antagonizes LUBAC signaling by specifically hydrolyzing Met1‐linked polyubiquitin. Cell 153: 1312–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, Sano S, Tokunaga F, Tanaka K, Iwai K (2006) A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J 25: 4877–4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovalenko A, Chable‐Bessia C, Cantarella G, Israel A, Wallach D, Courtois G (2003) The tumour suppressor CYLD negatively regulates NF‐kappaB signalling by deubiquitination. Nature 424: 801–805 [DOI] [PubMed] [Google Scholar]
- Kupka S, De Miguel D, Draber P, Martino L, Surinova S, Rittinger K, Walczak H (2016) SPATA2‐mediated binding of CYLD to HOIP enables CYLD recruitment to signaling complexes. Cell Rep 16: 2271–2280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manthiram K, Zhou Q, Aksentijevich I, Kastner DL (2017) The monogenic autoinflammatory diseases define new pathways in human innate immunity and inflammation. Nat Immunol 18: 832–842 [DOI] [PubMed] [Google Scholar]
- Matsumoto ML, Dong KC, Yu C, Phu L, Gao X, Hannoush RN, Hymowitz SG, Kirkpatrick DS, Dixit VM, Kelley RF (2012) Engineering and structural characterization of a linear polyubiquitin‐specific antibody. J Mol Biol 418: 134–144 [DOI] [PubMed] [Google Scholar]
- McLaren W, Gil L, Hunt SE, Riat HS, Ritchie GR, Thormann A, Flicek P, Cunningham F (2016) The ensembl variant effect predictor. Genome Biol 17: 122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer EA, Castellano RK, Diederich F (2003) Interactions with aromatic rings in chemical and biological recognition. Angew Chem Int Ed 42: 1210–1250 [DOI] [PubMed] [Google Scholar]
- Nabavi M, Shahrooei M, Rokni‐Zadeh H, Vrancken J, Changi‐Ashtiani M, Darabi K, Manian M, Seif F, Meyts I, Voet A et al (2019) Auto‐inflammation in a patient with a novel homozygous OTULIN mutation. J Clin Immunol 39: 138–141 [DOI] [PubMed] [Google Scholar]
- Nilsson J, Schoser B, Laforet P, Kalev O, Lindberg C, Romero NB, Dávila López M, Akman HO, Wahbi K, Iglseder S et al (2013) Polyglucosan body myopathy caused by defective ubiquitin ligase RBCK1. Ann Neurol 74: 914–919 [DOI] [PubMed] [Google Scholar]
- Oda H, Beck DB, Kuehn HS, Sampaio Moura N, Hoffmann P, Ibarra M, Stoddard J, Tsai WL, Gutierrez‐Cruz G, Gadina M et al (2019) Second case of HOIP deficiency expands clinical features and defines inflammatory transcriptome regulated by LUBAC. Front Immunol 10: 479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peltzer N, Darding M, Walczak H (2016) Holding RIPK1 on the ubiquitin leash in TNFR1 signaling. Trends Cell Biol 26: 445–461 [DOI] [PubMed] [Google Scholar]
- Rivkin E, Almeida SM, Ceccarelli DF, Juang Y‐C, MacLean TA, Srikumar T, Huang H, Dunham WH, Fukumura R, Xie G et al (2013) The linear ubiquitin‐specific deubiquitinase gumby regulates angiogenesis. Nature 498: 318–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard AD, Gundersen GW, Fernandez NF, Wang Z, Monteiro CD, McDermott MG, Ma'ayan A (2016) The harmonizome: a collection of processed datasets gathered to serve and mine knowledge about genes and proteins. Database 2016: baw100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicher L, Wissler M, Preiss F, Brauns‐Schubert P, Jakob C, Dumit V, Borner C, Dengjel J, Maurer U (2016) SPATA2 promotes CYLD activity and regulates TNF‐induced NF‐kappaB signaling and cell death. EMBO Rep 17: 1485–1497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu Y, Peltzer N, Sevko A, Lafont E, Sarr A, Draberova H, Walczak H (2017) The Linear ubiquitin chain assembly complex acts as a liver tumor suppressor and inhibits hepatocyte apoptosis and hepatitis. Hepatology 65: 1963–1978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim NL, Kumar P, Hu J, Henikoff S, Schneider G, Ng PC (2012) SIFT web server: predicting effects of amino acid substitutions on proteins. Nucleic Acids Res 40: W452–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spit M, Rieser E, Walczak H (2019) Linear ubiquitination at a glance. J Cell Sci 132: jcs208512. 10.1242/jcs208512 [DOI] [PubMed] [Google Scholar]
- Takashima K, Tateda K, Matsumoto T, Iizawa Y, Nakao M, Yamaguchi K (1997) Role of tumor necrosis factor alpha in pathogenesis of pneumococcal pneumonia in mice. Infect Immun 65: 257–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, Iwai K (2011) SHARPIN is a component of the NF‐kappaB‐activating linear ubiquitin chain assembly complex. Nature 471: 633–636 [DOI] [PubMed] [Google Scholar]
- Verboom L, Hoste E, van Loo G (2021) OTULIN in NF‐kappaB signaling, cell death, and disease. Trends Immunol 42: 590–603 [DOI] [PubMed] [Google Scholar]
- Verboom L, Martens A, Priem D, Hoste E, Sze M, Vikkula H, Van Hove L, Voet S, Roels J, Maelfait J et al (2020) OTULIN prevents liver inflammation and hepatocellular carcinoma by inhibiting FADD‐ and RIPK1 kinase‐mediated hepatocyte apoptosis. Cell Rep 30: 2237–2247.e6 [DOI] [PubMed] [Google Scholar]
- Wagner SA, Satpathy S, Beli P, Choudhary C (2016) SPATA2 links CYLD to the TNF‐alpha receptor signaling complex and modulates the receptor signaling outcomes. EMBO J 35: 1868–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Kim C, Bradfield J, Guo Y, Toskala E, Otieno FG, Hou C, Thomas K, Cardinale C, Lyon GJ et al (2013) Whole‐genome DNA/RNA sequencing identifies truncating mutations in RBCK1 in a novel Mendelian disease with neuromuscular and cardiac involvement. Genome Med 5: 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Li WD, Zhang CK, Wang Z, Glessner JT, Grant SF, Zhao H, Hakonarson H, Price RA (2011) A genome‐wide association study on obesity and obesity‐related traits. PLoS One 6: e18939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner ET, Jiang L, Adjei DN, Turman C, Gordon W, Wang L, Tamimi R, Kraft P, Lindstrom S (2021) A genome‐wide association study of childhood body fatness. Obesity 29: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TA P, Rempfer C, Bordoli L et al (2018) SWISS‐MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res 46: W296–W303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinelt N, van Wijk SJL (2021) Ubiquitin‐dependent and ‐independent functions of OTULIN in cell fate control and beyond. Cell Death Differ 28: 493–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellmer A, Gerber J, Ragheb J, Zysk G, Kunst T, Smirnov A, Bruck W, Nau R (2001) Effect of deficiency of tumor necrosis factor alpha or both of its receptors on Streptococcus pneumoniae central nervous system infection and peritonitis. Infect Immun 69: 6881–6886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk SJL, Fricke F, Herhaus L, Gupta J, Hotte K, Pampaloni F, Grumati P, Kaulich M, Sou YS, Komatsu M et al (2017) Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF‐kappaB and restricts bacterial proliferation. Nat Microbiol 2: 17066 [DOI] [PubMed] [Google Scholar]
- Wildermann C, Alosaimi M, Liebenehm S, Jacobsen E‐M, Barth TFE, Möller P, Debatin K‐M, Schulz A, Sirin M, Abosoudah IF et al (2021) Successful hematopoietic stem cell transplantation in a 4–1BB deficient patient with EBV‐induced lymphoproliferation. Clin Immunol 222: 108639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Luo Q, Liu Z (2020) Ubiquitination and deubiquitination of MCL1 in cancer: deciphering chemoresistance mechanisms and providing potential therapeutic options. Cell Death Dis 11: 556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii SR, Mizushima N (2017) Monitoring and measuring autophagy. Int J Mol Sci 18: 1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Yu X, Demirkaya E, Deuitch N, Stone D, Tsai WL, Kuehn HS, Wang H, Yang D, Park YH et al (2016) Biallelic hypomorphic mutations in a linear deubiquitinase define otulipenia, an early‐onset autoinflammatory disease. Proc Natl Acad Sci USA 113: 10127–10132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinngrebe J, Montinaro A, Peltzer N, Walczak H (2014) Ubiquitin in the immune system. EMBO Rep 15: 28–45 [DOI] [PMC free article] [PubMed] [Google Scholar]