

Abstract
Multiple myeloma (MM) is an incurable malignancy characterized by the uncontrolled expansion of plasma cells in the bone marrow. While proteasome inhibitors like bortezomib efficiently halt MM progression, drug resistance inevitably develop, and novel therapeutic approaches are needed. Here, we used a recently discovered Sec61 inhibitor, mycolactone, to assess the interest of disrupting MM proteostasis via protein translocation blockade. In human MM cell lines, mycolactone caused rapid defects in secretion of immunoglobulins and expression of pro‐survival interleukin (IL)‐6 receptor and CD40, whose activation stimulates IL‐6 production. Mycolactone also triggered pro‐apoptotic endoplasmic reticulum stress responses synergizing with bortezomib for induction of MM cell death and overriding acquired resistance to the proteasome inhibitor. Notably, the mycolactone–bortezomib combination rapidly killed patient‐derived MM cells ex vivo, but not normal mononuclear cells. In immunodeficient mice engrafted with MM cells, it demonstrated superior therapeutic efficacy over single drug treatments, without inducing toxic side effects. Collectively, these findings establish Sec61 blockers as novel anti‐MM agents and reveal the interest of targeting both the translocon and the proteasome in proteostasis‐addicted tumors.
Keywords: multiple myeloma, proteostatic stress, Sec61 translocon
Subject Categories: Cancer, Haematology
In this work, a pathogen‐derived inhibitor named mycolactone was used to demonstrate that the Sec61 translocon is a therapeutic target in Multiple Myeloma (MM).

The paper explained.
Problem
Multiple myeloma is an incurable blood cancer. While the majority of patients initially respond to combination chemotherapies, all eventually develop drug resistance or toxicity and their mean survival rate is only 6 years post‐diagnosis.
Results
Human cells interact with their environment via proteins that are either expressed on their surface or secreted into the extracellular environment. The process of delivering these proteins to the membrane wall or outside – the secretion pathway – uses a dedicated distribution network whose gateway is the Sec61 translocon. We recently found that mycolactone, a toxin produced by a bacterial pathogen, operates by blocking Sec61. Using mycolactone we demonstrate in the present study that blocking Sec61 is much more toxic to multiple myeloma cells than to normal cells in vitro and in vivo. Moreover, we show that mycolactone potentiates the activity of drugs forming the backbone of current multiple myeloma chemotherapies.
Impact
Our findings identify Sec61 as a therapeutic vulnerability in multiple myeloma, and potentially all other cancers needing an active Sec61 translocon to survive. They suggest that treating MM patients with agents blocking Sec61 will augment the efficacy of – and overcome resistance to – current chemotherapies.
Introduction
Multiple myeloma (MM) is a malignant disorder characterized by the uncontrolled expansion of clonal plasma cells in the bone marrow, eventually leading to organ dysfunction and death (Laubach et al, 2011). The global burden of MM has increased by 126% over the past 30 years, with highest incidences in developed countries (Cowan et al, 2018). The introduction of proteasome inhibitors (PIs) like bortezomib (BZ) in MM chemotherapies has revolutionized MM management, and combinations of BZ with immunomodulators (IMiDs) and dexamethasone have become the gold standard for MM treatment (Kouroukis et al, 2014). However, BZ can induce severe side effects such as peripheral neuropathy, requiring discontinuation of therapy (Lawasut et al, 2012). Moreover, many patients develop resistance to BZ whose molecular basis remains poorly understood, and less than half of them survive beyond 5 years post‐diagnosis. Second generation PIs were recently developed to treat patients who are resistant or intolerant to BZ (Gandolfi et al, 2017), but these drugs induce other types of side effects and do not prevent relapse. Therefore, despite significant therapeutic advances, MM remains an incurable disease and the identification of new therapeutic targets is critically needed.
The clinical efficacy of BZ is primarily attributed to its ability to induce the accumulation of misfolded proteins including immunoglobulins in the cytoplasm of MM cells, leading to lethal proteotoxic stress (Gandolfi et al, 2017). BZ also alters the survival and proliferation of MM cells in several other ways, such as inhibition of NF‐κB oncogenic signaling, suppression of pro‐adhesive cross‐talks with bone marrow stromal cells, and prevention of growth stimulation by cytokines like interleukin (IL)‐6 (Mahindra et al, 2010; Rosean et al, 2014). Among the different strategies that are currently explored to complement or replace PIs in MM chemotherapies are novel means to disrupt the homeostatic regulation of the secretory apparatus via proteolytic routes or endoplasmic reticulum (ER)‐associated protein degradation (ERAD) components other than the proteasome (Auner & Cenci, 2015). Despite its central importance in MM cell biology, the interest of targeting immunoglobulin transport into the ER has not yet been explored due to the lack of suitable inhibitor.
We reported recently that mycolactone, a diffusible lipid toxin secreted by the human pathogen Mycobacterium ulcerans, operates by inhibiting the mammalian translocon (Sec61) (Baron et al, 2016; Demangel & High, 2018). By targeting the central pore‐forming subunit of Sec61 (Sec61α), mycolactone prevents the import of newly synthesized secreted and transmembrane proteins into the ER, leading to their cytosolic degradation by the proteasome (Hall et al, 2014; McKenna et al, 2016, 2017). Contrary to the Sec61 inhibitor cotransin (Mackinnon et al, 2014), mycolactone is not substrate selective and blocks the translocation of the vast majority of Sec61 clients (Baron et al, 2016; Morel et al, 2018). The only substrates resisting its inhibitory action are transmembrane proteins belonging to the rare Type III subgroup (McKenna et al, 2016, 2017; Demangel & High, 2018; Morel et al, 2018). Within 1 h of treatment, mycolactone‐treated cells become defective for production of most secreted proteins and membrane‐anchored receptors. If sustained, mycolactone treatment triggers proteotoxic stress responses in cytosol and ER, ultimately inducing apoptosis (Morel et al, 2018; Ogbechi et al, 2018). Notably, a point mutation (R66G) in the Sec61α amino acid sequences preventing mycolactone binding without affecting the translocon functionality was sufficient to prevent stress responses and associated cytotoxicity, demonstrating that Sec61 inhibition by mycolactone is the molecular mechanism driving cell death (Baron et al, 2016).
Based on these findings, we hypothesized that Sec61 blockade may suppress survival and growth of MM cells in two ways: by preventing the expression of membrane receptors that are key to MM cell division and dissemination, and by generating lethal proteotoxic stress. The present study uses mycolactone as a prototypical Sec61 blocker to establish proof of concept, and evaluates the translational potential of Sec61 inhibitors in the treatment of MM.
Results
Sec61 blockade by mycolactone alters the biology and viability of MM cell lines
To assess the effect of mycolactone on MM cell viability, three human cell lines (MM.1S, JIM3, and KMS‐11) were treated with increasing concentrations of mycolactone for 24–72 h, and the induction of cell apoptosis was monitored by phosphatidylserine exposure (Annexin V staining) and loss of membrane integrity (PI staining) (Appendix Fig S1). While the three cell lines differed in sensitivity to mycolactone, MM.1S being the most resistant, a dose‐ and time‐dependent induction of apoptotic cell death was consistently observed after 48 h of treatment (Fig 1A). In all cell lines, initiation of apoptosis was preceded by a decrease in surface expression of the MM cell marker CD38 (Van De Donk et al, 2018) (Fig 1B). The plasma cell marker CD138 supports MM cell survival in the bone marrow by promoting growth factor signaling (Akhmetzyanova et al, 2020). While not detected on JIM3 and KMS‐11, CD138 expression by MM.1S was also dose dependently decreased by mycolactone after 24 h (Fig 1B). IL‐6 receptor (IL‐6R) and CD40 are two other MM cell markers whose signaling play a crucial role in MM development and dissemination (Tai et al, 2005; Rosean et al, 2014). When expressed by the MM cell lines, these receptors were also rapidly and potently downregulated by mycolactone treatment (Fig 1B).
Figure 1. Sec61 blockade by mycolactone strongly alters the biology and viability of MM cell lines.
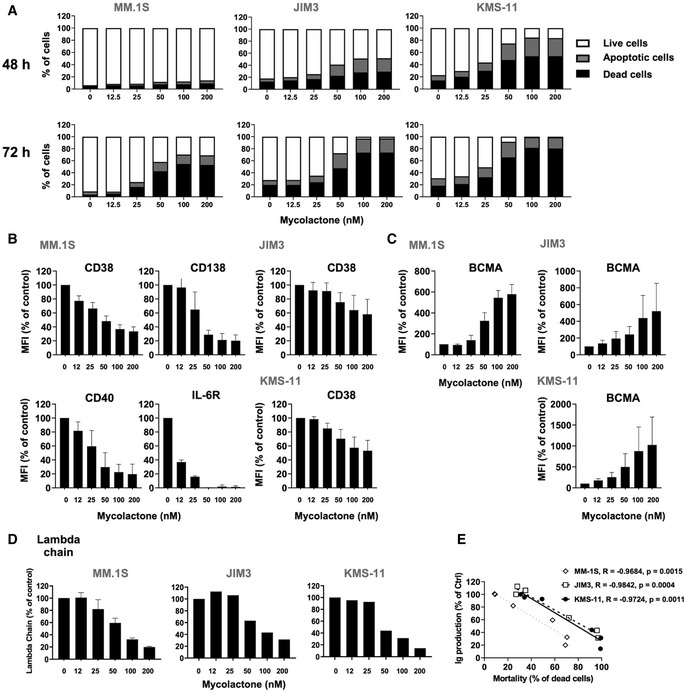
-
AEffect of mycolactone on MM.1S, JIM3, and KMS‐11 cell viability after 48 and 72 h of treatment, as measured by exposure of Annexin V and incorporation of PI. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, gated as in Appendix Fig S1, relative to total cells.
-
B, CInhibitory effect of mycolactone on cell expression of CD38 (Type II) and CD138, IL‐6R, and CD40 (Type I) transmembrane proteins (B). Stimulatory effect of mycolactone on cell expression of BCMA, a Type III transmembrane protein (C). Mean fluorescence intensities (MFIs) were measured in live cells 48 h after addition of mycolactone. Data are Mean MFIs ± SD from technical duplicates of two independent experiments, relative to vehicle‐treated controls (Ctrl).
-
DInhibitory effect of mycolactone on immunoglobulin (lambda chain) secretion, as quantified by ELISA in culture supernatants after 24 h of treatment. Data are Mean concentrations ± SD of secreted lambda chain (µg/ml) from technical duplicates of two independent experiments (MM.1S) or Mean of secreted lambda chain concentrations (µg/ml) from technical duplicates (JIM3 and KMS‐11), relative to vehicle‐treated controls.
-
ECorrelation between immunoglobulin lambda chain (Ig) secretion after 24 h (D) and MM cell line mortality after 72 h (A), in the three cell lines treated with 50 nM mycolactone. Slopes (R) and statistical significance (P values) are indicated.
Data information: In A (all cell lines) and D (JIM3 and KMS‐11), shown data are representative of two independent experiments with similar results.
In contrast to CD38, CD138, IL‐6R, and CD40, all Type I or II transmembrane proteins efficiently blocked in translocation by mycolactone; Type III transmembrane proteins are Sec61 substrates resisting mycolactone inhibition (Baron et al, 2016; McKenna et al, 2016, 2017; Morel et al, 2018). B‐cell maturation antigen (BCMA) is a Type III protein that is typically expressed by MM cells and the target of next‐generation immunotherapies (Cho et al, 2020). In all cell lines, mycolactone dose dependently increased cell surface expression of BCMA (Fig 1C). On the opposite, secreted proteins are Sec61 substrates that are efficiently blocked in translocation by mycolactone (Baron et al, 2016; McKenna et al, 2016, 2017; Morel et al, 2018). Figure 1D shows that Sec61 inhibition efficiently decreased MM cell line secretion of immunoglobulin light chains after only 24 h of treatment, and this reduction closely correlated with the onset of cell death after 72 h (Fig 1E). In conclusion, Sec61 blockade by mycolactone induces programmed cell death in MM preceded by phenotypic defects in expression of Type I/II membrane receptors and secretion of immunoglobulins.
Mycolactone synergizes with proteasome inhibitors for induction of MM cell apoptosis
The clinical efficacy of BZ is believed to result from its capacity to induce unresolvable proteotoxic stress in MM cells via accumulation of misfolded proteins in the cytoplasm (Gandolfi et al, 2017). Since Sec61 substrates blocked in translocation by mycolactone are diverted to the proteasome for degradation (Hall et al, 2014), we reasoned that mycolactone may potentiate the anti‐MM activity of BZ through generation of additional proteotoxic stress. While mycolactone‐mediated cytotoxicity did not manifest before 48 h of treatment (Fig 1A), BZ induced significant mortality in all MM cell lines after only 24 h (Fig 2A). Cell lines nevertheless differed in susceptibility to BZ cytotoxicity, JIM3 and KMS‐11 being relatively more resistant than MM.1S. To assess a potential synergy between mycolactone and BZ, MM cells were treated for 24 h with subtoxic doses of BZ together with increasing concentrations of mycolactone, and induction of apoptosis was assessed after 24 h. In all cell lines, the mycolactone–BZ combination induced more cell death than single drugs (Figs 2B and EV1). Heatmaps of synergy scores (Di Veroli et al, 2016) show that mycolactone synergized with BZ in all cell lines, irrespective of their basal resistance to BZ (Fig 2C). Similar findings were obtained with MM.1S cells treated with carfilzomib, a second‐generation PI that is proposed to BZ‐resistant MM patients (Fig 2D and E).
Figure 2. Mycolactone synergizes with PIs for induction of MM cell apoptosis.
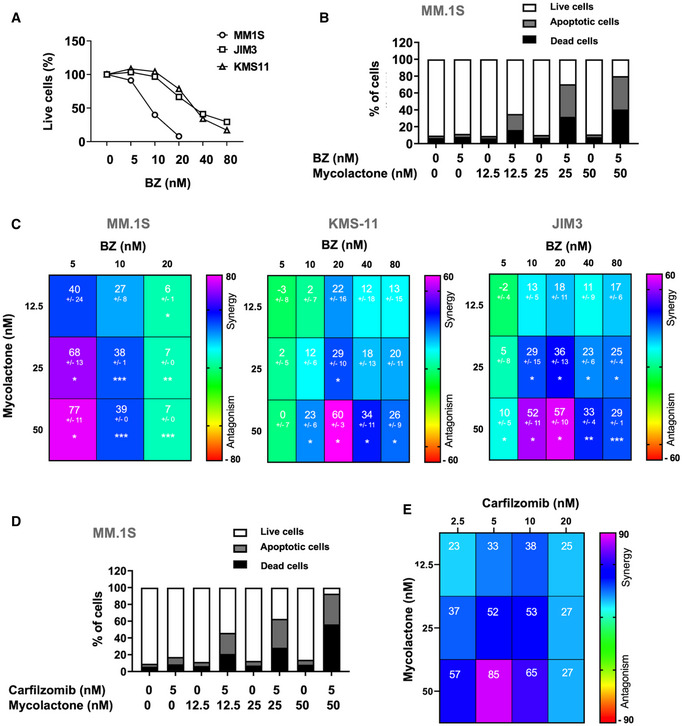
-
AMM.1S, JIM3, and KMS‐11 cells were incubated with increasing concentrations of BZ for 24 h. The proportion of live (Annexin V‐ PI‐) cells was then measured by flow cytometry as in Appendix Fig S1. Data are Mean % of live cells from technical duplicates, relative to vehicle controls (Ctrl).
-
BMM.1S were treated for 24 h with mycolactone and/or BZ at the indicated concentrations. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, gated as in Appendix Fig S1, relative to total cells.
-
CSynergy between mycolactone and BZ in the MM.1S, JIM3, and KMS‐11 cell lines, when treated as in (B). Data are Mean Loewe scores ± SD shown as heatmaps. N = 6 (cumulative data of three independent experiments with technical duplicates) for MM.1S and KMS‐11, and N = 4 (cumulative data of two independent experiments with technical duplicates) for JIM3. Statistical significance was assessed by Student’s t‐test: *P < 0.05; **P < 0.01; ***P < 0.001.
-
D, EMM.1S cell were treated as in (B) but using carfilzomib instead of BZ (D). Synergy scores (E) were calculated as in (C). Data are Mean Loewe scores of technical duplicates, shown as heatmaps.
Data information: In A and B, shown data are representative of two and three independent experiments with similar results, respectively.
Figure EV1. Mycolactone potentiates the effect of BZ in JIM3 and KMS‐11.
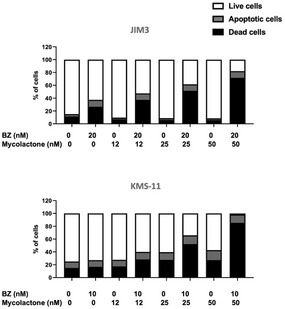
JIM3 and KMS‐11 cells were treated with mycolactone and/or BZ at the indicated concentrations for 24 h. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, gated as in Appendix Fig S1, relative to total cells. They are representative of two independent experiments with similar results.
The mycolactone–BZ combination activates pro‐apoptotic UPR in MM cells
We next sought to determine if the synergistic induction of apoptosis in MM cell lines treated with mycolactone and BZ correlated with enhanced ER stress. The unfolded protein response (UPR) is activated by three ER‐resident transmembrane sensor proteins: protein kinase RNA‐like ER kinase (PERK), inositol requiring enzyme 1 (IRE1), and activating transcription factor (ATF) 6 (Fig 3A) (Hetz & Papa, 2018). Activation of PERK leads to the phosphorylation of eIF2α, with p‐eIF2α reprogramming protein translation to promote the expression of stress response mRNAs such as ATF4. ATF4 subsequently drives the expression of genes alleviating stress such as the growth arrest and DNA damage‐inducible protein (GADD34), which promotes p‐eIF2α dephosphorylation in a negative feedback loop. Activation of IRE1 triggers the splicing of X‐box‐binding protein 1 (XBP1) mRNA into its transcriptionally active form (sXBP1), able to transcriptionally reprogram UPR target genes. Chronic stress promotes transition from adaptive to terminal UPR, marked by ATF4‐mediated induction of pro‐apoptotic C/EBP homology protein (CHOP) expression and accentuated by pro‐death signals originating from IRE1 signaling. Activation of ATF6 leads to its cleavage into an active form in the Golgi, which contributes to upregulate CHOP.
Figure 3. The mycolactone–BZ combination activates pro‐apoptotic UPR in MM.1S cells.
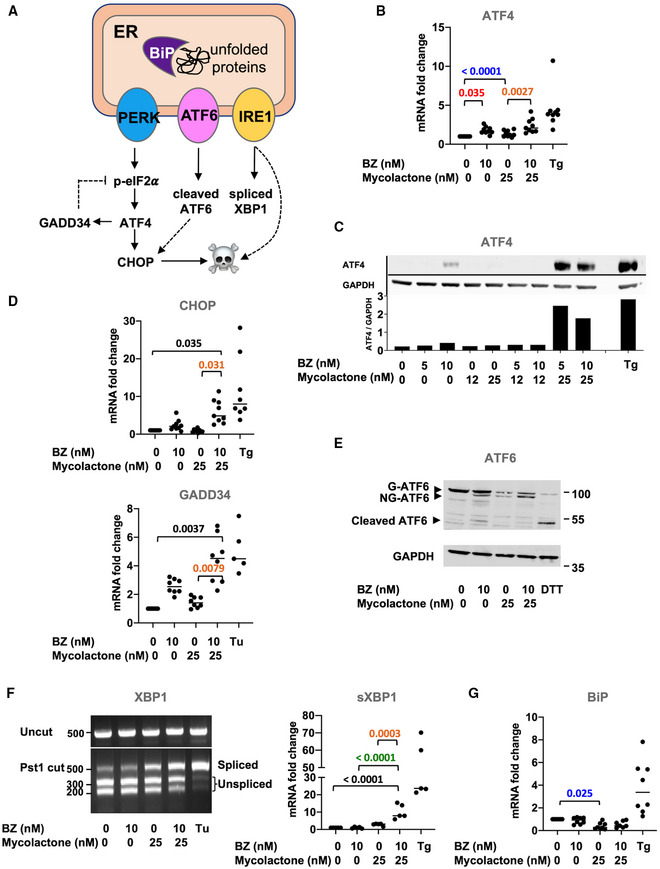
-
ADiagram illustrating the pro‐apoptotic pathways activated by UPR sensors PERK, ATF6, and IRE1 upon accumulation of unfolded proteins in the ER lumen.
-
B–GIn MM.1S cells treated with mycolactone and/or BZ at the indicated concentrations, or vehicle as control for 6 h: (B) ATF4 mRNA levels were quantified by qPCR; (C) ATF4 protein levels in cell lysates were assessed by Western blot (top panel) and quantified relatively to GADPH levels (lower panel); (D) CHOP and GADD34 mRNA levels were quantified by qPCR; (E) ATF6 and GAPDH protein levels in cell lysates were assessed by Western blot with molecular weight markers (kDa) indicated on the right; Bands corresponding to glycosylated (G‐ATF6), non‐glycosylated (NG‐ATF6), and cleaved ATF6 are indicated by arrows; (F) Total RNA was isolated and used as a template for RT‐PCR of XBP‐1 (upper panel), which was then digested with Pst1 and separated on a 2% agarose gel (lower panel) with molecular weight markers (bp) indicated on the left; spliced XBP1 (sXBP1) mRNA levels were also quantified by qPCR and normalized to total (spliced + unspliced) XBP1 mRNA levels, and (G) BiP mRNA level was quantified by qPCR.
Data information: Shown mRNA data are mean fold changes (2−ΔΔCT) ± SD, relative to untreated controls (cumulative data of at least two independent experiments with technical duplicates or triplicates), pairwise compared by nested one‐way ANOVA with Tukey’s multiple‐comparison test, exact P‐values indicated. Thapsigargin (Tg, 2 µM, 6 h), tunicamycin (Tu, 2 µM, 6 h), or DTT (4 mM, 2 h) were used as positive controls. In C, E, and F, shown data are representative of two independent experiments with similar results.
In all MM cell lines treated with subcytotoxic concentrations of mycolactone and BZ for 6 h, we detected an increased expression of ATF4, at both the mRNA and protein levels, compared to single drug treatments (Figs 3B and C, and EV2A). Elevated ATF4 correlated with enhanced expression of its CHOP target (Figs 3D and EV2B), reflecting the activation of pro‐apoptotic eIF2α/ATF4/CHOP signaling. It was also associated with the induction of ATF4 target GADD34 (Fig 3D), suggesting that dephosphorylation of p‐eIF2α is induced in treated MM cells. In MM.1S cells treated with BZ for 6 h, we also observed defects in ATF6 glycosylation and partial cleavage of the protein (Fig 3E). In agreement with previous reports (Ogbechi et al, 2018), mycolactone did not induce such ATF6 cleavage. However, defective glycosylation and depletion of full‐length ATF6 were observed, likely resulting from mycolactone‐mediated blockade of this Type II transmembrane protein in translocation. MM1.S cells exposed to the mycolactone–BZ combination displayed the sum of the alterations induced by each drug, namely depletion, defective glycosylation, and partial cleavage of ATF6. Finally, gel electrophoresis and qPCR analyses demonstrated an enhanced XBP1 splicing in MM1.S cells treated with the mycolactone/BZ combination, compared to single drugs (Fig 3F). A significant upregulation of sXBP1 mRNA levels was also observed in KMS‐11 cells exposed to the drug combination (Fig EV2B). From these data, we conclude that the mycolactone–BZ combination hyperactivates PERK and IRE1 signaling in MM cells, with potent activation of the ATF4/CHOP axis indicating a transition to terminal UPR. The ER‐resident chaperone BiP is a master regulator of the UPR that is normally induced by ER stress and determines the threshold for induction of apoptosis (Hetz & Papa, 2018). It is interesting to note that BiP expression was not augmented in MM cells treated with mycolactone, alone or combined to BZ (Figs 3G and EV2B).
Figure EV2. The mycolactone–BZ combination activates pro‐apoptotic UPR in KMS‐11 and JIM3 cells.
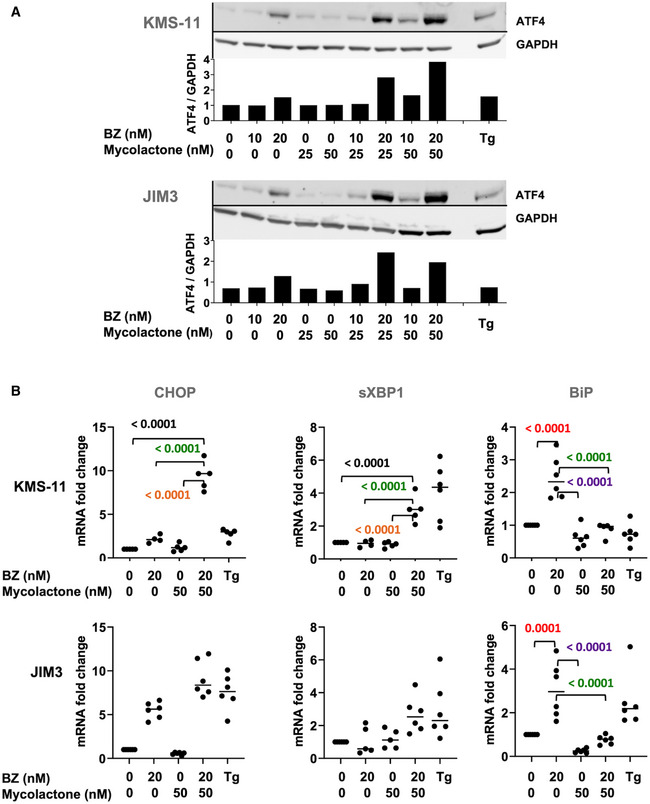
- KMS‐11 and JIM3 cells were treated with mycolactone and/or BZ, or vehicle as control, for 6 h. ATF4 protein levels were assessed in cell lysates by Western blot (top panel) and quantified relative to GADPH levels (lower panel). Data are representative of two independent experiments with similar results.
- CHOP, sXBP1, and BiP mRNA levels were quantified by qPCR in the two cell lines treated as in (A). sXBP1 mRNA levels were normalized to total (spliced + unspliced) XBP1 mRNA level. Data are Mean RNA fold changes (2−ΔΔCT) ± SD, relative to untreated controls. N ≥ 5 (cumulative data of two independent experiments with technical duplicates and triplicates), pairwise compared by nested one‐Way ANOVA with Tukey’s multiple‐comparison test, exact P‐values indicated. Thapsigargin (Tg, 2 µM, 6 h) was used as a positive control.
Source data are available online for this figure.
The cytotoxic synergy between mycolactone and BZ is maintained in BZ‐resistant MM.1S cells
The development of resistance to PIs is a major obstacle to successful treatment of MM patients (Lawasut et al, 2012). To determine if the cytotoxic synergy between mycolactone and BZ was maintained in BZ‐resistant MM cells, we generated a BZ‐resistant version of the MM.1S cell line (MM.1S BzR cells), with stable and > 15× increased resistance to BZ treatment (Fig 4A). Notably, MM.1S BzR cell susceptibility to mycolactone was identical to that of MM.1S BzS (Figs 4B and 1A), showing that acquired resistance to BZ does not confer cross‐resistance to mycolactone. As in MM.1S BzS cells (Fig 2C), mycolactone synergized with BZ for induction of MM.1S BzR cell death (Fig 4C and D) and engagement of apoptosis in MM.1S BzR cells exposed to the mycolactone–BZ combination correlated with activation of ER stress responses, reflected by elevated levels of ATF4, CHOP, and sXBP1 mRNAs (Fig 4E). Like MM.1S BzS cells, MM.1S BzR cells failed to upregulate BiP expression upon exposure to the mycolactone/BZ drug combination (Fig 4E). Together, these data demonstrate that mycolactone cytotoxicity and synergy with BZ override MM cell resistance to BZ. They consolidate the clinical relevance of Sec61 inhibitor‐based treatments for MM.
Figure 4. The cytotoxic synergy between mycolactone and BZ is maintained in BZ‐resistant cells.
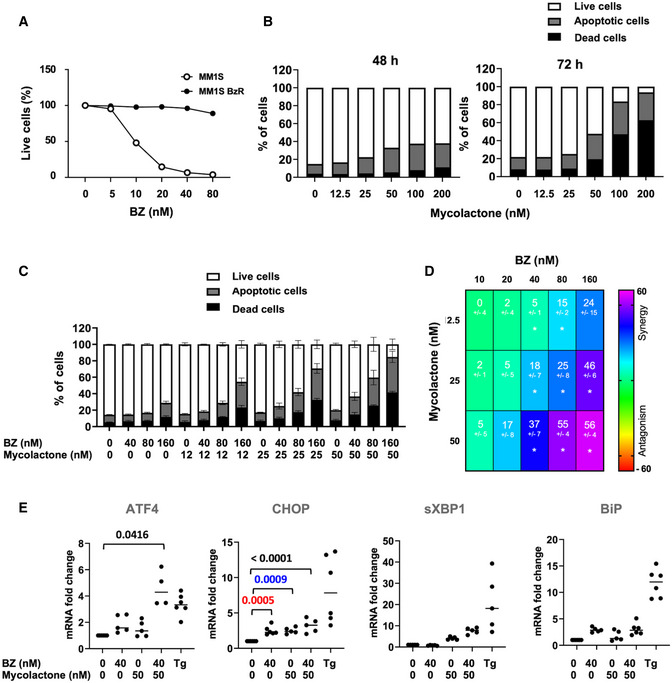
- Parental MM.1S (BzS) and MM.1S BzR cells were incubated with increasing concentrations of BZ for 24 h. The proportion of live (Annexin V‐ PI‐) cells was then measured by flow cytometry as in Appendix Fig S1. Data are Mean % of live cells from technical duplicates, relative to vehicle controls.
- Effect of mycolactone on MM.1S BzR cell viability after 48 and 72 h of treatment, as measured by exposure of Annexin V and incorporation of PI. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, gated as in Appendix Fig S1, relative to total cells.
- MM.1S BzR cells were treated for 24 h with mycolactone and/or BZ at the indicated concentrations, or vehicle as control. Data are Mean % ± SD of live, apoptotic, and dead cells, gated as in Appendix Fig S1, relative to total cells. N = 6 (cumulative data of three independent experiments with technical duplicates).
- Synergy between mycolactone and BZ in the MM.1S BzR lines, when treated as in (C). Data are Mean Loewe scores ± SD, shown as heatmaps. Differences between treated cells and controls by Student’s t‐test: *P < 0.05; N = 6 (cumulative data of three independent experiments with technical duplicates).
- ATF4, CHOP, sXBP1, and BiP mRNA levels were quantified by qPCR in MM.1s BzR cells treated as in (C). sXBP1 mRNA levels were normalized to total (spliced + unspliced) XBP1 mRNA level. Data are Mean RNA fold changes (2−ΔΔCT) ± SD, relative to untreated controls. N = 5 (cumulative data of two independent experiments with technical duplicates and triplicates), pairwise compared by nested one‐way ANOVA with Tukey’s multiple‐comparison test, exact P‐values indicated. Thapsigargin (Tg, 2 µM, 6 h) was used as a positive control.
Data information: In A and B, shown data are representative of two independent experiments with similar results.
The synergy between mycolactone and BZ cytotoxicity extends to mouse B‐cell lymphomas
Besides MM, PIs show promise in the treatment of other hematological malignancies such as acute leukemia (Cloos et al, 2017) and solid malignancies (Roeten et al, 2018). We tested the proteotoxic effects of mycolactone, alone and combined with BZ, in B‐cell acute lymphoblastic leukemia (B‐ALL) using mouse pro‐B‐cell lines generated by transformation of hematopoietic cells with the murine viral form of the Abelson oncogene (v‐abl). Mycolactone alone demonstrated potent ability to induce v‐abl cell apoptosis in vitro (Fig 5A). Moreover, as in human MM cell lines, it synergized with BZ for induction of v‐abl cell death (Fig 5B) and this correlated with activation of the ER stress‐associated ATF4/CHOP apoptotic pathway (Fig 5C and D). In addition to illustrating mycolactone’s ability to efficiently block mouse Sec61, these data revealed the potential interest of Sec61 inhibitors for the treatment of B‐ALL, and potentially other proteasome inhibition‐susceptible malignancies.
Figure 5. The synergy between mycolactone and BZ cytotoxicity extends to mouse B‐cell lymphomas.
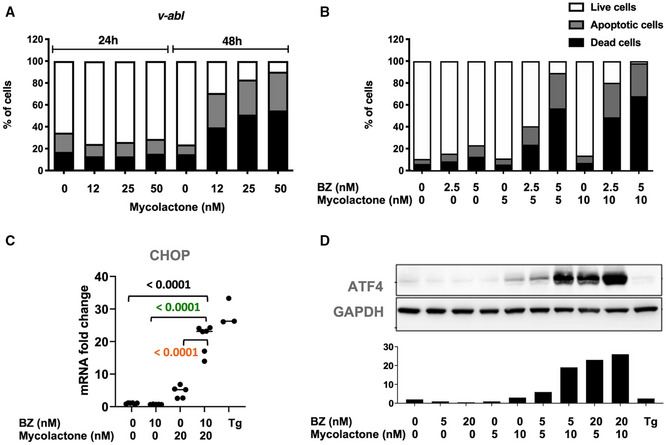
- Effect of mycolactone on v‐abl pro‐B‐cell viability after 24 and 48 h of mycolactone treatment, as measured by exposure of Annexin V and incorporation of PI. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, gated as in Appendix Fig S1, relative to total cells.
- v‐abl pro‐B cells were treated for 24 h with mycolactone and/or BZ at the indicated concentrations. The percentage of live, apoptotic, and dead cells was then measured by flow cytometry as in Appendix Fig S1. Data are Mean % from technical duplicates, relative to total cells.
- v‐abl pro‐B cells were treated with mycolactone and/or BZ at the indicated concentrations for 6 h. CHOP mRNA levels were quantified by qPCR. Data are Mean RNA fold changes (2−ΔΔCT) ± SD relative to untreated controls. N = 6 (cumulative data of three independent experiments with technical duplicates), pairwise compared by nested one‐way ANOVA with Tukey’s multiple‐comparison test, exact P‐values indicated.
- v‐abl pro‐B cells were treated for 6 h with mycolactone and/or BZ at the indicated concentrations. ATF4 protein levels were assessed in cell lysates by Western blot (top panel) and quantified relative to GADPH levels (lower panel).
Data information: Data shown in A, B, and D are representative of experiments performed with two independent v‐abl cell clones, with similar results. In C and D, thapsigargin (Tg, 2 µM, 6 h) was used as a positive control.
Patient‐derived MM tumors are highly susceptible to mycolactone toxicity and synergy with BZ
We next assessed the anti‐MM activity of mycolactone, alone and combined to BZ, in patient‐derived tumors. Mononuclear cells were isolated from bone marrow aspirates of four newly diagnosed MM patients, and two patients with relapsed MM after at least one line of treatment including PI and IMiDs (Table 1). Cells were placed in culture medium within 3 h post‐biopsy, then exposed to mycolactone and/or BZ for 18 h. Induction of apoptosis was determined in MM cells, gated as CD38+ CD138+/− plasma cells, following Annexin V/PI staining (Appendix Fig S2). Figure 6A shows the results as % of live MM cells and Fig EV3A as % of live/apoptotic/dead cells, for easier comparison with treatment effect on non‐cancerous lymphoid cells (Fig EV3B). MM cells from the six studied patients varied in susceptibility to BZ treatment, irrespective of their newly diagnosed or relapsed status (Figs 6A and EV3A). Tumor sensitivity to mycolactone was also variable, and not systematically associated with sensitivity to BZ (Figs 6A and EV3A). In all studied patients, significant cell death was achieved with 18 h exposure to ≥ 12 nM mycolactone, an anti‐MM activity globally equivalent to that of 10 nM BZ. Strikingly, mycolactone synergized with BZ to kill MM cells from all patients irrespective of their treatment naive or relapsed status and relative resistance to single drugs (Fig 6B), further demonstrating that mycolactone‐mediated cytotoxicity operates irrespective of MM cell resistance to BZ.
Table 1.
Characteristics of the MM patients included in this study.
| Patient | P2 | P5 | P7 | P9 | P12 | P13 |
|---|---|---|---|---|---|---|
| Age (years) | 57 | 72 | 72 | 67 | 72 | 66 |
| Gender (F/M) | F | F | F | F | M | F |
| Years since diagnosis (years) | – | – | 10 | – | 5 | – |
| Lines of treatment | – | – | 2 | – | 1 | – |
| Treatments | ||||||
| IMiDs | – | – | 2 | – | 1 | – |
| PI | – | – | 2 | – | 1 | – |
| Alkylating agent | – | – | 1 | – | 1 | – |
| HSCT a | – | – | 1 | – | – | – |
Hematopoietic stem cell transplantation.
Figure 6. MM primary tumors are highly susceptible to mycolactone toxicity and synergy with BZ.
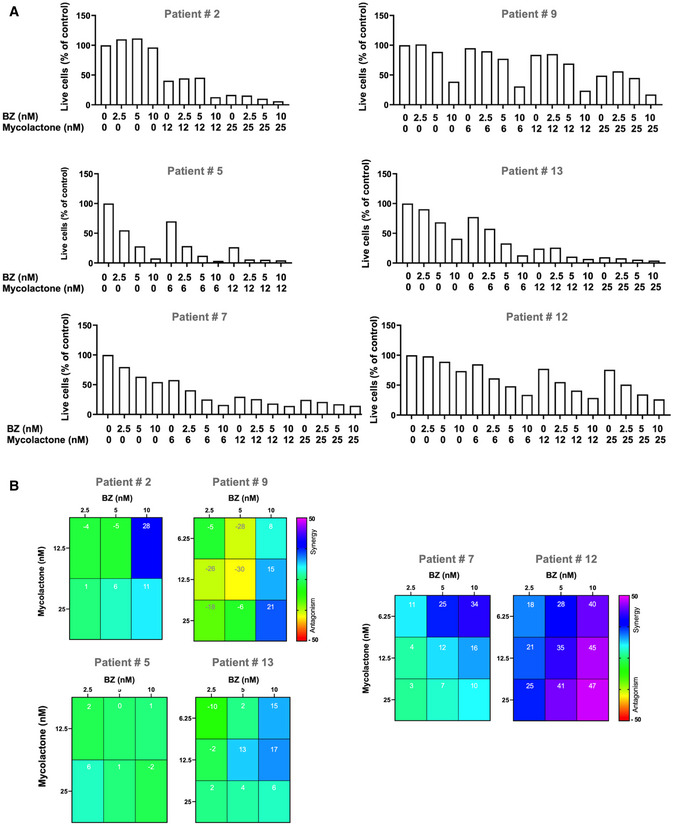
- Following treatment, induction of apoptosis/necrosis was measured by exposure of annexin V and PI incorporation. Data are Mean % of live cells from technical duplicates, relative to controls.
- Synergy between mycolactone and BZ in treated tumors. Data are Mean Loewe scores from technical duplicates, shown as heatmaps.
Figure EV3. Differential toxicity of mycolactone–BZ combination in MM cells and non‐cancerous lymphoid cells in bone marrow aspirates.
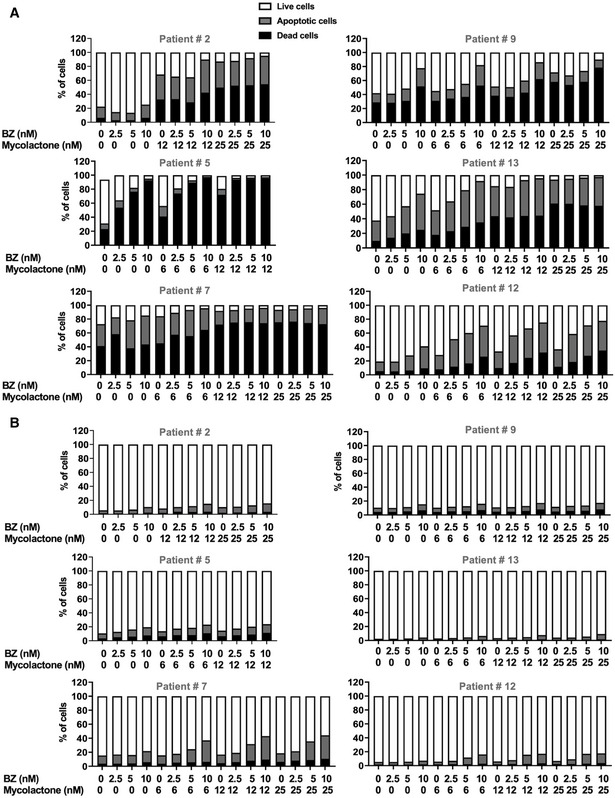
-
A, BIncidences of live, apoptotic, and dead cells within the MM cell (A) and the non‐cancerous lymphoid cell (B) subsets are compared in newly diagnosed (#2, #5, #9, and #13) or relapsed (#7 and #12) MM patients after an 18 h treatment with mycolactone and/or BZ. MM cells (A) and non‐cancerous lymphoid cells (B) were identified with the gating strategy depicted in Appendix Fig S2. Data are Mean % of live, apoptotic, and dead cells from technical duplicates, relative to total cells.
Importantly, the cytotoxicity of the mycolactone–BZ combination selectively affected MM cells, as minimal cell death was recorded in CD38‐ CD138‐ cells from the same bone marrow aspirates (Fig EV3A and B). It is noteworthy that bone marrow aspirates from the two relapsed patients contained a relatively higher incidence of monocytes/macrophages, as characterized by their SSC/FSC profile (as shown for Patient #7 in Appendix Fig S2). Although drugs alone had little effect, the mycolactone–BZ combination displayed some toxicity on this cell subset at the highest tested concentrations (Fig EV4A). To further assess the impact of mycolactone–BZ combination on immune cell viability, we subjected peripheral blood mononuclear cells (PBMCs) to the same drug treatments as patient‐derived tumors (Fig 6 Appendix Fig S3). Minimal or no viability loss was observed in total PBMCs, nor in gated T cells, B cells, natural killer cells, dendritic cells, and monocytes/macrophages exposed to mycolactone, BZ, or both drugs (Fig EV4B), indicating that the cytotoxicity of the mycolactone–BZ combination is selective of MM cells.
Figure EV4. Toxicity of the mycolactone–BZ combination in PBMCs and bone marrow monocytes/macrophages.
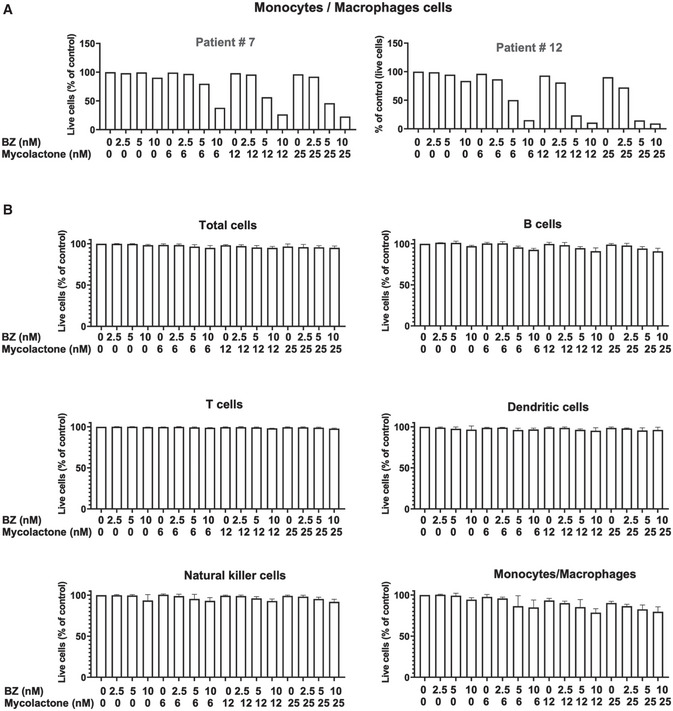
- The proportion of live (Annexin V‐ PI‐) cells within the monocyte/macrophage subset of mononuclear cells, after a 18 h treatment with mycolactone and/or BZ is shown for the two relapsed patients. Mononuclear cells from bone marrow aspirates were treated as in Fig 6. The monocyte/macrophage subset was identified using the gating strategy depicted in Appendix Fig S2. Data are Mean % of live cells, relative to vehicle controls.
- PBMCs from healthy donors were treated with mycolactone and BZ, alone or in combinations, at the indicated concentration for 18 h. Cells were then labelled with fluorophore‐conjugated anti‐CD3, anti‐CD19, and anti‐CD11c. Total cells, B cells, T cells, dendritic cells, NK cells, and monocytes/macrophages were identified by flow cytometry analysis, using the strategy depicted in Appendix Fig S3. Data are Mean % ± SD of live cells, relative to vehicle controls. N = 6, cumulative data from three donors with technical duplicates.
Combining mycolactone with BZ delays MM tumor growth in vivo
To evaluate the therapeutic efficacy of the mycolactone–BZ combination in vivo, we first analyzed its toxicity in mice. C57BL/6 mice were treated twice a week by intraperitoneal injection of BZ (0.5 mg/kg) alone and/or mycolactone (0.3 mg/kg) for 3 weeks, BZ and mycolactone treatments previously shown to induce anti‐MM activity (LeBlanc et al, 2002) and anti‐inflammatory effects in injected mice (Guenin‐Mace et al, 2015), respectively. No sign of distress or weight loss could be detected in mice receiving single drugs or the drug combination, and their blood cell counts remained unaltered throughout the experiment (Fig 7A), illustrating the safety of these treatment regimen. We next compared the anti‐MM activity of mono‐ and bi‐therapies in immunodeficient NOD/SCID/IL2rγnull (NSG) mice. Mice were subcutaneously engrafted with MM.1S cells, and 7 days later they were assigned to four treatment groups receiving (i) 0.5 mg/kg BZ, (ii) 0.3 mg/kg mycolactone, (iii) both drugs at these concentrations, or (iv) vehicle, twice weekly via the intraperitoneal route. Under these conditions, BZ and mycolactone both induced a minor yet significant delay in tumor growth, compared to vehicle controls (Fig 7B). Notably, the mycolactone–BZ combination was superior to single drug treatments in slowing down MM development. In addition to revealing a therapeutic window for Sec61 blockade in MM, these data confirmed the interest of combining inhibitors of translocon and proteasome in MM therapy.
Figure 7. Combining mycolactone with BZ delays MM tumor growth in vivo .
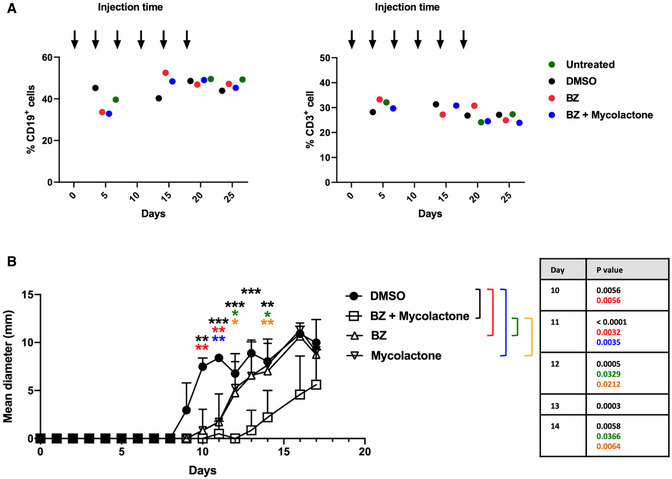
- C57BL/6 mice (N = 2/group) were injected intraperitoneally with BZ (0.5 mg/kg), BZ (0.5 mg/kg) + mycolactone (0.3 mg/kg), DMSO as vehicle controls every 3.5 days, or left untreated. Blood samples were analyzed 5, 15, 20, and 25 days after the first injection and percentages of CD3+ and CD19+ cells determined by flow cytometry. Results are expressed as % of positive cells among total mononuclear cells, each dot representing the Mean % from two mice.
- NSG mice (N = 9/group) were injected subcutaneously with 3 × 106 MM.1S cells at day 0. Seven days later, mice were injected with DMSO, BZ (0.5 mg/kg), and/or mycolactone (0.3 mg/kg) every 3.5 days intraperitoneally and tumor growth was followed by daily measurement of the tumor diameter. Data are Mean tumor diameters ± SD and represent cumulative data from three independent experiments with 3 mice/group. Difference between groups were analyzed with Tukey’s multiple‐comparison test using two‐way ANOVA, with mixed‐effect model for each time points: *P < 0.05; **P < 0.01; ***P < 0.001 (exact P‐values indicated). Only significant differences are shown.
Discussion
Although previous studies have suggested the interest of targeting Sec61 in Oncology, therapeutic indications and in vivo safety of Sec61 blockers as anti‐cancer agents have remained unexplored (Luesch & Paavilainen, 2020). The first identified Sec61 inhibitor, cotransin, was isolated by screens for molecules decreasing the expression of cell adhesion molecules by a vascular endothelial cell line (Garrison et al, 2005). The high substrate selectivity of cotransin was subsequently used to specifically block cancer cell expression of HER3 (Ruiz‐Saenz et al, 2015), a receptor activating oncogenic signaling. Here, we took the opposite approach and sought to exploit the broad, almost total lack of selectivity of mycolactone for Sec61 substrates (Morel et al, 2018). Having shown that Sec61 blockade by mycolactone elicits potent ER stress responses (Morel et al, 2018), we selected the MM cancer model for proof‐of‐concept because of its particularly high susceptibility to proteotoxic stress (Vincenz et al, 2013). Furthermore, we knew from previous studies that systemically delivered mycolactone diffuses broadly in injected organisms to accumulate in leukocytes of peripheral blood and lymphoid organs (Hong et al, 2008; Phillips et al, 2009), a distribution profile that was adapted to plasma cell targeting. Finally, we reasoned that mycolactone may cause defects in MM cell expression of membrane receptors critically contributing to its dissemination and growth, potentially providing an additional therapeutic benefit in vivo. Our in vitro investigations using MM cell lines validated these predictions. Moreover, our observations that mycolactone efficiently kills tumors from treatment naive and relapsed MM patients ex vivo and delays MM xenograft growth in a pre‐clinical murine model of disease establish Sec61 as a novel therapeutic vulnerability in MM.
Serum levels of paraproteins, the antibody fragments produced by MM, are used clinically to diagnose MM and monitor its progression (Tate, 2019). In MM cell lines, the level of immunoglobulin light chains released in culture supernatant was dose dependently decreased by mycolactone after 24 h, and this inhibition closely correlated with the onset of apoptosis at 72 h. This result is in line with our previous demonstration that inhibition of protein secretion on the one hand and induction of cell apoptosis on the other hand are early and late effects of mycolactone‐mediated Sec61 blockade, respectively (Guenin‐Macé et al, 2019; Demangel, 2021). This suggests that the prognostic significance of serum paraproteins would be conserved in MM patients treated with Sec61 blockers. Further, mycolactone‐mediated inhibition of paraprotein secretion may help limit their pathophysiological effects in treated patients (Cook & Macdonald, 2007).
The introduction of PIs in anti‐MM chemotherapies has significantly improved patient survival and PIs now form one of the backbones of treatment (Kouroukis et al, 2014). It was therefore important to determine whether Sec61 blockade interferes with the anti‐tumor activity of PIs. Our data obtained in MM cell lines revealed a synergistic effect between mycolactone and BZ for induction of terminal UPR, with combination of subtoxic concentrations of each drug triggering MM cell death within 24 h. Similar results were obtained with Carfilzomib, indicating that our findings obtained with BZ generalize to second‐generation PIs. The mycolactone–BZ combination was superior to single drugs in killing patient‐derived MM tumor cells ex vivo and delaying MM growth in mouse models of disease, demonstrating the interest of targeting both the translocon and the proteasome in MM. Most importantly, mycolactone cytotoxicity and synergy with BZ were observed in isogenic BZ‐susceptible and ‐resistant MM cells. This indicates that mycolactone and BZ resistance mechanisms do not overlap and suggests that MM patients developing resistance to PIs will respond to Sec61 blockade therapy.
Transition from adaptative to terminal UPR is primarily triggered by induction of ATF4/CHOP signaling (Hetz & Papa, 2018), which was potently activated by the BZ–mycolactone combination in all studied MM cell lines. Surprisingly, while expression of p‐eIF2α target ATF4 was clearly induced in MM.1S cells treated with the BZ–mycolactone combination, we could not demonstrate enhanced phosphorylation of eIF2α by Western blot analysis of cell lysates. Our observation that GADD34 expression was activated at the same time as CHOP indicates that drug treatment keeps p‐eIF2α dephosphorylation active but fails to inhibit the eIF2α/ATF4/CHOP pathway. In MM cells, high secretory activity associated with intracellular accumulation of misfolded immunoglobulin chains is believed to account for the limited ability of BiP to cope with PI‐mediated ER stress, and the particularly low threshold for induction of terminal UPR upon exposure to PIs in these cells (Obeng et al, 2006). In support of this hypothesis, recent genome‐wide meta‐analysis of differentially expressed genes indicated that Sec61A1 transcript levels are significantly upregulated in MM versus normal plasma cells (Katiyar et al, 2021). We reported previously that mycolactone‐mediated UPR differs from that triggered by typical ER stressors by the lack of BiP induction (Baron et al, 2016). In line with this finding, despite displaying clear marks of ER stress, MM cell lines failed to upregulate BiP expression upon exposure to mycolactone (Figs 3, 4 and EV2). In all cell lines, this defect was maintained in the presence of BZ. Therefore, we propose that mycolactone sensitizes MM cells to BZ‐driven apoptosis by preventing BiP induction.
Today, the gold standard of MM care is a combination of PIs, IMiDs, and corticosteroids. While the majority of newly diagnosed patients respond to this three‐drug combination, all eventually develop drug resistance. IMiDs such as lenalidomide operate primarily by targeting Cereblon (CRBN) (Zhu et al, 2019), a substrate receptor of the CRL4 ubiquitin ligase complex. IMiDs binding to CRBN leads to increased proteasomal degradation of pro‐survival transcription factors IKZF1/3, thereby promoting MM apoptosis. Notably, low CRBN expression is often associated with IMiD resistance and functional introduction of CRBN mutations in MM cells conferred resistance to lenalidomide in vitro (Kortüm et al, 2016). In both MM cell lines and primary tumors, mycolactone displayed cytotoxic effects irrespective of the cell basal resistance to BZ. It will be interesting to determine whether mycolactone also kills IMiD‐resistant MM and synergizes with IMiDs for induction of MM cell death.
Similar to PIs and IMiDs, the clinical use of Sec61 inhibitors raises questions about their potential toxicity in normal cells, and the risks to generate drug resistance in tumor cells. Mycolactone‐mediated Sec61 blockade provokes perturbations in protein homeostasis whose magnitude and ability to evolve toward apoptosis vary across cell types, depending on their functional biology (Demangel & High, 2018; Morel et al, 2018). Recent studies using computer simulations or lipid monolayers also suggested that mycolactone passage across cellular membranes may alter their integrity and dynamic properties (López et al, 2018; Nitenberg et al, 2018). Using primary human cells, we reported previously that contrary to macrophages, neutrophils and T lymphocytes are resistant to mycolactone‐mediated toxicity (Guenin‐Mace et al, 2015). The present study further indicates that MM cells are more susceptible to Sec61 blockade‐driven apoptosis than major PBMC subsets. In mice, repeated systemic administration of mycolactone significantly delayed MM growth without inducing toxic effects. Together, our in vitro and in vivo results thus define a therapeutic window for Sec61 blockers in MM treatment. Regarding the development of resistance in tumor cells, one can speculate that increased expression of Sec61α may reduce the inhibitor’s ability to saturate Sec61‐binding sites, thereby the induction of ER stress responses. Sec61α is a mycolactone‐sensitive Sec61 client that was mildly upregulated by mycolactone in previous proteomic studies using various cancer cell lines (Grotzke et al, 2017; Morel et al, 2018), suggesting that UPR‐mediated increase in Sec61α gene expression may override Sec61 blockade, at least to a limited extent. Mutations in the Sec61α gene sequence resulting in loss of inhibitor binding, such as those introduced experimentally in cancer cell lines in previous research (Baron et al, 2016; Gérard et al, 2020), may also confer MM tumors with resistance to Sec61 inhibitors. To our knowledge, none of these mutations has been reported in human clinical samples, suggesting that they may prevent the translocon from ensuring essential functions. Yet, it will be important to verify if such genetic adaptations arise in MM cells subjected to Sec61 blocker pressure in vitro and in animal models of disease. Cytogenetic abnormalities are found in most MM patients, among which translocation t(4;14) that has been associated with poor prognosis (Abdallah et al, 2020). It is interesting to note that mycolactone was relatively more toxic in t(4;14)‐positive JIM3 and KMS‐11 cells than in t(4;14)‐negative MM.1S cells (Fig 1A). While a direct comparison between isogenic t(4;14)‐positive and ‐negative cell lines would be required to conclude, this result suggests that gene deregulation caused by NSD2 overexpression and expansion of H3K36me2 in t(4;14) MM cells (Lhoumaud et al, 2019) may confer them with enhanced susceptibility to the pro‐death programs triggered by Sec61 blockade.
To address the toxicity and resistance issues associated with PI‐based treatments, novel approaches using antibody–drug conjugates, bispecific T‐cell engager antibodies, and chimeric antigen receptor (CAR)‐T cells are in test phase. Such strategies target membrane receptors that are specifically expressed by MM cells, such as BCMA (Shah et al, 2020). Inhibiting Sec61 with cotransin increased the surface expression of BCMA by MM cell lines, resulting in enhanced efficacy a BCMA‐targeting antibody–drug conjugate (Ramkumar et al, 2020). In line with this finding, mycolactone dose dependently increased BCMA expression in the MM cell lines tested in the present study (Fig 1). This argues that anti‐MM therapies targeting BCMA may benefit from combination with mycolactone.
Here, we used immunodeficient NSG mice to assess the translational potential of mycolactone. Although this setting is widely used to estimate the therapeutic index of drug candidates, it does not allow to evaluate their impact on immune responses. Since Sec61 inhibition strongly affects the functional biology of immune cells (Guenin‐Mace et al, 2015; Baron et al, 2016; Grotzke et al, 2017; Demangel & High, 2018; Guenin‐Macé et al, 2019; Demangel, 2021), it may suppress the development of anti‐tumor immunity. Further investigations using immunocompetent mouse models of MM and B‐ALL will be needed to address this hypothesis. Regardless of this limitation, this study reports the first attempt to combine inhibitors of both proteasome and translocon in Oncology. Our results establish Sec61 blockade as a novel therapeutic approach in MM, synergizing with proteasome inhibition. We used mycolactone for proof of concept, as it displays unprecedented Sec61 inhibition potency and a body‐wide distribution in vivo (Guenin‐Macé et al, 2019). However, mycolactone is a complex natural product whose chemical synthesis is a costly, multi‐step process making large‐scale industrial production challenging (Song et al, 2007; Gehringer & Altmann, 2017; Saint‐Auret et al, 2017). Structure–activity relationship studies have identified the minimal structural module retaining Sec61‐binding activity in mycolactone (Guenin‐Mace et al, 2015; Baron et al, 2016) and the tri‐dimensional structure of the Sec61 complex inhibited by mycolactone was recently resolved (Gérard et al, 2020). Our study thus paves the way to structure‐based design of drug‐like surrogates of mycolactone as novel therapeutics for MM, and potentially other proteostasis‐addicted cancers, such as non–small cell lung cancers and certain solid tumors.
Materials and Methods
Patients
Patients were seen at Saint‐Louis hospital, Paris, France, for a new or known diagnosis of MM. Characteristics of the patients are shown in Table 1. Bone marrow aspirates were performed in the Department of Immuno‐Hematology of the Saint‐Louis Hospital using standard procedures, as part of routine diagnostic work‐up. Written informed consent was obtained from all patients, and experiments conformed to the principles set out in the WMA Declaration of Helsinki and the Department of Health and Human Services Belmont Report. All samples containing measurable numbers of viable mononuclear cells were processed and included in data analysis. The protocol received approval of the ethical committee “Groupements hospitaliers universitaires” GHU Nord; No. 23798‐2020012709469025 # 121.
Reagents
Mycolactone was purified from M. ulcerans bacterial pellets (strain 1615, ATCC 35840), then quantified by spectrophotometry, and stored in ethanol at −20°C protected from light. For in vivo experiments, a 4 mM stock was diluted in a NaCl solution (0.9% w/v) immediately before injection in animals. For in vitro experiments, a 1,000× working solution was prepared by dilution of the ethanol stock in DMSO and stored at −20°C, then thawed and diluted in culture medium immediately before use. BZ purchased from Alfa Aesar (#J60378) was resuspended in DMSO to yield a 10 mM solution stored at −20°C. A 10 mM solution of carfilzomib in DMSO was purchased from ApexBio (#A1933) and stored at −20°C. BZ and carfilzomib were thawed and diluted in culture medium immediately before use. Thapsigargin (Tocris, #1138) and tunicamycin (Tocris, #3516) were resuspended in DMSO to yield a 10 mM solution stored at −20°C; and dithiothreitol (DTT, 1 M solution) was purchased from Invitrogen (#P2325) and stored at −20°C.
MM cell line cultures and drug treatment
The MM.1S and KMS‐11 cell lines were from J.‐C.B. (Fayon et al, 2020). JIM3 cells were kindly provided by Dr. MacLennan (Birmingham Medical School, Birmingham, UK). All cell lines were tested negative for mycoplasma. Cells were cultured in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS, Dominique Dutcher #S1810‐500), 100 Units/ml penicillin + 100 µg/ml streptomycin (Gibco, #15140‐122). A BZ‐resistant version of the MM.1S cell line (MM.1S BzR) was generated by serial subculture in the presence of increasing doses of BZ, starting from 0.1 nM and doubling BZ concentration every 2 weeks until 0.8 nM, and then increasing BZ concentration by 0.1 nM every 2 weeks until 15 nM. Compared to parental cells, the growth rate of MM.1S BzR cells was not reduced and their BZ‐resistant phenotype was stable upon removal of BZ from culture medium. Exponentially growing MM.1S, JIM3, and KMS‐11 cells were plated in 96‐well plates at a density of 1.5 × 105 cells/well (MM‐1S) or 105 cells/well (JIM3 and KMS‐11). Cells were then treated with mycolactone and/or BZ at the indicated concentrations for 24, 48, or 72 h at 37°C.
Lymphomas
Total bone marrow from 3‐ to 5‐week‐old female C57BL/6 mice was cultured and infected with a retrovirus encoding v‐abl kinase to generate immortalized pro‐B cell lines as previously described (Rosenberg et al, 1975; Lescale et al, 2016). v‐abl pro‐B cell lines were cultured in RPMI GlutaMAXTM supplemented with 12% heat‐inactivated FBS, 10 mM Hepes, and 50 µM 2‐mercaptoethanol. Cells were treated with mycolactone and/or BZ as above.
Flow cytometric analysis of MM cell lines
Following drug treatment, MM cells were pelleted and resuspended in flow cytometry buffer (1× PBS with 1% FBS) then incubated with human FcR blocking reagent (Miltenyi Biotec, # 130‐059‐901) before staining with diverse fluorescent dye‐conjugated monoclonal antibodies: anti‐CD126 (IL‐6Rα)/APC (#352806 Biolegend, dilution 1/50), anti‐CD38/BV421 (#303526 Biolegend, diluted 1/200), anti‐CD38/PE (#356604 Biolegend, diluted 1/200), anti‐CD138/APC (#130‐117‐395 Miltenyi Biotec, diluted 1/100), anti‐CD138/APC‐AF750 (#352316 Biolegend, diluted 1/100), anti‐CD40/FITC (#334306 Biolegend, diluted 1/25), and anti‐BCMA/PerCp‐Cy5.5 (#357510 Biolegend, diluted 1/25). Cells were stained for 15 min at 4°C, then washed and filtered before data acquisition by Cytoflex flow cytometer (Beckman Coulter). Data were analyzed with the FlowJo software (Tree Star, Ashland, OR). Mean fluorescence intensities (MFI) of isotype controls recommended by the Ab supplier were systematically subtracted to that of each stained sample. Cell viability was assessed by Annexin V exposure and PI incorporation using the FITC‐Annexin V/PI kit from Miltenyi Biotec (#130‐092‐052) following the manufacturer’s protocol. Live cells were characterized as Annexin V‐ PI‐, apoptotic cells as Annexin V+ PI−, and dead cells as Annexin V+ PI+ and Annexin V− PI+ as shown in Appendix Fig S1.
Detection of secreted immunoglobulin light chain by ELISA
Supernatants from MM cells cultures treated with mycolactone and/or BZ alone were collected after 24 h, and the concentration of free light chain was assessed using the SEBIA FLC lambda ELISA kit (SEBIA, 5101) according to the manufacturer’s recommendations.
Western blot analyses
Following a 6 h treatment with mycolactone and/or BZ at the indicated concentrations, 2 × 106 cells were harvested and solubilized at 108 cells/ml for 15 min in ice‐cold lysis buffer containing 1% Nonidet P‐40, 1% n‐dodecyl‐{beta}‐D‐maltoside, 20 mM Tris‐HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, and 1 mM EGTA in the presence of inhibitors of proteases and phosphatases (10 µg/ml leupeptin, 10 µg/ml aprotinin, 1 mM Pefabloc‐sc, 50 mM NaF, 10 mM Na4P2O7, and 1 mM NaVO4). For immunoblot analyses, loadings were normalized on the basis of the total amount of proteins per lane. Proteins were then separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS‐PAGE) using NuPAGE Bis‐Tris gels (Thermo Fisher Scientific) and transferred on nitrocellulose membranes (iBlot 2® gel transfer Stacks Nitrocellulose system from Invitrogen). Immune blotting was carried out with the primary antibodies anti‐ATF4 (Cell Signaling, #11815), anti‐GAPDH (Cell Signaling, #2118), and anti‐ATF6 (Biolegend, 853102) overnight at 4°C. After washing, the membranes were incubated with HRP‐conjugated anti‐rabbit, ‐mouse, or ‐rat IgG secondary antibodies (Santa Cruz, sc‐2004, Biolegend, 405405) for 45 min at room temperature. All antibodies were diluted 1/1,000 in PBS‐fat free milk (5% m/v). Detection of proteins was performed with the enhanced chemical luminescence (ECL) method using the ECL Prime Western Blotting Reagent, and image was acquired on an ImageQuant LAS 4000 Mini (GE Healthcare).
Gene expression analyses
Following a 6 h treatment with mycolactone and/or BZ at the indicated concentrations, 2 × 106 cells were harvested and lysed in Trizol (Qiagen). Chloroform was added to the trizol lysates, and the mix was then centrifuged for 15 min at 15,000 g and 4°C. After centrifugation, the aqueous phase was recovered and mixed with 1.5 volume of ethanol. Total mRNAs were then extracted using RNeasy Plus Mini Kit (Qiagen) according to the manufacturer’s procedure and reverse transcribed into cDNAs using High Capacity cDNA Reverse Transcription kit (BD Bioscience) from 1 µg total mRNA according to the manufacturer’s recommendations. The levels of transcription of the mRNAs coding for the genes of interest were assessed using SyberGreen (Power SYBR Green PCR Master Mix, applied biosystem ref: 4367659) and the following primers synthesized by Eurofins genomics: CHOP (forward), GCACCTCCCAGAGCCCTCACTCTCC; CHOP (reverse), GTCTACTCCAAGCCTTCCCCCTGCG; sXBP1 (forward), GGTCTGCTGAGTCCGCAGCAGG; sXBP1 (reverse), GGGCTTGGTATATATGTGG; XBP1total (forward), TGGCCGGGTCTGCTGAGTCCG; XBP1total (reverse), ATCCATGGGGAGATGTTCTGG; ATF4 (forward), CACCGCAACATGACCGAAAT; ATF4 (reverse), GACTGACCAACCCATCCACA; GADD34 (forward), GATGGCATGTATGGTGAGCG; GADD34 (reverse), GAGACAAGGCAGAAGTAGAG; BiP (forward), CGAGGAGGAGGACAAGAAGG; BiP (reverse), CACCTTGAACGGCAAGAACT. Quantitative PCR conditions used were as follows: 50°C 2 min, 95°C 10 min, 95°C 15 s (40 cycles), and 60°C for 1 min. The relative quantification was calculated by the 2−ΔΔCT method and the 18S mRNA was used as endogenous control.
RT‐PCR analysis
Total mRNA was extracted from MM.1S cells and cDNAs were synthetized as described above. PCR amplification was performed using GoTAQ Green Master Mix (Promega) and the following XBP‐1 primers: (forward) AAACAGAGTAGCAGCTCAGACTGC; (reverse) TCCTTCTGGGTAGACCTCTGGGAG. PCR products were digested using PstI (New England BioLabs) for 15 min at 37°C and separated on 2% agarose gel.
Flow cytometric analysis of MM patient‐derived samples and PBMCs
Patient‐derived mononuclear cells were isolated from bone marrow aspirate by density gradient centrifugation with Pancoll separating solution (PAN‐Biotech, DE). 2 × 105 cells were then plated in 96 wells plates in culture medium in the absence or the presence of mycolactone or BZ alone or in combination as described in the text for 18 h. Cells were then washed and stained with fluorescent‐conjugated anti‐CD38 and anti‐CD138 antibodies to gate plasma cells, in the presence of Annexin V‐FITC and PI for 15 min at room temperature. Cells were then acquired using a flow cytometer (Attune NXT, ThermoFisher) immediately after the staining and cell viability was assessed by analysis using FlowJo software (Tree Star, Ashland, OR). PBMCs were isolated from blood samples of healthy donors collected by the French Blood Establishment (EFS), by Pancoll density gradient centrifugation. 2 × 105 cells were plated in 96‐well plates and treated with mycolactone, BZ, alone or in combination, for 18 h. Cells were then washed and stained with anti‐CD3/PerCPCy5.5 (#332771 BD Bioscience), anti‐CD19/FITC (#302206 Biologend), anti‐CD56/APC (#341027 BD Bioscience), anti‐CD16/PE‐CF594 (#562293 BD Bioscience), and anti‐CD11c/PE (#333149 BD Bioscience). Dead cells were identified using LIVE/DEAD Fixable Near IR Dead Cell Stain Kit (#L34975 Invitrogen). Cells were then acquired using a flow cytometer (Cytoflex, Beckman Coulter).
Mice
Six‐ to eight‐week‐old female pathogen‐free C57BL/6J mice were purchased from Charles River Laboratories. The NOD. Cg‐Prkdcscid Il2rgtm1Wjl /SzJ (NSG, stock number: 005557) mice were purchased from the Jackson laboratory and were used between 6 and 12 weeks of age. All mice were housed at animal facilities of the Institut Pasteur under specific pathogen‐free conditions with food and water ad libitum. Mouse experiments were validated by CETEA Ethics Committee number 0068 (Institut Pasteur, Paris, France, DAP170027) and received the approval of the French Ministry of Higher Education and Research. They were performed in compliance with national guidelines and regulations.
Mouse experiments
In vivo toxicity assays were performed on 8‐week‐old C57BL/6J (B6) mice purchased from Charles River laboratories as follows. Mycolactone (0.3 mg/kg) and BZ (0.3 mg/kg) alone or in combination were diluted in NaCl solution (0.9% w/v) administrated every 3.5 days via the intraperitoneal route. The toxicity of the treatments was assessed by measuring the percentage of T and B cells in the blood sample collected in tubes containing EDTA (0.05 M) from the tail vein 5, 15, 20, and 25 days after the first injection. Red blood cells were lysed using red blood cells lysing buffer (SIGMA R7757). Leukocytes were stained with anti‐mouse CD3/PerCPcy5.5 (#45003182 eBioscience, diluted 1/100) and CD19/APC (#550992 BD Pharmingen, diluted 1/800) antibodies and fluorescence data were acquired by Cytoflex flow cytometer (Beckman Coulter). The percentages of T (CD3+) and B (CD19+) cells were determined using FlowJo software. Experiments assessing the effect of the treatments on the development of xenografted MM tumors were performed in 8‐ to 12‐week‐old NSG mice. 3 × 106 human MM‐1S cells were subcutaneously injected in the right flank of the animals in 200 µl of PBS. After 24 h, mice were randomly assigned to four groups and 6 days later, each group was randomly assigned a treatment. Mice were injected by intraperitoneal route with mycolactone (0.3 mg/kg and/or BZ (0.3 mg/kg), in 200 µl NaCL (0.9% w/v), or vehicle as control, every 3.5 days. Tumor growth was assessed daily by unblinded measurement of tumor size with a digital caliper. Data are presented as the average of two perpendicular diameters (millimeters). Mice were sacrificed when the tumor diameter reached 20 mm or whenever animals showed clinical signs of pain according to ethical guidelines. No animal was excluded from our analyses.
Synergy scores
Synergy between mycolactone and BZ was assessed with the combenefit software (Di Veroli et al, 2016), which calculates scores based on the Loewe additivity model using the dose response of each drug and P‐value ranges. Loewe synergy score is defined as SLoewe = Yobs ‐YLoewe, where Yobs is the observed effect of the combination and YLoewe is the theoretical effect of the combination. Therefore, a SLoewe > 0 shows that drugs act in synergy. On the contrary, SLoewe < 0 depicts an antagonist effect of the drugs. SLoewe were plotted as heatmaps and statistical significance was analyzed by one‐sample Student’s t‐test.
Statistical analyses
Other statistical treatments and graphical representations were performed with the Prism software (v8.4.3, GraphPad, La Jolla, CA) and values of P ≤ 0.05 were considered significant. Detailed information on the statistical test used and number of replicates is provided in figure legends. Positive controls were not considered in statistical analyses of qPCR data.
Author contributions
AD, CC, LB, and VM performed the experiments. AD, EP, and GD performed the statistical analyses. LD provided v‐abl B‐cell clones and consultation regarding lymphomas. BA and J‐CB provided bone marrow aspirates from MM patients and guidance with their handling and flow cytometric analysis. AD, GD, and CD designed the experiments. Data interpretation and writing of the manuscript were performed by AD, GD, and CD.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Expanded View
Source Data for Figure 3
Source Data for Figure 5
Acknowledgements
We would like to thank Jean‐David Morel (now at EPFL, Lausanne) for advice regarding the experiments using v‐abl cells, and the staff of Animal facility of Institut Pasteur for supervising mouse housing. This work was supported by the Cancéropôle Ile de France (CD and J‐CB; Emergence 2019), the Fondation Française pour la Recherche contre le Myélome et les Gammapathies (CD, FFRMG), and the Sanofi iAwards program (CD, 2020). CD also acknowledges core support from Institut Pasteur and INSERM (U1221). LB was recipient of a Roux‐Cantarini post‐doctoral fellowship (Institut Pasteur). AD is a BioSPC‐Université de Paris PhD student, recipient of a doctoral fellowship from the Ministère français de l’Enseignement Supérieur, de la Recherche et de l’Innovation.
Disclosure statement and competing interests
The authors declare that they have no conflict of interest.
EMBO Mol Med (2022) 14: e14740.
Contributor Information
Gilles Dadaglio, Email: gilles.dadaglio@pasteur.fr.
Caroline Demangel, Email: demangel@pasteur.fr.
Data availability
This study includes no data deposited in external repositories.
References
- Abdallah N, Rajkumar SV, Greipp P, Kapoor P, Gertz MA, Dispenzieri A, Baughn LB, Lacy MQ, Hayman SR, Buadi FK et al (2020) Cytogenetic abnormalities in multiple myeloma: association with disease characteristics and treatment response. Blood Cancer J 10: 82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmetzyanova I, McCarron MJ, Parekh S, Chesi M, Bergsagel PL, Fooksman DR (2020) Dynamic CD138 surface expression regulates switch between myeloma growth and dissemination. Leukemia 34: 245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auner HW, Cenci S (2015) Recent advances and future directions in targeting the secretory apparatus in multiple myeloma. Br J Haematol 68: 14–25 [DOI] [PubMed] [Google Scholar]
- Baron L, Paatero AO, Morel J‐D, Impens F, Guenin‐Macé L, Saint‐Auret S, Blanchard N, Dillmann R, Niang F, Pellegrini S et al (2016) Mycolactone subverts immunity by selectively blocking the Sec61 translocon. J Exp Med 213: 2885–2896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SF, Lin L, Xing L, Li Y, Yu T, Anderson KC, Tai YT (2020) BCMA‐targeting therapy: driving a new era of immunotherapy in multiple myeloma. Cancers 12: 1473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloos J, Roeten MS, Franke NE, van Meerloo J, Zweegman S, Kaspers GJ, Jansen G (2017) (Immuno)proteasomes as therapeutic target in acute leukemia. Cancer Metastasis Rev 36: 599–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook L, Macdonald DHC (2007) Management of paraproteinaemia. Postgrad Med J 83: 217–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AJ, Allen C, Barac A, Basaleem H, Bensenor I, Curado MP, Foreman K, Gupta R, Harvey J, Hosgood HD et al (2018) Global burden of multiple myeloma. JAMA Oncol 4: 1221–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demangel C (2021) Immunity against Mycobacterium ulcerans: the subversive role of mycolactone. Immunol Rev 301: 209–221 [DOI] [PubMed] [Google Scholar]
- Demangel C, High S (2018) Sec61 blockade by mycolactone: a central mechanism in Buruli ulcer disease. Biol Cell 110: 237–248 [DOI] [PubMed] [Google Scholar]
- Van De Donk NWCJ, Richardson PG, Malavasi F (2018) CD38 antibodies in multiple myeloma: back to the future. Blood 131: 13–29 [DOI] [PubMed] [Google Scholar]
- Fayon M, Martinez‐Cingolani C, Abecassis A, Roders N, Nelson E, Choisy C, Talbot A, Bensussan A, Fermand J‐P, Arnulf B et al (2020) Bi 38–3 is a novel CD38/CD3 bispecific T‐cell engager with low toxicities for the treatment of multiple myeloma. Haematologica 106: 1193–1197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandolfi S, Laubach JP, Hideshima T, Chauhan D, Anderson KC, Richardson PG (2017) The proteasome and proteasome inhibitors in multiple myeloma. Cancer Metastasis Rev 36: 561–584 [DOI] [PubMed] [Google Scholar]
- Garrison JL, Kunkel EJ, Hegde RS, Taunton J (2005) A substrate‐specific inhibitor of protein translocation into the endoplasmic reticulum. Nature 436: 285–289 [DOI] [PubMed] [Google Scholar]
- Gehringer M, Altmann KH (2017) The chemistry and biology of mycolactones. Beilstein J Org Chem 13: 1596–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gérard SF, Hall BS, Zaki AM, Corfield KA, Mayerhofer PU, Costa C, Whelligan DK, Biggin PC, Simmonds RE, Higgins MK (2020) Structure of the inhibited state of the sec translocon. Mol Cell 79: 406–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grotzke JE, Kozik P, Morel JD, Impens F, Pietrosemoli N, Cresswell P, Amigorena S, Demangel C (2017) Sec61 blockade by mycolactone inhibits antigen cross‐presentation independently of endosome‐to‐cytosol export. Proc Natll Acad Sci USA 114: E5910–E5919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenin‐Macé L, Baron L, Chany A‐C, Tresse C, Saint‐Auret S, Jönsson F, Le Chevalier F, Bruhns P, Bismuth G, Hidalgo‐Lucas S et al (2015) Shaping mycolactone for therapeutic use against inflammatory disorders. Sci Transl Med 7: 289ra85 [DOI] [PubMed] [Google Scholar]
- Guenin‐Macé L, Ruf M‐T, Pluschke G, Demangel C (2019) Mycolactone: more than just a cytotoxin, In Buruli ulcer, Pluschke G, Röltgen K (eds), pp 117–134. Cham: Springer; [PubMed] [Google Scholar]
- Hall BS, Hill K, McKenna M, Ogbechi J, High S, Willis AE, Simmonds RE (2014) The pathogenic mechanism of the Mycobacterium ulcerans virulence factor, mycolactone, depends on blockade of protein translocation into the ER. PLoS Pathog 10: e1004061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Papa FR (2018) The unfolded protein response and cell fate control. Mol Cell 69: 169–181 [DOI] [PubMed] [Google Scholar]
- Hong H, Coutanceau E, Leclerc M, Caleechurn L, Leadlay PF, Demangel C (2008) Mycolactone diffuses from Mycobacterium ulcerans‐Infected tissues and targets mononuclear cells in peripheral blood and lymphoid organs. PLoS Negl Trop Dis 2: e325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katiyar A, Kaur G, Rani L, Jena L, Singh H, Kumar L, Sharma A, Kaur P, Gupta R (2021) Genome‐wide identification of potential biomarkers in multiple myeloma using meta‐analysis of mRNA and miRNA expression data. Sci Rep 11: 10957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortüm KM, Mai EK, Hanafiah NH, Shi C‐X, Zhu Y‐X, Bruins L, Barrio S, Jedlowski P, Merz M, Xu J et al (2016) Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 128: 1226–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouroukis TC, Baldassarre FG, Haynes AE, Imrie K, Reece DE, Cheung MC (2014) Bortezomib in multiple myeloma: systematic review and clinical considerations. Curr Oncol 21: e573–603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laubach J, Richardson P, Anderson K (2011) Multiple myeloma. Annl Rev Med 62: 249–264 [DOI] [PubMed] [Google Scholar]
- Lawasut P, Chauhan D, Laubach J, Hayes C, Fabre C, Maglio M, Mitsiades C, Hideshima T, Anderson KC, Richardson PG (2012) New proteasome inhibitors in myeloma. Curr Hematol Malig Rep 7: 258–266 [DOI] [PubMed] [Google Scholar]
- LeBlanc R, Catley LP, Hideshima T, Lentzsch S, Mitsiades CS, Mitsiades N, Neuberg D, Goloubeva O, Pien CS, Adams J et al (2002) Proteasome inhibitor PS‐341 inhibits human myeloma cell growth in vivo and prolongs survival in a murine model. Cancer Res 62: 4996–5000 [PubMed] [Google Scholar]
- Lescale C, Abramowski V, Bedora‐Faure M, Murigneux V, Vera G, Roth DB, Revy P, De Villartay JP, Deriano L (2016) RAG2 and XLF/Cernunnos interplay reveals a novel role for the RAG complex in DNA repair. Nature Comm 7: 10529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lhoumaud P, Badri S, Rodriguez‐Hernaez J, Sakellaropoulos T, Sethia G, Kloetgen A, Cornwell M, Bhattacharyya S, Ay F, Bonneau R et al (2019) NSD2 overexpression drives clustered chromatin and transcriptional changes in a subset of insulated domains. Nature Comm 10: 4843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López CA, Unkefer CJ, Swanson BI, Swanson JMJ, Gnanakaran S (2018) Membrane perturbing properties of toxin mycolactone from Mycobacterium ulcerans . PLoS Comput Biol 14: e1005972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luesch H, Paavilainen VO (2020) Natural products as modulators of eukaryotic protein secretion. Nat Prod Rep 37: 717–736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackinnon AL, Paavilainen VO, Sharma A, Hegde RS, Taunton J (2014) An allosteric Sec61 inhibitor traps nascent transmembrane helices at the lateral gate. Elife 3: e01483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahindra A, Hideshima T, Anderson KC (2010) Multiple myeloma: biology of the disease. Blood Rev 24(Suppl 1): S5–11 [DOI] [PubMed] [Google Scholar]
- McKenna M, Simmonds RE, High S (2016) Mechanistic insights into the inhibition of Sec61‐dependent co‐ and post‐translational translocation by mycolactone. J Cell Sci 129: 1404–1415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna M, Simmonds RE, High S (2017) Mycolactone reveals the substrate‐driven complexity of Sec61‐dependent transmembrane protein biogenesis. J Cell Sci 130: 1307–1320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel JD, Paatero AO, Wei J, Yewdell JW, Guenin‐Macé L, Van Haver D, Impens F, Pietrosemoli N, Paavilainen VO, Demangel C (2018) Proteomics reveals scope of mycolactone‐mediated sec61 blockade and distinctive stress signature. Mol Cell Proteomics 17: 1750–1765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitenberg M, Benarouche A, Maniti O, Marion E, Marsollier L, Gean J, Dufourc EJ, Cavalier JF, Canaan S, Girard‐Egrot AP (2018) The potent effect of mycolactone on lipid membranes. PLoS Pathog 14: e1006814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr, Lee KP, Boise LH (2006) Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood 107: 4907–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogbechi J, Hall BS, Sbarrato T, Taunton J, Willis AE, Wek RC, Simmonds RE (2018) Inhibition of Sec61‐dependent translocation by mycolactone uncouples the integrated stress response from ER stress, driving cytotoxicity via translational activation of ATF4. Cell Death Dis 9: 397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips R, Sarfo FS, Guenin‐Mace L, Decalf J, Wansbrough‐Jones M, Albert ML, Demangel C (2009) Immunosuppressive signature of cutaneous Mycobacterium ulcerans infection in the peripheral blood of patients with Buruli ulcer disease. J Infect Dis 200: 1675–1684 [DOI] [PubMed] [Google Scholar]
- Ramkumar P, Abarientos AB, Tian R, Seyler M, Leong JT, Chen M, Choudhry P, Hechler T, Shah N, Wong SW et al (2020) CRISPR‐based screens uncover determinants of immunotherapy response in multiple myeloma. Blood Adv 4: 2899–2911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roeten MSF, Cloos J, Jansen G (2018) Positioning of proteasome inhibitors in therapy of solid malignancies. Cancer Chemother Pharmacol 81: 227–243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosean TR, Tompkins VS, Tricot G, Holman CJ, Olivier AK, Zhan F, Janz S (2014) Preclinical validation of interleukin 6 as a therapeutic target in multiple myeloma. Immunol Res 59: 188–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg N, Baltimore D, Scher CD (1975) In vitro transformation of lymphoid cells by Abelson murine leukemia virus. Proc Natl Acad Sci USA 72: 1932–1936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz‐Saenz A, Sandhu M, Carrasco Y, Maglathlin RL, Taunton J, Moasser MM (2015) Targeting HER3 by interfering with its Sec61‐mediated cotranslational insertion into the endoplasmic reticulum. Oncogene 34: 5288–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saint‐Auret S, Chany AC, Casarotto V, Tresse C, Parmentier L, Abdelkafi H, Blanchard N (2017) Total syntheses of mycolactone A/B and its analogues for the exploration of the biology of Buruli ulcer. Chimia 71: 836–840 [DOI] [PubMed] [Google Scholar]
- Shah N, Chari A, Scott E, Mezzi K, Usmani SZ (2020) B‐cell maturation antigen (BCMA) in multiple myeloma: rationale for targeting and current therapeutic approaches. Leukemia 34: 985–1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song F, Fidanze S, Benowitz AB, Kishi Y (2007) Total synthesis of mycolactones A and B. Tetrahedron 63: 5739–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai Y‐T, Li X, Tong X, Santos D, Otsuki T, Catley L, Tournilhac O, Podar K, Hideshima T, Schlossman R et al (2005) Human anti‐CD40 antagonist antibody triggers significant antitumor activity against human multiple myeloma. Cancer Res 65: 5898–5906 [DOI] [PubMed] [Google Scholar]
- Tate JR (2019) The paraprotein ‐ An enduring biomarker. Clin Biochem Rev 40: 5–22 [PMC free article] [PubMed] [Google Scholar]
- Di Veroli GY, Fornari C, Wang D, Mollard S, Bramhall JL, Richards FM, Jodrell DI (2016) Combenefit: an interactive platform for the analysis and visualization of drug combinations. Bioinformatics 32: 2866–2868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenz L, Jager R, O’Dwyer M, Samali A (2013) Endoplasmic reticulum stress and the unfolded protein response: targeting the achilles heel of multiple myeloma. Mol Cancer Ther 12: 831–843 [DOI] [PubMed] [Google Scholar]
- Zhu YX, Shi C‐X, Bruins LA, Wang X, Riggs DL, Porter B, Ahmann JM, de Campos CB, Braggio E, Bergsagel PL et al (2019) Identification of lenalidomide resistance pathways in myeloma and targeted resensitization using cereblon replacement, inhibition of STAT3 or targeting of IRF4. Blood Cancer J 9: 19 [DOI] [PMC free article] [PubMed] [Google Scholar]