

Abstract
Small nucleolar RNA host gene 15 (SNHG15) is upregulated in many malignancies and mediates the development of multiple cancers, including osteosarcoma (OS). However, data on the regulatory mechanisms and role of SNHG15 in the chemoresistance of OS remain scarce. Here, we show that p53 binds to the SNHG15 promoter, leading to decreased SNHG15 expression. Decreased SNHG15 expression promotes cisplatin-induced apoptosis and reactive oxygen species (ROS) accumulation in OS cells. Furthermore, SNHG15 sponges and inhibits the activity of endogenous miR-335-3p, leading to the upregulation of zinc finger protein 32 (ZNF32). Taken together, these findings reveal that p53 downregulates SNHG15 expression in OS. In addition, SNHG15 suppresses cisplatin-induced apoptosis and ROS accumulation through the miR-335-3p/ZNF32 pathway.
Keywords: SNHG15, p53, osteosarcoma, apoptosis
Introduction
Osteosarcoma (OS), a type of sarcoma that mainly originates from bone-forming mesenchymal cells, is the most common primary malignant bone tumor, with a peak incidence in adolescence [1,2]. The OS incidence is approximately 3/1,000,000 and occurs more often in men than in women [3]. Although the recent adoption of multidisciplinary treatment strategies for OS, such as surgery and neoadjuvant chemotherapy, has increased the 5-year survival rate of patients, the overall survival rate remains less than 70% [4,5]. In addition, drug resistance, especially to cisplatin (CDDP), has compounded challenges in the treatment of OS [6]. Therefore, an understanding of the molecular mechanisms of chemoresistance in OS is important to circumvent resistance.
Long noncoding RNAs (lncRNAs) mediate essential biological functions by interacting with DNA, RNA or proteins [7,8]. In addition, lncRNAs may serve as competing endogenous RNAs (ceRNAs) that sponge miRNAs [9]. Based on accumulating evidence, lncRNAs play a key role in diverse types of cancers, such as thyroid cancer [10], hepatocellular carcinoma [11], prostate cancer [12] and melanoma [13]. In addition, numerous lncRNAs, such as SNHG3 [14], AFAP1-AS1 [15] and CCAT2 [16], have been characterized as oncogenes, especially in OS. In contrast, some lncRNAs, such as NKILA [17], HIF2PUT [18] or MEG3 [19], serve as tumor suppressors.
SNHG15, which is located on chromosome 7p13, is a conserved, short-lived lncRNA with a size of 860 bp [20]. Previous studies documented that the upregulation of SNHG15 increases the occurrence, survival and metastasis of tumor cells, such as colorectal, lung and thyroid cancer cells [21]. Furthermore, Liu et al. showed that SNHG15 contributes to invasion, proliferation, migration, and autophagy in OS through the negative regulation of miR-141 [22]. In addition, a series of reports have described the role of SNHG15 in chemotherapeutic resistance [23,24]. Aberrant expression of SNHG15 also contributes to the resistance of lung adenocarcinoma cells and breast cancer cells to gefitinib and cisplatin, respectively [23,24]. However, limited data are available on the molecular regulatory mechanism of SNHG15 and its role in the chemoresistance of OS cells.
Here, we show that SNHG15 inhibits CDDP-induced apoptosis and ROS generation in OS. Furthermore, p53 downregulates SNHG15 expression by directly binding to the SNHG15 promoter in OS cells. In addition, SNHG15 sponges miR-335-3p, leading to the regulation of ZNF32 expression. Together, we report the importance of the p53-SNHG15-miR-335-3p-ZNF32 axis in mediating chemoresistance in OS.
Materials and methods
Cell culture and reagents
The human OS cancer cell lines 143B, U2OS and H1299 were provided by the American Type Culture Collection. We cultured 143B and U2OS cells in DMEM supplemented with 10% fetal bovine serum (FBS; ExCell Bio, Lot: FSP500), 2 mM L-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). H1299 cells were cultured in RPMI-1640 supplemented with 10% fetal bovine serum (FBS; ExCell Bio, Lot: FSP500), 2 mM L-glutamine, penicillin (100 U/ml), and streptomycin (100 μg/ml). The cells were maintained at 37°C in a humidified atmosphere with 5% CO2. The following antibodies and drugs were used in this study: PARP1 (Santa Cruz Biotechnology, SC-8007, 1:1000), p53 (Santa Cruz Biotechnology, SC-126, 1:10 for ChIP, 1:1000 for WB), GAPDH (Santa Cruz Biotechnology, SC-25778, 1:1000), ZNF32 (Proteintech, 14266-1-AP) and cisplatin (Sigma, P4394).
RNA interference
RNA interference was performed as previously described [25]. The shRNA was purchased from Sigma. In addition, the following sequences were used to target p53: p53 No. 1: CGGCGCACAGAGGAAGAGAA and No. 2: GTCCAGATGAAGCTCCCAGAA. We used siRNAs to silence SNHG15 expression using the following siRNA sequences: No. 1 5’-GCAGUCUUUGUCCAUGAAA-3’ and No. 2 5’-GCAAGCCUUGGCACCUUAA-3’.
MicroRNA mimics and inhibitors
The miRNA-335-3p mimics and inhibitors were synthesized by GenePharma Company (Shanghai, People’s Republic of China). For each transfection in a six-well plate, 100 nM miRNA mimics, scrambled miRNAs or miRNA inhibitor were used. OS cells were transfected using Oligofectamine (Invitrogen) according to the manufacturer’s instructions.
Long noncoding RNA sequencing analysis
RNA extraction, library construction, sequencing and data analysis were performed by BioMarker (Beijing, China).
Real-time RT-PCR and RT-PCR
Total RNA was extracted using TRIzol (Invitrogen). One microgram of total RNA was used for cDNA synthesis using the PrimeScriptTM RT reagent kit (Takara, RR047A) according to the manufacturer’s instructions. We used the following primers: actin: F: 5-GACCTGACTGACTACCTCATGAAGAT-3 and R: 5-GTCACACTTCATGATGGAGTTGAAGG-3; and SNHG15: F: 5-GTCTTCGGCAGTCTAGTCATC-3 and R: 5-CTCTTCCACTTTGAGACCGTC-3.
Promoter reporters and dual-luciferase assay
After transfecting the OS cells, luciferase activity was measured in a 1.5 ml Eppendorf tube using the Promega Dual-Luciferases Reporter Assay kit (Promega E1980) according to the manufacturer’s protocol. We normalized the relative Renilla luciferase activity to the firefly luciferase activity.
We inserted the SNHG15 promoter in the pGL3 basic vector to evaluate the potential regulation of this promoter by p53. We used the following primer sequences: P1 Up: gcGGTACCtaaaaactgtgacctcc; dn: gcCTCGACgtctctcgaccgccctt; P2 Up: gcGGTACCtccacccgcctccccg; dn: gcCTCGACgtctctcgaccgccctt; P3 Up: gcGGTACCatgaaacctctccac; dn: gcCTCGACgtctctcgaccgccctt; p4 gcGGTACCAtgaaacctctccaca; dn: gcCTCGACgtctctcgaccgccctt.
On the other hand, we inserted the SNHG15 and ZNF32 3’UTRs in the pSICHECK2 plasmid to evaluate whether miR-335-3p potentially regulated these sequences. We used the following primers: SNHG15 UP: gcGCGGCCGCCGGCGCAGCGCGCGGCGT, Dn: gcCTCGACCATATTTAAATCCATA; and ZNF32 3’UTR UP: gcGCGGCCGCCCACTTTCCTGAAGA, Dn: gcCTCGAC TAATGATAATAAACAAG.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation tests were performed using the Millipore ChIP kit (17-371RF) according to the manufacturer’s instructions. In addition, we used the following specific primers for RT-PCR to bind DNA fragments: BS Forward: 5’-taaaaactgtgacctcc-3’; Reverse: 5’-tcacctgaggtcaggaa-3’.
Cell viability assay
U2OS and 143B OS cells with or without SNHG15 overexpression were plated in 96-well plates at a density of 5000 cells in 200 µl of medium per well 24 hours before the experiment. Cells were treated with 10 μM cisplatin, and then cell viability was determined using the CCK-8 assay kit according to the manufacturer’s instructions.
Annexin V-FITC staining and FACS
We estimated cell death using the cell apoptosis assay (YEASEN) according to the staining protocol described by the manufacturer. Briefly, 5×105 cells were harvested by centrifugation at 1000 g for 5 minutes and then suspended cells in 100 μl of binding buffer. The cells were incubated for 10 minutes with 5 μl of Annexin V-Alexa Fluor 488 and 10 μl of PI at room temperature in the dark. After the addition of 400 μl of binding buffer, we performed a FACS (BD) analysis to detect apoptotic cells.
We also performed the ROS assay (Beyotime) using the staining protocol described by the manufacturer.
Statistical analysis
The statistical significance of the differences between various groups was determined using a one-tailed paired t test, and error bars represent the standard deviations of the means (s.d.).
Results
The tumor suppressor gene p53 downregulated the expression of the SNHG15 gene
The tumor suppressor gene p53 often undergoes inactivating mutations in many human cancers, including OS. We used H1299 cells, a p53-deficient lung adenocarcinoma cell line, to establish stable expression of wild-type p53 controlled by doxycycline as a method to assess whether wild-type p53 altered lncRNA expression. After an incubation with or without doxycycline, the cells were subjected to lncRNA sequencing, and then altered lncRNAs were identified (Figure 1A-C and Supplementary Table 1). Among other lncRNAs, p53 significantly suppressed the expression of SNHG15 (Figure 1D). We generated p53-overexpressing H1299 cells and confirmed the change in SNHG15 expression using qRT-PCR and RT-PCR to confirm that SNHG15 expression was suppressed. Our findings showed that p53 overexpression inhibited SNHG15 expression (Figure 1E-G). We subsequently overexpressed p53 in H1299 cells. Compared with the control cells, the ectopic expression of p53 downregulated SNHG15 expression in a dose-dependent manner (Figure 1H and 1I). Based on the decreased SNHG15 expression in H1299 cells expressing wild-type p53, we then analyzed the effects of p53 mutants (p53-R175H and p53-R273H) on SNHG15 expression. Intriguingly, unlike wild-type p53, the R175H and R273H mutants did not decrease SNHG15 expression (Figure 1J-L).
Figure 1.

p53 downregulates SNHG15 expression. (A-C) p53 Tet-on H1299 cells were treated with or without doxycycline for 24 h. Cells were then subjected to RNA sequencing analysis (A), and the differentially expressed genes are shown (B, C). (D) The expression level of SNHG15 was shown. (E-G) The expression of wild-type p53 was induced by doxycycline in H1299 cells, and SNHG15 expression was analyzed using RT-PCR and qRT-PCR (E, F). The expression of p53 was detected using Western blotting (G). (H, I) Wild-type p53 was introduced into H1299 cells, and then SNGH15 expression was analyzed using RT-PCR and qRT-PCR (H, I). (J-L) RT-PCR and qRT-PCR data showing SNHG15 expression in H1299 cells transfected with wild-type p53 or mutant p53 (R175H or R273H) constructs and induced by doxycycline (J, K). Western blot analysis showing the expression of p53 or its mutant protein (L). (M-O) RT-PCR and qRT-PCR data showing SNHG15 expression in U2OS cells with p53 knockdown (M, N). Western blot analysis showing the expression of p53 (O). (P, Q) U2OS cells with or without p53 knockdown were treated with cisplatin (CDDP). SNHG15 expression was analyzed using RT-PCR and qRT-PCR.
We silenced the p53 gene in U2OS cells using two different shRNAs to further confirm the downregulation of the SNHG15 gene by p53 in OS. Compared with the control cells, p53 silencing substantially increased SNHG15 expression (Figure 1M-O). We then treated control or p53 knockdown U2OS cells with 5 μM cisplatin. Cisplatin markedly decreased SNHG15 expression, a phenomenon that was abolished by silencing the p53 gene (Figure 1P and 1Q). Taken together, p53 downregulates SNHG15 expression.
p53 binds to the promoter of the SNHG15 gene
We determined the p53-binding sites in the SNHG15 gene by first inserting the upstream sequence of the SNHG15 gene as well as three different truncations into pGL3-based plasmids, which were referred to as P1-P4 (Figure 2A). The P1-P4 plasmids were separately transfected into 293T cells with or without p53 overexpression. Unlike P2-P4-transfected cells, P1 luciferase activity was reduced in cells overexpressing the p53 gene compared with control cells (Figure 2B). Thus, this finding implies that the region from -2000 to -1500 bp was critical for the regulation of SNHG15 expression by the p53 protein. We transfected the P1 plasmid into H1299 cells with or without p53 overexpression or U2OS cells with or without p53 knockdown to validate this observation. Overexpression of the p53 gene suppressed the luciferase activity of P1 in H1299 cells compared with the control group (Figure 2C). However, p53 knockdown increased the luciferase activity of cells expressing the P1 plasmid (Figure 2D).
Figure 2.
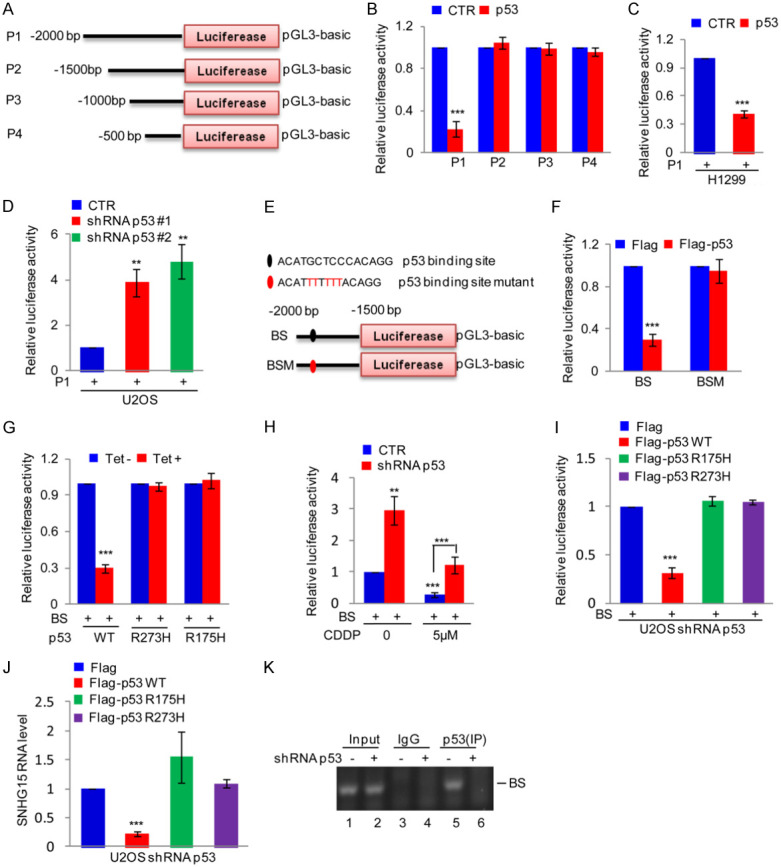
p53 binds the SNHG15 promoter. (A) Schematic illustration of the pGL3-based reporter constructs used to evaluate SNHG15 promoter activity. (B) The SNHG15 promoter constructs P1, P2, P3 and P4 were separately transfected into 293T cells with or without p53, and then the promoter activity was detected. Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (C) P1 was transfected into H1299 cells with or without p53 overexpression. The luciferase activity was measured. Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (D) P1 was transfected into p53 knockdown U2OS cells, and then SNHG15 promoter activity was measured. Data are presented as the means ± SD of triplicate measurements, **P<0.01. (E) Schematic illustration of the wild-type p53 binding site (BS) and the matching mutant site (BSM) in the SNHG15 promoter. (F) BS or BSM was transfected with or without p53 into HEK293T cells, and then the promoter activity of BS and BSM was measured. Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (G) BS was introduced into H1299 cells along with p53 or p53 mutants, and then the promoter activity was detected. Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (H) BS was introduced into p53 knockdown U2OS cells or control cells. Cells were then treated with or without 5 μM CDDP for 24 h before assaying for luciferase activity. Data are presented as the means ± SD of triplicate measurements, **P<0.01 and ***P<0.001. (I, J) The wild type p53 and mutants were introduced into p53 silenced U2OS cells together with BS. The activity of BS and SNHG15 expression were measured (I, J). Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (K) ChIP analysis showing the binding of p53 to the SNHG15 promoter in p53 knockdown U2OS cells or control cells.
An assessment of the P1 sequence using the JASPAR database showed a putative p53 binding site in the SNHG15 promoter. In addition, we validated the p53 binding site by employing two different pGL3-based luciferase reporter plasmids containing the wild-type binding site (BS) and binding site mutant (BSM) (Figure 2E). The BS and BSM plasmids were transfected into 293T cells with the p53 overexpression or control plasmid. Overexpression of the p53 gene strongly inhibited the activity of the luciferase reporter gene harboring BS but not BSM (Figure 2F). Subsequently, BS was transfected into H1299 cells with or without p53 or the mutant expression plasmid, and then the luciferase activity was assayed. Unlike the mutants, wild-type p53 downregulated the luciferase activities of BS (Figure 2G). In addition, we transfected BS into U2OS cells with or without p53 silencing. The cells were then treated with 5 μM cisplatin. This assay showed decreased luciferase activity in U2OS cells compared to the control group in response to cisplatin treatment. However, the decreased luciferase activity was reversed after 5 μM cisplatin treatment in cells with p53 knockdown (Figure 2H). To further confirm it, we transfected the p53 or the mutant expression plasmid together with BS into U2OS cells with p53 knockdown. The luciferase activity of BS and SNHG15 expression were detected. Similarly, wild-type p53 not the mutants downregulated the luciferase activities of BS and SNHG15 expression in osteosarcoma cells (Figure 2I and 2J). In addition, we performed a chromatin immunoprecipitation (ChIP) assay to determine the specificity of p53 binding to chromatin fragments containing BS using a p53 antibody for immunoprecipitation of samples from U2OS cells. The binding capacity of the p53 protein to the SNHG15 promoter was weakened in p53 knockdown cells (Figure 2K). Based on these results, the p53 protein binds to the SNHG15 promoter and downregulates the expression of the SNHG15 gene.
SNHG15 diminishes cisplatin-induced apoptosis and ROS accumulation in osteosarcoma
We overexpressed SNHG15 in U2OS and 143B cells to explore the functions of SNHG15 in OS cells. The cells were then treated with a gradual increase in cisplatin concentration. Cell apoptosis and viability were analyzed using Western blot and CCK-8 assays. Compared with control cells, SNHG15 overexpression reduced cisplatin-induced apoptosis, as evidenced by the decreased PARP cleavage and increased cell viability (Figure 3A-E). In addition, flow cytometry analyses indicated that SNHG15 suppressed cisplatin-induced apoptosis in OS cells (Figure 3F and 3G).
Figure 3.

SNHG15 decreased cisplatin-induced apoptosis and ROS levels in osteosarcoma cells. (A) SNHG15 was overexpressed in U2OS and 143B cells and assessed using RT-PCR. (B-G) U2OS and 143B cells with or without SNHG15 overexpression were treated with cisplatin. Cell apoptosis and cell viability were analyzed using Western blot (B and D), CCK-8 (C and E) and flow cytometry assays (F and G). The relative expression of cleaved PARP to GAPDH was quantified in (B and D). Data in (C, E and G) are presented as the means ± SD of triplicate measurements, **P<0.01 and ***P<0.001. (H) SNHG15 was silenced in U2OS cells using siRNAs, and its expression was analyzed using RT-PCR. (I-K) Cell apoptosis was analyzed using flow cytometry (I, J) and Western blot analysis (K). (L, M) ROS levels were analyzed using flow cytometry. Data in M are presented as the means ± SD of triplicate measurements, **P<0.01 and ***P<0.001. (N) p53 expression was silenced in U2OS cells with or without SNHG15 knockdown, and then the cells were treated with cisplatin. Cell apoptosis was detected using Western blotting.
We silenced SNHG15 using siRNAs in U2OS cells and then confirmed the knockdown efficiency using RT-PCR to further verify this observation. The data showed a significant reduction in SNHG15 expression (Figure 3H). Cells were treated with cisplatin, and cell apoptosis was measured using flow cytometry and Western blot analyses. SNHG15 knockdown facilitated cisplatin-induced cell apoptosis (Figure 3I-K). In addition, compared with the control group, overexpression of the SNHG15 gene diminished cisplatin-induced ROS accumulation in U2OS cells (Figure 3L and 3M).
Since our data showed that p53 inhibited SNHG15 expression, we sought to evaluate whether p53-mediated increase in cisplatin-induced cell apoptosis was mediated by SNHG15. We silenced the SNHG15 gene in U2OS cells with or without p53 depletion and then treated the cells with or without 5 μM cisplatin. Notably, p53 knockdown markedly decreased cisplatin-induced cell apoptosis. However, the decrease in apoptosis was reversed by SNHG15 knockdown (Figure 3N). Taken together, SNHG15 is a key regulator of chemoresistance in OS cells and mediates the effect of p53 on cisplatin-induced apoptosis.
SNHG15 suppresses cisplatin-induced apoptosis by functioning as a ceRNA of miR-335-3p
Based on the findings of a key role for SNHG15 in the suppression of cisplatin-induced apoptosis, we aimed to identify downstream effector proteins. Previous studies indicated that SNHG15 functions as a ceRNA with potential roles in posttranscriptional regulation [26]. We predicted the downstream miRNAs of SNHG15 using a bioinformatics analysis. Two candidate miRNAs (miR-7158-5p and miR-335-3p) were shown to bind SNHG15 (Figure 4A). A pSICHECK-2 reporter plasmid containing wild-type SNHG15 (SNHG15 WT) was transfected into U2OS and 143B cells with or without miR-7158-5p or miR-335-3p to confirm the in silico data. We showed that miR-7158-5p and miR-335-3p reduced the luciferase activity of SNHG15 WT. However, the downregulation of SNHG15 WT activity by miR-335-3p was more significant that than by miR-7158-5p in OS cells (Figure 4B and 4C). We then transfected SNHG15 WT into 143B cells with or without miR-7158-5p or miR-335-3p inhibitors. Measurement of the luciferase activity showed that, compared with the control group, the miR-335-3p inhibitor noticeably increased the luciferase activity of SNGH15 WT in 143B cells (Figure 4D).
Figure 4.
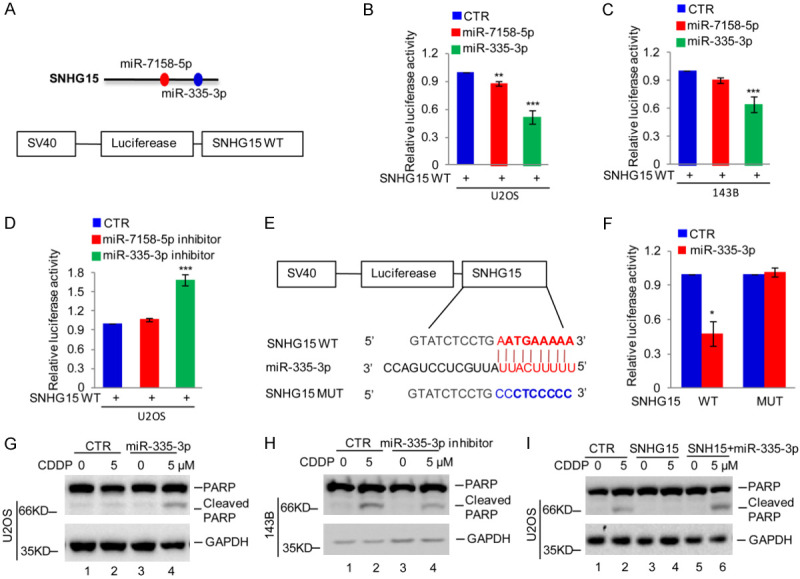
SNHG15 suppressed cisplatin-induced apoptosis by acting as a ceRNA of miR-335-3p. (A) The miRNAs binding SNHG15 were predicted by the miRDB database. (B, C) The pSICHECK-2 reporter plasmid containing wild-type SNHG15 (SNHG15 WT) was transfected into U2OS and 143B cells with or without miR-7158-5p or miR-335-3p, and then the activity of SNHG15 WT was detected. Data in (B and C) are presented as the means ± SD of triplicate measurements, **P<0.01 and ***P<0.001. (D) SNHG15 WT was transfected into U2OS cells with or without miR-7158-5p or miR-335-3p inhibitors, and then the activity of SNGH15 WT was detected. Data are presented as the means ± SD of triplicate measurements, ***P<0.001. (E) Schematic illustration of the pSICHECK-2 reporter plasmid containing the miR-335-3p wild-type binding site (BS) in SNHG15 and the matching mutant (BSM) that were used in luciferase assays. (F) SNHG15 WT or MUT was transfected into U2OS cells with or without miR-335-3p, and then the luciferase activity was measured. Data are presented as the means ± SD of triplicate measurements, *P<0.05. (G) U2OS cells with or without miR-335-3p overexpression were treated with cisplatin. Cell apoptosis was analyzed using Western blotting. (H) miR-335-3p was inhibited in 143B cells, and then the cells were treated with or without cisplatin. Cell apoptosis was analyzed using Western blotting. (I) U2OS cells with or without SNHG15 overexpression were transfected with miR-335-3p. The cells were then treated with cisplatin, and cell apoptosis was subsequently analyzed using Western blotting.
Thereafter, we inserted SNHG15 containing the miR-335-3p wild-type binding site (SNHG15 WT) or the matching mutant (SNHG15 MUT) into the pSICHECK-2 reporter plasmid (Figure 4E). These plasmids were transfected into U2OS cells with or without miR-335-3p overexpression. The luciferase activity was significantly suppressed by miR-335-3p in the cells expressing SNHG15 WT but not SNHG15 MUT (Figure 4F). Thus, SNHG15 functions as a ceRNA of miR-335-3p.
We evaluated whether SNHG15 inhibits cisplatin-induced apoptosis by sponging miR-335-3p by first investigating the effect of miR-335-3p on cisplatin-induced apoptosis and showed that miR-335-3p overexpression increased cisplatin-induced apoptosis in U2OS cells (Figure 4G). As expected, inhibition of miR-335-3p deceased cisplatin-induced apoptosis (Figure 4H). We overexpressed miR-335-3p in U2OS cells with or without SNHG15 overexpression and then treated them with or without 5 μM cisplatin. Notably, miR-335-3p overexpression diminished the effect of SNHG15 on cisplatin-induced cell apoptosis (Figure 4I). Taken together, SNHG15 inhibits cisplatin-induced apoptosis by sponging miR-335-3p.
SNHG15 decreases cisplatin-induced apoptosis and ROS accumulation by modulating the miR-335-3p-ZNF32 axis
Based on findings described above that SNHG15 suppresses cisplatin-induced apoptosis and ROS accumulation by sponging miR-335-3p, we hypothesized that miR-335-3p downstream genes might be key regulators of cell apoptosis and oxidative stress. ZNF32 was one of the predicted target genes of miR-335-3p, and it is involved in the regulation of cell oxidative stress and chemoresistance. We overexpressed miR-335-3p in U2OS or 143B cells to validate these in silico analyses. We then assayed ZNF32 expression using Western blot analysis. The data showed significant inhibition of ZNF32 expression by miR-335-3p (Figure 5A and 5B). In contrast, depletion of miR-335-3p increased ZNF32 expression (Figure 5C and 5D). Thereafter, we inserted the ZNF32 3’UTR containing the wild-type binding site (BS) or the matching mutant (BSM) into the pSICHEK2 plasmid (Figure 5E). BS and BSM were then transfected into U2OS cells with or without miR-335-3p overexpression, and then luciferase activity was measured. Compared with the control group, miR-335-3p significantly reduced the luciferase activity of the BS-containing cells but not in the cells transfected with BSM. Collectively, these data suggest that miR-335-3p targets ZNF32 in OS (Figure 5F).
Figure 5.

SNHG15 decreased cisplatin-induced apoptosis and ROS levels by regulating the miR-335-3p-ZNF32 axis. A, B. miR-335-3p was transfected into U2OS or 143B cells. We then analyzed ZNF32 expression using Western blotting. The relative expression of ZNF32 to GAPDH was quantified. C, D. The miR-335-3p inhibitor was transfected into U2OS or 143B cells, and then ZNF32 expression was analyzed using Western blotting. The relative expression of ZNF32 to GAPDH was quantified. E. The miR-335-3p binding site in the ZNF32 3’UTR was predicted by the TargetScan database. Schematic illustration of the pSICHECK-2 reporter plasmid containing the miR-335-3p wild-type binding site (BS) in the ZNF32 3’UTR or the matching mutant (BSM) used in the luciferase assays. F. ZNF32 3’UTR WT or MUT was transfected into U2OS cells with or without miR-335-3p overexpression, and then the luciferase activity was assessed. The data are presented as the means ± SD of triplicate measurements, ***P<0.001. G. SNHG15 expression was silenced in U2OS or 143B cells, and then ZNF32 levels were analyzed using Western blotting. H. The miR-335-3p inhibitor was transfected into U2OS cells with or without SNHG15 knockdown. ZNF32 expression was measured using Western blot analysis. I. U2OS cells with or without p53 knockdown were transfected with miR-335-3p, and then ZNF32 and p53 levels were analyzed using Western blotting. J. U2OS cells with or without ZNF32 overexpression were treated with cisplatin, and cell apoptosis was measured using Western blotting. The relative expression of cleaved PARP to GAPDH was quantified. K, L. U2OS cells with or without ZNF32 overexpression were treated with cisplatin, and then ROS levels were measured using flow cytometry. The data are presented as the means ± SD of triplicate measurements, ***P<0.001. M, N. SNHG15 was silenced in U2OS cells with or without ZNF32 overexpression and then cells were treated with cisplatin. Cell apoptosis was measured using flow cytometry. The data are presented as the means ± SD of triplicate measurements, ***P<0.001.
We silenced SNHG15 in U2OS or 143B cells to investigate the effects of SNHG15 and p53 on the miR-335-3p-specific expression of ZNF32. Compared with the control cells, SNHG15 knockdown reduced ZNF32 expression, but this change was reversed by the miR-335-3p inhibitor (Figure 5G and 5H). Similarly, p53 knockdown upregulated ZNF32 expression, a phenomenon that was abolished by overexpression of miR-335-3p (Figure 5I). Based on these data, the regulation of ZNF32 expression by SNHG15 or p53 was mediated by miR-335-3p.
In addition, we examined the effect of ZNF32 overexpression on cisplatin-induced cell apoptosis and ROS accumulation in OS cells. Compared with the control group, the upregulation of ZNF32 inhibited cisplatin-induced cell apoptosis and ROS accumulation (Figure 5J-L). In addition, the upregulation of ZNF32 reversed the increase in cell apoptosis induced by SNHG15 depletion (Figure 5M and 5N), thus indicating that SNGH15 suppressed cisplatin-induced OS cell apoptosis by regulating the miR-335-3P/ZNF32 axis.
Discussion
Although SNHG15 has been shown to mediate the development of multiple cancers, limited data are available on its role and regulatory mechanism in osteosarcoma. In the present study, we dissected the role of the lncRNA SNHG15 in cisplatin-induced OS cell apoptosis. We showed that p53 suppresses SNHG15 expression by directly binding to the SNHG15 promoter in OS cells. Furthermore, SNHG15 suppresses cisplatin-induced apoptosis and ROS accumulation by targeting the miR-335-3p-ZNF32 axis. Our study indicated that the p53-SNHG15-miR-335-3p-ZNF32 signaling pathway plays a key role in cisplatin-induced OS cell apoptosis.
Previous studies documented the upregulation of both SNHG15 and MYC in colorectal cancer, and SNHG15 was shown to increase cell proliferation, invasion and resistance to 5-FU [27]. Further investigation revealed that two E-box motifs in the SNHG15 sequence were bound by the MYC protein, implying that SNHG15 transcription is directly modulated by the MYC oncogene [27]. Here, we showed the significant suppression of SNHG15 expression in response to wild-type p53, indicating that SNHG15 might be downregulated by the tumor suppressor gene p53. Further studies confirmed that only WT p53, but not mutant p53, repressed SNHG15 expression by directly binding to its promoter in a transcription-dependent manner.
In addition, accumulating evidence has shown the upregulation of SNHG15 in hepatocellular carcinoma, pancreatic ductal adenocarcinoma and colorectal cancer [28-30]. In addition, SNHG15 is associated with chemoresistance in breast cancer by targeting miR-381 [24]; thus, SNHG15 might be a potential oncogene that induces drug resistance. Consistent with previous studies, SNHG15 decreased cisplatin-induced apoptosis and ROS accumulation in OS cells in the present study. SNHG15 overexpression decreased cisplatin-induced apoptosis and ROS accumulation and increased cell viability. In addition, SNHG15 ablation significantly facilitated cisplatin-induced cell apoptosis. On the other hand, p53 knockdown substantially repressed cisplatin-induced cell apoptosis, a phenomenon that was reversed by SNHG15 knockdown. Based on these data, SNHG15 contributes to the p53-mediated resistance of osteosarcoma cells to cisplatin.
We further studied the potential mechanisms of SNHG15 in the resistance of OS cells to cisplatin. Previous studies have shown that SNHG15 suppresses the expression of miRNAs by functioning as a ceRNA of miRNAs [26,31]. Consequently, using bioinformatics analysis, we predicted miR-335-3p as a putative SNHG15 target. In addition, luciferase reporter assays showed that miR-335-3p was a downstream target of SNHG15. On the other hand, miR-335-3p might function as a tumor suppressor. For example, Zhao et al. showed low expression of miR-335-3p in non-small cell lung cancer (NSCLC) tissues and cell lines compared with normal cells and this miRNA targeted S100 calcium-binding protein A14 (S100A14) to exert its effect as a tumor suppressor. Moreover, miR-355-3p expression is suppressed in OS, and inhibition of miR-355-3p increases the proliferation, migration and invasion of OS cells [32]. In our study, miR-335-3p overexpression enhanced cisplatin-induced apoptosis in U2OS cells. As expected, inhibition of miR-335-3p deceased cisplatin-induced apoptosis. Therefore, SNHG15 inhibits cisplatin-induced apoptosis by sponging miR-335-3p.
On the other hand, ZNF32, a crucial transcription factor belonging to the Kruppel-related zinc finger family, has been shown to contribute to the induction of multidrug resistance in lung cancer [33]. In our study, ZNF32 inhibited cisplatin-induced cell apoptosis and ROS accumulation. In addition, ZNF32 expression was inhibited by miR-355-3p by binding of the 3’UTR. In addition, further investigation suggested that SNHG15 induced ZNF32 expression by sponging miR-355-3p, thus decreasing CDDP-induced apoptosis in OS cells.
Taken together, SNHG15, a p53-responsive lncRNA, represses CDDP-induced apoptosis and ROS accumulation in human osteosarcoma cells through the miR-355-3p/ZNF32 axis.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (No. 31971275 to Hong Wang and No. 81902743 to Yuan Wang) and the Liaoning Provincial Natural Science Foundation of China (No. 2015020670 and LJKZ0827) to Xiaodong Li.
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Yang X, Yang P, Shen J, Osaka E, Choy E, Cote G, Harmon D, Zhang Z, Mankin H, Hornicek FJ, Duan Z. Prevention of multidrug resistance (MDR) in osteosarcoma by NSC23925. Br J Cancer. 2014;110:2896–2904. doi: 10.1038/bjc.2014.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cortini M, Avnet S, Baldini N. Mesenchymal stroma: role in osteosarcoma progression. Cancer Lett. 2017;405:90–99. doi: 10.1016/j.canlet.2017.07.024. [DOI] [PubMed] [Google Scholar]
- 3.Wang X, Lan Z, He J, Lai Q, Yao X, Li Q, Liu Y, Lai H, Gu C, Yan Q, Fang Y, Zhang Y, Li A, Liu S. LncRNA SNHG6 promotes chemoresistance through ULK1-induced autophagy by sponging miR-26a-5p in colorectal cancer cells. Cancer Cell Int. 2019;19:234. doi: 10.1186/s12935-019-0951-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao S, Su Y, Duan J, Qiu Q, Ge X, Wang A, Yin Y. Radiomics signature extracted from diffusion-weighted magnetic resonance imaging predicts outcomes in osteosarcoma. J Bone Oncol. 2019;19:100263. doi: 10.1016/j.jbo.2019.100263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang S, Chen H, Liu W, Fang L, Qian Z, Kong R, Zhang Q, Li J, Cao X. miR-766-3p Targeting BCL9L suppressed tumorigenesis, epithelial-mesenchymal transition, and metastasis through the β-catenin signaling pathway in osteosarcoma cells. Front Cell Dev Biol. 2020;8:594135. doi: 10.3389/fcell.2020.594135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wei X, Xu L, Jeddo SF, Li K, Li X, Li J. MARK2 enhances cisplatin resistance via PI3K/AKT/NF-κB signaling pathway in osteosarcoma cells. Am J Transl Res. 2020;12:1807–1823. [PMC free article] [PubMed] [Google Scholar]
- 7.Song EL, Xing L, Wang L, Song WT, Li DB, Wang Y, Gu YW, Liu MM, Ni WJ, Zhang P, Ma X, Zhang X, Yao J, Chen Y, An RH. LncRNA ADAMTS9-AS2 inhibits cell proliferation and decreases chemoresistance in clear cell renal cell carcinoma via the miR-27a-3p/FOXO1 axis. Aging (Albany NY) 2019;11:5705–5725. doi: 10.18632/aging.102154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kopp F, Mendell JT. Functional classification and experimental dissection of long noncoding RNAs. Cell. 2018;172:393–407. doi: 10.1016/j.cell.2018.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tay Y, Rinn J, Pandolfi PP. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Deng H, Zhao Y, Li C, Liang Y. LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor growth of thyroid cancer through MET-PI3K-AKT signaling. J Exp Clin Cancer Res. 2018;37:279. doi: 10.1186/s13046-018-0950-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen J, Huang X, Wang W, Xie H, Li J, Hu Z, Zheng Z, Li H, Teng L. LncRNA CDKN2BAS predicts poor prognosis in patients with hepatocellular carcinoma and promotes metastasis via the miR-153-5p/ARHGAP18 signaling axis. Aging (Albany NY) 2018;10:3371–3381. doi: 10.18632/aging.101645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lingadahalli S, Jadhao S, Sung YY, Chen M, Hu L, Chen X, Cheung E. Novel lncRNA LINC00844 regulates prostate cancer cell migration and invasion through AR signaling. Mol Cancer Res. 2018;16:1865–1878. doi: 10.1158/1541-7786.MCR-18-0087. [DOI] [PubMed] [Google Scholar]
- 13.Chen X, Gao J, Yu Y, Zhao Z, Pan Y. LncRNA FOXD3-AS1 promotes proliferation, invasion and migration of cutaneous malignant melanoma via regulating miR-325/MAP3K2. Biomed Pharmacother. 2019;120:109438. doi: 10.1016/j.biopha.2019.109438. [DOI] [PubMed] [Google Scholar]
- 14.Zheng S, Jiang F, Ge D, Tang J, Chen H, Yang J, Yao Y, Yan J, Qiu J, Yin Z, Ni Y, Zhao L, Chen X, Li H, Yang L. LncRNA SNHG3/miRNA-151a-3p/RAB22A axis regulates invasion and migration of osteosarcoma. Biomed Pharmacother. 2019;112:108695. doi: 10.1016/j.biopha.2019.108695. [DOI] [PubMed] [Google Scholar]
- 15.Shi D, Wu F, Mu S, Hu B, Zhong B, Gao F, Qing X, Liu J, Zhang Z, Shao Z. LncRNA AFAP1-AS1 promotes tumorigenesis and epithelial-mesenchymal transition of osteosarcoma through RhoC/ROCK1/p38MAPK/Twist1 signaling pathway. J Exp Clin Cancer Res. 2019;38:375. doi: 10.1186/s13046-019-1363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan L, Wu X, Yin X, Du F, Liu Y, Ding X. LncRNA CCAT2 promoted osteosarcoma cell proliferation and invasion. J Cell Mol Med. 2018;22:2592–2599. doi: 10.1111/jcmm.13518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang GD, Li Y, Liao GJ, Qiu HW. LncRNA NKILA inhibits invasion and migration of osteosarcoma cells via NF-κB/Snail signaling pathway. Eur Rev Med Pharmacol Sci. 2019;23:4118–4125. doi: 10.26355/eurrev_201905_17913. [DOI] [PubMed] [Google Scholar]
- 18.Zhao D, Wang S, Chu X, Han D. LncRNA HIF2PUT inhibited osteosarcoma stem cells proliferation, migration and invasion by regulating HIF2 expression. Artif Cells Nanomed Biotechnol. 2019;47:1342–1348. doi: 10.1080/21691401.2019.1596934. [DOI] [PubMed] [Google Scholar]
- 19.Chen L, Wang J, Li JW, Zhao XW, Tian LF. LncRNA MEG3 inhibits proliferation and promotes apoptosis of osteosarcoma cells through regulating notch signaling pathway. Eur Rev Med Pharmacol Sci. 2020;24:581–590. doi: 10.26355/eurrev_202001_20034. [DOI] [PubMed] [Google Scholar]
- 20.Tani H, Torimura M. Identification of short-lived long non-coding RNAs as surrogate indicators for chemical stress response. Biochem Biophys Res Commun. 2013;439:547–551. doi: 10.1016/j.bbrc.2013.09.006. [DOI] [PubMed] [Google Scholar]
- 21.Shuai Y, Ma Z, Lu J, Feng J. LncRNA SNHG15: a new budding star in human cancers. Cell Prolif. 2020;53:e12716. doi: 10.1111/cpr.12716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu K, Hou Y, Liu Y, Zheng J. LncRNA SNHG15 contributes to proliferation, invasion and autophagy in osteosarcoma cells by sponging miR-141. J Biomed Sci. 2017;24:46. doi: 10.1186/s12929-017-0353-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huang J, Pan B, Xia G, Zhu J, Li C, Feng J. LncRNA SNHG15 regulates EGFR-TKI acquired resistance in lung adenocarcinoma through sponging miR-451 to upregulate MDR-1. Cell Death Dis. 2020;11:525. doi: 10.1038/s41419-020-2683-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mi H, Wang X, Wang F, Li L, Zhu M, Wang N, Xiong Y, Gu Y. SNHG15 contributes to cisplatin resistance in breast cancer through sponging miR-381. Onco Targets Ther. 2020;13:657–666. doi: 10.2147/OTT.S223321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fu D, Lu C, Qu X, Li P, Chen K, Shan L, Zhu X. LncRNA TTN-AS1 regulates osteosarcoma cell apoptosis and drug resistance via the miR-134-5p/MBTD1 axis. Aging (Albany NY) 2019;11:8374–8385. doi: 10.18632/aging.102325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu DM, Wang S, Wen X, Han XR, Wang YJ, Shen M, Fan SH, Zhang ZF, Shan Q, Li MQ, Hu B, Lu J, Chen GQ, Zheng YL. LncRNA SNHG15 acts as a ceRNA to regulate YAP1-Hippo signaling pathway by sponging miR-200a-3p in papillary thyroid carcinoma. Cell Death Dis. 2018;9:947. doi: 10.1038/s41419-018-0975-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Saeinasab M, Bahrami AR, González J, Marchese FP, Martinez D, Mowla SJ, Matin MM, Huarte M. SNHG15 is a bifunctional MYC-regulated noncoding locus encoding a lncRNA that promotes cell proliferation, invasion and drug resistance in colorectal cancer by interacting with AIF. J Exp Clin Cancer Res. 2019;38:172. doi: 10.1186/s13046-019-1169-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang JH, Wei HW, Yang HG. Long noncoding RNA SNHG15, a potential prognostic biomarker for hepatocellular carcinoma. Eur Rev Med Pharmacol Sci. 2016;20:1720–1724. [PubMed] [Google Scholar]
- 29.Guo XB, Yin HS, Wang JY. Evaluating the diagnostic and prognostic value of long non-coding RNA SNHG15 in pancreatic ductal adenocarcinoma. Eur Rev Med Pharmacol Sci. 2018;22:5892–5898. doi: 10.26355/eurrev_201809_15917. [DOI] [PubMed] [Google Scholar]
- 30.Huang L, Lin H, Kang L, Huang P, Huang J, Cai J, Xian Z, Zhu P, Huang M, Wang L, Xian CJ, Wang J, Dong J. Aberrant expression of long noncoding RNA SNHG15 correlates with liver metastasis and poor survival in colorectal cancer. J Cell Physiol. 2019;234:7032–7039. doi: 10.1002/jcp.27456. [DOI] [PubMed] [Google Scholar]
- 31.Ye J, Tan L, Fu Y, Xu H, Wen L, Deng Y, Liu K. LncRNA SNHG15 promotes hepatocellular carcinoma progression by sponging miR-141-3p. J Cell Biochem. 2019;120:19775–19783. doi: 10.1002/jcb.29283. [DOI] [PubMed] [Google Scholar]
- 32.Yu Y, Wang L, Li Z, Zheng Y, Shi Z, Wang G. Long noncoding RNA CRNDE functions as a diagnostic and prognostic biomarker in osteosarcoma, as well as promotes its progression via inhibition of miR-335-3p. J Biochem Mol Toxicol. 2021;35:e22734. doi: 10.1002/jbt.22734. [DOI] [PubMed] [Google Scholar]
- 33.Li J, Ao J, Li K, Zhang J, Li Y, Zhang L, Wei Y, Gong D, Gao J, Tan W, Huang L, Liu L, Lin P, Wei Y. ZNF32 contributes to the induction of multidrug resistance by regulating TGF-β receptor 2 signaling in lung adenocarcinoma. Cell Death Dis. 2016;7:e2428. doi: 10.1038/cddis.2016.328. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.