

Abstract
Fasciculation and elongation protein zeta-1 (FEZ1) is a multifunctional kinesin adaptor involved in processes ranging from neurodegeneration to retrovirus and polyomavirus infection. Here, we show that, although modulating FEZ1 expression also impacts infection by large DNA viruses in human microglia, macrophages, and fibroblasts, this broad antiviral phenotype is associated with the pre-induction of interferon-stimulated genes (ISGs) in a STING-independent manner. We further reveal that S58, a key phosphorylation site in FEZ1’s kinesin regulatory domain, controls both binding to, and the nuclear-cytoplasmic localization of, heat shock protein 8 (HSPA8), as well as ISG expression. FEZ1- and HSPA8-induced changes in ISG expression further involved changes in DNA-dependent protein kinase (DNA-PK) accumulation in the nucleus. Moreover, phosphorylation of endogenous FEZ1 at S58 was reduced and HSPA8 and DNA-PK translocated to the nucleus in cells stimulated with DNA, suggesting that FEZ1 is a regulatory component of the recently identified HSPA8/DNA-PK innate immune pathway.
Graphical abstract
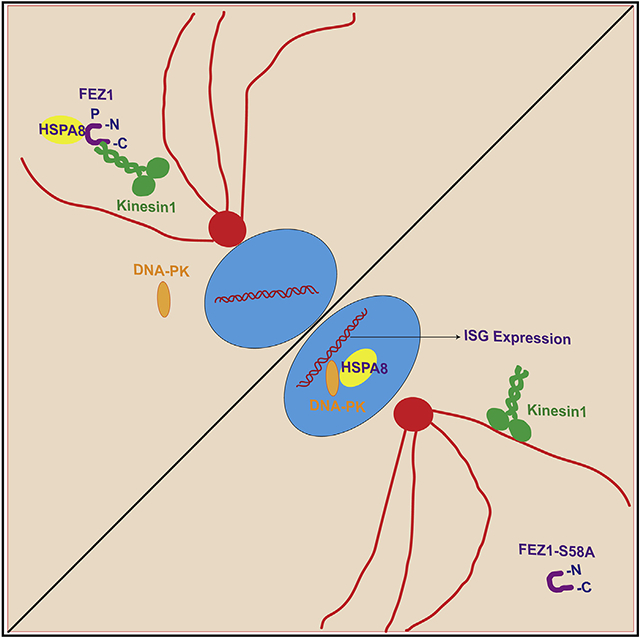
In brief
FEZ1 is a multifunctional adaptor involved in various biological processes, yet its broader role in pathology remains poorly understood. Malikov et al. reveal that Serine 58 in the kinesin regulatory domain of FEZ1 controls the localization of HSPA8 and DNA-PK and thereby functions in STING-independent interferon response pathways.
INTRODUCTION
FEZ1 is an adaptor for the outward-directed microtubule (MT) motor, kinesin-1, and a hub protein that interacts with a range of other proteins (Bloom and Horvitz, 1997) (reviewed in Maturana et al., 2010; Teixeira et al., 2019). These combined functions mean that FEZ1 is involved in various biological processes, including kinesin-mediated transport of vesicles and organelles along MTs in neurons (Fujita et al., 2007; Gindhart et al., 2003; Ikuta et al., 2007). Indeed, despite being expressed in many cell types, FEZ1 is highly expressed in neurons and both FEZ1 and its interacting partners have been linked to neurological diseases. FEZ1’s interaction with disrupted-in-schizophrenia (DISC1), a candidate gene for schizophrenia, is crucial for neurite outgrowth (Kang et al., 2011; Miyoshi et al., 2003), while its interaction with nuclear distribution element-like plays a role in regulating neurogenesis (Colantuoni et al., 2008). Disruption of FEZ1 activity can also cause cytoskeletal rearrangements resulting in interference with axonal outgrowth upon its interaction with necdin (Lee et al., 2005) and FEZ1 expression is reported to be reduced in patients with schizophrenia (Lipska et al., 2006; Vachev et al., 2015).
Recent work from both our group and others has also implicated FEZ1 in regulating viral infection in both neuronal and non-neuronal cell types. FEZ1 was found to block intracellular trafficking of the human neurotropic polyomavirus JC virus (JCV), while FEZ1-mediated inhibition of infection could be overcome by JCV agnoprotein (Suzuki et al., 2005). At the same time, our screens for host factors that regulate retroviral infection in non-neuronal cells found that FEZ1 regulates infection by retroviruses, such as murine leukemia virus and human immunodeficiency virus type 1 (HIV-1) (Gao and Goff, 1999; Haedicke et al., 2009; Naghavi et al., 2005). In the case of HIV-1, FEZ1 binds directly to the viral capsid (Huang et al., 2019) and, although it is a kinesin-1 adaptor, it regulates the balance of retrograde (inward) versus anterograde (outward) motility of HIV-1 particles to ensure net forward movement toward the nucleus for efficient infection (Malikov et al., 2015). This pro-viral function of FEZ1 is further regulated by MT-associated regulatory kinase 2, a host kinase that locally regulates FEZ1 phosphorylation on virus particles (Butkevich et al., 2016; Malikov and Naghavi, 2017). Indeed, phosphorylation of FEZ1 at Serine 58 (S58), which regulates its interaction with the kinesin-1 heavy chain, is required for trafficking and disassembly of incoming HIV-1 capsid during early infection (Chua et al., 2012; Malikov et al., 2015).
Cumulatively, these findings link FEZ1 to the diverse processes of RNA and small DNA virus infection, as well as neuronal development and neurological disorders, yet the underlying mechanisms by which FEZ1 influences physiological and pathological processes and its broader role in pathology remains poorly understood. As a result of testing its effects on infection by two distinct large DNA viruses, here we reveal that FEZ1 regulates stimulator of interferon genes (STING)-independent induction of IFN and ISG expression. Furthermore, we reveal that S58 in the N-terminal kinesin regulatory domain of FEZ1 controls the localization and activity of heat shock protein A8 (HSPA8) and DNA-PK, thereby positioning FEZ1 as a regulatory component of the HSPA8/DNA-PK arm of host innate immune response pathways.
RESULTS AND DISCUSSION
Depletion of FEZ1 induces ISG responses and inhibits DNA virus infection
Given that FEZ1 is known to regulate infection by RNA and small DNA viruses, we tested whether it could also influence infection by large DNA viruses. We did this by examining the effects of depleting FEZ1 in human immune cell lines and primary fibroblasts as these cells are central to infection by many types of viruses. We found that, compared with control non-targeting siRNAs, siRNA-mediated depletion of FEZ1 potently suppressed infection of either human microglia CHME3 cells or primary normal human dermal fibroblasts (NHDFs) with herpes simplex virus type 1 (HSV-1), as determined by reduced expression of early or intermediate infected cell proteins, ICP0, ICP4, or ICP5 in western blotting (WB) (Figures 1A and S1A, respectively). Similar to retroviruses or polyomaviruses, herpesviruses must reach the nucleus to establish infection. However, FEZ1 depletion also impaired infection of either CHME3 or NHDFs by vaccinia virus (VacV), a poxvirus that replicates entirely in the cytoplasm, as determined by reduced expression of a range of viral early, intermediate, and late proteins (I3, G8, and A25; Figures 1B and S1B, respectively).
Figure 1. FEZ1 regulates ISG expression in microglia cells.

(A and B) siRNA-mediated depletion of FEZ1 increases expression of ISGs (MxA, MxB, PKR, and ISG56) in CHME3s infected with HSV-1 (A) or VacV (B) as detected by the expression of viral proteins (HSV1: ICP4, ICP, ICP5; VacV: I3, G8, A25). L.E., long exposure; S.E., short exposure.
(C) KO of FEZ1 (FEZ1 1–4 or FEZ1 pooled), but not non-targeting (NT) gRNAs, increases ISG levels in CHME3s.
(D) Quantification of the ISG levels relative to HSPA8 in FEZ1 KO CHME3s from (C). Data are presented as the ratio to the difference between control and treatment groups.
(E) WB analysis showing effects of FEZ1-Flag or S58A-Flag on expression of ISGs in CHME3s.
(F) Quantification of ISG levels relative to HSPA8 in control, FEZ1-Flag, or FEZ1-S58A-Flag expressing CHME3s from (E). Statistical significance is presented as “A” when it was calculated for all groups using one-way ANOVA followed by Dunnett post-hoc test or as “t” if Student’s t test was applied to compare pairwisely Flag or FEZ1-S58A-Flag with FEZ1-Flag.
(G and H) Measurement of IFN-β levels in culture medium of CHME3 expressing Flag, FEZ1-Flag, and S58A-Flag by ELISA (G). Culture medium of CHME3s spiked with IFN-β was included as positive control. Note that samples (except for the spiked control) were concentrated in order to obtain readings within the range of the standard curve (H) and are divided accordingly to present the actual values shown. One-way ANOVA followed by Tukey post-hoc test was used to calculate statistical significance in (D and G). (D, F and G) n = 3; red line, mean; bars, SD.
See also Figure S1.
Given this broad inhibition of infection by viruses with very different modes of replication, we tested whether FEZ1 depletion perhaps pre-induced an antiviral state in these cells. WB analysis of either uninfected or infected cell lysates revealed that expression of several of ISGs, namely MxA, MxB, PKR, and ISG56, was increased in both CHME3 and NHDFs treated with FEZ1 siRNA compared with the control siRNA-treated cells (Figures 1A, 1B, S1A, and S1B). This increase in ISG expression was also observed in uninfected cells, suggesting that loss of FEZ1 induces an antiviral state before infection. Increases in ISG levels in response to FEZ1 depletion were observed with multiple different FEZ1 siRNAs (Figure S1C), ruling out off-target effects of the primary siRNA that we used. Intriguingly, FEZ1 levels were also decreased in control siRNA-treated CHME3 and NHDFs upon infection with HSV-1, and a moderate increase in MxA and ISG56 was detected (Figures 1A and S1A, respectively). It must be noted that viruses such as HSV-1 encode a wide range of proteins to modulate ISG expression, including targeting them for degradation, such that the robust induction of ISGs observed in uninfected cells in response to loss of FEZ1 is unlikely to be observed. However, compared with VacV wherein neither FEZ1 loss nor ISG induction were observed (Figures 1B and S1B), this raises the intriguing possibility that FEZ1 may be downregulated as part of a host response during infection with some viruses. These observations prompted us to explore the role of FEZ1 in regulating ISG expression in more detail, focusing on uninfected cells to avoid the broader complexity of ISG regulation by both host and virus during infection.
FEZ1 S58 is required for its function as a regulator of ISG responses
To further test the role of FEZ1 in ISG regulation we used two additional independent approaches, namely CRISPR-Cas9-mediated FEZ1 knockout (KO) and FEZ1 overexpression. To do this, we generated KO cells by electroporating CHME3 with CRISPR-Cas9 complexes loaded with either non-targeting (NT) or FEZ1-target single guide RNAs (sgRNAs). In line with our siRNA-based findings, compared with NT controls the loss of FEZ1 using either of four different sgRNAs (FEZ1 1–4) or with a pool of all four sgRNAs (FEZ1 pooled) resulted in a statistically significant increase in the ISGs (Figures 1C and 1D). Notably, while one NT sgRNA induced moderate expression of PKR and ISG56, which can occur in response to stress, little to no effect on MxA or MxB was observed compared with FEZ1 KO. This further suggested that FEZ1 functioned in innate immune and antiviral rather than stress response pathways.
In line with depletion or KO of FEZ1 resulting in increased ISG expression, transduction of either CHME3 or NHDFs with Flag-tagged FEZ1 resulted in reproducible reductions in ISG expression (Figures 1E, 1F, S1D, and S1E, respectively). Furthermore, expression of Flag-FEZ1 S58A, which blocks phosphorylation of FEZ1’s kinesin regulatory domain (Chua et al., 2012), resulted in reproducible increases in ISG expression in CHME3s (Figures 1E and 1F). While mean expression of ISGs were not all statistically significant across all three groups (one-way ANOVA, p > 0.05), overexpression of the FEZ1 wild-type (WT) versus S58A mutant resulted in significantly different expression levels in pairwise comparisons (Student’s t test, p < 0.05). By contrast, Flag-FEZ1 S58A did not increase ISG expression over controls in NHDFs (Figures S1D and S1E), demonstrating that S58A impairs the ISG-regulatory function of FEZ1 but that its ability to act in a dominant negative manner likely depends on cell type and differences in their basal level of FEZ1 activity and ISG production. The ability of exogenously expressed forms of FEZ1 to regulate ISG expression was moderate compared with the effects of robust FEZ1 depletion or KO. While we cannot exclude the possibility that tagging affects this function of FEZ1 to some extent, this is in line with the notion that endogenous FEZ1 already functions efficiently to regulate basal levels of ISG expression and is therefore only moderately affected by supplying more FEZ1 to the system. Regardless, findings using these overexpression approaches were broadly in line with findings using either RNAi- or CRISPR-mediated depletion or KO, respectively. Complementing WB-based observations, ELISA analysis of IFN levels in supernatants from CHME3s confirmed that Flag-FEZ1 decreased, while Flag-FEZ1 S58A increased, the secretion of IFN-β relative to controls (Figures 1G and 1H). As expected, IFN-α or IFN-γ were undetectable in microglia cells (data not shown). Together, these findings demonstrated that FEZ1 regulates ISG expression in a manner that is dependent upon S58.
FEZ1 regulates IRF activation in a STING-independent manner
To understand the innate immune pathway in which FEZ1 operates, we first tested if FEZ1 activity required STING. To do this, monocytic THP-1 cells that are genetically KO for STING and have been engineered to express distinct reporters for IRF and NF-κB activity were differentiated to macrophages (Figure S2A). Similar to our findings in CHME3 and NHDFs above, compared with control siRNA-treated samples FEZ1 depletion induced ISG expression in uninfected cells and blocked infection by either HSV-1 or VacV (Figures S2B and S2C, respectively). Furthermore, similar to other cell types, FEZ1 was downregulated in control siRNA-treated samples upon infection with HSV-1, and moderate increases in MxB and ISG56 were detected in these samples (Figures S2B). This both validated our core findings in other cell types and suggested that STING was not required for FEZ1-mediated ISG expression.
To determine which downstream effector(s) was activated by loss of FEZ1, we used these same THP-1 reporter cell lines that contain two stably integrated inducible reporters for detection of IRF and NF-κB activity. Comparing WT and STING KO THP-1-derived macrophages, reporter assays showed that, compared with control siRNA-treated samples, FEZ1 depletion resulted in an increase in IRF activity and, to a lesser extent, NF-κB activity, in either the presence or absence of STING (Figures S2D and S2E, respectively). As expected, loss of STING results in a significant drop in the basal level of IRF or NF-κB activity in KO cells, but equivalent increases in the activity of both reporters above each baseline was evident for both WT and STING KOs. In line with these observations in reporter THP-1s, we also observed that IRF9 levels in the nucleus were reduced in CHME3s expressing exogenous FEZ1 WT, but not FEZ1 S58A (Figures S2F and S2H), suggesting that FEZ1 regulates the basal IRF activity of these immune cells. Notably, effects on nuclear accumulation of NF-κB were modest (Figures S2I and S2J), in line with data in THP1 reporters above suggesting that IRF activity was predominant in this FEZ1-regulated STING-independent pathway.
FEZ1 interacts with HSPA8, a factor in STING-independent innate immune signaling
Given its function as a hub protein, we next investigated whether FEZ1-interacting proteins might provide clues to the specific innate response pathway in which it was operating. To identify FEZ1-interacting proteins, we generated CHME3s expressing GFP-tagged forms of FEZ1 WT, FEZ1 S58A, or GFP control alone. Soluble cell lysates were prepared and incubated with GFP-TRAP agarose, and isolated complexes were analyzed by mass spectrometry (MS). In line with our findings using STING KO cells, we failed to detect STING in our MS analyses but we detected peptides from HSPA8 as enriched in GFP-FEZ1 WT compared with empty or GFP-expressing controls, and even greater enrichment was observed for GFP-FEZ1 S58A (data not shown). This MS screen was only performed once to identify potential FEZ1-interacting proteins for further investigation, but HSPA8 was notable to us as it was recently identified as a phosphorylation target during DNA protein kinase (DNA-PK)-mediated DNA sensing pathways that operate independently of STING (SIDSP) (Bonam et al., 2019; Burleigh et al., 2020). Validating this interaction, WB analysis of GFP-pull-down samples and densitometry confirmed that FEZ1 binds endogenous HSPA8, along with increased recovery of HSPA8 in association with GFP-FEZ1 S58A (Figures 2A and 2B). Performing reciprocal GFP pulldowns from CHME3 lysates expressing GFP-tagged HSPA8 in the presence of Flag-FEZ1 WT or S58A independently showed that GFP-HSPA8 specifically bound Flag-FEZ1WT and, to a greater degree, Flag-FEZ1 S58A (Figures 2C and 2D). To confirm that endogenous forms of FEZ1 and HSPA8 interacted, anti-FEZ1 co-immunoprecipitation (coIP) was performed using lysates from control NT and FEZ1 KO CHME3s. In control cells, HSPA8 was detectable in FEZ1 coIP complexes, while recovery of HSPA8 was reduced proportionally to the level of residual FEZ1 expression and recovery in FEZ1 KO samples (Figures 2E and 2F), demonstrating the FEZ1-dependent recovery of HSPA8 using this approach. It is important to note that the enrichment of either binding partner compared with input levels in each approach was low, but this is to be expected for proteins such as HSPA8 that are highly expressed and function in several processes, meaning that only a small fraction of HSPA8 is bound to FEZ1. Despite this, these reciprocal approaches confirmed that FEZ1 interacts with HSPA8 and further showed that this interaction was regulated by S58 in FEZ1, the site that our earlier data showed also regulates FEZ1-mediated ISG expression.
Figure 2. HSPA8 interacts with FEZ1 and regulates ISG expression.

(A) WB confirmation of FEZ1 binding to endogenous HSPA8 in complexes isolated on GFP-TRAP agarose from CHME3s or CHME3s stably expressing GFP control, GFP-FEZ1, or GFP-S58A.
(B) Quantification of HSPA8 levels in protein complexes from (A).
(C) Reciprocal GFP pulldowns from CHME3 lysates expressing GFP or GFP-HSPA8 along with either FEZ1-Flag or S58A-Flag.
(D) Quantification of FEZ1 and FEZ1 S58A levels in protein complexes from (C).
(E) CoIP analysis showing interaction between endogenous FEZ1 and HSPA8 in CHME3 control or FEZ1 KO cells.
(F) Quantification of HSPA8 levels in protein complexes from (E).
(G) Representative WB (n = 2) showing decreases ISG levels in CHME3 expressing either GFP-HSPA8 (left panels) or HSPA8-Flag (right panels). Exo., exogenous; End., endogenous.
(H) KO of HSPA8 (HSPA8 1, 2, or 4, or HSPA8 pooled), but not non-targeting (NT) gRNAs, increases ISG levels in CHME3s.
(I) Quantification of ISG levels relative to HSPA8 in cells from (H). Data are presented as the ratio to the difference between control and treatment groups. A Student’s t test was used in (F) and one-way ANOVA followed by Tukey post-hoc test in (B, D, and I) to calculate statistical significance. (B, D, F, and I) n = 3; red line, mean; bars, SD.
See also Figure S2.
While HSPA8 has been shown to be phosphorylated in a DNA-PK-dependent manner, its functional contribution to this pathway remains unknown (Burleigh et al., 2020). In testing whether HSPA8 overexpression influenced expression of ISGs, we found that expression of either GFP-tagged or Flag-tagged HSPA8 reduced ISG levels compared with their respective GFP or Flag controls (Figure 2G). Complementing overexpression approaches, we found that CRISPR-Cas9 KO of HSPA8 using either of three different sgRNAs, as well as depletion of HSPA8 using a pool of all three sgRNAs, resulted in an increase in ISGs compared with control cells treated with NT sgRNAs (Figures 3H and 3I). Whether ISG induction after FEZ1 depletion is HSPA8 dependent remains an outstanding question that we hope to address in future studies.
Figure 3. FEZ1 regulates HSPA8 localization.

(A) Representative IF images of CHME3s expressing Flag control or HSPA8-Flag stained for total HSPA8 (red), exogenous HSPA8 (Flag, green), or the nucleus (Hoechst, blue). Scale bar, 10 μm.
(B, D, and F) Representative IF images of intracellular distribution of HSPA8 in either CHME3s treated with two different negative controls (NC1 and NC2) or FEZ1-specific siRNAs (B) or three different FEZ1 KO CHME3 pools (FEZ1 KO 1, 3, and 4) (D), or CHME3 expressing Flag control, FEZ1-Flag, or S58A-Flag (F). Nuclei were stained with Hoechst. Scale bar, 10 μm.
(C, E, and G) Quantitative analysis of HSPA8 staining in nuclei of CHME3s in (B, D, and F), respectively. Number of cells analyzed is indicated, n = 3; red line, mean; bars, SD. One-way ANOVA was used to calculate statistical significance with Tukey (C and G) and Dunnett (E) post-hoc tests.
(H and I) Representative (n = 2) WB analysis of nuclear and cytoplasmic fractions from control and FEZ1 KO CHME3s (cropped to remove irrelevant samples) (H) or Flag, FEZ1-Flag, and S58A-Flag expressing CHME3s (I).
See also Figure S3.
FEZ1 regulates HSPA8 localization
HSPA8 is a constitutively expressed protein that shuttles between the cytoplasm, where it is particularly abundant under normal conditions, and the nucleus, where it relocalizes during stresses, such as heat shock (Bonam et al., 2019). While signaling pathways involved in the nucleocytoplasmic trafficking of HSPA8 are not known, blocking its relocation into the cytoplasm impairs cell survival during stress recovery (Wang et al., 2018), suggesting a prominent role in the cytoplasm under normal homeostasis and nuclear functions in pathological states. As a transport protein, FEZ1’s binding to HSPA8 might affect its localization and/or activity. To test this, we first looked at HSPA8 localization in CHME3s. Immunofluorescence (IF) analysis in methanol-fixed CHME3 and HSPA8 KO cells showed the specificity of HSPA8 staining and that endogenous HSPA8 is distributed throughout the cytoplasm and in perinuclear regions under normal conditions, and translocates into the nucleus upon heat shock as expected (Figures S3A and S3B). Imaging also showed that exogenously expressed Flag-tagged HSPA8 localized in the cytoplasm (Figure 3A). Moreover, staining for HSPA8 demonstrated that overexpression of HSPA8 does not drive translocation of the endogenous protein into the nucleus. Given that exogenous expression of HSPA8 reduces basal ISG expression (Figure 2G), this suggests that similar to its functions during stress recovery, HSPA8 ensures low basal ISG levels through its functions in the cytoplasm. As a kinesin adaptor, FEZ1 may be required to maintain HSPA8 in the cytoplasm and sustain low ISG expression levels under normal conditions. To test this, we first measured the effects of FEZ1 depletion on the levels of HSPA8 in the nucleus. Because of the high levels of HSPA8 expression, its dynamic nucleocytoplasmic shuttling, its diverse functions in various processes, and the irregularity of cell size and shape, measurements of nuclear accumulation of HSPA8 were the simplest and most reliable readout for changes in the cytoplasmic versus nuclear pool of HSPA8 that is controlled by FEZ1, as opposed to other FEZ1-independent HSPA8 pools performing other functions in the cell. Imaging revealed that more HSPA8 was present in the nucleus of CHME3s treated with FEZ1-specific siRNA compared with either of the two different NT siRNA-treated controls (Figures 3B and 3C). Independently, levels of HSPA8 in the nucleus were also increased in three different FEZ1 KO CHME3 pools (Figures 3D and 3E). This occurred in the absence of changes in the total abundance of HSPA8 (Figure 1C). Complementing depletion approaches, expression of FEZ1-Flag in either CHME3 or NHDFs reduced HSPA8 levels in the nucleus of either cell type compared with control Flag-expressing cells, while Flag-FEZ1 S58A did not (Figures 3F, 3G, S3C, and S3D, respectively). Again, this occurred in the absence of changes in HSPA8 levels (Figure 1E), suggesting that these changes reflected differences in cytoplasmic versus nuclear localization of a subpopulation of HSPA8. Complementing imaging-based findings, nuclear fractionation of cell lysates showed more HSPA8 in the nuclear fractions recovered from FEZ1 KO cells compared with that of the control NT cells, while less HSPA8 was recovered in the nuclear fractions from CHME3s expressing FEZ1-Flag compared with that of the Flag-FEZ1 S58A (Figures 3H and 3I, respectively). Together, these findings suggest that FEZ1 and its phosphorylation at S58A is required to retain a subpopulation of HSPA8 in the cytoplasm to maintain low basal levels of ISG expression.
FEZ1 and HSPA8 regulate nuclear localization of DNA-PK
As a chaperone, HSPA8 regulates folding and degradation of many cellular proteins, while it has also been implicated in regulating the localization of many others (Bercovich et al., 1997; Bonam et al., 2019; Okuno et al., 1993; Stricher et al., 2013). Prompted by its recent connections with DNA-PK-mediated innate signaling pathways, we explored whether HSPA8 regulated DNA-PK nuclear localization. Probing our fractionated samples above we found that, similar to its effects on HSPA8, FEZ1 KO increased the levels of DNA-PK in nuclear fractions (Figure 3H). Conversely, and in line with broader effects on HSPA8 localization and ISG expression, FEZ1 overexpression decreased the levels of DNA-PK in nuclear fractions and this required S58 (Figure 3I). Complementing fractionation approaches, IF staining of cells revealed significantly more DNA-PK in the nuclei of FEZ1 KO CHME3 compared with that of the NT control (Figures 4A and 4B). By contrast, nuclear levels of DNA-PK were reduced in either CHME3 or NHDFs expressing FEZ1-Flag compared with control Flag-expressing cells, while nuclear DNA-PK levels were increased in cells expressing FEZ1 S58A-Flag (Figures 4C, 4D, S3E, and S3F, respectively). This suggests that FEZ1 prevents DNA-PK from entering the nucleus, potentially through retention of HSPA8 in the cytoplasm. Supporting this idea, staining of HSPA8 KO pools revealed a significant increase in DNA-PK levels in the nuclei of HSPA8 KO pools compared with that of the NT control (Figures 4E and 4F).
Figure 4. FEZ1 and HSPA8 regulate nuclear localization of DNA-PK.

(A, C, and E) Representative IF images of DNA-PK staining in three different FEZ1 KO CHME3 pools (FEZ1 KO 1, 3, and 4) (A), or CHME3s expressing Flag control, or FEZ1-Flag or S58A-Flag (C), or HSPA8 KO CHME3 pools (HSPA8 KO 1, 2, and 4) (E). Nuclei were stained with Hoechst (blue). Scale bar, 10 μm.
(B, D, and F) Quantitative analysis of nuclear DNA-PK staining in samples from (A, C, and E), respectively.
(G–L) Responses to CT-DNA in CHME3 microglia. (G) WB analysis showing decreased phosphorylated FEZ1 at S58 (pFEZ1) as well as increased levels of phosphorylated IRF3 (pIRF3) and ISGs (MxA, MxB, and ISG56) in CHME3s treated with CT-DNA. (H) Quantification of the pFEZ1 levels and ISGs from samples in (G) n = 3; bars, SD. (I–L) Representative images (I and K) and quantitative analysis (J and L) of HSPA8 (I and J) or DNA-PK (K and L) staining in nuclei of CHMEs either untreated or treated with CT-DNA. One-way ANOVA followed by Dunnett post-hoc test (B and F) or Tukey post-hoc test (D), and t test (J, H, and L) was used to calculate statistical significance. (B, D, F, J, and L) Number of cells analyzed is indicated; n = 3; red line, mean; bars, SD.
See also Figures S4 and S5.
These findings suggest that FEZ1 and HSPA8 prevent DNA-PK from accumulating in the nucleus and inducing ISG expression. Indeed, beyond links to SIDSP, DNA-PK is reported to phosphorylate and regulate IRF3 and broadly influence host immune responses (Karpova et al., 2002). We therefore tested whether DNA-PK was required for ISG expression in either FEZ1 or HSPA8 KO CHME3s. In both cell types, the DNA-PK inhibitor NU7441 significantly reduced the expression of several ISGs tested (Figures S4A and S4B). In line with inhibitor-based findings, CRISPR-based KO of DNA-PK similarly reduced ISG expression in either HSPA8 or FEZ1 KO cells (Figures S4C-S4E). Notably, the effects of DNA-PK inhibition or KO on ISG expression were constituent in trend but somewhat variable in magnitude across experiments. This is perhaps unsurprising in a complex biological response pathway and affects statistical significance at times, but also suggests that DNA-PK is likely not the only effector of HSPA8’s control over ISG expression. Indeed, HSPA8 has been shown to regulate the accumulation of several proteins in the nucleus (Banski et al., 2010; Wang et al., 2018) and ISG induction was found to be also highly dependent on IRFs (Figures S4F-S4H), in line with earlier data using THP1 reporter cell lines above.
FEZ1 phosphorylation is reduced during DNA-mediated IFN responses
The broad induction of IFN and ISG expression in cells where FEZ1 activity was experimentally modulated prompted us to test whether many of these phenotypes were attributable to IFN itself. Compared with untreated or control antibody-treated samples, treatment of FEZ1 or HSPA8 KO CHME3s with IFN-β-neutralizing antibody significantly reduced the expression of ISGs in both FEZ1 and HSPA8 KO CHME3 (Figures S5A-S5D). More moderate effects on ISG56 are perhaps not unexpected due to its high basal expression and responsiveness to a broader range of stimuli than other ISGs tested that are more tightly linked to IFN regulation. IF imaging also suggested that IFN-β neutralization blocked the translocation of IRF9, but not HSPA8 in CHME3s, although antibody cross-reactivity with internalized IFN-β-neutralizing antibody prevented us from reliably quantifying these effects (Figure S5E). However, in a reciprocal experiment, treatment of CHME3s with IFN-β revealed that IFN-β was sufficient to increase nuclear accumulation of IRF9 but not HSPA8 (Figures S5F-S5H). Combined with the fact that we have not detected changes in FEZ1 phosphorylation in IFN-β-treated cells (unpublished observation), this suggests that the induction of IRF9 translocation and ISG expression in cells depleted of FEZ1 or expressing inactive S58A FEZ1 is attributable to IFN production, but that production of this IFN initiates due to specific effects of FEZ1 perturbations on HSPA8 localization. Finally, we tested whether endogenous FEZ1 phosphorylation is altered in cells responding to CT-DNA, which induces SIDSP. Treatment of CHME3s with CT-DNA induced ISG expression and this was accompanied by a significant decrease in endogenous FEZ1 phosphorylation at S58 and activation of IRF3, a DNA-PK target (Karpova et al., 2002) (Figures 4G and 4H) as well as translocation of HSPA8 and DNA-PK to the nucleus (Figures 4I, 4J, 4K, and 4L, respectively). Given that IFN alone is not sufficient to cause HSPA8 translocation to the nucleus (Figure S5H), this suggests that regulating FEZ1 phosphorylation and HSPA8 localization is an important initiating event in broader IFN-based responses to stimuli such as CT-DNA.
HSPA8 and DNA-PK are central to host immune responses yet paradoxically, they play complex multifunctional roles in infection outcomes. Indeed, while HSPA8 functions in SIDSP, translocation of HSPA8 to the nucleus has also been suggested to promote lytic HSV-1 infection (Adlakha et al., 2020; Burch and Weller, 2004). Meanwhile, several viruses, including HSV-1, directly antagonize DNA-PK (Boyer et al., 1999; Burleigh et al., 2020; Ferguson et al., 2012; Lees-Miller et al., 1996; Parkinson et al., 1999; Peters et al., 2013; Scutts et al., 2018; Smith et al., 2014). Indeed, HSPA8 and DNA-PK play broad and extremely complex roles in infection by a variety of viruses (Bonam et al., 2019; Hristova et al., 2020; Lu and Zhang, 2020; Wang et al., 2020). It is now apparent that FEZ1 plays similarly complex roles in both host responses and infection. Intriguingly, we observe that HSV-1 infection results in loss of FEZ1 expression, which may contribute to HSPA8 re-localization that is reported to occur in HSV-1 infected cells. In the case of HIV-1, FEZ1 directly binds to viral particles and regulates their kinesin-based motility, yet our findings here reveal that FEZ1 also plays a role in regulating host ISG expression. As such, control of ISG expression may represent a previously unrecognized mechanism by which FEZ1 affects HIV-1 infection (Schoggins et al., 2011). A similar situation has been reported for the dynein adaptor, BICD2, which both mediates HIV-1 motility and host responses to infection (Dharan et al., 2017). As such, these combined data suggest that components of the SIDSP pathway, namely FEZ1, HSPA8, and DNA-PK play multiple and often opposing roles that can be challenging to disentangle in the complex context of infection.
Avoiding this complexity by focusing on uninfected cells, our findings show that FEZ1 phosphorylation functions as part of SIDSP, or a variation of this pathway that involves common components, such as HSPA8 and DNA-PK. In an apparent contradiction that may provide clues to the underlying process, FEZ1 S58A binds more robustly to HSPA8 than to WT FEZ1, yet more HSPA8 enters the nucleus in FEZ1 S58A-expressing cells. From this observation, and based on the high abundance of HSPA8, we propose that FEZ1 is a limiting factor and that phosphorylation enables it to cycle through the cellular pool of HSPA8 to retain it in the cytoplasm. Dephosphorylation of FEZ1 increases HSPA8 binding but this actually sequesters FEZ1 away from other HSPA8 molecules, allowing them to escape FEZ1-mediated retention in the cytosol and enter the nucleus. There is precedence for such a mechanism across multiple processes, including nucleotide sensing; PKR phosphorylates and inactivates the translation factor, eIF2, in response to dsRNA (Jan et al., 2016). Phosphorylation-based inactivation functions by causing eIF2 to bind more tightly to eIF2B, a lower abundance guanine nucleotide exchange factor that cycles across the larger eIF2 population to maintain the active eIF2-GTP state. Effectively, eIF2B’s target also acts to sequester it from the broader eIF2 population when it becomes more tightly bound to phosphorylated eIF2. A similar mechanism is likely used by HSPA8 to sequester FEZ1 upon dephosphorylation and allow translocation of other HSPA8 molecules to the nucleus during host responses. While much of the underlying mechanism remains to be elucidated in future studies, our findings here show that FEZ1 plays a central role in regulating host innate immune signaling by regulating the localization of HSPA8 and DNA-PK, adding insights to our understanding of this newly emerging STING-independent pathway.
Limitations of the study
Beyond the specific limitations discussed above, it remains unknown and an avenue for future study to determine whether modulating FEZ1 activity affects infection by the DNA viruses tested here through its ability to regulate ISG expression or whether FEZ1 functions directly, or both, similar to the complexity of its functions during early HIV-1 infection.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Please direct any requests for further information and reagents to the lead contact, Mojgan H. Naghavi (mojgan.naghavi@northwestern.edu).
Materials availability
All plasmids and cell lines generated in this study will be made available upon request from the lead author.
Data and code availability
All data reported in this study will be shared by the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells and culture conditions
Human immortalized microglial cell line clone 3 (CHME3) was described previously (Janabi et al., 1995). Primary Normal Human Dermal Fibroblasts (NHDFs) were purchased from Lonza (CC-2509). The cell lines were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Fisher Scientific) containing 2 mM L-Glutamine, 1 mM Sodium Pyruvate, 100 U/mL penicillin, 100 μg/mL Streptomycin and 10% or 5% Nu-Serum culture supplement (Corning) for CHME3 and NHDFs, respectively. HEK-293T (293T) cells were obtained from ATCC (CLR-3216) and maintained in DMEM supplemented with 10% Fetal Bovine Serum (FBS, Atlanta Biologicals), 2 mM L-Glutamine, 100 U/mL penicillin, and 100 μg/mL Streptomycin. BSC-40 and VERO cells (female) were a gift from Dr. Ian Mohr, NYU and cultured as described previously (Walsh and Mohr, 2004). THP1-Dual (thpd-nfis) and THP1-Dual KO-STING (thpd-kostg) cell lines stably expressing inducible NF-κB-SEAP and IRF-Lucia luciferase reporters were purchased from InvivoGen. Cells were kept in RPMI 1640 medium (Gibco) containing 10% FBS, 2 mM L-Glutamine, 100 U/mL penicillin, and 100 μg/mL Streptomycin. Expression of the reporters were maintained with 10 μg/mL blasticidin and 100 μg/mL Zeocin. THP-1 monocytes were differentiated into macrophages before infection experiments. Cells were plated in 12-well plates at 5 × 105 cells/well and incubated with 30 ng/mL phorbol 12- myristate 13-acetate (PMA, Sigma, P8139) for 48 hrs, followed by maintenance for additional 24 hrs in medium omitting PMA. All cell lines were tested negative for mycoplasma with PlasmoTest mycoplasma detection kit (InvivoGen) and kept in a cell culture incubator in 5% CO2 at 37°C.
Generation of stable cell pools
A plasmid encoding eGFP was generated by inserting amplified eGFP PCR product into pQCXIN vector at SbfI and Not-I restriction sites. Constructs encoding human FEZ1-Flag and FEZ1(S58A)-Flag (S58A-Flag) proteins tagged to eGFP at the N-terminus were generated through re-cloning from pQCXIN-FEZ1-Flag and pQCXIN-FEZ1-S58A-Flag (Malikov et al., 2015) into pQCXIN-eGFP vector at Not-I and EcoRI restriction sites. HSPA8 was amplified by PCR reaction from pPM-C-HA-HSPA8 plasmid (Applied Biological Materials) and inserted into pQCXIN-eGFP to produce eGFP-tagged protein or into pQCXIN to create pQCXIN-HSPA8-Flag construct using primers containing Not-I and EcoRI restriction sites. In addition, Flag sequences were included in front of EcoRI in the reverse primer for generation of C-terminus Flag-tagged HSPA8. Several E. coli clones for each plasmid were amplified, the plasmids were purified with QIAprep spin miniprep kits (Qiagen) and inserts were sequenced at ACGT, Inc. using universal primers provided by the company. Plasmids with verified sequences of inserts were amplified with QIAfilter plasmid maxi kits (Qiagen). Murine leukemia virus (MuLV)-based retroviruses pseudotyped with vesicular stomatitis virus G envelope protein (VSV-G) were produced using 293T cells. Confluent 10 cm dishes were split at 1:4 ratio and the next day were transfected with 2.85 μg of each pCMV-intron and pVSV-G along with 4.3 μg of a transducing vector. The plasmids were mixed with 22.5 μL of 50 μg/mL Polyethylenimine (PEI, Polysciences, #23966-1) in 1 mL of Opti-Mem medium, incubated for 10 min at room temperature (RT) and applied onto cells. Growth medium was changed the next morning and supernatants were collected 48 hrs post transfection, clarified through 0.45 μm filters, aliquoted and stored at −80°C. CHME3 and NHDF cell pools stably expressing Flag, FEZ1-Flag and FEZ1(S58A)-Flag (S58A-Flag), eGFP, eGFP-FEZ1, eGFP-FEZ1S58A (GFP-S58A), eGFP-HSPA8 or HSPA8-Flag proteins were generated by transduction of the cell lines with the appropriate retroviruses described above. Low passage cells were split in 6-well plates at 10-20% confluency and the next day were transduced with the viral vector of interest in 1 mL regular growth medium containing 10 μg/mL polybrene. 1 mL of medium was added to each well following overnight incubation. Selection of expressing cells was started 48 h post transduction by changing medium to growth medium containing 1 mg/mL G-418 and selection continued until all non-transduced control cells were dead. WB analysis was used to confirm expression of the protein of interest after selected cell lines were established.
Generation of CRISPR-Cas9 knock-out in CHME3 cell line
CRISPR-Cas9 ribonucleoprotein complexes (crRNPs) were prepared as previously published (Hultquist et al., 2019). Briefly, lyophilized FEZ1 and HSPA8 guide RNA (gRNA) and tracrRNA (Dharmacon, U-002005-50) were suspended at a concentration of 160 μM in sterile 10 mM Tris-HCL, 150 mM KCl, pH 7.4.1 μL of 160 μM gRNA was mixed with 1 μL of 160 μM tracrRNA and incubated for 30 min at 37°C. gRNA:tracrRNA complexes were mixed gently with 2 μL of 40 μM Cas9 (UC-Berkeley Macrolab) and incubated for 15 min at 37°C to form crRNPs. 4 μL crRNP aliquots were stored frozen in Lo-Bind 96-well V-bottom plates (E&K Scientific) at −80°C.
To generate knock-out cells, CHME3 cells were trypsinized, re-suspended in culture medium and counted. Immediately prior to electroporation, 250,000 cells were centrifuged at 400 g for 3 min, supernatant was removed by aspiration, and the pellet was resuspended in 20 μL of room-temperature electroporation buffer prepared by combining 16 μL of SF Nucleofector solution with 4 μL of supplement (Lonza). Then, cell suspension was gently mixed with 4 μL of each crRNP and aliquoted into a 96-well electroporation cuvette for nucleofection with the 4-D Nucleofector X-Unit (Lonza) using pulse code CM-158. Immediately after electroporation, 100 μL of pre-warmed culture media was added to each well and cells were allowed to rest for 30 min in a 37°C cell culture incubator. Cells were subsequently moved to 12-well flat-bottomed culture plates pre-filled with 500 μL pre-warmed media. After 48 hrs in culture, cells were split in 10 cm dishes and their aliquots were lysed with Laemmli buffer to validate KO efficiencies by Western blot (WB).
HSPA8 and FEZ1 were targeted by 4 gRNA delivered either independently or as a single multiplexed pool. Non-targeting (NT) gRNA (Dharmacon, U-007504-20) was delivered in parallel as a non-cutting, negative control. To generate the double knock-out cell lines, the CHME3 cells previously treated with HSPA8-targeting gRNA #4 or FEZ1-targeting gRNA #3 were nucleofected for a second time with a multiplex pool of 5 gRNA targeting PRKDC (DNA-PKcs) or IRF3. To generate the triple knock-out cell lines, the FEZ1/IRF3 and HSPA8/IRF3 double knock-out cell lines from above were nucleofected for a third time with a multiplex pool of 5 gRNA targeting IRF7. All gRNA were derived from the Dharmacon pre-designed Edit-R library for gene knock-out (refer to Table S1 for sequences).
METHOD DETAILS
Generation of viruses and infections
VacV and HSV-1 virus stocks were grown and titrated using BSC40 cells. Briefly, cultures were infected at multiplicity of infection (MOI) 0.01 and once 90– 100% CPE was observed, virus was harvested by three rounds of freeze-thaw. Cell debris was removed by centrifugation and virus titer was determined by serial dilution and plaque assay (Walsh et al., 2008). In experimental set-ups, VacV or HSV-1 infections were performed at MOI 5 for the indicated times.
Detection of NF-κB and IRF reporters
The secreted embryonic alkaline phosphatase (SEAP) and Lucia luciferase reporters were detected in cell culture media using QUANTI-Blue and QUANTI-Luc (InvivoGen) reagents according to the manufacturer instructions. Briefly, 20 μL of cell supernatant or control samples were mixed with 180 μL of QUANTI-Blue in 96-well plate, incubated for 1 hr at 37°C and SEAP activity were measured in a microplate reader at 655 nm. Lucia luciferase levels were determined in a luminometer set to end-point measurement with a 4 s start time, 0.1 s reading time and 50 μL of injection. The instrument was primed with QUANTI-Luc solution and measurements were made in an opaque 96-well plate with pre-loaded 20 μL of cell supernatant or control samples.
WB analysis and measurement of protein quantities on membranes
Samples for WB were produced by lysing cells in wells with freshly made 1xLaemmli buffer (62.5 mM Tris-HCl at pH6.8, 2% SDS, 10% glycerol, 0.7 M β-mercaptoethanol) followed by boiling for 5 min. Sample proteins were resolved in 10% or 15% SDS PAGE at 150 V followed by transfer to a nitrocellulose membrane at 70 V for 1 hr. Membranes were incubated in blocking buffer (3% non-fat milk in TBS-T) for 1 hr on a rotary shaker, rinsed and washed for 5 min in TBS-T. Proteins were bound by primary antibodies during overnight incubation at +4°C. Proteins bands were visualized with Pierce ECL or Femto High Sensitivity Western Blotting Substrates (Thermo Fisher Scientific) after 1 hr incubation at RT with appropriate HRP-conjugated secondary antibody (GE Healthcare UK) at 1:10,000 dilution in the blocking buffer. The following primary antibodies at 1:1,000 dilution in 3% BSA in TBS-T were used: anti-HSPA8 (1B5, #ADI-SPA-815-J) from Enzo, anti-GFP (ab13970), anti-pIRF3 (phospho S386, ab76493), anti-Tubulin (YL1/2, ab6160), anti-HSV-1 ICP0 (ab6513), and anti-ICP4 (ab6514) from Abcam, anti-FEZ1 (#42480), anti-PKR (#12297), anti-MxA (#37849), anti-MxB (#43924), anti-ISG56 (#14769), anti-IRF3 (#11904), anti-IRF7 (D8V1J, #72073), anti- DNA-PK (3H6, #12311), anti-Lamin B1 (D9V6H, #13435) from Cell Signaling, anti-GAPDH (sc-25778) from Santa Cruz.Antibodies. Anti-phospho-S58-FEZ1 antibody was custom made (Synaptic Systems). Antibodies against VacV proteins were a kind gift of Dr. Yan Xiang, University of Texas Health Sciences Center, San Antonio, Dr. Paula Traktman, Medical University of South Carolina and Dr. David Evans, University of Alberta. Relative intensities of WB bands were calculated in Fiji software. Grayscale WB film scans were inverted in the software, protein bands were demarcated with rectangular selection tool of same size for all bands and areas and integrated densities of each selection along with background were measured. Data were transferred into Microsoft Excel, where all values underwent background subtraction. Then, data points were directly plotted on the scattered graphs with dots representing protein level in WB bands. If controls values were significantly varied between repeats, the ratios of WB data to absolute values of the differences between controls and treatment groups were plotted. These ratios were presented as relative protein level in WB bands.
GFP-pulldowns and mass-spectrometry
CHME3 stable pools expressing eGFP and eGFP-HSPA8 described above, were transiently transfected with pQCXIN-FEZ1-Flag or pQCXIN-FEZ1-S58A-Flag. The pools were plated into 10 cm dishes at 25% confluency and the next day they were transfected with 10 μg plasmid using the PEI method as described above. 48 hrs post transfection each dish was washed with 10 mL cold PBS and lysed in a 1 mL NP-40 lysis buffer (50 mM HEPES pH 7.4, 150 mM NaCl, 0.5 mM MgCl2, 2 mM EDTA, 2 mM Na3VO4, 25 mM glycerophosphate, 1.5% NP-40) completed with mini EDTA-free protease inhibitor cocktail (Roche). To compensate for differences in proteins quantities due to low expression, lysates from eight dishes of eGFP-HSPA8 pool were combined at this stage and treated further as a lysate from one dish. Lysates were rocked for 40 min followed by centrifugation at 10,000 g for 10 min. Supernatants were separated from pellets and 30 μL were retained as input samples. Remaining supernatants were incubated with 3.5 μL (dry volume) GFP-Trap agarose (ChromoTek; cat. #gta-100) pre-equilibrated with NP-40 lysis buffer and rocked for 4 hrs. Agarose beads with bound proteins were spun down at 2,000 rpm for 1 min and washed 3 times for 5 min in 0.5 mL NP-40 lysis buffer. Each wash was followed by a brief centrifugation at 2,000 rpm for 1 min. Final pellets were re-suspended and boiled for 3 min in 30 μL of 2x Laemmli buffer. All procedures were carried out at 4°C. CHME3 stable pools expressing eGFP, eGFP-FEZ1 oreGFP-S58A were processed equally. GFP-pulldown samples for mass-spectrometry identification were re-suspended and boiled in 30 μL 2x Laemmli buffer omitting Bromophenol Blue dye and centrifuged at 2,000 rpm for 1 min. Supernatants were transferred to fresh epi-tubes and sample proteins were precipitated with Trichloroacetic acid (TCA) as described (Link and LaBaer, 2011). Briefly, 7.5 μL of TCA was added to each sample, incubated for 10 min at +4°C and precipitate was spun down at 14,000 rpm for 5 min. Pellet was washed twice with 200 μL of ice-cold acetone and dried up in +95°C heat block for 5 min. The precipitated proteins were incubated in 50 μL of 8 M urea in 50 mm ammonium bicarbonate for 1 hr and then in 50 μL of 0.2% ProteaseMAX (Promega; Cat# V207A) for 1 hr. Protein extracts were reduced and alkylated with 1 μL of 500 mM TCEP for 1 hr and after that in 2 μL of 500 mM iodoacetamide for 20 min in the dark. The reaction was stopped with 5 μl of 500 mM TCEP. After the addition of 215 μL 50 mM ammonium bicarbonate, 2.5 μL 1% ProteaseMAX and 1.0 μg Trypsin Gold (Promega; Cat# V528A), samples were digested overnight at 37°C. The digestion reaction was quenched with formic acid and purified using C18 resin in Pierce Spin Columns (Thermo Fisher Scientific, cat. #89879). The peptides were quantified using a microBCA protein assay kit (Thermo Fisher Scientific; cat. #23235), separated on nanoViper trap column (Thermo Fisher Scientific; cat. #164535) coupled to nanoViper analytical column (Thermo Fisher Scientific; cat. #164942) and analyzed on an Orbitrap Fusion Tribrid mass spectrometer set with the parameters described previously (DiGiuseppe et al., 2020). Spectrum raw files were obtained with the in-house program RawConverter (http://fields.scripps.edu/downloads.php), and the tandem mass spectra were searched against UniProt human database (downloaded on 25 March 2014). The parameters used for the search were previously described (DiGiuseppe et al., 2020).
Co-immunoprecipitation (co-IP) analysis
Co-IP’s were performed as described previously (Walsh and Mohr, 2006). Briefly, lysates of CHME3 NT KO and FEZ1 KO pool #3 were prepared as for GFP-pulldowns described above. Following centrifugation at 10,000 g for 10 min, supernatants were pre-cleared by incubating with 25 μL of 10% G-sepharose from GE Healthcare (#17-0618-01) rocking for 1hr at 4°C. Sepharose was then sedimented by centrifugation, input samples were taken and the remaining pre-cleared lysates were rocked with 10 μL anti-FEZ1 antibody from Abnova (#H00009638-B02P) for 2 hrs at 4°C followed by overnight incubation with 40 μL 50% G-sepharose. Beads were then washed 3 times with 700 μL of NP-40 buffer for 5 min and boiled in 30 μL of Laemmli buffer.
Inhibitor, antibody neutralization and CT-DNA treatment
CHME3 cells were plated in 12-well plates at 80% confluency and transfected with a mixture of 4 μl Lipofectamine 2000 and 4 μg CT DNA purchased from Sigma (#D4764). Cells were lysed 16 h post transfection and analyzed by WB. For immunofluorescence analysis cells were plated on glass coverslips in 24-well plates and transfected with 2 μg CT-DNA using 2 μl Lipofectamine 2000 for 16 h. Cells were fixed with ice-cold methanol and stained and quantified as described below. For inhibition of DNA-PK, CHME3 FEZ1 KO pool #3 and HSPA8 KO pool #4 were treated overnight with 0.25 μM Nu-7441 DNA-PK inhibitor (SelleckChem, #S2638). Neutralization of IFNβ was performed by adding anti-IFNβ antibody (Millipore-Sigma; ab1431) at a concentration of 7.5 μl/200 ml to the cell culture medium overnight.
Knockdowns
Transient siRNAs transfections were carried out as described (Haedicke et al., 2009). CHME3 and NHDF cells were plated in 12-well plates in normal growth medium omitting antibiotics at 80,000 cells/well or at 1/25 cell number of a confluent 10 cm dish, respectively. The next day growth media were substituted for 0.5 mL pre-warmed Opti-Mem medium (Gibco) and cells were transfected with 50 μL of transfection mixture per a well of 12-well plate for 4 hrs. At the end of incubation 0.5 mL of normal growth medium omitting antibiotics were added to each well. Transfection mixture was prepared by combining 17 μL Opti-Mem medium with 1.34 μL of RNAiMax reagent and 33 μL of Opti-Mem with 1 μL of 100 pmol/μL siRNA stock followed by 20 min incubation before adding onto the cells. 24 hrs post transfection media was replaced with normal growth medium with antibiotics and in-well whole cell lysates were prepared for a WB analysis 48 hrs after transfections. For immunofluorescence staining cells were split onto 3-4 gelatin-treated coverslips placed in 24-well plates. Remnants of cells were plated back in 12-well plates to further check for knockdown efficiency. The following pre-designed siRNAs from Ambion (Thermo Fisher Scientific) were used: ID# AM4635 (Negative Control #1 siRNA), ID# AM4637 (Negative Control #2 siRNA), ID# 15759 (FEZ1-A), ID# 45012 (FEZ1-B), ID# 45101 (FEZ1-C).
Immunofluorescence (IF) and analysis of proteins quantities
Cells were plated at 70-80% confluency on gelatin coated coverslips a day before they were fixed. Fixation was performed with 3.7% paraformaldehyde (PFA) for 15 min or with chilled down to −20°C methanol for 5 min. PFA-fixed cells were permeabilized with 0.1% Triton X-100 in PBS for 30 min, rinsed twice and washed for 5 min with PBS before blocking. Both methanol- and PFA-fixed cells were blocked with 10% donkey normal serum in PBS containing 0.25% saponin for 1 hr as described (Malikov et al., 2015). Coverslips were incubated with primary antibodies at 4 °C overnight. Anti-HSPA8 (1B5, #ADI-SPA-815-J) from Enzo, anti-ICP4 (ab6514) from Abcam, anti-A27 (J97Q) from Life Technologies, anti-DNA-PK (3H6, #12311), anti-IRF9 (D2T8M, #76684) or anti-Flag (D6W5B, #86861) from Cell Signaling were diluted at 1:400 in antibody buffer (PBS supplemented with 10% donkey normal serum and 0.025% saponin). Coverslips were washed 3 times for 5 min with wash buffer (PBS supplemented with 0.025% saponin) and incubated for 1 hr at RT with the appropriate Alexa 647-conjugated secondary antibody diluted at 1:400 in antibody buffer. After a rinse and 5 min wash, nuclei were stained with Hoechst 33342 for 20 min. 10 mg/mL Hoechst 33342 stock solution in water was diluted at 1:500 in wash buffer. Coverslips were washed 3 times in wash buffer for 5 min, rinsed with water, partially dried up at RT and mounted onto glass slides with FluorSave reagent (Calbiochem). Widefield images were acquired in Metamorph imaging software using Leica DMI 6000B motorized microscope equipped with Photometrics Prime 95B camera. All acquisitions settings were kept strictly equal for the images used in quantifications. Calculations of protein quantities in nuclei were performed in Metamorph imaging software. Cell nuclei on Hoechst 33342 images were selected with “auto threshold for light objects” function and subsequently converted into regions. The regions were transferred onto HSPA8 images and their areas and integrated fluorescence intensities were measured by region measurement tool. All images were calibrated using pre-determined calibrate distances function of the software before measurements.
Enzyme-linked immunosorbent assay (ELISA)
To measure concentration of interferons (IFN) alpha, beta and gamma, culture media of CHME3 pools stably expressing Flag, FEZ1-Flag or S58A-Flag were collected, filtered through 0.45 μm filter and stored at −80°C. On the day of procedure, culture media were thawed and concentrated 10-fold using Amicon Ultra-15 Ultracel-3K centrifugal filters (Millipore). Culture media of CHME3 Flag pool supplemented with 1,000 U/mL of IFNα, IFNβ or 100 ng/mL of IFNγ were collected in parallel as positive controls. Fresh culture medium was measured to determine baselines and subtracted from values in samples. ELISA was performed on 150 μL samples according to the instructions by the manufacturers. VeriKine Human IFN alpha (#411001) and Human IFN beta (#41100-1) ELISA kits were purchased form PBL Assay Sciences, and interferon gamma Human ELISA kit (KHC4021) was obtained from Invitrogen. O.D. values were measured using a microplate reader (Bio-Rad, iMARK plate reader), actual concentrations of interferons were calculated from a linear plot of standards provided by the kit manufacturer.
Fractionation assay
Fractionation of cytoplasmic and nuclear fractions was performed with NE-PER nuclear and cytoplasmic extraction reagents from Thermo Scientific (#78833) in accordance with the manufacturer’s instructions. Briefly, 106 cells were washed with PBS, centrifuged at 500 g for 5 min, and then re-suspended in 100 μl of buffer CER I. After 15 sec vortexing followed by 10 minutes incubation on ice and addition of 5.5 μl of buffer CER II, the cytoplasmic fraction was isolated by centrifugation at 21,130 g for 5 min. The pelleted nuclear fraction was prepared by incubation on ice for 40 minutes with 50 μl buffer NER. The fractions were mixed with equal volumes of Laemmli buffer, boiled, and analyzed by WB. Anti-Tubulin and anti-Lamin B1 antibodies were used to evaluate cross-contamination of the fractions.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was calculated in GraphPad Prizm software version 8.4.3 using either a Student’s t-test for two groups or one-way analysis of variance (ANOVA) with Tukey’s honestly significant difference (HSD), Dunnett or Sidak’s post hoc tests for groups of three or more groups. Results are expressed as means ± standard deviations (SD). Statistical significance is represented by ns, *, ** and *** over the bars representing a p-value >0.05, ≤0.05, ≤0.01 and ≤0.001, respectively.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-HSV-1 ICP4 Immediate Early Protein [10F1], mouse monoclonal | Abcam | cat #ab6514; RRID:AB_305537 |
| anti-HSV-1 ICP0 [5H7], mouse monoclonal | Abcam | cat #ab6513; RRID:AB_305536 |
| anti-HSV ICP5, mouse monoclonal | Virusys Corporation | cat #HA018; RRID:AB_2713935 |
| anti-VSV-G tag [P5D4], mouse monoclonal | Abcam | cat #ab50549; RRID:AB_883494 |
| anti-VacV I3 | Dr. David Evans (University of Alberta, Edmonton, Canada) | N/A |
| anti-VacV G8 | Dr. Paula Traktman (Medical University of South Carolina, Columbia, SC, USA) | N/A |
| anti-VacV A25 | Dr. Yan Xiang (University of Texas Health Sciences Center, Austin, TX, USA) | N/A |
| anti-VacV A27 | Life Technologies | cat #J97Q |
| anti-IFNβ antibody | Millipore-Sigma | cat #ab1431; RRID:AB_90638 |
| anti-Flag DYKDDDDK Tag [D6W5B] for IF, rabbit monoclonal | Cell Signaling Technology | cat #15009; RRID:AB_2798687 |
| anti-DNA-PK, mouse monoclonal | Cell Signaling Technology | cat #12311, 3H6; RRID:AB_2797881 |
| anti-IRF9 | Cell Signaling Technology | cat #76684, D2T8M; RRID:AB_2799885 |
| anti-HSPA8, rat | Enzo | cat #ADI-SPA-815-J, 1B5 |
| anti-tyrosinated tubulin, rat | Dr. Gregg Gunderson (Columbia University, New York, NY, USA) | N/A |
| anti-GFP | Abcam | cat #ab13970; RRID:AB_300798 |
| anti-pIRF3, phospho S386 | Abcam | cat #ab76493: RRID:AB_1523836 |
| anti-FEZ1, rabbit monoclonal | Cell Signaling Technology | cat #42480, D9R8Q; RRID:AB_2799222 |
| anti-FEZ1, mouse monoclonal | Abnova | cat #H00009638-B02P |
| anti-PKR | Cell Signaling Technology | cat #12297, D7F7; RRID:AB_2665515 |
| anti-MxA | Cell Signaling Technology | cat #37849, D3W7I; RRID:AB_2799122 |
| anti-MxB | Cell Signaling Technology | cat #43924, E7Y8H |
| anti-ISG56 | Cell Signaling Technology | cat #14769, D2X9Z; RRID:AB_2783869 |
| anti-IRF3 | Cell Signaling Technology | cat #11904, D6I4C; RRID:AB_2722521 |
| anti-IRF7 | Cell Signaling Technology | cat# 72073, D8V1J |
| anti-Lamin B1 | Cell Signaling Technology | cat #13435, D9V6H; RRID:AB_2737428 |
| anti-NF-kB | Cell Signaling Technology | cat #8242, D14E12; RRID:AB_10859369 |
| anti-GAPDH | Santa Cruz.Antibodies | cat #sc-25778; RRID:AB_10167668 |
| anti-phospho-S58-FEZ1 | Dr. John Jia En Chua (Max Planck Institute for Biophysical Chemistry, Gottingen, Germany) | N/A |
| anti Rabbit IgG, HRP | GE Healthcare UK | cat #NA934V |
| anti Mouse IgG, HRP | GE Healthcare UK | cat #NA931V |
| anti Rat IgG, HRP | Invitrogen | cat #PA1-28664 |
| anti-Rat secondary, Alexa 647 | Jackson ImmunoResearch | cat #712-605-150; RRID:AB_2340693 |
| anti-Mouse secondary, Alexa 488 | Life Technologies | cat #A21202; RRID:AB_141607 |
| anti-Rabbit secondary, Alexa 488 | Life Technologies | cat #A21206; RRID:AB_2535792 |
| Bacterial and virus strains | ||
| Escherichia coli (E. coli) DH5α strain | Thermo Fisher Scientific | cat #18265017 |
| HSV-1 | Dr. Ian Mohr (NYU-Lagone, New York, NY, USA) | N/A |
| VACV (WR) | Dr. Stewart Shuman (Sloan-Kettering Institute, New York NY, USA) | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| ProteaseMAX | Promega | cat #V207A |
| Trypsin Gold | Promega | cat #V528A |
| Pierce Spin Columns with C18 resin | Thermo Fisher Scientific | cat #89879 |
| microBCA protein assay kit | Thermo Fisher Scientific | cat #23235 |
| nanoViper trap column | Thermo Fisher Scientific | cat #164535 |
| nanoViper analytical column | Thermo Fisher Scientific | cat #164942 |
| G-sepharose | GE Healthcare | cat #17-0618-01 |
| Lipofectamine 2000 Transfection Reagent | Thermo Fisher Scientific | cat # 11668019 |
| CT DNA | Sigma | cat #D4764 |
| Nu-7441 DNA-PK inhibitor | SelleckChem | cat #S2638 |
| Polybrene | Santa Cruz Biotechnology | cat #sc-134220 |
| FluroSave Reagent | MilliporeSigma | cat #345789 |
| Hoechst 33342 | Thermo Fisher Scientific | cat #62249 |
| PlasmoTest mycoplasma detection kit | InvivoGen | Cat #rep-pt1 |
| Lipofectamine RNAiMAX Transfection Reagent | Thermo Fisher Scientific | cat #13778030 |
| QUANTI-Blue | InvivoGen | cat # rep-qbs |
| QUANTI-Luc | InvivoGen | cat #rep-qlc1 |
| Cas9 protein | UC-Berkeley Macrolab | N/A |
| SF Nucleofector solution | Lonza | cat #V4XC-2012 |
| GFP-Trap agarose | ChromoTek | cat #gta-100 |
| Opti-Mem medium | Fisher Scientific | cat #31985070 |
| DMEM | Fisher Scientific | cat #MT15013CV |
| RPMI 1640 | Fisher Scientific | cat #11875093 |
| 0.1% Gelatin solution | Millipore | cat #ES-006-B |
| Amicon Ultra-15 Ultracel-3K centrifugal filters | Millipore | cat #UFC500324 |
| Human IFN beta ELISA kit | PBL Assay Sciences | cat #41100-1 |
| Human IFN-b | EMD Millipore, Millipore Sigma | cat #IF014 |
| NE-PER nuclear and cytoplasmic extraction reagent | Thermo Fisher Scientific | cat #78833 |
| EcoRI-HF | New England BioLabs | cat #R3101S |
| SbfI-HF | New England BioLabs | cat # R3642S |
| NotI-HF | New England BioLabs | cat #R3189S |
| Pierce Protease Inhibitor Mini Tablets, EDTA-free | Thermo Fisher Scientific | cat #A32955 |
| Experimental models: Cell lines | ||
| CHME3 | Dr. Marc Tardieu (Universite Paris Sud, France) | N/A |
| Human: Normal Human Dermal Fibroblasts (NHDFs) | Lonza | cat #CC-2509 |
| African green monkey: BSC-40 | Dr. Ian Mohr (NYU-Lagone, New York, NY, USA) | N/A |
| African green monkey: Vero | Dr. Ian Mohr (NYU-Lagone, New York, NY, USA) | N/A |
| HEK-293T (293T) | ATCC | cat #CLR-3216 |
| THP1-Dual cell line | InvivoGen | cat #thpd-nfis |
| THP1-Dual KO-STING cell line | InvivoGen | cat #thpd-kostg |
| Oligonucleotides | ||
| See Table S2 for oligonucleotide information | N/A | |
| Recombinant DNA | ||
| pQCXIN | Clontech | cat #631514 |
| pQCXIN-Flag | (Malikov et al., 2015) | N/A |
| pQCXIN-FEZ1-Flag | (Malikov et al., 2015) | N/A |
| pQCXIN-FEZ1-S58A-Flag | (Malikov et al., 2015) | N/A |
| pQCXIN-eGFP | This paper | N/A |
| pQCXIN-eGFP-FEZ1 | This paper | N/A |
| pQCXIN-eGFP-FEZ1-S58A | This paper | N/A |
| pPM-C-HA-HSPA8 | Applied Biological Materials | Cat # BC016179 |
| pQCXIN-HSPA8-Flag | This paper | N/A |
| pQCXIN-eGFP-HSPA8 | This paper | N/A |
| pCMV-intron | Dr. Stephen P. Goff (Columbia University, New York, NY, USA) | N/A |
| pVSV-G | Dr. Stephen P. Goff (Columbia University, New York, NY, USA) | N/A |
| Software and algorithms | ||
| FIJI (image J installation) | Open source | http://fiji.sc RRID:SCR_002285 |
| MetaMorph Microscopy Automation and Image Analysis Software | Molecular Devices | RRID:SCR_002368 |
| GraphPad Prism (Version 7) | Graphpad | RRID:SCR_002798 |
Highlights.
FEZ1 is a regulator of STING-independent induction of interferon and ISG expression
FEZ1’s function as a regulator of ISG responses requires Serine 58 phosphorylation
FEZ1 Serine 58 controls both binding to, and localization of, HSPA8
FEZ1 and HSPA8 regulate nuclear accumulation of DNA-PK and ISG responses
ACKNOWLEDGMENTS
We thank Jeffrey Savas for performing mass spectrometry, and Yan Xiang, Paula Traktman, and David Evans for antibodies against VacV proteins. This work was supported by NIH grant R01 GM101975 (to M.H.N.), NIH grant R01 AI150559 (to M.H.N.), the Gilead Sciences Research Scholars Program in HIV (to J.F.H.), NIH grant K22 AI136691 (to J.F.H.), NIH grant R01 AI150998 (to J.F.H.), the NIH-supported Third Coast CFAR P30 AI117943 (to J.F.H.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110396.
DECLARATION OF INTERESTS
The authors declare conflicting interests.
REFERENCES
- Adlakha M, Livingston CM, Bezsonova I, and Weller SK (2020). The herpes simplex virus 1 immediate early protein ICP22 is a functional mimic of a cellular J protein. J. Virol 94, e01564–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banski P, Mahboubi H, Kodiha M, Shrivastava S, Kanagaratham C, and Stochaj U (2010). Nucleolar targeting of the chaperone hsc70 is regulated by stress, cell signaling, and a composite targeting signal which is controlled by autoinhibition. J. Biol. Chem 285, 21858–21867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bercovich B, Stancovski I, Mayer A, Blumenfeld N, Laszlo A, Schwartz AL, and Ciechanover A (1997). Ubiquitin-dependent degradation of certain protein substrates in vitro requires the molecular chaperone Hsc70. J. Biol. Chem 272, 9002–9010. [DOI] [PubMed] [Google Scholar]
- Bloom L, and Horvitz HR (1997). The Caenorhabditis elegans gene unc-76 and its human homologs define a new gene family involved in axonal outgrowth and fasciculation. Proc. Natl. Acad. Sci. U S A 94, 3414–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonam SR, Ruff M, and Muller S (2019). HSPA8/HSC70 in immune disorders: a molecular rheostat that adjusts chaperone-mediated autophagy substrates. Cells 8, 849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyer J, Rohleder K, and Ketner G (1999). Adenovirus E4 34k and E4 11k inhibit double strand break repair and are physically associated with the cellular DNA-dependent protein kinase. Virology 263, 307–312. [DOI] [PubMed] [Google Scholar]
- Burch AD, and Weller SK (2004). Nuclear sequestration of cellular chaperone and proteasomal machinery during herpes simplex virus type 1 infection. J. Virol 78, 7175–7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burleigh K, Maltbaek JH, Cambier S, Green R, Gale M Jr., James RC, and Stetson DB (2020). Human DNA-PK activates a STING-independent DNA sensing pathway. Sci. Immunol 5, eaba4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkevich E, Hartig W, Nikolov M, Erck C, Grosche J, Urlaub H, Schmidt CF, Klopfenstein DR, and Chua JJ (2016). Phosphorylation of FEZ1 by microtubule affinity regulating kinases regulates its function in presynaptic protein trafficking. Sci. Rep 6, 26965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chua JJ, Butkevich E, Worseck JM, Kittelmann M, Gronborg M, Behrmann E, Stelzl U, Pavlos NJ, Lalowski MM, Eimer S, et al. (2012). Phosphorylation-regulated axonal dependent transport of syntaxin 1 is mediated by a Kinesin-1 adapter. Proc. Natl. Acad. Sci. U S A 109, 5862–5867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colantuoni C, Hyde TM, Mitkus S, Joseph A, Sartorius L, Aguirre C, Creswell J, Johnson E, Deep-Soboslay A, Herman MM, et al. (2008). Age-related changes in the expression of schizophrenia susceptibility genes in the human prefrontal cortex. Brain Struct. Funct 213, 255–271. [DOI] [PubMed] [Google Scholar]
- Dharan A, Opp S, Abdel-Rahim O, Keceli SK, Imam S, Diaz-Griffero F, and Campbell EM (2017). Bicaudal D2 facilitates the cytoplasmic trafficking and nuclear import of HIV-1 genomes during infection. Proc. Natl. Acad. Sci. U S A 114, E10707–E10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGiuseppe S, Rollins MG, Astar H, Khalatyan N, Savas JN, and Walsh D (2020). Proteomic and mechanistic dissection of the poxvirus-customized ribosome. J. Cell Sci 134, jcs246603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson BJ, Mansur DS, Peters NE, Ren H, and Smith GL (2012). DNA-PK is a DNA sensor for IRF-3-dependent innate immunity. Elife 1, e00047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Maturana AD, Ikuta J, Hamada J, Walchli S, Suzuki T, Sawa H, Wooten MW, Okajima T, Tatematsu K, et al. (2007). Axonal guidance protein FEZ1 associates with tubulin and kinesin motor protein to transport mitochondria in neurites of NGF-stimulated PC12 cells. Biochem. Biophys. Res. Commun 361, 605–610. [DOI] [PubMed] [Google Scholar]
- Gao G, and Goff SP (1999). Somatic cell mutants resistant to retrovirus replication: intracellular blocks during the early stages of infection. Mol. Biol. Cell 10, 1705–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindhart JG, Chen J, Faulkner M, Gandhi R, Doerner K, Wisniewski T, and Nandlestadt A (2003). The kinesin-associated protein UNC-76 is required for axonal transport in the Drosophila nervous system. Mol. Biol. Cell 14, 3356–3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haedicke J, Brown C, and Naghavi MH (2009). The brain-specific factor FEZ1 is a determinant of neuronal susceptibility to HIV-1 infection. Proc. Natl. Acad. Sci. U S A 106, 14040–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hristova DB, Lauer KB, and Ferguson BJ (2020). Viral interactions with non-homologous end-joining: a game of hide-and-seek. J. Gen. Virol 101, 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PT, Summers BJ, Xu C, Perilla JR, Malikov V, Naghavi MH, and Xiong Y (2019). FEZ1 is recruited to a conserved cofactor site on capsid to promote HIV-1 trafficking. Cell Rep. 28, 2373–2385.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hultquist JF, Hiatt J, Schumann K, McGregor MJ, Roth TL, Haas P, Doudna JA, Marson A, and Krogan NJ (2019). CRISPR-Cas9 genome engineering of primary CD4(+) T cells for the interrogation of HIV-host factor interactions. Nat. Protoc 14, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuta J, Maturana A, Fujita T, Okajima T, Tatematsu K, Tanizawa K, and Kuroda S (2007). Fasciculation and elongation protein zeta-1 (FEZ1) participates in the polarization of hippocampal neuron by controlling the mitochondrial motility. Biochem. Biophys. Res. Commun 353, 127–132. [DOI] [PubMed] [Google Scholar]
- Jan E, Mohr I, and Walsh D (2016).A cap-to-tail guide to mRNA translation strategies in virus-infected cells. Annu. Rev. Virol 3, 283–307. [DOI] [PubMed] [Google Scholar]
- Janabi N, Peudenier S, Heron B, Ng KH, and Tardieu M (1995). Establishment of human microglial cell lines after transfection of primary cultures of embryonic microglial cells with the SV40 large T antigen. Neurosci. Lett 195, 105–108. [DOI] [PubMed] [Google Scholar]
- Kang E, Burdick KE, Kim JY, Duan X, Guo JU, Sailor KA, Jung DE, Ganesan S, Choi S, Pradhan D, et al. (2011). Interaction between FEZ1 and DISC1 in regulation of neuronal development and risk for schizophrenia. Neuron 72, 559–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpova AY, Trost M, Murray JM, Cantley LC, and Howley PM (2002). Interferon regulatory factor-3 is an in vivo target of DNA-PK. Proc. Natl. Acad. Sci. U S A 99, 2818–2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Walker CL, Karten B, Kuny SL, Tennese AA, O’Neill MA, and Wevrick R (2005). Essential role for the Prader-Willi syndrome protein necdin in axonal outgrowth. Hum. Mol. Genet 14, 627–637. [DOI] [PubMed] [Google Scholar]
- Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, and Spencer CA (1996). Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol 70, 7471–7477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link AJ, and LaBaer J (2011). Trichloroacetic acid (TCA) precipitation of proteins. Cold Spring Harb. Protoc 2011, 993–994. [DOI] [PubMed] [Google Scholar]
- Lipska BK, Mitkus SN, Mathew SV, Fatula R, Hyde TM, Weinberger DR, and Kleinman JE (2006). Functional genomics in postmortem human brain: abnormalities in a DISC1 molecular pathway in schizophrenia. Dialogues Clin. Neurosci 8, 353–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, and Zhang L (2020). DNA-sensing antiviral innate immunity in poxvirus infection. Front. Immunol 11, 1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikov V, da Silva ES, Jovasevic V, Bennett G, de Souza Aranha Vieira DA, Schulte B, Diaz-Griffero F, Walsh D, and Naghavi MH (2015). HIV-1 capsids bind and exploit the kinesin-1 adaptor FEZ1 for inward movement to the nucleus. Nat. Commun 6, 6660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malikov V, and Naghavi MH (2017). Localized phosphorylation of a kinesin-1 adaptor by a capsid-associated kinase regulates HIV-1 motility and uncoating. Cell Rep. 20, 2792–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maturana AD, Fujita T, and Kuroda S (2010). Functions of fasciculation and elongation protein zeta-1 (FEZ1) in the brain. Sci. World J 10, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyoshi K, Honda A, Baba K, Taniguchi M, Oono K, Fujita T, Kuroda S, Katayama T, and Tohyama M (2003). Disrupted-In-Schizophrenia 1, a candidate gene for schizophrenia, participates in neurite outgrowth. Mol. Psychiatry 8, 685–694. [DOI] [PubMed] [Google Scholar]
- Naghavi MH, Hatziioannou T, Gao G, and Goff SP (2005). Overexpression of fasciculation and elongation protein zeta-1 (FEZ1) induces a post-entry block to retroviruses in cultured cells. Genes Dev. 19, 1105–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno Y, Imamoto N, and Yoneda Y (1993). 70-kDa heat-shock cognate protein colocalizes with karyophilic proteins into the nucleus during their transport in vitro. Exp. Cell Res 206, 134–142. [DOI] [PubMed] [Google Scholar]
- Parkinson J, Lees-Miller SP, and Everett RD (1999). Herpes simplex virus type 1 immediate-early protein vmw110 induces the proteasome-dependent degradation of the catalytic subunit of DNA-dependent protein kinase. J. Virol 73, 650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters NE, Ferguson BJ, Mazzon M, Fahy AS, Krysztofinska E, Arribas-Bosacoma R, Pearl LH, Ren H, and Smith GL (2013). A mechanism for the inhibition of DNA-PK-mediated DNA sensing by a virus. PLoS Pathog. 9, e1003649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, and Rice CM (2011). A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scutts SR, Ember SW, Ren H, Ye C, Lovejoy CA, Mazzon M, Veyer DL, Sumner RP, and Smith GL (2018). DNA-PK is targeted by multiple Vaccinia virus proteins to inhibit DNA sensing. Cell Rep. 25, 1953–1965.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith S, Reuven N, Mohni KN, Schumacher AJ, and Weller SK (2014). Structure of the herpes simplex virus 1 genome: manipulation of nicks and gaps can abrogate infectivity and alter the cellular DNA damage response. J. Virol 88, 10146–10156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricher F, Macri C, Ruff M, and Muller S (2013). HSPA8/HSC70 chaperone protein: structure, function, and chemical targeting. Autophagy 9, 1937–1954. [DOI] [PubMed] [Google Scholar]
- Suzuki T, Okada Y, Semba S, Orba Y, Yamanouchi S, Endo S, Tanaka S, Fujita T, Kuroda S, Nagashima K, et al. (2005). Identification of FEZ1 as a protein that interacts with JC virus agnoprotein and microtubules: role of agnoprotein-induced dissociation of FEZ1 from microtubules in viral propagation. J. Biol. Chem 280, 24948–24956. [DOI] [PubMed] [Google Scholar]
- Teixeira MB, Alborghetti MR, and Kobarg J (2019). Fasciculation and elongation zeta proteins 1 and 2: from structural flexibility to functional diversity. World J. Biol. Chem 10, 28–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachev TI, Stoyanova VK, Ivanov HY, Minkov IN, and Popov NT (2015). Investigation of fasciculation and elongation protein zeta-1 (FEZ1) in peripheral blood reveals differences in gene expression in patients with schizophrenia. Balkan J. Med. Genet 18, 31–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, Arias C, Perez C, Halladin D, Escandon M, Ueda T, Watanabe-Fukunaga R, Fukunaga R, and Mohr I (2008). Eukaryotic translation initiation factor 4F architectural alterations accompany translation initiation factor redistribution in poxvirus-infected cells. Mol. Cell Biol 28, 2648–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, and Mohr I (2004). Phosphorylation of eIF4E by Mnk-1 enhances HSV-1 translation and replication in quiescent cells. Genes Dev. 18, 660–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh D, and Mohr I (2006). Assembly of an active translation initiation factor complex by a viral protein. Genes Dev. 20, 461–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F, Bonam SR, Schall N, Kuhn L, Hammann P, Chaloin O, Madinier JB, Briand JP, Page N, and Muller S (2018). Blocking nuclear export of HSPA8 after heat shock stress severely alters cell survival. Sci. Rep 8, 16820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Li Y, Yang X, Zhao J, Cheng Y, and Wang J (2020). Mechanism and complex roles of HSC70 in viral infections. Front. Microbiol 11, 1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this study will be shared by the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this work is available from the lead contact upon request.