

Abstract
Purpose:
Immune checkpoint inhibitors (ICIs) have shown clinical benefit in many types of metastatic cancers with only a few predictive biomarkers identified so far. CDKN2A is commonly altered in human cancers, but prior studies have provided conflicting evidence regarding the association between CDKN2A genomic alterations (GAs) and response to ICIs. Herein, we examined the impact of loss-of-function CDKN2A alterations on response and survival in patients treated with ICIs.
Experimental design:
We studied the association between loss-of-function CDKN2A alterations and the response to ICIs in two independent cohorts of six different cancer types. 789 patients treated at Dana-Farber Cancer Institute (DFCI) and 1250 patients treated at Memorial Sloan-Kettering Cancer Center (MSKCC) were included in the final analysis. Patients’ tumors were sequenced using Oncopanel or MSK-IMPACT. RNA-seq data from TCGA and IMvigor210 were used to investigate differences in the tumor microenvironment.
Results:
In the DFCI cohort, CDKN2A GAs were associated with poor response and survival in urothelial carcinoma patients treated with ICIs, but not those treated with platinum-based therapy. Similarly, CDKN2A GAs were associated with worse outcomes in the MSKCC urothelial carcinoma cohort treated with ICI. There was no association of CDKN2A status with ICI treatment outcome in five other cancers: esophagogastric, head and neck, non-small cell lung, renal cell carcinomas, and melanoma. Immuno-inflammatory pathways were significantly reduced in expression in CDKN2A altered tumors.
Conclusion:
Our data show that CDKN2A GAs were associated with reduced benefit from ICI therapy in urothelial carcinoma as well as changes in the tumor immune microenvironment.
Keywords: biomarkers, immunotherapy, genomics
Introduction
Immune checkpoint inhibition (ICI) using anti-PD-1/PD-L1 or anti-CTLA4 antibodies has proven to be beneficial in the treatment of multiple cancers(1–5). Given the variability in response rates to immunotherapy agents, efforts have focused on identifying predictors of response to therapy, using cancer-specific as well as pan-cancer approaches. Expression of PD-L1 on cancer cells, high microsatellite instability, and tumor mutation burden (TMB) are the main predictors of response that have been identified, although the microbiome and a variety of immune, transcriptomic, metabolomic, and genomic biomarkers have been studied(4,6–20). Identification of such biomarkers is both of clinical utility in choice and prioritization of treatment, and may provide insight into immune regulatory mechanisms and novel treatment approaches.
CDKN2A is a tumor suppressor gene on chromosome 9p21.3 that encodes two different proteins – p14ARF and p16INK4a. Translated in alternative reading frames, p14ARF and p16INK4a each act as regulators of the cell cycle. p14ARF interacts with MDM2 and degrades it, preventing the inactivation of p53 through ubiquitin-mediated proteolysis or transcriptional silencing(21). p16INK4a binds CDK4 and CDK6 to prevent phosphorylation of the RB protein. Loss of function mutations or homozygous deletion of CDKN2A leads to loss of both proteins, releasing G1/S and G2/M cell cycle checkpoints and resulting in uninhibited cell proliferation and tumor formation(22).
Along with TP53, CDKN2A is one of the most commonly altered genes in human cancers(22,23). In the Cancer Genome Atlas (TCGA) studies, genomic alterations in CDKN2A were found in about 50% of head and neck cancers, 47% of pancreatic and esophageal cancers, 37% of melanomas and bladder cancers, 35% of gliomas and 30% of non-small cell lung cancers (NSCLC) and gastroesophageal junction and gastric adenocarcinomas, and 3% of renal cell carcinoma(24–31). Three different mechanisms can lead to inactivation of the gene: genomic deletion, point mutations, and CpG-island hypermethylation at the promoter region of CDKN2A. Genomic deletion, so-called ‘deep deletion’ or homozygous loss, which typically extends to include other genes on 9p21.3, is the most common alteration affecting CDKN2A(32).
Multiple studies have examined the impact of CDKN2A alterations on the tumor immune microenvironment and on the response to ICI(33,34). In melanoma, immune checkpoint blockade is less effective when CDKN2A is deleted, thought to be due in part to loss of senescence induced by CDKN2A deletion(35,36). In a study of 909 NSCLC tumor specimens, CDKN2A/CDKN2B homozygous loss was significantly associated with negative PD-L1 expression by immunohistochemistry (IHC)(37). In pancreatic cancer and glioma, tumors harboring deletions or mutations in CDKN2A/CDKN2B had lower immune cytolytic activity, as defined by a cytolytic index that considers the expression signature of GZMA and PRF-1(38,39).
There is conflicting evidence regarding the association between CDKN2A alterations and ICI response. For example, in patients with advanced renal cell carcinoma (RCC) infiltrated with CD8+ T-cells, deletions in 9p21.3 (affecting CDKN2A) were associated with reduced clinical benefit and inferior survival when treated with ICI after tyrosine kinase inhibitor treatment(40). Similarly, in urothelial carcinoma (UC), CDKN2B homozygous deletions (often concomitant with CDKN2A deletions) were associated with worse objective response rates and worse survival in the setting of ICI treatment(41). However, in melanoma, the evidence is less consistent, with some studies showing ICI resistance(36,42,43), and others demonstrating no effect of CDKN2A alteration on tumor response(44). In other tumor subtypes, associations between CDKN2A alterations and ICI response have not been examined in detail.
Herein, we assessed the association between loss-of-function variants in CDKN2A and clinical outcomes in patients treated with ICIs in two large independent cohorts including six different cancer types.
Methods
Study design and patient cohorts
DFCI and MSKCC clinical cohorts
The study included patients diagnosed with a solid cancer type for which ICIs were FDA-approved: esophagogastric adenocarcinoma (EGC), head and neck squamous cell carcinoma (HNSCC), melanoma, non-small cell lung cancer (NSCLC), RCC, and urothelial carcinoma. Two independent cohorts were analyzed: An internal cohort of 789 patients treated at the Dana-Farber Cancer Institute (DFCI, Boston MA) and an external cohort of 1250 patients treated at Memorial Sloan Kettering Cancer Center (MSKCC, New York, NY, Supplementary Figure S1). All DFCI patients received an anti–PD-1/PD-L1 or anti–CTLA-4 agent in the metastatic setting and had next-generation targeted sequencing (NGS) of their tumor tissue performed (as described below). Patients were excluded if they were lost to follow-up, had no measurable disease, or had clinical deterioration within one week of the first ICI dose. Patients on combined treatment, e.g. mutation-targeted or chemotherapy combined with non-ICI therapy were excluded. Among the DFCI cohort, formalin-fixed paraffin-embedded (FFPE) specimens were collected between 2012 and 2020.
The study was conducted in accordance with the ethical guidelines of the Declaration of Helsinki. The Institutional Review Board (IRB) of DFCI (Boston, MA) approved this work and informed written consent for cancer genetic analysis was obtained from each subject.
For the MSKCC cohort, we obtained de-identified publicly available data on 1250 patients(13) (Supplementary Figure S1). Specimens were collected between 2013 and 2017.
IMvigor210 cohort
We analyzed a publicly available dataset, IMvigor210(33), which was a single arm Phase II study investigating atezolizumab (1200mg 3 times weekly) in mUC patients (NCT02108652, NCT02951767). All 236 patients included had available tumor tissue taken in the 2 years prior to study entry. This tissue was used for PD-L1 analysis (SP142 antibody using the VENTANA platform, see below), immune cell infiltration assessment, and targeted next generation sequencing using the FoundationOne® panel (Foundation Medicine, Cambridge, MA) as described previously(33). We excluded patients without FoundationOne sequencing, RNA-seq, PD-L1 staining or immune cell infiltration assessment by IHC. We also excluded patients with CDKN2A alterations of unknown significance (missense mutations, synonymous mutations and single-copy losses).
The Cancer Genome Atlas (TCGA) Bladder Cancer cohort
For the TCGA BLCA cohort(26), we focused our survival analysis on patients with stage IV muscle-invasive bladder cancer (MIBC, n=135 patients). For transcriptomic analysis, we included MIBC tumors across all stages (II-IV) for a total of 406 tumor samples with available bulk RNA sequencing data.
Data collection
Data about gender, cancer type, age at initiation of ICI therapy, type and class of ICI-based regimen were collected for both the DFCI and MSKCC cohorts. Additional clinical variables available for the DFCI cohort included lines of therapy prior to ICIs and site of lesion subjected to targeted sequencing.
Genomic analysis
Details of the tissue collection, DNA extraction, and tumor targeted sequencing using the Oncopanel/PROFILE and MSK-IMPACT for DFCI and MSKCC cohorts, respectively, were previously described in detail(45–48). The Oncopanel gene panels include capture probes for 275–447 cancer-associated genes, as well as intronic portions of 60 genes for rearrangement detection(46). As previously described(13), MSK-IMPACT includes 3 versions and identifies somatic exonic mutations in a predefined subset of genes ranging between 341–468. The MSK-IMPACT analysis included both tumor- derived and matched germline normal DNA, so that somatic mutations were certain.
For the purposes of this study, we focused our mutational and copy number variation (CNV) analyses on CDKN2A. TMB was defined as the number of exonic, nonsynonymous SNVs, and indel mutations per megabase of genome examined.
Variant assessment
For the DFCI Oncopanel data, we excluded putative germline variants if they were previously found in either 1) a panel of historical normal samples sequenced via Oncopanel, 2) the exome sequencing project(49) (http://evs.gs.washington.edu/EVS/; accessed May 2013), or 3) in the gnomAD(50) at a population allele frequency ≥0.1%, unless the variant was also reported in COSMIC(45). SNVs and indels were called by the Oncopanel pipeline using MuTect v.1 0.27200 (https://confluence.broadinstitute.org/display/CGATools/MuTect; accessed May 2013) and annotated using Oncotator (http://www.broadinstitute.org/oncotator; accessed May 2013).
For the MSKCC cohort, germline variants were eliminated through the use of patient-matched blood DNA(47). SNVs were called using MuTect, VarDict(51), and Somatic indel detector(52), and reported for >5% allele frequency (novel variants) or >2% allele frequency (recurrent hotspots). Copy number alterations were called using a custom pipeline and reported for fold-change >2(47). Structural rearrangements were called using Delly(53).
In the IMvigor210 cohort, putative germline variants were filtered out using the gnomAD(50) at a frequency ≥0.1%, unless the variant was also reported in COSMIC(45).
For the DFCI, MSKCC, IMvigor210, and TCGA UC cohorts, LOF variants in CDKN2A were defined as either nonsense SNVs, out-of-frame and in-frame indels, splice-site variants affecting consensus nucleotides; or homozygous deletions. Missense SNVs were not included as we could not assess functional effects with confidence. For the DFCI cohort, CNVs were identified using a custom R-based tool (VisCap-Cancer(54)) that compares read-depth at all genomic regions assayed among different samples. For all four cohorts, we focused on homozygous deletion CNVs in this analysis, and excluded heterozygous deletions, as the latter have uncertain functional effects, and can be confounded by background noise on both Oncopanel and MSK-IMPACT sequencing platforms(46,47).
RNA-sequencing
We obtained publicly available bulk RNA-sequencing data from the TCGA BLCA(55) and IMvigor210 cohorts(33). For each specimen, the transcripts-per-million (TPM) counts were calculated and the data was upper-quartile normalized using the R package edgeR. TIMER (cistrome.shinyapps.io/timer) was used as previously described to investigate the molecular characterization of tumor-immune interactions as a function of CDKN2A genomic alteration status(56). GSEA (http://software.broadinstitute.org/gsea/index.jsp) was performed on the TCGA dataset (n=406 tumors) to test whether any biologically-relevant gene sets (Hallmark, C6-oncogenic signatures, C7-immunologic signatures, C2-canonical pathways) were differentially expressed between CDKN2A altered versus WT tumors.
Immunohistochemistry staining
IMvigor210
We obtained data on PD-L1 and CD8 IHC staining from the IMvigor210 cohort of metastatic urothelial carcinoma(33). In this prior study, samples were scored for PD-L1 expression on tumor-infiltrating ICs, as IHC IC0, IC1, or IC2+, if <1%, ≥1% but <5%, ≥5% of IC were PD-L1 positive, respectively. Tumor CD8 IHC was used to categorize tumors as “desert” when the prevalence of CD8+ cells was low (<10 CD8+ cells per 10 200x fields); “immune excluded” when CD8+ cells were present exclusively in tumor stroma; and inflammatory when CD8+ cells were in direct contact with malignant epithelial cells.
Statistical analysis
Statistical analyses were performed using R version 3.6.2. Statistical tests included x2 or Fisher exact tests for categorical variables and the Wilcoxon Rank-Sum test (two-group comparisons) or the Kruskal–Wallis exact test (three-group comparisons) for continuous variables.
Clinical outcomes included overall survival (OS) for the DFCI, MSKCC, and TCGA cohorts. For the DFCI and MSKCC cohorts, OS was calculated from the date of ICI initiation to the date of death. For the TCGA cohort, OS was calculated from the date of diagnosis to the date of death and progression-free survival (PFS) was calculated from the date of diagnosis to the date of progression or death. Alive patients were censored at the date of last follow-up. For the DFCI cohort, time-to-treatment failure (TTF), and overall response rate (ORR) were also available. TTF was calculated from the start date of ICI therapy to the start date of the next treatment or death. Patients alive and not started on next line were censored at the date of last follow-up. Response was investigator-assessed. ORR was defined as complete response (CR) or partial response (PR).
The distributions of OS and TTF were estimated with the Kaplan–Meier method along with 95% confidence intervals (95% CI), and their associations with genomic alterations were examined with the Wald x2 test from the Cox regression, adjusted for treatment type (single vs. combination) and number of lines of therapy prior to ICI for the DFCI cohort, and treatment type for the MSKCC cohort. For cohorts with significant associations, we further adjusted for TMB as a continuous variable.
The effects of CDKN2A alterations on ORR are presented as odds ratios (OR) estimated from logistic regression models, adjusted for prior lines of therapy, treatment type and TMB in the DFCI cohort. All comparisons were conducted separately for each tumor histology. Two-sided P values are reported. Multiple comparison adjustment was not performed as these studies were exploratory in nature.
Results
Spectrum of genetic alterations (GAs) in CDKN2A
Of 25,881 patients with available Oncopanel sequencing results at the Dana-Farber Cancer Institute (DFCI, Boston, MA) between 9/2012 and 10/2020, we identified 1,485 patients who received ICI treatment and did not harbor any single nucleotide variant (SNV) of unknown significance in CDKN2A, across the six included tumor types (Materials and Methods; Supplementary Figure S1, Supplementary Table S1). From the Memorial Sloan Kettering Cancer Center (MSKCC, New York, NY) cohort, we identified 1250 patients who fit the same criteria (Supplementary Figure S1, Supplementary Table S2).
The frequency of loss-of-function genomic alterations (GAs) in CDKN2A was similar in the DFCI and MSKCC cohorts (Figure 1, Supplementary Figure S1, Supplementary Table S1, S3–S4), but differed by histology. Head and neck squamous cell carcinoma (HNSCC) and melanoma had the highest rate of CDKN2A GAs (HNSCC: 30/77, 39%, melanoma: 124/375, 33.1%) in the DFCI cohort. Homozygous deletions were the most common CDKN2A GA for all cancer types in both cohorts except for HNSCC in the DFCI cohort (Figure 1, Supplementary Table S1, S2, S3).
Figure 1. Landscape of CDKN2A alterations in the DFCI and MSKCC cancer cohorts.
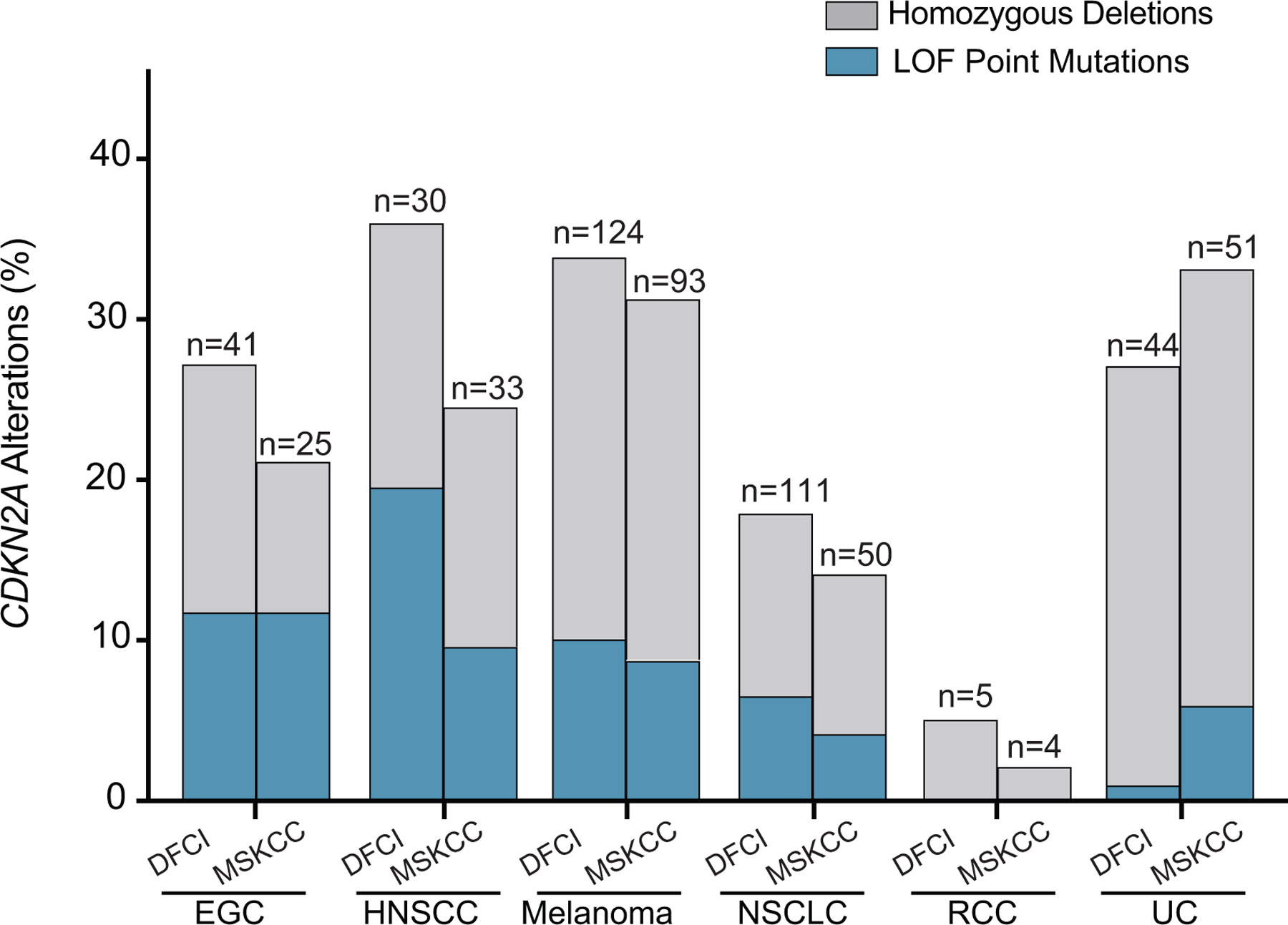
Frequency of homozygous deletions and loss-of-function CDKN2A GAs across all six cancer types in the DFCI and MSKCC cohorts, grouped by cancer type. EGC: esophagogastric cancer, NSCLC: non-small cell lung cancer; HNSCC: head and neck cancer; UC: urothelial carcinoma.
Clinical cohorts and patient characteristics
DFCI cohort patients were excluded if they had any of: heterozygous loss of CDKN2A, received ICIs with concurrent chemo- or other therapy, received ICIs in the adjuvant setting, or deteriorated within a week of ICI initiation (Materials and Methods, Supplementary Figure S1). 789 patients met the eligibility criteria and constituted the DFCI clinical cohort (Supplementary Figure S1; Supplementary Tables S5, S6, and S7). For the MSKCC cohort, these exclusion criteria could not be applied because: 1) heterozygous loss data was not available; and 2) clinical data was limited. Thus, the entire cohort of 1,250 MSKCC patients was used for comparative analyses (Supplementary Tables S1 and S2).
Median follow-up after initiation of ICI therapy was 26.1 months (95% CI: 22.3–31.1, table 1) and 18 months (95% CI: 17.0–19.0) for the DFCI and MSKCC cohorts, respectively (Table 1, Supplementary Table S2 and S5). The most common tumors in the DFCI and MSKCC cohorts are shown in Supplementary Figure S2.
Table 1.
Baseline clinical characteristics of the overall population.
| DFCI | MSKCC | |||
|---|---|---|---|---|
| N (median) | % (range) | N (median) | % (range) | |
| Age at ICI start | 65 | 25–93 | NA | NA |
| Tumor Type | ||||
| EGC | 61 | 7.7 | 120 | 9.6 |
| HNSCC | 44 | 5.6 | 136 | 10.9 |
| Melanoma | 162 | 20.6 | 296 | 23.7 |
| NSCLC | 357 | 45.2 | 341 | 27.3 |
| RCC | 56 | 7.1 | 151 | 12.1 |
| UC | 109 | 13.8 | 206 | 16.5 |
| Site of specimen sequenced | ||||
| Primary | 394 | 50.0 | NA | NA |
| Metastatic | 395 | 50.0 | NA | NA |
| ICI type | ||||
| Single | 690 | 87.4 | 1032 | 82.6 |
| Combination | 99 | 12.5 | 218 | 17.4 |
| ICI class | ||||
| Anti-PD-1/PD-L1 | 674 | 85.4 | 963 | 77.0 |
| Anti-CTLA4 | 16 | 2.0 | 69 | 5.5 |
| Anti-PD-1/PD-L1 + anti-CTLA4 | 99 | 12.5 | 218 | 17.4 |
| Number of prior lines | ||||
| 0 | 364 | 46.1 | NA | NA |
| 1 | 305 | 38.7 | NA | NA |
| ≥2 | 120 | 15.2 | NA | NA |
Median age at first dose of ICI was 65 (range: 25–93) years for the DFCI cohort. 33% of patients from MSKCC were aged 61–70 years (N=412). Nearly all of the patients received anti–PD-1– or anti–PD-L1–based therapy (DFCI: N=773, 98%; MSKCC: N=1181, 95%, Table 1), most commonly as single agents (DFCI: N=690, 87.4%; MSKCC: N=963, 77%; Table 1; Supplementary Tables S2 and S5). Among 789 patients treated with ICI at DFCI, 71 (9%) had their tumor sequencing performed on biopsies obtained after initiation of ICI.
CDKN2A GAs are associated with worse clinical outcomes in patients with urothelial carcinoma (UC) and melanoma
On multivariable analysis of the DFCI cohort (see methods), UC patients with loss-of-function (LOF) CDKN2A GAs treated with ICI had significantly shorter overall survival (OS) compared to those without such alterations (median OS 8.8 months vs. 25.2 months, respectively; Hazard ratio (HR)=1.8; 95% CI=1.1–3.1, p=0.019, Figure 2A). Similarly, melanoma patients whose tumors had LOF CDKN2A GAs also had worse OS compared to those with wild-type (WT) CDKN2A (median OS 27.2 months vs. not reached, respectively; HR=1.8; 95% CI=1.1–3.1, p=0.026, Figure 2B). Similarly, in both the UC and melanoma cohorts, CDKN2A alterations were associated with shorter time to treatment failure (TTF): for UC, median TTF 4.2 months vs. 8.4 months, respectively, HR=2.0; 95% CI=1.3–3.1, p=0.002 (Figure 2C); for melanoma, median TTF 10.0 months vs. 20.1 months, respectively, HR=1.8; 95% CI=1.2–2.9, p=0.003 (Figure 2D). In contrast, there was no association between CDKN2A mutation status and overall survival or TTF for EGC, HNSCC, NSCLC, and RCC in the DFCI cohort (Supplementary Figures S3, S4A and B). However, all of these DFCI cohorts were much smaller than the DFCI UC and melanoma cohorts with the exception of NSCLC, and therefore had lower power of detection. We also examined the possibility that the type of CDKN2A alteration would have an effect on ICI response. Although this comparison was underpowered by the small number of cancers with one type of alteration vs. the other for several cancer types, every cancer type showed similar OS and TTF for patients with homozygous deletions in comparison to those with LOF point mutations (all p-values > 0.05).
Figure 2. Impact of CDKN2A GAs on overall survival (OS) and time to treatment failure (TTF) in ICI-treated patients with melanoma and urothelial carcinoma.
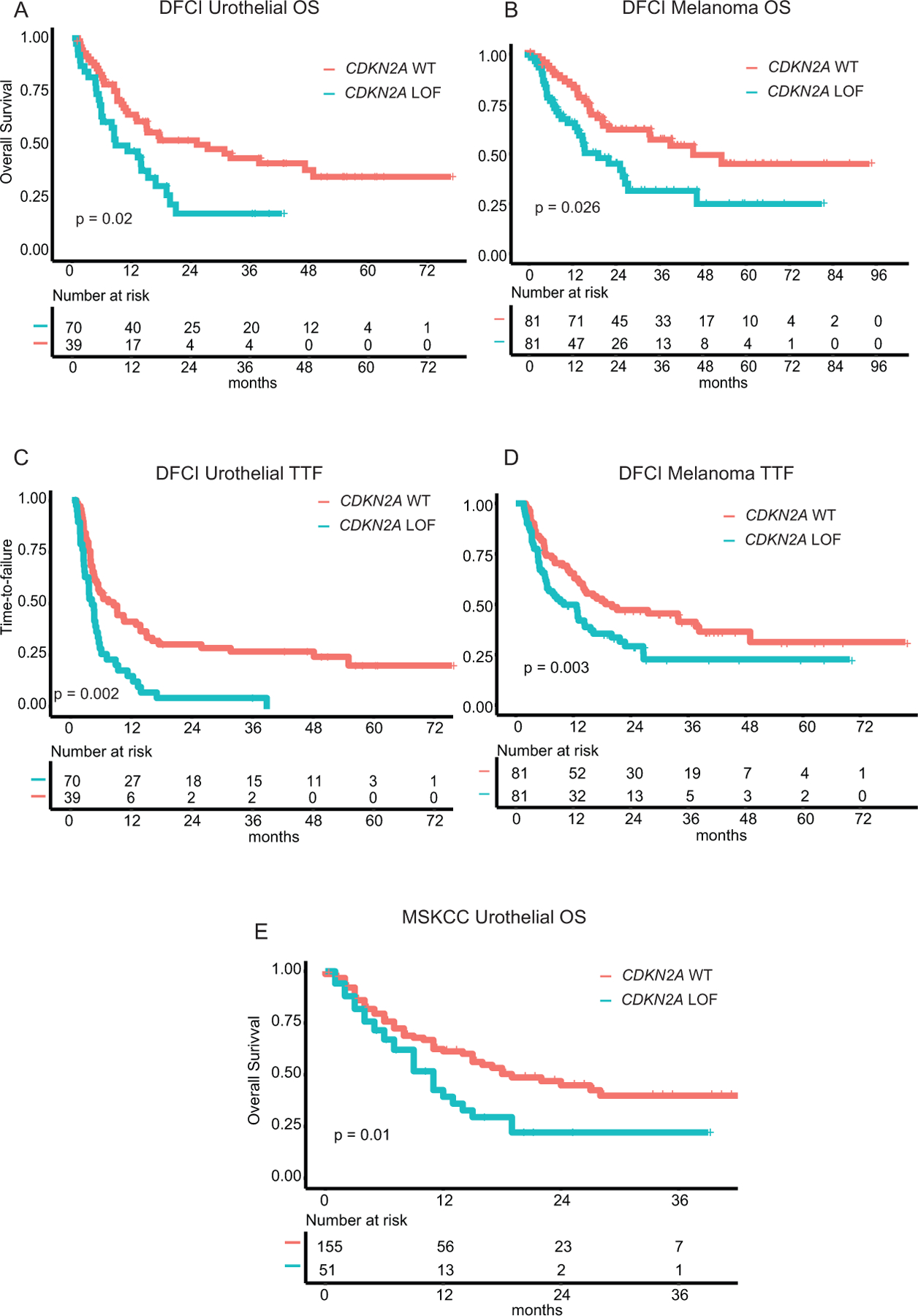
A. Overall survival in DFCI UC cohort. B. Time-to-failure in DFCI UC cohort. C. Overall survival in DFCI melanoma cohort. D. Time-to-failure in DFCI melanoma cohort. E. Overall survival in MSKCC UC cohort. All reported p-values are of the Wald x2 test from the Cox regression analysis, adjusted as detailed in the methods section. WT: wild type
In the MSKCC UC cohort (N=206), there also was a significantly shorter OS among tumors with LOF CDKN2A alterations vs. WT CDKN2A after adjusting for TMB and treatment type (see methods, median OS=11 months vs. OS=19 months, respectively, HR=1.7, 95% CI=1.1–2.6, p=0.017; Figure 2E). We also noted a significant association between LOF CDKN2A GAs and worse OS in both the MSKCC RCC and EGC cohorts (RCC: median OS of 13 months vs. 50 months, respectively, HR=5, 95% CI=1.8–14.1, p=0.002; EGC: median OS of 8 months vs. 17 months, respectively, HR=2.4, 95% CI=1.3–4.6, p=0.006, Figures S3C, S4A, E). We could not detect any significant association between LOF CDKN2A alterations and OS (after adjusting for covariates, see methods) among MSKCC patients with HNSCC, melanoma, or NSCLC (Supplementary Figures S4C and S5). Notably, for the MSKCC cohort, we were unable to adjust for other clinical variables such as lines of therapy prior to ICIs as this data was not available. Similarly, TTF and response data were not available in the MSKCC cohort.
In the UC and melanoma DFCI cohorts, tumors harboring CDKN2A alterations were less likely to respond compared to WT tumors, but the associations were only statistically significant for UC after adjusting for TMB, type of therapy, and prior lines of systemic treatment (see methods, UC: OR=4.0, 95% CI=1.2–18.5; p=0.04; melanoma: OR=2.0, 95% CI=0.9–4.1; p=0.06). CDKN2A alterations were not associated with objective response rates (ORR) in any of the other 5 cancer types (Supplementary Figure S4D).
In the DFCI UC cohort, PD-L1 IHC was performed for 22/109 (20%) patients treated with ICI. Using a cut-off of ≥1% for positivity, 10 were PD-L1 positive and 12 were negative. No association between tumor cell PD-L1 status and OS, TTF or ORR to ICI therapy was seen in this very small subset. Moreover, prior reports have shown that PD-L1 expression on tumor cells was not reported to be associated with response to ICI in UC(33).
We were also interested in examining the potential association between heterozygous loss of CDKN2A and ICI response in the DFCI UC cohort. Twenty-six UC patients with heterozygous loss of CDKN2A who were treated with ICI were compared to the original CDKN2A-altered and WT groups. Overall survival was slightly longer for patients with heterozygous loss of CDKN2A in comparison to CDKN2A WT patients, but neither comparison was statistically significant (adjusted p-values = 0.16 and 0.82 for OS and TTF respectively, Supplementary Figure S6, Supplementary Table S8). Moreover, UC patients with the earlier definition of LOF alteration in CDKN2A had significantly worse OS and TTF compared to tumors with heterozygous deletions in CDKN2A (OS p-value = 0.0003; TTF p-value = 0.009).
CDKN2A alterations are predictive of response to ICI treatment and not prognostic
To assess whether the association between CDKN2A alterations and clinical outcomes in the UC cohort treated with ICIs was a general prognostic feature, we analyzed a DFCI cohort composed of 56 patients with UC treated with platinum-based therapy (38 CDKN2A WT and 18 CDKN2A altered, Supplementary Table S9). Although the number of subjects was relatively small, there was no association with OS in CDKN2A-altered versus WT tumors among UC patients treated with platinum-based therapy (CDKN2A-altered median OS=14.9 months; CDKN2A WT median OS=13.4 months; HR=0.78; 95% CI=0.66–2.5; p=0.5, Figure 3A). To examine this further, we analyzed the TCGA data for 131 patients with stage IV UC (87 CDKN2A WT and 44 CDKN2A LOF). In this patient population as well, there was no significant association between CDKN2A status and OS (Figure 3B, Supplementary Table S10) or progression-free survival (Figure 3C, Supplementary Table S11). Note that the TCGA tumors were collected over 5 years ago, prior to the use of ICI therapy for UC.
Figure 3. Survival outcomes of urothelial carcinoma patient according to CDKN2A GAs, not treated with ICI therapy.
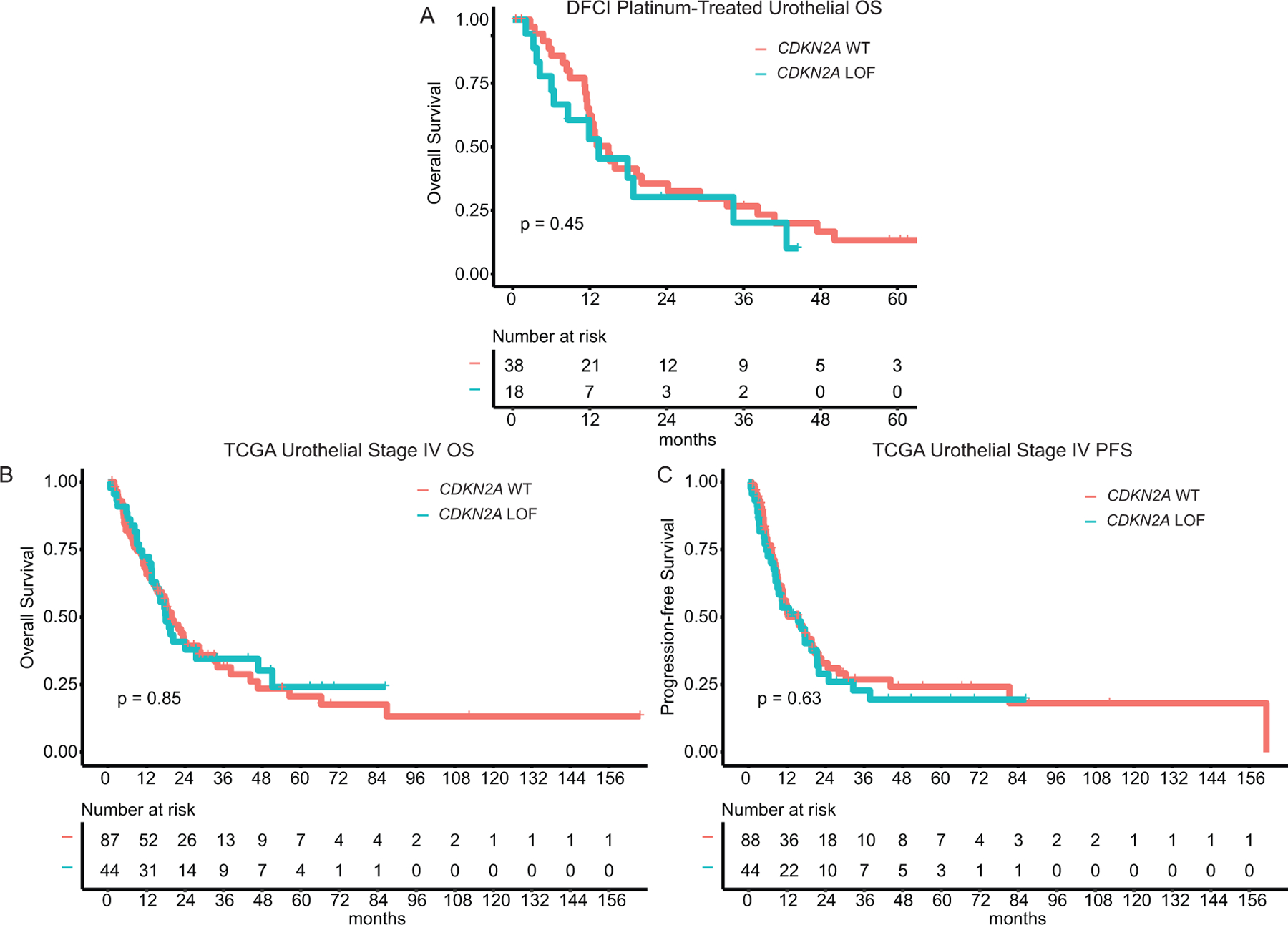
A. Overall survival of DFCI platinum-treated mUC cohort. B. Overall survival of TCGA mUC patients. C. Progression-free survival in TCGA mUC patients.
CDKN2A alterations and the tumor microenvironment in UC
We examined the possibility that CDKN2A GAs were associated with differences in the tumor microenvironment in UC. Bulk RNA-seq data was obtained for two independent UC cohorts (IMvigor210 and TCGA, see methods) at baseline and used for this purpose. First, gene set enrichment analysis (GSEA,http://software.broadinstitute.org/gsea/index.jsp) was performed, comparing the CDKN2A LOF UC subset (used as reference) with WT UC samples in the IMvigor210 cohort (CDKN2A altered n=43; CDKN2A WT n=193; Supplementary Table S12). Antigen processing, interferon-gamma and T-cell receptor signaling pathways, and other immune/inflammatory pathways were reduced in expression in CDKN2A altered tumors (FDR-corrected p<0.1 for T-cell receptor, antigen processing, and interferon gamma response, Figure 4A, Supplementary Figure S7, Supplementary Table S13). Similar analysis performed using the TCGA BLCA RNA-Seq data (CDKN2A altered n=142; CDKN2A WT n=264), was not statistically significant (Figure 4A).
Figure 4.
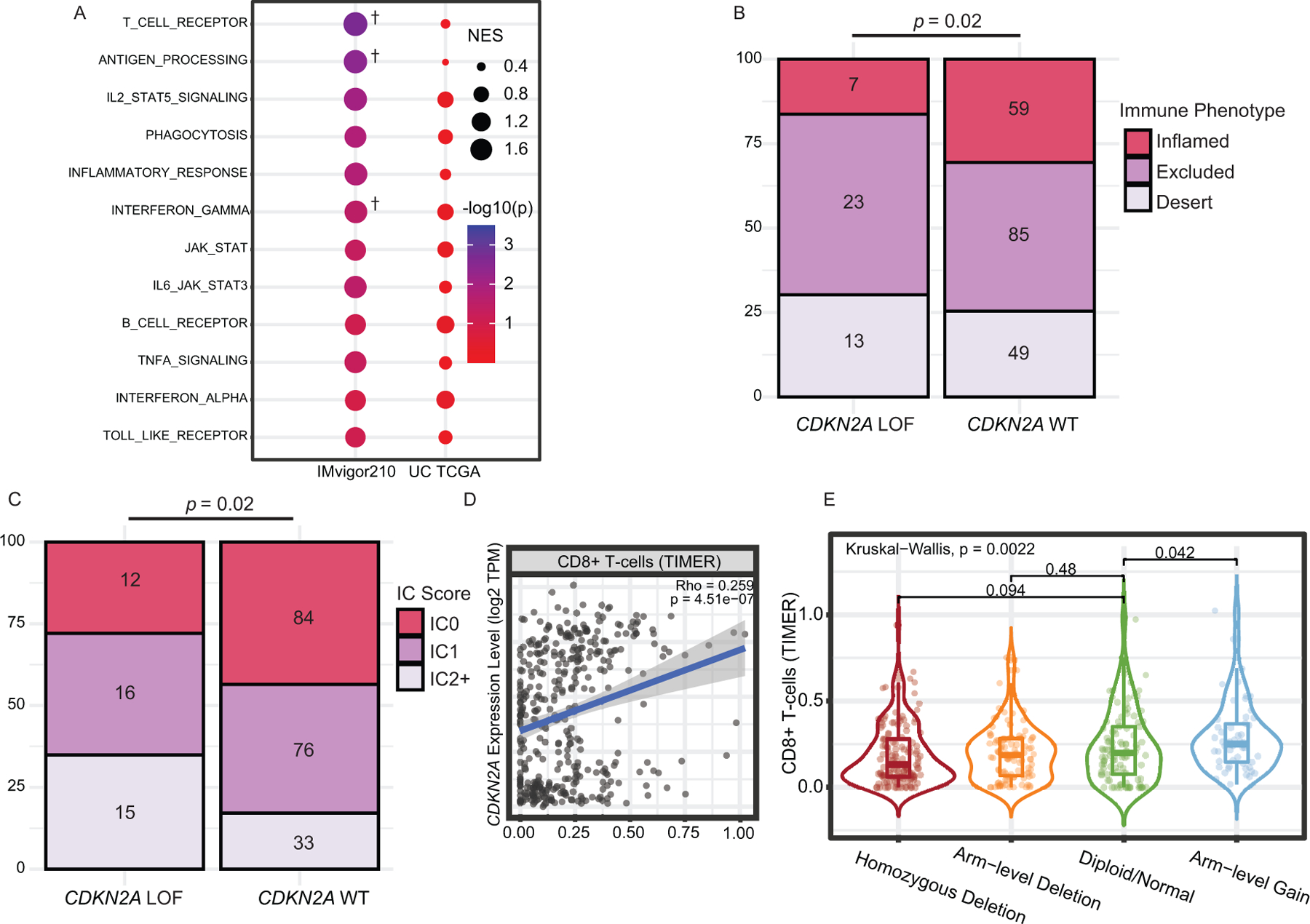
CDKN2A genomic alterations and the Tumor Immune Microenvironment in urothelial carcinoma (UC). A. Gene Set Enrichment Analysis of CDKN2A WT vs. LOF in the IMvigor210 and TCGA UC cohorts. Gene set pathways enriched in the WT samples are sorted by their nominal p-values in the IMvigor210 cohort. CDKN2A LOF alterations were used as reference. †: FDR-corrected p-value < 0.1. B. Spearman correlation between CDKN2A expression in TCGA UC samples and degree of CD8+ T-cell infiltration, computed using TIMER. C. Degree of CD8+ T-cell infiltration as a function of the number of CDKN2A gene copies in TCGA data. D. Association between CDKN2A genomic alteration status and immune phenotype in the IMvigor210 cohort. E. Immune-cell (IC) PD-L1 Expression in CDKN2A WT versus altered UC in the IMvigor210 cohort. Specimens were scored as IHC IC0, IC1, or IC2+, if <1%, ≥1% but <5%, ≥5% of IC were PD-L1 positive, respectively.
Next, we used a 5-gene expression signature described by Oh et. al (ABCB1, APBA2, GPR18, PEG10 and SLAMF7) to define CD4+ cytotoxic T-cells, a subset of T-cells thought to be highly active in the immune response to infiltrated bladder tumors(57). Out of 57 inflamed bladder samples for which signature scores were computed and available in the IMvigor210 cohort, CDKN2A WT samples were found to have a higher composite signature score than CDKN2A GA samples (Wilcoxon Rank-Sum Test p-value = 0.06, Supplementary Figure S8, Supplementary Table S12).
Finally, we checked tumor-infiltrating immune cell (IC) status and PD-L1 IHC on both IC and tumor cell (TC) expression in the IMvigor210 cohort. Out of 236 analyzed tumors, 66 had an inflammatory immune phenotype. IC1 (1–5%) and IC2+ (≥5% cut-off) PD-L1 expression was observed in 92 of 236 (39%) and 96 of 236 (40.7%) patients, respectively. Patients with CDKN2A alterations were less likely to have an inflammatory immune phenotype and less likely to express PD-L1 on ICs compared to the CDKN2A WT group (p=0.02 for both analyses, Figures 4B and 4C, Supplementary Table S12). However, we found no association between CDKN2A alteration status and PD-L1 expression on TCs (not shown).
Since IHC data was not available on TCGA UC samples, we used Tumor Immune Estimation Resource (TIMER) immune deconvolution to check for associations between CDKN2A and immune infiltration. We showed that CDKN2A expression in UC samples correlated significantly with CD8+ T-cell infiltration (Spearman’s correlation coefficient Rho= 0.259, p-value = 4E-7; Figure 4D). A higher copy number of CDKN2A was also associated with higher CD8+ infiltration (Kruskal-Wallis p-value = 0.0022; Figure 4E).
Discussion
In this work, we evaluated the effect of LOF GAs in CDKN2A on clinical outcomes of ICI treatment across different cancer types in two independent cohorts from DFCI and MSKCC. CDKN2A GAs were consistently associated with poorer outcomes in the DFCI and MSKCC UC cohorts treated with ICI. This is in accordance with prior reports suggesting that loss of CDKN2B, a neighboring gene located 25 kb centromeric to CDKN2A, was associated with unfavorable prognosis(41). Notably, CDKN2A GAs were not associated with poor clinical outcomes in TCGA patients with metastatic UC (mUC), or in a cisplatin-treated mUC cohort from DFCI. This supports the idea that CDKN2A status is a predictive rather than a prognostic biomarker in this patient population.
Poor outcomes to ICI in RCC patients with deletion of 9p21.3, a chromosomal region that encompasses the CDKN2A gene, have recently been described(40). In contrast, no prior report has assessed the impact of CDKN2A alterations on clinical outcomes in patients with EGC treated with ICIs. In our dataset, CDKN2A alterations were associated with worse OS in both the MSKCC RCC and EGC cohorts but not the corresponding DFCI cohorts (Supplementary Figures S3G–H and 5E). For EGC, the discrepancy may be due to the breakdown of samples by site along the upper gastrointestinal tract between the MSKCC and DFCI cohorts. A very small number of RCC patients had CDKN2A alterations (inactivating mutations or homozygous copy number loss) in both DFCI (4/56) and MSKCC (4/151) cohorts which limits the power to detect any association. In contrast, the analysis by Braun et al.(40) included patients with heterozygous copy-number losses in 9p21.3 where the association with worse clinical outcomes was only found in infiltrated tumors. For EGC, further study is required to explain the discordant results.
Prior clinical reports have led to conflicting results regarding the association of genomic aberrations in CDKN2A with clinical outcomes in ICI-treated patients with melanoma(34–36,43). In our study, findings in melanoma patients were also inconsistent in these two cohorts. DFCI melanoma patients harboring CDKN2A LOF GAs responded poorly to ICIs when compared to patients with no CDKN2A GAs. In the MSKCC cohort however, there was no appreciable difference between the two groups. One possible explanation is that the MSKCC cohort is more heterogeneous, a mix of different disease stages at the time of treatment, including patients being treated with adjuvant ICI therapy. This could clearly confound this analysis.
In mUC, high expression of PD-L1 on tumor-infiltrating immune cells is associated with enhanced ICI efficacy(33,58). In the IMvigor210 cohort, we were able to show that patients harboring CDKN2A GAs were more likely to have lower PD-L1 expression on tumor-infiltrating immune cells and a less inflammatory immune microenvironment. This is in contrast with RCC, where tumors with 9p21.3 loss were found to have a stronger immune infiltration(40). CDKN2A-altered UCs also showed depletion of the T-cell receptor, antigen processing, and interferon gamma pathways, replicating the findings seen in melanoma(42) although we were unable to confirm these findings in the TCGA BLCA cohort.
The association between CDKN2A GAs and response to ICI treatment of UC raises the question of potential mechanisms of this effect. CDK4, a critical cell cycle kinase, is normally restrained by p16INK4a (the protein product of CDKN2A). In the absence of p16INK4a, active CDK4-cyclin D1 phosphorylates SPOP, stabilizing it and protecting it from degradation by the anaphase-promoting complex activator FZR1. Phospho-SPOP is an adaptor protein that enhances the activity of Cullin 3-based ubiquitin ligases to enhance PD-L1 ubiquitination and ultimately degradation. Hence, it has been reported that CDKN2A activity leads to reduced PD-L1 surface expression on cancer cells. However, we observed no correlation between CDKN2A loss and PD-L1 expression on tumor cells by IHC analysis in the IMvigor210 cohort. This suggests that this mechanism is not operative in UC, and further study is required to elucidate how CDKN2A loss reduces ICI treatment effect independent of PD-L1 expression in UC. We note that the majority of CDKN2A GAs are homozygous gene deletions in UC. In the TCGA BLCA cohort, 43% of samples with homozygous deletion of CDKN2A have co-deletion of part or all of a nearby interferon gene cluster on 9p21.3. We hypothesize that this co-deletion event may contribute in part to the poor ICI response in UC with CDKN2A GAs. Unfortunately, neither the Oncopanel nor the MSK-Impact analyses include analysis of this interferon gene cluster, so a direct association with response cannot be examined.
One limitation to our study is that it is a retrospective analysis of clinical outcomes in select patient cohorts treated at two tertiary care centers. However, we included several variables that are thought to influence response to ICI, such as the number of prior treatment regimens, single versus combination therapy and TMB, and all of our analyses were multivariate, taking into account these other factors. Second, targeted panel sequencing (OncoPanel and MSK-IMPACT) was used to identify mutations and copy-number events in tumor samples. While IMPACT detects germline variants by sequencing peripheral blood or normal adjacent tissue, OncoPanel does not; this makes it possible that some of the GAs captured in the DFCI cohort are germline rather than somatic. However, all variants that were observed at a frequency >0.1% in the Genome Aggregation database (gnomAD) were excluded, and germline mutations in CDKN2A are very rare. We also excluded samples with only single-copy losses, missense and synonymous mutations as their effect on protein function is variable. Also, promoter hypermethylation, a mechanism of CDKN2A expression silencing, was not assessed in either cohort. Hence, a subset of our “wild-type” patient cohorts could, in fact, have aberrant p16 expression due to promoter methylation. Finally, the modest level of data granularity (genomic and clinical) available to us for the MSKCC dataset prevented exclusion of tumors harboring heterozygous deletions in CDKN2A, and adjustment for important clinical variables that may have impacted the results such as systemic lines of therapy prior to ICI. These issues may explain in part the differences in results between the DFCI cohort and the MSK cohort.
In conclusion, CDKN2A GAs were found to be associated with reduced benefit from ICI therapy in UC, but not consistently in any of the five other cancer types. Significant heterogeneity of response association was seen in these two cohorts, which is likely partially due to the different criteria applied to identify CDKN2A GAs, as well as differences in the size and composition of the patient populations at the two sites.
Supplementary Material
Translational relevance:
CDKN2A is one of the most commonly altered genes in human cancer. Prior work investigated the impact of CDKN2A alterations on the tumor immune microenvironment and on the response to immune checkpoint inhibition, but conflicting results were noted. Herein, we tested the effect of CDKN2A genomic alterations in two large cohorts of patients treated with ICIs. We assessed the association between loss-of-function variants in CDKN2A and outcomes of cancer patients treated with ICIs across six cancer types. Our work elucidates genomic alterations in CDKN2A as predictive of clinical outcomes in patients with urothelial carcinoma. In addition, immune-inflammatory pathways were significantly reduced in expression in CDKN2A altered tumors. This cancer-specific finding necessitates further investigation in both the clinical and lab-based settings to better understand the basis of the genomic-immune interaction in urothelial carcinoma and other cancer types.
Acknowledgments:
D.A.B. acknowledges support by the DF/HCC Kidney Cancer SPORE Career Enhancement Program (P50CA101942–15), DOD CDMRP (KC170216, KC190130), and the DOD Academy of Kidney Cancer Investigators (KC190128).
S.A.S. acknowledges support by the NCI (R50 RCA211482A).
A.G. acknowledges support by the NCI (R01 CA227237, R01 CA244569) and the Louis B. Mayer Foundation.
Footnotes
Conflict of interest statement:
David A. Braun reports nonfinancial support from Bristol-Myers Squibb, honoraria from LM Education/Exchange Services, and personal fees from Octane Global, Defined Health, Dedham Group, Adept Field Solutions, Slingshot Insights, Blueprint Partnerships, Charles River Associates, Trinity Group, and Insight Strategy, outside of the submitted work.
Sachet A. Shukla previously advised and has received consulting fees from Neon Therapeutics. S.A.S. reports nonfinancial support from Bristol-Myers Squibb, and equity in Agenus Inc., Agios Pharmaceuticals, Breakbio Corp., and Lumos Pharma, outside the submitted work.
Marios Giannakis receives research funding from Bristol-Myers Squibb, Merck and Servier.
- Research (Institutional and personal): AstraZeneca, Alexion, Bayer, Bristol Myers-Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Ipsen, Tracon, Genentech, Roche, Roche Products Limited, F. Hoffmann-La Roche, GlaxoSmithKline, Lilly, Merck, Novartis, Peloton, Pfizer, Prometheus Labs, Corvus, Calithera, Analysis Group, Sanofi/Aventis, Takeda.
- Honoraria: AstraZeneca, Alexion, Sanofi/Aventis, Bayer, Bristol Myers-Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Genentech, Roche, Roche Products Limited, F. Hoffmann-La Roche, GlaxoSmithKline, Merck, Novartis, Peloton, Pfizer, EMD Serono, Prometheus Labs, Corvus, Ipsen, Up-to-Date, NCCN, Analysis Group, NCCN, Michael J. Hennessy (MJH) Associates, Inc (Healthcare Communications Company with several brands such as OnClive, PeerView and PER), Research to Practice, L-path, Kidney Cancer Journal, Clinical Care Options, Platform Q, Navinata Healthcare, Harborside Press, American Society of Medical Oncology, NEJM, Lancet Oncology, Heron Therapeutics, Lilly
- Consulting or Advisory Role: AstraZeneca, Alexion, Sanofi/Aventis, Bayer, Bristol Myers-Squibb/ER Squibb and sons LLC, Cerulean, Eisai, Foundation Medicine Inc., Exelixis, Genentech, Heron Therapeutics, Lilly, Roche, GlaxoSmithKline, Merck, Novartis, Peloton, Pfizer, EMD Serono, Prometheus Labs, Corvus, Ipsen, Up-to-Date, NCCN, Analysis Group, Pionyr, Tempest.
- No speaker’s bureau
- Stock ownership: Pionyr, Tempest.
- No leadership or employment in for-profit companies. Other present or past leadership roles: Director of GU Oncology Division at Dana-Farber and past President of medical Staff at Dana-Farber), member of NCCN Kidney panel and the GU Steering Committee, past chairman of the Kidney Cancer Association Medical and Scientific Steering Committee)
- Patents, royalties or other intellectual properties:
- International Patent Application No. PCT/US2018/12209, entitled “PBRM1 Biomarkers Predictive of Anti-Immune Checkpoint Response,” filed January 3, 2018, claiming priority to U.S. Provisional Patent Application No. 62/445,094, filed January 11, 2017
- International Patent Application No. PCT/US2018/058430, entitled “Biomarkers of Clinical Response and Benefit to Immune Checkpoint Inhibitor Therapy,” filed October 31, 2018, claiming priority to U.S. Provisional Patent Application No. 62/581,175, filed November 3, 2017
- Travel, accommodations, expenses, in relation to consulting, advisory roles, or honoraria
- Medical writing and editorial assistance support may have been funded by Communications companies funded by pharmaceutical companies (ClinicalThinking, Envision Pharma Group, Fishawack Group of Companies, Health Interactions, Parexel, Oxford PharmaGenesis, and others).
- The institution (Dana-Farber Cancer Institute) may have received additional independent funding of drug companies or/and royalties potentially involved in research around the subject matter.
- CV provided upon request for scope of clinical practice and research
- Mentored several non-US citizens on research projects with potential funding (in part) from non-US sources/Foreign Components
- Asmar Wood S.A.L. is a private company based in Beirut, Lebanon that provided a total of $100,000 in salary support to Dr. Sarah Abou Alaiwi from 7/1/2018 to 7/1/2020 during her post-doctoral research fellowship at DFCI.
David Kwiatkowski receives research support from Genentech and Revolution Medicines; and is a consultant to Novartis, Genentech, and AADi.
All other authors declare no potential conflicts of interest.
Code Availability:
Algorithms used for data analysis are all publicly available from the indicated references in the Methods section. Any other queries about the custom code used in this study should be directed to the corresponding authors of this study.
Data availability:
All relevant correlative data are available from the authors and/or are included with the manuscript. Clinical and genomic data for the DFCI and MSKCC cohorts are available in Supplementary Tables S1–S13. While targeted sequencing raw data were not deposited in a public repository since full sequencing data are not consented to be shared, the data that support the findings of this study are available from the corresponding author upon request.
All clinical and correlative data from the TCGA BLCA and IMvigor 210 clinical trial are publicly available. All intermediate data from the RNA-seq analyses of the IMvigor 210 and TCGA cohorts are publicly available. Any other queries about the data used in this study should be directed to the corresponding authors of this study.
References
- 1.Larkin J, Hodi FS, Wolchok JD. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 2015;373(13):1270–1 doi 10.1056/NEJMc1509660. [DOI] [PubMed] [Google Scholar]
- 2.Hellmann MD, Paz-Ares L, Bernabe Caro R, Zurawski B, Kim SW, Carcereny Costa E, et al. Nivolumab plus Ipilimumab in Advanced Non-Small-Cell Lung Cancer. N Engl J Med 2019;381(21):2020–31 doi 10.1056/NEJMoa1910231. [DOI] [PubMed] [Google Scholar]
- 3.Motzer RJ, Tannir NM, McDermott DF, Aren Frontera O, Melichar B, Choueiri TK, et al. Nivolumab plus Ipilimumab versus Sunitinib in Advanced Renal-Cell Carcinoma. N Engl J Med 2018;378(14):1277–90 doi 10.1056/NEJMoa1712126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reck M, Rodriguez-Abreu D, Robinson AG, Hui R, Csoszi T, Fulop A, et al. Pembrolizumab versus Chemotherapy for PD-L1-Positive Non-Small-Cell Lung Cancer. N Engl J Med 2016;375(19):1823–33 doi 10.1056/NEJMoa1606774. [DOI] [PubMed] [Google Scholar]
- 5.Motzer RJ, Escudier B, McDermott DF, George S, Hammers HJ, Srinivas S, et al. Nivolumab versus Everolimus in Advanced Renal-Cell Carcinoma. N Engl J Med 2015;373(19):1803–13 doi 10.1056/NEJMoa1510665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Havel JJ, Chowell D, Chan TA. The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat Rev Cancer 2019;19(3):133–50 doi 10.1038/s41568-019-0116-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti–PD-1 antibody in cancer. New England Journal of Medicine 2012;366(26):2443–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shukla SA, Bachireddy P, Schilling B, Galonska C, Zhan Q, Bango C, et al. Cancer-germline antigen expression discriminates clinical outcome to CTLA-4 blockade. Cell 2018;173(3):624–33. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. New England Journal of Medicine 2014;371(23):2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ott P, Bang Y, Razak A, Bennouna J, Soria J, Rugo H, et al. 84PDRelationship of PD-L1 and a T-cell inflamed gene expression profile (GEP) to clinical response in a multicohort trial of solid tumors (KEYNOTE [KN] 028). Annals of Oncology 2017;28(suppl_5). [Google Scholar]
- 11.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 2015;350(6257):207–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, et al. Tumor mutational burden and efficacy of nivolumab monotherapy and in combination with ipilimumab in small-cell lung cancer. Cancer cell 2018;33(5):853–61. e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Samstein RM, Lee CH, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet 2019;51(2):202–6 doi 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Legrand FA, Gandara DR, Mariathasan S, Powles T, He X, Zhang W, et al. Association of high tissue TMB and atezolizumab efficacy across multiple tumor types. American Society of Clinical Oncology; 2018.
- 15.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell 2017;171(4):934–49. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, et al. PD-1 blockade in tumors with mismatch-repair deficiency. New England Journal of Medicine 2015;372(26):2509–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science 2017;355(6322). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li H, Bullock K, Gurjao C, Braun D, Shukla SA, Bosse D, et al. Metabolomic adaptations and correlates of survival to immune checkpoint blockade. Nat Commun 2019;10(1):4346 doi 10.1038/s41467-019-12361-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti-PD-1 immunotherapy in melanoma patients. Science 2018;359(6371):97–103 doi 10.1126/science.aan4236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pomerantz J, Schreiber-Agus N, Liegeois NJ, Silverman A, Alland L, Chin L, et al. The Ink4a tumor suppressor gene product, p19Arf, interacts with MDM2 and neutralizes MDM2’s inhibition of p53. Cell 1998;92(6):713–23 doi 10.1016/s0092-8674(00)81400-2. [DOI] [PubMed] [Google Scholar]
- 22.Zhao R, Choi BY, Lee MH, Bode AM, Dong Z. Implications of Genetic and Epigenetic Alterations of CDKN2A (p16(INK4a)) in Cancer. EBioMedicine 2016;8:30–9 doi 10.1016/j.ebiom.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018;173(2):371–85 e18 doi 10.1016/j.cell.2018.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Network CGAR. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489(7417):519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Network CGAR. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014;511(7511):543–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2017;171(3):540–56 e25 doi 10.1016/j.cell.2017.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Network CGA. Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517(7536):576–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Network CGAR. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513(7517):202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Network CGAR. Integrated genomic characterization of oesophageal carcinoma. Nature 2017;541(7636):169–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Raphael BJ, Hruban RH, Aguirre AJ, Moffitt RA, Yeh JJ, Stewart C, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer cell 2017;32(2):185–203. e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, et al. The somatic genomic landscape of glioblastoma. Cell 2013;155(2):462–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tam KW, Zhang W, Soh J, Stastny V, Chen M, Sun H, et al. CDKN2A/p16 inactivation mechanisms and their relationship to smoke exposure and molecular features in non-small-cell lung cancer. J Thorac Oncol 2013;8(11):1378–88 doi 10.1097/JTO.0b013e3182a46c0c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8 doi 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.DeLeon TT, Almquist DR, Kipp BR, Langlais BT, Mangold A, Winters JL, et al. Assessment of clinical outcomes with immune checkpoint inhibitor therapy in melanoma patients with CDKN2A and TP53 pathogenic mutations. PLoS One 2020;15(3):e0230306 doi 10.1371/journal.pone.0230306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brenner E, Schorg BF, Ahmetlic F, Wieder T, Hilke FJ, Simon N, et al. Cancer immune control needs senescence induction by interferon-dependent cell cycle regulator pathways in tumours. Nat Commun 2020;11(1):1335 doi 10.1038/s41467-020-14987-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hilke FJ, Sinnberg T, Gschwind A, Niessner H, Demidov G, Amaral T, et al. Distinct Mutation Patterns Reveal Melanoma Subtypes and Influence Immunotherapy Response in Advanced Melanoma Patients. Cancers (Basel) 2020;12(9) doi 10.3390/cancers12092359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamberti G, Spurr LF, Li Y, Ricciuti B, Recondo G, Umeton R, et al. Clinicopathological and genomic correlates of programmed cell death ligand 1 (PD-L1) expression in nonsquamous non-small-cell lung cancer. Ann Oncol 2020. doi 10.1016/j.annonc.2020.02.017. [DOI] [PubMed] [Google Scholar]
- 38.Balli D, Rech AJ, Stanger BZ, Vonderheide RH. Immune Cytolytic Activity Stratifies Molecular Subsets of Human Pancreatic Cancer. Clin Cancer Res 2017;23(12):3129–38 doi 10.1158/1078-0432.CCR-16-2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang ZL, Wang Z, Li GZ, Wang QW, Bao ZS, Zhang CB, et al. Immune Cytolytic Activity Is Associated With Genetic and Clinical Properties of Glioma. Front Immunol 2019;10:1756 doi 10.3389/fimmu.2019.01756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Braun DA, Hou Y, Bakouny Z, Ficial M, Sant’ Angelo M, Forman J, et al. Interplay of somatic alterations and immune infiltration modulates response to PD-1 blockade in advanced clear cell renal cell carcinoma. Nat Med 2020;26(6):909–18 doi 10.1038/s41591-020-0839-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nassar AH, Mouw KW, Jegede O, Shinagare AB, Kim J, Liu CJ, et al. A model combining clinical and genomic factors to predict response to PD-1/PD-L1 blockade in advanced urothelial carcinoma. Br J Cancer 2020;122(4):555–63 doi 10.1038/s41416-019-0686-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yu J, Yan J, Guo Q, Chi Z, Tang B, Zheng B, et al. Genetic Aberrations in the CDK4 Pathway Are Associated with Innate Resistance to PD-1 Blockade in Chinese Patients with Non-Cutaneous Melanoma. Clin Cancer Res 2019;25(21):6511–23 doi 10.1158/1078-0432.CCR-19-0475. [DOI] [PubMed] [Google Scholar]
- 43.Horn S, Leonardelli S, Sucker A, Schadendorf D, Griewank KG, Paschen A. Tumor CDKN2A-Associated JAK2 Loss and Susceptibility to Immunotherapy Resistance. J Natl Cancer Inst 2018;110(6):677–81 doi 10.1093/jnci/djx271. [DOI] [PubMed] [Google Scholar]
- 44.Morrison C, Pabla S, Conroy JM, Nesline MK, Glenn ST, Dressman D, et al. Predicting response to checkpoint inhibitors in melanoma beyond PD-L1 and mutational burden. J Immunother Cancer 2018;6(1):32 doi 10.1186/s40425-018-0344-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Garcia EP, Minkovsky A, Jia Y, Ducar MD, Shivdasani P, Gong X, et al. Validation of OncoPanel: A Targeted Next-Generation Sequencing Assay for the Detection of Somatic Variants in Cancer. Arch Pathol Lab Med 2017;141(6):751–8 doi 10.5858/arpa.2016-0527-OA. [DOI] [PubMed] [Google Scholar]
- 46.Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, et al. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight 2016;1(19):e87062 doi 10.1172/jci.insight.87062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat Med 2017;23(6):703–13 doi 10.1038/nm.4333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abou Alaiwi S, Nassar AH, Xie W, Bakouny Z, Berchuck JE, Braun DA, et al. Mammalian SWI/SNF Complex Genomic Alterations and Immune Checkpoint Blockade in Solid Tumors. Cancer Immunol Res 2020;8(8):1075–84 doi 10.1158/2326-6066.CIR-19-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Fu W, O’Connor TD, Jun G, Kang HM, Abecasis G, Leal SM, et al. Analysis of 6,515 exomes reveals the recent origin of most human protein-coding variants. Nature 2013;493(7431):216–20 doi 10.1038/nature11690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alfoldi J, Wang Q, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature 2020;581(7809):434–43 doi 10.1038/s41586-020-2308-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lai Z, Markovets A, Ahdesmaki M, Chapman B, Hofmann O, McEwen R, et al. VarDict: a novel and versatile variant caller for next-generation sequencing in cancer research. Nucleic Acids Res 2016;44(11):e108 doi 10.1093/nar/gkw227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res 2010;20(9):1297–303 doi 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rausch T, Zichner T, Schlattl A, Stutz AM, Benes V, Korbel JO. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 2012;28(18):i333–i9 doi 10.1093/bioinformatics/bts378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pugh TJ, Amr SS, Bowser MJ, Gowrisankar S, Hynes E, Mahanta LM, et al. VisCap: inference and visualization of germ-line copy-number variants from targeted clinical sequencing data. Genet Med 2016;18(7):712–9 doi 10.1038/gim.2015.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Robertson AG, Kim J, Al-Ahmadie H, Bellmunt J, Guo G, Cherniack AD, et al. Comprehensive Molecular Characterization of Muscle-Invasive Bladder Cancer. Cell 2018;174(4):1033 doi 10.1016/j.cell.2018.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li T, Fan J, Wang B, Traugh N, Chen Q, Liu JS, et al. TIMER: a web server for comprehensive analysis of tumor-infiltrating immune cells. Cancer research 2017;77(21):e108–e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Oh DY, Kwek SS, Raju SS, Li T, McCarthy E, Chow E, et al. Intratumoral CD4(+) T Cells Mediate Anti-tumor Cytotoxicity in Human Bladder Cancer. Cell 2020;181(7):1612–25 e13 doi 10.1016/j.cell.2020.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bellmunt J, Mullane SA, Werner L, Fay AP, Callea M, Leow JJ, et al. Association of PD-L1 expression on tumor-infiltrating mononuclear cells and overall survival in patients with urothelial carcinoma. Ann Oncol 2015;26(4):812–7 doi 10.1093/annonc/mdv009. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All relevant correlative data are available from the authors and/or are included with the manuscript. Clinical and genomic data for the DFCI and MSKCC cohorts are available in Supplementary Tables S1–S13. While targeted sequencing raw data were not deposited in a public repository since full sequencing data are not consented to be shared, the data that support the findings of this study are available from the corresponding author upon request.
All clinical and correlative data from the TCGA BLCA and IMvigor 210 clinical trial are publicly available. All intermediate data from the RNA-seq analyses of the IMvigor 210 and TCGA cohorts are publicly available. Any other queries about the data used in this study should be directed to the corresponding authors of this study.