

Abstract
Objective:
Defects in the human solute carrier family 26 member 4 (SLC26A4) gene are reported to be one of the causes of congenital hypothyroidism (CH). We aimed to identify SLC26A4 mutations in Chinese patients with CH and analyze the function of the mutations.
Methods:
Patients with primary CH were screened for 21 CH candidate genes mutations by targeted next-generation sequencing. All the exons and exon-intron boundaries of SLC26A4 were identified and analyzed. The function of six missense mutation in SLC26A4 were further investigated in vitro.
Results:
Among 273 patients with CH, seven distinct SLC26A4 heterozygous mutations (p.S49R, p.I363L, p.R409H, p.T485M, p.D661E, p.H723R, c.919-2A>G) were identified in 10 patients (3.66%, 10/273). In vitro experiments showed that mutation p.I363L, p.R409H, p.H723R affect the membrane location and ion transport of SLC26A4, while p.S49R did not. Mutation p.T485M and p.D661E only affected ion transport, but had no effect on the membrane location.
Conclusion:
The prevalence of SLC26A4 mutations was 3.66% in Chinese patients with CH. Five mutations (p.I363L, p.R409H, p.T485M, p.D661E and p.H723R) impaired the membrane location or ion transport function of SLC26A4, suggesting important roles for Ile363, Arg409, Thr485, Asp661, and His723 residues in SLC26A4 function. As all variants identified were heterozygous, the pathogenesis of these patients cannot be explained, and the pathogenesis of these patients needs further study.
Keywords: Congenital hypothyroidism, next-generation sequencing, SLC26A4, cell location, ion transport
What is already known on this topic?
Defects in the human SLC26A4 gene are reported to be one of the causes of congenital hypothyroidism (CH). SLC26A4 encodes the apical iodide transporter protein, pendrin.
What this study adds?
We identified seven distinct variants of SLC26A4, including one novel mutation, in a cohort of Chinese patients with CH. Functional studies showed that five out of six missense mutations had different effects on gene function, including ion transport and/or membrane location of the SLC26A4 protein, Pendrin. These results provide an important basis for future mechanism research.
Introduction
Congenital hypothyroidism (CH) is a common neonatal endocrine disorder. Unless treated in the first few months of life, severe CH can lead to growth retardation and permanent intellectual disability (1). The incidence of CH was reported to be about 1:4000 in 1970, and the incidence of the disease has increased to 1:2000 in the past decades (2). About 85% of cases of CH are caused by abnormal thyroid development (thyroid dysgenesis), but genetic associations with thyroid dysgenesis have only been identified in 2-5% of cases. These pathogenic genes leading to dysgenesis include thyroid stimulating hormone (TSH) receptor (TSHR), PAX8, GLIS3, NKX2.1 and FOXE1 (3). The remaining 15% of cases of CH are due to defects of thyroid hormone biosynthesis (dyshormonogenesis). Dyshormonogenesis is often caused by mutations in genes that are involved in the pathway of thyroid hormone synthesis, such as thyroperoxidase (TPO), dual oxidase 2 (DUOX2), sodium-iodide symporter (NIS; SLC5A5) and the apical iodide transporter, pendrin (PDS; SLC26A4) (4).
SLC26A4 encodes pendrin, a multi transmembrane (TM) protein composed of 780 amino acids, consisting of 12-14 TM segments and a segment of intracellular STAS (Sulfate Transporter and Anti-Sigma factor antagonist) functional domain (5,6,7,8,9). Pendrin is an anion exchanger that is highly expressed in thyroid, inner ear and kidneys. In thyroid, pendrin is expressed at the apical membrane of thyroid follicular cells. It acts as a chloride-iodide exchanger, transporting iodide from the cell to the follicular lumen, where thyroid hormone is synthesized (10). Although previous studies have shown that SLC26A4 biallelic mutation may result in CH (11), biallelic mutation of SLC26A4 have not been found in Chinese patients with CH. In order to evaluate the role of SLC26A4 in the pathogenesis of the Chinese CH patients, our study identified SLC26A4 mutations in a cohort of Chinese patients with CH and analyzed the function of any identified mutations in vitro.
Methods
Clinical Subjects
We enrolled Chinese CH patients through newborn screening. Newborn screening was done with filter-paper blood spots between 3 and 5 days after birth. Blood samples were collected from the heel and TSH level was measured by time-resolved fluorescence assay (PerkinElmer, USA). Subjects with increased TSH (TSH ≥10 mU/L) levels observed during neonatal screening were recalled for further evaluation. The levels of TSH, total triiodothyronine (T3), total thyroxine (T4), free T3, and free T4 (fT4) in serum were determined by performing an immuno-chemiluminometric assay (UniCel DxI 800, Beckman, USA). The details of the diagnostic criteria to establish permanent CH in patients were from our previous study (12); briefly these included i) elevated TSH levels, ii) T4 or fT4 levels less than the reference range, and iii) restoration of normal thyroid parameters after receiving replacement therapy with L-thyroxine, but, after stopping treatment, a rise in TSH and a drop in fT4 were observed again. In addition, some patients were recruited from outpatient clinics who were on L-thyroxine replacement therapy. Although these patients lack initial diagnostic data, they have a definite history of CH. A written consent was obtained from the parents of the CH patients, and the study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated to Shanghai JiaoTong University School of Medicine (decision no: 2016-76-T33, date: 2016-08-03). Informed consent was obtained from all patients or their legal guardians, and all unaffected family members who participated in the study.
Next-generation Sequencing
Genomic DNA was extracted from the peripheral blood using the Quick Gene DNA Whole Blood Kit L (Kurabo, Japan) according to the manufacturer’s protocol (13). Twenty-one previously reported possible causative genes for CH, including TPO (GenBank reference sequence: NM_000547), SLC5A5 (NM_000453), thyroglobulin (TG) (NM_003235), TSHR (NM_000369), DUOX2 (NM_014080), DUOXA2 (NM_207581), SLC26A4 (NM_000441), FOXE1 (NM_004473), PAX8 (NM_013952), NKX2-1 (NM_001079668), NKX2-5 (NM_004387), IYD (NM_001164694), DIO1 (NM_000792), DIO2 (NM_000793), THRA (NM_001190918), THRB (NM_00125263), DUOX1 (NM_017434), DUOXA1 (NM_001276268), GNAS (NM_016592), SLC16A2 (NM_006517) and HHEX (NM_002729) were analyzed in this study (16). All the exons and exon-intron boundaries of these genes were amplified by performing multiplex polymerase chain reaction (PCR) using a 48×48 Access Array™ microfluidic platform (Fluidigm, USA) according to the manufacturer’s protocol. The primers were designed using iPLEX Assay Design software (Sequenom, USA). The HiSeq 3000 platform (Illumina, San Diego, CA, USA) was used to perform deep sequencing of these amplicon libraries. The target sequences were amplified and deep sequenced in duplicate for each sample to avoid base pair (bp) variants caused by multiplex PCR.
Calling of SLC26A4 Variants from Next-generation Sequencing Data and Verification Using Sanger Sequencing
Raw sequence data was analyzed in fastq format and the quality scores were obtained, as previously described (14,15). Credible variants were selected according to the following criteria: (i) the quality scores of variants with ≥30 bps; (ii) mapping the quality scores of variants with ≥50 bps; (iii) sequencing to estimate the depth of variants with ≥20 bps; (iv) variant allele frequency ≥30%; (v) variants with read depth ≥5; and (vi) the presence of mutation on both the DNA strands (16). Variants with frequencies >1% in the dbSNP 135 and ESP6500 v2 databases were filtered out and the focus of the study was on the functional (protein altering) variants after removal of intergenic and 3’/5’ UTR variants, nonsplice related intronic variants, synonymous variants identified in duplicate samples. Then the remaining variants were selected for validation by Sanger sequencing.
Construction of Plasmid
Human wild-type (WT) cDNA of SLC26A4 was cloned into p-enhanced green fluorescent protein (EGFP)-N2 plasmid (TransGen Biotech, China). Identified missense mutations were introduced into the SLC26A4-pEGFP-N2 WT plasmid by Fast Mutagenesis System kit (TransGen Biotech, China) according to the manufacturer’s protocol. Meanwhile, human NIS cDNA was cloned into a eukaryotic expression vector pcDNA3.1. All the plasmid constructs were validated by Sanger sequencing.
Cell Culture and Transfections
293T cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM)/high-glucose medium (Gibco, USA) supplemented with 10% fetal bovine serum (Sigma Aldrich, USA) at 37 oC in a humidified atmosphere containing 5% CO2. Transfections were performed on cells by Lipofectamine™ 2000 Transfection Reagent (Invitrogen™, USA) following the manufacturer’s instructions. Cells were plated in 20 mm glass bottom cell culture dish (NEST), transfected with 1 µg plasmid DNA to detected the cell localization of the WT or mutants of SLC26A4 plasmids. Iodide efflux assays were performed on 293T cells, cultured in 12 well plates, co-transfected with 0.5 µg pcDNA3.1-NIS and 0.5 µg WT or mutant SLC26A4-pEGFP-N2 plasmids.
The Assays for the Cell Localization of WT or Mutants of SLC26A4
Forty-eight hours after transfecting with WT or mutants of SLC26A4-pEGFP-N2 plasmids, 293T cells were washed twice in PBS (1X). Then cells were fixed in 4% paraformaldehyde for 30 minutes. After washing with PBS (1X), cells were stained with the membrane probe DiI (Beyotime, Haimen, China) at 37 oC for 5-10 min, then nucleii were stained with DAPI (Beyotime Biotech, Haimen, China) at room temperature for 5 minutes. Confocal imaging for cells was carried out on Nikon A1 confocal microscope using the 40x objective (Nikon A1 Microsystems, Japan).
Iodide Efflux Assay for WT or Mutants of SLC26A4 in 293T Cells
The iodide efflux assay was performed as described previously (17). In brief, forty-eight hours after co-transfecting with 0.5 µg pcDNA3.1-NIS and 0.5 µg WT or mutant SLC26A4-pEGFP-N2 plasmids, 293T cells were washed once in serum-free DMEM medium and incubated for 1 hour in 1 mL serum-free medium containing 131I at 5 KBq/mL as the only source of iodide. The cells were then washed briefly in HBSS buffer and then incubated with 1 mL HBSS for 5 minutes after which HBSS was removed. The cells were solubilized by the addition of 1 mL 1 N NaOH and the radioactivity measured using a γ counter (GC1200, Anhui, China). All experiments were carried out three times on triplicate cultures. Statistical significance of the iodide efflux assay results was determined by use of t-test.
Statistical Analysis
All data are expressed as mean±standard deviation. Data analysis is mainly processed by Excel, elisacalc and Statistical Package for the Social Sciences 19.0 statistical software. P<0.05 is considered to be statistically significant. The images in this paper are mainly processed and produced by Photoshop software, image J and Graphpad prism 6. The gene sequence retrieval website National Center for Biotechnology Information used in this paper: http://www.ncbi.nlm.nih.gov/; University of California, Santa Cruz: http://genome.ucsc.edu/
Results
Clinical Characteristics of Patients with CH
The cohort of CH patients enrolled consisted of 273 patients, including 141 (51.6%) females. The median value of serum TSH and serum fT4 level were 54.075 uIU/mL and 0.718 ng/dL, respectively. All of them had normal hearing. Whether these children had enlarged vestibular aqueduct (EVA) is unknown because the examination was unnecessary and the patient’s family refused.
Screening the Missense Mutations of SLC26A4 in the Chinese Patients with CH
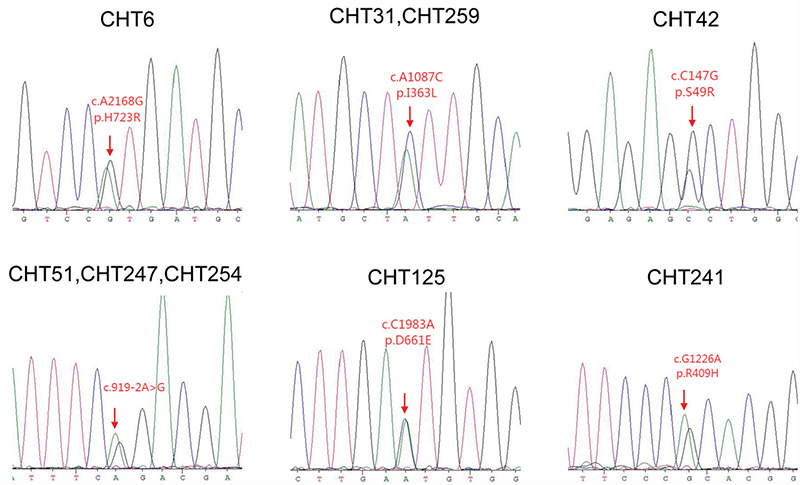
All the exons and exon-intron boundaries were amplified by performing multiplex PCR using customized primers designed to generate 200-250bp amplicons. After the quality control assessment, the average coverage of SLC26A4 with sequencing depth ≥20x was 89.04%. Seven heterozygous mutations in SLC26A4 were identified in 10 patients, including one novel mutation (p.I363L). Interestingly, 8 of these 10 patients also carried mutations in other candidate gene for CH (Table 1). All mutation sites were verified by Sanger sequencing (Figure 1), with the exception of one patient. In patient 190 it was not possible to obtain a Sanger sequencing result because of DNA sample damage and patient refusal to provide a repeat sample. The frequency of SLC26A4 mutation in Chinese patients diagnosed with CH was 3.66% (10/273). Among the seven mutations, p.S49R was located in the N-terminal intracellular region, p.D661E and p.H723R were located in the STAS domain of the C-terminal intracellular region which plays a key role in the membrane location of SLC26A4. The remaining mutation sites were scattered in the 12 TM regions of SLC26A4 (Figure 2).
Table 1. The clinical data and genetic characteristics of the 10 congenital hypothyroidism patients with mutation of SLC26A4.
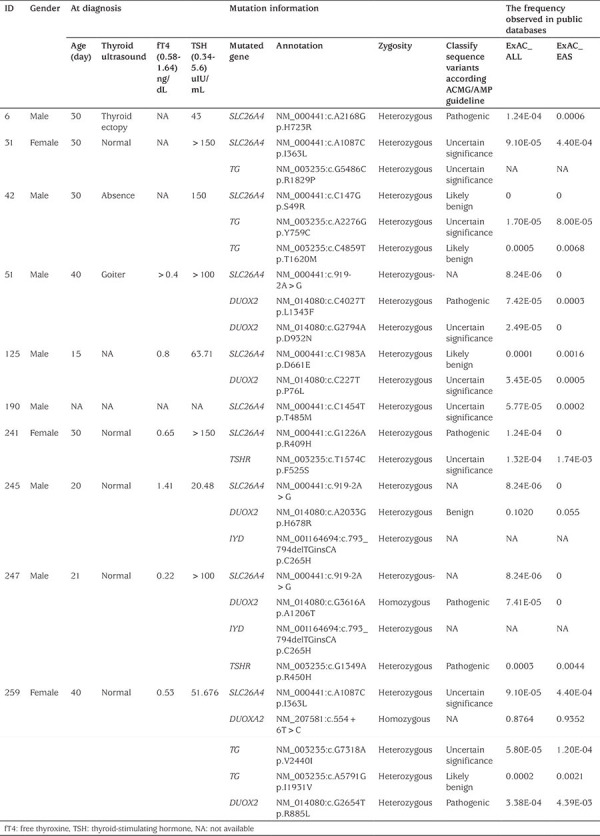
Figure 1.
Sanger sequencing of SLC26A4 mutation
Figure 2.
The mutations, identified from our patients with congenital hypothyroidism, located in the protein domain of SLC26A4. The mutation p.S49R is located in the N-terminal intracellular region, p.D661E and p.H723R are located in the STAS domain of the C-terminal intracellular region of the SLC26A4. The remainer 3 mutations were scattered in 12 transmembrane domains of the SLC26A4
Cellular Localization of SLC26A4 Mutants
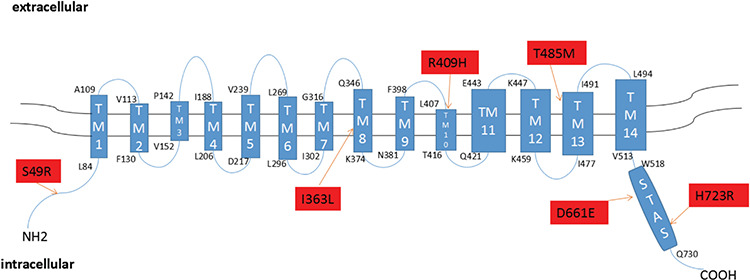
SLC26A4 has been shown, by immunohistochemical analysis, to be located at the apical membrane of thyroid follicular cells (18). To assess the effect of mutations on membrane location of SLC26A4, we expressed WT and mutants of the SLC26A4-pEGFP-N2 plasmid in 293T cells and observed location using a confocal fluorescence microscope. Although these cells lack the polarization of thyroid follicular cells, WT SLC26A4 was clearly present at the cell membrane and significant co-localization with marker of cell membrane. Mutant p.S49R showed a cell membrane protein distribution similar to that of the WT SLC26A4. Mutants p.R409H and p.H723R did not express on the cell membrane obviously. Novel mutant p.I363L showed partly express in the cytoplasm, but was mostly expressed at the cell membrane. The remaining mutants, p.T485M, p.D661E, and p.S49R had no effect on cell membrane localization (Figure 3).
Figure 3.
Cellular localization of the six mutants of SLC26A4 in 293T cells detected by confocal microscopy. Mutant p.S49R, p.T485M, p.D661E of SLC26A4 show strong membrane fluorescence in 293T cells, which is similar to the WT of SLC26A4 expressed in the 293T cells, and there was no obvious cytoplasmic retention. Mutants p.I363L, p.R409H, and p.H723R reduce the localization on the cell membrane, and p.R409H, p.H723R show obvious cytoplasmic retention, p.I363L is less. All mutant plasmids were homozygous. Scale: 10 μm
WT: wild-type
Assessment of Iodide Transport in the WT and Identified SLC26A4 Mutants
It has been suggested that SLC26A4 mediates iodide efflux at the apical membrane of thyroid follicular cell (10). In order to assess the effect of mutations on the ability of iodide transport, we co-expressed NIS with WT or mutant SLC26A4-pEGFP-N2 plasmids in 293T cells to provide a cell model by which cells could uptake iodide from culture medium. The 293T cells transfected with NIS only showed an accumulation of radioiodide (131I) in the cells. In contrast, only a small amount of 131I was retained in the 293T cells after co-transfection with NIS and WT SLC26A4 plasmids. However, compared to the 293T cells co-transfected with NIS and WT SLC26A4 plasmids, with one exception (p.S49R), cells co-transfected with NIS and the identified SLC26A4 mutants resulted in significantly decreased iodine efflux, indicating that these mutants lead to a decrease in cellular ability of iodide transport in 293T cells (Figure 4).
Figure 4.
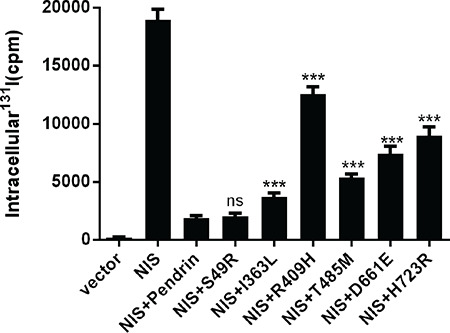
The effect of the mutations in SLC26A4 on iodine transport capacity in 293T cells. Intracellular iodide accumulation in 293T cells after co-transfection with sodium-iodide symporter (NIS) and wild-type (WT) or the mutant SLC26A4-pEGFP-N2 plasmids were detected by γ-counter. Intracellular iodide accumulation in 293T cells after co-transfection with NIS and mutants palsmids from the p.I363L, p.R409H, p.T485M, p.D661E and p.H723R variants were significantly increased, compared to those co-transfected with NIS and WT (pendrin) SLC26A4-pEGFP-N2 plasmid, indicating that these mutants reduced iodide efflux mediated by SLC26A4. All mutant plasmids were homozygous. The data are shown as mean±standard error for three independent experiments. Statistical analysis used Welch’s t-test
ns: No statistical difference, ***p<0.001
Discussion
CH is a relatively common endocrine disease with a prevalence ranging from 1:2000 to 1:4000 in newborns (19). Most cases are dysgenetic although a substantial proportion are due to dyshormonogenesis. There is a close association between genetic abnormalities and dyshormonogenesis but the search for genetic mechanisms in dysgenesis has identified <5% to have a genetic pathogensis. Therefore, it is important to expand the spectrum of the pathogenic genes in patients with CH, given the relatively common occurrence. In this study, mutations in SLC26A4 were investigated in Chinese patients with CH and seven different heterozygous variants in 10 individuals (10/273, 3.66%) were found. The prevalence of SLC26A4 mutations in our study was similar to a previous study that screened SLC26A4 mutation in CH patients from Guangxi Zhuang Autonomous Region, China (20). Fu et al (20) reported that all the mutations detected were heterozygous mutations, and thus cannot be assumed to be pathogenetic. These findings suggested that SLC26A4 might be an uncommon pathogenic gene for CH in the Chinese population.
SLC26A4 is a member of the SLC26 anion transporter family that encodes the pendrin protein which was originally predicted to contain 12 TM domains (21). This has since been shown to be incorrect as 14 TM domains were subsequently confirmed by Gorbunov et al (22). These regions contain many anion-binding sites or substrate-binding sites, including TM1, TM3, and TM10, which would affect the function of this protein (5). The protein also has a STAS domain in the cytosol, which is critical for membrane targeting of many SLC26 anion transporters, and STAS domain mutations are associated with at least three human recessive diseases (22). In our study, a total of seven mutation sites in SLC26A4 were identified, including a novel variant which, to the best of our knowledge, has not previously been reported.
The variant S49R, located in the N-terminal intracellular segment, was shown by functional experiments to have no effect on membrane localization or ion transport. It may be that the site is not an ion binding site, so the mutation has little effect on gene function. The novel variant, I363L was located in TM8, which is an anion-binding domain (5). Our study confirmed that mutation I363L affects the membrane localization of SLC26A4 slightly and reduced its ability to transport iodine ions by about 53%. We speculate that because TM8 is an ion binding region, mutations in this region may affect the overall ion binding ability, thus affecting gene function. R409H, located in TM10, significantly reduced the membrane localization and iodine transport capacity by 83.7%, a result which is consistent with previous reports. Related studies have shown that this site is an anion-binding site, the mutation directly affect the anion-binding site would have a great impact on gene function. His723 is a conserved site that is located in the STAS domain. The mutation H723R would disrupt the π-cation interaction and polar contact between Tyr530 and His723, again affecting protein function (5). D661E and T485M are located in the STAS domain and TM13, respectively. The two mutation sites have no effect on the localization of the pendrin protein to the cell membrane, but have a significant impact on ion transport, reducing it by about 74% and 67% respectively. There are mutants that have been characterized as having an effect at the cell surface but with reduced function, such as G209V, F335L, M775T. So, the mechanism of the effect of D661E and T485M on the protein may be similar to these loci, which needs further study (5,17,23).
SLC26A4 is expressed in the inner ear and thyroid (17). In the inner ear, SLC26A4 functions as a Cl-/HCO3- exchanger and regulates the balance of endolymphatic ions, thus affecting the function of the inner ear (24). Homozygous mutations in SLC26A4 lead to EVA, which is the most common inner ear malformation associated with sensorineural deafness in children (25). However, some patients with EVA carried a heterozygous mutation in SLC26A4, rather than a biallelic mutation, suggesting that there are other genetic factors involved in the occurrence of EVA. A study conducted by Yang et al (26) confirmed this hypothesis. They identified heterozygous mutations in SLC26A4 and KCNJ10 from one patients with EVA. In our study, through follow-up, we found that these 10 patients with CH did not have deafness, but they refused to carry out imaging examination of the inner ear, and thus we were unable to determine whether there was EVA.
Pathogenic mutations in SLC26A4 are well recognized as being the pathogenic mutation in Pendred syndrome (PS), which is an autosomal recessive disorder characterized by sensorineural hearing loss, and goiter and some cases may be identified in the neonatal period with CH (27). However, the thyroid phenotype of PS patients is not clearly defined. In 2014, Ladsous et al (28) found that about 30% of PS patients will present with CH and 78.9% patients have goiter in PS patients with biallelic mutations of SLC26A4. The researchers speculated that these differences in PS thyroid phenotype in patients with biallelic mutations of SLC26A4 might be due to different iodine intakes, as most of the patients with PS presented with hypothyroidism in a moderately iodine deficient region in France, but PS patients from Japan and Korea, regions with high iodine intake, were euthyroid (28,29). Although the thyroid phenotype in human patients with PS seemed to be related to iodine intake, lower iodine intake did not lead to goiter and hypothyroidism in the SLC26A4 knockout mice (30), indicating that other genetic factors or environmental factors might be involved in the pathogentic mechanism resulting in goiter and hypothyroidism in humans with SLC26A4 mutation. Indeed, among the 10 patients with SLC26A4 heterozygous mutation, eight patients carried other mutations in genes associated with CH in our cohort (Table 1). Patient 42 and 259 carried compound heterozygous mutations in the TG gene, which is the key gene in the thyroid hormone synthesis and was the pathogenic gene in these two patients. Patient 51 and 247 carried biallelic mutation in DUOX2, which is also involved in thyroid hormone synthesis, but the association between DUOX2 variants and CH is less definite and thus the DUOX2 variants in our patients may be pathogenetic. Four of the remaining six patients carried at least one heterozygous mutation in another candidate gene for CH, while the remaining two patients were found to only have heterozygous mutation in SLC26A4 amongst the 21 CH-associated genes tested. Although we confirmed that the mutations in SLC26A4 in our patients with CH could decreased the ability of the iodide transport in vitro, none of the parents of these patients, some of whom were also carriers, did not have hypothyroidism, suggesting that these heterozygous mutations in SLC26A4 are probably not the pathogenic gene for our patients who had all been diagnosed with CH and that there are other genetic or environmental factors which might lead to CH. As EVA could be caused by the heterozygous mutation in SLC26A4 combining with the heterozygous mutation of KCNJ10, and a previous study has reported that heterozygous mutations in DUOXA2 and DUOX2 might lead to CH in a 4-year-old patient (31), it cannot be excluded that those patients with monoallelic mutation of SLC26A4 may combine with other, unidentified, gene variants to cause hypothyroidism.
Study Limitations
The sample size of this study was limited, and no individuals with a homozygous mutation in SLC26A4 were found. In addition, we could not further elucidate the pathogenesis of SLC26A4. Furthermore, the candidate gene panel used in this study did not include SLC26A7, which can also lead to hypothyroidism.
Conclusion
In this study, we identified seven distinct variants of SLC26A4 in 10 patients from a cohort of 273 Chinese patients with CH. Functional studies showed that five out of six missense mutations in SLC26A4, including one novel mutation, p.I363L, have variable effects on protein function. However, because these mutations were all heterozygous mutations, and 8 out of ten patients also carried variants in other CH candidate genes, the pathogenesis of CH in these patients cannot be explained by these SLC26A4 variants. The pathogenesis of CH in these patients needs further study.
Acknowledgments
We would like to thank all patients and their families for the participation in our study and for allowing us to publish their cases.
Footnotes
Ethics
Ethics Committee Approval: The study was approved by the Ethics Committee of Shanghai Ninth People’s Hospital affiliated to Shanghai JiaoTong University School of Medicine (decision no: 2016-76-T33, date: 2016-08-03).
Informed Consent: Informed consent was obtained from all patients or their legal guardians, and all unaffected family members who participated in the study.
Peer-review: Externally peer-reviewed.
Authorship Contributions
Surgical and Medical Practices: Cao-Xu Zhang, Feng Sun, Wen-Jiao Zhu, Rui-Jia Zhang, Ya Fang, Chen-Yan Yan, Concept: Shuang-Xia Zhao, Huai-Dong Song, Design: Chang-Run Zhang, Data Collection or Processing: Chang-Run Zhang, Qian-Yue Zhang, Ying-Xia Ying, Analysis or Interpretation: Chang-Run Zhang, Yuan-Ping Shi, Literature Search: Chang-Run Zhang, Writing: Chang-Run Zhang, Shuang-Xia Zhao.
Financial Disclosure: This work was supported by National Key R&D Program of China (2017YFC1001801) and the National Natural Science Foundation of China (81770786, 81661168016, 81870537).
References
- 1.Wassner AJ, Brown RS. Hypothyroidism in the newborn period. Curr Opin Endocrinol Diabetes Obes. 2013;20:449–454. doi: 10.1097/01.med.0000433063.78799.c2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Diaz A, Lipman Diaz EG. Hypothyroidism. Pediatr Rev. 2014;35:348–349. doi: 10.1542/pir.35-8-336. [DOI] [PubMed] [Google Scholar]
- 3.Wassner AJ. Congenital Hypothyroidism. Clin Perinatol. 2018;45:1–18. doi: 10.1016/j.clp.2017.10.004. [DOI] [PubMed] [Google Scholar]
- 4.Szinnai G. Clinical genetics of congenital hypothyroidism. Endocr Dev. 2014;26:60–78. doi: 10.1159/000363156. [DOI] [PubMed] [Google Scholar]
- 5.Rapp C, Bai X, Reithmeier RAF. Molecular analysis of human solute carrier SLC26 anion transporter disease-causing mutations using 3-dimensional homology modeling. Biochim Biophys Acta Biomembr. 2017;1859:2420–2434. doi: 10.1016/j.bbamem.2017.09.016. [DOI] [PubMed] [Google Scholar]
- 6.Sharma AK, Rigby AC, Alper SL. STAS domain structure and function. Cell Physiol Biochem. 2011;28:407–422. doi: 10.1159/000335104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dossena S, Rodighiero S, Vezzoli V, Nofziger C, Salvioni E, Boccazzi M, Grabmayer E, Bottà G, Meyer G, Fugazzola L, Beck-Peccoz P, Paulmichl M. Functional characterization of wild-type and mutated pendrin (SLC26A4), the anion transporter involved in Pendred syndrome. J Mol Endocrinol. 2009;43:93–103. doi: 10.1677/JME-08-0175. [DOI] [PubMed] [Google Scholar]
- 8.Gillam MP, Sidhaye AR, Lee EJ, Rutishauser J, Stephan CW, Kopp P. Functional characterization of pendrin in a polarized cell system. Evidence for pendrin-mediated apical iodide efflux. J Biol Chem. 2004;279:13004–13010. doi: 10.1074/jbc.M313648200. [DOI] [PubMed] [Google Scholar]
- 9.Royaux IE, Suzuki K, Mori A, Katoh R, Everett LA, Kohn LD, Green ED. Pendrin, the protein encoded by the Pendred syndrome gene (PDS), is an apical porter of iodide in the thyroid and is regulated by thyroglobulin in FRTL-5 cells. Endocrinology. 2000;141:839–845. doi: 10.1210/endo.141.2.7303. [DOI] [PubMed] [Google Scholar]
- 10.Dossena S, Nofziger C, Tamma G, Bernardinelli E, Vanoni S, Nowak C, Grabmayer E, Kössler S, Stephan S, Patsch W, Paulmichl M. Molecular and functional characterization of human pendrin and its allelic variants. Cell Physiol Biochem. 2011;28:451–466. doi: 10.1159/000335107. [DOI] [PubMed] [Google Scholar]
- 11.Kühnen P, Turan S, Fröhler S, Güran T, Abali S, Biebermann H, Bereket A, Grüters A, Chen W, Krude H. Identification of PENDRIN (SLC26A4) mutations in patients with congenital hypothyroidism and “apparent” thyroid dysgenesis. J Clin Endocrinol Metab. 2014;99:169–176. doi: 10.1210/jc.2013-2619. [DOI] [PubMed] [Google Scholar]
- 12.Fang Y, Sun F, Zhang RJ, Zhang CR, Yan CY, Zhou Z, Zhang QY, Li L, Ying YX, Zhao SX, Liang J, Song HD. Mutation screening of the TSHR gene in 220 Chinese patients with congenital hypothyroidism. Clin Chim Acta. 2019;497:147–152. doi: 10.1016/j.cca.2019.07.031. [DOI] [PubMed] [Google Scholar]
- 13.Zhan M, Chen G, Pan CM, Gu ZH, Zhao SX, Liu W, Wang HN, Ye XP, Xie HJ, Yu SS, Liang J, Gao GQ, Yuan GY, Zhang XM, Zuo CL, Su B, Huang W, Ning G, Chen SJ, Chen JL, Song HD; China Consortium for Genetics of Autoimmune Thyroid Disease. Genome-wide association study identifies a novel susceptibility gene for serum TSH levels in Chinese populations. Hum Mol Genet. 2014;23:5505–5517. doi: 10.1093/hmg/ddu250. [DOI] [PubMed] [Google Scholar]
- 14.Zhang LL, Kan M, Zhang MM, Yu SS, Xie HJ, Gu ZH, Wang HN, Zhao SX, Zhou GB, Song HD, Zheng CX. Multiregion sequencing reveals the intratumor heterogeneity of driver mutations in TP53-driven non-small cell lung cancer. Int J Cancer. 2017;140:103–108. doi: 10.1002/ijc.30437. [DOI] [PubMed] [Google Scholar]
- 15.Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y, Shi JY, Zhu YM, Tang L, Zhang XW, Liang WX, Mi JQ, Song HD, Li KQ, Chen Z, Chen SJ. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nature genetics. 2011;43:309–315. doi: 10.1038/ng.788. [DOI] [PubMed] [Google Scholar]
- 16.Sun F, Zhang JX, Yang CY, Gao GQ, Zhu WB, Han B, Zhang LL, Wan YY, Ye XP, Ma YR, Zhang MM, Yang L, Zhang QY, Liu W, Guo CC, Chen G, Zhao SX, Song KY, Song HD. The genetic characteristics of congenital hypothyroidism in China by comprehensive screening of 21 candidate genes. Eur J Endocrinol. 2018;178:623–633. doi: 10.1530/EJE-17-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor JP, Metcalfe RA, Watson PF, Weetman AP, Trembath RC. Mutations of the PDS gene, encoding pendrin, are associated with protein mislocalization and loss of iodide efflux: implications for thyroid dysfunction in Pendred syndrome. J Clin Endocrinol Metab. 2002;87:1778–1784. doi: 10.1210/jcem.87.4.8435. [DOI] [PubMed] [Google Scholar]
- 18.Bidart JM, Mian C, Lazar V, Russo D, Filetti S, Caillou B, Schlumberger M. Expression of pendrin and the Pendred syndrome (PDS) gene in human thyroid tissues. J Clin Endocrinol Metab. 2000;85:2028–2033. doi: 10.1210/jcem.85.5.6519. [DOI] [PubMed] [Google Scholar]
- 19.Chiesa A, Prieto L, Mendez V, Papendieck P, Calcagno Mde L, Gruñeiro- Papendieck L. Prevalence and etiology of congenital hypothyroidism detected through an argentine neonatal screening program (1997-2010) Horm Res Paediatr. 2013;80:185–192. doi: 10.1159/000354409. [DOI] [PubMed] [Google Scholar]
- 20.Fu C, Zheng H, Zhang S, Chen Y, Su J, Wang J, Xie B, Hu X, Fan X, Luo J, Li C, Chen R, Shen Y, Chen S. Mutation screening of the SLC26A4 gene in a cohort of 192 Chinese patients with congenital hypothyroidism. Arch Endocrinol Metab. 2016;60:323–327. doi: 10.1590/2359-3997000000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cordat E, Reithmeier RA. Structure, function, and trafficking of SLC4 and SLC26 anion transporters. Curr Top Membr. 2014;73:1–67. doi: 10.1016/B978-0-12-800223-0.00001-3. [DOI] [PubMed] [Google Scholar]
- 22.Gorbunov D, Sturlese M, Nies F, Kluge M, Bellanda M, Battistutta R, Oliver D. Molecular architecture and the structural basis for anion interaction in prestin and SLC26 transporters. Nature Commun. 2014;5:3622. doi: 10.1038/ncomms4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi BY, Stewart AK, Madeo AC, Pryor SP, Lenhard S, Kittles R, Eisenman D, Kim HJ, Niparko J, Thomsen J, Arnos KS, Nance WE, King KA, Zalewski CK, Brewer CC, Shawker T, Reynolds JC, Butman JA, Karniski LP, Alper SL, Griffith AJ. Hypo-functional SLC26A4 variants associated with nonsyndromic hearing loss and enlargement of the vestibular aqueduct: genotype-phenotype correlation or coincidental polymorphisms? Hum Mutat. 2009;30:599–608. doi: 10.1002/humu.20884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen Z, Zhu H, Li Z, Zhang S, Zhang A, Zhang T, Fu X, Sun D, Zhang J, Gao J. A knock-in mouse model of Pendred syndrome with Slc26a4 L236P mutation. Biochem Biophys Res Commun. 2019;515:359–365. doi: 10.1016/j.bbrc.2019.05.157. [DOI] [PubMed] [Google Scholar]
- 25.Usami S, Abe S, Weston MD, Shinkawa H, Van Camp G, Kimberling WJ. Non-syndromic hearing loss associated with enlarged vestibular aqueduct is caused by PDS mutations. Hum Genet. 1999;104:188–192. doi: 10.1007/s004390050933. [DOI] [PubMed] [Google Scholar]
- 26.Yang T, Gurrola JG, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wémeau JL, Kopp P. Pendred syndrome. Best Pract Res Clin Endocrinol Metab. 2017;31:213–224. doi: 10.1016/j.beem.2017.04.011. [DOI] [PubMed] [Google Scholar]
- 28.Ladsous M, Vlaeminck-Guillem V, Dumur V, Vincent C, Dubrulle F, Dhaenens CM, Wémeau JL. Analysis of the thyroid phenotype in 42 patients with Pendred syndrome and nonsyndromic enlargement of the vestibular aqueduct. Thyroid. 2014;24:639–648. doi: 10.1089/thy.2013.0164. [DOI] [PubMed] [Google Scholar]
- 29.Tsukamoto K, Suzuki H, Harada D, Namba A, Abe S, Usami S. Distribution and frequencies of PDS (SLC26A4) mutations in Pendred syndrome and nonsyndromic hearing loss associated with enlarged vestibular aqueduct: a unique spectrum of mutations in Japanese. Eur J Hum Genet. 2003;11:916–922. doi: 10.1038/sj.ejhg.5201073. [DOI] [PubMed] [Google Scholar]
- 30.Calebiro D, Porazzi P, Bonomi M, Lisi S, Grindati A, De Nittis D, Fugazzola L, Marinò M, Bottà G, Persani L. Absence of primary hypothyroidism and goiter in Slc26a4 (-/-) mice fed on a low iodine diet. J Endocrinol Invest. 2011;34:593–598. doi: 10.3275/7262. [DOI] [PubMed] [Google Scholar]
- 31.Zheng X, Ma SG, Qiu YL, Guo ML, Shao XJ. A Novel c.554+5C>T Mutation in the DUOXA2 Gene Combined with p.R885Q Mutation in the DUOX2 Gene Causing Congenital Hypothyroidism. J Clin Res Pediatr Endocrinol. 2016;8:224–227. doi: 10.4274/jcrpe.2380. [DOI] [PMC free article] [PubMed] [Google Scholar]