

Abstract
BCL-XL and BCL-2 are important targets for cancer treatment. BCL-XL specific PROTACs have been developed to circumvent the on-target platelet toxicity associated with BCL-XL inhibition. However, they have minimal effects on cancer cells that are dependent on BCL-2 or both BCL-XL and BCL-2. Here we report a new series of BCL-PROTACs. The lead PZ703b exhibits high potency in inducing BCL-XL degradation and in inhibiting but not degrading BCL-2, showing a hybrid dual-targeting mechanism of action that is unprecedented in a PROTAC molecule. As a result, PZ703b is highly potent in killing BCL-XL dependent, BCL-2 dependent and BCL-XL/BCL-2 dual-dependent cells, in an E3 ligase (VHL)-dependent fashion. We further found that PZ703b forms stable {BCL-2:PROTAC:VCB} ternary complexes in live cells which likely contributes to the enhanced BCL-2 inhibition by PZ703b. With further optimization, analogues of PZ703b could potentially be developed as effective antitumor agents by co-targeting BCL-XL and BCL-2.
Graphical Abstract
INTRODUCTION
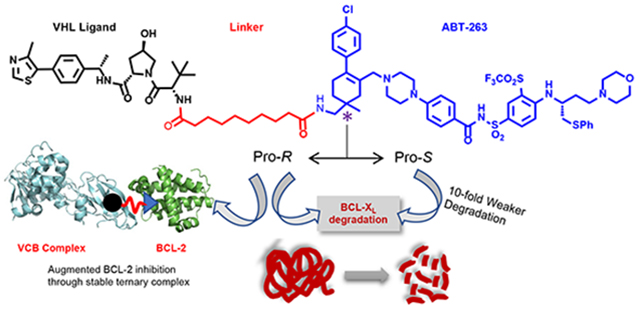
Evasion of cellular apoptosis is a hallmark of cancer, largely mediated by the dysregulation in the ratio of pro- and anti-apoptotic proteins.1 The anti-apoptotic BCL-2 family proteins including BCL-XL, BCL-2 and MCL-1 are well-validated cancer targets.2,3 They protect cells from apoptosis by sequestering pro-apoptotic BCL-2 family proteins followed by preventing activation of effector proapoptotic proteins (BAX and BAK) and the subsequent release of cytochrome c from mitochondria. This whole series of events prevents the activation of caspase-cascade and inhibits apoptosis. The apoptosis evasion induced by the upregulation of anti-apoptotic BCL-2 family proteins lead to tumor initiation, progression, and resistance to chemo- and targeted therapies.4 Numerous small molecules that can directly inhibit these anti-apoptotic proteins have been developed as potential cancer treatments.5 Among them, venetoclax, a BCL-2 selective inhibitor, has been approved by the FDA for the treatment of chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML);6 thus validating the translational relevance of targeting anti-apoptotic BCL-2 family proteins. Compared with its predecessor navitoclax (ABT-263), a BCL-2/BCL-XL dual inhibitor, venetoclax is not effective against cancer cells that rely on BCL-XL or both BCL-2 and BCL-XL for survival. Targeting BCL-XL might have a broader application in cancer therapy as it is the most common BCL-2 family member overexpressed in solid tumors, as well as in a subset of leukemia and lymphoma cells.7 However, the clinical applications of BCL-XL-selective or BCL-2/BCL-XL dual inhibitors are greatly limited by their on-target thrombocytopenia (or platelet toxicity) because platelets are dependent on BCL-XL for survival.8 To address this issue, we have been developing platelet-sparing BCL-XL proteolysis-targeting chimeras (PROTACs) that can induce ubiquitination and proteasomal degradation of BCL-XL.9 These PROTACs are bifunctional small-molecules with a BCL-XL binding moiety (warhead) linked to a binding moiety for an E3 ubiquitin ligase. We have shown that these PROTACs, which target BCL-XL to the von Hippel-Lindau (VHL),10a,10b cereblon (CRBN),10b,10c or inhibitor of apoptosis proteins (IAPs) E3 ligase,10d are capable of degrading BCL-XL protein in cancer cells but have minimal or no effect on the BCL-XL levels in human platelets. This is because platelets express minimal levels of VHL, CRBN, and IAPs. Thus, we have demonstrated that the PROTAC technology could be used to achieve tissue/cell selectivity by targeting E3 ligases that are differentially expressed in different tissues/cells. It is worth to note that several other strategies have also been successfully implemented to minimize platelet toxicity of BCL-XL inhibitors, including the development of BCL-2/XL dual inhibitor prodrug palcitoclax (APG-1252),11 dendrimer conjugate AZD0466,12 and antibody-drug conjugate ABBV-155.13
Our previous studies have identified DT2216 (Figure 1A),10a a VHL-recruiting BCL- XL specific PROTAC recently entered clinical phase. DT2216 is 4-fold more potent than its warhead ABT-263 against MOLT-4, a human T-cell acute lymphoblastic leukemia (ALL) cell line dependent on BCL-XL for survival and exhibited ~200-fold selectivity for MOLT-4 over human platelets while ABT-263 was equally toxic to MOLT-4 cells and platelets.10a Although, DT2216 could effectively degrade BCL-XL but not BCL-2 even though it has a relatively higher binding affinity for BCL-2 than BCL-XL. The specific degradation for BCL-XL over BCL-2 unfortunately narrows the anticancer spectrum of DT2216 as it is ineffective in cancer cells that are dependent on BCL-2 and is moderately effective against cancer cells that are dependent on both BCL-2/BCL-XL for survival.
Figure 1.
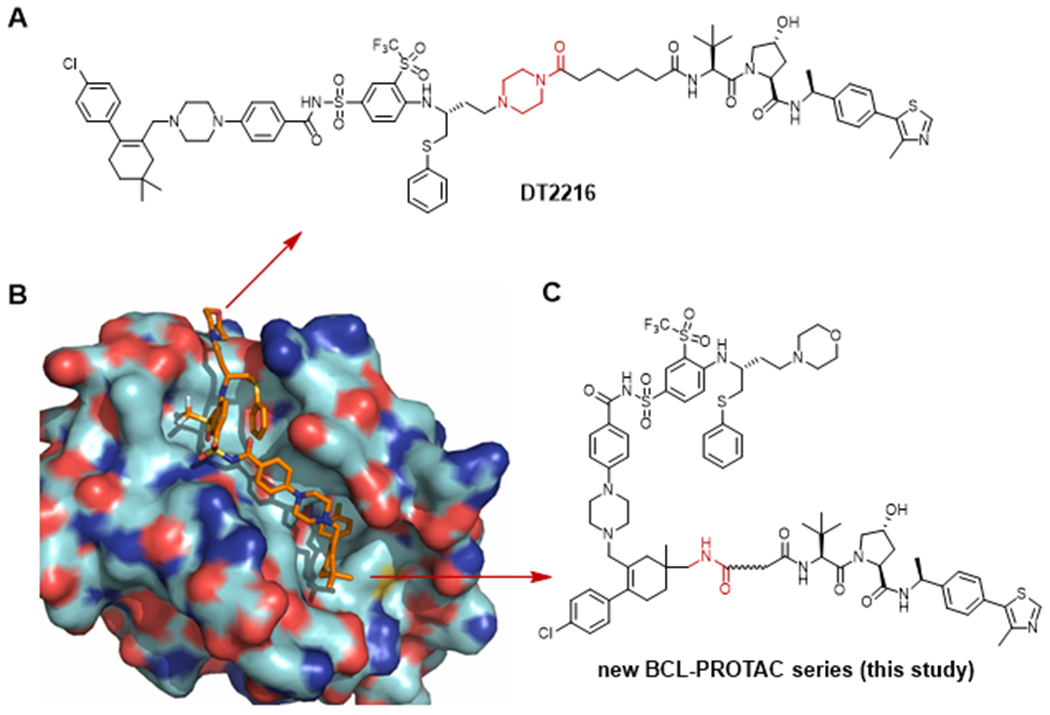
Design of ABT-263-based PROTACs. (A) Structure of DT2216; (B) Co-crystal structure of BCL-XL with ABT-263 (PDB code 4QNQ); (C) General structure of newly explored BCL-2/BCL-XL-targeting PROTACs. Arrows indicate the linking position.
Our mechanistic studies revealed that BCL-XL, but not BCL-2, can form stable ternary complexes with DT2216 and VHL-EloC-EloB (VCB) in live cells even though DT2216 can engage both proteins in cell-free AlphaLISA assay and a cellular thermal-shift assay.10a Thus, lack of the ability of forming a stable ternary complex, which is essential for inducing target ubiquitination, is the potential reason that DT2216 cannot induce BCL-2 degradation.
DT2216 was designed by connecting ABT-263 to a VHL ligand via attachment to the solvent-exposed morpholine ring, which was replaced by a bioisosteric piperazine group as a synthetic handle. Inspection of the co-crystal structures of ABT-263 in complex with BCL-XL (PDB code 4QNQ)14a and BCL-2 (PDB code 6QGH)14b revealed that the two methyl groups on the cyclohexene ring of ABT-263 are also solvent-exposed, and thus can be considered as suitable linker attachment points (Figure 1B). Linking from a different position of either the ligand for the protein of interest (POI) or the ligand for E3 ligase changes the interaction surfaces of the POI and E3 ligase when they form ternary complexes, which could alter the target degradation efficiency and specificity.15 We envisioned that PROTACs with linkers attached to the two methyl groups on the cyclohexene ring of ABT-263 might favor the ternary complex formation as BCL-XL/BCL-2 could have larger potential protein-protein interaction (PPI) surface in this region compared to the PPI surface induced by DT2216.
Herein, we describe the synthesis and biological evaluation of the new generation of BCL-targeting PROTACs that are constructed via attachment of linker and VHL ligand to one of the two methyl groups on the cyclohexene ring of ABT-263.
RESULTS AND DISCUSSION
To validate the new linker tethering position and identify the optimal linker length, we started our investigation by synthesizing a small set of new PROTACs that vary in linker length. A chiral quaternary carbon center on the cyclohexene ring of ABT-263 will be formed when one of the two methyl groups is functionalized for linker attachment. It is apparent from the co-crystal structures of ABT-263 in complex with BCL-XL that both methyl groups are solvent-exposed. To simplify the synthesis, we first prepared the “PROTAC-ready” warhead as an epimeric mixture for the construction of this series of PROTACs. Therefore, our synthetic sequence was commenced with the known compound 1 where the desired attachment point was disguised as tert-butyl ester. α-Methoxycarbonylation of 1 was achieved by using NaH and Me2CO3 to afford the β-keto ester 2. Converting 2 to the triflate intermediate by treatment with diisopropylethylamine (DIPEA) and trifluoromethanesulfonic anhydride followed by Suzuki coupling with 4-chlorophenyl boronic acid gave the diester 3. The ester groups of 3 were reduced with LAH to afford diol 4. NCS/Me2S mediated chemoselective chlorination of the allylic alcohol of 4 followed by nucleophilic substitution with ethyl 4-(piperazin-1-yl) benzoate generated alcohol 5 in good yield. Mesylation of the primary alcohol 5 followed by treatment with NaN3 generated an azide intermediate, which was converted to amine by Staudinger reduction and subsequent Boc-protection in the same pot to afford compound 6 in high yield. Ester hydrolysis of 6 followed by coupling with sulfonamine 7, prepared by following a reported method with minor modifications,16 and the removal of the Boc protection yielded amine 8. PROTACs PP1-PP8 were prepared by coupling 8 with the corresponding acids 9a-h (see Supporting Information) (Scheme 1).
Scheme 1.
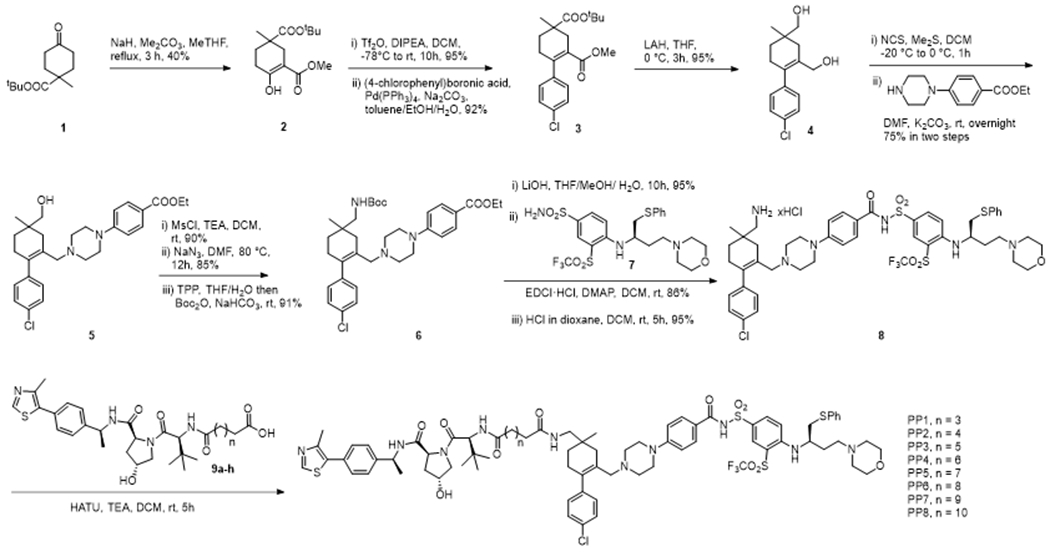
Synthesis of PP1-PP8
We tested PROTACs in cell viability assays using MOLT-4 and RS4;11 cells, with ABT-263 and DT2216 as the positive controls (Tables 1). Unlike DT2216 and its homologues, which have diminished potency in killing RS4;11 cells in comparison with ABT-263,10b the new PROTACs exhibited balanced cell killing potency in both MOLT-4 and RS4;11 cells. The improved killing potency of RS4;11 indicates a significant change with the underlying mechanism of action of the new PROTACs. It is well established that the linker length plays a major role in governing the bioactivity of PROTACs.17 We found that minimal deviation of the alkane-linker length in this series of PROTACs significantly changed their cytotoxicity against MOLT-4 and RS4;11 cells. The 8-carbon methylene linker PROTAC (PP5) exhibited the highest potency in killing both MOLT-4 and RS4;11 cells, with an IC50 of 32.1 nM and 23.3 nM, respectively (Table 1). PP5 was also found to be an efficient BCL-XL degrader with a half-degrading concentration (DC50) of 27.2 nM in MOLT-4 cells (Figure 2A and 2B). As expected, the optimal linker length for the new PROTAC series is different from the DT2216 series, due to the completely different PPI interface.
Table 1.
Cell-Killing Potency of PP1-PP8 in MOLT-4 and RS4;11 Cellsa
| Compound | MOLT-4 | RS4;11 | Compound | MOLT-4 | RS4;11 |
|---|---|---|---|---|---|
| ABT-263 | 212.3 ± 26.0 | 42.6 ± 5.7 | PP6 | 74.6 ± 14.6 | 29.2 ± 4.9 |
| DT2216 | 75.3 ± 9.1 | 211.7 ± 27.4 | PP7 | 111.8 ± 9.3 | 96.3 ± 15.3 |
| PP1 | 340.2 ± 29.4 | 303.6 ± 32.8 | PP8 | 333.6 ± 25.8 | 409.8 ± 38.6 |
| PP2 | 306.8 ± 31.9 | 234.2 ± 21.6 | PZ703a | 170.4 ± 19.6 | 418.3 ± 45.4 |
| PP3 | 219.4 ± 18.6 | 108.4 ± 12.9 | PZ703b | 15.9 ± 2.7 | 11.3 ± 2.2 |
| PP4 | 124.7 ± 11.3 | 43.7 ± 5.1 | PZ703b-NC | 820.6 ± 112.5 | 328.0 ± 48.5 |
| PP5 | 32.1 ± 3.7 | 23.3 ± 4.0 |
IC50 values (in nM) are reported as the mean ± standard deviation from three independent experiments performed in triplicates. Cells were treated for 48 h.
Figure 2.
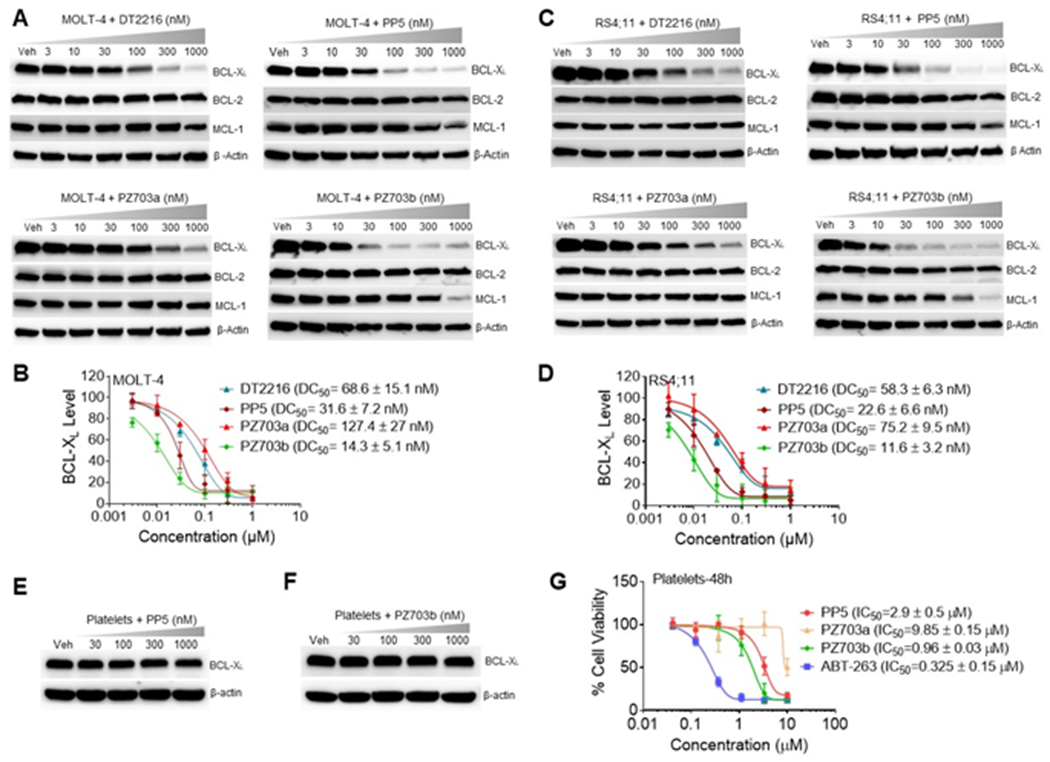
Degradation of BCL-XL by PROTACs in MOLT-4, RS4;11, and human platelets. (A,C) Representative western blotting analysis of BCL-XL, BCL-2, and MCL-1 protein levels in MOLT-4 (A) or RS4;11 cells (C) after treatment with indicated concentrations of DT2216, PP5, PZ703a, PZ703b for 16 h. (B,D) Densitometric analysis of BCL-XL expression in MOLT-4 (B) and RS4;11 cells (D). (E,F) Representative western blotting analysis of BCL-XL protein levels in human platelets after 16 h treatment with indicated concentrations of PP5 (E) or PZ703b (F). (G) Platelets were treated with increasing concentrations of ABT-263, PP5, PZ703a, PZ703b for 48 h. Cell viability was measured using MTS assay. (B,D,G) Data are mean ± SD of three independent experiments performed in triplicates.
The initial proof-of-concept studies indicated that tethering the linker from the cyclohexene ring of ABT-263 resulted in PROTACs with a distinct pharmacological profile. As both the epimeric methyl groups on the cyclohexene ring are solvent-exposed but clearly with different protruding angles (Figure 1B), we next sought to investigate the difference between these two tethering points by synthesizing both epimers of PP5. To access both epimers rapidly, we planned to separate the enantiomers of a suitable intermediate via attaching to an appropriate chiral auxiliary. After prolonged investigation, we found that diester 3 can be selectively hydrolyzed to acid 10, which can be attached to Evan’s chiral oxazolidinone via amidation (Scheme 2). To our delight, the resulting diastereomers 11a and 11b were separable through silica gel column chromatography. Reduction of the said diastereomers separately with LAH afforded the enantio-pure diols 4a and 4b. Enantiopurity of the diacetates of the corresponding diols (4a and 4b) were analyzed by chiral HPLC (Supporting Information) and found to be > 99.0%. Diols 4a/4b were converted to the corresponding amines 8a/8b by following the same synthetic protocol used to convert 4 to 8 (Scheme 1). Diastereo-pure amines 8a/8b were then maneuvered to the corresponding epimers of PP5, PZ703a and PZ703b, respectively. PZ703b exhibited 10-fold and 37-fold higher cytotoxicity than PZ703a against MOLT-4 and RS4;11 cells, respectively (Table 1). This clearly indicates that the linking position plays a crucial role in generating an efficient PROTAC. From structural analysis, the pro-R methyl group will likely give a better PPI interface. However, attempts to generate single crystals for determining the absolute configuration of enantiomers 11a/11b by X-ray diffraction or using NMR spectroscopy were unsuccessful. Hence, the absolute configuration of PZ703a/PZ703b remained elusive at this point. In addition, although the synthetic route delineated in Scheme 2 gave us access to the two epimeric PROTACs PZ703a/PZ703b, it suffers from the low overall yield for the potential active epimer as we had to sacrifice half of the intermediate in the diastereomer separation stage. Therefore, we devised an asymmetric synthetic route to prepare intermediate 4b as the stereochemistry resembles with our anticipated active PROTAC epimer PZ703b.
Scheme 2.
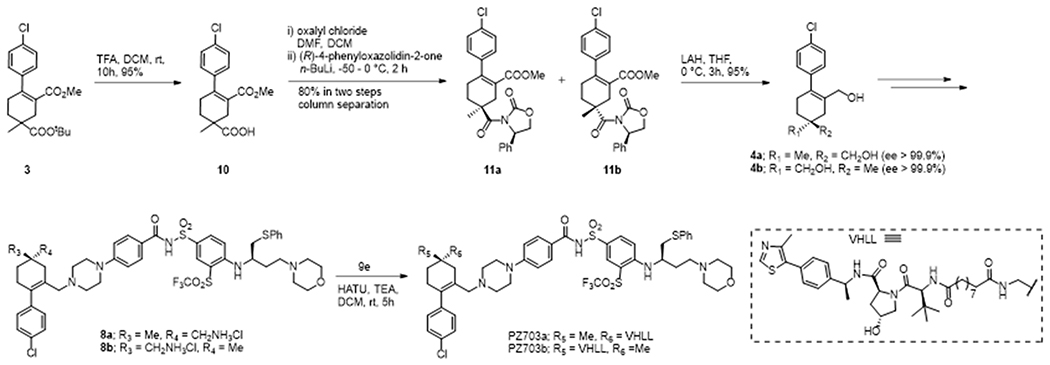
Synthesis of epimeric PROTACs PZ703a and PZ703b
We selected the (S)-isomer of 4,4-disubstituted cyclohexenone 13 as our starting material that can be accessed in four steps via a chiral amine driven Diels-Alder reaction by following Rawal’s protocol.18 TBS protection of the primary alcohol of 13 followed by installation of an ester group at the α-position of the carbonyl and subsequent reduction of the α,β-unsaturation afforded β-keto ester 14 in gratifying yield. Compound 14 was treated with triflic anhydride to form an enol triflate intermediate which upon Suzuki coupling with 4-chlorophenyl boronic acid afforded compound 15. Reduction of the ester with DIBAL-H and subsequent TBS ether deprotection afforded diol 4b (Scheme 3). Enantiomeric excess of 4b was determined by chiral HPLC via converting to the corresponding diacetate and was found to be 92.8%. The absolute configuration of the diols 4a and 4b from Scheme 2 was determined via comparing their corresponding diacetates with the diacetate of the enantio-selectively prepared 4b (see Supporting Information). Diol 4b was converted to 8b by following the same synthetic protocol used for the conversion of racemic diol 4 to 8. PZ703b was synthesized from 8b by using the same synthetic protocol described in Scheme 2.
Scheme 3.
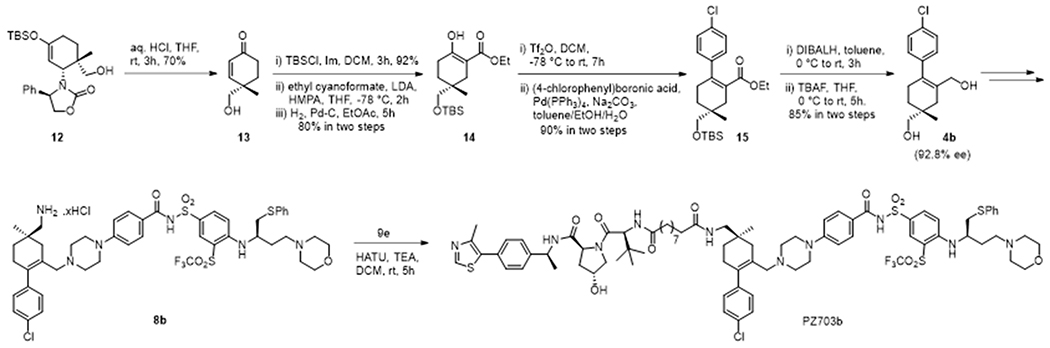
Enantioselective synthesis of PZ703b
We first compared PP5, PZ703a, and PZ703b, along with DT2216 and ABT-263 for their cytotoxicity in MOLT-4 and RS4;11 cells using MTS cell viability assay (Table 1), followed by examining their ability to induce BCL-XL/BCL-2/MCL-1 degradation in these cells using western blotting (Figure 2). In this assay, we found that PZ703b was significantly more potent than its S-epimer PZ703a in killing MOLT-4 and RS4;11 cells. Consistently, PZ703b was ~2-fold more potent than the diastereomeric mixture PP5 in these two cell lines. More importantly, PZ703b was also more potent than DT2216 and ABT-263 in these cells with IC50 values of 15.9 nM, 75.3 nM, and 212.3 nM, respectively, for MOLT-4; and IC50 values of 11.3 nM, 211.7 nM, and 42.6 nM, respectively for RS4;11 (Table 1). We also prepared a negative control of PZ703b, i.e., PZ703b-NC (see Supporting Information), by the inversion of the stereocenters on the hydroxyproline moiety of the VHL ligand; which causes the loss of binding affinity to VHL.19 As expected, PZ703b-NC had minimal effects on MOLT-4 and RS4;11 cells.
The cell viability data in MOLT-4 were well correlated with the BCL-XL protein degradation data when comparing the DC50 values among DT2216, PP5, PZ703a, PZ703b (Figure 2A & 2B) and PZ703b-NC (Supporting Information, Figure S1A). Similar DC50 values for BCL-XL were also observed in RS4;11 cells (Figure 2C & 2D). Notably, MCL-1 levels were also decreased when cells were treated with PZ703b or PP5 at high concentrations (Figure 2A & 2C). As we previously observed with cells that are highly sensitive to BCL-XL degradation,20 the MCL-1 downregulation is caused by apoptosis-induced caspase cleavage.21 To further confirm that the MCL-1 degradation is apoptosis dependent, we tested PZ703b in HEK 293T cells which are known to be insensitive to BCL-XL/BCL-2 inhibition. Although PZ703b can effectively induce BCL-XL degradation in these cells, no MCL-1 decrease was observed due to the absence of apoptosis as indicted by the lack of cleaved PARP (Supporting Information, Figure S2). Next, we evaluated the effects of PZ703b on human platelets. As expected, PZ703b did not cause significant changes on BCL-XL protein levels in platelets, which translated into its increased selectivity for MOLT-4 cells over platelets (100-fold) compared to the warhead ABT-263 (Figure 2F & 2G).
PZ703b induced rapid and durable BCL-XL degradation in MOLT-4 cells (Figure 3A & 3B). To demonstrate that PZ703b-induced BCL-XL degradation occurs through E3 ligase-dependent, proteasomal degradation, MOLT-4 cells were treated with proteasome inhibitor MG-132 or VHL ligand VHL-032.22 Both MG-132 and VHL-032 could effectively block the BCL-XL degradation induced by PZ703b (Figure 3C). In addition, PZ703b-NC did not induce BCL-XL degradation (Supporting Information, Figure S1A & S1B). Thus, PZ703b acts as a PROTAC. In addition, PZ703b potently induced apoptosis in MOLT-4 cells, which is caspase-3 mediated and can be blocked by pan-caspase inhibitor QVD (Figure 3D–3F). When compared with DT2216, PZ703b possesses ~3-fold higher binding affinity to BCL-XL in both binary (without VCB) and ternary binding (with VCB) affinity assays (Table 2), which correlates well with the increased cellular potency in MOLT-4 cells. However, the largely increased potency of PZ703b than its epimer PZ703a in inducing BCL-XL degradation and killing MOLT-4 cells cannot be attributed to higher binary or ternary BCL-XL binding affinity, as they are equally potent in binding to the protein (Table 2). Even more striking, although PZ703b is nearly 4-, 19-, and 37-fold more potent than ABT-263, DT2216, and PZ703a, respectively, in killing RS4;11 cells (Table 1); we did not observe significant BCL-2 degradation with PZ703b treatment (Figure 2). Only small differences (< 3-fold) in binding affinity to BCL-2 were observed among these compounds (Table 2), indicating that their cellular activity in RS4;11 cannot be explained by BCL-2 binding alone.
Figure 3.
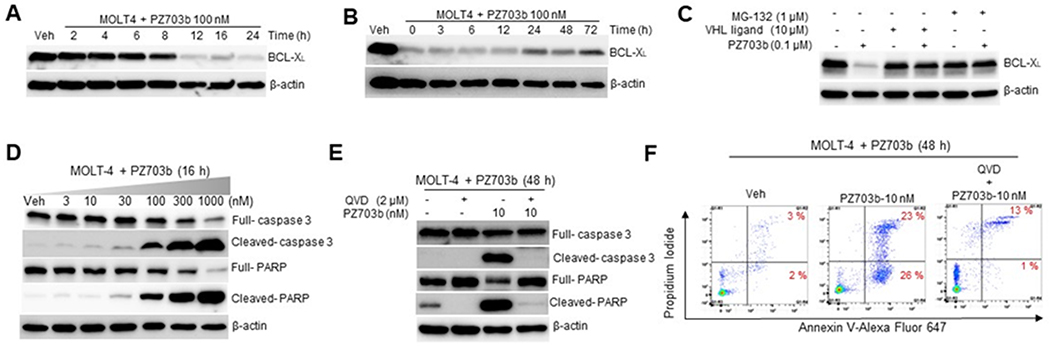
PZ703b induces rapid, sustained and VHL- and proteasome-dependent BCL-XL degradation and triggers apoptosis in cancer cells. (A) MOLT-4 cells were treated with vehicle (Veh) or 100 nM PZ703b for indicated time points. (B) Washout experiment: MOLT-4 cells were treated with 100 nM PZ703b for 16 h, followed by removal of PZ703b. Indicated times are after PZ703b was removed. (C) Degradation of BCL-XL is via PROTAC mechanism. MOLT-4 cells were pre-treated with 10 μM VHL-032 or 1 μM MG-132 followed by 12 h treatment with PZ703b. (A-C) Cells were lysed and protein samples were subjected to immunoblotting with antibodies against BCL-XL and β-actin. (D) MOLT-4 cells were treated with indicated concentrations of PZ703b for 16 h and immunoblotting was performed for cleaved-caspase-3, full-caspase-3, cleaved-PARP, full-PARP, and β-actin. (E) MOLT-4 cells pretreated with or without 2 μM QVD for 4 h and then were treated with 10 nM PZ703b for 48 h. Immunoblotting was performed for cleaved-caspase-3, full-caspase-3, cleaved-PARP, full-PARP, and β-actin. (F) MOLT-4 cells pretreated with or without 2 μM QVD for 4 h and then were treated with 10 nM PZ703b for 48 h before apoptosis was analyzed using Annexin-V/propidium iodide staining and flow cytometer. All immunoblotting data are representative of three independent experiments.
Table 2.
BCL-XL and BCL-2 Binding Affinity of Selected Compounds without or with VHL a
| BCL-XL | BCL-2 | |||
|---|---|---|---|---|
| Compound | w/o VHL | w VHL | w/o VHL | w VHL |
| ABT-263 | 2.07 ± 0.48 | NDb | 0.65 ± 0.36 | NDb |
| DT2216 | 7.58 ± 0.62 | 2.18 ± 0.51 | 2.25 ± 0.33 | 1.46 ± 0.41 |
| PZ703a | 2.75 ± 0.55 | 0.64 ± 0.18 | 1.47 ± 0.25 | 0.55 ± 0.18 |
| PZ703b | 2.59 ± 0.16 | 0.65 ± 0.19 | 1.22 ± 0.61 | 0.53 ± 0.19 |
Kd values (in nM) are reported as the mean ± SD of at least two individual experiments performed in triplicates.
not determined.
To gain more understanding of the cellular activity among DT2216, PZ703a, and PZ703b, we first measured the in vitro formation of {BCL-XL, BCL-2, or MCL-1:compound:VCB} ternary complexes using the cell-free AlphaLISA assay.23 Similar to DT2216,10a both PZ703a and PZ703b can form stable ternary complexes with either BCL-XL or BCL-2 with VCB (Figure 4A & 4B). PZ703b gave strongest signals among these compounds, which is consistent with cellular activity. As expected, none of these compounds can form stable ternary complexes with MCL-1 and VCB (Figure 4C). We further evaluated the ternary complex formation using the NanoBRET assay, a bioluminescence resonance energy transfer (BRET)-based assay to measure the interaction of two binding proteins in live cells.24 Previously, we found that DT2216 was able to form stable {BCL-2:DT2216:VCB} ternary complexes in vitro but not in live cells as determined by NanoBRET assay.10a Here, we found that PP5, PZ703a and PZ703b were able to form {BCL-2: compound:VCB} ternary complexes in live cells whereas DT2216 and PZ703b-NC could not (Figure 4E and Supporting Information Figure S1C & S1D). PZ703b induced more prominent {BCL-2:PZ703b:VCB} ternary complex formation compared to PZ703a. These results correlate well with the cellular activity among these compounds. Next, we conducted a series of competition assays with pre-treatment of RS4;11 with VHL ligand VHL-032 to block the formation of ternary complexes. As shown in Figure 5A, a nearly 20-fold activity loss for both PP5 and PZ703b and a smaller loss (2.6-fold) for PZ703a were observed in RS4;11 cells, whereas DT2216 and PZ703b-NC (Supporting Information Figure S1E) had no significant IC50 value changes after pre-treatment with VHL-032. Further, as a BCL-2 inhibitor, ABT-263 has been reported to disrupt the interaction of BCL-2 with anti-apoptotic protein Bim in RS4;11 cells.25 Similarly, PZ703b also functions as a BCL-2 inhibitor that can inhibit BCL-2:Bim interaction (Figure 5B). However, pre-treatment of RS4;11 with VHL-032 can block PZ703b but not ABT-263 from disrupting BCL-2:Bim interaction (Figure 5C). As expected, DT2216 and PZ703b-NC had minimal effect on BCL-2:Bim interaction with or without VHL-032 treatment (Figure 5B & 5C). These results are in agreement with the data from the NanoBRET assay; and further confirm that formation of stable {BCL-2:PZ703b:VCB} ternary complexes increased the inhibition of PZ703b to BCL-2 protein and enabled efficient killing of RS4;11 cells.
Figure 4.
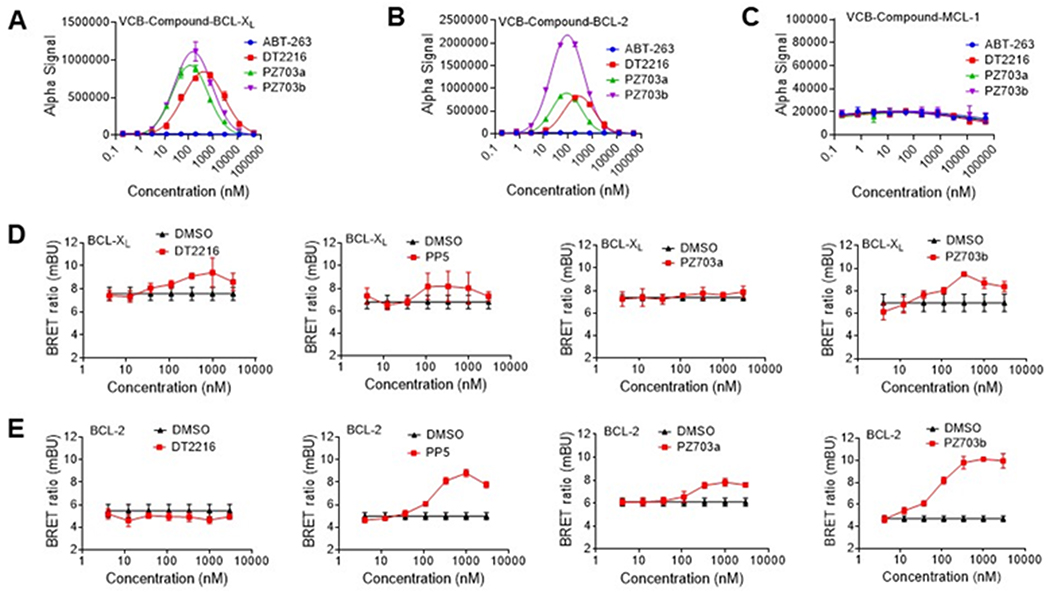
Ternary complex formation studies. (A-C) Cell-free ternary complex formation assay with recombinant His-tagged BCL-XL protein (A), His-tagged BCL-2 protein (B), and His-tagged MCL-1 protein (C) using AlphaLISA assay. (D) NanoBRET ternary complex formation with BCL-XL and VCB. (E) NanoBRET ternary complex formation with BCL-2 and VCB. Ternary complex formation was determined in 293T cells after they transiently expressed HiBit-BCL-XL/HiBit-BCL-2, LgBit and HaloTag-VHL and then treated with a serial dilution of DT2216, PP5, PZ703a, or PZ703b. Data are expressed as mean ± s.e.m. of two independent experiments performed in triplicates.
Figure 5.
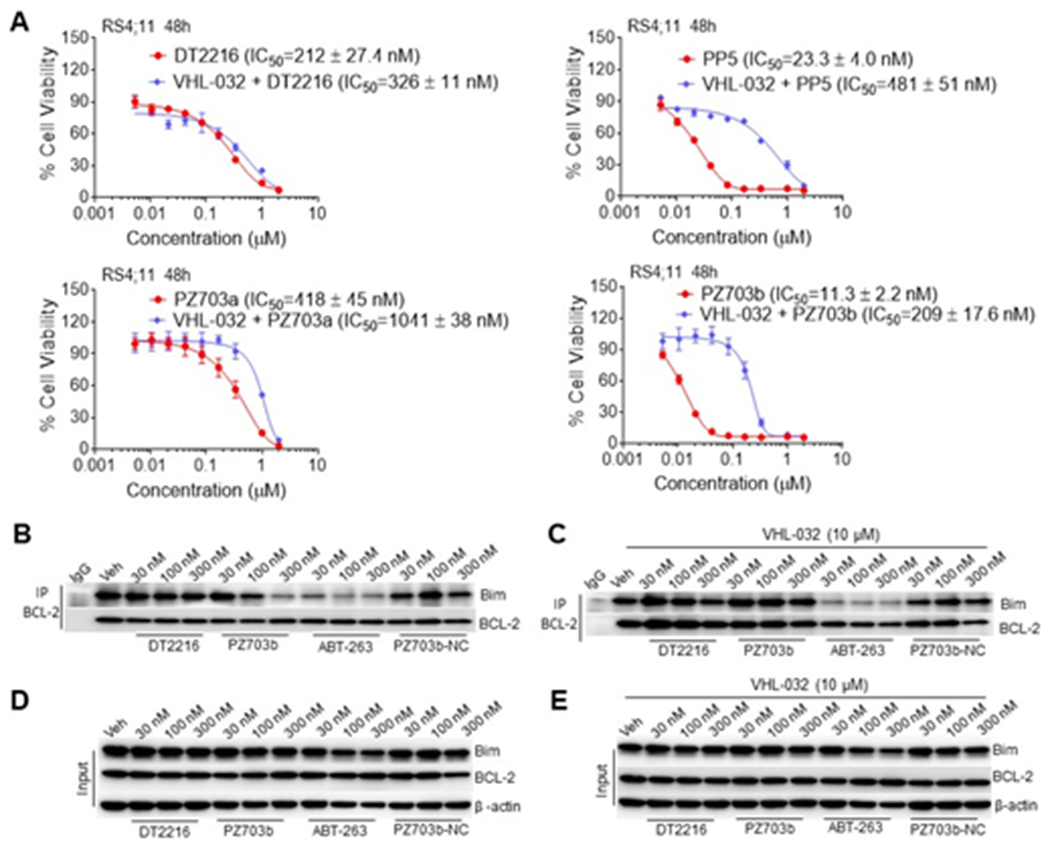
Cell viability and immunoprecipitation assays in RS4;11 with compound treatment in the presence or absence of VHL ligand VHL-032. (A) Viability of RS4;11 cells was determined after they were treated with DT2216, PP5, PZ703a, or PZ703b for 48 h with or without pre-treatment with VHL-032 (10 μM). Cell viability was measured using MTS assay. Data are representative of mean of three independent experiments performed in triplicates. (B,C) RS4;11 cells were treated with DT2216, PZ703b, ABT-263, or PZ703b-NC for 12 h with or without VHL-032. Immunoblots of Bim after immunoprecipitation with BCL-2 in the absence of VHL-032 (B) and in the presence of VHL-032 (C). (D,E) Bim, BCL-2, β-actin in whole-cell lysates (Input) are shown from two independent experiments.
We further observed that the improved BCL-XL degradation along with the increased BCL-2 inhibition through formation of stable ternary complexes also enabled PZ703b to efficiently kill BCL-2/BCL-XL dual-dependent NCI-H146 and NCI-H211 cells (Supporting Information Figure S3). Thus, we demonstrated that linking from the methyl group on the cyclohexene ring of ABT-263 is a valid approach for the generation of novel BCL-XL PROTACs with improved inhibitory activity against BCL-2 by promoting the formation of the {BCL-2:PROTAC:VCB} complex.
CONCLUSION
We have designed and synthesized a novel BCL-targeting PROTAC (PZ703b) based on BCL-XL/BCL-2 dual inhibitor ABT-263 by tethering the pro-R methyl group on the cyclohexene ring of ABT-263. PZ703b is remarkably more potent than its epimer PZ703a and predecessor PROTAC DT2216 in inducing BCL-XL degradation. PZ703b also appears to potently inhibit BCL-2 through the formation of stable {BCL-2:PZ703b:VCB} ternary complexes in live cells, which is distinct from DT2216. Thus, PZ703b possesses a unique mechanism of action (MOA) in inhibiting anti-apoptotic BCL-2 proteins, i.e., potent degradation of BCL-XL and simultaneously enhanced inhibition of BCL-2, that enables its high potency against BCL-XL dependent, BCL-2 dependent, and BCL-XL/BCL-2 dual-dependent cancer cells. To the best of our knowledge, this is the first time that such hybrid mechanism has been observed in a PROTAC molecule. Underneath this study lies a great scope of opportunity to uncover the expansion of the target coverage for degradation and inhibition by PROTAC technology. However, further understanding of the specific mechanism that govern the ternary complex formation and the enhanced target inhibition is needed to facilitate the rational design. Moreover, it will be interesting to find out if PROTACs with such hybrid MOA can be effective in vivo as degraders and inhibitors have distinct pharmacokinetic-pharmacodynamic profiles. In this regard, BCL-XL/BCL-2 dual-degrader, if can be identified, could be a better choice for clinical development.15b,26
EXPERIMENTAL SECTION
Chemistry General Procedures.
All chemicals obtained from commercial suppliers were used as purchased without further purification. Water was purified with a Milli-Q Simplicity 185 Water Purification System (Merck Millipore). All reactions with water- and/or air-sensitive starting materials were carried out in pre-dried glass wares under argon atmosphere with standard procedure. THF, DCM, and DMF were obtained via a solvent purification system by filtering through two columns packed with activated alumina and 4 Å molecular sieve, respectively. Flash chromatography was performed using silica gel (230–400 mesh) as the stationary phase. Reaction progress was monitored by thin layer chromatography (silica coated glass plates) and visualized by UV light, and/or by LC-MS. NMR spectra were recorded in CDCl3 or CD3OD at 600 MHz. Chemical shifts δ are given in ppm using tetramethylsilane as an internal standard. Multiplicities of NMR signals are designated as singlet (s), broad singlet (br s), doublet (d), doublet of doublets (dd), triplet (t), quartet (q), and multiplet (m). All final compounds for biological testing were of ≥95.0% purity as analyzed by LC–MS, performed on an Advion AVANT LC system with the expression CMS using a Thermo Accucore™ Vanquish™ C18+ UHPLC Column (1.5 μm, 50 x 2.1 mm) at 40 °C. Gradient elution was used for UHPLC with a mobile phase of acetonitrile and water containing 0.1% formic acid. High resolution mass spectra (HRMS) were recorded on a Bruker Impact II QTOF mass spectrometer.
1-(tert-Butyl) 3-methyl 4-hydroxy-1-methylcyclohex-3-ene-1,3-dicarboxylate (2).
To a stirring suspension of NaH (9.43 g, 235 mmol) in MeTHF (300 mL) was added a solution of compound 1 (20 g, 94 mmol) in MeTHF (50 mL) dropwise at rt. To this mixture Me2CO3 (39.5 mL, 470 mmol) was added and the resulting mixture was refluxed for 5 h. After consumption of the starting material, the reaction mixture was cooled to 0 °C and then carefully quenched with ice-cold water until effervescence stopped. The organic portion was collected, and the pH of the aqueous part was adjusted to 5.0. The aqueous part was further extracted with EtOAc. The combined organic portion was washed with brine solution and then dried over Na2SO4. The solvent was evaporated in reduced pressure and crude was purified by silica gel column flash chromatography (2.5% EtOAc in hexane) to afford the desired compound 2 (10.1 g, 37.6 mmol) as a clear oil in 40% yield. 1H NMR (600 MHz, Chloroform-d) δ 12.12 (s, 1H), 3.76 (s, 3H), 2.76 (d, J = 15.8 Hz, 1H), 2.42 – 2.34 (m, 1H), 2.32 – 2.25 (m, 1H), 2.03 (d, J = 16.2 Hz, 2H), 1.61 – 1.54 (m, 1H), 1.42 (s, 9H), 1.20 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 175.9, 172.9, 171.0, 96.3, 80.4, 51.6, 41.9, 31.8, 30.7, 28.1, 26.8, 24.8. HRMS (ESI) m/z = 293.1365 calcd. for C14H22NaO5 [M + Na]+, found: 293.1349.
4-(tert-Butyl) 2-methyl 4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2,4-dicarboxylate (3).
Step one: synthesis of (1-(tert-butyl) 3-methyl 1-methyl-4-(((trifluoromethyl)sulfonyl)oxy)cyclohex-3-ene-1,3-dicarboxylate).
To a stirring solution of the compound 2 (10.1 g, 37.6 mmol) in DCM (120 mL) was added DIPEA (32.7 mL,188 mmol). The mixture was cooled to −78 °C and Tf2O (7.6 mL, 45.1 mmol) was added dropwise. The temperature of the reaction was allowed to rise to the rt and stirred for 8 h. Upon completion, the reaction was quenched with water. The organic part was washed with saturated aqueous NaHCO3 solution followed by brine, dried over Na2SO4 and the volatiles were removed under reduced pressure. To the crude mixture was added 10% EtOAc in hexane solution and it was stirred over 1 h. The mixture was filtered and filter cake was washed repeatedly by the 10% EtOAc in hexane solution. The combined filtrate was collected and evaporated under reduced pressure to give the dark brown crude. The crude was purified by silica gel flash column chromatography (3% EtOAc in hexane) to afford the triflate intermediate (14.4 g, 35.7 mmol) as an oil in 95% yield. 1H NMR (600 MHz, Chloroform-d) δ 3.78 (d, J = 2.3 Hz, 3H), 2.99 (dq, J = 17.6, 1.8 Hz, 1H), 2.52 (ddt, J = 14.9, 5.9, 3.0 Hz, 1H), 2.44 – 2.31 (m, 1H), 2.25 (d, J = 17.6 Hz, 1H), 2.11 (ddd, J = 8.8, 4.3, 1.8 Hz, 1H), 1.62 (dq, J = 13.4, 6.8 Hz, 1H), 1.41 (d, J = 1.7 Hz, 9H), 1.26 – 1.17 (m, 3H). 13C NMR (151 MHz, Chloroform-d) δ 174.7, 164.8, 151.1, 121.7, 121.6, 119.6, 117.5, 81.4, 52.4, 41.4, 35.3, 31.3, 26.7, 24.6. HRMS (ESI) m/z = 425.0858 calcd. for C15H21F3NaO7S [M + Na]+, found: 425.0833.
Step two: synthesis of compound 3.
To a stirring solution of the triflate from above (14.4 g, 35.7 mmol) in toluene (70 mL) and EtOH (35 mL) was added 2N Na2CO3 solution (35 mL). The above mixture was purged with argon for 15 min and 4-chlorophenylboronic acid (6.68 g, 42.8 mmol) and Pd(PPh3)4 (824 mg, 0.714 mmol) was added. The mixture was heated to 90 °C for 7 h. Organic solvents was removed under vacuo and the reaction was diluted with EtOAc (150 mL), washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The crude material was purified by silica gel flash chromatography (hexanes/EtOAc = 10:1) to afford diester 3 (11.95 g, 32.84 mmol) in 92% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.27 (d, J = 8.5 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 3.47 (s, 3H), 2.93 (dd, J = 17.5, 1.7 Hz, 1H), 2.45 (dddt, J = 17.0, 8.5, 6.0, 3.0 Hz, 1H), 2.37 (dtd, J = 17.3, 6.0, 2.8 Hz, 1H), 2.22 (dt, J = 17.5, 2.6 Hz, 1H), 2.09 – 2.04 (m, 1H), 1.62 (ddd, J = 13.9, 8.3, 6.3 Hz, 1H), 1.45 (s, 9H), 1.26 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 175.9, 169.2, 144.4, 141.4, 133.0, 128.4, 128.2, 126.8, 80.5, 51.5, 41.5, 35.9, 31.5, 31.1, 28.1, 24.7. HRMS (ESI) m/z = 387.1339 calcd. for C20H25ClNaO4 [M + Na]+, found: 387.1318.
(4’-Chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2,4-diyl)dimethanol (4).
To a stirring suspension of LAH (2.6 g, 68.5 mmol) in THF (100 mL) at 0 °C was added a solution of compound 3 (10 g, 27.4 mmol) in THF (35 mL) dropwise. After stirring at same temperature for 3 h, water (2.6 mL) was added slowly followed by addition of 15% NaOH solution (2.6 mL). After stirring for 1 h, 7.8 mL water was added. The mixture was stirred for 30 min at rt before added solid Na2SO4 and stirred for 1 h. The mixture was filtered through a pad of celite and washed several times with EtOAc. The organic portion was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was washed with 10% DCM in diethyl ether solution 3 times to give the diol 4 (6.9 g, 26 mmol) as a white powder in 95% yield. 1H NMR (600 MHz, Methanol-d4) δ 7.33 (d, J = 8.4 Hz, 2H), 7.16 (d, J = 8.4 Hz, 2H), 3.87 (s, 2H), 3.43 (d, J = 10.8 Hz, 1H), 3.37 (d, J = 10.9 Hz, 1H), 2.33 (t, J = 6.3 Hz, 2H), 2.18 (d, J = 17.5 Hz, 1H), 1.98 (d, J = 17.3 Hz, 1H), 1.65 (dd, J = 13.4, 6.7 Hz, 1H), 1.48 (dt, J = 12.1, 5.6 Hz, 1H), 1.01 (s, 3H). 13C NMR (151 MHz, Methanol-d4) δ 142.6, 135.1, 133.4, 133.0, 130.8, 129.2, 71.2, 63.9, 36.8, 34.9, 31.6, 30.3, 22.7. HRMS (ESI) m/z = 289.0971 calcd. for C15H19ClNaO2 [M + Na]+, found: 289.0955.
Ethyl 4-(4-((4’-chloro-4-(hydroxymethyl)-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (5).
To a stirring solution of 4 (5 g, 18.8 mmol) in DCM (130 mL) was added NCS (2.87 g, 21.6 mmol). The mixure was cooled to −30 °C and Me2S (1.58 mL, 21.6 mmol) was added dropwise. After 1 h the reaction was diluted with DCM and then quenched with water. The organic part was washed with brine, dried over anhydrous Na2SO4, filtered, and and concentrated under vacuum. The crude product was used in the next step without further purification.
The crude product from above was dissolved in DMF (50 mL) followed by the successive addition of ethyl 4-(piperazin-1-yl)benzoate (5.72 g, 24.4 mmol) and K2CO3 (5.18 g, 37.6 mmol). The mixture was stirred at rt for 7 h, upon consumption of the starting material the reaction was diluted with EtOAc. The organic part was washed with water several times followed by brine. The organic portion was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by column chromatography (hexanes/EtOAc = 50/50) to afford 5 (6.8 g, 14.1 mmol) as an off-white powder in 75% yield in two steps. 1H NMR (600 MHz, Chloroform-d) δ 7.89 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.4 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 4.31 (q, J = 7.1 Hz, 2H), 3.46 (d, J = 2.4 Hz, 2H), 3.25 (t, J = 5.1 Hz, 4H), 2.86 – 2.76 (m, 2H), 2.36 (qt, J = 11.0, 4.9 Hz, 4H), 2.29 (s, 2H), 2.16 (d, J = 17.4 Hz, 1H), 2.00 (d, J = 17.3 Hz, 1H), 1.62 (dd, J = 13.4, 6.8 Hz, 1H), 1.50 (dt, J = 12.5, 5.9 Hz, 1H), 1.36 (t, J = 7.1 Hz, 3H), 1.01 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 166.9, 154.3, 141.7, 135.1, 132.3, 131.3, 129.9, 129.1, 128.5, 120.0, 113.7, 71.2, 60.7, 60.5, 52.7, 47.7, 36.6, 34.2, 30.7, 30.2, 22.5, 14.6. HRMS (ESI) m/z = 483.2414 calcd. for C28H36ClN2O3 [M + H]+, found: 483.2393.
Ethyl 4-(4-((4-(((tert-butoxycarbonyl)amino)methyl)-4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate (6).
Step one: synthesis of ethyl 4-(4-((4’-chloro-4-methyl-4-(((methylsulfonyl)oxy)methyl)-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate.
To a stirring solution of 5 (2 g, 4.14 mmol) in THF (12 mL) was added TEA (1.74 mL,12.4 mmol) and the mixture was cooled to 0 °C. MsCl (0.48 mL, 6.21 mmol) was added at the same temperature and the mixture was stirred at rt for 8 h. Upon consumption of the starting material, the mixture was diluted with DCM and the organic portion was washed with saturated aqueous NaHCO3 solution followed by brine. The organic part was dried over anhydrous Na2SO4, filtered, and concentrated under vacuum. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 3:1) to afford the desired compound (2.1 g, 3.7 mmol) in 90% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.89 (d, J = 9.0 Hz, 2H), 7.29 (d, J = 8.3 Hz, 2H), 6.99 (d, J = 8.4 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 4.31 (q, J = 7.1 Hz, 2H), 4.05 (q, J = 9.3 Hz, 2H), 3.25 (t, J = 5.1 Hz, 4H), 3.04 (s, 3H), 2.81 (s, 2H), 2.35 (t, J = 5.1 Hz, 4H), 2.30 (d, J = 6.2 Hz, 2H), 2.20 (d, J = 17.7 Hz, 1H), 2.09 (d, J = 17.7 Hz, 1H), 1.69 (dd, J = 13.3, 6.7 Hz, 1H), 1.57 (d, J = 6.7 Hz, 1H), 1.36 (t, J = 7.1 Hz, 3H), 1.09 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 166.7, 154.1, 141.0, 134.8, 132.4, 131.1, 129.6, 128.4, 128.4, 119.9, 113.5, 60.3, 53.4, 52.5, 47.6, 37.2, 36.1, 33.0, 31.5, 30.4, 29.6, 22.5, 14.5. HRMS (ESI) m/z = 561.2190 calcd. for C29H38ClN2O5S [M + H]+, found: 561.2168.
Step two: synthesis of ethyl 4-(4-((4-(azidomethyl)-4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoate.
To a stirring solution of the produce from above (2.1 g, 3.7 mmol) in DMF (10 mL) was added NaN3 (481 mg, 7.4 mmol) and catalytic amount TBAI. The reaction mixture was heated at 100 ºC for 24 h. Upon completion, the mixture was diluted with EtOAc and washed with brine. The organic part was dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 90:10) to afford the desired compound (1.68 g, 3.33 mmol) in 85% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.90 (d, J = 9.0 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 6.99 (d, J = 8.4 Hz, 2H), 6.81 (d, J = 9.0 Hz, 2H), 4.32 (q, J = 7.1 Hz, 2H), 3.28 – 3.20 (m, 6H), 2.81 (s, 2H), 2.38 – 2.32 (m, 4H), 2.26 (ddd, J = 24.3, 17.2, 6.4 Hz, 2H), 2.15 (d, J = 17.7 Hz, 1H), 2.03 (d, J = 16.3 Hz, 1H), 1.62 (dt, J = 13.7, 6.9 Hz, 1H), 1.51 (dt, J = 12.4, 5.9 Hz, 1H), 1.36 (t, J = 7.1 Hz, 3H), 1.04 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 166.8, 154.3, 141.4, 134.9, 132.4, 131.2, 129.8, 128.9, 128.5, 120.0, 113.7, 61.8, 60.6, 60.5, 52.6, 47.7, 37.4, 34.1, 31.5, 30.1, 23.4, 14.6. HRMS (ESI) m/z = 508.2479 calcd. for C28H35ClN5O2 [M + H]+, found: 508.2462.
Step three: synthesis of compound 6.
To a stirring solution of the azide from above (1.68 g, 3.33 mmol) in THF/H2O (12 mL/1 mL) was added TPP (1.7 g, 6.66 mmol) at rt and the mixture was stirred for 2 h. Once the azide consumed, Boc2O (937 mg, 4.3 mmol) and NaHCO3 (697 mg, 8.3 mmol) were added sequencially to the above mixture. The mixture was stirred for another 7 h and THF was removed under reduced pressure. The residue was diluted with EtOAc followed by washing with water and brine. The organic part was dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 90:10) to afford compound 6 (1.53 g, 3 mmol) in 91% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.89 (d, J = 8.9 Hz, 2H), 7.27 (d, J = 8.3 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 9.0 Hz, 2H), 4.74 (s, 1H), 4.31 (q, J = 7.1 Hz, 2H), 3.25 (t, J = 5.1 Hz, 4H), 3.13 (dd, J = 13.5, 7.0 Hz, 1H), 3.05 (dd, J = 13.5, 6.0 Hz, 1H), 2.79 (t, J = 10.4 Hz, 2H), 2.40 – 2.25 (m, 6H), 2.10 (d, J = 17.4 Hz, 1H), 1.98 (d, J = 17.4 Hz, 1H), 1.57 – 1.49 (m, 2H), 1.43 (s, 9H), 1.36 (t, J = 7.1 Hz, 3H), 0.97 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 166.8, 156.5, 154.2, 141.5, 135.0, 132.3, 131.2, 129.9, 128.9, 128.4, 120.0, 113.6, 79.2, 60.6, 60.4, 52.7, 49.3, 47.6, 37.6, 33.3, 31.8, 30.2, 28.6, 23.6, 14.6. HRMS (ESI) m/z = 582.3099 calcd. for C33H45ClN3O4 [M + H]+, found: 582.3072.
4-(4-((4-(Aminomethyl)-4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)-N-((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)benzamide hydrochloric salt (8).
Step one: synthesis of 4-(4-((4-(((tert-Butoxycarbonyl)amino)methyl)-4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-yl)methyl)piperazin-1-yl)benzoic acid.
To a stirring solution of ester 6 (1.53 g, 3 mmol) in THF/MeOH (10 mL/3 mL) was added a solution of LiOH∙H2O (252 mg, 6 mmol) in water (3 mL). The mixture was stirred at 50 °C for 10 h. Upon completion, solvents were removed under reduced pressure and the pH was adjusted to 7 with 1N HCl. The mixture was diluted with EtOAc (150 mL). The organic portion was washed with water and brine, dried over anhydrous NaSO4. filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 60:40) to afford the desired acid (1.57 g, 2.85 mmol) as a white powder in 95% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.93 (d, J = 8.6 Hz, 2H), 7.28 (d, J = 8.4 Hz, 2H), 7.00 (d, J = 8.1 Hz, 2H), 6.80 (d, J = 8.6 Hz, 2H), 4.75 (d, J = 6.2 Hz, 1H), 3.28 (t, J = 5.1 Hz, 4H), 3.13 (dd, J = 13.5, 7.0 Hz, 1H), 3.05 (dd, J = 13.5, 5.9 Hz, 1H), 2.87 – 2.77 (m, 2H), 2.42 – 2.21 (m, 6H), 2.12 (d, J = 17.0 Hz, 1H), 1.99 (d, J = 17.4 Hz, 1H), 1.72 – 1.63 (m, 1H), 1.59 – 1.52 (m, 1H), 1.43 (s, 9H), 0.97 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 156.6, 154.5, 141.5, 135.7, 132.4, 131.9, 129.9, 128.5, 128.4, 119.5, 113.6, 79.3, 72.9, 62.8, 60.5, 58.7, 52.6, 49.4, 47.3, 37.6, 33.3, 31.7, 30.3, 30.3, 28.6, 26.9, 23.6. HRMS (ESI) m/z = 554.2786 calcd. for C31H41ClN3O4 [M + H]+, found: 554.2764.
Step two: synthesis of tert-Butyl ((4’-chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl) piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)carbamate.
To a stirring solution of the acid from above (663 mg, 1.2 mmol) in DCM (5 mL) was successively added compound 7 (553 mg, 1 mmol), EDCI∙HCl (558 mg, 3.6 mmol), DMAP (122 mg, 1 mmol) and TEA (0.28 mL, 2 mmol). The resulting mixture was stirred at rt for 7 h. Upon completion, DCM was evaporated under reduced pressure and the crude product was purified by gradient silica gel flash column chromatography (DCM/MeOH = 100:1 to 10:1) to afford the desired compound (935 mg, 0.86 mmol) in 86% yield. 1H NMR (600 MHz, Acetone-d6) δ 8.32 (s, 1H), 8.10 (d, J = 8.1 Hz, 1H), 7.86 (d, J = 8.9 Hz, 2H), 7.41 (d, J = 7.7 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 7.31 (t, J = 7.7 Hz, 2H), 7.22 (t, J = 7.4 Hz, 1H), 7.19 (d, J = 8.4 Hz, 2H), 7.00 (dd, J = 25.6, 7.8 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 6.00 (t, J = 5.9 Hz, 1H), 4.21 (s, 1H), 3.55 (ddd, J =17.5, 8.6, 5.6 Hz, 4H), 3.36 (qd, J = 14.0, 6.0 Hz, 2H), 3.30 – 3.26 (m, 4H), 3.22 (td, J = 13.3, 6.2 Hz, 2H), 3.12 (dd, J = 13.5, 6.8 Hz, 1H), 3.05 (dd, J = 13.5, 6.3 Hz, 1H), 2.84 (q, J = 12.4 Hz, 4H), 2.40 – 2.36 (m, 6H), 2.32 – 2.21 (m, 3H), 2.19 – 2.10 (m, 2H), 1.82 (td, J = 13.7, 5.2 Hz, 1H), 1.61 (dt, J = 13.1, 6.6 Hz, 1H), 1.50 (dt, J = 13.4, 6.5 Hz, 1H), 1.41 (s, 9H), 0.99 (s, 3H). HRMS (ESI) m/z = 1089.3667 calcd. for C52H65ClF3N6O8S3 [M + H]+, found: 1089.3637.
Step three: synthesis of compound 8.
To a stirring solution of the compound from above (500 mg, 0.46 mmol) in DCM (5 mL) was added HCl in dioxane (1.1 mL, 4.6 mmol) and the resultant mixture was stirred at rt for 5 h. Once the starting material was consumed, the volatiles were removed under reduced pressure and the resultant solid was washed with diethyl ether two times to afford the desired compound 8 (480 mg) as an off-white solid powder, which was used in the next step without further purification. MS (ESI) m/z = 989.3 calcd. for C47H57ClF3N6O6S3 [M + H]+, found: 989.2.
General procedure for the synthesis of acids 9d-9h.
To a stirring solution of compound (2S,4R)-1-((S)-2-amino-3,3-dimethylbutanoyl)-4-hydroxy-N-((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)pyrrolidine-2-carboxamide14 (1 mmol, salt form) in DCM (5 mL) was added TEA (5 mmol) and the mixture was stirred at rt. In a separate reaction vessle HATU (1.1 mmol) was added to a solution of corresponding dicarboxylic acid (1 mmol) and TEA (3 mmol) in DCM (10 mL) at 0 °C and the mixture was stirred for 1 h. The previous amine solution was added dropwise to this solution at 0 °C and the reaction was stirred for 5 h at rt. Once the reaction was complete, the volatiles were removed under reduced pressure and the crude product was purified by silica gel flash column chromatography (DCM/MeOH/TEA = 100:1:1 to 100:15:1). Yields was found to be around 60%. Synthetic protocol of acids 9a-9c can be found in ref. 8d.
9-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-9-oxononanoic acid (9d).
1H NMR (600 MHz, Chloroform-d) δ 8.69 (s, 1H), 7.56 (d, J = 7.6 Hz, 1H), 7.42 – 7.35 (m, 4H), 6.78 (d, J = 9.1 Hz, 1H), 5.09 – 5.04 (m, 1H), 4.65 – 4.52 (m, 2H), 4.47 (s, 1H), 3.98 (d, J = 11.4 Hz, 1H), 3.63 (dd, J = 11.4, 3.4 Hz, 1H), 3.40 (dt, J = 3.2, 1.6 Hz, 1H), 2.52 (s, 3H), 2.29 (t, J = 7.4 Hz, 2H), 2.26 – 2.20 (m, 3H), 2.18 – 2.12 (m, 1H), 1.65 – 1.57 (m, 4H), 1.50 (d, J = 7.0 Hz, 3H), 1.36 – 1.29 (m, 6H), 1.03 (s, 9H). MS (ESI) m/z = 615.3 calcd. for C32H47N4O6S [M + H]+, found: 615.2.
10-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-10-oxodecanoic acid (9e).
1H NMR (600 MHz, Chloroform-d) δ 8.65 (s, 1H), 7.60 (d, J = 7.7 Hz, 1H), 7.37 – 7.30 (m, 4H), 6.90 (d, J = 8.8 Hz, 1H), 5.02 (p, J = 7.0 Hz, 1H), 4.60 (t, J = 8.3 Hz, 1H), 4.49 (d, J = 8.8 Hz, 1H), 4.45 (s, 1H), 3.98 (d, J = 11.5 Hz, 1H), 3.58 (dd, J = 11.5, 3.3 Hz, 1H), 2.48 (s, 3H), 2.29 – 2.12 (m, 6H), 1.60 – 1.53 (m, 4H), 1.46 (d, J = 7.0 Hz, 3H), 1.27 (s, 9H), 0.99 (s, 9H). 13C NMR (151 MHz, Chloroform-d) δ 177.1, 175.1, 171.5, 170.9, 150.9, 148.1, 143.7, 132.1, 130.4, 129.5, 126.5, 70.0, 59.2, 58.1, 56.8, 46.6, 36.7, 36.1, 35.3, 34.2, 28.9, 28.8, 28.8, 26.4, 25.6, 24.7, 22.1, 15.7, 8.7. MS (ESI) m/z = 629.3 calcd. for C33H49N4O6S [M + H]+, found: 629.2.
11-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-11-oxoundecanoic acid (9f).
1H NMR (600 MHz, Chloroform-d) δ 8.68 (s, 1H), 7.43 – 7.35 (m, 5H), 6.57 (d, J = 8.9 Hz, 1H), 5.12 – 5.06 (m, 1H), 4.70 (t, J = 8.0 Hz, 1H), 4.61 (d, J = 8.9 Hz, 1H), 4.50 (s, 1H), 4.11 (d, J = 11.4 Hz, 1H), 3.61 (dd, J = 11.3, 3.6 Hz, 1H), 2.53 (s, 3H), 2.48 – 2.42 (m, 1H), 2.27 (t, J = 7.3 Hz, 2H), 2.24 – 2.13 (m, 2H), 2.12 – 2.06 (m, 1H), 1.64 – 1.55 (m, 4H), 1.48 (d, J = 6.9 Hz, 3H), 1.34 – 1.26 (m, 10H), 1.04 (s, 9H). MS (ESI) m/z = 643.3 calcd. for C33H51N4O6S [M + H]+, found: 643.2.
12-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-12-oxododecanoic acid (9g).
1H NMR (600 MHz, Chloroform-d) δ 8.71 (s, 1H), 7.42 (d, J = 8.2 Hz, 2H), 7.39 (d, J = 8.2 Hz, 2H), 7.30 – 7.28 (m, 1H), 7.04 (d, J = 9.1 Hz, 1H), 5.14 – 5.08 (m, 1H), 4.69 (dd, J = 17.2, 8.7 Hz, 2H), 4.54 (s, 1H), 4.16 (d, J = 11.5 Hz, 1H), 3.66 (dd, J = 11.3, 3.5 Hz, 1H), 2.48 (s, 3H), 2.46 (ddd, J = 12.8, 7.9, 4.5 Hz, 1H), 2.38 – 2.32 (m, 2H), 2.23 (dt, J = 8.5, 6.3 Hz, 2H), 2.12 (dd, J = 13.4, 8.0 Hz, 1H), 1.68 – 1.55 (m, 4H), 1.50 (d, J = 6.9 Hz, 3H), 1.40 – 1.24 (m, 13H), 1.05 (s, 9H). MS (ESI) m/z = 657.3 calcd. for C33H53N4O6S [M + H]+, found: 657.2.
13-(((S)-1-((2S,4R)-4-Hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl) pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)amino)-13-oxotridecanoic acid (9h).
1H NMR (600 MHz, Chloroform-d) δ 8.69 (s, 1H), 7.41 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.1 Hz, 2H), 7.29 (d, J = 7.8 Hz, 1H), 6.74 (d, J = 8.6 Hz, 1H), 5.09 (p, J = 6.8 Hz, 1H), 4.71 (t, J = 7.9 Hz, 1H), 4.63 (d, J = 9.0 Hz, 1H), 4.52 (s, 1H), 4.18 (d, J = 11.4 Hz, 1H), 3.61 (dd, J = 11.3, 3.2 Hz, 1H), 2.53 (s, 3H), 2.50 (dt, J = 8.0, 4.8 Hz, 1H), 2.34 (q, J = 6.9 Hz, 2H), 2.21 (dt, J = 15.4, 8.1 Hz, 2H), 2.14 – 2.07 (m, 1H), 1.62 (ddd, J = 19.9, 12.9, 5.6 Hz, 5H), 1.48 (d, J = 6.8 Hz, 3H), 1.36 – 1.21 (m, 14H), 1.04 (s, 9H). MS (ESI) m/z = 670.3 calcd. for C33H55N4O6S [M + H]+, found: 670.2.
General procedure for the synthesis of PROTACs PP1 to PP8.
To a stirring solution of compound 8 (1 equiv.) in DCM was successively added acid 9 (1.2 equiv.) with HATU (1.2 equiv.) and TEA (10 equiv.). Upon consumption of amine 8, the mixture was diluted with DCM. The mixture was washed with saturated NH4Cl (2 times), followed by water and brine. The organic portion was dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure. The crude product was purified by silica gel flash column chromatography (DCM/MeOH = 100:1 to 12:1). The obtained mass was further purified by PLC and the average yield of PP1 to PP8 was found to be 52% with respect to the amine 8.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N6-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)adipamide (PP1).
1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.33 (d, J = 2.2 Hz, 1H), 8.06 (d, J = 9.4 Hz, 1H), 7.69 (dd, J = 8.7, 6.3 Hz, 2H), 7.47 – 7.44 (m, 1H), 7.39 – 7.35 (m, 6H), 7.33 – 7.28 (m, 4H), 7.25 – 7.22 (m, 1H), 7.01 (dd, J = 8.3, 1.8 Hz, 2H), 6.96 (d, J = 8.6 Hz, 1H), 6.62 (d, J = 9.2 Hz, 3H), 5.09 (t, J = 7.2 Hz, 1H), 4.74 (q, J = 8.4 Hz, 1H), 4.61 (dd, J = 9.0, 2.9 Hz, 1H), 4.47 (s, 1H), 4.09 (t, J = 9.3 Hz, 1H), 3.90 (s, 1H), 3.65 (d, J = 10.1 Hz, 5H), 3.57 (d, J = 11.1 Hz, 1H), 3.35 (s, 6H), 3.11 (dd, J = 13.8, 5.1 Hz, 2H), 3.03 – 2.99 (m, 1H), 2.49 (d, J = 2.1 Hz, 3H), 2.44 (d, J = 5.4 Hz, 3H), 2.35 (d, J = 15.4 Hz, 7H), 2.20 – 2.14 (m, 3H), 2.14 – 2.06 (m, 5H), 2.00 (d, J = 7.4 Hz, 2H), 1.67 (dd, J = 14.3, 7.6 Hz, 2H), 1.54 (t, J = 6.9 Hz, 5H), 1.46 (d, J = 7.0 Hz, 5H), 1.29 – 1.23 (m, 3H), 1.03 (s, 9H), 0.99 (d, J = 2.9 Hz, 3H). HRMS (ESI) m/z = 1543.5705 calcd. for C76H95ClF3N10O11S4 [M+H]+, found: 1543.5731.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N7-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)heptanediamide (PP2).
1H NMR (600 MHz, Chloroform-d) δ 8.70 (d, J = 1.3 Hz, 1H), 8.36 (t, J = 2.5 Hz, 1H), 8.10 (ddd, J = 9.3, 4.9, 2.3 Hz, 1H), 7.72 (dd, J = 14.0, 8.6 Hz, 2H), 7.46 (d, J = 2.8 Hz, 1H), 7.43 – 7.38 (m, 6H), 7.33 (dd, J = 3.1, 1.7 Hz, 3H), 7.28 (s, 1H), 7.05 – 7.02 (m, 2H), 7.02 – 6.98 (m, 1H), 6.65 (d, J = 9.6 Hz, 3H), 6.36 (s, 1H), 5.12 (t, J = 7.2 Hz, 1H), 4.77 (dd, J = 8.2, 5.6 Hz, 1H), 4.70 (d, J = 8.9 Hz, 1H), 4.66 (d, J = 8.8 Hz, 1H), 4.52 (s, 1H), 4.13 (q, J = 8.8, 6.6 Hz, 1H), 3.93 (s, 1H), 3.71 – 3.65 (m, 4H), 3.62 – 3.59 (m, 1H), 3.31 (d, J = 14.6 Hz, 6H), 3.13 (dd, J = 13.8, 5.1 Hz, 2H), 3.05 – 3.01 (m, 1H), 2.53 (s, 3H), 2.45 (s, 4H), 2.41 – 2.32 (m, 7H), 2.23 – 2.16 (m, 4H), 2.13 (s, 4H), 2.08 – 2.00 (m, 3H), 1.73 – 1.66 (m, 2H), 1.57 (d, J = 7.6 Hz, 4H), 1.50 (dd, J = 6.9, 3.4 Hz, 5H), 1.36 – 1.30 (m, 2H), 1.24 – 1.19 (m, 2H), 1.06 (d, J = 3.9 Hz, 9H), 1.01 (d, J = 7.5 Hz, 3H). HRMS (ESI) m/z = 1557.5862 calcd. for C77H97ClF3N10O11S4 [M+H]+, found: 1557.5873.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N8-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)octanediamide (PP3).
1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.32 – 8.29 (m, 1H), 8.11 – 8.07 (m, 1H), 7.75 (d, J = 8.6 Hz, 1H), 7.72 (d, J = 8.6 Hz, 1H), 7.41 – 7.36 (m, 6H), 7.32 – 7.29 (m, 3H), 7.24 (s, 1H), 7.00 (dd, J = 8.4, 1.6 Hz, 4H), 6.72 (d, J = 8.5 Hz, 2H), 6.62 (d, J = 9.1 Hz, 1H), 6.30 (s, 1H), 6.24 (s, 1H), 5.11 – 5.09 (m, 1H), 4.74 (t, J = 7.5 Hz, 2H), 4.69 (d, J = 8.9 Hz, 1H), 4.51 (s, 1H), 4.15 (d, J = 11.5 Hz, 1H), 3.90 (s, 1H), 3.65 (s, 4H), 3.58 (d, J = 11.4 Hz, 1H), 3.22 (s, 6H), 3.13 – 3.08 (m, 2H), 3.02 (dd, J = 13.9, 7.1 Hz, 2H), 2.51 (d, J = 4.8 Hz, 4H), 2.42 (s, 3H), 2.35 (s, 3H), 2.30 (s, 4H), 2.14 – 2.08 (m, 4H), 2.04 (s, 3H), 1.63 (s, 7H), 1.48 (d, J = 7.0 Hz, 4H), 1.41 (d, J = 7.2 Hz, 3H), 1.19 – 1.11 (m, 5H), 1.05 (d, J = 3.6 Hz, 9H), 0.99 (d, J = 3.1 Hz, 3H). HRMS (ESI) m/z = 1571.6018 calcd. for C78H99ClF3N10O11S4 [M+H]+, found: 1571.6026.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N9-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)nonanediamide (PP4).
1H NMR (600 MHz, Chloroform-d) δ 8.70 (s, 1H), 8.33 (dd, J = 5.2, 2.2 Hz, 1H), 8.12 – 8.09 (m, 1H), 7.77 (dd, J = 13.2, 8.6 Hz, 2H), 7.48 – 7.45 (m, 1H), 7.43 – 7.38 (m, 6H), 7.34 – 7.31 (m, 4H), 7.04 – 7.01 (m, 3H), 6.71 (s, 2H), 6.64 (dd, J = 9.7, 3.6 Hz, 1H), 6.35 (d, J = 9.5 Hz, 1H), 5.12 (td, J = 7.2, 4.0 Hz, 1H), 4.78 – 4.73 (m, 2H), 4.53 (s, 1H), 4.15 (d, J = 10.3 Hz, 1H), 3.93 (s, 2H), 3.67 (d, J = 7.9 Hz, 5H), 3.62 (d, J = 11.4 Hz, 1H), 3.25 (s, 6H), 3.13 (dd, J = 13.8, 5.0 Hz, 2H), 3.04 (dd, J = 13.9, 7.2 Hz, 2H), 2.54 (s, 3H), 2.46 (d, J = 8.7 Hz, 4H), 2.35 (s, 6H), 2.19 – 2.11 (m, 5H), 1.70 (d, J = 6.8 Hz, 5H), 1.57 (d, J = 8.9 Hz, 6H), 1.53 – 1.48 (m, 3H), 1.45 – 1.39 (m, 3H), 1.24 – 1.19 (m, 4H), 1.13 (s, 5H), 1.07 (d, J = 2.0 Hz, 9H), 1.01 (s, 3H). HRMS (ESI) m/z = 1585.6175 calcd. for C79H101ClF3N10O11S4 [M+H]+, found: 1585.6179.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N10-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)decanediamide (PP5).
1H NMR (600 MHz, Chloroform-d) δ 8.70 (s, 1H), 8.33 (d, J = 1.9 Hz, 1H), 8.11 – 8.09 (m, 1H), 7.77 – 7.73 (m, 2H), 7.43 – 7.37 (m, 7H), 7.34 – 7.31 (m, 4H), 7.04 – 7.00 (m, 3H), 6.70 (d, J = 8.5 Hz, 2H), 6.64 (d, J = 9.2 Hz, 1H), 6.32 (dd, J = 14.7, 8.9 Hz, 1H), 5.11 (td, J = 7.3, 3.9 Hz, 1H), 4.78 – 4.68 (m, 3H), 4.53 (s, 1H), 4.16 (d, J = 11.5 Hz, 1H), 3.92 (s, 1H), 3.68 (q, J = 5.8, 5.3 Hz, 4H), 3.64 – 3.60 (m, 1H), 3.32 (s, 1H), 3.25 (s, 5H), 3.13 (dd, J = 13.8, 5.0 Hz, 3H), 3.04 (dd, J = 13.8, 7.2 Hz, 2H), 2.54 (s, 4H), 2.45 (s, 3H), 2.35 (s, 6H), 2.22 – 2.17 (m, 3H), 2.17 – 2.10 (m, 5H), 1.69 (dd, J = 14.5, 8.0 Hz, 3H), 1.58 (dt, J = 22.9, 8.2 Hz, 5H), 1.50 (t, J = 6.5 Hz, 4H), 1.45 (d, J = 8.0 Hz, 1H), 1.23 (s, 4H), 1.15 (dd, J = 14.4, 6.8 Hz, 6H), 1.08 (s, 9H), 1.01 (d, J = 4.1 Hz, 3H). HRMS (ESI) m/z = 1599.6331 calcd. for C80H103ClF3N10O11S4 [M+H]+, found: 1599.6338.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N11-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)undecanediamide (PP6).
1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.30 (d, J = 3.4 Hz, 1H), 8.10 (dd, J = 9.4, 2.3 Hz, 1H), 7.74 (dd, J = 15.9, 8.6 Hz, 3H), 7.41 – 7.35 (m, 6H), 7.30 (ddt, J = 10.8, 6.3, 4.5 Hz, 6H), 7.17 (d, J = 7.9 Hz, 1H), 7.00 (dd, J = 8.2, 1.4 Hz, 4H), 6.71 (t, J = 8.0 Hz, 2H), 6.62 (d, J = 9.3 Hz, 1H), 6.38 (s, 1H), 6.31 (d, J = 8.9 Hz, 1H), 5.08 (dt, J = 10.8, 7.1 Hz, 2H), 4.73 – 4.68 (m, 3H), 4.52 (s, 1H), 4.15 – 4.10 (m, 2H), 3.90 (s, 2H), 3.66 (m, 5H), 3.60 (dd, J = 11.5, 3.5 Hz, 2H), 3.23 (m, 7H), 3.12 – 3.08 (m, 2H), 3.02 (dd, J = 13.9, 7.2 Hz, 2H), 2.51 (d, J = 1.8 Hz, 4H), 2.42 (s, 4H), 2.30 (s, 9H), 2.22 – 2.16 (m, 4H), 2.13 – 2.07 (m, 3H), 1.48 – 1.39 (m, 8H), 1.16 (m, 4H), 1.05 (d, J = 1.6 Hz, 9H), 0.99 (s, 3H). HRMS (ESI) m/z = 1613.6488 calcd for C81H105ClF3N10O11S4 [M+H]+, found: 1613.6500.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N12-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)dodecanediamide (PP7).
1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.31 (t, J = 2.7 Hz, 1H), 8.10 – 8.08 (m, 1H), 7.74 (d, J = 8.5 Hz, 2H), 7.40 – 7.33 (m, 6H), 7.32 – 7.26 (m, 6H), 7.04 – 6.98 (m, 3H), 6.69 (d, J = 8.6 Hz, 2H), 6.62 (d, J = 9.3 Hz, 1H), 6.33 (s, 1H), 5.11 – 5.05 (m, 2H), 4.70 (ddd, J = 12.5, 8.4, 4.4 Hz, 3H), 4.51 (s, 1H), 4.17 – 4.09 (m, 2H), 3.91 (s, 1H), 3.66 (s, 4H), 3.60 (dd, J = 11.5, 3.5 Hz, 2H), 3.32 – 3.17 (m, 7H), 3.12 – 3.07 (m, 2H), 3.02 (dd, J = 13.9, 7.2 Hz, 2H), 2.51 (d, J = 1.2 Hz, 4H), 2.43 (s, 3H), 2.34 (d, J = 22.9 Hz, 6H), 2.18 (d, J = 9.6 Hz, 5H), 2.09 (t, J = 10.1 Hz, 4H), 1.61 (d, J = 7.2 Hz, 5H), 1.51 (t, J = 9.0 Hz, 5H), 1.46 (dd, J = 7.0, 1.5 Hz, 3H), 1.41 (t, J = 7.3 Hz, 2H),1.18 – 1.11 (m, 7H), 1.05 (s, 9H), 0.99 (s, 3H). HRMS (ESI) m/z = 1627.6644 calcd. for C82H107ClF3N10O11S4 [M+H]+, found: 1627.6654.
N1-((4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N13-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)tridecanediamide (PP8).
1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.32 (d, J = 2.0 Hz, 1H), 8.08 (dt, J = 9.3, 2.1 Hz, 1H), 7.72 (d, J = 8.5 Hz, 2H), 7.41 – 7.33 (m, 7H), 7.32 – 7.27 (m, 5H), 7.00 (d, J = 7.7 Hz, 3H), 6.67 (d, J = 8.5 Hz, 2H), 6.62 (d, J = 9.3 Hz, 1H), 6.29 (d, J = 8.8 Hz, 1H), 5.07 (t, J = 7.2 Hz, 1H), 4.72 – 4.69 (m, 1H), 4.65 (dd, J = 8.9, 1.7 Hz, 1H), 4.51 (d, J = 4.0 Hz, 1H), 4.13 (d, J = 11.4 Hz, 1H), 3.90 (s, 1H), 3.66 (s, 4H), 3.62 – 3.58 (m, 1H), 3.27 (s, 4H), 3.18 (s, 2H), 3.10 (dd, J = 13.9, 5.1 Hz, 2H), 3.02 (dd, J = 13.8, 7.2 Hz, 2H), 2.51 (m, 6H), 2.38 (m,8H), 2.18 (m, 8H), 2.09 – 2.02 (m, 3H), 1.68 (s, 2H), 1.60 (d, J = 6.2 Hz, 3H), 1.53 (d, J = 7.7 Hz, 4H), 1.46 (dd, J = 6.9, 1.9 Hz, 3H), 1.23 (d, J = 7.2 Hz, 2H), 1.15 (d, J = 7.2 Hz, 12H), 1.05 (s, 9H), 1.01 – 0.96 (m, 3H). HRMS (ESI) m/z = 1641.6801 calcd. for C83H109ClF3N10O11S4 [M+H]+, found: 1641.6807.
4’-Chloro-6-(methoxycarbonyl)-4-methyl-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-carboxylic acid (10).
To a stirring solution of compoud 3 (10 g, 27.4 mmol) in DCM (60 mL) was added TFA (30 mL) and the mixture was stirred at rt for 12 h. After completion of the reaction, the volatiles were removed under reduced pressure and the crude product (8 g, 26.1 mmol) was used directly in the next step without further purification. 1H NMR (600 MHz, Chloroform-d) δ 7.28 (d, J = 8.4 Hz, 2H), 7.04 (d, J = 8.4 Hz, 2H), 3.48 (s, 3H), 3.01 – 2.94 (m, 1H), 2.52 (ddd, J = 18.6, 8.7, 6.1 Hz, 1H), 2.44 – 2.32 (m, 2H), 2.12 (dt, J = 12.9, 5.8 Hz, 1H), 1.72 (dt, J = 13.5, 6.6 Hz, 1H), 1.36 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 183.6, 169.1, 144.8, 141.1, 133.3, 128.5, 128.3, 126.1, 51.7, 40.9, 35.5, 31.0, 30.8, 24.4. HRMS (ESI) m/z = 331.0713 calcd. for C16H17ClNaO4 [M+Na]+, found: 331.0694.
Methyl (S)-4’-chloro-4-methyl-4-((R)-2-oxo-4-phenyloxazolidine-3-carbonyl)-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-carboxylate (11a) and Methyl (R)-4’-chloro-4-methyl-4-((R)-2-oxo-4-phenyloxazolidine-3-carbonyl)-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-carboxylate (11b).
To a stirring solution of 10 (10 g, 32.4 mmol) in DCM (150 mL) was added oxalyl chloride (4.1 mL, 49 mmol) dropwise at 0 °C followed by the addition of two drops of DMF. After stirring at rt for 8 h, the volatiles were removed under reduced pressure and the crude product was used in the next step without further purification.
To a stirring solution of (R)-4-phenyloxazolidin-2-one (6 g, 37 mmol) in THF (150 mL) at −78 °C was added n-BuLi (2.5 M solution in hexane) (15.6 mL, 39 mmol) dropwise. After stirring for 30 min at the same temperature, a solution of the crude acid chloride obtained above in THF (30 mL) was added dropwise. The reaction temperature was allowed to rise to rt and the reaction was slowly quenched with sat. NH4Cl solution. THF was evaporated under reduced pressure and the residue was diluted with EtOAc. The organic part was washed with water and brine, dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure to give the crude mixture of the two diasteromers. The diastereomers were separated through gradient silica gel flash column chromatography (hexanes/EtOAc = 90:10 to 70:30) to afford 4.1 g 11a and 3.5 g 11b along with 4 g mixture. The mixture was separated by repeating the same chromatography procedure. In combination, 5.6 g of 11a and 5.8 g of 11b were obtained. 11a: 1H NMR (600 MHz, Chloroform-d) δ 7.35 – 7.32 (m, 3H), 7.30 (dd, J = 6.6, 3.1 Hz, 2H), 7.20 (d, J = 8.5 Hz, 2H), 6.75 (d, J = 8.5 Hz, 2H), 5.54 (dd, J = 8.9, 5.5 Hz, 1H), 4.69 (t, J = 8.9 Hz, 1H), 4.22 (dd, J = 8.9, 5.5 Hz, 1H), 3.50 (s, 3H), 3.40 (d, J = 18.0 Hz, 1H), 2.43 (d, J = 18.0 Hz, 1H), 2.35 – 2.26 (m, 3H), 1.79 (dd, J = 12.1, 7.3 Hz, 1H), 1.53 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 176.3, 169.1, 153.0, 144.2, 140.8, 139.1, 133.0, 129.4, 128.7, 128.3, 128.2, 126.1, 126.1, 70.1, 59.8, 51.6, 43.8, 35.2, 32.0, 30.8, 22.2. HRMS (ESI) m/z = 454.1421 calcd. for C25H25ClNO5 [M+H]+, found: 454.1402. 1H NMR of 11b (600 MHz, Chloroform-d) δ 7.37 – 7.30 (m, 5H), 7.19 (d, J = 8.5 Hz, 2H), 6.72 (d, J = 8.5 Hz, 2H), 5.50 (dd, J = 8.7, 3.9 Hz, 1H), 4.72 (t, J = 8.8 Hz, 1H), 4.29 (dd, J = 8.9, 3.9 Hz, 1H), 3.45 (s, 3H), 3.08 (dd, J = 17.6, 1.7 Hz, 1H), 2.58 (dtd, J = 13.2, 5.8, 1.6 Hz, 1H), 2.40 (dt, J = 17.6, 2.7 Hz, 1H), 2.29 (dt, J = 19.4, 5.6 Hz, 1H), 2.12 (dddt, J = 19.3, 8.4, 5.5, 2.4 Hz, 1H), 1.80 (ddd, J = 14.1, 8.6, 6.1 Hz, 1H), 1.53 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 175.6, 168.7, 152.9, 144.8, 141.0, 139.5, 132.9, 129.3, 128.8, 128.2, 128.1, 126.4, 126.2, 70.1, 59.8, 51.5, 44.0, 36.8, 31.2, 30.1, 21.5. HRMS (ESI) m/z = 454.1421 calcd. for C25H25ClNO5 [M+H]+, found: 454.1402.
(S)-(4’-Chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2,4-diyl)dimethanol (4a) and (R)-(4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2,4-diyl)dimethanol (4b).
To a stirring suspension of LAH (3.1 g, 82 mmol) in THF (120 mL) was added a solution of 11b (10.6 g, 23 mmol) in THF (50 mL) dropwise at 0 °C. The reaction stirred at the same temperature for 3 h and then water (3 mL) was added followed by 15% NaOH solution (3 mL). After stirring for 1 h, water (6 mL) was added and stirred for another 2 h. Solid Na2SO4 (3 g) was added to the mixture and stirred for another 1 h. The mixture was filtered through a pad of celite and the solid was washed with EtOAc several times. The combined organic portion was dried over anhydrous NaSO4, filtered, and concentrated under reduced pressure to afford the crude solid. 10% DCM in diethyl ether (25 mL) was added to it and the mixture was stirred for 15 min and then the solvent decanted. This washing was repeated 4 times which leaves the desired diol as a white powder. The combined washed organic portions was evaporated under reduced pressure and the residue was purified by gradient silica gel flash column chromatography (hexanes/EtOAc = 70:30 to 30:70) to afford 4b (5.8 g) in 95% yield. Diol 4a was synthesized from 11a in 93% isolated yield by following the same procedure described for 4b. NMR and MS data of the diols 4a and 4b are in consistent with the racemic compound 4.
tert-Butyl (((R)-4’-chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)carbamate (8b).
Compoud 4b was converted to the corresponding compound 8b by following the same synthetic protocol used to convert diol 4 to compound 8. In this synthetic sequence, all the spectral data of the intermidiates are in consistent with the corresponding recemic intermidiates synthesized en route to compound 8. 1H NMR (600 MHz, Chloroform-d) δ 8.35 (d, J = 2.2 Hz, 1H), 8.09 (dd, J = 9.4, 2.3 Hz, 1H), 7.66 (d, J = 8.6 Hz, 2H), 7.38 – 7.35 (m, 2H), 7.32 – 7.26 (m, 5H), 7.04 (d, J = 8.6 Hz, 1H), 7.02 – 6.97 (m, 2H), 6.74 (d, J = 8.6 Hz, 2H), 6.60 (d, J = 9.4 Hz, 1H), 4.81 (s, 1H), 3.90 (dt, J = 8.8, 4.4 Hz, 1H), 3.72 – 3.61 (m, 4H), 3.27 (t, J = 5.2 Hz, 4H), 3.15 – 3.07 (m, 2H), 3.02 (dt, J = 13.9, 7.4 Hz, 2H), 2.88 (s, 2H), 2.37 (ddddd, J = 40.9, 35.7, 23.8, 17.7, 11.2 Hz, 12H), 2.16 – 2.08 (m, 2H), 1.99 (d, J = 17.4 Hz, 1H), 1.68 (ddt, J = 14.2, 8.3, 5.6 Hz, 1H), 1.55 (dt, J = 13.4, 6.7 Hz, 1H), 1.49 (d, J = 6.6 Hz, 2H), 1.42 (s, 9H), 0.96 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 167.5, 156.6, 153.8, 151.5, 141.1, 137.7, 137.1, 134.8, 134.5, 132.67, 131.2, 130.2, 129.7, 129.4, 128.9, 128.7, 127.5, 127.2, 123.5, 123.0, 121.3, 119.2, 117.0, 113.8, 113.0, 108.6, 79.2, 66.7, 60.4, 54.7, 53.7, 52.5, 50.8, 49.3, 46.8, 39.1, 37.3, 33.3, 31.6, 30.3, 30.1, 28.6, 23.4. HRMS (ESI) m/z = 1089.3667 calcd. for C52H65ClF3N6O8S3 [M + H]+, found: 1089.3647.
tert-butyl (((S)-4’-chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)carbamate (8a).
Compoud 4a was converted to the corresponding compound 8a by following the same synthetic protocol used to convert diol 4 to compound 8. In this synthetic sequence, all the spectral data of the intermidiates are in consistent with the corresponding recemic intermidiates synthesized en route to compound 8. 1H NMR (600 MHz, Chloroform-d) δ 8.35 (d, J = 2.3 Hz, 1H), 8.09 (dd, J = 9.2, 2.4 Hz, 1H), 7.66 (t, J = 7.5 Hz, 2H), 7.39 – 7.34 (m, 2H), 7.32 – 7.26 (m, 5H), 7.04 (d, J = 8.5 Hz, 1H), 6.99 (d, J = 8.5 Hz, 2H), 6.74 (d, J = 8.8 Hz, 2H), 6.59 (d, J = 9.4 Hz, 1H), 4.83 (s, 1H), 3.90 (qt, J = 8.2, 4.4 Hz, 1H), 3.71 – 3.61 (m, 4H), 3.27 (t, J = 5.2 Hz, 4H), 3.10 (dd, J = 13.8, 5.0 Hz, 2H), 3.03 (td, J = 13.8, 11.7, 6.5 Hz, 2H), 2.87 (d, J = 23.8 Hz, 2H), 2.47 – 2.25 (m, 12H), 2.17 – 2.08 (m, 2H), 2.02 – 1.95 (m, 1H), 1.68 (td, J = 8.6, 4.3 Hz, 1H), 1.55 (dt, J = 13.4, 6.7 Hz, 1H), 1.50 (s, 2H), 1.42 (d, J = 2.9 Hz, 9H), 0.95 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 167.9, 156.6, 153.6, 151.4, 141.0, 137.6, 137.3, 134.8, 134.3, 132.7, 131.1, 130.2, 129.7, 129.4, 129.2, 128.6, 127.4, 126.9, 123.5, 123.4, 121.3, 119.1, 117.0, 113.8, 113.0, 108.5, 79.1, 66.6, 60.3, 54.7, 53.6, 52.5, 50.7, 49.2, 46.7, 39.1, 37.3, 33.3, 31.6, 30.3, 30.0, 28.6, 23.4. HRMS (ESI) m/z = 1089.3667 calcd. for C52H65ClF3N6O8S3 [M + H]+, found: 1089.3654.
N1-(((S)-4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N10-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)decanediamide (PZ703a) and N1-(((R)-4’-Chloro-4-methyl-6-((4-(4-(((4-(((R)-4-morpholino-1-(phenylthio)butan-2-yl)amino)-3-((trifluoromethyl)sulfonyl)phenyl)sulfonyl)carbamoyl)phenyl)piperazin-1-yl)methyl)-2,3,4,5-tetrahydro-[1,1’-biphenyl]-4-yl)methyl)-N10-((S)-1-((2S,4R)-4-hydroxy-2-(((S)-1-(4-(4-methylthiazol-5-yl)phenyl)ethyl)carbamoyl)pyrrolidin-1-yl)-3,3-dimethyl-1-oxobutan-2-yl)decanediamide (PZ703b).
Amine salt 8a/8b was converted to the corresponding PROTACs PZ703a/PZ703b by following the same general synthetic procedure for the synthesis of PROTACs PP1 to PP8. PZ703a: 1H NMR (600 MHz, Chloroform-d) δ 8.69 (s, 1H), 8.33 (s, 1H), 8.10 (ddd, J = 9.0, 6.2, 2.3 Hz, 1H), 7.78 – 7.71 (m, 2H), 7.43 – 7.35 (m, 7H), 7.33 (t, J = 7.4 Hz, 4H), 7.03 (d, J = 8.2 Hz, 3H), 6.67 (dd, J = 32.4, 10.7 Hz, 3H), 6.31 (d, J = 8.8 Hz, 1H), 5.11 (p, J = 7.0 Hz, 1H), 4.73 (dt, J = 24.0, 8.9 Hz, 2H), 4.54 (s, 1H), 4.14 (t, J = 10.8 Hz, 1H), 3.97 – 3.90 (m, 1H), 3.71 – 3.60 (m, 5H), 3.35 – 3.21 (m, 5H), 3.08 (ddd, J = 53.3, 13.8, 6.2 Hz, 5H), 2.64 (s, 2H), 2.54 – 2.53 (m, 3H), 2.50 (dd, J = 13.7, 5.2 Hz, 2H), 2.47 – 2.43 (m, 2H), 2.41 – 2.29 (m, 7H), 2.24 (s, 1H), 2.24 – 2.17 (m, 3H), 2.11 (dt, J = 13.8, 6.9 Hz, 5H), 1.70 (dt, J = 13.6, 6.4 Hz, 1H), 1.56 (dq, J = 21.1, 7.0 Hz, 5H), 1.48 (dd, J = 14.6, 7.2 Hz, 5H), 1.23 – 1.11 (m, 8H), 1.07 (s, 9H), 1.02 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 174.2, 173.9, 172.1, 170.1, 153.3, 151.4, 150.5, 148.6, 143.5, 140.6, 137.4, 134.9, 134.0, 133.1, 131.8, 131.2, 131.0, 130.4, 129.7, 129.6, 129.4, 129.0, 127.5, 126.6, 123.6, 121.4, 119.2, 117.1, 114.1, 113.0, 108.6, 70.1, 66.9, 60.2, 58.9, 57.6, 57.1, 54.7, 53.8, 52.4, 50.8, 49.0, 48.5, 46.3, 39.2, 37.0, 36.8, 36.6, 36.1, 35.6, 33.6, 32.2, 30.4, 30.3, 29.3, 29.3, 29.2, 29.1, 26.7, 26.1, 25.7, 22.4, 22.4, 16.3. HRMS (ESI) m/z = 1599.6331 calcd. for C80H103ClF3N10O11S4 [M+H]+, found: 1599.6303. PZ703b: 1H NMR (600 MHz, Chloroform-d) δ 8.67 (s, 1H), 8.31 (d, J = 1.9 Hz, 1H), 8.07 (d, J = 8.9 Hz, 1H), 7.72 (d, J = 8.8 Hz, 2H), 7.38 (dd, J = 13.3, 7.8 Hz, 7H), 7.32 – 7.28 (m, 4H), 6.99 (dd, J = 17.0, 8.4 Hz, 3H), 6.69 – 6.58 (m, 3H), 6.27 (d, J = 8.7 Hz, 1H), 5.09 (p, J = 7.2 Hz, 1H), 4.72 (t, J = 8.0 Hz, 1H), 4.66 (d, J = 8.9 Hz, 1H), 4.50 (s, 1H), 4.15 – 4.08 (m, 1H), 3.90 (dt, J = 9.0, 4.7 Hz, 1H), 3.70 – 3.62 (m, 4H), 3.60 (dd, J = 11.4, 3.6 Hz, 1H), 3.33 (t, J = 7.1 Hz, 1H), 3.23 (s, 4H), 3.10 (dd, J = 13.8, 5.1 Hz, 3H), 3.01 (dd, J = 13.8, 7.2 Hz, 1H), 2.58 (d, J = 43.5 Hz, 2H), 2.51 (d, J = 1.4 Hz, 3H), 2.50 – 2.27 (m, 11H), 2.17 (t, J = 7.6 Hz, 3H), 2.10 (dd, J = 11.9, 8.0 Hz, 4H), 1.70 – 1.63 (m, 1H), 1.58 – 1.51 (m, 4H), 1.47 (dd, J = 6.9, 1.9 Hz, 3H), 1.43 (d, J = 7.4 Hz, 2H), 1.33 – 1.06 (m, 12H), 1.05 (d, J = 2.8 Hz, 9H), 0.98 (s, 3H). 13C NMR (151 MHz, Chloroform-d) δ 174.2, 173.9, 172.1, 170.0, 153.4, 151.5, 150.5, 148.6, 143.4, 140.6, 137.5, 134.9, 133.1, 131.8, 131.2, 131.0, 130.4, 129.7, 129.6, 129.4, 129.0, 127.5, 126.6, 123.6, 121.4, 119.2, 117.1, 114.0, 113.0, 108.6, 70.1, 67.0, 60.2, 58.8, 57.6, 57.1, 54.7, 53.8, 52.4, 50.8, 49.0, 48.6, 46.3, 39.3, 37.0, 36.9, 36.6, 35.9, 35.54, 33.53, 32.2, 30.4, 30.4, 29.3, 29.3, 29.19, 29.17, 26.7, 26.1, 25.7, 22.6, 22.4, 16.3. HRMS (ESI) m/z = 1599.6331 calcd. for C80H103ClF3N10O11S4 [M+H]+, found: 1599.6296.
(S)-4-(Hydroxymethyl)-4-methylcyclohex-2-en-1-one (13).
To a stirring solution of compound 1213 (10 g, 24 mmol) in THF (120 mL) was added 2N HCl (24 mL) and the mixture was stirred at rt for 10 h. Upon consumption of the starting material, the reaction was quenched with the solid Na2CO3 powder and then THF was removed under redcuced pressure. The crude product was dissolved in EtOAc (200 mL) and then washed with water and brine. The organic part was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The crude oil was purified by silica gel flash column chromatography (hexanes/EtOAc = 70:30) to afford the desired alcohol 13 (2.35 g, 16.8 mmol) as a clear oil in 95% yield. 1H NMR (600 MHz, Chloroform-d) δ 7.26 (s, 0H), 6.73 (d, J = 10.2 Hz, 2H), 5.98 (d, J = 10.2 Hz, 2H), 3.58 (d, J = 10.7 Hz, 2H), 3.51 (d, J = 10.7 Hz, 2H), 2.53 – 2.46 (m, 4H), 2.13 – 2.03 (m, 3H), 1.76 (dt, J = 12.5, 5.7 Hz, 3H), 1.15 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 200.3, 156.8, 129.0, 69.8, 38.4, 34.0, 30.9, 22.0. HRMS (ESI) m/z = 141.0916 calcd. for C8H13O2 [M+H]+, found: 141.0904.
Ethyl (R)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2-hydroxy-5-methylcyclohex-1-ene-1-carboxylate (14).
Step one: synthesis of (S)-4-(((tert-Butyldimethylsilyl)oxy)methyl)-4-methylcyclohex-2-en-1-one.
To a stirring solution of alcohol 13 (2.35 g, 16.8 mmol) in DCM (65 mL) was added imidazole (2.28 g, 33.6 mmol) and TBSCl (3 g, 20.16 mmol) at 0 °C. After stirring the reaction at rt for 3 h, the mixture was diluted with DCM (100 mL) and was washed with saturated NaHCO3 and brine. The organic part was dried over anhydrous NaSO4, filtered, and concentrated to dryness. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 90:10) to afford the desired TBS-ether (3.9 g, 15.4 mmol) as a clear oil in 92% yield. 1H NMR (600 MHz, Chloroform-d) δ 6.71 (d, J = 10.2 Hz, 1H), 5.93 (d, J = 10.2 Hz, 1H), 3.49 – 3.38 (m, 2H), 2.52 – 2.40 (m, 2H), 2.05 – 1.97 (m, 1H), 1.76 – 1.67 (m, 1H), 1.12 (s, 3H), 0.89 (s, 9H), 0.04 (s, 6H). 13C NMR (151 MHz, Chloroform-d) δ 199.9, 156.6, 128.7, 70.2, 38.5, 34.2, 31.3, 26.0, 25.9, 22.3, 18.4, −5.37, −5.38. HRMS (ESI) m/z = 255.1780 calcd. for C14H27O2Si [M+H]+, found: 255.1765.
Step two: synthesis of ethyl (S)-5-(((tert-butyldimethylsilyl)oxy)methyl)-2-hydroxy-5-methylcyclohexa-1,3-diene-1-carboxylate.
To a stirring solution of the TBS-ether from above (3.9 g, 15.4 mmol) in THF (40 mL) and HMPA (4.0 mL, 23.1 mmol) was added 1N LDA in THF (18.5 mL) dropwise at −78 °C. To the mixture ethyl cyanoformate (9.1 mL, 92.4 mmol) was added at the same temperature. The reaction was slowly allowed to worm to 0 °C and was then quenched with saturated NH4Cl solution. THF was removed under reduced pressure and the residue was diluted with EtOAc (200 mL). The organic portion was washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The crude product was used in the next step without further purification. The aqueous part was mixed with excess 30% H2O2 solution and kept overnight to neutralize toxic cyanide and then discarded.
Step three: synthesis of compound 14.
The crude product from above was mixed with EtOAc (60 mL) and to this mixture 10% Pd/C (195 mg) was added. The mixture was purged with H2 gas for 10 min and then the mixture was stirred under H2 balloon for 5 h. After disapprence of the starting material, the reaction mxture was filtered through a pad of celite and washed with EtOAc. The filtrate was evaporated under reduced pressure to afford the crude product which was purified by silica gel flash column chromatography (hexanes/EtOAc = 90:10) to afford the desired enol ether 14 (4 g, 12.32 mmol) in 80% yield in two steps. 1H NMR (600 MHz, Chloroform-d) δ 12.23 (s, 1H), 4.20 (qd, J = 7.1, 2.8 Hz, 2H), 3.35 (d, J = 9.5 Hz, 1H), 3.29 (d, J = 9.5 Hz, 1H), 2.27 (t, J = 6.5 Hz, 2H), 2.11 (d, J = 16.0 Hz, 1H), 1.96 – 1.89 (m, 1H), 1.61 (dt, J = 14.2, 7.3 Hz, 1H), 1.44 – 1.38 (m, 1H), 1.30 (t, J = 7.1 Hz, 3H), 0.91 (s, 3H), 0.89 (s, 9H), 0.02 (d, J = 1.0 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 173.1, 171.4, 96.2, 70.5, 60.3, 34.3, 31.3, 29.5, 26.2, 26.1, 22.7, 18.5, 14.4, −5.3, −5.4. HRMS (ESI) m/z = 329.2148 calcd. for C17H33O4Si [M+H]+, found: 329.2131.
Ethyl (R)-4-(((tert-butyldimethylsilyl)oxy)methyl)-4’-chloro-4-methyl-3,4,5,6-tetrahydro-[1,1’-biphenyl]-2-carboxylate (15).
To a stirring solution of compound 14 (4 g, 12.32 mmol) in DCM (50 mL) was added DIPEA (10.7 mL, 61.6 mmol). The mixture was cooled to −78 °C and Tf2O (2.7 mL, 16 mmol) was added dropwise. The temperature of the reaction was allowed to rise to rt. After stirred for 8 h, the reaction was quenched with water. The organic part was washed with saturated NaHCO3 solution followed by brine. The mixture was dried over Na2SO4 and the volatiles were removed under reduced pressure. To the crude mixture was added 10% EtOAc in hexanes solution and stirred over 1 h. The mixture was filtered and the filter cake was washed repeatedly with 10% EtOAc in hexanes solution. The combined filtrate was collected and evaporated under reduced pressure to give a dark brown crude which was used in the next step without further purification.
To the stirring solution of the crude product above in toluene (25 mL) and EtOH (12 mL) was added 2N Na2CO3 solution (12 mL). The mixture was purged with argon for 15 min and 4-chlorophenylboronic acid (2.3 g, 14.7 mmol) and Pd(PPh3)4 (284 mg, 0.24 mmol) was added. The mixture was heated to 90 °C and the reaction was completed in 7 h. Organic solvents were removed under reduced pressure and the residue was diluted with EtOAc (150 mL). The above mixture was washed with water and brine, dried over anhydrous Na2SO4, filtered, and concentrate under reduced pressure to dryness. The crude product was purified by silica gel flash column chromatography (hexanes/EtOAc = 10:1) to afford the desired compound 15 (4.67 g, 11 mmol) in 90% yield in two steps. 1H NMR (600 MHz, Chloroform-d) δ 7.27 (d, J = 8.5 Hz, 2H), 7.06 (d, J = 8.4 Hz, 2H), 3.90 (q, J = 7.1 Hz, 2H), 3.43 – 3.32 (m, 2H), 2.38 – 2.30 (m, 3H), 2.14 – 2.09 (m, 1H), 1.67 (dd, J = 13.6, 6.7 Hz, 1H), 1.46 – 1.39 (m, 1H), 0.96 (s, 3H), 0.90 (s, 9H), 0.05 (d, J = 3.3 Hz, 6H). 13C NMR (151 MHz, Chloroform-d) δ 169.6, 143.8, 141.9, 132.9, 128.4, 128.3, 127.5, 70.8, 60.3, 35.4, 33.9, 30.4, 30.1, 26.1, 22.3, 18.5, 13.8, −5.3, −5.3. HRMS (ESI) m/z = 423.2122 calcd. for C23H36ClO3Si [M+H]+, found: 423.2102.
(R)-(4′-Chloro-4-methyl-3,4,5,6-tetrahydro-[1,1′-biphenyl]-2,4-diyl)dimethanol (4b).
To a stirring solution of compound 15 (4.67 g, 11 mmol) in toluene (50 mL) at 0 °C was added 1M DIBALH in THF (24 mL) dropwise and the reaction was stirred at rt for 3 h. After completion, the reaction temperature was again lowered to 0 °C and quenched with saturated Rochelle’s salt solution. The mixture was diluted with diethyl ether and then filtered through a pad of celite. The filtrate was evaporated under reduced pressure and the crude product was used in the next step without further purication.
The crude product from above was dissolved in THF (40 mL) followed by the addition of 1M TBAF solution in THF (13.2 mL) at 0 °C. The mixture was stirred at rt for 5 h and then was quenched with saturated NH4Cl solution. Organic solvent was removed under vacuum and the residue was diluted with EtOAc (150 mL), washed with water and brine. The organic portion was dried over anhydrous Na2SO4, filtered, and concentrated to dryness. The solid crude was stirred with 10% DCM in diethyl ether solution and then filtered. The filter cake was washed with same solution for another 3 times to get the pure white powder 4b (2.48 g ,9.35 mmol) in 85% yield in two steps. Analytical data was in consistent with the 4b prepared from 11b.
Cell lines and culture.
T-ALL MOLT-4 (Cat. No. CRL-1582), B-ALL RS4;11 (RS4, Cat# CRL-1873), and SCLC NCI-H146 (H146, Cat. No. HTB-173) cell lines were recently purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). SCLC NCI-H211 cell line was kindly gifted from Dr. Fredrick J Kaye’s lab (UF Health Medical Oncology, Gainesville, FL, USA). All the cell lines were cultured in RPMI 1640 media (Life Technologies, Carlsbad, CA, USA), supplemented with 10% FBS (Atlanta Biologicals, Flowery Branch, GA, USA) and 1% penicillin-streptomycin solution (Thermo Fisher Scientific, Waltham, MA, USA). Epithelial kidney HEK 293T (293T, Cat. No. ACS-4500) cell line was purchased from ATCC, and cultured in complete Dulbecco’s modified Eagle medium (DMEM, Cat. no. 12430054, Thermo Fisher Scientific) supplemented with 10% FBS, 1 % penicillin streptomycin solutions. All cell lines were maintained at 37 °C in a humidified 5% CO2 atmosphere.
Cell viability assay.
Cancer cells were seeded in 96 well plates in 100 μL of complete medium (100,000 MOLT-4, RS4;11 cells, and 50,000 NCI-H146, NCI-H211 cells). Celle were treated with compounds after 30 min of seeding. Compound’s dilutions were prepared in complete medium and 100 μL of 2X medium was added to the wells. Each compound was tested with nine concentration in three replicates. Cell viability was measured after 48 h treatment using tetrazolium-based MTS reagent (Cat. No. G1111, Promega, Madison, WI, USA) according to the manufacturer’s instructions. The data were expressed as average percentage cell viability and fitted in non-linear regression curves using GraphPad Prism 7 (GraphPad Software, La Jolla, CA, USA).
Immunoblotting.
Cells were collected and lysed in Lysis buffer (Cat. No. BP-115DG, Boston Bio Products, Ashland, MA, USA) containing protease and phosphatase inhibitor cocktails (Cat. No. PPC1010, Sigma-Aldrich, St. Louis, MO, USA). The equal amount of protein lysates was separated on pre-casted 4-20% Tris-Glycine gels (Mini-PROTEAN TGX, Cat. No. 456-1094, Bio-Rad, Hercules, CA, USA). Thereafter, the proteins were transferred to PVDF membranes (Cat. No. LC2002, MilliporeSigma, Billerica, MA, USA). The membranes were blocked with 5% w/v non-fat dry milk in TBS + Tween-20 (0.1% v/v), and then probed with primary antibodies for overnight at 4 °C. Next day, the membranes were washed and incubated with appropriate horse radish peroxidase (HRP)-conjugated secondary antibodies. Primary antibodies against BCL-XL (Cat. No. 2762), BCL-2 (Cat. No. 4223), MCL-1 (Cat. No. 5453), Bim (Cat. No. 2933), caspase-3 (Cat. No. 9662), cleaved caspase-3 (Cat. No. 9661), cleaved- PARP (Cat. No. 5625), PARP (Cat. No. 9532), β-actin (Cat. No. 4970), and the secondary HRP-linked antibody (Cat. No. 7074) were purchased from Cell Signaling Technology (Danvers, MA, USA). The signal was detected using ECL substrate (Cat. No. WBKLS0500, MilliporeSigma, Billerica, MA, USA) and captured on ChemiDoc MP Imaging System (Bio-Rad, Hercules, CA, USA). The band intensities were calculated on ImageJ software and normalized to equal loading control β-actin.
Human platelet isolation and viability assays.
Platelet rich plasma (PRP) was purchased from Life South Community Blood Centers (Gainesville, FL, USA). PRP was transferred into a 50 mL tube containing 5 mL acid citrate buffer (Cat. No. sc-214744, Santa Cruz Biotechnology). To prevent clotting, prostaglandin E1 (PGE1, Cat. No. sc-201223A, Santa Cruz Biotechnology, Dallas, TX, USA) and apyrase (Cat. No. A6237, Sigma-Aldrich, St. Louis, MO, USA) were added to final concentrations of 1 μM and 0.2 units/mL, respectively. After gently mixing the solution, platelets were pelleted by centrifugation at 1200 g for 10 min without break. Pelleted platelets were gently washed without disrupting platelets in 2 mL HEPES Tyrode’s buffer (Cat. No. PY-921WB, Boston BioProducts, Ashland, MA, USA) containing 1 μM PGE1 and 0.2 units/mL apyrase. After washing, pellets were slowly suspended in 10 mL HEPES Tyrode’s buffer containing 1 μM PGE1 and 0.2 units/mL apyrase. Then platelets number was counted using a HEMAVET 950FS hematology analyzer (Drew Scientific, Inc., Oxford, CT, USA). For viability assays, platelet number was adjusted to 2×108/mL in HEPES Tyrode’s buffer containing 1 μM PGE1 and 0.2 units/mL apyrase. Each treatment was given in 2 mL platelet suspension in 15 mL polypropylene tubes. The tubes were placed on rotating platform at room temperature and the viability of platelets was measured after 48 h treatment by using the MTS reagent (Cat. No. G1111, Promega, Madison, WI, USA). The data were analyzed by GraphPad Prism 7 software for the calculation of IC50 values.
Annexin-V/PI apoptosis assay using flow cytometry.
MOLT-4 cells were seeded in 12-well plates at a density of 5×104 cells per well and were pretreated with or without QVD for 4 h. After pretreatment, cells were treated with PZ703b (10 nM) for 48 h. After incubation, cells were stained with Annexin V-Alexa Fluor 647 (1:50, Cat. No. 640912, BioLegend, San Diego, CA, USA) and propidium iodide (PI, 10 μg/mL, Cat. No. 421301, BioLegend, San Diego, CA, USA) at room temperature for 30 min in dark condition and were analyzed using a Cytek Aurora flow cytometer and the Spectraflo software (Cytek, Fremont, CA, USA).
AlphaScreen for the determination of binary and ternary binding affinities.
To test the binary/ternary binding affinities of compounds to BCL-2/BCL-XL, AlphaScreen displacement binding assay was performed to test the capabilities of testing compounds to displace a Biotin-tagged Bim (Biotin-MRPEIWIAQELRRIGDEFNA) or Bad (Biotin-LWAAQRYGRELRRMSDEFEGSFKGL) from BCL-2 or BCL-XL for BCL-2 and BCL-XL binding affinities, respectively. All reagents were diluted in assay buffer of 25 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% BSA, and 0.005% tween 20 prior incubation. To a 96-well PCR plate (VWR, Cat. No. 82006-636) was added 5 μL Biotin-tagged Bim (80 nM) or Bad (16 nM), 4 μL series diluted compounds solution, 16 μL buffer (for binary binding) or 16 μL 100 μM GST-VCB [GST-tagged VHL–elongin C–elongin B–Cul2 complex (Cat. No. 029641, US Biological) for ternary binding] and 5 μL 6×His tagged protein [8 nM BCL-2 (Sigma, Cat. No. SRP0186) or 4 nM BCL-XL (Sigma, Cat. No. SRP0187)]. After incubating at room temperature for 2 h, 5 μL of 160 μg/mL of anti-6×His acceptor beads (Cat. No. AL128M, PerkinElmer) was added and the mixture was incubated at room temperature for 1 h. 5 μL of 160 μg/mL of streptavidin donor beads (Cat. No. 6760002, PerkinElmer) was then added under subdued light and the mixture was incubated at dark for 30 min before being transferred to two adjacent wells (17 μL each) of 384-well white OptiPlate (Cat. No. 6008280, PerkinElmer). The luminescence signal was detected on a Biotek’s Synergy Neo2 multi-mode plate reader installed with an AphaScreen filter cube. The Ki values of each compounds were calculated by fitting data into “Binding-competitive, One site-Fit Ki” function in GraphPad Prism based on concentrations of Biotin-peptide used and experimentally determined Kd values of each 6×His protein and Biotin-Peptide pair.
AlphaLISA assay for evaluating the ternary complex formation.
AlphaLISA assay was used to evaluate the in vitro ternary complex formation between potential target proteins [BCL-XL (His-tagged, Cat. No. SRP0187, Sigma-Aldrich), BCL-2 (His-tagged, Cat. No. SRP0186, Sigma-Aldrich),10a or MCL-1 (His-tagged, Cat. No. 40742, Bioscience; dialyzed to remove imidazole)], PROTAC and VCB complex [(GST-tagged VHL–elongin B–elongin C–Cul2 complex (Cat. No. 029641, US Biological)]. Assays were performed at room temperature. All reagents were diluted in assay buffer of 25 mM HEPES, pH 7.5, 100 mM NaCl, 0.1% BSA, and 0.005% tween 20. To a 96-well PCR plate was added 10 μl 6×His tagged protein (BCL-XL, BCL-2, or MCL-1, 20 nM, 10 μL series diluted compounds solution (4× dilution) and 10 μL of GST-VCB (20 nM). After incubating at room temperature for 30 min, 5 μL of alpha glutathione-donor beads (160 μg/mL, Cat. No. 6765300, PerkinElmer) were added to each well under subdued light and the mixture was incubated for 15 min in the dark. Then added 5 μL of anti-6×His acceptor beads (160 μg/mL, Cat. No. AL128M, PerkinElmer) to each well and incubated for additional 45 min in the dark before being transferred to two adjacent wells (17 μL each) of 384-well white OptiPlate (Cat. No. 6008280, PerkinElmer). The luminescence signal was detected on a Biotek’s Synergy Neo2 multi-mode plate reader installed with an AphaScreen filter cube. Intensity values were plotted in Graphpad Prism (9.0) with PROTAC concentration values represented on a log10 scale.
NanoBRET ternary complex formation assay.
The ternary complex formation was evaluated by NanoBRET assay as described in previous studies10a,24 with some modifications. Briefly, HEK 293T cells (8×105) were transfected with lipofectamine (Life Technologies) and 1 μg HaloTag-VHL, 10 ng HiBit-BCL-XL and 10 ng LgBit or 1 μg HaloTag-VHL, 10 ng HiBit-BCL-2 and 10 ng LgBit. After 24 h, 2× 104 transfected cells were seeded into white 96-well tissue culture plates in Gibco™ Opti-MEM™ I Reduced Serum Medium, No Phenol Red (Cat. No. 11-058-021, Fisher) containing 4% FBS with or without HaloTag NanoBRET 618 Ligand (Cat. No. PRN1662, Promega) and incubated overnight at 37 °C, 5% CO2. The following day, different doses of compounds were added into the medium and plates were incubated at 37 °C, 5% CO2, for 6 h. After treatment, NanoBRET™ Nano-Glo® Substrate (Cat. No. N1662, Promega) were added into the medium and the signals were measured as described previously.10a mBRET ratios were calculated following the NanoBRET™ Nano-Glo® Detection System (Cat. No. N1662, Promega).
Co-immunoprecipitation.
Cell pellets were lysed in Pierce IP Lysis Buffer supplemented with protease and phosphatase inhibitors (Cat. No. PPC1010; Sigma). After lysis samples were centrifuged at 15000 rpm for 10 min. The supernatants were collected, and aliquots of 1 mg were precleared with 1 μg of mouse IgG (cat. no. sc-2025; Santa Cruz Biotechnology) and 25 μl of protein A/G-PLUS agarose beads (25% vol/vol; Cat. No. sc-2003; Santa Cruz Biotechnology) for 1 h at 4 °C. After incubation, samples were centrifuged at 3000 rpm for 30 sec and supernatants were collected. To the supernatants 2 μg of anti-BCL-2 (Cat. No. sc-7382; Santa Cruz Biotechnology) or anti-IgG was added and incubated for overnight at 4 °C then samples were incubated with 25 μl of protein A/G-PLUS agarose beads for additional 2 h. The Immunoprecipitates were collected by centrifugation and washed with Pierce IP lysis buffer for 3 times. Samples were prepared using Laemmli’s SDS buffer. Denatured samples were analyzed using SDS-PAGE and immunoblotting.
Supplementary Material
ACKNOWLEDGMENT
This study was supported in part by NIH grants R01CA241191 and R01CA242003.
ABBREVIATIONS
- PROTAC
proteolysis targeting chimera
- BCL-XL
B-cell lymphoma-extra large
- BCL-2
B-cell lymphoma 2
- UPS
ubiquitin-proteasome system
- VHL
von Hippel-Lindau
- VCB
the protein complex of VHL, Elongin C (EloC), and Elongin B (EloB)
- CRBN
cereblon
- ALL
acute lymphoblastic leukemia
- SCLC
small cell lung cancer
- HATU
hexafluorophosphate azabenzotriazole tetramethyluranium
- DIPEA
N,N′-diisopropylethylamine
- TEA
triethylamine
- EDCI
N-ethyl-N’-(3-dimethylaminopropyl)carbodiimide hydrochloride
- NaH
sodium hydride
- MsCl
methane sulfonyl chloride
Footnotes
Supporting Information
Molecular formula strings of all compounds (CSV); Effect of negative control PROTAC on BCL-XL degradation and ternary complex formation; Effect of PZ703b on BCL-XL, BCL-2, and MCL-1 in HEK 293T cells; Effect of PZ703b on NCI-H211 and NCI-H146 cells; Detailed scheme for the synthesis of compound 8, acids 9d-9h, compound 4b, amine 8a/8b, and PZ703-NC; HPLC data; Proton and carbon NMR spectra.
The authors declare the following competing financial interest(s): P.P., D.T., D.L., X.L., P.Z., W.H., Y.Y., X.Z., D.Z., and G.Z. are co-inventors for BCL-XL PROTACs disclosed in this study. D.Z. and G.Z. are co-founders and shareholders of Dialectic Therapeutics, a company that is developing BCL-XL PROTACs to treat cancers.
REFERENCES
- (1).Hanahan D; Weinberg RA Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [DOI] [PubMed] [Google Scholar]
- (2).Singh R; Letai A; Sarosiek K Regulation of apoptosis in health and disease: the balancing act of BCL-2 family proteins Nat. Rev. Mol. Cell Biol 2019, 20, 175–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Igney FH; Krammer PH Death and anti-death: tumour resistance to apoptosis Nat. Rev. Cancer, 2002, 2, 277–288. [DOI] [PubMed] [Google Scholar]
- (4).a) Jan R; Chaudhry GS Adv. Pharm. Bull, 2019, 9, 205–218. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Elmore S Apoptosis: a review of programmed cell death. Toxicol. Pathol, 2007, 35, 495–516. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tang D; Kang R; Berghe TV; Vandenabeele P; Kroemer G The molecular machinery of regulated cell death. Cell Res, 2019, 29, 347–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ashkenazi A; Fairbrother WJ; Leverson JD; Souers AJ Nat. Rev. Drug Discov, From basic apoptosis discoveries to advanced selective BCL-2 family inhibitors. 2017, 16, 273–284. [DOI] [PubMed] [Google Scholar]
- (6).a) Roberts AW; Davids MS; Pagel JM; Kahl BS; Puvvada SD; Gerecitano JF; Kipps TJ; Anderson MA; Brown JR; Gressick L; Wong S; Dunbar M; Zhu M; Desai MB; Cerri E; Enschede SH; Humerickhouse RA; Wierda WG; Seymour JF Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. N. Engl. J. Med, 2016, 4, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jones JA; Mato AR; Wierda WG; Davids MS.; Choi M; Cheson BD; Furman RR; Lamanna N; Barr PM; Zhou L; Chyla B; Salem AH; Verdugo M; Humerickhouse RA; Potluri J; Coutre S; Woyach J; Byrd JC Venetoclax for chronic lymphocytic leukaemia progressing after ibrutinib: an interim analysis of a multicentre, open-label, phase 2 trial. Lancet Oncol, 2018, 19, 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) DiNardo CD; Pratz K; Pullarkat V; Jonas BA; Arellano M; Becker PS; Frankfurt O; Konopleva M; Wei AH; Kantarjian HM; Xu T; Hong WJ; Chyla B; Potluri J; Pollyea DA; Letai A Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood, 2019, 133, 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Trisciuoglio D; Tupone MG; Desideri M; Martile MD; Gabellini C; Buglioni S; Pallocca M; Alessandrini G; D’Aguanno S; Bufalo DD BCL-XL overexpression promotes tumor progression-associated properties. Cell Death Dis, 2017, 8, 3216–3231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).a) Mason KD; Carpinelli MR; Fletcher JI; Collinge JE; Hilton AA; Ellis S; Kelly PN; Ekert PG; Metcalf D; Roberts AW; Huang DCS; Kile BT Programmed anuclear cell death delimits platelet life span. Cell. 2007, 128, 1173–1186. [DOI] [PubMed] [Google Scholar]; b) Zhang H; Nimmer PM; Tahir SK; Chen J; Fryer RM; Hahn KR; Iciek LA; Morgan SJ; Nasarre MC; Nelson R; Preusser LC; Reinhart GA; Smith ML; Rosenberg SH; Elmore SW; Tse C Bcl-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [DOI] [PubMed] [Google Scholar]
- (9).Zhang P; Zhang X; Liu X; Khan S; Zhou D; Zheng G PROTACs are effective in addressing the platelet toxicity associated with BCL-XL inhibitors. Explor. Target Antitumor Ther 2020, 1, 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).a) Khan S; Zhang X; Lv D; Zhang Q; He Y; Zhang P; Liu X; Thummuri D; Yuan Y; Wiegand JS; Pei J; Zhang W; Sharma A; McCurdy CR; Kuruvilla VM; Baran N; Ferrando AA; Kim Y; Rogojina A; Houghton PJ; Huang G; Hromas R; Konopleva M; Zheng G; Zhou D A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity Nat. Med, 2019, 25, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang X; Thummuri D; Liu X; Hu W; Zhang P; Khan S; Yuan Y; Zhou D; Zheng G Discovery of PROTAC BCL-X L degraders as potent anticancer agents with low on-target platelet toxicity. Eur. J. Med. Chem, 2020, 192, 112186–112210. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhang X; Thummuri D; He Y; Liu X; Zhang P; Zhou D; Zheng G Utilizing PROTAC technology to address the on-target platelet toxicity associated with inhibition of BCL-XL. Chem. Commun, 2019, 55, 1476–14768. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang X; He Y; Zhang P; Budamagunta V; Lv D; Thummuri D; Yang Y; Pei J; Yuan Y; Zhou D; Zheng G Discovery of IAP-recruiting BCL-XL PROTACs as potent degraders across multiple cancer cell lines. Eur. J. Med. Chem, 2020, 199, 112397–112408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Lakhani NJ; Rasco DW; Zeng Q; Tang Y; Liang Z; Wang H; Lu M; Chen J; Fu L; Wang C; Yang D; Zhai Y First-in-human study of palcitoclax (APG-1252), a novel dual Bcl-2/Bcl-xL inhibitor, demonstrated advantages in platelet safety while maintaining anticancer effect in U.S. patients with metastatic solid tumors. Am. J. Clin. Oncol, 2020, 38, suppl 3509. [Google Scholar]
- (12).Patterson CM; Balachander SB; Grant I; Pop-Damkov P; Kelly B; McCoull W; Parker J; Giannis M; Hill KJ; Gibbons FD; Hennessy EJ; Kemmitt P; Harmer AR; Gales S; Purbrick S; Redmond S; Skinner M; Graham L; Secrist JP; Schuller AG; Wen S; Adam A; Reimer C; Cidado J; Wild M; Gangl E; Fawell SE; Saeh J; Davies BR; Owen DJ; Ashford MB Commun. Biol 2021, 4, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Tolcher AW; Carneiro BA; Dowlati A; Razak ARA; Chae YK; Villella JA; Coppola S; Englert S; Phillips AC; Souers AJ; Salman Z; Penugonda S; Powderly JD; LoRusso P Am. J. Clin. Oncol, 2021, 39, suppl 3015. [Google Scholar]
- (14).a) Souers AJ; Leverson JD; Boghaert ER; Ackler SL; Catron ND; Chen J; Dayton BD; Ding H; Enschede SH; Fairbrother WJ; Huang DCS; Hymowitz SG; Jin S; Khaw SL; Kovar PJ; Lam LT; Lee J; Maecker HL; Marsh KC; Mason KD; Mitten MJ; Nimmer PM; Oleksijew A; Park CH; Park CM; Phillips DC; Roberts AW; Sampath D; Seymour JF; Smith ML; Sullivan GM; Tahir SK; Tse C; Wendt MD; Xiao Y; Xue JC; Zhang H; Humerickhouse RA; Rosenberg SH; Elmore SW ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med, 2013, 19, 202–208. [DOI] [PubMed] [Google Scholar]; b) Murray JB; Davidson J; Chen I; Davis B; Dokurno P; Graham CJ; Harris R; Jordan A; Matassova N; Pedder C; Ray S; Roughley SD; Smith J; Walmsley C; Wang Y; Whitehead N; Williamson DS; Casara P; Diguarher TL; Hickman J; Stark J; Kotschy A; Geneste O; Hubbard RE Establishing Drug Discovery and Identification of Hit Series for the Anti-apoptotic Proteins, Bcl-2 and Mcl-1. ACS Omega, 2019, 4, 8892–8906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).a) Smith BE; Wang SL; Jaime-Figueroa S; Harbin A; Wang; Hamman BD; Crews CM Differential PROTAC substrate specificity dictated by orientation of recruited E3 ligase. Nat. Commun, 2019, 10, 131–144. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zoppi V; Hughes SJ; Maniaci C; Testa A; Gmaschitz T; Wieshofer C; Koegl M; Riching KM; Daniels DL; Spallarossa A; Ciulli A Iterative Design and Optimization of Initially Inactive Proteolysis Targeting Chimeras (PROTACs) Identify VZ185 as a Potent, Fast, and Selective von Hippel–Lindau (VHL) Based Dual Degrader Probe of BRD9 and BRD7. J. Med. Chem, 2019, 62, 699–726. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wu H; Yang K; Zhang Z; Leisten ED; Li Z; Xie H; Liu J; Smith KA; Novakova Z; Barinka C; Tang W Development of Multifunctional Histone Deacetylase 6 Degraders with Potent Antimyeloma Activity. J. Med. Chem, 2019, 62, 7042–7057. [DOI] [PubMed] [Google Scholar]
- (16).Wang G; Zhang H; Zhou J; Ha C; Pei D; Ding K An Efficient Synthesis of ABT-263, a Novel Inhibitor of Antiapoptotic Bcl-2 Proteins. Synthesis, 2008, 15, 2398–2404. [Google Scholar]
- (17).Schapira M; Calabrese MF; Bullock AN; Crews CM Targeted protein degradation: expanding the toolbox. Nat. Rev. Drug. Discov, 2019, 18, 949–963. [DOI] [PubMed] [Google Scholar]
- (18).Janey JM; Iwama T; Kozmin SA; Rawal VH Racemic and Asymmetric Diels−Alder Reactions of 1-(2-Oxazolidinon-3-yl)-3-siloxy-1,3-butadienes. J. Org. Chem, 2000, 65, 9059–9068. [DOI] [PubMed] [Google Scholar]
- (19).Raina K; Lu J; Qian Y; Altieri M; Gordon D; Rossi AMK; Wang J; Chen X; Dong H; Siu K; Winkler JD; Crew AP; Crews CM; Coleman KG PROTAC-induced BET protein degradation as a therapy for castration-resistant prostate cancer. Proc. Natl Acad. Sci. USA, 2016, 113, 7124–7129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).He Y; Koch R; Budamagunta V; Zhang P; Zhang X(uan).; Khan S; Thummuri D; Ortiz YT; Zhang X(in).; Lv D; Wiegand JS; Li W; Palmer AC; Zheng G; Weinstock DM; Zhou D DT2216-a Bcl-xL-specific degrader is highly active against Bcl-xL-dependent T cell lymphomas. J. Hematol. Oncol. 2020, 13, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Herrant M; Jacquel A; Marchetti S; Belhacene N; Colosetti P; Luciano F; Auberger P Cleavage of Mcl-1 by caspases impaired its ability to counteract Bim-induced apoptosis. Oncogene, 2004, 23, 7863–7873. [DOI] [PubMed] [Google Scholar]
- (22).Galdeano C; Gadd MS; Soares P; Scaffidi S; Molle IV; Birced I; Hewitt S; Dias DM; Ciulli A Structure-Guided Design and Optimization of Small Molecules Targeting the Protein–Protein Interaction between the von Hippel–Lindau (VHL) E3 Ubiquitin Ligase and the Hypoxia Inducible Factor (HIF) Alpha Subunit with in Vitro Nanomolar Affinities. J. Med. Chem, 2014, 57, 8657–8663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Gadd MS; Testa A; Lucas X; Chan K-H; Chen W; Lamont DJ; Zengerle M; Ciulli A Structural Basis of PROTAC Cooperative Recognition for Selective Protein Degradation. Nat. Chem. Biol, 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Riching KM; Mahan S; Corona CR; McDougall M; Vasta JD; Robers MB; Urh M; Daniels DL Quantitative Live-Cell Kinetic Degradation and Mechanistic Profiling of PROTAC Mode of Action. ACS Chem. Biol, 2018, 13, 2758–2770. [DOI] [PubMed] [Google Scholar]
- (25).Alford SE; Kothari A; Loeff FC; Eichhorn JM; Sakurikar N; Goselink HM; Saylors RL; Jedema I; Falkenburg JHF; Chambers TC BH3 Inhibitor Sensitivity and Bcl-2 Dependence in Primary Acute Lymphoblastic Leukemia Cells. Cancer Res, 2015, 75, 1366–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Examples of PROTAC dual-degraders:; a) Teng M; Jiang J; He Z; Kwiatkowski NP; Donovan KA; Mills CE; Victor C; Hatcher JM; Fischer ES; Sorger PK; Zhang T; Gray NS Development of CDK2 and CDK5 Dual Degrader TMX-2172. Angew. Chem. Int. Ed 2020, 59, 13865–13870. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jiang B; Wang ES; Donovan KA; Liang Y; Fischer ES; Zhang T; Gray NS Development of Dual and Selective Degraders of Cyclin-Dependent Kinases 4 and 6. Angew. Chem. Int. Ed, 2019, 58, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.