

Abstract
A subset of Ras proteins, including N-Ras, depend on a palmitoylation/depalmitoylation cycle to regulate their subcellular trafficking and oncogenicity. General lipase inhibitors such as Palmostatin M block N-Ras depalmitoylation, but lack specificity and target several enzymes displaying depalmitoylase activity. Here, we describe ABD957, a potent and selective covalent inhibitor of the ABHD17 family of depalmitoylases, and show that this compound impairs N-Ras depalmitoylation in human acute myeloid leukemia (AML) cells. ABD957 produced partial effects on N-Ras palmitoylation compared to Palmostatin M, but was much more selective across the proteome, reflecting a plasma membrane-delineated action on dynamically palmitoylated proteins. Finally, ABD957 impaired N-Ras signaling and the growth of NRAS-mutant AML cells in a manner that synergizes with MEK inhibition. Our findings uncover a surprisingly restricted role for ABHD17 enzymes as regulators of the N-Ras palmitoylation cycle and suggest that ABHD17 inhibitors may have value as targeted therapies for NRAS-mutant cancers.
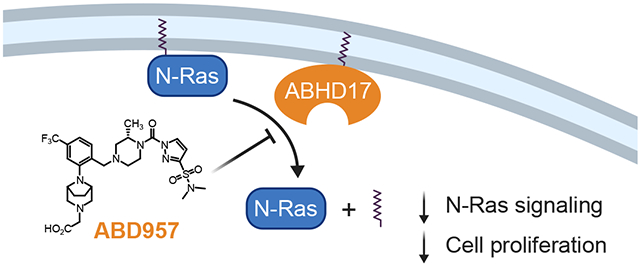
Graphical Abstract
RAS genes are the most common targets of dominant mutations in human cancer1. There are three different RAS genes, which encode four highly homologous proteins (H-Ras, N-Ras, K-Ras4a, and K-Ras4b)1. An elegant approach for directly targeting Ras oncoproteins involves designing covalent inhibitors targeting cysteine 12 of K-RasG12C 2,3. However, it is more challenging to apply this approach to oncogenic amino acid substitutions that introduce side chains lacking reactive nucleophiles.
Ras proteins share high homology throughout most of their protein sequence with the exception of the C-terminal “hypervariable region” (HVR), which contains signals that specify post-translational modifications required for proper subcellular localization4. The HVR of all four isoforms terminates with a CAAX motif, where the cysteine is prenylated by farnesyltransferases. This lipid modification provides weak membrane binding affinity that is stabilized by a second signal motif. For K-Ras4b, this is provided by a polybasic lysine domain5. By contrast, H-Ras, N-Ras, and K-Ras4a are S-palmitoylated at cysteine(s) adjacent to the CAAX motif4,5, and a dynamic cycle of palmitoylation and depalmitoylation mediated by palmitoyl acyl transferase (PAT) and depalmitoylase enzymes, respectively, has been shown to regulate H- and N-Ras trafficking, subcellular localization, and function6,7.
The enzymes involved in depalmitoylating H- and N-Ras are presumed to be serine hydrolases based on studies showing that N-Ras depalmitoylation is blocked by the beta-lactone small molecules palmostatin B (1) and M (2) (Palm B and M)7,8 or hexadecylfluorophosphonate (HDFP, 3)9, as well as earlier work demonstrating that the yeast serine hydrolase APT1 (or, in humans, LYPLA1) can depalmitoylate H-Ras10. While LYPLA1 and LYPLA2 are inhibited by Palm B and M11, selective inhibitors or genetic knockdown of these serine hydrolases do not affect the palmitoylation dynamics of N-Ras in cancer cells12, pointing to other enzymes being involved in this process. Activity-based protein profiling (ABPP) methods were recently used to identify ABHD17 enzymes, a poorly characterized group of three sequence-related serine hydrolases (ABHD17A, B, and C, or the ABHD17s), as additional targets of Palm M and HDFP and showed that these enzymes can depalmitoylate N-Ras12. The ABHD17s are themselves palmitoylated on their N-termini, which promotes localization to the plasma membrane of cells13. Importantly, overexpression of ABHD17A was found to enhance N-Ras depalmitoylation and altered N-Ras subcellular localization in COS-7 cells, and, conversely, triple knockdown of ABHD17A/B/C by RNA interference impaired N-Ras palmitoylation turnover, albeit less effectively than Palm B, in HEK293T cells12.
Previous findings, taken together, support a potential role for ABHD17s in regulating N-Ras palmitoylation. However, ABHD17A, B, and C are all broadly expressed in most cell types, which makes characterization of their collective contribution to N-Ras palmitoylation challenging using genetic methods. Additionally, the studies performed to date relating ABHD17s to N-Ras depalmitoylation have occurred in heterologous cell types that do not depend on N-Ras as an oncogenic driver. Accordingly, it remains unknown whether ABHD17s regulate N-Ras palmitoylation in relevant biological systems and if disrupting these enzymes will affect the growth of NRAS mutant cancer cells. Finally, the broader potential substrate scope of ABHD17 enzymes beyond N-Ras and a handful of other palmitoylated proteins (e.g. PSD-9512,14) remains unknown.
To address the aforementioned knowledge gaps, we describe herein a potent, selective, and cell-active covalent inhibitor of ABHD17s. We find that this compound – termed ABD957 (4) – inhibits all three ABHD17s and partially impairs N-Ras depalmitoylation, ERK phosphorylation, and the growth of NRAS mutant cancer cells. ABD957 stabilizes N-Ras palmitoylation less completely than Palm M, but is much more selective across the proteome, being restricted in its effects to plasma membrane-associated, dynamically palmitoylated proteins. ABD957 and Palm M also differentially impacted the subcellular distribution of N-Ras, leading to the accumulation of this protein at the plasma membrane and intracellular membranes, respectively. Finally, we show that ABD957 synergizes with MEK inhibition to block NRAS mutant cancer cell growth and that ABD957 pharmacological effects are abrogated in cells lacking ABHD17A and ABHD17B. These data, taken together, indicate that ABHD17s perform a specialized function as plasma membrane-delineated S-depalmitoylases and that the chemical inhibition of these enzymes attenuates dynamic N-Ras depalmitoylation, resulting in impaired signaling and growth of NRAS mutant cancer cells.
Results
Discovery and characterization of ABHD17 inhibitors.
The palmostatins (Palm B and Palm M)7,8 and HDFP9 (Fig. 1a) are useful pharmacological tools for studying protein palmitoylation through blockade of depalmitoylase enzymes and, to date, remain the only reported inhibitors of ABHD17A/B/C12. HDFP is a broad-spectrum serine hydrolase inhibitor, targeting multiple depalmitoylases including LYPLA1/2, PPT1, ABHD10 and ABHD17A/B/C9. This compound9,15,16, along with other long-chain FPs17,18, have been shown to preserve the palmitoylation state of several dynamically palmitoylated proteins, including Ras proteins9,12, in mammalian cells. Palm B and M also inhibit several other serine hydrolases11,12 and this lack of selectivity, along with the presence of a metabolically labile beta-lactone, has limited the broad utility of these compounds in biological systems. Such factors motivated us to develop a more advanced chemical probe that selectively inhibits ABHD17 enzymes.
Figure 1. Discovery and characterization of ABD957 - a potent and selective inhibitor of the ABHD17 enzymes.
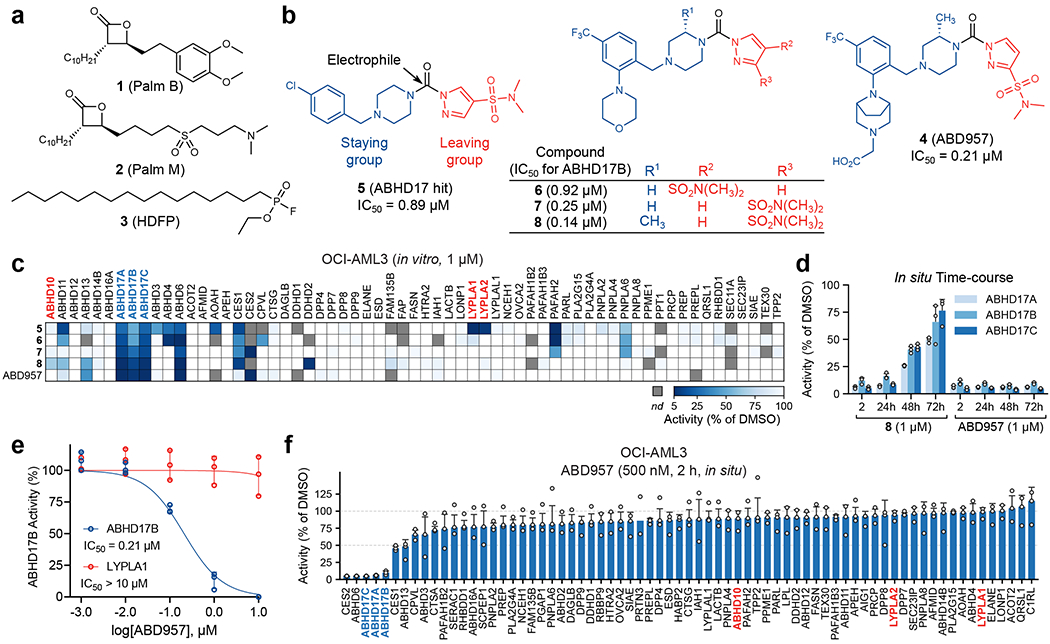
a, Structures of broad-spectrum serine hydrolase inhibitors Palm B, Palm M and HDFP. b, Structures of pyrazole urea class of ABHD17 inhibitors discovered herein, highlighting proposed site of covalent reactivity with the active-site serine residue of ABHD17 enzymes. c, MS-ABPP data of serine hydrolase activities in the particulate fraction of OCI-AML3 proteomes treated with compounds 5-8 and ABD957 (1 μM, 30 min). See Extended Data Fig. 1c for MS-ABPP data of compounds tested at 10 μM and Supplementary Dataset 1 for detailed proteomic data. MS-ABPP data are from single experiments performed at the indicated concentrations for each compound. d, Targeted MS-ABPP data for an in situ time-course of ABHD17A/B/C inhibition by compounds 8 and ABD957 (1 μM) in ONK cells, revealing persistent inhibition of ABHD17A/B/C over 72 h in cells treated with ABD957, but not with compound 8. Data are plotted from biological replicates, and error bars represent s.d. and center around the average (n = 3 independent experiments). e, IC50 curves for ABD957 inhibition of human ABHD17B (recombinantly expressed) and LYPLA1 (endogenous) activity in lysates of HEK293T cells (in vitro) measured by gel-ABPP. Data represent average values ± s.d. (n = 3 independent experiments). f, In situ MS-ABPP data for ABD957 (500 nM, 2 h) in OCI-AML3 cells confirming ABHD17A/B/C inhibition and selectivity across the majority of quantified serine hydrolases, including LYPLA1, LYPLA2, and ABHD10. Data plotted represent the median from biological replicates, and error bars represent s.d. (n = 3 independent experiments).
HDFP and palmostatins feature reactive electrophilic centers that inhibit serine hydrolases through covalent modification of the catalytic serine residue. While this feature likely contributes to a lack of specificity across the serine hydrolase class, more tempered electrophiles, such as carbamates and ureas, have been shown to covalently inhibit individual serine hydrolases with excellent potency and selectivity19–21. The evaluation of candidate inhibitors of serine hydrolases has also benefited from the chemical proteomic technology activity-based protein profiling (ABPP), wherein activity-based probes showing broad reactivity with serine hydrolases are used to measure target engagement and selectivity of inhibitors in native biological systems22. We accordingly screened a serine hydrolase-directed compound library internally developed at Lundbeck La Jolla Research Center, Inc. (~5,000 compounds screened in total) by gel-based ABPP in native mouse brain proteomes, where ABHD17 enzymes and other serine hydrolases can be visualized using a fluorescently (rhodamine) tagged fluorophosphonate (FP) activity-based probe (Supplementary Table 1). This screen furnished a piperazine-based pyrazole urea hit (compound 5, Fig. 1b) that displayed moderate potency (IC50 [95% CI] = 0.89 [0.71-1.1] μM, Supplementary Table 2 and Extended Data Fig. 1a, b) against human ABHD17B, assayed by gel-based ABPP in stably transduced HEK293T cells.
Compound 5 represented an attractive starting point due to its simple, chemically tractable core and the presence of a moderately electrophilic pyrazole urea group resembling those found in other advanced chemical probes that covalently inhibit serine hydrolases19,21,23. We next used a biotinylated FP probe24 and quantitative, mass spectrometry (MS)-based ABPP to assess the selectivity of 5 in the proteome of the human NRAS mutant acute myeloid leukemia (AML) cell line OCI-AML3 (Supplementary Dataset 1). Experiments performed with 1 and 10 μM of 5 confirmed inhibition of the ABHD17s, as well as several additional serine hydrolases, including LYPLA1 and LYPLA2 (Fig. 1c and Extended Data Fig. 1c). Through iterative ABPP-guided medicinal chemistry, we eliminated LYPLA1/2 activity while simultaneously improving inhibitory potency and selectivity for ABHD17s. Introduction of a morpholino group on the benzyl moiety and replacement of the chlorine with a trifluoromethyl group (compound 6, Fig. 1b), followed by shifting the sulfonamide from the 4- to 3-position on the pyrazole leaving group, decreased potency for LYPLA1 and concurrently enhanced ABHD17B potency (compound 7, Fig. 1b; IC50 [95% CI] = 0.25 [0.20-0.32] μM) (Supplementary Table 2). MS-based ABPP experiments revealed that these modifications also had a favorable impact on overall selectivity, removing off-targets such as ABHD4 and ABHD11 (Fig. 1c and Extended Data Fig. 1c). Reasoning that increasing the steric demand around the electrophile could further improve selectivity20,21, we introduced a methyl substituent on the piperazine staying group adjacent to the urea (compound 8, Fig. 1b), which boosted potency (IC50 [95% CI] = 0.14 [0.11-0.16] μM, Supplementary Table 2) and shifted the selectivity profile - eliminating PAFAH2, but introducing ABHD10, a recently identified mitochondrial depalmitoylase18, as an off-target (Fig. 1c and Extended Data Fig. 1c). Furthermore, 8 suffered from poor metabolic stability in human liver microsomes (480 μL/min/mg, Supplementary Table 2) and, when tested in cells, did not produce sustained inhibition of ABHD17s over 72 h (Fig. 1d), a common endpoint used for cell-based assays that measure, for instance, proliferation.
Additional modifications to 8 furnished the more advanced chemical probe ABD957 (compound 4, Fig. 1b) that displayed much lower microsomal clearance (4.8 μL/min/mg, Supplementary Table 2) while maintaining good potency for ABHD17B (IC50 [95% CI] = 0.21 [0.16-0.28] μM, Fig. 1e, Extended Data Fig. 1b, and Supplementary Table 2) and selectivity, including avoidance of ABHD10 as an off-target (Fig. 1c and Extended Data Fig. 1c). We also performed MS-based ABPP experiments on OCI-AML3 cells treated in situ with ABD957 (500 nM) for 2 h (Fig. 1f), which revealed 90%+ blockade of ABHD17s with good overall selectivity, including no cross-reactivity with LYPLA1, LYPLA2, or ABHD10. While ABD957 inhibited a handful of other serine hydrolases, including CES1/2, ABHD6, and ABHD13, the global selectivity was markedly improved compared to previous ABHD17 inhibitors, such as Palm M and HDFP, which, consistent with previous studies9,12, caused widespread serine hydrolase inhibition in cells (Extended Data Fig. 1d). Also, we identified inactive control compounds – JJH254 (9)25 and ABD298 (10) – that inhibited the majority of off-targets of ABD957, while leaving the ABHD17 enzymes unperturbed (Extended Data Fig. 2a–d). Importantly, ABD957 (1 μM) produced sustained cellular inhibition of ABHD17A/B/C for 72 h (Fig. 1d), indicating that this compound was well-suited for a diverse array of cell-based experiments.
ABD957 attenuates N-Ras depalmitoylation in AML cells.
To assess the impact of ABHD17 inhibition on N-Ras palmitoylation, we adapted a pulse-chase assay for measuring dynamic protein palmitoylation9. We first generated sublines of OCI-AML3 cells in which we used RNA interference to reduce the expression of endogenous human N-Ras and then introduced into these cells either i) a murine GFP-N-RasG12D protein, or ii) a murine GFP-N-RasG12D variant where the palmitoylated HVR sequence was replaced with the non-palmitoylated HVR sequence of K-Ras4b (Supplementary Fig. 1). The two engineered cell lines expressing GFP-N-RasG12D and GFP-N-RasG12D, KRAS HVR proteins – termed “ON” and “ONK”, respectively – were expected to retain (ON) or lose (ONK) dependency on the palmitoylation of mutant N-Ras for signaling and growth (as we have shown in previous studies that an oncogenic form of N-Ras loses transforming activity when the C181 palmitoylation site is mutated26). We next evaluated the effect of ABD957 and other compounds on N-Ras palmitoylation dynamics by pre-incubating ON or ONK cells with compounds or DMSO control for 1 h, then treating cells with the palmitate analog 17-octadecynoic acid (17-ODYA13,27; 20 μM) for 1 h, followed by a 1 h chase period where the media was changed to remove 17-ODYA and supplemented with fresh inhibitor (Extended Data Fig. 3, left and middle panels). The palmitoylation state of N-Ras was then determined by anti-GFP immunoprecipitation, on-bead copper-catalyzed azide-alkyne cycloaddition (CuAAC)28 to a rhodamine-azide (Rh-N3) reporter tag, and SDS-PAGE and in-gel fluorescence scanning (Extended Data Fig. 3, right lower panel). We also used a similar experimental protocol to survey the proteome-wide effects of candidate depalmitoylation inhibitors by performing CuAAC with Rh-N3 on lysates from 17-ODYA-treated cells (Extended Data Fig. 3, right upper panel).
Consistent with previous studies7–9,12, we found that the promiscuous lipase inhibitors Palm M and HDFP near-completely preserved N-Ras palmitoylation following the chase period (Fig. 2a, b). ABD957 also protected N-Ras from depalmitoylation, but to a lesser degree than Palm M or HDFP (Fig. 2a, b). In contrast, the control compounds JJH254 and ABD298 did not affect N-Ras palmitoylation dynamics, either alone or in combination with ABD957 (Fig 2a, b). As expected, palmitoylation signals were not observed in ONK cells (Fig. 2a, c), indicating that the GFP-N-RasG12D, KRAS HVR protein does not undergo palmitoylation.
Figure 2. Effects of inhibitor treatment on the dynamic palmitoylation state of N-Ras.
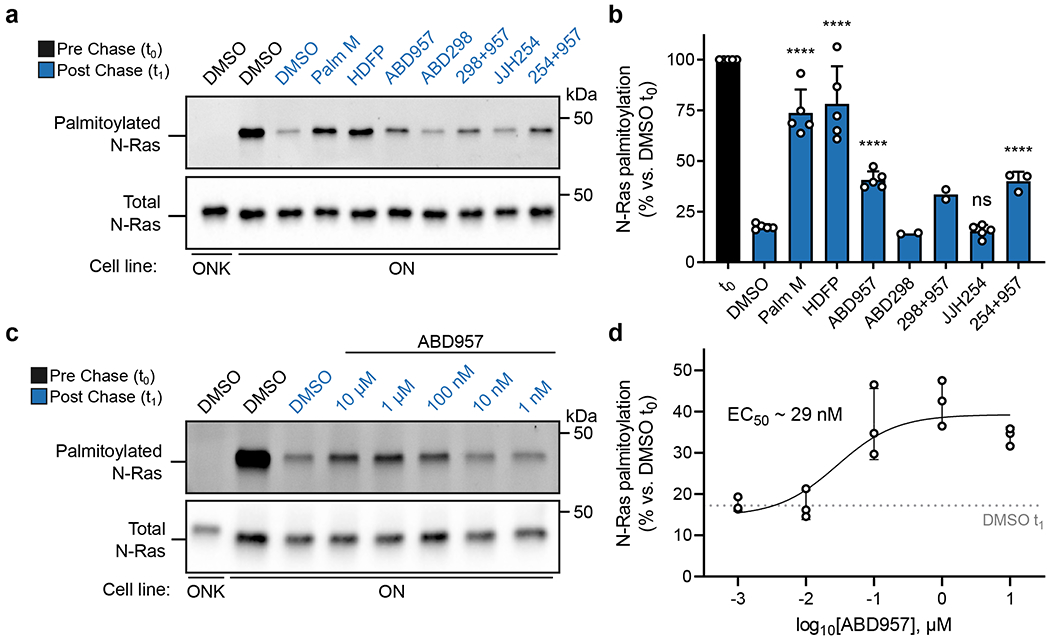
a, Representative gel measuring N-Ras palmitoylation by 17-ODYA comparing effect of treatment with Palm M (10 μM), HDFP (20 μM), ABD957 (500 nM), ABD298 (500 nM), and JJH254 (1 μM) in OCI-AML3 cells stably expressing GFP-N-RasG12D (ON) with GFP-N-RasG12D-KRAS-HVR (ONK) as a control (upper panel). Samples co-treated with ABD957 and ABD298 or JJH254 are abbreviated as 298+957 and 254+957 respectively. Gel is representative of 5 independent experiments (only one including ABD298, three including JJH254, and 5 for all other conditions). N-Ras was immunoprecipitated via GFP and the degree of palmitoylation visualized by rhodamine attached via CuAAC to the alkyne of 17-ODYA. Total N-Ras content was measured by Western blotting of GFP enrichments (lower panel). b, Quantification of inhibitor effects on dynamic palmitoylation. Data represent average values ± s.d. (biological replicates; n = 2 for ABD298 and ABD298 + ABD957 samples, n = 3 for JJH254 + ABD957 samples, and n = 5 for all others). Statistical significance was calculated using unpaired two-tailed Student’s t-test with equal variance, ****P<0.0001 represents significant increase compared to DMSO t1. P values were 4.9×10−6 (Palm M), 8.3×10−5 (HDFP), 2.5×10−6 (ABD957), 0.19 (JJH254), 3.9×10−5 (JJH254 + ABD957). c, Representative gel (from 3 independent experiments) measuring N-Ras palmitoylation by 17-ODYA in the presence of varying concentrations of ABD957 (upper panel). Dotted line represents mean of residual post-chase DMSO (t1) signal. N-Ras was enriched and visualized as described in a. Total N-Ras content was measured by Western blotting of GFP enrichments (lower panel). d, Quantification of concentration-dependent effects of ABD957 on N-Ras palmitoylation. Data are plotted from biological replicates, and error bars represent s.d. and center around the mean (n = 3 independent experiments).
The partial stabilization of N-Ras palmitoylation observed with ABD957 was concentration-dependent with an EC50 value of 29 nM (Fig. 2c, d), which is consistent with the 90%+ inhibition of ABHD17s in cells treated with 500 nM of this compound (Fig. 1f). Initial proteome-wide surveys of palmitoylation dynamics by SDS-PAGE suggested that ABD957 had a more restricted impact on dynamically palmitoylated proteins compared to Palm M or HDFP (Supplementary Fig. 2). Intrigued by these distinct profiles, we next evaluated compounds for effects on global protein palmitoylation by MS-based proteomics.
ABHD17s regulates plasma membrane palmitoylation.
We evaluated the impact of compounds on global palmitoylation dynamics using a similar pulse-chase protocol to that previously described9 and shown in Extended Data Fig. 3, where 17-ODYA-labeled proteins were conjugated by CuAAC to a biotin-N3 tag for streptavidin enrichment followed by quantitative, multiplexed MS-based proteomics using tandem mass tagging (TMT). 17-ODYA-labeled proteins were required to show sensitivity in their enrichment to hydroxylamine treatment (Supplementary Dataset 2) for designation as palmitoylated proteins29. In total, 1227 palmitoylated proteins were identified in OCI-AML3 cells (Supplementary Dataset 2). Consistent with past studies9,16 only a modest subset of these proteins (22 in total), which included N-Ras, displayed clear evidence of dynamic palmitoylation, as reflected by at least three-fold reductions in 17-ODYA modification following the 1 h chase period (t0/t1 ≥ 3) (Fig. 3a, b, green and red proteins rightward of vertical dashed red line; and Supplementary Dataset 3). The palmitoylation states of most of these proteins were stabilized by treatment with Palm M (10 μM) (Fig. 3a, red proteins; and Supplementary Dataset 3). In contrast, ABD957 (500 nM) preserved the palmitoylation state of only a select few proteins, including N-Ras (Fig. 3b, red proteins). Palm M, but not ABD957, also increased the apparent palmitoylation state of an additional set of proteins that did not show evidence of dynamic palmitoylation (Fig. 3a, blue proteins; and Extended Data Fig. 4a). We are unsure of the mechanistic basis for these changes, but they point to a much broader effect of Palm M on the palmitoylated proteome compared to ABD957.
Figure 3. Global palmitoylation effects of Palm M and ABD957 in leukemia cells.
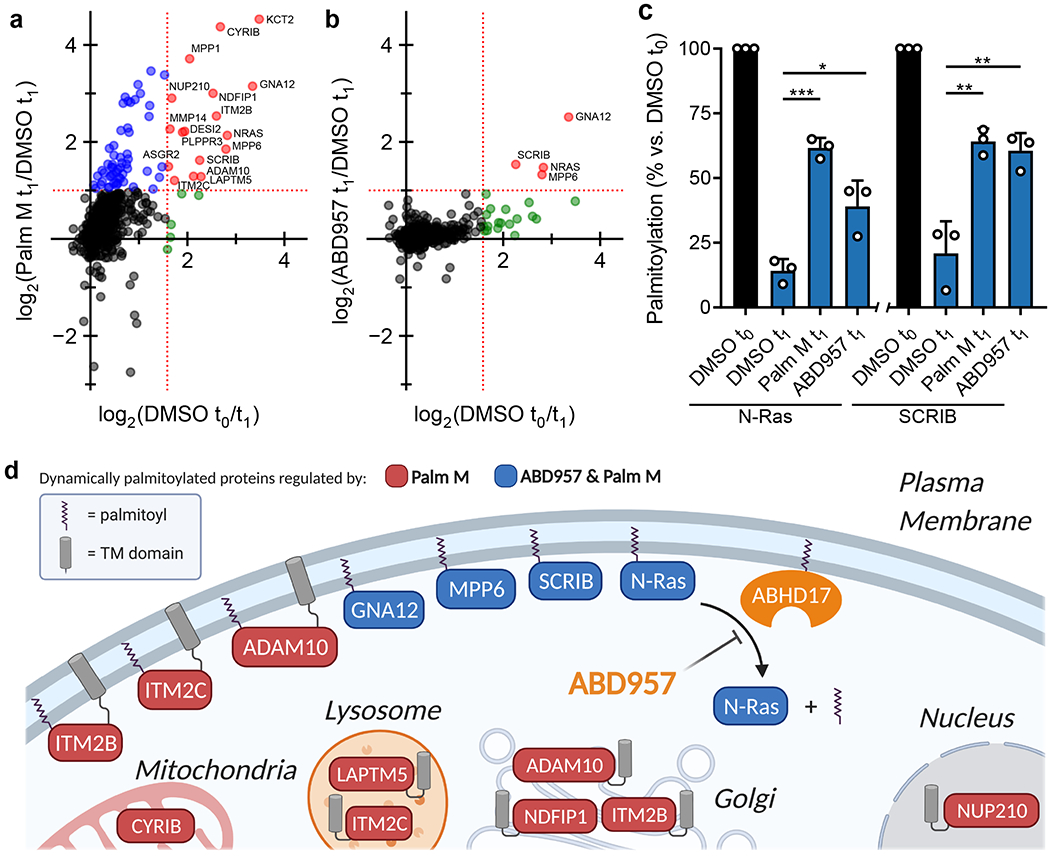
a, b, Mass spectrometry (MS)-based profiling of OCI-AML3 cells as described in Extended Data Fig. 3. Cells were preincubated with Palm M (10 μM) or ABD957 (500 nM) for 1 h, metabolically labeled with 20 μM 17-ODYA for 1 h (t0) and chased with media lacking 17-ODYA for 1 h (t1) containing Palm M (10 μM, a), ABD957 (500 nM, b), or DMSO control. Scatter plots compare log2 ratios of palmitoylated proteins in experiments measuring dynamic palmitoyation (DMSO to/t1; x-axis) versus the effect of inhibitor treatment (inhibitor t1/DMSO t1; y-axis). Proteins shown were designated as palmitoylated based on their sensitivity to hydroxylamine (≥75% reduction in enrichment following hydroxylamine treatment) as reported in Supplementary Dataset 2. Proteins in red are both dynamically palmitoylated (DMSO t0/DMSO t1 ≥ 3 fold) and preserved in their palmitoylation state by inhibitor treatment (inhibitor t1/DMSO t1 ≥ 2 fold), green are dynamically palmitoylated, but not preserved by inhibitor treatment (inhibitor t1/DMSO t1 < 2 fold), and blue are proteins that did not display evidence of dynamic palmitoylation (DMSO t0/DMSO t1 < 3 fold), but showed higher palmitoylation signals following inhibitor treatment (inhibitor t1/DMSO t1 ≥ 2 fold). Red dotted lines represent 2-fold and 3-fold ratio values for y and x-axes respectively. Data represent average values (n = 3 biological replicates). HLA proteins not shown. c, Bar graphs quantifying N-Ras (left) and SCRIB (right) palmitoylation from MS-based proteomic experiments. Data represent average values relative to DMSO t0 ± s.d. (n = 3 biological replicates). Statistical significance was calculated with unpaired two-tailed Student’s t-test with equal variance, *P<0.05, **P<0.01, ***P<0.001 represent significant increase compared to DMSO t1. P values were 1.6×10−4 (Palm M N-Ras), 0.0173 (ABD957 N-Ras), 0.0050 (Palm M SCRIB), 0.0084 (ABD957 SCRIB). d, Cartoon diagram depicting the subcellular localization of selected proteins showing dynamic palmitoylation that was inhibited by Palm M (red) or both Palm M and ABD957 (blue) (created with BioRender.com). Transmembrane domains (TM) are annotated by PSORT II48.
In agreement with our focused palmitoylation assays on N-Ras (Fig. 2), the palmitoylation state of this protein was preserved by both ABD957 and Palm M, with the latter compound showing a greater effect (Fig. 3c). Concordant changes in N-Ras signals were found in both OCI-AML3 and ON cells using either peptides that are unique to N-Ras (relative to other Ras isoforms; for both OCI-AML3 and ON cells) or peptides from the fused GFP protein as a surrogate (for ON cells) (Fig. 3b,c, Extended Data Fig. 4b–d, and Supplementary Dataset 3). We also confirmed using an MS-based proteomic pulse-chase stable isotope labeling protocol30 that, at the time points where ABD957 stabilized N-Ras palmitoylation, the cellular abundance of endogenous N-Ras was unaltered (Extended Data Fig. 5 and Supplementary Dataset 4).
When considering possible reasons why ABD957 did not impact other palmitoylated proteins regulated by Palm M, we noted that the four main proteins affected by ABD957 all reside at the plasma membrane (Fig. 3d) including, in addition to N-Ras, SCRIB (Fig. 3b, c), a scaffolding protein that depends on palmitoylation for plasma membrane localization and function31, MPP6, a palmitoylated guanylate kinase that has been shown to localize to the basolateral plasma membrane of intestinal epithelial cells32, and GNA12, a palmitoylated guanine nucleotide-binding protein that plays a role in Rho signaling33. In contrast, dynamically palmitoylated proteins affected by Palm M, but not ABD957, localized to other subcellular compartments (e.g., Golgi (NDFIP1), nucleus (NUP210), lysosome (LAPTM5), mitochondria (CYRIB)) (Fig. 3d), where they may not interface with plasma membrane-localized ABHD17s. Some possible exceptions included a handful of Palm M-regulated transmembrane proteins (e.g., ADAM10, ITM2B/C) that localize to both intracellular membrane compartments and the plasma membrane34,35. Finally, we also analyzed the effects of depalmitoylation inhibitors on the dynamically palmitoylated proteome of the K-Ras mutant cancer cell line NB-4, which revealed that ABD957 also controlled SCRIB palmitoylation in these cells (Extended Data Fig. 6a and Supplementary Dataset 3). Notably, the dynamic palmitoylation state of wild type N-Ras was only modestly sensitive to ABD957 in NB-4 cells, despite still being fully preserved by Palm M (Extended Data Fig. 6b and Supplementary Dataset 3). Our quantitative proteomic data thus indicate that the ABHD17 enzymes regulate the dynamic palmitoylation state of mutant N-Ras and a discrete set of plasma membrane-localized proteins in leukemia cells.
ABD957 impacts N-Ras localization in leukemia cells.
Previous studies have shown that Palm B and M alter the subcellular localization of N-Ras, leading to redistribution of this protein from the plasma membrane to endomembranes7,8,36. This effect was also shown to depend on the presence of the N-Ras HVR in transduced primary hematopoietic cells36. Using live-cell confocal microscopy, we monitored GFP-N-Ras localization in ON cells following treatment with Palm M or ABD957. Consistent with previous studies7, 16, we found that Palm M promoted the time-dependent accumulation of N-Ras at endomembranes that overlapped with the Golgi marker N-acetylgalactosaminyltransferase (GALNT2) (Fig. 4a, b and Supplementary Fig. 3). ABD957, on the other hand, did not cause intracellular accumulation of GFP-N-Ras (Fig. 4a, b), but instead promoted N-Ras retention at the plasma membrane, as reflected in sustained overlapping distribution with the plasma membrane marker protein wheat germ agglutinin (Fig. 4c–e and Supplementary Video 1a–c). The effect of ABD957 was selective for palmitoylated N-Ras, as it was observed in ON, but not ONK cells (Fig. 4c–e). These cell imaging results indicate that ABHD17 inhibition, in addition to partially protecting the palmitoylation state of N-Ras, leads to retention of this protein at the plasma membrane, where ABHD17s are themselves localized13.
Figure 4. Effect of inhibitor treatment on GFP-N-Ras localization in ON and ONK cells.
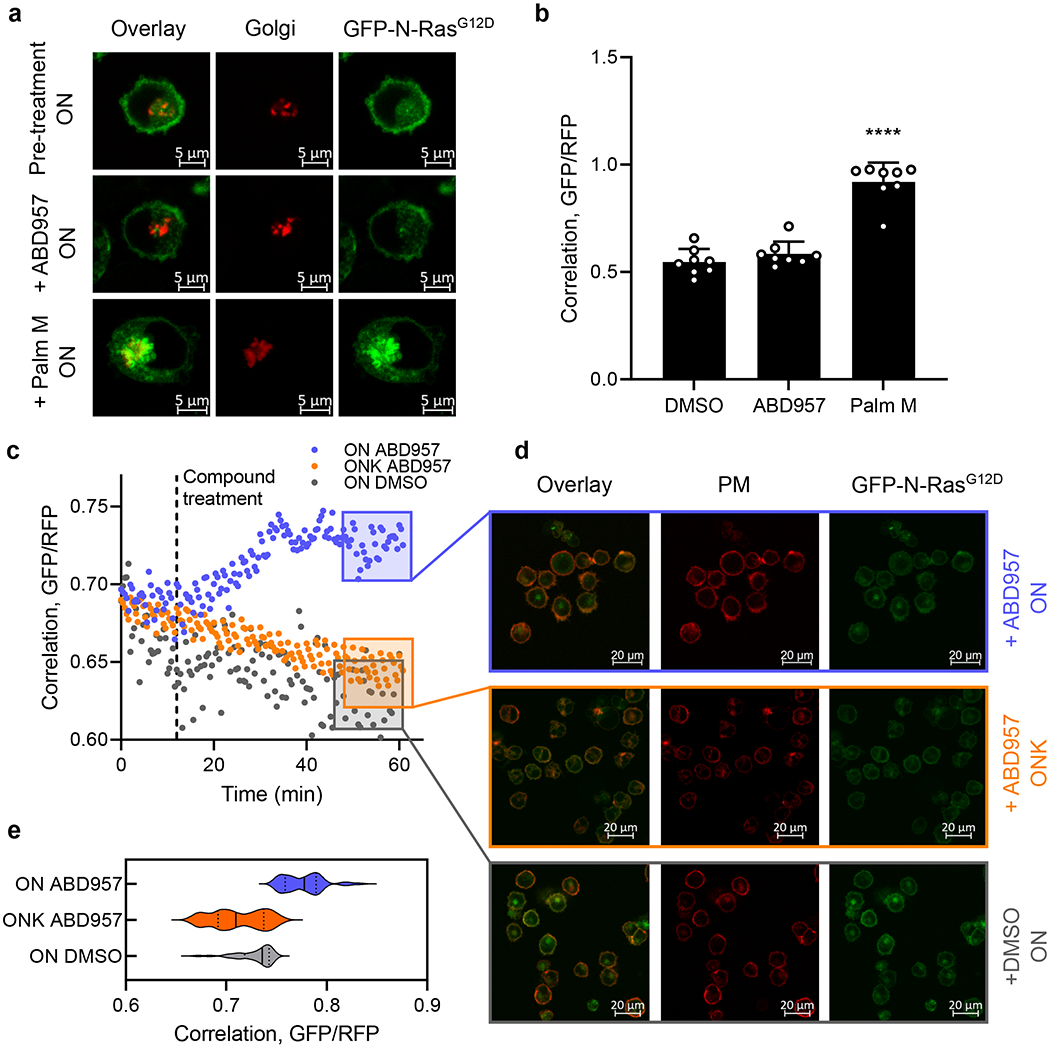
a, Representative images from eight biological replicates of cells co-stained with the Golgi marker RFP-N-acetylgalactosaminyltransferase (RFP-GALNT2) before and after treatment with Palm M (10 μM) or ABD957 (500 nM). Red channel shows Golgi marker (middle), green channel shows GFP-N-Ras (right), and overlay of the two markers (left). Scale bar, 5 μm. b, Change in N-Ras colocalization to the Golgi, as a representative endomembrane compartment, measured by co-staining with RFP-GALNT2 after 10-minute compound treatment. Colocalization is quantified using the Mander’s correlation coefficient between GFP-N-Ras and RFP-GALNT2 signal. Data represent average values ± s.d. (n = 8 individual cells per group). Statistical significance was calculated with unpaired two-tailed Student’s t-test with equal variance, ****P<0.0001 represents significant increase compared to DMSO. P value 1.4×10−7. c, Real-time quantification of colocalization of GFP-N-Ras signal intensity and the plasma membrane marker Alexa-647-conjugated wheat germ agglutinin (WGA) in ON (purple) and ONK cells (orange) treated with ABD957 (1 μM at t = 10 minutes), in comparison to ON cells treated with DMSO (grey). Colocalization between GFP and RFP was quantified as in b and the correlation coefficient is shown on the y-axis. Images were acquired every 2 minutes for 10 minutes, then medium was infused with ABD957 and images collected for an additional 50 minutes (see Methods for details). Data shown are representative of three biological replicates. d, Representative images taken from c at t = 60 minutes (purple, yellow and grey boxes). Red channel shows plasma membrane staining with Alexa-647-conjugated WGA (middle), green channel shows GFP-N-Ras (right) and the overlay of the two markers is also shown (left). Treatment with ABD957 in ON (top) and ONK (middle) or DMSO in ON cells (bottom). Scale bar, 20 μm. e, Violin plot of cumulative colocalization between GFP-N-Ras and Alexa-647-conjugated WGA at t = 60 minutes after treatment with ABD957 or DMSO in ON or ONK cells is shown across three biological replicates of time-course experiments as shown in c. Median values represented by solid lines, lower and upper quartiles indicated by dotted lines. Also see Supplementary Video 1a–c.
ABD957 blocks NRAS-mutant cancer cell signaling and growth.
We next investigated N-Ras signaling, specifically ERK phosphorylation, which we found was substantially blocked by both Palm M and ABD957 in NRAS mutant OCI-AML3 cells, but not in KRAS mutant NB-4 cells (Fig. 5a, b). In contrast, the MEK inhibitor, PD901, which functions downstream of Ras signaling, blocked ERK phosphorylation in both cell lines (Fig. 5a, b). The effect of Palm M on ERK phosphorylation was greater than ABD957, mirroring their respective impacts on N-Ras palmitoylation (Fig. 2 and 3).
Figure 5. ABHD17 inhibition selectively impairs the signaling and proliferation of NRAS mutant leukemia cells.
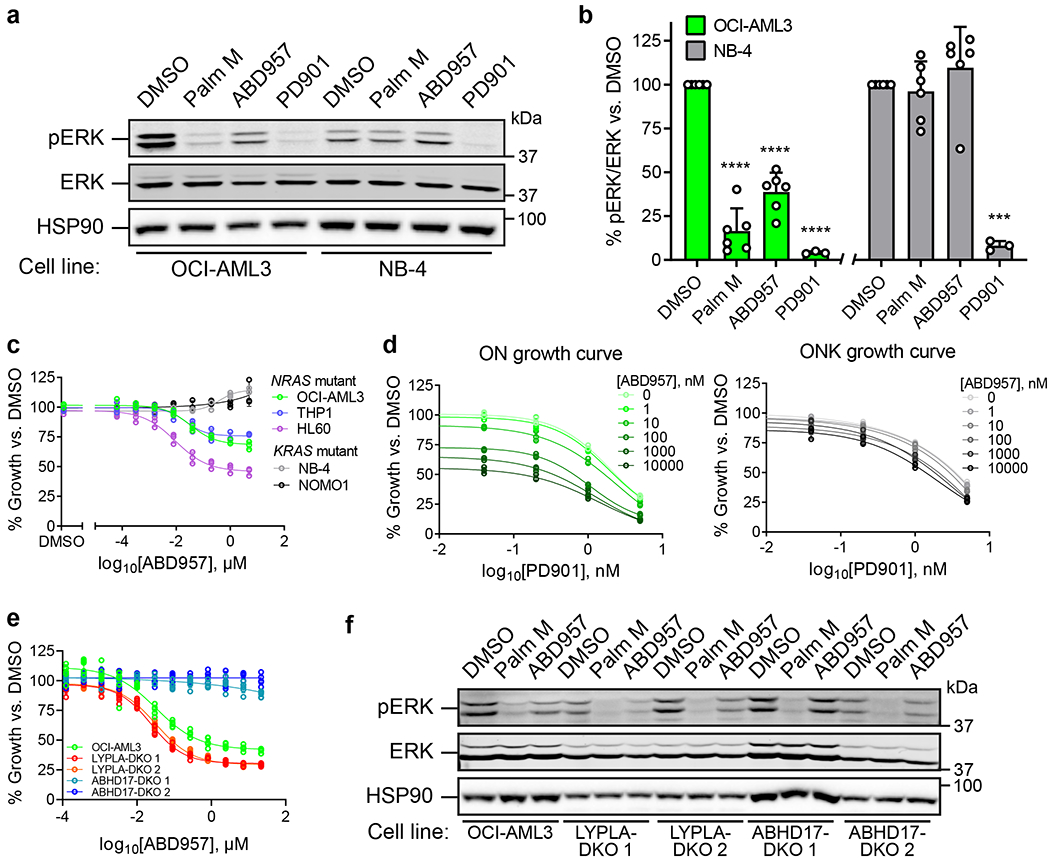
a, Representative Western blot for phosphorylated ERK (pERK) following exposure to Palm M (10 μM), ABD957 (500 nM), or PD901 (10 nM) for 4 h in NRAS mutant OCI-AML3 cells or KRAS mutant NB-4 cells. b, Quantification of data shown in a; average values ± s.d. (n = 6 independent experiments except for PD901, where n = 3 independent experiments). Statistical significance was calculated with unpaired two-tailed Student’s t-test with unequal variance compared to DMSO control, ***P<0.001, ****P<0.0001. P values were 1.9×10−5 (Palm M OCI-AML3), 3.9×10−5 (ABD957 OCI-AML3), 2.2×10−5 (PD901 OCI-AML3), 0.00028 (PD901 NB-4). c, d, Growth of the indicated human AML cell lines (c) and of isogenic ON and ONK cells (d) treated with varying concentrations of ABD957 for 72 h. Cell growth was measured by CellTiter Glo. Data represent average values ± s.d. (n = 3 biological replicates). e, Growth of the indicated OCI-AML3 parental and KO cell lines treated with varying concentrations of ABD957 for 72 h. Data represent average values ± s.d. (n = 6 biological replicates). f, Representative Western blot measuring pERK effects of compounds in the indicated OCI-AML3 parental and KO cell lines, performed as described in a.
We als found that ABD957 reduced the growth of NRAS mutant AML cell lines (OCI-AML3, THP1, and HL60), but not KRAS mutant cell lines (NB-4 and NOMO1) (Fig. 5c). The growth inhibitory effects of ABD957 were partial and plateaued at ~500 nM (Fig. 5c) consistent with near-complete inhibition of ABHD17s (see Fig. 1f). We next explored the impact of combining ABD957 with PD901 on the growth of ON and ONK cells. Using the Bliss Independence Model, which assesses drug synergy by comparing observed combination drug responses to predicted combination responses (where predicted responses assume no effect from drug-drug interactions)37, we found that the combination of ABD957 and PD901 was substantially synergistic in ON cells, but not ONK cells (Fig. 5d, Supplementary Fig. 4). This analysis identifies ABHD17 inhibition as a possible way to augment the therapeutic activity of MEK inhibitors in N-Ras-dependent cancers.
Finally, we generated OCI-AML3 cell lines lacking both ABHD17A and ABHD17B (ABHD17-DKO) or both LYPLA1 and LYPLA2 (LYPLA-DKO) using CRISPR/Cas9 genome editing methods and confirmed selective loss of the knocked-out serine hydrolases in each line by MS-ABPP (Supplementary Fig. 5a and Supplementary Dataset 1). Signals for ABHD17C were low in OCI-AML3 cells, but we did detect a unique peptide for this enzyme, indicating its maintained presence in ABHD17-DKO and LYPLA-DKO cells (Supplementary Fig. 5a and Supplementary Dataset 1). Interestingly, despite not showing an obvious growth defect compared to parental OCI-AML3 or LYPLA-DKO control lines (Supplementary Fig. 5b), ABHD17-DKO cells were insensitive to the anti-proliferative effects of ABD957 (Fig. 5e). Additionally, ABD957 no longer suppressed ERK phosphorylation in ABHD17-DKO cells, while Palm M maintained similar activity in these cells compared to control cells (Fig. 5f, Extended Data Fig. 7). Finally, dynamic palmitoylation proteomic experiments revealed that: i) ABD957-regulated proteins, including N-Ras, were partially protected from depalmitoylation in ABHD17-DKO cells compared to parental OCI-AML3 cells; and ii) the palmitoylation state of ABD957-regulated proteins was no longer sensitive to ABD957 treatment in ABHD17-DKO cells (Extended Data Fig. 8a,b and Supplementary Dataset 3). We interpret these data as strong support that ABD957 produces its effects on protein palmitoylation, N-Ras signaling, and cancer growth through inhibiting ABHD17s, with genetic loss of ABHD17A and ABHD17B leading to adapations in N-Ras mutant AML cells that subvert, by an unknown mechanism, dependency on ABHD17s, while maintaining sensitivity to the more promiscuous depalmitoylation inhibitor Palm M.
Discussion
The fundamental role that C-terminal lipid modifications play in regulating Ras signaling has stimulated diverse efforts to target the post-translational processing of Ras onco-proteins38–41. Biochemical and pharmacological studies with first-generation inhibitors of N-Ras depalmitoylation have implicated several enzymes in this process7, with more recent studies revealing a possible key contribution from the ABHD17 sub-group of serine hydrolases12. The high sequence identity shared by ABHD17A, ABHD17B, and ABHD17C (>70%) and their co-expression in most cell types presents technical challenges for studying the net contribution of these enzymes to N-Ras palmitoylation. Using a pan-ABHD17 inhibitor ABD957, we have shown here that the ABHD17s make a substantial, but partial contribution to N-Ras depalmitoylation in NRAS-mutant AML cells. Whether other targets of the more promiscuous inhibitor Palm M account for the apparent ABHD17-independent N-Ras depalmitoylation activity remains unknown. We also note that our broader proteomic investigations of Palm M identified several proteins with heightened palmitoylation, including those that were not dynamically palmitoylated over the time course of our pulse-chase experiments. This result suggests that Palm M may affect other cellular metabolic processes beyond protein depalmitoylation that in turn impact the net palmitoylation state of proteins in cells.
One of our most striking findings was the selectivity that ABD957 displayed for N-Ras across the broader palmitoylated proteome. Unlike Palm M, which impacted most dynamically palmitoylated proteins, ABD957 only preserved the palmitoylation state of a handful of plasma membrane-associated proteins. Considering that other dynamically palmitoylated proteins affected by Palm M, but not ABD957, localize to intracellular membrane compartments (Fig. 3d), our data support a model where ABHD17s regulate palmitoylated proteins exclusively at the plasma membrane. Additional factors may influence whether palmitoylated proteins are substrates for ABHD17s, as we noted that transmembrane proteins bearing dynamic palmitoylation events were not affected by ABHD17 inhibition, even when predicted to localize, at least in part, to the plasma membrane (e.g., ADAM10, ITM2B/C). We also cannot exclude the possibility that, as more substrates for ABHD17s are identified, some may reside at other locations beyond the plasma membrane in the cell. Conversely, we do not know yet whether individual ABHD17s (A, B, or C) show different substrate specificities in cells, but, regardless, it is likely that the substrate scope for these enzymes will expand as more cell types are examined with ABHD17 inhibitors. For instance, PSD-95 (DLG4), a palmitoylated plasma membrane-associated protein that is predominantly expressed in neurons and was not detected in the AML cells examined herein, has also been shown to be regulated by ABHD17s12,14. Conversely, SCRIB has been found, in other cell types, to be regulated by the depalmitoylase LYPLA242, indicating the potential for cellular context to also restrict the substrate scope of ABHD17s. With regards to enzymes that may be responsible for regulating intracellular protein palmitoylation, ABHD10 was recently identified as a depalmitoylase localized to the mitochondria18, and LYPLA1 localizes to the Golgi43 and mitochondria44. Emerging data thus point to the existence of multiple S-depalmitoylases that individually regulate dynamic protein palmitoylation in specific organelles of mammalian cells.
The general strategy of inhibiting the palmitoylation/depalmitoylation cycle of N-Ras has the advantage of selectively targeting cancer cells that depend on this oncoprotein for growth while preserving the functions of K-Ras4b in normal tissues. Our data indicate that ABHD17 inhibitors offer one potential way to achieve this objective. However, several important mechanistic questions about the interactions between ABHD17s and N-Ras palmitoylation and signaling remain to be answered. How, for instance, does ABD957-induced stabilization of palmitoylated N-Ras at the plasma membrane lead to perturbation in N-Ras signaling? Might, for instance, persistently palmitoylated N-Ras mislocalize, over time, to plasma membrane microdomains that are less productive for signal transduction, as has been postulated45? Understanding more generally how ABHD17-mediated N-Ras depalmitoylation interfaces with other established modes of regulating N-Ras trafficking and signaling represents another important future direction. For instance, retention of N-Ras in the cytoplasm by chaperones such as PDE6δ and VPS3546,47 may protect this oncoprotein from depalmitoylation by ABHD17 enzymes, and, conversely, the stabilization of N-Ras palmitoylation in ABHD17-inhibited cells may decrease the ability of PDE6δ to extract N-Ras from membranes. We also wonder whether the partial blockade of mutant N-Ras signaling and cancer growth caused by ABHD17 inhibitors reflects maximal possible effects of the plasma membrane-delineated stabilization of mutant N-Ras palmitoylation and if such incomplete activity could facilitate resistance mechanisms, as apparently observed in ABHD17-DKO cells. Considering that Palm M more greatly affected N-Ras signaling (as measured by ERK phosphorylation), it is possible that disrupting mutant N-Ras signaling on intracellular membranes may be required to more completely block oncogenic activity, an outcome that, based on our data, may not be achievable with ABHD17 inhibition alone. On the other hand, the restricted impact of ABHD17 inhibitors on the palmitoylated proteome suggests that these compounds may perturb N-Ras signaling without gross effects on normal cell physiology. Such a profile may enable ABHD17 inhibitors to safely augment the activity of other anti-cancer agents, as we have shown for the MEK inhibitor PD901, to provide a compelling path for the targeted therapy of NRAS mutant cancers.
Methods
Cell lines and tissue culture
OCI-AML3 (DSMZ: ACC-582) cells were grown in RPMI supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), and 50 μM β-mercaptoethanol and grown at densities between 0.3e6 and 2e6 cells/mL. NB-4 (DSMZ: ACC-207), NOMO1 (DSMZ: ACC-542), HL60 (DSMZ: ACC-3), and THP1 (ATCC: TIB-202) were grown in RPMI, HEK293T (ATCC: CRL-3216) were grown in DMEM, and PC3 (ATCC: CRL-1435) were grown in F-12K, all supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/mL), and streptomycin (100 μg/mL). All cells were maintained at 37 °C with 5% CO2.
hABHD17B (MHS1010-202726047, 4748883) and mABHD13 (MMM1013-202768782, 5148187) clones in pCMD-SPORT6 vector were purchased from GE Dharmacon (now known as Dharmacon Inc).
hCES2 in pcDNA3.1 vector was a custom order from GenScript.
The following primers were used for subcloning into an untagged pCL vector for viral transfection: 5’-TTT AAG CTT GCC ACC ATG AAT AAT CTT TCA TTT AGT G-3’ (forward; FAM108B1_F_pCL_HindIII), 5’- AAA CTC GAG TTA CAA ATT TAC CAG TTC CTG-3’ (reverse; FAM108B1_R_pCL_XhoI).
Generation of HEK293T transgenic lines
HEK293T cells stably expressing various constructs were made as follows. HEK293T cells (1.8e6) were plated in 6 cm plates and allowed to settle overnight. To 200 μL of serum-free DMEM the following were added: 1 μg plasmid containing gene of interest in lentivirus vector, 900 ng ΔVPR, 100 ng VSV-G and 6 μL of XtremeGene HP transfection reagent. Reagents were flicked to mix, and after 15 minutes, the transfection mixture was added dropwise to plates containing cells. Virus containing supernatants were collected 48 hours later and concentrated using a Lenti-X Concentrator (Clontech), and then used to infect HEK293T cells (plated the day prior at 0.25e6 per well of 12-well plate) in the presence of 10 μg/mL Polybrene (Santa Cruz). The media was replaced 24 hours later, cells were allowed to recover for an additional 24 hours, and then puromycin was added for selection.
Gel-based activity-based protein profiling
Inhibitor potency (IC50 values) against hABHD17B, hLYPLA1, mABHD13 and hABHD6 was determined by competitive gel-based ABPP using FP-Rhodamine49 (hABHD17B, hLYPLA1 and mABHD13) or JW91250 (hABHD6) competition. Endogenous hLYPLA1 and hABHD6 activity were measured in HEK293T and PC3 proteomes, respectively, whereas hABHD17B and mABHD13 activity was measured in HEK293T cell proteomes overexpressing this protein (see above).
Cell lysates were fractionated by ultra-centrifugation (100,000g for 45 min at 4°C) and membrane pellets were resuspended in PBS and diluted to a final protein concentration of 1.0 mg/mL for HEK293T or 2.0 mg/mL for PC3. Cell proteomes (50 μg) were treated with inhibitor (0.001-10 μM) or DMSO for 30 min at 37°C and subsequently treated with FP-Rh or JW912 (1.0 μM) for an additional 30 min at room temperature. Reactions were quenched with 4X SDS-PAGE loading buffer and FP-Rh-labeled enzymes were resolved by SDS-PAGE (10% acrylamide). In-gel fluorescence was visualized using a Bio-Rad ChemiDoc™ XRS imager. Fluorescence is shown in gray scale. Quantification of enzyme activities was performed by densitometric analysis using ImageJ software (NIH). Integrated peak intensities were generated for bands corresponding to hABHD17B, hLYPLA1, mABHD13 and hABHD6. IC50 values were calculated through curve fitting semi-log-transformed data (x-axis) by non-linear regression with the log(inhibitor) vs. normalized response function in Prism software (GraphPad).
In vitro competitive substrate hydrolysis activity assay for hCES2
Inhibitor potency (IC50 values) against hCES2 was determined by measuring the rate of enzyme hydrolysis of the substrate 4-nitrophenyl acetate (pNPA) (Sigma, Cat: N8130, CAS: 830-03-5) based on a previously described method51.
HEK293T cell lysates expressing recombinant hCES2 and pNPA substrate were diluted separately in 50 mM HEPES (pH 7.0) containing 100 mM NaCl. Lysates (50 μL, ~2.5 μg total protein) were preincubated with varying concentrations of inhibitors (0.03-2 μM) or DMSO for 30 min at 25 °C. Subsequently, 2X pNPA substrate (50 μL, 2.5 mM) was added and the rate of substrate turnover was monitored by measuring the increase in absorbance at wavelength 405 nm for 20 minutes at 25 °C using a Biotek Neo2 plate reader. The mean velocity was converted to percent enzyme activity following background subtraction of non-specific substrate turnover and normalization to DMSO controls. The mean reaction velocity (Vmean, ΔAbs / min•μg protein) in mock- versus hCES2-transfected proteomes was 8.72 ± 1.86 min−1 μg−1 and 30.9 ± 1.16 min−1 μg−1, respectively. IC50 values were calculated through curve fitting semi-log-transformed data (x-axis) by non-linear regression with the log(inhibitor) vs. normalized response function in Prism software (GraphPad).
Liver Microsomal Stability
A master solution was first prepared by mixing pooled human liver microsomes (50 μL, 5 mg/mL, Corning – Cat# 452117), phosphate buffer (250 μL, 200 mM), ultrapure water (95 μL) and MgCl2 solution (50 μL, 50 mM). A solution of NADPH (50 μL, 10 mM) or ultrapure water (50 μL, negative control) was then added to the master solution and the mixture was warmed to 37 °C for 5 min. To begin the reaction, test compounds (5 μL, 200 uM) were then added to the mixture (t = 0). Aliquots (50 μL) were removed from the reaction solution at 0, 15, 30, 45 and 60 minutes, and immediately quenched with ACN (200 μL), diluted with ultrapure water (90 μL) and analyzed by LC-MS/MS. Extracted ion chromatograms were integrated and peak areas were plotted against the incubation time to determine in vitro t1/2. Intrinsic clearance (Clint) was calculated using the following equation:
MS-ABPP sample preparation
For in situ treatments, OCI-AML3 or ONK cells were resuspended in fresh media at 2e6 cells/mL and treated with DMSO, Palm M (10 μM), HDFP (20 μM), JJH254 (1 μM), ABD298 (500 nM) or ABD957 (500 nM or 1 μM) and incubated for the indicated times. Cells were pelleted at 500 RCF by centrifugation, washed with PBS, pelleted again and snap frozen.
Frozen cell pellets were thawed, diluted in PBS and subsequently lysed by probe sonication. The membrane fraction of each cell sample was isolated using ultra-centrifugation (100,000g for 45 min at 4°C) and membrane pellets were resuspended in PBS and diluted to 2.0 mg/mL. Membrane proteomes (2 mg/mL in 1 mL of PBS) were labeled with FP-biotin (10 μM) for 1 h at room temperature while rotating. After labeling, the proteomes were denatured and precipitated using 4:1 MeOH/CHCl3, resuspended in 0.5 mL of 6 M urea in PBS, reduced using tris(2-carboxyethyl)phosphine (TCEP, 10 mM) for 30 min at 37 °C, and then alkylated using iodoacetamide (40 mM) for 30 min at room temperature in the dark. The biotinylated proteins were enriched with PBS-washed avidin-agarose beads (100 μL; Sigma-Aldrich) by rotating at room temperature for 1.5 h in PBS with 0.2% SDS (6 mL). The beads were then washed sequentially with 5 mL 0.2% SDS in PBS (3x), 5 mL PBS (3x) and 5 mL H2O (3x). On-bead digestion was performed using sequencing-grade trypsin (2 μg; Promega) in 2 M urea in 100 mM triethylammonium bicarbonate buffer with 2 mM CaCl2 for 12-14 h at 37 °C (200 μL).
Samples from the time-course study with compound 7 and ABD957 were desalted using SOLAμ™ SPE plates (HRP 2mg / 1mL) and dried by centrifugal evaporation and stored at −80 °C until analysis by parallel reaction monitoring.
For all other samples, duplex reductive dimethylation (ReDiMe) was performed as previously described52. Briefly, for duplex ReDiMe of WT versus KO (or DMSO vs. inhibitor treated) cells either 13CD2O (heavy) or CH2O (light) was added to each sample (0.15%) followed by addition of NaBH3CN (22.2 mM). Vehicle- or inhibitor-treated samples were labeled using a triplex ReDiMe protocol where control samples (i.e. vehicle-treated) were labeled with CH2O (light) and inhibitor-treated samples labeled with 13CD2O (heavy). Light samples were then treated with NaBH3CN whereas heavy samples were treated with NaBD3CN. Following a 1 h incubation period at room temperature, the reaction was quenched by addition of NH4OH (0.23%) and formic acid (0.5%). The samples were then combined, desalted using SOLAμ™ SPE plates (HRP 2mg / 1mL) and dried by centrifugal evaporation and stored at −80 °C until analysis using data-dependent acquisition LC/MS/MS methods.
MS-ABPP data analysis
Data-dependent Acquisition:
Nanoflow LC-MS/MS measurements were performed on a Dionex RSLC nanoLC (Thermo Fisher Scientific) interfaced with an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Scientific) via an EASY-Spray source (Thermo Scientific). Data were collected using Xcalibur (version 2.2). Dry peptide samples were reconstituted in a water:ACN (95:5) mixture containing 0.1% formic acid (30 μL) and 20 μL were injected on an EASY-Spray™ C18 column (2 μm particle size, 75 μm × 50 cm; Thermo Scientific, ES801) heated to 55 °C using a flow rate of 400 nl/min. The compositions of LC solvents were A: water and 0.1 % formic acid, and B: 95% acetonitrile, 5% water and 0.1% formic acid. Peptides were eluted over 4 hours using the linear gradient, 2.5-215 min 3-35% B, 215-230 min 25-40% B, 230-231 min 45-70% B, 231-233 min 70-90% B, 233-234 min 5-70% B, 234-236 min 70-90% B, 236-240 min 3% B.
MS data were acquired in data dependent mode (top 20 dependent scans). MS1 profile scans were acquired in the Orbitrap (resolution: 120,000, scan range: 375–1500 m/z, AGC target: 4.0e5, maximum injection time: 50 ms). Monoisotopic peak determination was set to “peptide”. Only charge states 2-5 were included. Dynamic exclusion was enabled (repeat count, n: 1, exclusion duration: 60 s, mass tolerance: ppm, low: 10 and high: 10, excluding isotopes). An intensity threshold of 5e3 was set. MS2 spectra were acquired in centroid mode. Precursor ions were isolated using the quadrupole (isolation window: 1.6 m/z), fragmented using CID (collision energy: 35%, activation time: 10 ms, activation Q: 0.25), and detected in the ion trap (scan range mode: auto m/z normal, scan rate: rapid, AGC target: 4.0e3, maximum injection time: 300 ms, injecting ions for all available parallelizable time).
Spectrum raw files were extracted into MS2 files using RawConverter (version 1.1.0.22 from http://fields.scripps.edu/rawconv/) and searched using the ProLuCID (version 1.4) algorithm53 against a human reverse concatenated nonredundant Uniprot database (2016 version), with static modifications for cysteine residues to account for alkylation by iodoacetamide (+57.0215 m/z), and standard static modifications for reductive dimethylation: lysine and N-terminus (+28.0313 m/z for light, +34.06312 m/z for heavy). Data was assembled using DTASelect version 2.0,54 and ratio quantification was performed using in-house CIMAGE software (see GitHub repository linked in Data Availability section). Peptides were required to be fully tryptic, unique, to have an envelope correlation score of R2 ≥ 0.5, and ratios were capped to a maximum value of 20. Ratios (R) for each protein were calculated from the median of all quantified peptide ratios. In cases where a protein had exactly one peptide with a ratio of 20, and at least one other peptide with a ratio below 2, the 20 value was discarded. Peptides with a ratio of 20 were also manually reviewed and filtered.
For in vitro datasets (n = 1), peptides with ratios of 20 and only a single MS2 event triggered during the elution of the parent ion were discarded. Proteins were required to have at least two quantified peptides.
For in situ datasets (n = 3), peptides with ratios of 20 and only a single MS2 event triggered during the elution of the parent ion were discarded. Proteins were required to be quantified in at least 2 biological replicates, and to have at least 2 unique quantified peptides across all replicates. Additionally, for proteins with R = 20 in one experimental replicate and R≤2 in one or more replicates, the 20 value was discarded. The median ratio was calculated and reported across experimental replicates.
For CRISPR knockout datasets (n = 1). Chromatograms showing evidence of knockdown were manually selected.
Parallel reaction monitoring:
Dry peptide samples were reconstituted in a water:ACN (95:5) mixture containing 0.1% formic acid (30 μL) and 5 μL were injected onto an EASY-Spray™ C18 column (2 μm particle size, 75 μm × 50 cm; Thermo Scientific, ES801) using a Dionex RSLCnano LC (Thermo Fisher Scientific). Peptides were separated over a 60 min gradient of 0 to 40% acetonitrile (0.1% formic acid) and analyzed on an Orbitrap Fusion Lumos (Thermo Fisher Scientific) operated using a parallel reaction monitoring (PRM) method for two distinct peptides for each serine hydrolase (ABHD17A, ABHD17B, ABHD17C, LYPLA1, LYPLA2) and ten additional peptides from endogenously biotinylated proteins (PCCA and MCCC1) as controls. Selected ions were isolated and fragmented by high energy collision dissociation (HCD) and fragments were detected in the Orbitrap at 15,000 resolution. Peptide sequences and targeting parameters can be found in Supplementary Table 3.
Raw data files were uploaded analyzed in Skyline (v4.2.0.19009) to determine the abundance of each peptide in vehicle-treated samples relative to inhibitor-treated samples. Peptide quantification was performed by calculating the sum of the peak areas corresponding to 6 fragment ions from each peptide. The peptides and fragment ions were pre-selected from in-house reference spectral libraries acquired in data-dependent acquisition mode to identify authentic spectra for each peptide.
Syngeneic ON/ONK cells
A pCDH-LMN-GFP lentiviral vector was obtained by cloning the miR30-PGK-NeoR-IRES-GFP cassette from LMN-GFP55 into a pCDH Expression Lentivector (System Biosciences). A miR30-based shRNA targeting human NRAS (sense: 5’- CAGGGTGTTGAAGATGCTTTT -3’) was cloned into the vector. The coding sequence of NrasG12D was cloned downstream of GFP to create a N-terminal GFP-fused N-RasG12D expression construct with N-Ras endogenous HVR (N-HVR). Alternatively, a chimeric version was cloned were the sequence corresponding to amino acids 166-188 from N-Ras was replaced with that of K-Ras-4B (K-HVR). Lentiviral vector production and transduction of OCI-AML3 cells were performed as previously described56. ON and ONK cells were validated by Western Blot (Supplementary Fig. 1).
Dynamic palmitoylation assay
Dynamic palmitoylation assay was modeled after earlier work in our lab9. OCI-AML3 cells were grown as described above, then spun down (3 min, 500 RCF) and re-suspended in fresh medium at a density of 2e6 cells/ml (10e6 cells per sample for gel-based assay and 20e6 cells per sample for mass spectrometry-based assay). Cells were pre-incubated with inhibitor or DMSO for 1 hour, then 17-ODYA (20 μM) was added for 1 hour. Samples were pelleted and snap frozen immediately following the 17-ODYA “pulse” and designated t0, or re-suspended in pre-warmed chase medium which consisted of OCI-AML3 growth media, supplemented with DMSO or inhibitor at the same concentration as in the pre-incubation step and incubated for 1 hour. Cells were then centrifuged, placed on ice, washed in cold PBS, snap frozen, and designated t1.
Immunoprecipitation
Cell pellets were re-suspended in cold lysis buffer which consisted of: 1% Triton-X PBS with 1 mM PMSF, 0.2 mM HDSF, 20 μM HDFP and protease inhibitors (complete Ultra EDTA-free mini tablets, 5892791001 Roche), sonicated with a microtip probe sonicator (7 times, 50% rate, power 4), and placed on an end-over-end rotator for 30 minutes at 4 °C. Samples were then hard spun (16,300 RCF, 5 minutes) and the supernatant was transferred to fresh tubes on ice. Protein concentration was measured using a DC assay kit from Bio-Rad and adjusted to 1 mg/mL. Input samples were taken at this point and stored at −80°C.
Enrichment was performed with anti-GFP Sepharose beads (20 μL 50:50 slurry, ab69314, abcam). Antibody conjugated beads were spun down (500 RCF), storage buffer was aspirated with a 26-gauge needle, and beads were washed three times with 1% Triton-X PBS. To each sample, 20 μL of washed bead slurry was added using a cut pipette tip, and samples were placed on an end-over-end rotator for 3 hours at 4 °C. Next, washes were performed by centrifugation/aspiration (3x, 500 RCF) with cold 1% Triton-X PBS containing 500 mM NaCl. After the last wash, supernatant was removed, and a 26-gauge needle was quickly inserted into the bead slurry to remove all remaining liquid. GFP beads were then re-suspended in 50 μL wash buffer. At this point, IP samples were either stored at −80°C or processed further for analysis by SDS-PAGE gel.
Click chemistry and processing for SDS-PAGE gel
On-bead click chemistry was performed to conjugate a rhodamine fluorophore reporter to 17-ODYA labeled proteins. To each 50 μL sample, 6 μL of a click chemistry reaction mixture was added. Click reaction mixture was freshly prepared as follows (amounts given are per sample): 1 μL 50 mM CuSO4 (in water; final concentration during reaction of 1 mM), 3 μL 1.7 mM TBTA (4:1 t-BuOH:DMSO; 100 μM final), 1 μL 50 mM TCEP (freshly made in PBS; 1 mM final) and 1 μL Rh-N3 (DMSO; 25 μM final). Reactions were allowed to proceed for 1 hour at room temperature. Samples were then quenched with 4x SDS loading buffer containing 1% BME (described below), boiled for 5 minutes to effect elution and finally 17-ODYA proteins were resolved and imaged on gel.
Loading buffer was prepared as follows (for 100 mL): 3.02 g Tris base was added to 40 mL water. 40 mL of glycerol was added slowly, and the mixture was stirred using a stir bar while the pH was brought to 6.75 using concentrated HCl. 8 g of SDS were then added followed by 20 mL of water and a pinch of bromophenol blue. Loading buffer was stored at room temperature, and 1% (10 μL per 1 mL buffer) β-mercaptoethanol (BME) was added immediately before quenching.
Finally, samples were loaded onto a 10% acrylamide gel, resolved by SDS-PAGE (300V for approx. 2.5 hours), and in-gel fluorescence was visualized using a Bio-Rad ChemiDoc MP flatbed fluorescence scanner.
Preparation of tandem mass tag (TMT) labeled 17-ODYA enrichment samples
Samples were prepared as previously described9, with the following modifications. 17-ODYA labeled ON cells were lysed in DPBS containing protease inhibitors (Roche) and 1 mM PMSF using a Branson Sonifier probe sonicator (3 rounds of 8 pulses, 40% duty cycle, output setting = 4). Protein concentration was determined by DC assay on a microplate reader, and 500 μL of whole cell lysate at 2 mg/mL was added to a mixture of TBTA (30 μL/sample, 1.7 mM in 1:4 DMSO:t-BuOH), CuSO4 (10 μL/sample, 50mM in H2O), TCEP (10 μL/sample, 50mM in DPBS) and Biotin-N3 (10 μL/sample, 10 mM in DMSO). After 1 h, samples were MeOH/CHCl3 precipitated. Hydroxylamine-sensitivity was performed as previously described from ON cells metabolically labeled with 17-ODYA for 2 h9. Following precipitation, each sample was solubilized in 1.2% SDS in DPBS via sonication and diluted to 0.2% SDS for enrichment. Streptavidin beads (Thermo Scientific) 100 μL slurry was added to each sample and rotated at room temperature for 2 h, washed three times with 0.2% SDS, three times with DPBS, and three times with water. Beads were resuspended in 6 M urea in 200 mM EPPS, reduced with 10 mM neutral TCEP for 30 min at 37°C followed by addition of 20 mM iodoacetamide for 30 min at room temperature. Samples were diluted to 2 M urea in 200 mM EPPS, pelleted, and resuspended with 200 μL 2 M urea in 200 mM EPPS containing 2 μg sequence grade porcine trypsin (Promega) and 1 mM CaCl2 and digested overnight at 37°C. Digests were centrifuged to remove the beads, and acetonitrile was added to reach 30% final volume. 6 μL (20 μg/μL) of respective 6-plex, 10-plex, or 16-plex TMT tag (Thermo Scientific) was added to digests and incubated at room temperature for 1 h with occasional vortexing. Labeling reaction was quenched with 6 μL of 5% hydroxylamine for 15 minutes, followed by acidification by 15 μL formic acid. Samples were combined and vacuum-centrifuged to dryness to remove acetonitrile, reconstituted in Buffer A (95% H2O, 5% acetonitrile, 0.1% formic acid) and stored at −80°C until MS analysis by MudPIT or off-line fractionated using Pierce High pH Reversed-Phase Peptide Fractionation Kit (Thermo), separating samples into 9 fractions concatenated to 3.
Mass spectrometry analysis of tandem mass tag (TMT) labeled peptides
Labeled peptides were prepared for MS analysis as previously described57. Briefly, labeled peptides were pressure loaded onto a 250 μm (inner diameter) fused silica capillary column packed with 4 cm C18 resin (Phenomenex, Aqua 5 μm). Samples were analyzed on an Orbitrap Fusion mass spectrometer (Thermo Scientific) coupled to an UltiMate 3000 Series Rapid Separation LC system and autosampler (Thermo Scientific Dionex). Data were collected using Xcalibur (version 2.2). Peptides were either separated on a 100 μm inner diameter capillary column with a 5 μm tip packed with 10 cm C18 (Phenomenex, Aqua 5 μm) and 3 cm strong cation exchange resin (SCX, Phenomenex)] using a 5-step ‘MudPIT’ protocol that injects 5 μL 0%, 20%, 50%, 80%, 100% salt bumps of ammonium acetate (500mM) in buffer A (100% H2O, 0.1% formic acid) followed by an increasing gradient of buffer B (100% acetonitrile, 0.1% formic acid) in Buffer A in each step, or the peptides were eluted onto a capillary column (75 μm inner diameter fused silica, packed with C18 (Waters, Acquity BEH C18, 1.7 μm, 25 cm) and separated at a flow rate of 0.25 μL/min using the following gradient: 5% buffer B in buffer A from 0-15 min, 5%–35% buffer B from 15-155 min, 35%–95% buffer B from 155-160 min, 95% buffer B from 160-169 min, 95%–5% buffer B from 169-170 min, and 5% buffer B from 170-200 min (buffer A: 95% H2O, 5% acetonitrile, 0.1% FA; buffer B: 5% H2O, 95% acetonitrile, 0.1% FA). The voltage applied to the nano-LC electrospray ionization source was 1.9 kV. A MS3-based TMT method was used for data acquisition. MS1 full scan spectrum (resolution: 120,000, scan range: 400–1700 m/z, RF lens 60%, AGC target 2e5, maximum injection time 50 ms, centroid mode) with dynamic exclusion enabled (repeat count 1, duration 15s) was performed. The top ten precursors were then selected for MS2/MS3 analysis. MS2 analysis consisted of quadrupole ion trap analysis, AGC 1.8e4, CID collision energy 35%, Activation Q 0.25, maximum injection time 120 ms, and isolation window at 0.7 m/z. Synchronous Precursor Selection (SPS) was enabled to include up to 10 MS2 fragment ions for the MS3 spectrum. MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (resolution: 15,000 for 6-plex and 50,000 for 10-plex and 16-plex, collision energy 55%, AGC 1.5e5, maximum injection time 120 ms). For MS3 analysis, charge state–dependent isolation windows were used. For charge state z = 2, the MS isolation window was set at 1.2 m/z; for z = 3-6, the MS isolation window was set at 0.7 m/z.
Raw files were processed using Integrated Proteomics Pipeline (IP2 v6.5.5) using the Homo sapiens reviewed Uniprot FASTA database (July 2020) with N-Ras substituted for N-RasQ61L for OCI-AML3 samples, or GFP-N-RasG12D for the syngeneic ON samples, with FDR <1% at the peptide level, and removed identifications from reverse and contaminants database. A minimum of 2 unique peptides per MS experiment were required. A minimum MS3 reporter ion intensity of 5,000 per control channel (DMSO t0) for each peptide spectrum match (PSM) was required, and in cases where there were multiple control channels (hydroxylamine experiments and 10-plex time-course experiments), PSMs were discarded if they had a coefficient of variation (CV) ≥ 0.5 for the control channels. Relative protein abundance was then averaged across all PSMs.
For hydroxylamine datasets, proteins were required to be quantified in either NB-4 (n=1), ON (n=1), or at least 2 experiments for OCI-AML3 studies (n=2).
For time-course studies, PSMs were required to have non-zero intensities in all t0 channels. Additionally, outlier PSMs, having a CV ≥ 0.5 for percent of control values on a per channel basis for a particular protein (having 3 or more PSMs), per experiment, were removed. Proteins were required to be quantified in at least 2 experiments for OCI-AML3 (n=3 individual experiments) and ON studies (n=4). For NB-4 studies where 6-plex and 10-plex data were combined, proteins were required to have a standard deviation (across percent control values) of < 100 in all studied conditions, and to have been quantified in at least 4 DMSO t1 biological replicates (channels).
Stable Isotope Labeling and SILAC-TMT data processing for protein turnover
OCI-AML3 cells were grown and passaged six times in SILAC RPMI supplemented with 10% fetal bovine serum (FBS), L-glutamine (2 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), 50 μM β-mercaptoethanol, and L-lysine⋅HCl and L-arginine⋅HCl (100 μg/mL each) or [13C6, 15N2]-L-lysine and [13C6, 15N4]-L-arginine (100 μg/mL each). Cells in ‘light’ media were pelleted and resuspended at 0.5e6 cells/mL in ‘heavy’ media and treated with DMSO or 500 nM ABD957. Cells were counted to determine growth rates and harvested at 3, 6, 24, and 72 hours in triplicate (doubling times of 29 hr and 34 hr for DMSO and ABD957 treated cells respectively). Cells pellets were lysed in DPBS containing protease inhibitors (Roche) and 1 mM PMSF using a Branson Sonifier probe sonicator (3 rounds of 8 pulses, 40% duty cycle, output setting = 4). Protein concentration was determined by DC assay on a microplate reader, and 200 μg was precipitated with 1 mL ice cold acetone followed by centrifugation at 16,000 g for 10 minutes, and protein pellets washed with 1 mL ice cold acetone. Proteomes were resuspended with 50 μL 8 M urea in 200 mM EPPS (pH 8), reduced with 10 mM DTT incubated at 65 C for 15 minutes. Samples were cooled to room temperature and alkylated with 20 mM IA incubated at 37 C for 30 minutes. Samples were diluted to 2 M urea by addition of 145 μL 200 mM EPPS (pH 8) and digested overnight at 37 C with 4 μg sequence grade porcine trypsin (Promega) and 1 mM CaCl2. Peptide concentrations from digests were determined by microBCA assay (Thermo Scientific). TMT labeling and mass spectrometry were performed as previously described58. In brief, for each sample, 25 μg of peptides was transferred to a new Eppendorf tube, and brought up to 35 μL with 200 mM EPPS (pH 8), diluted with 9 μL acetonitrile, and incubated with 5 μL (20 μg/μL) of the corresponding 10-plex TMT tag (Thermo Scientific) at room temperature for 1 h with occasional vortexing. Labeling reaction was quenched with 5 μL of 5% hydroxylamine for 15 minutes, followed by acidification by 2.5 μL formic acid. Samples were combined and vacuum-centrifuged to dryness to remove acetonitrile, reconstituted in Buffer A (95% H2O, 5% acetonitrile, 0.1% formic acid) and desalted by passing through Sep-Pak C18 cartridges (55-105 μm, Waters). The eluent was evaporated to dryness, resuspended in Buffer A (95% H2O, 5% acetonitrile, 0.1% formic acid) and fractionated into a 96 deep-well plate using HPLC (Agilent), for offline high pH fractionation. The peptides were eluted onto a capillary column (ZORBAX 300Extend-C18, 3.5 μm) and separated at a flow rate of 0.5 mL/min. The plate was evaporated to dryness and every 12th fraction concatenated for 12 final fractions (combining every column).
Raw files were processed using Integrated Proteomics Pipeline (IP2 v6.5.5) using the Homo sapiens reviewed Uniprot FASTA database (July 2020) with N-Ras substituted for N-RasQ61L. Samples were simultaneously searched for both light and heavy isotopes specifying the heavy labels as lysine (+8.0142) and arginine (+10.0083). Cysteine carbamidomethylation (+57.02146), and TMT labels to N-terminus and lysine (229.1629) were static modifications, and methionine oxidation (+15.994915) was differential allowing one per peptide. Proteins required two unique peptides and FDR <1% at the peptide level. Data normalization and processing were performed as previously described30. In brief, peptides were required to be unique fully-tryptic and non-terminal. Light and heavy peptide spectrum matches (PSMs) were required to have non-zero intensities in the light and heavy channels respectively. For each individual experiment, intensities for heavy and light peptides were separately row-wise normalized and total sum normalization was then applied. Heavy and light PSMs were required to have a normalized intensity ≥ 2500 in heavy and light control channels respectively. Fractional ratios for each time point versus the corresponding control channel were then calculated. Then, light and heavy fractional ratios at the PSM level were fitted to exponential equations modeling protein synthesis and degradation:
PSM were filtered to require the following fitted parameters ranges: K: 0-5, A: 0.67-1.5, B: 0-0.3, and were required to have a coefficient of determination R2 ≥ 0.7. PSM ratios across experiments were then grouped per protein and curve fitting and filtering was repeated on the protein level. Half-lives for each protein were calculated after taking into account the different rates of cell doubling (kcd) for each treatment condition using the following equation:
Proliferation Assay
A 40mM stock solution of ABD957 was serially diluted in DMSO to obtain 2000X concentrations. Next, twice-concentrated medium (2X) was obtained by diluting the drugs 1000-fold in culture medium. A solvent control was obtained by diluting DMSO 1000-fold in culture medium. To test the effect of ABD957 on cellular growth, cells were resuspended in fresh culture medium to a concentration of 6e5 cells/ml and seeded in a 96-well plate (75 μL/plate), or 5,000 cells/well in 50 μL. Subsequently, equal volume of 2X drugged medium or control was added. Each treatment was performed in triplicate. After 72 h in culture, 50 μL of each well were transferred to a white, flat-bottom 96 well plate and an equal volume of CellTiter Glo reagent was added. After 30 min of shaking at room temperature, luminescence was analyzed using a Tecan Infinite M200PRO or CLARIOstar (BMG Labtech) plate reader. Raw values were normalized using Prism GraphPad software.
Western Blot Analysis
A minimum of 2e6 cells per condition were pelleted and resuspended in fresh medium at 1e6 cells/mL containing compounds at the indicated concentration. Compound containing media were obtained by diluting a 1000-fold concentrated drug stock or vehicle (DMSO control) in fresh medium. Unless otherwise indicated, cells were treated for 4 hours, and then pelleted by centrifugation (500 RCF), washed in PBS, and frozen. Cells were resuspended in RIPA buffer (Thermo Scientific) supplemented with PhosSTOP (Sigma-Aldrich) and protease inhibitors (Roche) and lysed using a Branson Sonifier probe sonicator (3 rounds of 8 pulses, 40% duty cycle, output setting = 4). Lysates were separated on SDS-PAGE TG gels, and transferred to nitrocellulose membranes at 350 mA for 90 minutes, block and probe with primary and secondary antibodies in 5% milk in 1% TBS-T. Antibodies for Western blotting included anti-pan-Ras (Cell Signaling, 3965, 1:500), anti-N-RAS (Santa Cruz, sc-31, 1:1000), anti-Ras (G12D specific mutant) (Cell Signaling, 14429, 1:1000), anti-pERK (Cell Signaling, 4370, 1:1000), anti-ERK (Cell Signaling, 9107, 1:2500), anti-HSP90-HRP (Cell Signaling, 79641, 1:5000), anti-mouse HRP (Cell Signaling, 7076, 1:10,000), anti-rabbit HRP (Cell Signaling 7074 or Santa Cruz sc2030, 1:10,000), Li-cor IRDye 800CW Donkey anti-rabbit (1:10,000), Li-cor IRDye 680RD Goat anti-mouse (1:10,000). Blots were imaged on a Li-cor Odyssey (Model 9120) (pERK and ERK) or detected using chemiluminescence on a Bio-Rad ChemiDoc™ XRS imager. Densitometry analysis was performed using ImageJ software (NIH).
Live-cell imaging and Co-localization Analysis
2e5 ON or ONK control cells were plated onto 12 or 27 mm Nunc Glass Bottom Dishes the night prior to imaging and diluted with 400 μL complete RPMI medium so that the entire glass bottom was covered. For co-localization analysis between GFP-N-RasG12D and RFP-GALNT2 cells were treated with Cell Light Golgi RFP 2.0 reagent (ThermoFisher, cat# C10593) and incubated overnight and imaged the following day. For co-localization between GFP-N-RasG12D and plasma membrane, the day following plating, cells were diluted to 2 mL with complete RPMI containing Alexa-647 conjugated wheat germ agglutinin (ThermoFisher, cat# W32466) so that the final concentration was 5 μg/mL and transferred to a ZEISS LSM 880 laser scanning confocal microscope equipped with an incubator equilibrated to 37°C and 5% CO2 for 10 minutes prior to imaging. Cells were imaged using a C-Apochromat 63x/1.2 W autocorr M27 Oil objective with image size of ~200×200 μm. GFP and RFP were excited using a 633 and 488 nm laser lines and spectral filtering of emission bands was limited to 493-598 nm for GFP and 639-755 nm for RFP. For plasma membrane co-localization, images were acquired over 5 z-planes (~5 μm) every two minutes using sequential line scanning for 10 minutes prior to compound infusion. After 10 minutes, medium was infused with ABD957 to the indicated concentration and images collected for an additional 30 minutes using the same acquisition parameters. For Golgi co-localization, images were acquired at focal planes based on medial Golgi signal and imaged by sequential line scanning. In addition to sampling every 2 minutes, GFP only images were acquired by sampling every minute over five z-planes (~1 uM) using the similar acquisition parameters. Post-acquisition, images were exported as 16-bit tif files using zen blue and analyzed using the custom cell profiler pipeline “Correlation_Ras_Lectin_8-19-20.cppipe” or “Correlation_Ras_Golgi_8-19-20” generated in version 3.1.9. Briefly, cellular objects were identified using minimum cross entropy and offset invariant Pearson correlation between GFP and RFP plasma membrane signal within cellular objects was used for time-course analysis for each optical section collected, using a global threshold value of 15% of maximum59. Basal correlation was normalized for direct comparison of change in correlation with time across experiments. Aggregation of biological replicates of time course experiments was conducted from frames collected at 60 minutes post compound exposure for each optical section. For Golgi co-localization, cellular objects were selected by minimum cross entropy and Mander’s correlation coefficient between the fraction of total intracellular GFP signal that overlapped with the RFP-Golgi signal was calculated for each optical section after global thresholding at 15% of maximum intensity.
CRISPR/Cas9 KO lines
The following sgRNA sequenced were cloned in the pLX-sgRNA vector (Addgene plasmid # 50662): 5’-GTCAGAAATACTCACGCATGC-3’, targeting LYPLA1 (LYPLA1_4); 5’-GACAGCTGCTCACCTGGCACC-3’, targeting ABHD17A (ABHD17A_1). OCI-AML3 cells were transduced with either pLX-LYPLA1_4 or pLX-ABHD17A_1, as previously described,56 and selected with blasticidin. The lentiCRISPR v2 vector (Addgene plasmid # 52961) was modified to replace the puromycin resistance gene with a mCherry marker. Subsequently, the following sgRNA sequenced were cloned in lentiCRISPRv2-mCherry: 5’-GGTCCAGTTCAAGACATACCC-3’, targeting LYPLA2 (LYPLA2_3); 5’-GCATGGCACTGGAGTATGGCC-3’, targeting LYPLA2 (LYPLA2_4); 5’-GCGAGCAGCAAGATCCACAGA, targeting ABHD17B (ABHD17B_4). OCI-AML3 cells transduced with pLX-LYPLA1_4 were transduced with either LYPLA2_3 or LYPLA2_4; OCI-AML3 cells transduced with pLX-ABHD17A_1 were transduced with either ABHD17B_1or ABHD17B_4. mCherry+ cells were sorted and diluted to 4 cells/ml to allow isolation of single cell clones. After expansion, genomic regions around the sgRNA were amplified, cloned into a Zero Blunt TOPO PCR vector (Thermo Fisher) and sequenced to verify the presence of frameshift-disrupting insertions/deletions. The following knock-out clones were identified: LYPLA-DKO 1 (LYPLA1_4+LYPLA2_3), LYPLA-DKO 2 (LYPLA1_4+LYPLA2_4), ABHD17-DKO 1 (ABHD17A_1+ABHD17B_4), ABHD17-DKO 2 (ABHD17A_1+ABHD17B_4). For all clones, ABPP analysis was used to functionally verify the knock-out of respective serine hydrolases (Supplementary Fig. 7a).
Statistical analysis
Unless otherwise stated, quantitative data are expressed in bar and line graphs with mean ± s.d. (standard deviation, shown as an error bar) shown. Differences between two groups were examined using an unpaired two-tailed Student’s t-test with equal or unequal variance as noted. Significant P values were indicated (*P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001).
Extended Data
Extended Data Fig. 1. Characterization of ABHD17 inhibitors.
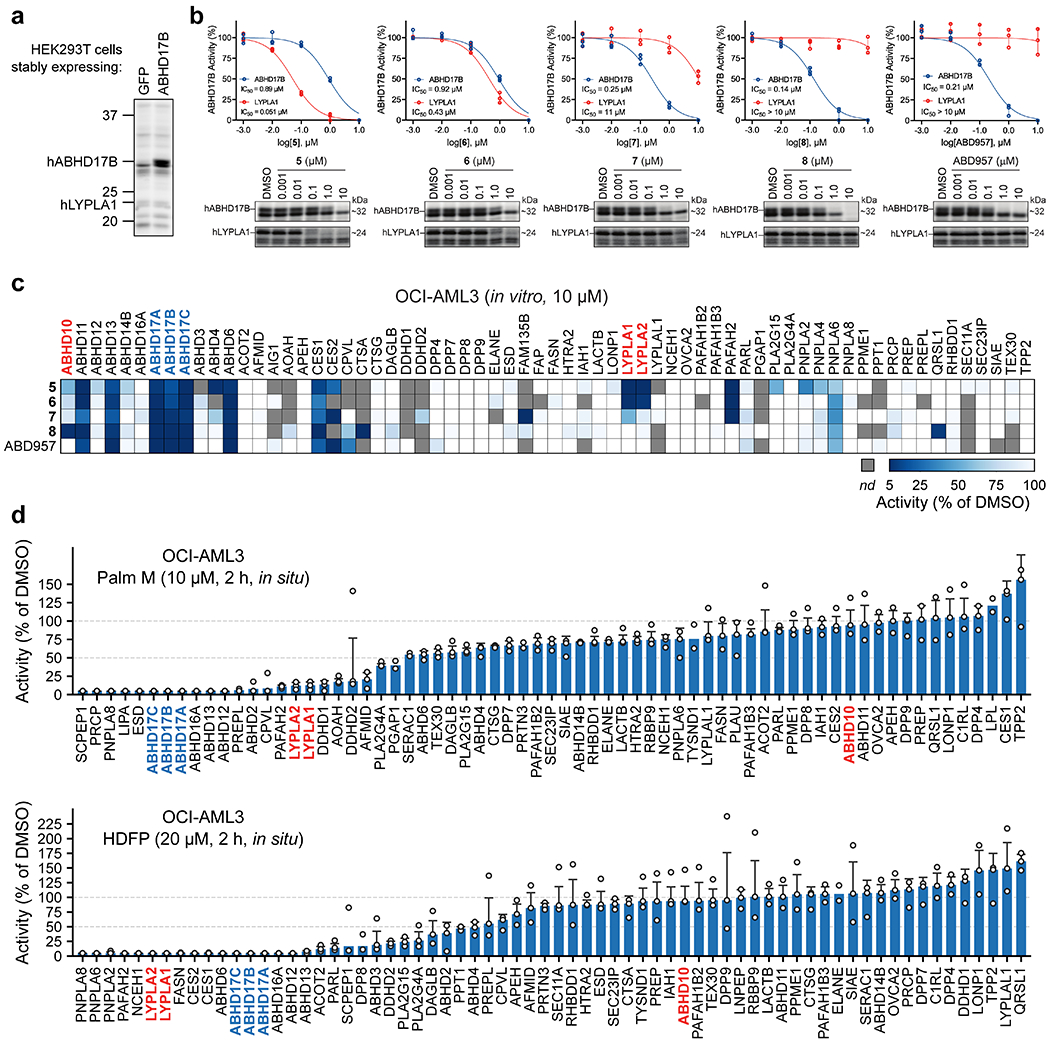
a, Representative gel-ABPP image showing FP-rhodamine labeling of recombinant human ABHD17B (hABHD17B) in proteomic lysates of stably transfected HEK293T cells. b, IC50 curves and representative gel-ABPP images of human ABHD17B and LYPLA1 activity in HEK293T cell lysate treated with compounds 5-8 and ABD957 for 30 min. Data represent average values ± s.d. (n = 3 independent experiments). c, MS-ABPP data of serine hydrolase activities in the particulate fraction of OCI-AML3 proteomes treated with compounds 5-8 and ABD957 for 30 min (10 μM). Data are from single experiments performed at the indicated concentrations for each compound. d, MS-ABPP data of serine hydrolase activities in OCI-AML3 cells treated in situ with Palm M (10 μM) or HDFP (20 μM) for 2 h. Data represent the median from three experiments corresponding to independent treatments of cells with compound (Palm M or HDFP) and error bars represent s.d.
Extended Data Fig. 2. Analysis of off-targets of ABD957 and control probes accounting for these serine hydrolases.
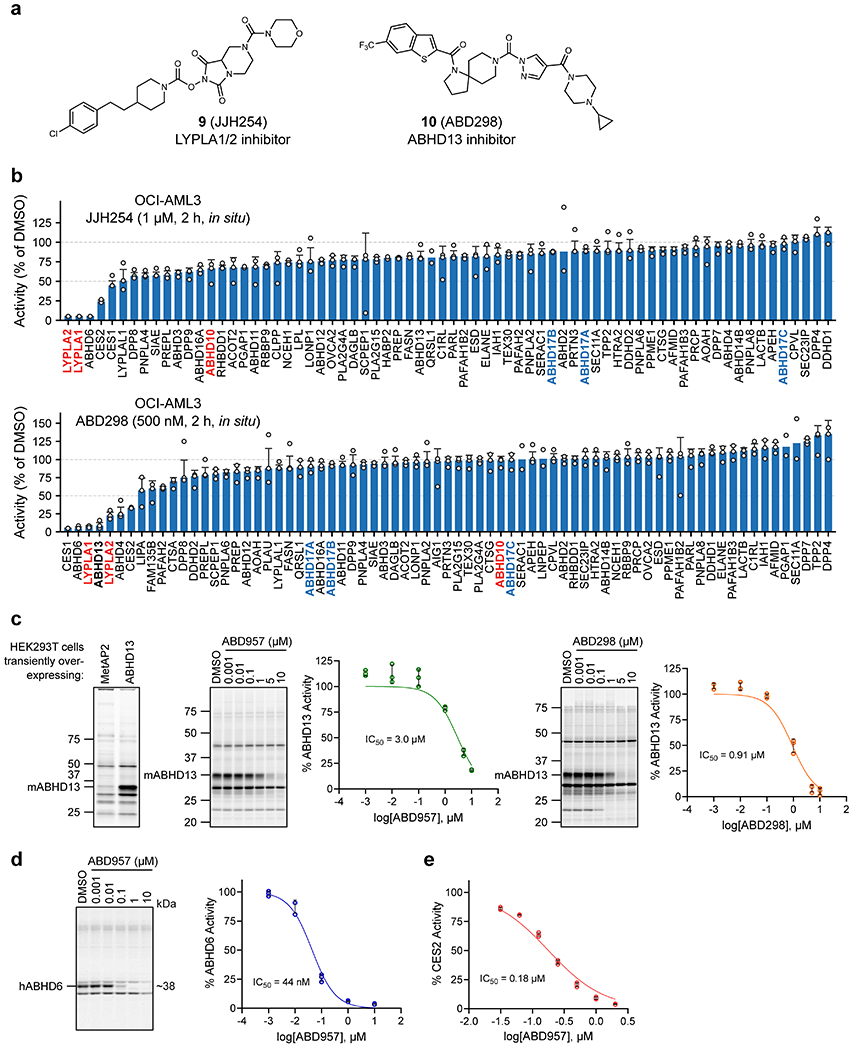
a, Structures of ABD298, an ABHD13 inhibitor, and JJH254, a previously reported LYPLA1/2 inhibitor25. b, MS-ABPP data of serine hydrolase activities in OCI-AML3 cells treated in situ with JJH254 (1 μM) or ABD298 (500 nM) for 2 h. Data represent the median from three experiments corresponding to independent treatments of cells with each compound (JJH254 or ABD298) and error bars represent standard deviation. c, IC50 curves and representative gel-ABPP images for ABD957 and ABD298 determined in proteomes of HEK293T cells expressing recombinant mouse ABHD13.
Lysates were treated in vitro with inhibitor for 30 minutes at 37 °C, followed by incubation with FP-Rh for 30 minutes, RT. Experiments were performed in triplicate. Error bars represent s.d. and center around the mean. d, IC50 curve and representative gel-ABPP image for ABD957 determined for endogenous human ABHD6 in PC3 cell proteomes following a 30 min inhibitor treatment. Experiments were performed in triplicate. Error bars represent s.d. and center around the mean. e, IC50 curve for ABD957 derived by measuring the CES2-dependent rate of hydrolysis (background substracted) of the substrate 4-nitrophenyl acetate (pNPA) in proteomes of HEK293T cells expressing recombinant human CES2 following a 30 min inhibitor treatment. The mean reaction velocity (Vmean, ΔAbs / min•μg protein) in mock- versus CES2- transfected proteomes was 8.7 ± 1.9 min−1•μg−1 and 31 ± 1.2 min−1•μg−1, respectively. Data presented for all IC50 determinations represent average values ± s.d. (n = 3 independent experiments).
Extended Data Fig. 3. Schematic of a dynamic palmitoylation assay using metabolic labeling with 17-ODYA.
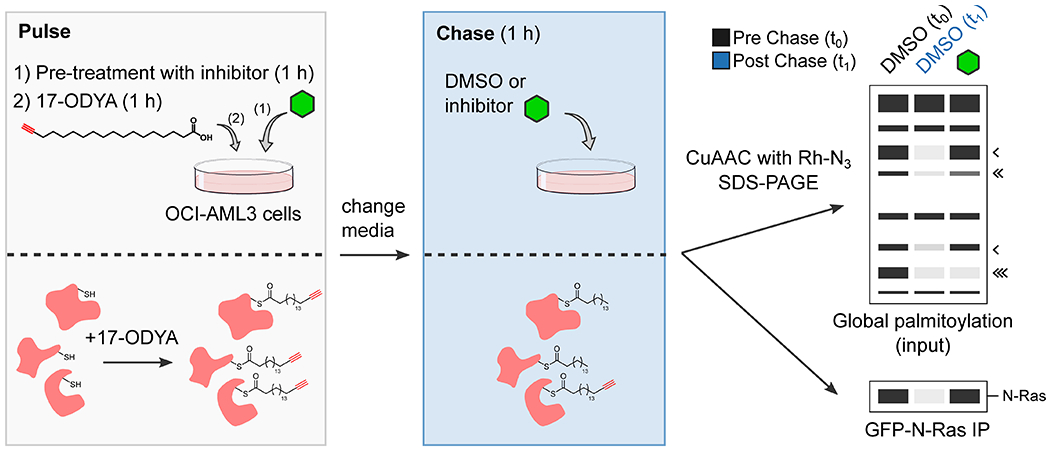
Black arrowheads next to mock gel mark proteins that show dynamic palmitoylation fully (one arrowhead), partially (two arrowheads), or not (three arrowheads) preserved by serine hydrolase inhibition.
Extended Data Fig. 4. Global palmitoylation effects of Palm M and ABD957 in leukemia cells.
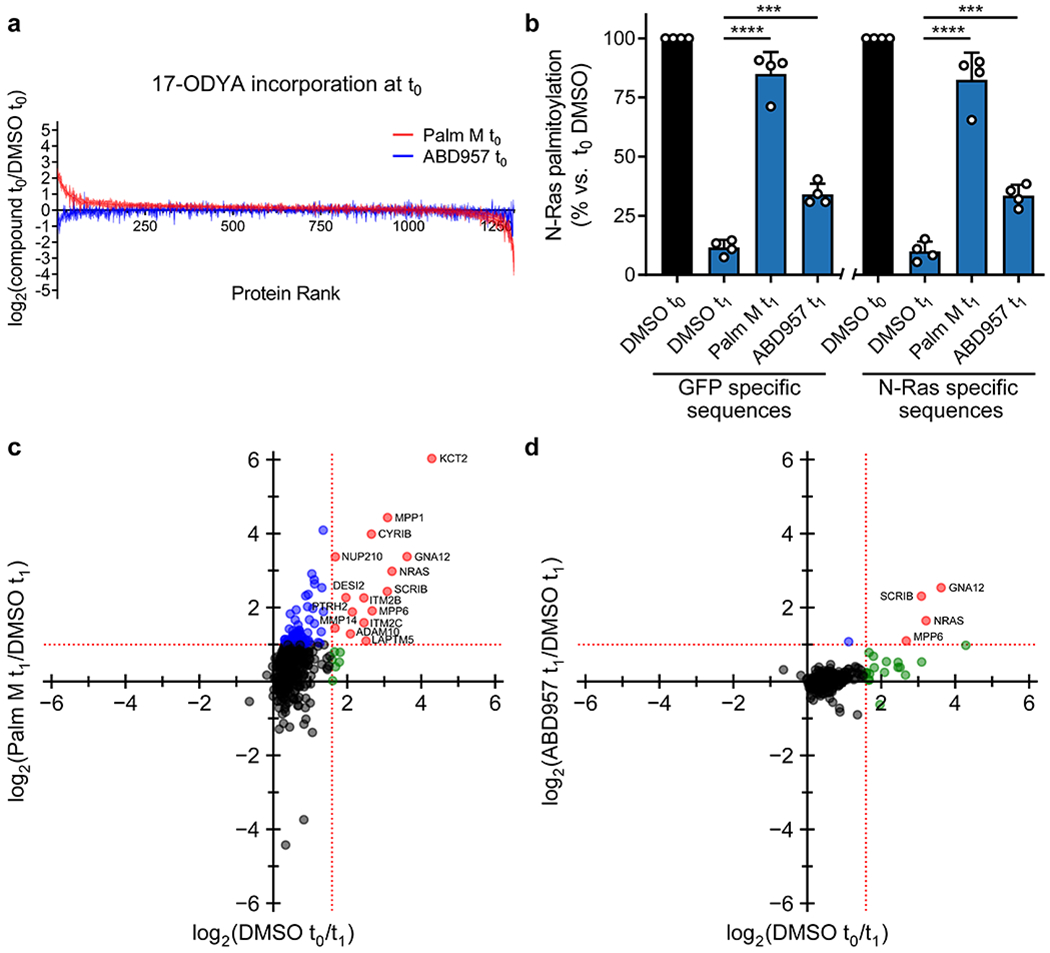
a, MS-based proteomics in OCI-AML3 cells showing the 17- ODYA labeling at t0. The results indicate that Palm M increases the apparent palmitoylation state of several proteins prior to the chase period and, as shown in Fig. 3a, some of these proteins are not dynamically palmitoylated (blue proteins in Fig. 3a). b, Bar graph quantifiying different categories of N-Ras peptides from MS-based proteomics experiments in ON cells. Data represent average values relative to DMSO t0 ± s.d. (n = 4 from two biological replicates). Statistical significance was calculated with unpaired two-tailed Student’s t-test with equal variance, ***P<0.001, ****P<0.0001 represent significant increase compared to DMSO t1 (also see Supplementary Dataset 3). P values were 5.5×10−5 (Palm M GFP specific), 1.8×10−4 (ABD957 GFP specific), 2.1×10−5 (Palm M N-Ras specific), 2.7×10−4 (ABD957 N-Ras specific). c, d, Scatter plots as described in Fig. 3a in ON cells.
Extended Data Fig. 5. Effects of ABD957 treatment on N-Ras turnover in OCI-AML3 cells.
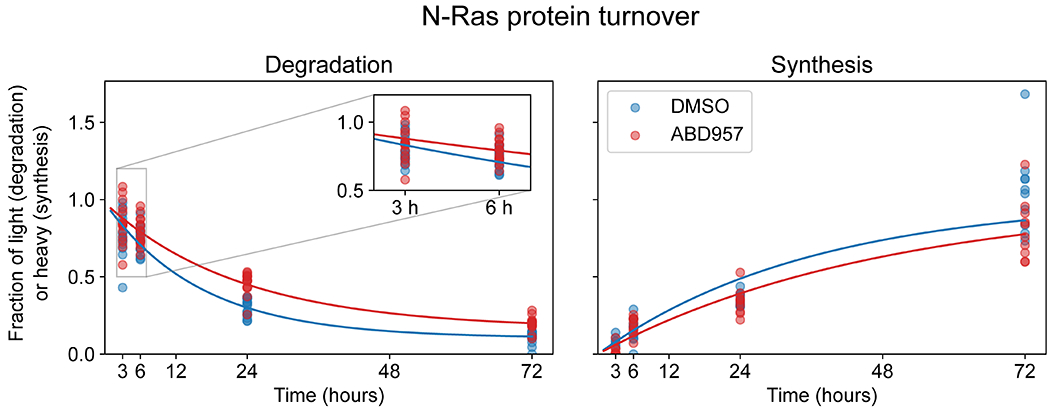
a, Degradation and synthesis curves for N-Ras in OCI-AML3 cells treated with DMSO or ABD957 (500 nM). Each dot represents a peptide spectrum match (PSM) from one of three biological replicates. OCI-AML3 cells grown in light SILAC media were pelleted and resuspended in heavy SILAC media containing DMSO or ABD957 and harvested at the indicated times. Also see Supplementary Dataset 4.
Extended Data Fig. 6. Global palmitoylation effects of Palm M and ABD957 in NB-4 cells.
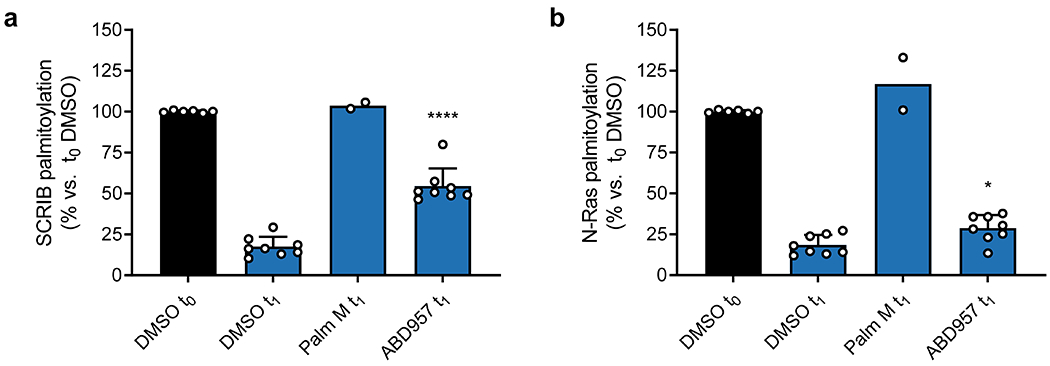
a, b, Bar graphs quantifying SCRIB and N-Ras palmitoylation from MS-based proteomic experiments in NB-4 cells. Data represent average values relative to DMSO t0 ± s.d. (biological replicates; n=2 for Palm M t1, n=8 for all other conditions).
Statistical significance was calculated with unpaired two-tailed Student’s t-test with equal variance, *P<0.05, ****P<0.0001 represent significant increase compared to DMSO t1 (also see Supplementary Dataset 2 and 3). P values were 6.5×10−7 (SCRIB), 0.013 (N-Ras).
Extended Data Fig. 7. Quantification of data shown in Figure 5f; average values ± s.d. (n = 4 independent experiments).
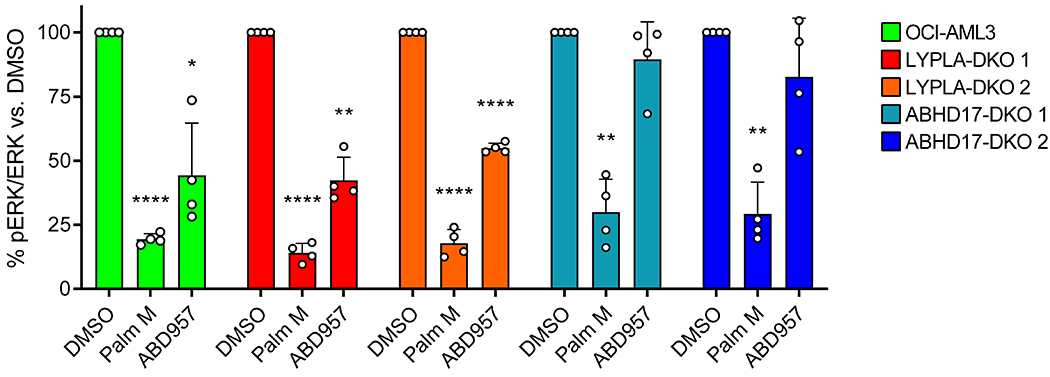
Statistical significance was calculated with unpaired two-tailed Student’s t-test with unequal variance compared to DMSO control, *P<0.05, **P<0.01, ****P<0.0001. P values were 4.8×10−6 (Palm M OCI-AML3), 0.012 (ABD957 OCI-AML3), 0.0016 (Palm M ABHD17-DKO 1), 0.0014 (Palm M ABHD17-DKO 2), 2.2×10−5 (Palm M LYPLA-DKO 1), 0.0010 (ABD957 LYPLA-DKO 1), 7.4×10−5 (Palm M LYPLA-DKO 2), 2.1×10−5 (ABD957 LYPLA-DKO 2).
Extended Data Fig. 8. MS-based proteomic analysis of dynamic palmitoylation in parental and ABHD17-DKO cells.
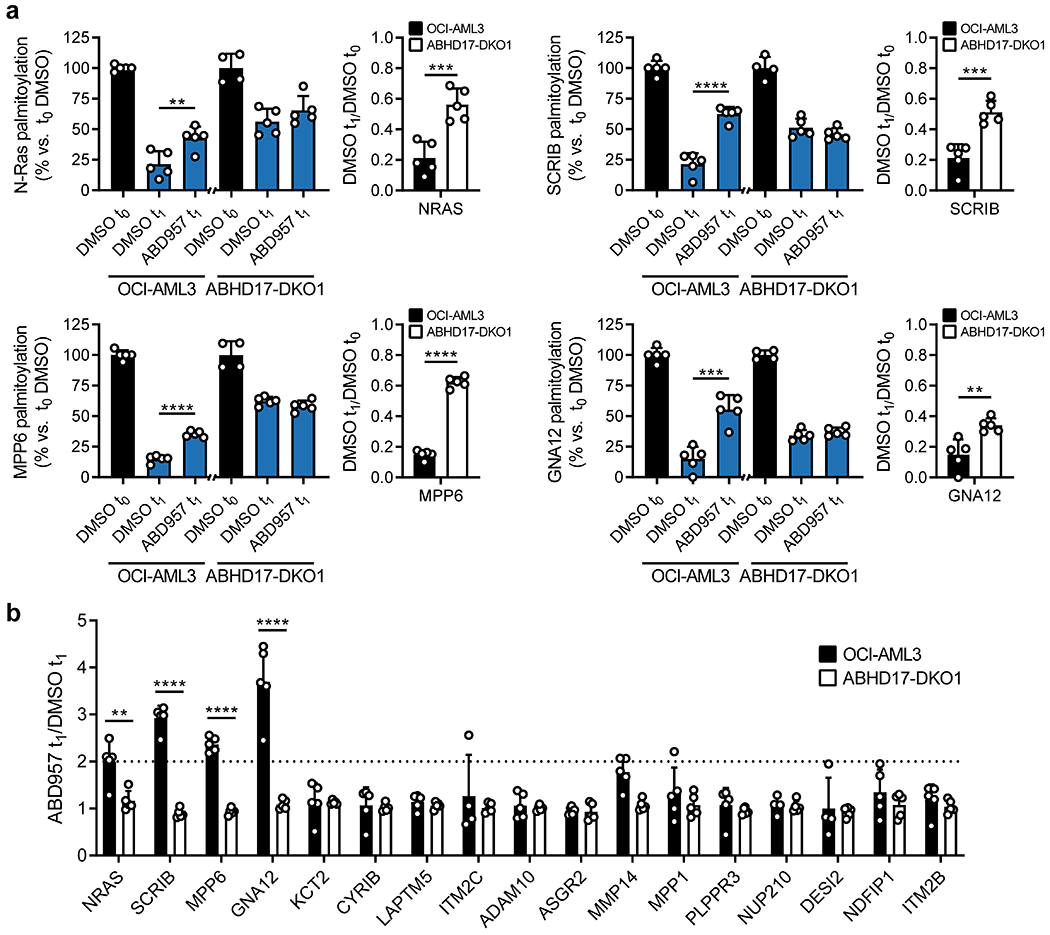
a, Profiles for ABD957-regulated palmitoylated proteins from mass spectrometry (MS)-based experiments of OCI-AML3 and ABHD17-DKO 1 cells performed as described in Extended Data Fig. 3. Cells were preincubated with ABD957 (500 nM) or DMSO for 1 h, metabolically labeled with 20 μM 17-ODYA for 1 h (t0) and chased with media lacking 17-ODYA for 1 h (t1) containing ABD957 (500 nM) or DMSO control. Data represent average values relative to each cell lines corresponding DMSO t0 control ± s.d. (biological replicates, n = 4 for DMSO t0 values, n=5 for all others). Statistical significance was calculated with unpaired two-tailed Student’s t-test with equal variance, *P<0.05, **P<0.01, ***P<0.001, ****P<0.0001, compared to DMSO t1 for left panels or compared to DMSO t1/t0 ratio between OCI-AML3 and ABHD17-DKO1 cells for right panels. P values were 0.0096 (OCI-AML3 ABD957 N-Ras), 7.8×10−4 (OCI-AML3 DMSO t1/t0 N-Ras), 2.4×10−5 (OCI-AML3 ABD957 SCRIB), 4.4×10−4 (OCI-AML3 DMSO t1/t0 SCRIB), 1.9×10−6 (OCI-AML3 ABD957 MPP6), 1.2×10−8 (OCI-AML3 DMSO t1/t0 MPP6), 3.9×10−4 (OCI-AML3 ABD957 GNA12), 0.0044 (OCI-AML3 DMSO t1/t0 GNA12). b, Effect of ABD957 on dynamically palmitoylated proteins in OCI-AML3 and ABHD17-DKO1 cells. Data represent average values ± s.d. (n = 5 biological replicates). Statistical significance was calculated for proteins with >2-fold increases in ABD957-treated OCI-AML3 cells with unpaired two-tailed Student’s t-test with equal variance, **P<0.01, ****P<0.0001 compared to ABHD17-DKO1 cells. P values were 0.0049 (N-Ras), 2.3×10−7 (SCRIB), 7.6×10−8 (MPP6), 8.1×10−5 (GNA12).
Supplementary Material
MS-ABPP studies for target profiling
Data from mass spectrometry based studies of hydroxylamine-sensitive 17-ODYA labeled proteins
Data from mass spectrometry based studies of dynamic Palmitoylation
Data from SILAC-TMT mass spectrometry studies to assess protein turnover
Effect of inhibitor treatment on GFP-N-Ras localization in ON cells. Representative time-lapse videos from ON cells before and after treatment with Palm M (10 μM) (a), ABD957 (500 nM) (b), or DMSO control (c). Green channel shows GFP-N-Ras signal. Frames were acquired every 90s over 680x 680 pixels for 10 frames prior to compound infusion and a total of 35 frames is shown, with each frame requiring 13.37 seconds to scan. Relative acquisition time is shown in red text in the upper left. Frames where compound infusion begins are captioned in the upper left in red text with the name of compound used for the experiment. Data shown are a representative of three biological replicates. One medial z-plane over a 400 x 400-pixel area is shown for clarity. Scale bar, 10 μm.
Acknowledgements.
This work was supported by grants from the NIH (CA193994, CA231991, and CA72614), an American Cancer Society postdoctoral fellowship PF-18-217-01-CDD (J.R.R.), the Leukemia and Lymphoma Society (LLS Fellowship 5465-18 to N.A.Z.), and the Damon Runyon Cancer Research Foundation (Fellowship DRG-2149-13 to A.J.F). We thank Scott Henderson for assistance with the microscopy studies, and Titus Jung and Robin Park for assistance with processing the SILAC-TMT proteomic data.
Footnotes
Competing interests. The authors declare no competing interests.
Supplementary Information. Included are Supplementary Figures 1–5, Supplementary Datasets 1–4, Supplementary Note – Chemistry Methods, Supplementary Tables 1–3, and Supplementary Video 1.
Code availability
Code used to process mass spectrometric and imaging data are available on GitHub.
Data availability
All mass spectrometry data are available via ProteomeXchange with identifier PXD023932. All other data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Schubbert S, Shannon K & Bollag G Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer 7, 295–308 (2007). [DOI] [PubMed] [Google Scholar]
- 2.Ostrem JM, Peters U, Sos ML, Wells JA & Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–51 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Canon J et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 575, 217–223 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Omerovic J, Laude AJ & Prior IA Ras proteins: paradigms for compartmentalised and isoform-specific signalling. Cellular and molecular life sciences : CMLS 64, 2575–89 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hancock JF, Paterson H & Marshall CJ A polybasic domain or palmitoylation is required in addition to the CAAX motif to localize p21ras to the plasma membrane. Cell 63, 133–139 (1990). [DOI] [PubMed] [Google Scholar]
- 6.Rocks O et al. An acylation cycle regulates localization and activity of palmitoylated Ras isoforms. Science 307, 1746–52 (2005). [DOI] [PubMed] [Google Scholar]
- 7.Dekker FJ et al. Small-molecule inhibition of APT1 affects Ras localization and signaling. Nat Chem Biol 6, 449–56 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Hedberg C et al. Development of highly potent inhibitors of the Ras-targeting human acyl protein thioesterases based on substrate similarity design. Angew Chem Int Ed Engl 50, 9832–7 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Martin BR, Wang C, Adibekian A, Tully SE & Cravatt BF Global profiling of dynamic protein palmitoylation. Nat Methods 9, 84–9 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duncan JA & Gilman AG A cytoplasmic acyl-protein thioesterase that removes palmitate from G protein alpha subunits and p21(RAS). J Biol Chem 273, 15830–7 (1998). [DOI] [PubMed] [Google Scholar]
- 11.Rusch M et al. Identification of acyl protein thioesterases 1 and 2 as the cellular targets of the Ras-signaling modulators palmostatin B and M. Angew Chem Int Ed Engl 50, 9838–42 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Lin DT & Conibear E ABHD17 proteins are novel protein depalmitoylases that regulate N-Ras palmitate turnover and subcellular localization. Elife 4, e11306 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martin BR & Cravatt BF Large-scale profiling of protein palmitoylation in mammalian cells. Nat Methods 6, 135–8 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yokoi N et al. Identification of PSD-95 Depalmitoylating Enzymes. J Neurosci 36, 6431–44 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jia L et al. A mechanism regulating G protein-coupled receptor signaling that requires cycles of protein palmitoylation and depalmitoylation. J Biol Chem 289, 6249–57 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Won SJ & Martin BR Temporal Profiling Establishes a Dynamic S-Palmitoylation Cycle. ACS Chem Biol 13, 1560–1568 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang MM, Tsou LK, Charron G, Raghavan AS & Hang HC Tandem fluorescence imaging of dynamic S-acylation and protein turnover. Proc Natl Acad Sci U S A 107, 8627–32 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cao Y et al. ABHD10 is an S-depalmitoylase affecting redox homeostasis through peroxiredoxin-5. Nat Chem Biol 15, 1232–1240 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adibekian A et al. Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat Chem Biol 7, 469–78 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chang JW, Nomura DK & Cravatt BF A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem Biol 18, 476–84 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsu KL et al. DAGLbeta inhibition perturbs a lipid network involved in macrophage inflammatory responses. Nat Chem Biol 8, 999–1007 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Niphakis MJ & Cravatt BF Enzyme inhibitor discovery by activity-based protein profiling. Annu Rev Biochem 83, 341–77 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Otrubova K, Chatterjee S, Ghimire S, Cravatt BF & Boger DL N-Acyl pyrazoles: Effective and tunable inhibitors of serine hydrolases. Bioorg Med Chem 27, 1693–1703 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Patricelli MP & Cravatt BF Activity-based protein profiling: the serine hydrolases. Proc Natl Acad Sci U S A 96, 14694–9 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cognetta AB 3rd et al. Selective N-Hydroxyhydantoin Carbamate Inhibitors of Mammalian Serine Hydrolases. Chem Biol 22, 928–37 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zambetti NA et al. Genetic disruption of N-RasG12D palmitoylation perturbs hematopoiesis and prevents myeloid transformation in mice. Blood 135, 1772–1782 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Charron G et al. Robust fluorescent detection of protein fatty-acylation with chemical reporters. J Am Chem Soc 131, 4967–75 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Rostovtsev VV, Green LG, Fokin VV & Sharpless KB A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew Chem Int Ed Engl 41, 2596–9 (2002). [DOI] [PubMed] [Google Scholar]
- 29.Drisdel RC & Green WN Labeling and quantifying sites of protein palmitoylation. Biotechniques 36, 276–85 (2004). [DOI] [PubMed] [Google Scholar]
- 30.Zecha J et al. Peptide Level Turnover Measurements Enable the Study of Proteoform Dynamics. Mol Cell Proteomics 17, 974–992 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen B et al. ZDHHC7-mediated S-palmitoylation of Scribble regulates cell polarity. Nat Chem Biol 12, 686–93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kamijo A, Saitoh Y, Ohno N, Ohno S & Terada N Immunohistochemical study of the membrane skeletal protein, membrane protein palmitoylated 6 (MPP6), in the mouse small intestine. Histochem Cell Biol 145, 81–92 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Jones TL & Gutkind JS Galpha12 requires acylation for its transforming activity. Biochemistry 37, 3196–202 (1998). [DOI] [PubMed] [Google Scholar]
- 34.Saraceno C et al. SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis 5, e1547 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Choi SI, Vidal R, Frangione B & Levy E Axonal transport of British and Danish amyloid peptides via secretory vesicles. FASEB J 18, 373–5 (2004). [DOI] [PubMed] [Google Scholar]
- 36.Xu J et al. Inhibiting the palmitoylation/depalmitoylation cycle selectively reduces the growth of hematopoietic cells expressing oncogenic Nras. Blood 119, 1032–5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao W et al. A New Bliss Independence Model to Analyze Drug Combination Data. J Biomol Screen 19, 817–21 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Ahearn IM, Haigis K, Bar-Sagi D & Philips MR Regulating the regulator: post-translational modification of RAS. Nat Rev Mol Cell Biol 13, 39–51 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ryan MB & Corcoran RB Therapeutic strategies to target RAS-mutant cancers. Nat Rev Clin Oncol 15, 709–720 (2018). [DOI] [PubMed] [Google Scholar]
- 40.Cox AD, Der CJ & Philips MR Targeting RAS Membrane Association: Back to the Future for Anti-RAS Drug Discovery? Clin Cancer Res 21, 1819–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cox AD, Fesik SW, Kimmelman AC, Luo J & Der CJ Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 13, 828–51 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hernandez JL et al. APT2 Inhibition Restores Scribble Localization and S-Palmitoylation in Snail-Transformed Cells. Cell Chem Biol 24, 87–97 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vartak N et al. The autodepalmitoylating activity of APT maintains the spatial organization of palmitoylated membrane proteins. Biophys J 106, 93–105 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kathayat RS et al. Active and dynamic mitochondrial S-depalmitoylation revealed by targeted fluorescent probes. Nat Commun 9, 334 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Levental I, Lingwood D, Grzybek M, Coskun U & Simons K Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci U S A 107, 22050–4 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chandra A et al. The GDI-like solubilizing factor PDEdelta sustains the spatial organization and signalling of Ras family proteins. Nat Cell Biol 14, 148–58 (2011). [DOI] [PubMed] [Google Scholar]
- 47.Zhou M et al. VPS35 binds farnesylated N-Ras in the cytosol to regulate N-Ras trafficking. J Cell Biol 214, 445–58 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nakai K & Horton P PSORT: a program for detecting sorting signals in proteins and predicting their subcellular localization. Trends Biochem Sci 24, 34–6 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Patricelli MP, Giang DK, Stamp LM & Burbaum JJ Direct visualization of serine hydrolase activities in complex proteomes using fluorescent active site-directed probes. Proteomics 1, 1067–71 (2001). [DOI] [PubMed] [Google Scholar]
- 50.Chang JW, Cognetta AB 3rd, Niphakis MJ & Cravatt BF Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem Biol 8, 1590–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hatfield MJ et al. Biochemical and molecular analysis of carboxylesterase-mediated hydrolysis of cocaine and heroin. Br J Pharmacol 160, 1916–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Inloes JM et al. The hereditary spastic paraplegia-related enzyme DDHD2 is a principal brain triglyceride lipase. Proc Natl Acad Sci U S A 111, 14924–9 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xu T et al. ProLuCID: An improved SEQUEST-like algorithm with enhanced sensitivity and specificity. J Proteomics 129, 16–24 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cociorva D, D LT & Yates JR Validation of tandem mass spectrometry database search results using DTASelect. Curr Protoc Bioinformatics Chapter 13, Unit 13 4 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Zuber J et al. An integrated approach to dissecting oncogene addiction implicates a Myb-coordinated self-renewal program as essential for leukemia maintenance. Genes Dev 25, 1628–40 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Burgess MR et al. Preclinical efficacy of MEK inhibition in Nras-mutant AML. Blood 124, 3947–55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang Y et al. Expedited mapping of the ligandable proteome using fully functionalized enantiomeric probe pairs. Nat Chem 11, 1113–1123 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vinogradova EV et al. An Activity-Guided Map of Electrophile-Cysteine Interactions in Primary Human T Cells. Cell 182, 1009–1026 e29 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adler J & Parmryd I Quantifying Colocalization by Correlation: The Pearson Correlation Coefficient is Superior to the Mander’s Overlap Coefficient. Cytometry Part A 77a, 733–742 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
MS-ABPP studies for target profiling
Data from mass spectrometry based studies of hydroxylamine-sensitive 17-ODYA labeled proteins
Data from mass spectrometry based studies of dynamic Palmitoylation
Data from SILAC-TMT mass spectrometry studies to assess protein turnover
Effect of inhibitor treatment on GFP-N-Ras localization in ON cells. Representative time-lapse videos from ON cells before and after treatment with Palm M (10 μM) (a), ABD957 (500 nM) (b), or DMSO control (c). Green channel shows GFP-N-Ras signal. Frames were acquired every 90s over 680x 680 pixels for 10 frames prior to compound infusion and a total of 35 frames is shown, with each frame requiring 13.37 seconds to scan. Relative acquisition time is shown in red text in the upper left. Frames where compound infusion begins are captioned in the upper left in red text with the name of compound used for the experiment. Data shown are a representative of three biological replicates. One medial z-plane over a 400 x 400-pixel area is shown for clarity. Scale bar, 10 μm.
Data Availability Statement
All mass spectrometry data are available via ProteomeXchange with identifier PXD023932. All other data that support the findings of this study are available from the corresponding authors upon reasonable request.