

Summary:
Organizing a rational treatment strategy for patients with multifocal structural brain injuries and disorders of consciousness (DOC) is an important and challenging clinical goal. Among potential clinical end points, restoring elements of communication to DOC patients can support improved patient care, caregiver satisfaction, and patients’ quality of life. Over the past decade, several studies have considered the use of the anterior forebrain mesocircuit model to approach this problem because this model proposes a supervening circuit-level impairment arising across DOC of varying etiologies. We review both the conceptual foundation of the mesocircuit model and studies of mechanisms underlying DOC that test predictions of this model. We consider how this model can guide therapeutic interventions and discuss a proposed treatment algorithm based on these ideas. Although the approach reviewed originates in the evaluation of patients with chronic DOC, we consider some emerging implications for patients in acute and subacute settings.
Keywords: Mesocircuit, Brain injury, Stimulants, Disorders of consciousness, Pharmacology
This review addresses the clinician’s need to understand the theoretical mechanisms underlying a wide range of neuromodulation approaches used to support recovery from disorders of consciousness (DOC) arising after structural brain injuries. Patients with severe structural brain injuries producing DOC are typically not given systematic empirical time trials of pharmacotherapeutic agents. On the one hand, only limited, emerging, evidence-based recommendations for DOC treatments are available based on small numbers of existing studies.1,2 Moreover, the marked heterogeneity of structural brain injuries arising from varying etiologies presents a considerable challenge to approaching the individual patient. Developing principles to engage a rational polypharmacy for patients in the chronic care setting has been largely lacking and consideration of any extensions of the approaches taken in chronic DOC patients to potential interventions in the acute phase of coma or other DOC is also lacking. Here, we organize an approach to short-time pharmacological challenges in patients with chronic DOC based on the anterior forebrain mesocircuit model. The potential use and limitations of some neuromodulation approaches in the context of treatment of acute and subacute DOC are considered with important caveats reflecting the very limited knowledge of advancing any treatments efforts for DOC patients in these settings. We explain the foundations in neuroanatomy, neurophysiology, and molecular neuroscience underpinning our empirical strategy and organize an approach to sequential time trials with review of side effects, drug interactions, unknowns, and future directions.
Management of patients with chronic DOC is often vexing for physicians who often feel left without a sensible framework for clinical decision making and systematic efforts to engage patients and their surrogates over time. Although patients with DOC are typically not given empirical time trials for therapeutics after structural brain injuries, most patients reaching the level of minimally conscious state (MCS) track a slow recovery over time.3,4 In this context, specific goals of care can be considered particularly recovery of communication, which is independent of severe disability or total physical dependency.5 Late recovery past 1 year of interactive communication and varying elements of executive function is not uncommon in MCS.3 Although such transitions may not change a categorization of severe disability and a lack of physical independence, they can fundamentally alter the patient’s interaction with the world around them, their caregiver’s and family’s sense of their personhood, and improve the ability to meet their needs and preferences.5 Keeping these goals of care in mind can guide the physician’s efforts to approach each DOC patient with short-time pharmacological challenges. Appropriate use of measurement tools for behavioral assessments of function in DOC is outside the scope of this review.6
We present a strategy based on the published literature and an empirically organized algorithm for short-time trials of pharmacologic agents in patients with complex brain injuries based on our collective and, some instance, joint experience with DOC. The underlying approach is informed by a proposed mesocircuit model that integrates the circuit-level mechanisms that arise as common aspects of pathophysiology of severe brain injuries,7–9 with the known mechanisms and responses observed with stimulants,10,11 and common mechanisms and effects of anesthetic agents.12
MESOCIRCUIT MODEL
The anterior forebrain mesocircuit model for recovery of consciousness (or “mesocircuit model”) provides an organizing framework for understanding mechanisms underlying DOC following structural brain injuries and the process of recovery from complex structural brain injuries6,13–15 (Fig. 1). The model further suggests a set of principles for rationalizing therapeutic approaches that use pharmacologic and electrical stimulation techniques.13,16,17 We focus primarily on known pharmacological strategies and the potential role of polypharmacy approaches that might provide synergistic interactions of neuromodulators and medications, providing more direct support for long-range excitatory pathways that crucially determine functional recovery in DOC.
FIG. 1.
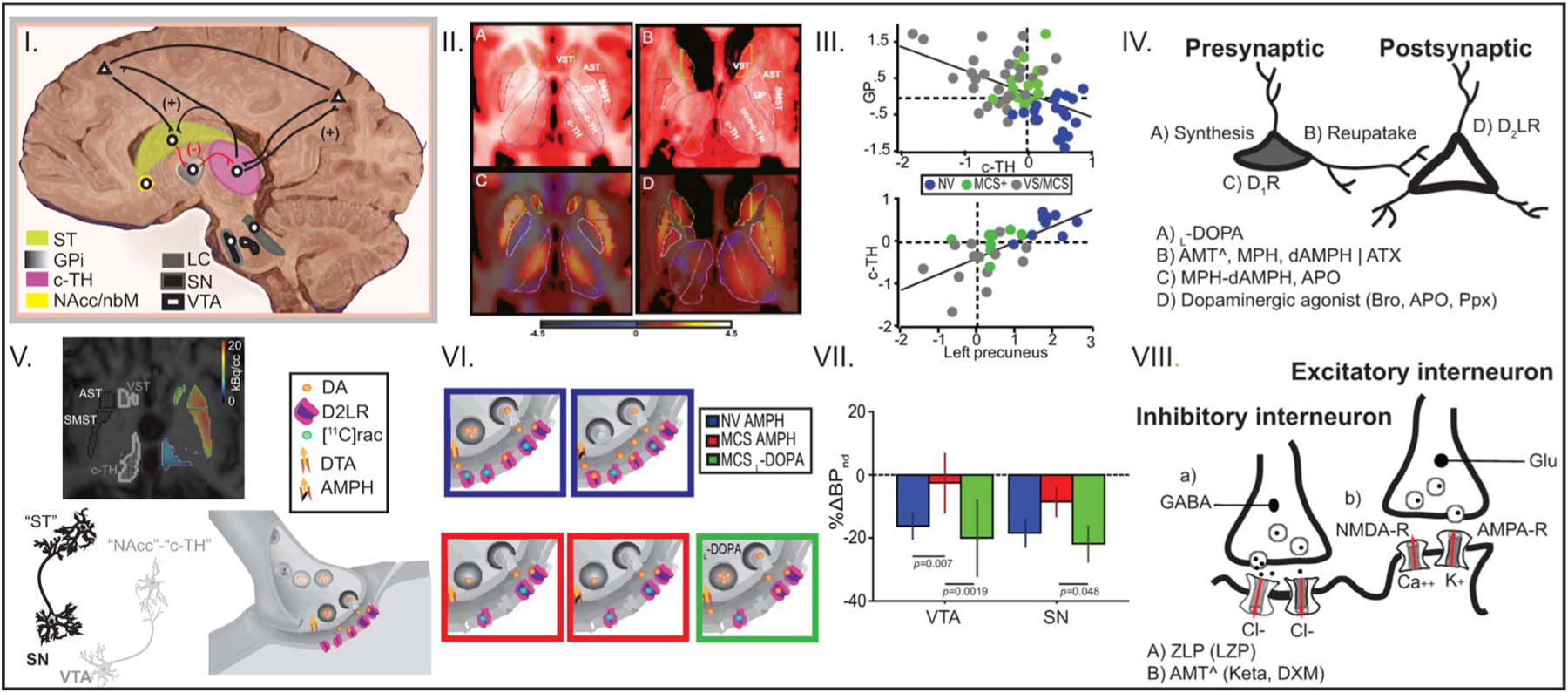
I, Anterior forebrain “mesocircuit.” Sagittal illustration displaying key cortical and subcortical components of the anterior forebrain mesocircuit vulnerable to effects of severe brain injuries and widespread cerebral deafferentation. Lines in black and red represent the direction of the projections for branching axons. (+) denotes “excitatory” projections and (−) denotes “inhibitory” projections. Following multifocal brain injuries that produce widespread deafferentation and neuronal cell loss, a functional disfacilitation of the c-TH reduces activity across thalamocortical projections from this structure to the frontal cortex, posterior medial parietal cortex, and striatum. Downregulation of neuronal activity across these structures is further generated by the release of inhibition of the globus pallidus producing an active thalamic inhibition. Collectively, an overall result of marked reduction of activity across the entire mesocircuit is expected following different mechanisms of brain injury. c-TH, central thalamus; GPi, globus pallidus; LC, locus coeruleus; NAcc/nbM, nucleus accumbens/nucleus basalis of Meynert; SN, substantia nigra pars compacta; ST, striatum; VTA, ventral tegmental area. II, Mesocircuit component downregulation. A and B, Axial anatomical high-resolution MRI in representative normal volunteer (NV) and brain-injured subjects (BI), respectively, showing locations and manual delimitation for ventral, associative, and sensorimotor striatum (VST, AST, and SMST, respectively), globus pallidus (GP), central and nonc-TH (c-TH and non-c-TH, respectively), and color scheme for regions of interest (VST, yellow; AST, green; SMST, red; GP, white; c-TH, magenta; non-c-TH, violet). C and D, [18F]FDG-PET, T1-MRI fusion in representative NV and BI subjects, respectively. NV demonstrates symmetric pattern of relatively increased c-TH metabolism compared with non–c-TH. A marked asymmetry of thalamic metabolism with loss of contrast in c-TH compared with non–c-TH metabolism is evident in BI subject. III, Mesocircuit downregulation correlates with clinical impairment in DOC. Group data displaying mean normalized uptake values of glucose metabolism in deep brain structures measured with [18F]-FDG PET in NV and BI subjects. Above, the bivariate scatter plot demonstrates an inverse linear correlation between glucose metabolic rate of the c-TH (x-axis) and the GP (y-axis), P < 0.001. Below, the bivariate scatter plot demonstrates a linear correlation between glucose metabolic rate of the left precuneus (x-axis) and the c-TH (y-axis) (P < 0.001). c-TH, central thalamus; GP, globus pallidus; MCS, minimally conscious state; MCS(1), MCS “plus”; NV, normal volunteer; VS, vegetative state. IV, Dopaminergic (and noradrenergic, NA) pharmacological neuromodulation of the mesocircuit. Bodies of neurons located in the SNc and VTA (presynaptic level) project to target postsynaptic neurons in the sensorimotor and limbic striatum, prefrontal cortex, and c-TH. Dopaminergic neuromodulation (NA as well) can be achieved using drugs that target the selective mechanisms at the presynaptic level (A, B, C), and/or with drugs targeting the postsynaptic level (D). AMT, amantadine; APO, apomorphine; ATX, atomoxetine; Bro, bromocriptine; dAMPH, dextroamphetamine; MPH, methylphenidate; Ppx, pramipexole. V and VI, Tested model of mesocircuit pharmacological dopaminergic neuromodulation. Above, [11C]raclopride-PET-MRI fusion of MCS1 displaying ventral, associative, and sensorimotor striatum (VST, AST, and SMST, respectively) and c-TH. Below, monosynaptic nigrostriatal pathway from substantia nigra (SN) and mesolimbic and mesothalamic pathways from ventral tegmentum (VT) to target nuclei, respectively; a dopaminergic terminal at rest in target structures shows background tonic dopamine (DA) leaving unoccupied D2-like receptors (D2LRs). In NV, [11C]raclopride binds at rest to the unoccupied D2LR (shown in 1st blue square in the upper panel). Dopamine transporter reuptake (DTA) blockade with AMPH in NV initiates a pharmacologically induced phasic responses that increases DA levels at the synaptic cleft, reducing D2LR occupancy by [11C]raclopride (second blue square in the upper panel). Lack of this physiologic reduction following AMPH is a marker of a presynaptic deficit as predicted to occur in MCS (see first and second red squares in the lower panel for rest and post-AMPH conditions, respectively). In this setting, reversal of the presynaptic dopaminergic deficit is expected with administration of a single, high dose of the dopamine precursor L-DOPA (see green square in the lower panel). VII, Mesocircuit dopamine response with L-DOPA administration. A, Bars show mean delta percentage change in binding potential nondisplaceable (%ΔBPnd) and 95% confidence interval in 11 NV using AMPH, 6 MCS patients using AMPH, and 4 MCS using L-DOPA. A clear reversibility of the presynaptic deficit from VT and SN follows a single dose of L-DOPA, reaching physiological values obtained from NV. VIII, Theoretical framework for mesocircuit dopaminergic/glutamatergic interplay. In a context of a presynaptic dopaminergic deficit, a GABAergic modulation prevalence over glutamatergic modulation is proposed. An excess of GABA release induces hyperpolarization of the pyramidal cell. GABAergic activation impedes removal of the Mg++ block in the NMDA-R, blocking Ca++ entry. Lack of Ca++ influx disrupts signaling, resulting in the lower expression of AMPA-R and perpetuating pyramidal cell hyperpolarization that disrupts NMDA receptor–induced synaptic plasticity. NMDA-R antagonists may not be effective enough to restore glutamatergic neurotransmission unless a background dopamine function is restored. AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; AMPA-R, AMPA receptor; AMPH, amphetamine; AST, associative striatum; FDG-PET, fluorodeoxyglucose positron emission tomography; NMDA-R, N-methyl-D-aspartate receptor; SMST, somatosensory striatum; VST, ventral striatum.
The core idea of the mesocircuit model is that the final common mechanism underlying varying etiologies of structural brain injuries producing DOC is a broad downregulation of background synaptic activity across primary components of the anterior forebrain mesocircuit: frontal cortical regions (most importantly, medial frontal cortices and anterior cingulate cortex), central thalamus (c-TH, most importantly, the central lateral nucleus), and the striatum. These components support brain arousal regulation through a variety of specializations of local circuit modulations at the cortical and striatal levels and receive direct neuromodulatory control from brain stem and basal forebrain “arousal regulation” systems.18–20 Neurons within central regions of the thalamus project broadly across the frontal–striatal system and have more widespread but selective anatomical connectivity across the entire forebrain.20,21 A “central thalamus” is more a physiological, functional distinction than one that is defined by sharp anatomical boundaries.22–24 Central thalamic neurons have primary projection to supragranular and infragranular layers of the cerebral cortex,25 wide arborization over rostral striatum,26 and participate in a range of frontal executive functions such as sustained attention and working memory.22–24 All forms of severe brain injuries resulting in DOC are associated with marked deafferentation of connections from the c-TH and its striatal and cortical targets.8,9,27 For multifocal brain injuries, this correlation arises from the wide point-to-point connectivity of the c-TH with the entire corticothalamic system,28 leading to integration of loss of inputs of central thalamic neurons from many distant sources; in the case of DOC produced by focal injuries to the c-TH, both disfacilitation29 of cortical and striatal targets as a result of loss of excitatory thalamocortical and thalamostriatal projections and loss of the primary arousal regulation functions of medial frontal cortical regions (anterior cingulate and medial frontal,30,31 receiving strong monosynaptic input from the central lateral nucleus) play a key role. In either type of pathology, the injuries lead to marked reduction of thalamocortical and thalamostriatal outflow. The resulting reduction of outputs from the central thalamic neurons is expected to produce at least two crucial consequences: (1) a drop-off of this long-range excitation withdraws important afferent drive to medial frontal cortical regions critical to arousal regulation; these frontal cortical regions have direct innervation of brain stem and basal forebrain “arousal systems,” and (2) reduction of both corticostriatal and thalamostriatal input to the medium spiny neurons of the striatum can be expected to produce a sharp drop in firing rates from the medium spiny neurons. These neurons require high rates of background synaptic activity to remain sufficiently depolarized to fire action potentials.32 With broad decreases in background activity provided by corticostriatal and thalamostriatal inputs, the medium spiny neurons may fail to reach firing thresholds, and the loss of this active inhibition from the striatum consequently allows neurons of the globus pallidus interna to tonically fire and provide active inhibition to their synaptic targets including relay neurons of components within the already strongly disfacilitated c-TH.13
Relationship of Mesocircuit and Frontoparietal Network
Clinical and experimental studies have also demonstrated that the anterior forebrain mesocircuit has an interactive role in supporting activity within the frontoparietal network. Graded activity within the frontoparietal network has been demonstrated to correlate with outcomes across DOC.6,17,33 Reactivation of the posterior parietal complex along with the c-TH correlates with the restoration of consciousness in healthy human subjects injected with the acetylcholinesterase inhibitor physostigmine.34 In experimental studies in nonhuman primates, direct electrical stimulation of the central lateral thalamus effective to elicit a limited arousal from general anesthesia coactivates the frontal and parietal cortices.35,36 Studies in patients with DOC provide support for the functional interaction of the anterior forebrain mesocircuit with the frontoparietal network. Fridman et al.14 examined detailed metabolic profiles of functional components of the striatum and c-TH in severely brain-injured patients with DOC and compared them with results obtained in healthy controls: in DOC patients, metabolic activity in the c-TH and globus pallidus showed an inverse correlation and the ratio obtained combining them (i.e., globus pallidus/c-TH, Fig. 1, II and III) predicted that following the mesocircuit model, those patients who could not follow commands (i.e., vegetative state [VS] and MCS2) showed depressed central thalamic activity compared with those with behavioral evidence of command following (i.e., MCS “+,” and confusional state). Healthy controls showed a normalization of this ratio; in the same study, a positive correlation of central thalamic metabolism with the precuneus of the dominant hemisphere correlated with higher levels of behavioral recovery. This observation is consistent with linkage of the anterior forebrain mesocircuit and frontoparietal network.6,17,33 This positive correlation likely derives from the innervation of the posterior medial complex by afferents from the central lateral thalamus37 that presumably underlie parietal cortical activation with electrical stimulation of the central lateral nucleus.35 Recovery from MCS to the level of confusional state (characterized by alert, interactive behavior but disorientation) is generally correlated with a persistent abnormality of parietal cortical function measured in the EEG by reduction of posterior parietal–occipital spectral power in the alpha (8–12 Hz) compared with the delta (1–4 Hz) frequency range. This alpha:delta ratio abnormality grades with depth of confusional state symptoms of confusion and disorientation as measured using a standard assessment tool, the Confusion Assessment Protocol.38
NEUROMODULATION OF THE CONSCIOUS STATE IN DOC
Many medications and approaches have been used to promote consciousness recovery in patients with severe DOC; primarily, stimulants acting via selective neuromodulator receptor systems distributed in the forebrain mesocircuit including dopaminergic, noradrenergic, glutamatergic, and cholinergic agents are used. However, paradoxical responses to strong agonists of the gamma-aminobutyric acid (GABA) receptor, such as zolpidem, have also proven effective in a subset of patients39 and evidence for direct electrical stimulation of the central nervous system.6,17,33
Mesocircuit Considerations in Organizing Pharmacological Strategies in DOC
Dopaminergic drugs have been most commonly used to attempt to improve level of consciousness in patients with DOC; however, it is only recently that evidence for the general efficacy of any pharmacologic agent has emerged. In a large, double-blind, placebo controlled, randomized clinical trial in post-traumatic VS and MCS patients, the drug amantadine (AMT) was demonstrated to accelerate the speed and rate of recovery of patients within the initial 3 months after a severe traumatic brain injury (TBI). Using a 4-week treatment period, recovery was significantly faster in the AMT group than in the placebo group, as measured by the Disability Rating Score.40 In this study, the positive effect of AMT was proposed to derive from the neuromodulatory effect on the nigrostriatal, mesolimbic, and frontostriatal dopaminergic systems. Amantadine has since been prescribed as the drug of choice and is now considered a level-B treatment recommendation for subacute posttraumatic VS and MCS patients.1,2 Of note, in this study, AMT modestly improved the primary outcome measure when compared with placebo (i.e., 1.4-point difference in a 30-point scale), and almost 60% of the patients treated with AMT did not demonstrate consistent command following, object recognition, or functional object use at trial end.40 There are many possible reasons to explain the mild effect of AMT in subacute posttraumatic DOC that may be intrinsic to the target population (i.e., heterogeneity, severity, etc.) and/or related to the design, which targeted a period of rapidly advancing baseline recovery in all subjects. Below, we focus our review around potential mechanisms of action of AMT within the mesocircuit and its singular role as a proven therapy for DOC. We highlight the pharmacodynamic aspects of modulation of the mesocircuit to consider alternative approaches for DOC patients who did not improve or show a sustained effect using AMT and potential design of future research strategies.
Cell bodies of dopaminergic neurons located in the substantia nigra pars compacta and ventral tegmental area project to the frontal cortex and striatum through the nigrostriatal,41 mesolimbic, and mesocortical42 dopaminergic pathways. Additionally, dopaminergic neurons within the ventral tegmental area project to the c-TH.43 The c-TH demonstrates high levels of dopamine approximating those observed in substantia nigra44 and a preponderant distribution of “D2-Like” –type receptors45,46; thus, dopaminergic modulation could directly facilitate increased neuronal activity within the anterior forebrain mesocircuit via direct activation at c-TH and striatum.13
At the presynaptic dopaminergic level, AMT may increase the synthesis of endogenous dopamine by blocking N-methyl-D-aspartate (NMDA) receptors that have a positive effect on the enzyme tyrosine hydroxylase increasing the bioconversion of L-DOPA into dopamine.47 However, in DOC patients, there is an uncharacterized posttraumatic defect of the enzyme tyrosine hydroxylase that could impede this presynaptic dopamine role of AMT.46 Unlike AMT, L-DOPA bypasses the defect of the enzyme tyrosine hydroxylase, allowing the final bioconversion of L-DOPA into dopamine. Unfortunately, there are not yet controlled clinical studies using the dopamine precursor L-DOPA, and evidence for its use is only supported by a few small case series of DOC patients who were treated off-label.46,48–50 A recently published phase-0 clinical trial (i.e., pharmacodynamics), however, supports the proposed L-DOPA mechanism of action in DOC.46 In brief, using molecular neuroimaging combining [11C]raclopride, a D2-like receptor antagonist, and pharmacological challenges with dextroamphetamine and L-DOPA (to block dopamine transporter reuptake and to increase dopamine synthesis, respectively; Fig. 1, VII) it was shown that chronic severe TBI patients had a presynaptic dopamine deficit in substantia nigra and ventral tegmentum that could not be reversed by only blocking the dopamine transporter with dextroamphetamine. Reversal was achieved after administration of the dopamine precursor L-DOPA (Fig. 1, I–VII).46 These findings point to a presynaptic dopaminergic deficit affecting the first step of the biosynthesis of dopamine, most likely, a posttraumatic-induced deficit of the enzyme tyrosine hydroxylase.46,51 Finally, at the presynaptic dopaminergic level, blockade of the dopamine transporter using the mild blocker AMT52 or stronger blockers, such as methylphenidate,53 may increase dopamine availability at the synaptic cleft if the dopamine biosynthesis is not severely affected.46 For instance, methylphenidate in a nontraumatic animal model of coma (i.e., emergence from anesthetic coma) accelerates the emergence from coma by enhancing dopamine response from the ventral tegmental area.54 On the other hand, methylphenidate did not show a clinically meaningful effect in posttraumatic patients in VS or MCS.55 Importantly, methylphenidate increases attention in a lesser affected population of patients with posttraumatic attention deficit disorders.56 These contrasting results using methylphenidate suggest that in patients with severe posttraumatic DOC, blockade of the dopamine transporter alone cannot increase dopamine background activity as a result of the presynaptic deficit in dopamine biosynthesis.46
At the postsynaptic dopaminergic level, AMT does not play a major role. However, it is suggested that AMT slightly increases the expression of D2-like dopamine receptors.57,58 In comparison, a pool of more powerful dopaminergic agonists that enhance the membrane excitability59 were tested in a small cohort of cases. For instance, 5 TBI-VS patients emerged from a VS into a MCS and regained functional status with bromocriptine up to 2.5 mg 2 times a day.60 In addition, 8 posttraumatic VS/MCS patients prospectively recruited into a phase-IIa clinical trial of apomorphine (nonrandomized and open-label) showed improvement in the primary outcomes. Awakening was seen as rapidly as within the first 24 hours of drug administration and as late as 4 weeks. Seven of the patients completely recovered consciousness; all improvements were sustained for at least 1 year, even after apomorphine was discontinued.61,62 A phase-IIb clinical trial to further test apomorphine efficacy in posttraumatic VS-MCS has been submitted to the European Medicines Agency.63 Another study of 10 children and adolescents in posttraumatic MCS sustained at least 1 month earlier tested pramipexole and found significant improvement in all tested subjects in this small cohort.64
Less is known about the pharmacodynamics of the noradrenergic system in DOC. Terminals of noradrenergic cells and norepinephrine transporter are found in the c-TH65 and are widespread across neocortical, frontal, and parietal cortices.66 Thus, pharmacological modulation of the noradrenergic system may be another critical target as well in severe DOC. In vitro studies demonstrate that AMT can strongly block the noradrenaline transporter,52 suggesting a possible additional mechanism of action that could underlie its efficacy in severe DOC. Furthermore, it raises the possibility that use of other agents producing strong reuptake blockade of the noradrenergic receptor (e.g., atomoxetine) might also have a potential use in DOC.
Pharmacological modulation of the glutamatergic system using NMDA receptor antagonists has an overall facilitatory effect (Fig. 1, VIII). Blockade of NMDA-receptor on tonically firing GABAergic interneurons leads to glutamatergic disinhibition and increase of glutamate in the prefrontal cortex that subsequently increases a-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor activation.67 It was previously proposed that NMDA antagonism is the main mechanism of action of AMT in severe DOC46; however, direct evidence confirming this theory is lacking. Nonetheless, paradoxical excitation of the cerebral cortex and striatum also arises with the NMDA antagonist ketamine when used as an anesthesia,12 and at subsedative doses for the treatment of resistant depression, ketamine induces activation of the frontal cortex.68
No studies have systematically examined the role of the cholinergic system in the treatment of severe DOC. Indirect evidence arises from a nontraumatic model of coma: human subjects receiving a systemic cholinergic agonist to initiate emergence from anesthetic coma increased the blood flow in both the thalamus and precuneus correlating with recovery of consciousness.34 Importantly, recent detailed studies of emergence from generalized anesthesia show that coactivation of cholinergic and glutamatergic receptors within the Layer V pyramidal cell is required for maintenance of the conscious state; these observations provide a rational foundation for exploring the combined use of cholinergic and glutamatergic support in DOC.69
In DOC subjects with structural brain injuries, paradoxical excitation of the GABAergic system with sedative agents such as zolpidem (a GABA-A alpha subunit positive allosteric modulator that binds to many of the same site as benzodiazepines) has been demonstrated to facilitate behavior in a small percentage of randomly selected and prospectively studied subjects.39 Quantitative EEG investigations in a small number of such zolpidem responsive DOC patients showed that an initial burst of EEG activity consistent with effects in normal subjects (increased 15–30 Hz activity over frontocentral cortical regions) evolved into more narrowband activity lasting hours and replacing dominant low-frequency EEG in subjects when behavioral improvements were observed70; in some instances, transient restoration of a posterior dominant alpha rhythm emerged. Zolpidem may directly bind to GABA-A alpha 1 receptor subtypes in the neocortex to increase thalamocortical and thalamostriatal outflow indirectly as a result of activation of cortical inhibitory interneuron networks.71 In addition, zolpidem may activate striatal GABA-A currents and support alpha and beta (~8–30 Hz) rhythms within the striatum and normalize medium spiny neuron function.72 An important additional proposed activating effect of zolpidem is suppression of increased firing of the GPi via a direct effect of zolpidem on the globus pallidus interna, which is hypothesized to be overactive in the setting of the structurally deafferented brain, as noted above.73
Electrical Stimulation Approaches for Treatment of DOC
Direct neuromodulation of the outflow from the c-TH has been demonstrated to improve level of consciousness and goaldirected behaviors in a proof-of-concept study in a human subject after 6 years remaining in MCS.74 Electrical activation of the c-TH was causally linked in this study to the subjects transition from MCS to confusional state. Additionally, in nonhuman primates, c-TH-DBS robustly regulated arousal and enhanced cognitive performance.75 As a noninvasive method, transcranial direct current stimulation has been shown to improve function in chronic DOC patients.76 For a more comprehensive review of electrical stimulation approaches see the study by Thibaut et al.17
ALGORITHM FOR SHORT-TIME TRIALS OF PHARMACOLOGICAL AGENTS IN DOC PATIENTS
Clinicians caring for DOC patients first need to assume that data from clinical trials for each agent potentially useful in this patient population will never canvass the variety of mixed etiologies (e.g., trauma, hypoxic, inflammatory, postinfectious, autoimmune, and other processes), age, and specific patterns of structural brain injuries that may be present in any particular patient. For patients in VS or MCS between 16 and 65 years with post-traumatic etiology who are between 4 and 16 weeks of injury, guidelines indicate prescribing AMT 100 to 200 mg twice daily over a period of 4 weeks (level B).1 The empirical algorithm shown in Fig. 2 is thus based on our own clinical assessment of DOC patients over time and is suggested as an approach if
FIG. 2.
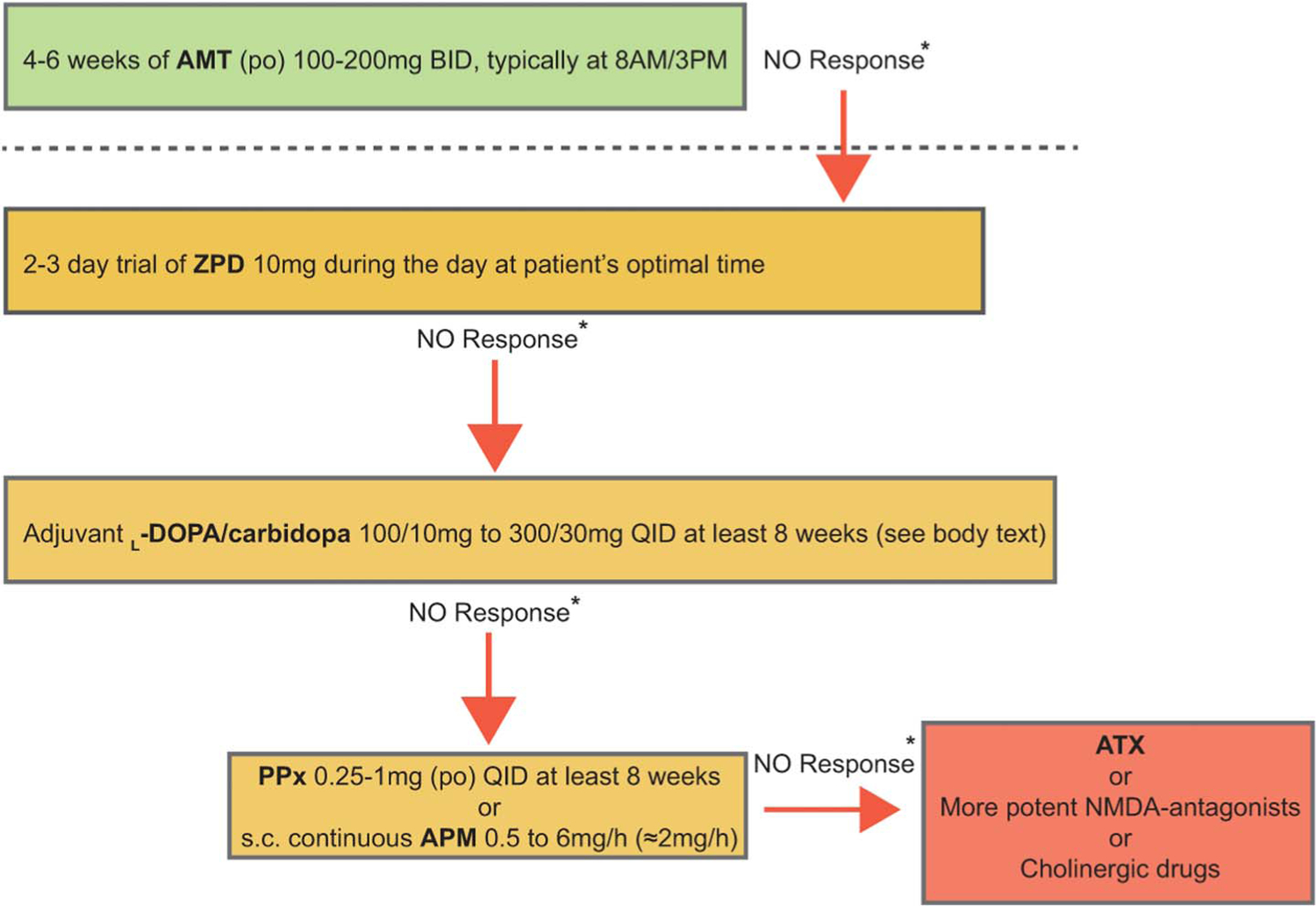
Algorithm for short-time pharmacologic challenges in patients with chronic disorders of consciousness. See text for rationale and linkage to anterior forebrain mesocircuit model. AMT, amantadine; ATX, atomoxetine; BID, twice daily; NMDA, N-methyl- D-aspartate; PPx, pramipexole; QID, four times daily; ZPD, zolpidem.
a patient is excluded from the current treatment recommendation 14 of the AAN/ACRM/NIDILRR Guidelines1,2;
a patient* who after receiving AMT for more than 4 weeks remains ≥ 4 weeks on a neurological plateau based on longitudinal behavioral changes observed in the Coma Recovery Scale-Revised without counting changes on reflexive behaviors (i.e., used to rate acute coma). For the sake of simplicity, we will refer to them as “refractory”;
a patient* who is receiving AMT and shows behavioral regression (i.e., after demonstrating higher level of brain function outside the period of acute injury and early convalescence), without having suffered a new neurological insult.
*In the absence of clinical (i.e., urinary tract infection, pneumonia, electrolyte misbalance, pharmacological interactions) or neurological complications associated with severe DOC (i.e., seizures, hydrocephalus, moderate to severe spasticity).
The sequence and structure of this algorithm derives from considerations of practicality, and the general circuit-level model (shown in Fig. 1) of most common anticipated underlying mechanisms of DOC reviewed above, to provide a framework for considering a wide range of agents for the activation of the anterior forebrain mesocircuit.
As widespread cerebral deafferentation is the most typical originating cause of a chronic DOC, the initial suggested trials of pharmacologic agents are aimed at agents that will broadly activate the anterior forebrain mesocircuit. The suggested algorithm begins with a very brief time trial of zolpidem; a test dose of 10 mg orally is suggested to be given at least 2 times a day during 2 consecutive days, at a point of maximal alertness to support the possible elaboration of emergent behaviors. Although zolpidem response is very rare, with <5% of patients with chronic DOC in prospective study demonstrating a measureable effect,39 effects are clear when clinically meaningful. In responsive patients, long-term use of the medication can produce idiosyncratic results. Some patients may remain consistently responsive for years, others may quickly show tolerance effects.39,70 Although zolpidem has shown the most consistent value in our experience in producing paradoxical behavioral facilitation, other sedative medications have in individual instances shown similar effects. We have observed, and the literature supports, instances of similar paradoxical responses to other sedative agents (including but not limited to midazolam, diazepam, and lorazepam). Notably, clinical experience also demonstrates that some such patients will only respond paradoxically to GABAergic agonists when delivered parentally (examples include midazolam, lorazepam, and sodium pentothal).10
Following this step, a trial with adjuvant L-DOPA should be prescribed because the release of frontostriatal-mediated behaviors by dopamine plays a facilitatory role modulating the neocortex via NMDA antagonism. In other words, DOC patients with a synaptic dopamine background activity deficit may restore membrane depolarization across neocortical and striatal neurons shifting their dynamics when provided with L-DOPA. L-DOPA is the only existing drug acting directly on this impaired molecular biosynthesis pathway, and its adjuvant role will need further clinical testing. At least a single 8-week full cycle using adjuvant L-DOPA/carbidopa to AMT should be tested. Starting from daily dose ranging from 400/40 mg to 1200/120 mg of L-DOPA/carbidopa divided in QID. On days 1 and 2, single doses of L-DOPA/carbidopa 100 mg/10 mg at 8 AM, 11 AM, 3 PM, and 6 PM; on days 3 and 4, single doses of L-DOPA/carbidopa 200/20 mg at 8 AM, 11 AM, 3 PM, and 6 PM; from day 5, L-DOPA/carbidopa 300/30 mg QID at 8 AM, 11 AM, 3 PM, and 6 PM.46 Because L-DOPA absorption is reduced by concomitant protein intake,77 feeding must be stopped ~30´ before dose administration and remain discontinued for another ~30´. L-DOPA is the most widely used medication to treat patients with Parkinson disease. The most common side effect of using it in this population of patients is gastrointestinal intolerance (i.e., nausea most likely; vomiting rarely). There is little information of long-term safety using L-DOPA in DOC patients. However, in our personal experience, L-DOPA use for less than a year at doses below 2000 mg/day has not been associated with the known long-term complications described in patients with Parkinson disease (i.e., dyskinesias).
If no response to adjuvant L-DOPA is observed or the initial response (i.e., behavioral improvement) flattens and vanishes, other medications can be tested. For instance, it is possible to modulate dopaminergic neurotransmission using D2-like receptor agonists such as pramipexole (using increasing doses from 0.25 mg QID up to 1 mg QID PO) or the strongest existing dopamine agonist, apomorphine. Apomorphine requires use of a subcutaneous continuous infusion. The infusion rhythm for this compound must be calibrated according to the patient response and to the presence of adverse reactions to the drug because apomorphine is a well-known proemetic compound. Further expected adverse events of apomorphine are somnolence, drooling, and hypotension. If apomorphine is chosen for testing, the initial infusion rhythm should be 0.5 mg/hour (sc) for 8 to 12 hours/day, to mimic the circadian rhythm.61,62 Although apomorphine can be raised up to 8 mg/hour for 8 to 12 hours/day, intolerance must be close monitored above 3 mg/hour.
Atomoxetine, a norepinephrine reuptake inhibitor, can be considered if no sustained effect of any of the previously described approaches is identified. Initial doses are in between 16 and 20 mg/day (po) and can be raised up to 80 mg/day. An important caveat to the use of all medications targeting the noradrenergic system is the potential to induce seizures or exacerbate underlying seizure disorders. Cholinergic medications such acetylcholinesterase inhibitors may also be considered; as noted above, comodulation of glutamatergic and cholinergic tone may facilitate restoring cortical background activity essential to maintain the wakeful state.69
Considerations for Introducing Treatment of DOC in the Acute Injury Phase
At present, insufficient knowledge exists to guide specific interventions with pharmacologic agents in the acute care setting. On the one hand, promoting increased excitatory neurotransmission may create worsening effects of seizures or direct excitotoxicity associated with trauma or ischemic injury.78 Although seizures have been reported in patients with TBI-related DOC, incidence rates were comparable to placebo in randomized trials.40,79 In a recent retrospective evaluation of 608 clinical charts, only 7.9% of patients with TBI (n = 42) received pharmacological stimulant therapy during the acute phase (median, 11 days). Of those 42 patients in intensive care unit, 85.4% were initiated on AMT, whereas only 14.6% received modafinil.80 It worth noting that acute intervention with stimulants was prescribed for the low arousal response. Alternatively, one recent study raises some possible directions of exploration in the acute setting. Building on observations of a small cohort of three patients with prolonged coma after cardiac arrest, a recent study proposed a mechanism for rare late recovery of independent function that depended on cellular energy resources.81 Two conclusions of this model could guide future studies: (1) maintaining sufficient metabolic substrate (hydration, input/ output balances) in prolonged acute DOC may be important to support reemergence of brain function in settings of severe functional deafferentation from loss of excitatory neurotransmission. Although no process akin to a “stunning” of neurons similar to the cardiac myocyte is known, an existing model suggests that network activity in the form of burst suppression may act in a similar fashion.82 If this model is predictive, supporting metabolic needs more aggressively may support recovery in some patients; (2) in one patient, introduction of AMT after 14 days postinjury may have accelerated coma emergence. The safety of such interventions aimed at activating the anterior forebrain mesocircuit is unknown, and depending on the etiology of brain injury, the clearance of active processes such as inflammation may be required for the initiation of efforts in this direction. The fundamental rationale would be to overcome a functional disfacilitation of this network secondary to multifocal neuronal injuries or altered intracellular function. Future work examining the safety and potential efficacy of such an approach in the acute setting will require disease-specific studies and careful consideration of cofactors and their time course of evolution in the acute care context.
ACKNOWLEDGMENTS
The authors acknowledge the support and significant input of Dr. Jerome B. Posner who contributed to the overall conception and organization of the initial treatment algorithm. An earlier version of this algorithm appears in Posner J, Saper C, Schiff N, Claassen J, editors. 5th edition of Plum and Posner’s Diagnosis of Stupor and Coma; 2019.
Supported by the NIH-NINDS UH3NS095554, R21NS093268, R21NS109697, The James S. McDonnell Foundation, the Jerold B. Katz and Lenny C. Katz Foundations.
Footnotes
N. D. Schiff is an advisor to EnspireDBS, Inc, a Cleveland, Ohio, company developing an electrical stimulation therapy for stroke that is unrelated to this article (cerebellar stimulation approach for motor function). The other author has no conflicts of interest to disclose.
REFERENCES
- 1.Giacino JT, Katz DI, Schiff ND, et al. Practice guideline update recommendations summary: disorders of consciousness. Neurology 2018;91:450–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kondziella D, Bender A, Diserens K, et al. European Academy of Neurology guideline on the diagnosis of coma and other disorders of consciousness. Eur J Neurol 2020;27:741–756. [DOI] [PubMed] [Google Scholar]
- 3.Estraneo A, Moretta P, Loreto V, Lanzillo B, Santoro L, Trojano L. Late recovery after traumatic, anoxic, or hemorrhagic long-lasting vegetative state. Neurology 2010;75:239–245. [DOI] [PubMed] [Google Scholar]
- 4.Nakase-Richardson R, Whyte J, Giacino JT, et al. Longitudinal outcome of patients with disordered consciousness in the NIDRR TBI Model Systems Programs. J Neurotrauma 2012;29:59–65. [DOI] [PubMed] [Google Scholar]
- 5.Fins J. Rights come to mind Cambridge: Cambridge University Press, 2015. [Google Scholar]
- 6.Giacino JT, Fins J, Laureys S, Schiff ND. Disorders of consciousness after acquired brain injury: the state of the science. Nat Rev Neurol 2014;10:99–114. [DOI] [PubMed] [Google Scholar]
- 7.Schiff ND, Plum F. The role of arousal and “gating” systems in the neurology of impaired consciousness. J Clin Neurophysiol 2000;17:438–452. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell WL, MacKinnon MA, Smith DH, McIntosh TK, Graham DI. Thalamic nuclei after human blunt head injury. J Neuropathol Exp Neurol 2006;65:478–488. [DOI] [PubMed] [Google Scholar]
- 9.Adams J, Graham DI, Jennett B. The neuropathology of the vegetative state after an acute brain insult. Brain 2000;123:1327–1338. [DOI] [PubMed] [Google Scholar]
- 10.Posner JB, Saper CB, Schiff ND, Claassen J. Stupor and coma. In Plum and Posner’s diagnosis and treatment of stupor and coma 5th ed. Oxford: Oxford University Press, 2019. [Google Scholar]
- 11.Zafonte R, Hammond F, Dennison A, Chew E, Pharmacotherapy to enhance arousal: what is known and what is not. Prog Brain Res 2009;177:293–316. [DOI] [PubMed] [Google Scholar]
- 12.Brown E, Lydic R, Schiff ND. General anesthesia, sleep, and coma. N Engl J Med 2010;363:2638–2650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schiff ND. Recovery of consciousness after brain injury: a mesocircuit hypothesis. Trends Neurosci 2010;33:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fridman EA, Beattie BJ, Broft A, Laureys S, Schiff ND. Regional cerebral metabolic patterns demonstrate the role of anterior forebrain mesocircuit dysfunction in the severely injured brain. Proc Natl Acad Sci U S A 2014;111:6473–6478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chatelle C, Thibaut A, Gosseries O, et al. Changes in cerebral metabolism in patients with a minimally conscious state responding to zolpidem. Front Hum Neurosci 2014;8:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridman EA, Schiff ND. Neuromodulation of the conscious state following severe brain injuries. Curr Opin Neurobiol 2014;29:172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thibaut A, Schiff N, Giacino J, Laureys S, Gosseries O. Therapeutic interventions in patients with prolonged disorders of consciousness. Lancet Neurol 2019;18:600–614. [DOI] [PubMed] [Google Scholar]
- 18.Pace-Schott EF, Hobson JA. The neurobiology of sleep: genetics, cellular physiology and subcortical networks. Nat Rev Neurosci 2002;3:591–605. [DOI] [PubMed] [Google Scholar]
- 19.Saper CB, Lu J, Chou TC, Gooley J. The hypothalamic integrator for circadian rhythms. Trends Neurosci 2005;28:152–157. [DOI] [PubMed] [Google Scholar]
- 20.Schiff ND. Central thalamic contributions to arousal regulation and neurological disorders of consciousness. Ann N Y Acad Sci 2008;1129:105–118. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Lee HJ, Weitz AJ, et al. Frequency-selective control of cortical and subcortical networks by central thalamus. Elife 2015;4:e09215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schiff ND, Shah SA, Hudson AE, Nauvel T, Kalik SF, Purpura KP. Gating of attentional effort through the central thalamus. J Neurophysiol 2012;109:1152–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wyder MT, Massoglia DP, Stanford TR. Contextual modulation of central thalamic delay-period activity: representation of visual and saccadic goals. J Neurophysiol 2004;91:2628–2648. [DOI] [PubMed] [Google Scholar]
- 24.Schlag J, Schlag-Rey M. Visuomotor functions of central thalamus in monkey. II. Unit activity related to visual events, targeting, and fixation. J Neurophysiol 1984;51:1175–1195. [DOI] [PubMed] [Google Scholar]
- 25.Purpura K, Schiff ND. The thalamic intralaminar nuclei: a role in visual awareness. Neuroscientist 1997;3:8–15. [Google Scholar]
- 26.Deschenes M, Bourassa J, Parent A. Striatal and cortical projections of single neurons from the central lateral thalamic nucleus in the rat. Neuroscience 1996;72:679–687. [DOI] [PubMed] [Google Scholar]
- 27.Castaigne P, Lhermitte F, Buge A, Escourolle R, Hauw JJ, Lyon-Caen O. Paramedian thalamic and midbrain infarcts: clinical and neuropathological study. Ann Neurol 1981;10:127–148. [DOI] [PubMed] [Google Scholar]
- 28.Scannell JW, Burns GAPC, Hilgetag CC, O’Neil MA, Young MP. The connectional organization of the cortico-thalamic system of the cat. Cereb Cortex 1999;9:277–299. [DOI] [PubMed] [Google Scholar]
- 29.Gold L, Lauritzen M. Neuronal deactivation explains decreased cerebellar blood flow in response to focal cerebral ischemia or suppressed neocortical function. Proc Natl Acad Sci U S A 2002;99:7699–7704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paus T, Zatorre RJ, Hofle N, Caramanos Z, Gotman J, Petrides M, Evans AC. Time-related changes in neural systems underlying attention and arousal during the performance of and auditory vigilance task. J Cogn Neurosci 1997;9:392–408. [DOI] [PubMed] [Google Scholar]
- 31.Narayanan NS, Cavanagh JF, Frank MJ, Laubach M. Common medial frontal mechanisms of adaptive control in humans and rodents. Nat Neurosci 2013;16:1888–1895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grillner S, Hellgren J, Ménard A, Saitoh K, Wikström M. Mechanisms for selection of basic motor programs–roles for the striatum and pallidum. Trends Neurosci 2005;28:364–370. [DOI] [PubMed] [Google Scholar]
- 33.Laureys S, Schiff ND. Coma and consciousness: paradigms (re)framed by neuroimaging. Neuroimage 2012;61:478–491. [DOI] [PubMed] [Google Scholar]
- 34.Xie G, Deschamps A, Backman SB, et al. Critical involvement of the thalamus and precuneus during restoration of consciousness with physostigmine in humans during propofol anaesthesia: a positron emission tomography study. Br J Anaesth 2011;106:548–557. [DOI] [PubMed] [Google Scholar]
- 35.Redinbaugh MJ, Phillips JM, Kambi NA, et al. Thalamus modulates consciousness via layer-specific control of cortex. Neuron 2020;106:66–75.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schiff ND. Central lateral thalamic nucleus stimulation awakens cortex via modulation of cross-regional, laminar-specific activity during general anesthesia. Neuron 2020;106:1–3. [DOI] [PubMed] [Google Scholar]
- 37.Schiff ND. Posterior medial corticothalamic connectivity and consciousness. Ann Neurol 2015;72:305–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shah SA, Mohamadpour M, Askin G, et al. Focal electroencephalographic changes index post-traumatic confusion and outcome. J Neurotrauma 2017;34:2691–2699. [DOI] [PubMed] [Google Scholar]
- 39.Whyte J, Rajan R, Rosenbaum A, et al. Zolpidem and restoration of consciousness. Am J Phys Med Rehabil 2014;93:101–113. [DOI] [PubMed] [Google Scholar]
- 40.Giacino JT, Whyte J, Bagiella E, et al. Placebo-controlled trial of amantadine for severe traumatic brain injury. N Engl J Med 2012;366:819–826. [DOI] [PubMed] [Google Scholar]
- 41.Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci 2011;34:441–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sesack SR, Grace A. Cortico-Basal Ganglia reward network: microcircuitry. Neuropsychopharmacology 2010;35:27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Volkow ND, Wang GJ, Ma Y, et al. Activation of orbital and medial prefrontal cortex by methylphenidate in cocaine-addicted subjects but not in controls: relevance to addiction. J Neurosci 2005;25:3932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hornykiewicz O. Dopamine (3-hydroxytyramine) and brain function. Pharmacol Rev 1996;18:925–964. [PubMed] [Google Scholar]
- 45.Rieck RW, Ansari MS, Whetsell WO, Deutch AY, Kessler RM. Distribution of dopamine D2-like receptors in the human thalamus: autoradiographic and PET studies. Neuropsychopharmacology 2004;29:362–372. [DOI] [PubMed] [Google Scholar]
- 46.Fridman EA, Osborne JR, Mozley PD, Victor JD, Schiff ND. Presynaptic dopamine deficit in minimally conscious state patients following traumatic brain injury. Brain 2019;142:1887–1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Deep P, Dagher A, Sadikot A, Gjedde A, Cumming P. Stimulation of dopa decarboxylase activity in striatum of healthy human brain secondary to NMDA receptor antagonism with a low dose of amantadine. Synapse 1999;34:313–318. [DOI] [PubMed] [Google Scholar]
- 48.Matsuda W, Matsumura A, Komatsu Y, Yanaka K, Nose T. Awakenings from persistent vegetative state: report of three cases with parkinsonism and brain stem lesions on MRI. J Neurol Neurosurg Psychiatry 2003;74:1571–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krimchansky B, Keren O, Sazbon L, Groswasser Z. Differential time and related appearance of signs, indicating improvement in the state of consciousness in vegetative state traumatic brain injury (VS-TBI) patients after. Brain Inj 2004;18:1099–1105. [DOI] [PubMed] [Google Scholar]
- 50.Ugoya SO, Akinyemi RO. The place of L-dopa/carbidopa in persistent vegetative state. Clin Neuropharmacol 2010;33:279–284. [DOI] [PubMed] [Google Scholar]
- 51.Liu M, Bachstetter AD, Cass WA, Lifshitz J, Bing G. Pioglitazone attenuates neuroinflammation and promotes dopaminergic neuronal survival in the nigrostriatal system of rats after diffuse brain injury. J Neurotrauma 2017;34:414–422. [DOI] [PubMed] [Google Scholar]
- 52.Sommerauer C, Rebernik P, Reither H, Nanoff C, Pifl C. The noradrenaline transporter as site of action for the anti-Parkinson drug amantadine. Neuropharmacology 2012;62:1708–1716. [DOI] [PubMed] [Google Scholar]
- 53.Chemali JJ, Van Dort CJ, Brown EN, Solt K. Active emergence from propofol general anesthesia is induced by methylphenidate. Anesthesiology 2012;116:998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Taylor N, Chemali J, Brown E, Solt K. Activation of D1 dopamine receptors induces emergence from isoflurane general anesthesia. Anesthesiology 2013;118:30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Martin RT, Whyte J. The effects of methylphenidate on command following and yes/no communication in persons with severe disorders of consciousness: a meta-analysis of n-of-1 studies. Am J Phys Med Rehabil 2007;86:613–620. [DOI] [PubMed] [Google Scholar]
- 56.Willmott C, Ponsford J. Efficacy of methylphenidate in the rehabilitation of attention following traumatic brain injury: a randomised, crossover, double blind, placebo controlled inpatient trial. J Neurol Neurosurg Psychiatry 2009;80:552–557. [DOI] [PubMed] [Google Scholar]
- 57.Volonté MA, Moresco RM, Gobbo C, et al. A PET study with [11-C] raclopride in Parkinson’s disease: preliminary results on the effect of amantadine on the dopaminergic system. Neurol Sci 2001;22:107–108. [DOI] [PubMed] [Google Scholar]
- 58.Gianutsos G, Chute S, Dunn J. Pharmacological changes in dopaminergic systems induced by long-term administration of amantadine. Eur J Pharmacol 1985;110:357–361. [DOI] [PubMed] [Google Scholar]
- 59.Lavin A, Grace AA. Dopamine modulates the responsivity of mediodorsal thalamic cells recorded in vitro. J Neurosci 1998;18:10566–10578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Passler M, Riggs R. Positive outcomes in traumatic brain injury-vegetative state: patients treated with bromocriptine. Arch Phys Med Rehabil 2001;82:311–315. [DOI] [PubMed] [Google Scholar]
- 61.Fridman E, Krimchansky BZ, Bonetto M, et al. Continuous subcutaneous apomorphine for severe disorders of consciousness after traumatic brain injury. Brain Inj 2010;24:636–641. [DOI] [PubMed] [Google Scholar]
- 62.Fridman E, Calvar J, Bonetto M, et al. Fast awakening from minimally conscious state with apomorphine. Brain Inj 2009;23:172–177. [DOI] [PubMed] [Google Scholar]
- 63.Sanz LRD, Lejeune N, Blandiaux S, et al. Treating disorders of consciousness with apomorphine: protocol for a double-blind randomized controlled trial using multimodal assessments. Front Neurol 2019;10:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Patrick P, Blackman J, Mabry J, Buck ML, Gurka MJ, Conaway MR. Dopamine agonist therapy in low-response children following traumatic brain injury. J Child Neurol 2006;21:879–885. [DOI] [PubMed] [Google Scholar]
- 65.Gallezot JD, Weinzimmer D, Nabulsi N, et al. Evaluation of [(11)C] MRB for assessment of occupancy of norepinephrine transporters: studies with atomoxetine in non-human primates. Neuroimage 2011;56:268–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Arnsten AFT. Catecholamine influences on dorsolateral prefrontal cortical networks. BPS 2011;69:e89–e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Esterlis I, DellaGioia N, Pietrzak RH, et al. Ketamine-induced reduction in mGluR5 availability is associated with an antidepressant response: an [11C]ABP688 and PET imaging study in depression. Mol Psychiatry 2018;23:824–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Långsjö JW, Salmi E, Kaisti KK, et al. Effects of subanesthetic ketamine on regional cerebral glucose metabolism in humans. Anesthesiology 2020;100:1065–1071. [DOI] [PubMed] [Google Scholar]
- 69.Suzuki M, Larkum ME. General anesthesia decouples cortical pyramidal neurons. Cell 2020;180:666–676.e13. [DOI] [PubMed] [Google Scholar]
- 70.Williams ST, Conte MM, Goldfine AM, et al. Common resting brain dynamics indicate a possible mechanism underlying zolpidem response in severe brain injury. Elife 2013;2:e01157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.McCarthy MM, Brown EN, Kopell N. Potential network mechanisms mediating electroencephalographic beta rhythm changes during propofolinduced paradoxical excitation. J Neurosci 2008;28:13488–13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.McCarthy MM, Moore-Kochlacs C, Gu X, Boyden ES, Han X, Kopell N. Striatal origin of the pathologic beta oscillations in Parkinson’s disease. Proc Natl Acad Sci USA 2011;108:11620–11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schiff ND, Posner J. Another “awakenings.” Ann Neurol 2007;62:5–7. [DOI] [PubMed] [Google Scholar]
- 74.Schiff ND, Giacino JT, Kalmar K, et al. Behavioural improvements with thalamic stimulation after severe traumatic brain injury. Nature 2007;448:3–7. [DOI] [PubMed] [Google Scholar]
- 75.Baker JL, Ryou JW, Wei XF, Butson CR, Schiff ND, Purpura KP. Robust modulation of arousal regulation, performance and frontostriatal activity through central thalamic deep brain stimulation in healthy non-human primates. J Neurophysiol 2016;116:2383–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Thibaut A, Di Perri C, Chatelle C, et al. Clinical response to tDCS depends on residual brain metabolism and grey matter integrity in patients with minimally conscious state. Brain Stimul 2015;8:1116–1123. [DOI] [PubMed] [Google Scholar]
- 77.Nutt JG, Woodward WR, Hammerstad JP, Carter JH, Anderson JL. The “on-off” phenomenon in Parkinson’s disease. N Engl J Med 1984;310:483–488. [DOI] [PubMed] [Google Scholar]
- 78.Obrenovitch TP, Urenjak J. Is high extracellular glutamate the key to excitotoxicity in traumatic brain injury? J Neurotrauma 1997;14:677–698. [DOI] [PubMed] [Google Scholar]
- 79.Moein H, Khalili HA, Keramatian K. Effect of methylphenidate on ICU and hospital length of stay in patients with severe and moderate traumatic brain injury. Clin Neurol Neurosurg 2006;108:539–542. [DOI] [PubMed] [Google Scholar]
- 80.Barra ME, Izzy S, Sarro-Schwartz A, Hirschberg RE, Mazwi N, Edlow BL. Stimulant therapy in acute traumatic brain injury: prescribing patterns and adverse event rates at 2 level 1 trauma centers. J Intensive Care Med 2019. doi: 10.1177/0885066619841603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Forgacs PB, Devinsky O, Schiff ND. Independent functional outcomes after prolonged coma following cardiac arrest: a mechanistic hypothesis. Ann Neurol 2020;87:618–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ching S, Purdon PL, Vijayan S, Kopell NJ, Brown EN. A neurophysiological-metabolic model for burst suppression. Proc Natl Acad Sci U S A 2012;109:3095–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]