

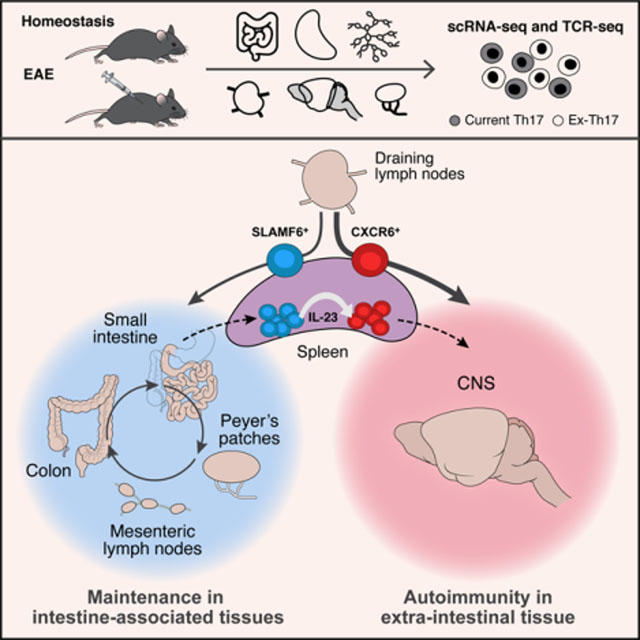
Summary
While intestinal Th17 cells are critical for maintaining tissue homeostasis, recent studies have implicated their roles in the development of extra-intestinal autoimmune diseases including multiple sclerosis. However, the mechanisms by which tissue Th17 cells mediate these dichotomous functions remain unknown. Here, we characterized the heterogeneity, plasticity, and migratory phenotypes of tissue Th17 cells in vivo by combined fate mapping with profiling of the transcriptomes and TCR clonotypes of over 84,000 Th17 cells at homeostasis and during CNS autoimmune inflammation. Inter- and intra-organ single-cell analyses revealed a homeostatic, stem-like TCF1+ IL-17+ SLAMF6+ population that traffics to the intestine where it is maintained by the microbiota, providing a ready-reservoir for the IL-23-driven generation of encephalitogenic GM-CSF+ IFNγ+ CXCR6+ T cells. Our study defines a direct in vivo relationship between IL-17+ non-pathogenic and GM-CSF+ and IFNγ+ pathogenic Th17 populations and provides a mechanism by which homeostatic intestinal Th17 cells direct extra-intestinal autoimmune disease.
Graphical Abstract
In Brief
Stem-like Th17 cells that exert homeostatic effects in the intestine form a reservoir from which pathogenic Th17 cells can differentiate and contribute to neuroinflammatory pathology in experimental autoimmune encephalitis.
Introduction
IL-17-producing CD4+ T cells (Th17 cells) play an important role in multiple autoimmune diseases (Weaver et al., 2013, Korn et al., 2009, Kebir et al., 2009). This has been supported experimentally in pre-clinical models of human autoimmune diseases, in which in vitro differentiated Th17 cells with specificity for self-antigens induce tissue inflammation and autoimmunity (Ghoreschi et al., 2010, Lee et al., 2012). However, recent studies propose that IL-17 and Th17 cells are not critical for driving autoimmune tissue inflammation (Haak et al., 2009, McGinley et al., 2020), whereas GM-CSF-producing T cells (ThGM) were proposed as the primary inducers of autoimmune tissue inflammation (Komuczki et al., 2019, Rasouli et al., 2020, Codarri et al., 2011, Galli et al., 2019). It has been difficult to reconcile some of these opposing data and it is unclear whether these two effector T-cell populations are related to one another or are independent subsets with distinct origins.
A related, open question concerns the homeostatic vs. pathogenic functions of Th17 cells. Th17 cells, besides being highly pathogenic and inducing autoimmunity, have also been implicated in mediating tissue homeostatic functions and in regulating tissue integrity. Th17 cells mediate host defense against extracellular bacterial and fungal infections (Hernandez-Santos and Gaffen, 2012, Bedoya et al., 2013). In the intestine, where most Th17 cells are present during homeostasis, Th17 cells contribute to intestinal barrier integrity and limit microbial invasion (Blaschitz and Raffatellu, 2010, Kinugasa et al., 2000, Cao et al., 2012, Hirota et al., 2013). Finally, intestinal Th17 cells have been suggested to influence extra-intestinal autoimmune diseases, such that their microbiota-specific induction led to exacerbated autoimmune disease in extra-intestinal sites in multiple disease models (Lee et al., 2011, Wu et al., 2010, Krebs et al., 2016). Nonetheless, the mechanisms by which tissue Th17 cells mediate such divergent functions, i.e., promoting intestinal homeostasis while contributing to the development of autoimmune tissue inflammation, remain unclear.
Recent studies suggest that Th17 cell function is affected by environmental changes such as circadian rhythm, specific metabolites, and the microbiota (Yu et al., 2013, Wu et al., 2013, Kleinewietfeld et al., 2013, Hang et al., 2019, Ivanov et al., 2009, Tan et al., 2016, Wagner et al., 2021). However, whether Th17 cells attain different phenotypes and functions in distinct tissue microenvironments as do, for example, FoxP3+ Tregs (Panduro et al., 2016) and tissue-resident macrophages (Lavin et al., 2014), remains unknown. Furthermore, Th17 cells comprise a highly heterogeneous and plastic cell population in vivo, giving rise to multiple different effector and regulatory T-cell populations (Gaublomme et al., 2015, Hirota et al., 2011, Gagliani et al., 2015, Zhang et al., 2020). However, it remains unclear how tissue Th17 cells integrate such diverse signals into a set of cellular programs that allow them to maintain tissue homeostasis yet become primary drivers of tissue inflammation.
Here, we combined single-cell RNA- and TCR-seq (scRNA/TCR-seq) with fate mapping studies to profile 84,124 tissue Th17 cells and characterize their heterogeneity, plasticity, and migration at homeostasis and during central nervous system (CNS) autoimmunity. Tissue Th17 cells exhibited both tissue-specific signatures and intra-tissue heterogeneity. Upon induction of EAE, we discovered a stem-like, intestinal TCF1+ SLAMF6+ IL-17+ population, that gives rise to a second pathogenic GM-CSF+ IFNγ+ CXCR6+ Th17 cell population that migrates specifically to the CNS. Our work identifies the transition of stem-like intestinal Th17 cells to pathogenic Th17 cells and explains the controversy over IL-17+ vs. GM-CSF+ IFNγ+ pathogenic T cells during EAE, a concept, that is likely applicable to other autoimmune and inflammatory diseases.
Results
scRNA/TCR-seq of current Th17 and ex-Th17 cells from multiple tissues
To characterize the diversity of Th17 cells within and across tissues during homeostasis, we profiled both current Th17 cells and ex-Th17 cells, obtained from multiple tissues of naïve reporter mice, including spleen (SPL), mesenteric lymph nodes (MLN), Peyer’s patches (PP), small intestine (SI), and colon (COL), by droplet-based single-cell RNA-seq or scRNA/TCR-seq (Figure 1A, Table S1). To identify instances of Th17 plasticity, we included all CD4+ T cells that ever-produced IL-17A, by crossing the active IL-17A-GFP reporter mouse (Il17aGFP) to the IL-17A fate-reporter mouse (Il17aCreRosa26tdTomato) (Hirota et al., 2011) generating Il17aCreGFP X Rosa26tdTomato mice and performing single-cell profiling of all CD4+ T cells that express tdTomato+ (Figure 1A, Figure S1A,B). In this setting, tdTomato+ GFP+ cells are current Th17 cells, whereas tdTomato+ GFP− cells are ex-Th17 cells. To account for biological and technical variation, we profiled cells in 9 batches, each spanning multiple tissues, such that each tissue was represented in at least 4 of the batches (Table S1). We distinguished ex-Th17 cells (GFP−) from current Th17 cells (GFP+) by the expression of the Il17aGFP transcript in each cell (Figure S1C). Intestinal tissues (SI and COL) had a higher frequency of tdTomato+ cells and a higher ratio of current:ex-Th17 cells than the spleen and lymphoid tissues, indicating an increased presence of current Th17 cells in the intestinal sites, especially the colon (Figure 1B,C).
Figure 1: Single-cell RNA sequencing identifies tissue-specific Th17 signatures.
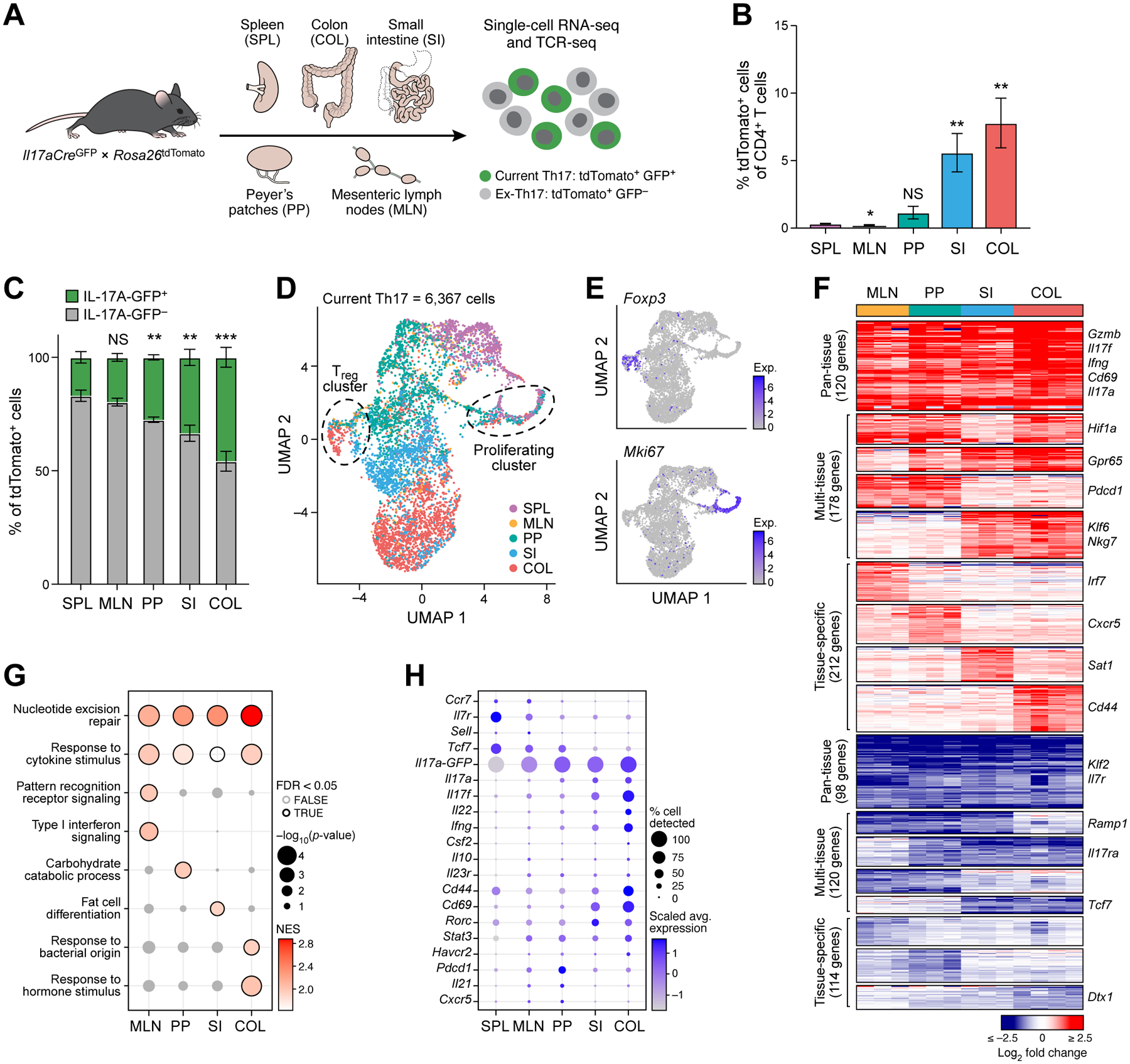
(A) Experimental workflow.
(B, C) Frequencies of tdTomato+ cells (B) or IL-17A-GFP+ and IL-17A-GFP− cells (C) in tissues profiled (n=5–7). Statistical significance determined from comparison to spleen.
(D, E) UMAP of all current Th17 cells (GFP+) at homeostasis. Colored by tissue of origin (D) or transcript expression (log2(CP10k + 1)) of Foxp3 and Mki67 (E).
(F) Up- and down-regulated genes (FDR <0.05, fold change ≥1.5) in tissue vs. splenic current Th17 cells. Columns represent batches. Small multi-tissue gene sets (<20 genes) not shown.
(G) Selected GO terms from gene set enrichment analysis of tissue vs. splenic current Th17 cells. Only positively enriched results were shown.
(H) Selected Th17 effector genes in the tissues at homeostasis.
See also Figures S1 and S2, and Tables S1 and S2.
Th17 cells exhibit tissue-specific signatures
Clustering analysis divided 6,367 current Th17 cells (GFP+ tdTomato+) into 9 subsets (Figure 1D, Figure S1D) with distinct expression signatures (Figure S1E). Most clusters were dominated by cells from a single tissue type (50–89%; Figure S1F). The remaining clusters included Il17aGFP-expressing Foxp3+ regulatory T cells (Tregs, cluster 6) and Mki67+ proliferating Th17 cells (cluster 7), containing cells from all tissues (Figure 1D,E, Figure S1D–F). Notably, these distinctions were not driven by experimental batch effects (Figure S1G). We concluded that tissue-of-origin is a major determinant of variance of the transcriptional states of Th17 cells.
We identified transcriptional signatures that distinguished all tissue Th17 cells (pan-tissue residency signature), Th17 cells from some tissues (multi-tissue signature), and Th17 cells from single tissues (tissue-specific signatures) by comparing the expression profiles of Th17 cells from each tissue to those of splenic Th17 cells (Figure 1F, Table S2). Overall, 943 genes were significantly up- or down-regulated vs. spleen (FDR <0.05, fold change ≥1.5) in at least one tissue population, 120 forming a pan-tissue residency signature, and 212 in one of the tissue-specific signatures. Colonic Th17 cells had the largest tissue-specific signature (62 up-regulated and 31 down-regulated genes, Figure 1F) and were the most distant from splenic Th17 cell profiles on a principal components space (Figure S1H).
The pan-tissue signature included up-regulated genes encoding Th17 effector molecules, such as Il17a, Il17f, Ifng, Cd69, and Gzmb, and down-regulated genes known to antagonize the generation of effector T cells, such as Klf2 and Il7r (Sebzda et al., 2008, Tan et al., 2001), and was enriched in pathways involved in the responses to cytokines (Figure 1F,G). Tissue-specific Th17 signatures were enriched in processes highlighting functional differences of Th17 cells in different tissues (Figure 1G), for example enrichment for response to bacteria in the colon-specific signature genes.
Notably, genes associated with a stem-like T-cell state (Ccr7, Il7r, Sell, Tcf7) were highly expressed in the spleen and lymphoid tissues, while genes associated with activation of Th17 cells (Cd44, Cd69, Il17a, Il17f, Il22, Ifng) were highly expressed in intestinal tissues (Figure 1H). Thus, intestinal Th17 cells had a more activated state than SPL, MLN, and PP populations. We observed similar expression signatures when considering the entire set of 37,773 tdTomato+ profiled cells (ex-Th17 and current Th17 cells, Figure S2A–E). Differential gene expression analysis of ex- vs. current Th17 cells showed relatively modest differences, with the top up-regulated genes in current Th17 cells including, as expected, Il17a-GFP, Il17a, and Il17f, and genes involved in T-cell activation (e.g., Lag3, Pdcd1, Ctla4, Furin, Nr4a1, Figure S2F).
Common features of intra-tissue variation in Th17 cells across tissues
We next examined the variation between all Th17 cells (ex- and current Th17) within each tissue, clustering all cells (GFP+ and GFP−) from each tissue separately (Figure 2A) and quantifying the transcriptional similarity between each pair of clusters within and across tissues (Figure 2B).
Figure 2: Intra-tissue heterogeneity of tissue Th17 cells revealed with single-cell analysis.
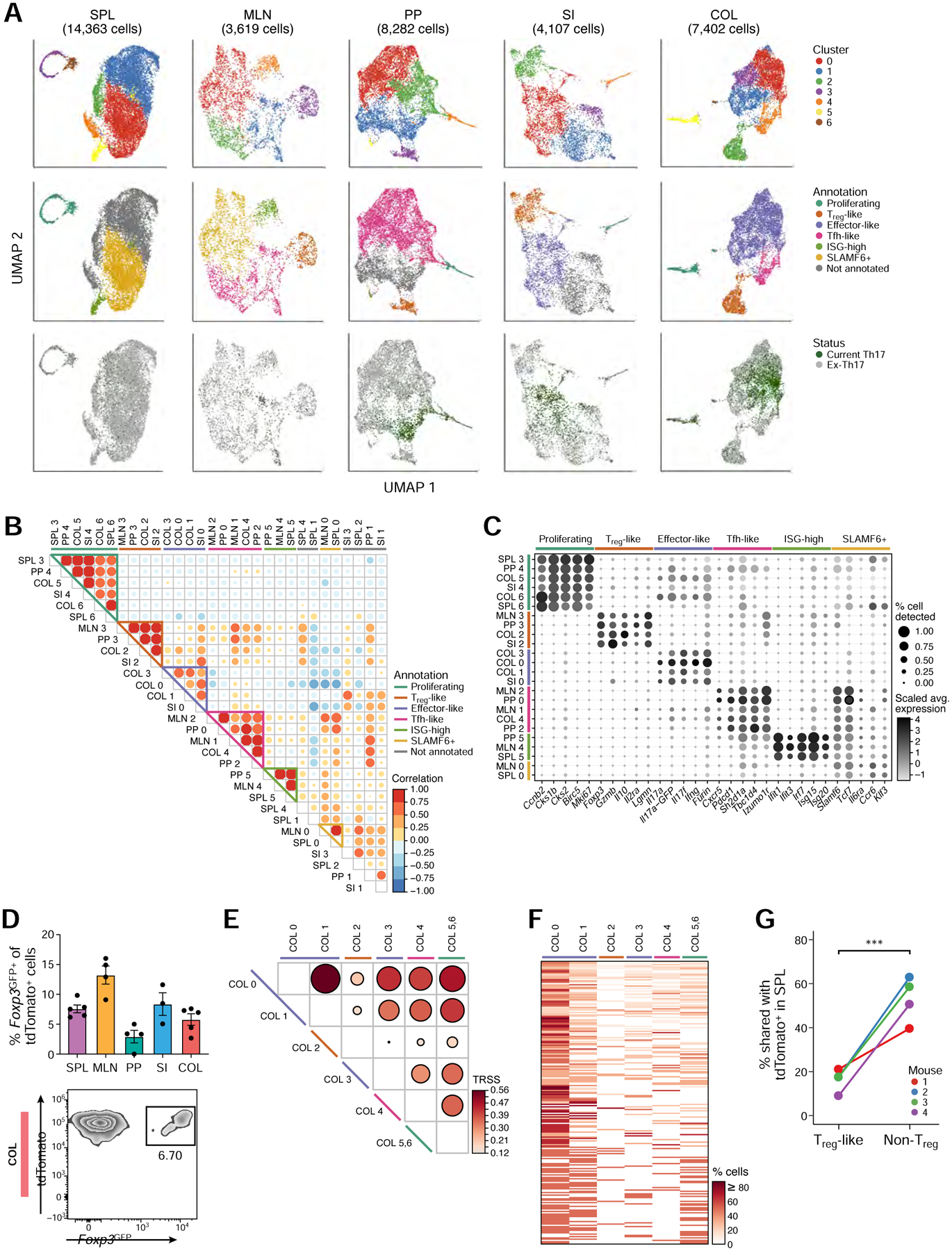
(A) UMAP visualization of all current (GFP+) and ex-Th17 cells (GFP−) at homeostasis, colored by cluster assignment (top row), cluster function annotation (middle row), and Th17 status (bottom row).
(B) Correlation of transcriptomic landscape for all pairs of intra-tissue clusters.
(C) Expression of selected genes used to define the intra-tissue cluster functional annotations.
(D) Frequency of Foxp3GFP+ of tdTomato+ cells by flow cytometry in each tissue (top, n=3–5). Representative flow cytometry plot of the colonic population (bottom).
(E) TCR repertoire similarity analysis across the colon tissue clusters. The two proliferating clusters (COL5 and COL6) were combined in this analysis.
(F) Clonotype sharing across colon clusters. All clonotypes (rows) found in at least two colon clusters (columns) were included.
(G) Percentage of Treg-like (cluster COL2) and non-Treg colonic tdTomato+ cells (all other clusters) that share their TCR sequences with splenic tdTomato+ cells (n=4).
Interestingly, sets of clusters from different tissues were transcriptionally similar, indicating the presence of Th17 cells of similar functional subpopulations in different tissue sites. We annotated each set of clusters by the genes that were differentially up-regulated within each cluster-group relative to the other clusters in the tissue (Figure 2C). The shared clusters included an effector-like set (SI and COL; Il17a, Il17f, Ifng, and Furin), comprising of most current Th17 cells in these tissues (Figure 2A); Tfh-like clusters (MLN, PP, and COL; Cxcr5, Pdcd1, Sh2d1a, Tbc1d4, Izumo1r), as previously described for Th17 cells in the PP (Hirota et al., 2013); stem-like clusters (SPL and MLN; Slamf6, Tcf7, Il6ra, Ccr6, and Klf3), as well as a proliferating set (all tissues except MLN; Ccnb2, Cks1b, Cks2, Birc5, Mki67) and a Treg set (all tissues except spleen; Foxp3, Gzmb, Il10, Il2ra, Lgmn).
In particular, we identified small clusters of Th17 cells in the SPL, MLN, and PP that expressed high levels of interferon-stimulated genes (ISG-high; Ifit1, Ifit3, Irf7, Isg15, Isg20), similar to previously described ISG-high subpopulations (Arazi et al., 2019, Hemmers et al., 2019, Tibbitt et al., 2019, Kiner et al., 2021). COMET (Delaney et al., 2019) analysis predicted PD-L1 (CD274) as a potential cell surface marker for ISG-high cells in the SPL (ranked 7) and in the MLN (ranked 3) (Figure S3A). We validated this prediction, confirming the presence of PD-L1+ tdTomato+ cells in the SPL and MLN (Figure S3B), that expressed elevated levels of ISGs (Figure S3C). PD-L1 was also more frequently detected in the ISG-high T cells in other published data sets (Arazi et al., 2019, Hemmers et al., 2019, Tibbitt et al., 2019) (Figure S3D). Based on our scTCR-seq data, cells in the ISG-high clusters tended to have higher TCR repertoire similarity with clusters in their own tissue than those in other tissues (Figure S3E), indicating that the ISG-high cell population could be clonally related to cells in the same tissue.
tdTomato+ Foxp3+ Treg cells display low TCR similarity with tissue Th17 cells
Multiple previous studies described the presence of IL-17+ and/or RORγt+ FoxP3+ Treg cells at homeostasis and during inflammatory diseases (Yang et al., 2008, Komatsu et al., 2014, Downs-Canner et al., 2017, Pratama et al., 2020, Sefik et al., 2015). However, the relationship of this Th17-like Treg population to cells in the Treg lineage or the Th17 lineage has not been determined.
To explore this, we first crossed the Il17aCre X Rosa26tdTomato mice to Foxp3GFP reporter mice (generating Foxp3GFP X Il17aCretdTomato mice) and validated distinct populations of tdTomato+ Foxp3GFP+ cells by flow cytometry in each tissue (Figure 2D, Figure S3F,G). We then assessed the cell lineage (clonal) and cell fate relationships between tdTomato+ Foxp3+ Treg cells and other Th17 cell subpopulations in tissues by the extent of TCR sharing. We identified a total of 3,706 different clonotypes across 4 mice, with only 12 clonotypes shared by 2 mice, and none by ≥3 mice (Table S3A,B).
Several lines of evidence suggested that Tregs and non-Treg tissue Th17 cells have separate clonal origins. Firstly, within a given tissue (e.g., COL), the TCR sequences in the tdTomato+ Treg clusters were highly distinct from those of non-Treg Th17 clusters, while the non-Treg Th17 clusters displayed high TCR similarity with each other (Figure 2E,F, Figure S3H). Furthermore, non-Treg tissue Th17 cells shared many TCR clonotypes with Th17 cells in the SPL (e.g., 40–63% shared SPL and COL non-Treg), while the corresponding tdTomato+ Treg cells displayed significantly less TCR similarity (9–21%) with cells in the SPL (Figure 2G, Figure S3I). Thus, our paired scRNA/TCR-seq profiles indicate that at homeostasis, tdTomato+ Treg cells are likely more frequently derived from a non-Th17 cell source, presumably tdTomato− Treg cells (Figure S3J), whereas different non-Treg tissue Th17 cells have high clonal similarities within a tissue and show intra-tissue plasticity and transitions between subpopulations.
Heterogeneity of encephalitogenic Th17 cells
Since Th17 cells are drivers of multiple autoimmune diseases (Weaver et al., 2013, Korn et al., 2009), we next analyzed tissue Th17 cell heterogeneity in an autoimmune setting in EAE, a mouse model for MS induced by immunization with the encephalitogenic MOG35–55 peptide. Using scRNA/TCR-seq, we profiled 46,351 tdTomato+ tissue Th17 cells (current and ex-Th17) isolated at onset of disease from the SPL, MLN, PP, COL, SI, the inguinal draining lymph nodes (DLN) of the EAE immunization site and the CNS (Figure 3A, Table S1).
Figure 3: Identification of functionally distinct encephalitogenic Th17 cell subpopulations.
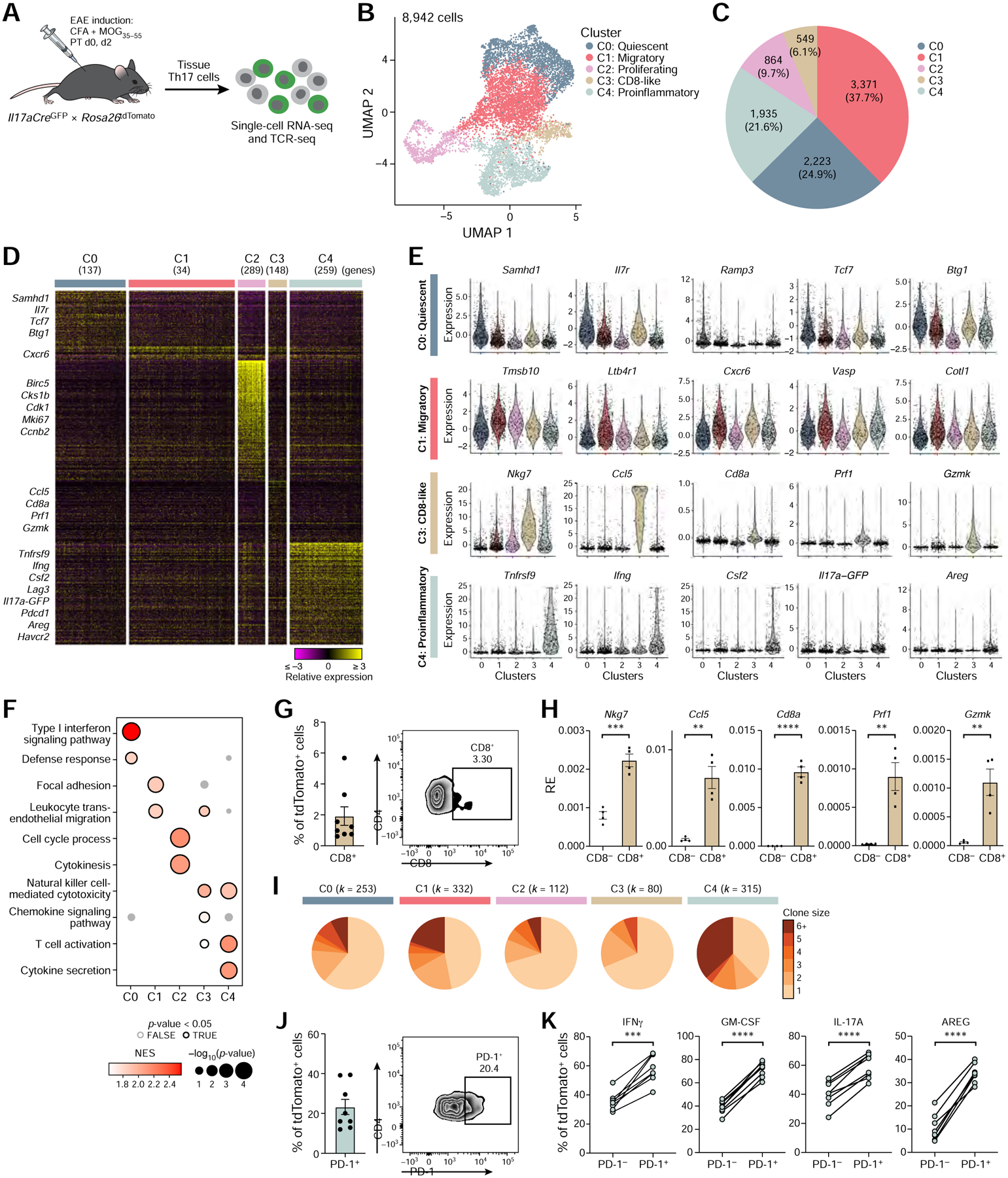
(A) Schematic of the experimental setup.
(B) UMAP representation of unsupervised clustering of current (GFP+) and ex-Th17 cells (GFP−) from the CNS of EAE mice at disease onset.
(C) Cell numbers and relative frequencies of each cluster.
(D) Relative expression of genes uniquely up-regulated in each cluster (Wilcoxon rank-sum test; FDR <0.05, fold change ≥1.5). Data are shown for a random sample of 1,000 cells.
(E) Expression of a selected set of cluster markers. Extreme expression values (<0.5 or >99.5 percentiles) were excluded and points were shown for randomly selected 20% of the cells.
(F) Selected GO terms and KEGG pathways from cluster-specific gene set enrichment analysis. Only positively enriched results were shown.
(G) CD8α expression by flow cytometry on viable TCRβ+ CD4+ tdTomato+ CNS cells (n=8). Quantification (left) and a representative flow cytometry plot (right).
(H) qPCR gene expression analysis of cluster 3 marker genes comparing CD8α− to CD8α+ tdTomato+ CNS cells (n=4). RE = Relative expression.
(I) Clonal expansion for each CNS cluster. Size of a pie slice represents the proportion of cells that belong to clones of the color-indicated size computed within each cluster. Number of cells is shown on the top. Clonal expansion scores for clusters (0–4) are 0.04, 0.07, 0.03, 0.03 and 0.13, respectively.
(J) PD-1 expression by flow cytometry on viable TCRβ+ CD4+ tdTomato+ CNS cells (n=8). Quantification (left) and a representative flow cytometry plot (right).
(K) Effector cytokine expression by flow cytometry in PD-1− and PD-1+ tdTomato+ CNS cells (n=8). Paired two-tailed t-tests were performed.
First, we focused on CNS-infiltrating T cells. The 8,942 Th17 (tdTomato+) CNS-derived cells partitioned into 5 clusters (Figure 3B,C), which we annotated by differential gene signatures as quiescent (cluster 0), migratory (cluster 1), proliferating (cluster 2), CD8-like (cluster 3) and proinflammatory (cluster 4) (Figure 3D–F). The cells were ordered in low dimensional space from quiescent to migratory, and then split from migratory cells into proliferating cells (following the cell cycle; Figure S4A), CD8-like cells, and proinflammatory cells (Figure 3B). Cells in quiescent cluster 0 highly expressed genes associated with T-cell quiescence (e.g., Samhd1, Il7r, Tcf7, and Btg1 (Hwang et al., 2020)) and had the lowest number of detected transcripts per cell, supporting a quiescent cell state (Figure S4B). Cells in migratory cluster 1 highly expressed genes involved in cell migration (Ltb4r1, Cxcr6, Vasp) and morphology (Tmsb10, Cotl1), suggesting these cells recently migrated into the CNS. Proliferating cluster 2 cells highly expressed cell cycle genes (Birc5, Cks1b, Cdk1, Mki67).
Interestingly, CD8-like cells (cluster 3) expressed Cd4 and CD8+ T cell markers (Nkg7, Ccl5, Cd8a) and cytotoxic (Prf1, Gzmk) genes. Flow cytometry confirmed the presence of a small TCRβ+ CD4+ tdTomato+ CD8+ population that expressed CD8-like and cytotoxic genes (Figure 3G,H). Notably, we and others have recently reported such CD8+ IL17-expressing cells in inflammatory bowel disease in humans (Tom et al., 2016, Smillie et al., 2019). Thus, emergence of CD8-like Th17 cells might be a common phenomenon in T-cell driven autoimmune tissue inflammation.
Most notably, we identified a cell subset (cluster 4) of highly proinflammatory, activated, and expanded cells, enriched for current Th17 cells in the CNS. Specifically, cells in proinflammatory cluster 4 expressed the highest levels of genes encoding proinflammatory cytokines and activation markers including co-inhibitory receptors (Tnfrsf9, Ifng, Csf2, Lag3, Il17a, Pdcd1) (Schnell et al., 2020) (Figure 3D,E), were the most clonally expanded (by TCR analysis) (Figure 3I), had the highest proportion of current Th17 cells (GFP+) (Figure S4C), the highest Il17aGFP expression (FDR <0.0001), and the largest number of transcripts per cells (except for proliferating cluster 2 cells) (Figure S4B). To confirm the presence of the cluster 4 cell population in the CNS during EAE, we predicted (by COMET (Delaney et al., 2019)) and validated PD-1 as a highly expressed surface marker in cluster 4 cells (Figure S4D), consistent with observations that PD-1 is highly expressed on activated effector cells (Riley, 2009). PD-1 marked highly proinflammatory Th17 cells that produced high levels of IL-17A, IFNγ, and GM-CSF, cytokines associated with encephalitogenicity (Figure 3J,K).
Two distinct clonally related cross-tissue communities of Th17 cells during EAE
To study the mechanism and role of tissue Th17 cells in the induction of EAE, we focused on Th17 cells in peripheral tissues and examined all 84,124 tissue Th17 cells from both naïve and EAE mice. Similar to our analyses at homeostasis, control and EAE cells grouped together primarily by tissue-origin rather than treatment, suggesting that the tissue Th17 signatures are stronger discriminants than expression changes induced upon EAE immunization (Figure 4A,B and Figure S4E,F). While cells of intestinal tissues from naïve or EAE mice were well-mixed and had only few differentially expressed genes between the conditions (SI (COL) 30 (10) genes up-regulated, 23 (32) genes down-regulated; FDR <0.05, fold change ≥1.5), splenic Th17 cells were clearly more distinguishable between the two conditions (Figure 4B–D, Figure S4G–I), with a larger number of differentially expressed genes (58 up- and 58 down-regulated, Figure S4H).
Figure 4: Single-cell profiling of tissue Th17 cells during EAE.
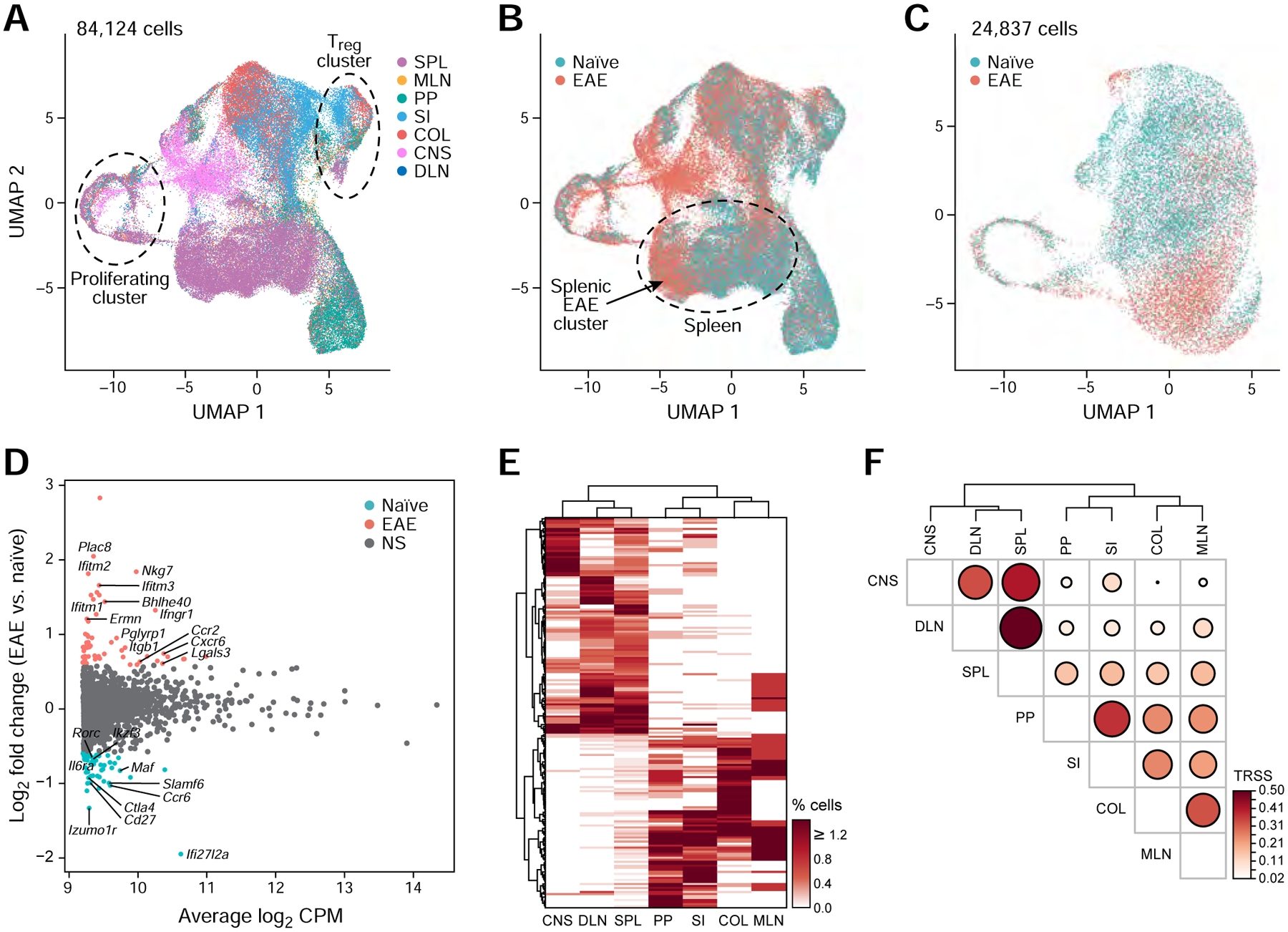
(A,B) UMAP of all current (GFP+) and ex-Th17 cells (GFP−) from naïve and EAE-diseased mice. Cells are colored by tissue origin (A) or by treatment condition (B).
(C) UMAP of all splenic Th17 cells. Cells are colored by treatment condition.
(D) MA plot of differentially expressed genes comparing EAE vs. naïve splenic Th17 cells (FDR <0.05 and fold change ≥1.5).
(E) Th17 clonotype sharing across tissues during EAE. Tissue-specific or small (<10 cells) clonotypes were excluded.
(F) Tissue-level TCR repertoire similarity analysis for Th17 cells during EAE.
Next, we used TCR sequences of the Th17 cells in EAE-diseased mice to understand the relationship between Th17 cells across all tissues and the encephalitogenic Th17 population. Only a handful of clonotypes were shared across mice during EAE (12 of 5,278 (0.2%) clonotypes; only 1 clonotype in 2 mice in the CNS; Table S3A,C), whereas 5–35% of the clonotypes were shared between tissues in the same mouse. TCR repertoire similarity analysis across tissues (Figure 4E,F) revealed a Th17 population with high clonal similarity between cells from the DLN, SPL, and CNS, indicating that these clonotypes may be the main disease-driving population, and a Th17 population with high clonal similarity in the intestinal tissues (MLN, PP, SI, COL). Recent studies highlight the role of the “gut-brain axis”, in which intestinal Th17 cells have been suggested, by an unknown mechanism, to regulate CNS autoimmune inflammation (Lee et al., 2011, Miyauchi et al., 2020, Cosorich et al., 2017). However, our results suggest a model where Th17 cells are induced in the DLN, and then traffic to the CNS via SPL.
To address the potential role of the intestinal Th17 population in EAE disease, we compared the TCR sequences of intestinal Th17 cells to other tissue Th17 cells. Interestingly, we identified a minor population of Th17 cells in the spleen that shared TCR clonotypic structures with gut-associated Th17 cells in the MLN, PP, SI, and COL (Figure 4E,F). We concluded that there may be two different clonally related Th17 communities in the SPL, one that shared clonotypes with DLN and CNS cells (26.2% of the SPL cells), and a second, smaller, community in the SPL (10.6% of the SPL cells) that shared clonotypes with Th17 cells from intestinal tissues. We thus hypothesized that the SPL is potentially a key hub, serving as a source for disseminating both intestinal Th17 cells and encephalitogenic Th17 cells.
A SLAMF6+ homeostatic stem-like population and a CXCR6+ pathogenic population of Th17 cells in the spleen
Further analysis of splenic Th17 cell heterogeneity during EAE showed that most cells (85%) were in one of two clusters, resembling either homeostatic non-pathogenic Th17 cells (SPL0) or pathogenic Th17 cells (SPL1) (Figure 5A–C), based on expression of known genes and signatures (Lee et al., 2012, Gaublomme et al., 2015), with pathogenic-like SPL1 cells substantially more clonally expanded (Figure 5D). Among the genes up-regulated in the homeostatic Th17 cells (Figure 5E) were both those previously associated with non-pathogenic Th17 cells (Ccr6, Il6ra, Ikzf3, Maf) (Figure 5B), and genes expressed in stem-like CD8+ T cells (Tcf7, Cxcr5) described in tumors and during LCMV-infections (Im et al., 2016, Siddiqui et al., 2019, Hudson et al., 2019). Conversely, SPL1 cells expressed high levels of genes that we and others have previously associated with pathogenic Th17 cells (Ifng, Tbx21, Bhlhe40, Csf2) (Gaublomme et al., 2015, Lee et al., 2012).
Figure 5: Discovery of a homeostatic and a pathogenic Th17 population in the spleen.
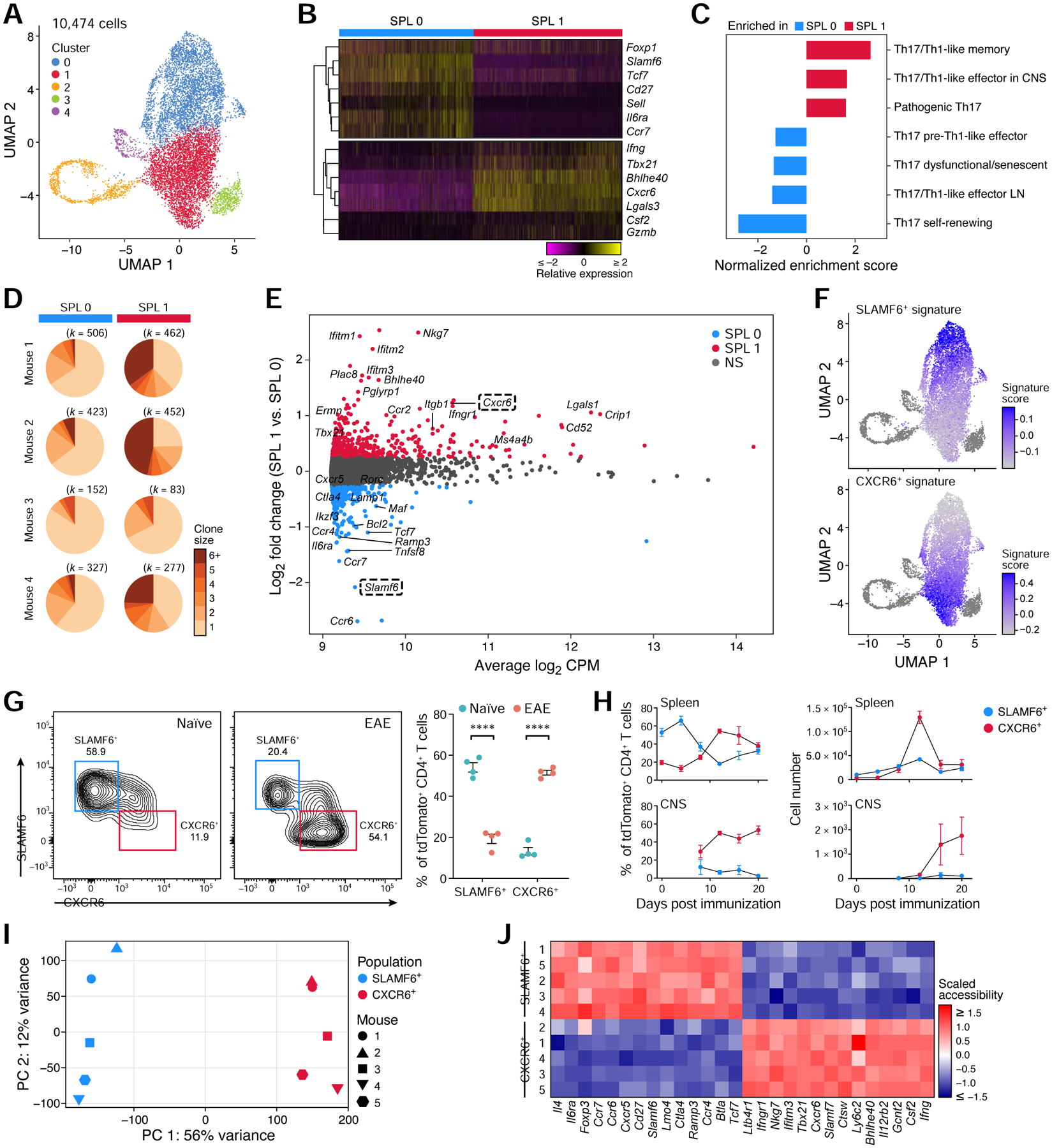
(A) UMAP of all splenic current (GFP+) and ex-Th17 cells (GFP−) during EAE.
(B) Relative expression of selected homeostatic and pathogenic genes that are differentially expressed (FDR <0.05) between SPL0 and SPL1.
(C) Gene set enrichment analysis of published Th17 pathway signatures (Lee et al., 2012, Gaublomme et al., 2015) in SPL1 vs. SPL0.
(D) Clonal expansion of SPL0 and SPL1 cells in each mouse. The number of cells is shown above. Clonal expansion scores are 0.018, 0.026, 0.016, and 0.022 for each mouse (1–4) in SPL0, and 0.071, 0.116, 0.024, and 0.080 in SPL1.
(E) MA plot of differentially expressed genes between SPL1 and SPL0 (FDR <0.05, fold change ≥1.5).
(F) Overlay of the bulk RNA-seq derived CXCR6+ and SLAMF6+ gene signatures (Table S4) on the splenic UMAP.
(G) Frequency of the CXCR6+ and SLAMF6+ spleen populations in naïve and EAE-diseased mice analyzed by flow cytometry (n=4). Representative flow cytometry plots (left) and quantification (right).
(H) Frequencies (left) and number (right) of SLAMF6+ and CXCR6+ cells in the spleen (top) and CNS (bottom) over the EAE disease time-course (n=3–4). CNS cells were sampled starting at day 8.
(I, J) ATAC-seq of the SLAMF6+ and CXCR6+ populations. (I) Scatter plot of samples based on the top two PCs. (J) Normalized chromatin accessibility for a selected list of genes from the bulk RNA-seq derived SLAMF6+ and CXCR6+ gene signatures.
We identified SLAMF6+ and CXCR6+ as two cell surface molecules that could distinguish the SPL0 and SPL1 populations (Figure 5E), and validated them as markers of SPL0 and SPL1 cells, respectively, by sorting of SLAMF6+ and CXCR6+ tdTomato+ CD4+ T cells from the spleens of EAE-diseased mice followed by bulk RNA-seq, showing that their associated signatures (Table S4) were expressed in SPL0 and SPL1 cells, respectively (Figure 5F). SLAMF6+ cells are present in the spleen at high proportion during homeostasis and reduced in EAE, whereas CXCR6+ cells increase dramatically in the spleen and CNS during EAE disease, with a temporary peak in the spleen, followed by a steady rise in the CNS (Figure 5G,H). The bulk CXCR6+ signature was positively correlated with scRNA-seq profiles of pathogenic encephalitogenic T cells from cerebrospinal fluid (CSF) from MS patients (Schafflick et al., 2020) (Figure S5A–C). Notably, the SLAMF6+ and CXCR6+ signatures also marked distinct cell populations in scRNA-seq profiles of tdTomato+ CD4+ T cells from mice with DSS-induced colitis, suggesting potential broader implications of our findings to other inflammatory conditions (Figure S5D).
Splenic SLAMF6+ and CXCR6+ populations had distinct chromatin accessibility landscapes (by ATAC-Seq), with 17,449 differentially accessible regions (FDR <0.05, fold change ≥2) (Figure 5I), supporting their separate identity. Gene loci involved in the homeostatic and stem-like Th17 cell phenotype (Il6ra, Ccr7, Ccr6, Slamf6, Tcf7, Cxcr5) were specifically accessible in the SLAMF6+ population, and loci involved in Th17 pathogenicity (Ifngr1, Nkg7, Tbx21, Cxcr6, Bhlhe40, Csf2, Ifng) were specifically accessible in the CXCR6+ population (Figure 5J).
Thus, we identified two splenic Th17 cell populations with distinct expression and chromatin accessibility profiles that resemble a homeostatic stem-like and a pathogenic Th17 cell population and are marked by the surface markers SLAMF6 and CXCR6, respectively.
Distinct in vivo migratory behavior of the homeostatic and pathogenic splenic Th17 populations
Based on our discovery of two clonally related Th17 communities, we hypothesized that SPL0 (homoeostatic) and SPL1 (pathogenic) cells represent gut-associated and encephalitogenic Th17 communities in the spleen, respectively. Indeed, the SPL0 cells showed TCR repertoire similarity to Th17 cells in all tissues, whereas SPL1 cells shared TCRs almost exclusively with Th17 cells in the DLN and the CNS (Figure 6A,B). SPL0 cells highly expressed genes involved in immune cell migration and retention in lymphoid tissues (Cxcr5, Ccr7, Sell) and to the intestine (Ccr9, Itga4), whereas SPL1 cells expressed genes involved in immune cell trafficking to sites of inflammation (Ccr5, Itgb1, Crip1, Cxcr6, Ccr2) (Figure 6C).
Figure 6: The homeostatic and pathogenic splenic Th17 populations display distinct migratory behavior in vivo.
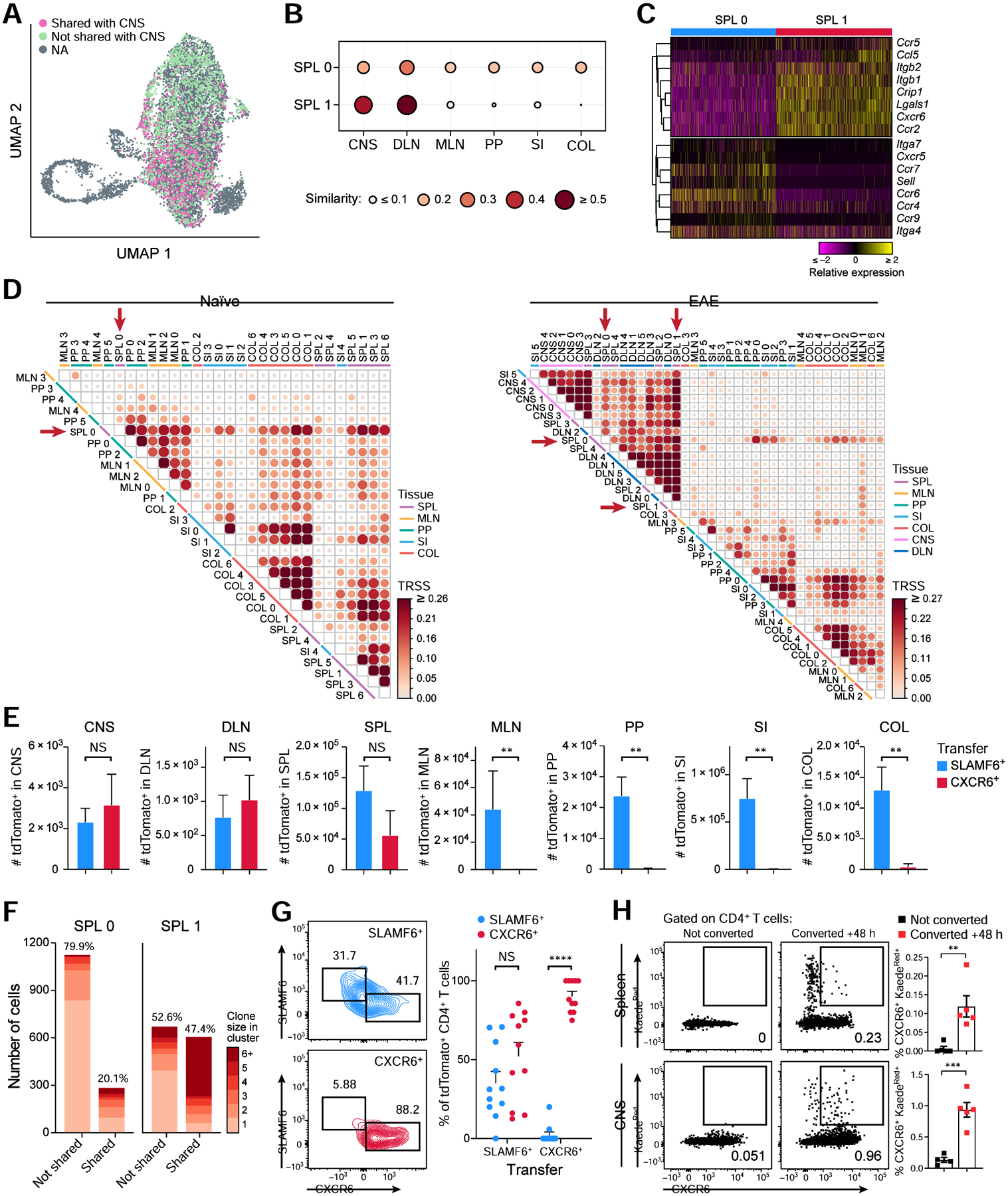
(A) UMAP of the SPL0 and SPL1 cells colored by TCR sharing (pink) or not sharing (green) with Th17 cells in the CNS.
(B) TCR repertoire similarity analysis of SPL0 and SPL1 with cells in all other tissues.
(C) Relative expression of selected genes involved in immune cell trafficking that are differentially expressed (FDR <0.05) in clusters SPL0 and SPL1.
(D) TCR repertoire similarity analysis of intra-tissue clusters at homeostasis (left) and during EAE (right). Clusters were defined as in Figure 2A and Figure S6B.
(E) Cell numbers of tdTomato+ cells in each tissue following SLAMF6+ or CXCR6+ tdTomato+ cell transfer into EAE-bearing recipients (n=5). Two-tailed Mann Whitney test was performed.
(F) Number of cells in SPL0 and SPL1 whose TCRs were not shared (left) or shared (right) with the other cluster. Proportions are annotated on the top. Clonal expansion scores are 0.01 and 0.05 for not shared and shared clones in SPL0, and 0.03 and 0.10 in SPL1.
(G) SLAMF6 and CXCR6 expression on transferred tdTomato+ cells in the spleen. SLAMF6+ or CXCR6+ tdTomato+ cells were transferred into EAE-bearing recipient mice and spleens were harvested 7 days post transfer (n=11). Representative flow cytometry plots (left) and quantification (right) are shown.
(H) Frequencies of photoconverted KaedeRed+ CXCR6+ cells in the spleen (top) and CNS (bottom) of EAE-bearing mice 48 h after photoconversion of intestinal cells (n=4–5). See also Figures S6 and S7.
TCR similarity analysis across all tissue clusters at homeostasis and EAE revealed patterns of clonotype sharing (Figure 6D). Specifically, at homeostasis, cell clusters from the same tissue shared clonotypes, indicating high clonality (and plasticity) within each tissue and that TCR specificity is a main factor dictating the homing and expansion of tissue Th17 cells. Using the GLIPH2 algorithm (Huang et al., 2020), which groups TCRs that may recognize the same epitope based on shared similarity in the CDR3 regions, we identified 190 tissue-specific TCR groups (Figure S6A), indicating that different Th17 clonotypes in a tissue recognize the same antigen. Homeostatic SLAMF6+ SPL0 cells had high clonal similarities with clusters across all tissues (Figure 6D). In EAE, comparing clonotypes across the cell clusters defined in each tissue (Figure S6B–D), there were high similarities between clonotypes in SPL, DLN, and CNS cell clusters, and (separately) between SPL, MLN, PP, SI, and COL cell clusters (Figure 6D), reflecting the homeostatic (gut-associated) and the disease-driving communities (Figure 4E,F) of tissue Th17 cells. Strikingly, only SPL0 cells shared considerable TCR repertoires with both communities in EAE, whereas SPL1 cells exhibited close TCR sharing with all clusters from the SPL, DLN, and CNS. SPL1 exhibited strong TCR sharing with all CNS clusters, indicating that SPL1 is connected to all Th17 cell phenotypes in the CNS (Figure 3). Notably, one small SI cluster (SI5) grouped with the disease-driving ecosystem (Figure 6D) and highly expressed migratory genes (Figure S6E,F). A small proportion of similar cells expressing the migratory signature were detected in every tissue except SPL during EAE (Figure S6G,H), suggesting these cells are a systemic migratory Th17 subpopulation present in all tissues following EAE induction.
Based on these findings, we hypothesized that CXCR6+ SPL1 Th17 cells migrate to the CNS following induction of EAE. Supporting our hypothesis, we detected CXCR6+ tdTomato+ cells in the blood of mice immunized for EAE, and their frequency increased over time post immunization (Figure S7A). Moreover, the expression of Cxcl16, encoding the ligand for CXCR6, was significantly higher in immune cells in the CNS than in the spleen during EAE, supporting a model in which the recruitment of CXCR6+ cells from the spleen to the CNS via the blood is driven by a CXCL16-gradient (Figure S7B).
To validate that SPL0 and SPL1 cells have distinct migratory behaviors in vivo during EAE, we sorted and adoptively transferred SLAMF6+ and CXCR6+ SPL cells obtained from mice with activate EAE into congenically marked mice that were immunized for EAE 7 days prior to the transfer and harvested the cells from each tissue 7 days post transfer (14 days post EAE immunization). The CXCR6+ transferred population was only found in the CNS, DLN, and SPL, while the SLAMF6+ population was present in all tissues including intestinal tissues, confirming the distinct migratory profile of SPL0 and SPL1 in vivo during EAE (Figure 6E).
The homeostatic SLAMF6+ Th17 population gives rise to the pathogenic CXCR6+ Th17 population
Based on previous studies indicating that intestinal Th17 cells can influence the development of EAE (Lee et al., 2011, Haghikia et al., 2015, Miyauchi et al., 2020, Cosorich et al., 2017), we hypothesized that the stem-like, intestinal TCF1+ SLAMF6+ SPL0 cells could give rise to pathogenic SPL1 cells and replenish them during EAE.
To test our hypothesis, we first analyzed the extent of TCR repertoire sharing between SPL0 and SPL1 cells and found that there is substantial sharing of clones between them (Figure 6F), spanning ~20% of SPL0 cells and ~50% of SPL1 cells. SPL0 cells spanned a diverse TCR repertoire with fewer expanded clones, consistent with a homeostatic population induced by diverse agents (such as microbiota or EAE immunization), whereas SPL1 had a less diverse but more expanded repertoire. The shared clonotypes were highly expanded in SPL1 compared to SPL0 cells. Indeed, a higher proliferation signature is observed in SPL1 vs. SPL0 cells (Figure S7C).
To test for in vivo conversion of SPL0 to SPL1 cells, we transferred either SLAMF6+ or CXCR6+ cells from mice with EAE into congenically marked immunized recipient mice, as above (Figure 6G). Following transfer of SLAMF6+ donor cells, we recovered both SLAMF6+ and CXCR6+ cells, confirming the ability of SLAMF6+ cells to give rise to CXCR6+ cells in vivo. In contrast, following transfer of CXCR6+ donor cells, we recovered only CXCR6+ cells in recipients’ spleens, but little to no SLAMF6+ cells, suggesting that the CXCR6+ cells exhibit a terminal effector cell phenotype. The transfer of CXCR6+ cells exacerbated EAE disease (Figure S7D), supporting CXCR6+ cells as the pathogenic population during EAE.
To test whether the SLAMF6+ population may be maintained by the gut microbiota, we eliminated intestinal microbes with a broad-spectrum antibiotic cocktail (VMNA) (Wu et al., 2010). Antibiotics (Abx)-treatment led on average to a ~9-fold reduction of tdTomato+ Th17 cells in all tissues, including a corresponding proportional reduction of splenic SLAMF6+ cells (Figure S7E,F), suggesting that the homeostatic SLAMF6+ population is maintained by the presence of the gut microbiota. Mice that were Abx-treated and immunized for EAE were resistant to EAE (Figure S7G) and showed a substantial reduction of both SLAMF6+ and CXCR6+ cells in the spleens (Figure S7H), suggesting that lack of SLAMF6+ to CXCR6+ conversion may contribute to EAE resistance in gut-microbiota depleted mice.
Finally, we directly validated the migration and conversion of intestinal cells to CXCR6+ cells in the spleen and CNS by performing photoconversion experiments using Kaede mice, which ubiquitously express a photoconvertible fluorescent protein (Tomura et al., 2008). After photoconversion of intestinal cells in immunized mice, we detected photoconverted CXCR6+ CD4+ T cells in the spleen and CNS (Figure 6H), that expressed IFNγ and GM-CSF (data not shown), confirming that intestinal cells are the reservoir for pathogenic CXCR6+ cells that migrate to the CNS to drive neuroinflammation.
The generation of GM-CSF+ IFNγ+ CXCR6+ pathogenic Th17 cells from IL-17A+ SLAMF6+ homeostatic Th17 cells
It is currently unclear whether IL-17-producing Th17 cells or a unique subset of GM-CSF- and IFNγ-producing T cells are the critical pathogenic cells driving autoimmune tissue inflammation (Ghoreschi et al., 2010, Lee et al., 2012, Haak et al., 2009, McGinley et al., 2020, Komuczki et al., 2019, Rasouli et al., 2020, Codarri et al., 2011, Galli et al., 2019). To shed light on this question, we examined the expression patterns of genes encoding for key cytokines in scRNA-seq of the homeostatic SLAMF6+ and the pathogenic CXCR6+ cells.
Interestingly, a significantly higher proportion of SPL0 cells expressed Il17a, Il17f, and Il2, whereas a higher proportion of SPL1 cells expressed Csf2 and Ifng (Figure 7A). This cytokine gene expression was confirmed as antigen-specific by MOG recall assay, as SLAMF6+ cells exhibited increased IL-17A and IL-2 positivity after ex vivo stimulation with MOG35–55 peptide, while CXCR6+ cells displayed a dramatically increase in GM-CSF and IFNγ positivity and expression level after peptide stimulation (Figure 7B, Figure S7I). This suggests a role for both IL-17-producing Th17 cells and GM-CSF- and IFNγ-producing T cells in induction of EAE, such that a homeostatic IL-17+ population gives rise to GM-CSF+ IFNγ+ T cells. Moreover, there is a higher fraction of IFNGR1-expressing proinflammatory CXCR6+ SPL1 cells (Figure 7C), suggesting an autocrine positive feedback loop mediated by IFNγ in expanding or maintaining the SPL1 phenotype (Schneider et al., 2014).
Figure 7: IL-23R signaling is driving the generation of GM-CSF+ IFNγ+ CXCR6+ cells from IL-17+ SLAMF6+ cells.
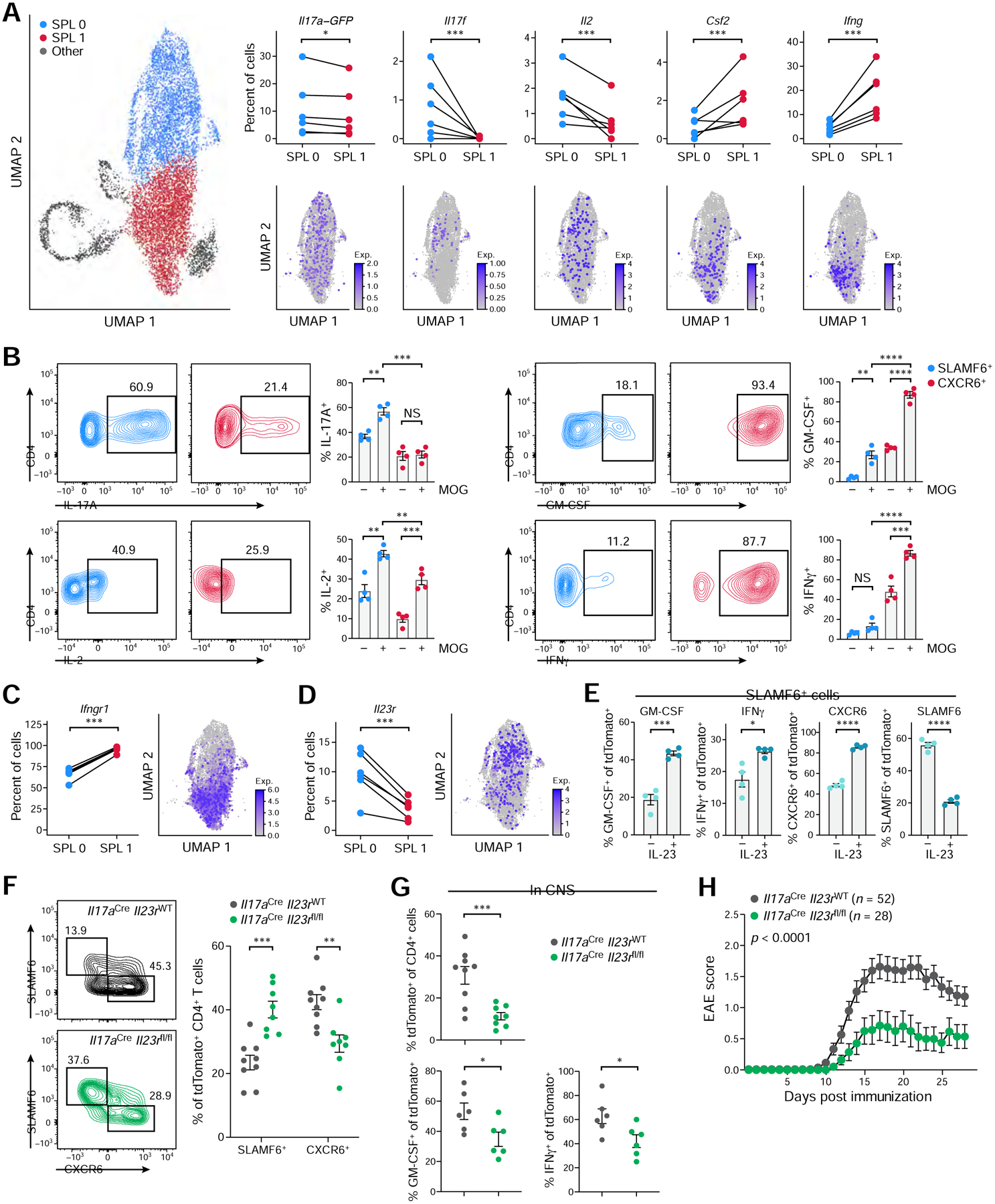
(A) Cytokine expression in SPL0 and SPL1 clusters. Percent of cells with UMI count ≥1 by sample (top, n=6) and normalized expression on the UMAP (bottom).
(B) Cytokine expression in SLAMF6+ and CXCR6+ cells during recall assay with or without MOG peptide (n=4). Representative flow cytometry plots (left) and quantification (right).
(C-D) Ifngr1 (C) and Il23r (D) expression in SPL0 and SPL1 clusters. Percent of cells with UMI count ≥1 by sample (left, n=6) and normalized expression on the UMAP (right).
(E) Phenotype of the SLAMF6+ population co-cultured for 3 days with CD4-deplected splenocytes with or without IL-23 by flow cytometry (n=4).
(F) Frequencies of splenic SLAMF6+ and CXCR6+ tdTomato+ CD4+ T cells in immunized Il17aCreIl23rWT and Il17aCreIl23rfl/fl mice analyzed at EAE onset (n=8–9). Representative flow cytometry plots (left) and quantification (right) are shown.
(G) tdTomato+ CD4+ T cell frequencies (top) and GM-CSF- and IFNγ- expression (bottom) in the CNS of immunized Il17aCreIl23rWT and Il17aCreIl23rfl/fl mice at disease onset (n=6–9).
(H) EAE disease curve in Il17aCreIl23rWT (n=52) and Il17aCreIl23rfl/fl (n=28) mice. Data are mean from four independent experiments.
IL-23R-driven generation of encephalitogenic CXCR6+ T cells from SLAMF6+ T cells
IL-23 signaling plays a pivotal role in autoimmune pathogenesis (Cua et al., 2003, Murphy et al., 2003) and is required for the generation of pathogenic T cells (Langrish et al., 2005, Lee et al., 2012), but the mechanism by which it drives the generation of pathogenic T cells remains unclear. Interestingly, the receptor for IL-23 (IL-23R) is expressed in a higher proportion of SPL0 than SPL1 cells (Figure 7D), while there was a higher expression of the IL-23 signature in SPL1 cells and higher chromatin accessibility at gene loci downstream of IL-23R signaling in CXCR6+ vs. SLAMF6+ cells (Figure S7J,K), suggesting greater IL-23R signaling in CXCR6+ cells. We thus hypothesized that IL-23R signaling may mediate the conversion of SLAMF6+ to CXCR6+ cells. To test our hypothesis, we co-cultured SLAMF6+ cells with splenocytes in vitro and treated them with IL-23 (Figure 7E). The proportion of SLAMF6+ cells decreased in the presence of IL-23, whereas the proportion of GM-CSF+, IFNγ+, and CXCR6+ cells increased, suggesting an IL-23R signaling-mediated conversion of homeostatic stem-like Th17 cells into pathogenic CXCR6+ cells. As putative sources of IL-23 during EAE, we identified dendritic cells in the spleen and monocytes, macrophages, neutrophils, and dendritic cells in the CNS as the immune populations expressing highest levels of Il12b and Il23a transcripts (Figure S7L,M).
Finally, to characterize the effect of IL-23R-signaling on the generation of pathogenic CXCR6+ cells in vivo, we generated IL-23R conditional knockout mice (Il23rfl/fl; STAR Methods) and induced EAE in Il17aCreIl23rfl/fl mice, where IL-23R is specifically deleted in Th17 cells. We found an increased frequency of SLAMF6+ cells and significant reduction in CXCR6+ cells in the spleen compared to wild-type littermate controls (Figure 7F), and, consistently, fewer tdTomato+ CD4+ T cells in the CNS, fewer of those cells expressed GM-CSF and IFNγ (Figure 7G), and there was resistance of the mice to the induction of EAE (Figure 7H). Bulk RNA-seq of Il17aCreIl23rfl/fl CXCR6+ cells revealed substantial expression changes in the CXCR6+ population without IL-23R-signaling (34 genes up-regulated, 120 genes down-regulated; FDR <0.05; Table S5), including down-regulation of gene signatures involved in Th17 pathogenicity (Figure S7N,O). In conclusion, these data suggest that IL-23R-signaling mediates the generation of pathogenic CXCR6+ cells from SLAMF6+ cells.
Discussion
In this study we analyzed the expression landscape and connectivity of tissue Th17 cells during homeostasis and CNS autoimmunity and provide a mechanism by which Th17 cells from the intestine contribute to CNS pathogenesis.
The homeostatic SLAMF6+ cell population shows multiple parallels to previously described stem-like CD8+ T cells in tumors and during LCMV-infections (Im et al., 2016, Siddiqui et al., 2019, Hudson et al., 2019) in that SLAMF6+ cells highly express Tcf7, Cxcr5, Slamf6, Ccr7, and Sell and are present in multiple lymphoid organs. Moreover, stem-like CD8+ T cells are characterized by their ability to undergo self-renewal and to differentiate into effector and terminally exhausted CD8+ T cells (Im et al., 2016, Siddiqui et al., 2019, Escobar et al., 2020, Hudson et al., 2019). Analogously, we found that stem-like SLAMF6+ population can give rise to pathogenic CXCR6+ Th17 cells. Hence, it is tempting to speculate that in CD4+ T cell-driven autoimmune diseases, stem-like SLAMF6+ cells act as a tissue-dwelling reservoir for the generation of numerous pathogenic T cells, contributing to and further amplifying tissue inflammation.
Recent studies have highlighted a critical role of the intestinal microbiota in the development of extra-intestinal autoimmune diseases (Berer et al., 2011, Haghikia et al., 2015, Miyauchi et al., 2020, Berer et al., 2017), and the induction of intestinal Th17 cells by gut microbes has been suggested as the mechanism (Lee et al., 2011, Miyauchi et al., 2020, Cosorich et al., 2017). The conversion of the SLAMF6+ Th17 cell population that is maintained by gut microbiota to the CXCR6+ cells might provide a potential connection between the gut microbiota-induced Th17 population and the pathogenic Th17 population in the CNS. Building on previous work demonstrating cross-reactivity of microbiota-induced Th17 cells to autoantigens like MOG (Miyauchi et al., 2020, Bradley et al., 2017), it is possible that microbiota-induced SLAMF6+ cells display cross-reactivity to self-antigens during EAE, allowing them to proliferate and give rise to the clonally expanded disease-inducing CXCR6+ population. Notably, since our study was focused on intestinal tissues, we cannot exclude the possibility that SLAMF6+ cells might migrate to other mucosal sites (lungs) or other tissues where host microbiota can maintain them.
One open question in the field concerns the relative roles of IL-17-producing T cells vs. GM-CSF-producing T cells in driving autoimmune tissue inflammation (Ghoreschi et al., 2010, Lee et al., 2012, Haak et al., 2009, McGinley et al., 2020, Komuczki et al., 2019, Rasouli et al., 2020, Codarri et al., 2011, Galli et al., 2019). We discovered the conversion of SLAMF6+ to CXCR6+ cells to be accompanied by down-regulation of an IL-17-program and up-regulation of a pathogenic GM-CSF- and IFNγ- program. Thus, our results suggest that IL-17-producing T cells are the progenitors of GM-CSF- and IFNγ-producing T cells, directly relating the two effector T-cell populations in a model where both are sequentially involved in the development of autoimmune disease.
In conclusion, our study provides mechanistic insights that may apply to various autoimmune contexts and targeting the stem-like intestinal Th17 population and IL-23R signaling could allow for better therapeutic control over the chronicity and relapses observed across many human autoimmune conditions (Rosenblum et al., 2015).
Limitations of the study
The TCR analysis statistics in this study, including clonal expansion and clonal similarity score, may have residual dependencies on the number of cells even after normalization. Concerning the study design, our study is focused on the Th17 lineage including all CD4+ T cells that ever-expressed Il17a and therefore does not account for other T cell lineages that are likely to play important roles in autoimmune disorders, including Th1 cells and Treg cells.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Vijay K. Kuchroo (vkuchroo@evergrande.hms.harvard.edu).
Materials availability
There are no newly generated materials in this paper. Il23rfl/fl mice can be shared upon request.
Data and code availability
scRNA/TCR-seq, bulk RNA-seq, and bulk ATAC-seq data have been deposited at GEO under accession number GSE188322 and are publicly available as of the date of publication. Detailed accession numbers are listed in the key resources table.
All original code has been deposited at GitHub (https://github.com/lhuang1/Th17_single_cell) and is publicly available as of the date of publication. The GitHub information is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 APC-Cy7 (clone 3-F11) | Biolegend | Cat# 103116, RRID: AB_312981 |
| Anti-mouse CD4 APC (clone RM4-5) | Biolegend | Cat# 100516, RRID: AB_312719 |
| Anti-mouse CD4 APC-Cy7 (clone RM4-5) | Biolegend | Cat# 100526, RRID: AB_312727 |
| Anti-mouse TCRp PE-Cy7 (clone H57-597) | Biolegend | Cat# 109222, RRID: AB_893625 |
| Anti-mouse PD-L1 APC (clone 10F.9G2) | Biolegend | Cat# 124312, RRID: AB_10612741 |
| Anti-mouse CXCR6 APC (clone SA051D1) | Biolegend | Cat# 151106, RRID: AB_2572143 |
| Anti-mouse SLAMF6 Pacific Blue (clone 330-AJ) | Biolegend | Cat# 134608, RRID: AB_2188093 |
| Anti-mouse CD8a PE-Cy7 (clone 53-6.7) | Biolegend | Cat# 100722, RRID: AB_312761 |
| Anti-mouse PD-1 Brilliant Violet 711 (clone 29F.1A12) | Biolegend | Cat# 135231, RRID: AB_2566158 |
| Anti-mouse NK1.1 Brilliant Violet 421 (clone PK136) | Biolegend | Cat# 108732, RRID: AB_2562218 |
| Anti-mouse Ly-6C Brilliant Violet 605 (clone HK1.4) | Biolegend | Cat# 128036, RRID: AB_2562353 |
| Anti-mouse I-A/E Brilliant Violet 785 (clone M5/114.15.2) | Biolegend | Cat# 107645, RRID: AB_2565977 |
| Anti-mouse CD64 PE (clone X54-5/7.1) | Biolegend | Cat# 139304, RRID: AB_10612740 |
| Anti-mouse/human CD11b PE-Cy7 (clone M1/70) | Biolegend | Cat# 101216, RRID: AB_312799 |
| Anti-mouse CD19 APC-Cy7 (clone 6D5) | Biolegend | Cat# 115530, RRID: AB_830707 |
| Anti-mouse Gr1 APC (clone RB6-8C5) | Biolegend | Cat# 108412, RRID: AB_313377 |
| Anti-mouse/human CD44 PerCP/Cy5.5 (clone IM7) | Biolegend | Cat# 103032, RRID: AB_2076204 |
| Anti-mouse IFNγ APC-Cy7 (clone XMG1.2) | Biolegend | Cat# 505850, RRID: AB_2616698 |
| Anti-mouse GM-CSF PE-Cy7 (clone MP1-22E9) | Biolegend | Cat# 505412, RRID: AB_2721682 |
| Anti-mouse IL-17A FITC (clone TC11-18H10.1) | Biolegend | Cat# 506908, RRID: AB_536010 |
| Anti-mouse IL-2 Brilliant Violet 605 (clone JES6-5H4) | Biolegend | Cat# 503829, RRID: AB_11204084 |
| Anti-mouse CD11c PE-CF594 (clone N418) | BD Biosciences | Cat# 565591, RRID: AB_2869692 |
| Anti-mouse AREG antibody (clone 206220) | R&D Systems | Cat# MAB989, RRID: RRID: AB_2060661 |
| Chemicals, peptides, and recombinant proteins | ||
| MOG peptide | Genemed Synthesis | Cat# MOG3555-P2-5 |
| M. tuberculosis H37 Ra, desiccated | Becton Dickinson | Cat# 231141 |
| Adjuvant, Complete H37 Ra | Becton Dickinson | Cat# 231131 |
| Pertussis toxin | List Biological Laboratories | Cat# 181 |
| Ampicillin sodium salt | Sigma Aldrich | Cat# A9518 |
| Metronidazole | Acros Organics | Cat# AC210340050 |
| Vancomycin hydrochloride | Research products international | Cat# V06500-5.0 |
| Neomycin sulfate | Thermo Fischer Scientific | Cat# BP26695 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-14 |
| DNase I | Sigma Aldrich | Cat# 11284932001 |
| Collagenase Type VIII | Sigma Aldrich | Cat# C2139 |
| CaCl2 | Sigma Aldrich | Cat# C5670 |
| MgCl2 | Sigma Aldrich | Cat# 63069 |
| DTT (dithiothreitol) | Thermo Fischer Scientific | Cat# R0861 |
| RPMI 1640 medium | Thermo Fischer Scientific | Cat# A1049101 |
| EDTA | Thermo Fischer Scientific | Cat# AM9261 |
| Collagenase D | Roche Diagnostics | Cat# 11088882001 |
| Percoll | Cytiva | Cat# 17-0891-01 |
| 2-mercaptoethanol | Sigma Aldrich | Cat# M6250 |
| Digitonin | Promega | Cat# G9441 |
| Dextran Sulfate Sodium Salt (DSS) | Affymetrix | Cat# 14489-100-GM |
| Critical commercial assays | ||
| Pacific Blue Antibody Labeling Kit | Molecular Probes | Cat# P30013 |
| ACK lysis buffer | Lonza | Cat# 10-548E |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat# 00-4975-93 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Cat# 130117043 |
| Recombinant Mouse IL-23 Protein | R&D Systems | Cat# 1887-ML-010 |
| LegendPlex Mouse Inflammation Panel | Biolegend | Cat# 740446 |
| PicoPure™ RNA Isolation Kit | Applied Biosystems | Cat# KIT0204 |
| SuperScript™ VILO™ cDNA Synthesis Kit | Invitrogen | Cat# 11754050 |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher Scientific | Cat# 4444557 |
| HiSeq X Ten Reagent Kit v2.5 | Illumina | Cat# FC-501-2501 |
| TCL buffer | QIAGEN | Cat# 1031576 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fischer Scientific | Cat# 1102-200 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1024 |
| NextSeq 500 High Output v2 Kit (75 cycles) | Illumina | Cat# FC-404-2005 |
| BAMBANKER freeze medium | LYMPHOTEC Inc | Cat# NC9582225 |
| Illumina Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | Cat# 20034198 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| NEBNext® High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541L |
| SYBR™ Green I Nucleic Acid Gel Stain | Invitrogen | Cat# S7585 |
| KAPA Library Quantification Kit | KAPA Biosystems | Cat# KK4835 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Chromium Single Cell 3’ Reagent Kit (v2 Chemistry) | 10x Genomics | Cat# 120237 |
| Chromium Single Cell 5’ Library & Gel Bead Kit 16 rxn | 10x Genomics | Cat# 1000006 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10x Genomics | Cat# 1000071 |
| Deposited data | ||
| Mouse reference genome NCBI build 38, GRCm38 (mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Public human kidney immune cell scRNA-seq data | (Arazi et al., 2019) | https://www.immport.org/shared/study/SDY997 |
| Public mouse thymus immune cell scRNA-seq data | (Hemmers et al., 2019) | GEO: GSE134902 |
| Public mouse T helper cell scRNA-seq data | (Tibbitt et al., 2019) | GEO: GSE131935 |
| Public human PBMC and CSF immune cell scRNA-seq data | (Schafflick et al., 2020) | GEO: GSE138266 |
| Public mouse CNS immune cell scRNA-seq data | (Wheeler et al., 2020) | GEO: GSE130119 |
| Bulk RNA-seq and ATAC-seq of SLAMF6+ and CXCR6+ Th17 cells | This paper | GEO: GSE188319 |
| scRNA/TCR-seq of tissue Th17 cells in homeostasis and EAE | This paper | GEO: GSE188320 |
| scRNA-seq of colonic Th17 cells in colitis | This paper | GEO: GSE188321 |
| Experimental models: Mice | ||
| C57BL/6-Il17atmlBcgen/J (Il17aGFP) | Jackson Laboratory | JAX: 018472 |
| Il17atm1.1(icre)Stck/J (Il17aCre) | Jackson Laboratory | JAX: 016879 |
| B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Rosa26tdTomato) | Jackson Laboratory | JAX: 007905 |
| B6.SJL-PtprcaPepcb/BoyJ (CD45.1) | Jackson Laboratory | JAX: 002014 |
| Foxp3tm1Kuch (Foxp3GFP) | (Bettelli et al., 2006) | N/A |
| Il23r fl/fl | This paper | |
| Tg(CAG-Kaede)15Kgwa (Kaede) | RIKEN BRC, Japan | N/A |
| Oligonucleotides | ||
| Eukaryotic 18S rRNA Endogenous Control | Thermo Fischer Scientific | Cat# 4352930E |
| Mouse ACTB (Actin, Beta) Endogenous Control | Thermo Fischer Scientific | Cat# 4352341E |
| Ifit1 (Mm00515153_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Ifit3 (Mm01704846_s1) | Thermo Fischer Scientific | Cat# 4448892 |
| Irf7 (Mm00516793_g1) | Thermo Fischer Scientific | Cat# 4448892 |
| Isg15 (Mm01705338_s1) | Thermo Fischer Scientific | Cat# 453320 |
| Isg20 (Mm00469585_m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Zbp1 (Mm01247052_m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Nkg7 (Cat# Mm01205900_g1) | Thermo Fischer Scientific | Cat# 4448892 |
| Ccl5 (Cat# Mm01302427_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Cd8a (Cat# Mm01182107_g1) | Thermo Fischer Scientific | Cat# 453320 |
| Prf1 (Cat# Mm00812512_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Gzmk (Cat# Mm00492530 m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Cxcl16 (Cat# Mm00469712_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Il12b (Cat# Mm01288989_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Il23a (Cat# Mm00518984_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Ad1 primer: AATGATACGGCGACCACCGAGATCTACACXXXXXXXXTCGTCGGCAGCGTCAGATGTGTAT | IDT | N/A |
| Ad2 primer: CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTCTCGTGGGCTCGGAGATGTG | IDT | N/A |
| Software and algorithms | ||
| FlowJo | TreeStar | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| R v3.6.2 | R core | https://www.r-project.org/ |
| CellRanger v3.0.2 | (Zheng et al., 2017) | https://support.10xgenomics.com/single-cell-gene-expression/software |
| DemuxEM v0.1.5 | (Gaublomme et al., 2019) | https://demuxem.readthedocs.io/ |
| Seurat v3.2.2 | (Stuart et al., 2019) | https://satijalab.org/seurat/ |
| edgeR | (Robinson et al., 2010, McCarthy et al., 2012) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| fGSEA | (Korotkevich et al., 2019) | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| COMET | (Delaney et al., 2019) | http://www.cometsc.com/index |
| GLIPH2 | (Huang et al., 2020) | http://50.255.35.37:8080/ |
| HISAT2 | (Kim et al., 2019) | http://daehwankimlab.github.io/hisat2/ |
| RSEM | (Li and Dewey, 2011) | https://deweylab.github.io/RSEM/ |
| ATAC-seq data analyses pipeline | (Lee et al., 2016) | https://github.com/kundajelab/atac_dnase_pipelines |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 v2.1.2 | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| Bedtools v2.26.1 | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| GREAT v4.0.4 | (McLean et al., 2010) | http://great.stanford.edu/public/html/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Analysis code | This paper | https://github.com/lhuang1/Th17_single_cell |
| Other | ||
| 200 mW Electra Pro portable laser | Laserglow Technologies | GEL200XXX |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6-Il17atm1Bcgen/J named Il17aGFP), Il17atm1.1(icre)Stck/J (named Il17aCre), B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (named Rosa26tdTomato), and B6.SJL-PtprcaPepcb/BoyJ (named CD45.1) mice were obtained from the Jackson Laboratory. Foxp3tm1Kuch (named Foxp3GFP) mice were generated in the V.K.K. laboratory (Bettelli et al., 2006). Tg(CAG-Kaede)15Kgwa mice (named Kaede, (Tomura et al., 2008)) on C57Bl/6J background were obtained from RIKEN BRC, Japan. To generate conditional IL-23 receptor knockout mice (named Il23rfl/fl) we took advantage of a recently established ES cell resource (Skarnes et al., 2011). ES cell clone EPD0812_2_C11 with a targeted IL-23 receptor allele (containing a neomycin selectable marker flanked by frt sites) with conditional potential was acquired from EuMMCR (European Mouse Mutant Cell Repository; www.eummcr.org) within EUCOMM (European Conditional Mouse Mutagenesis Program). This ES cell clone was used for blastocyst injections. Faithful targeting of the IL-23 receptor allele was confirmed in the ES clone prior to blastocyst injections by Southern blot analysis. Chimeric mice were bred to C57Bl/6 mice and offspring screened for germline transmission by PCR for the neomycin selectable marker cassette. Offspring harboring the targeted IL-23 receptor neo+ allele were then bred to Flpo deleter mice (Kranz et al., 2010). Offspring carrying the final conditional IL-23 receptor allele (2lox) were bred to C57Bl/6 to remove the Flpo transgene. The IL-23 receptor conditional allele was bred to homozygosity and then to various Cre strains to obtain tissue/cell-type specific deletion of IL-23 receptor. The primers TCCAGAGACGCCTGTACTCA and CACCAAGCTTTTCTCCTCCTC are used for routine genotyping (wt=414bp and 2lox=577bp). For experiments, mice were sex- and age- matched (8–12 weeks old) and littermates were used as controls.
Mice were housed under specific-pathogen-free conditions (SPF) at the Brigham and Women’s Hospital mouse facility in Boston. All experiments that involved the use of mice were performed in accordance with the guidelines outlined by the Brigham and Women’s Institutional Animal Care and Use Committee (IACUC) in Boston.
METHOD DETAILS
Experimental autoimmune encephalomyelitis (EAE)
For active induction of EAE, mice were immunized subcutaneously in the flanks with the myelin oligodendrocyte glycoprotein (MOG) peptide MOG35–55 (100 μg/mouse, Genemed Synthesis), emulsified in complete Freund’s adjuvant (CFA) (200 μl/mouse, Becton Dickinson), and supplemented with Mycobacterium tuberculosis H37Ra extract (5 mg/ml, Becton Dickinson). Pertussis toxin (100 ng/mouse, List Biological Laboratories) was administered intravenously into the tail vein on day 0 and day 2 post immunization. Disease course was monitored daily, and mice were assigned a clinical score as follows: 0, healthy; 1, limp tail; 2, hind limb weakness or impaired righting reflex; 3, hind limb paralysis; 4, hind and front limb paralysis; 5, moribund or dead. Mice with a clinical score >4 were euthanized.
DSS-induced colitis
Colitis was induced by adding 2% DSS (Affymetrix) to the drinking water on days 0–7, followed by a two-day recovery period with normal drinking water. The disease progression was monitored by measurement of the body weight.
Antibiotic treatment
Immediately after weaning, 3- to 4- week-old littermates were administrated weekly with 1 g/L of Ampicillin sodium salt (Sigma Aldrich), 1 g/L of Metronidazole (Acros Organics), 0.5 g/L Vancomycin hydrochloride (Research products international), and 1 g/l of Neomycin sulfate (Thermo Fischer Scientific) in the drinking water as previously described (Wu et al., 2010). Sweetener (Equal) was added to the water (2.5 g/L) of the experimental group and the control group. For analysis at homeostasis, mice were treated with antibiotics for 6 weeks. For analysis during EAE, mice were treated with antibiotics starting 2-weeks prior to immunization and through the disease course.
Photoconversion of intestinal cells in Kaede mice
For photoconversion of intestinal tissues (MLN, PP, COL, and SI) mice were anaesthetized and a small cut was made into the peritoneal cavity. The intestinal tissues were carefully extended and exposed to 405 nm violet light using a 200 mW Electra Pro portable laser (Laserglow Technologies) for a total of 4 mins (one min each tissue) with continuous wetting of the tissues with PBS. The surrounding tissues were covered with sterile aluminum foil to protect against unspecific photoconversion. After returning the intestinal tissues into the peritoneal cavity, the peritoneum was sutured, and the skin closed with surgical staples. Mice were euthanized 48 h after the procedure and the biodistribution of the photoconverted red protein was analyzed by flow cytometry.
Isolation of tissue Th17 cell populations
For tissue harvest, mice were perfused intracardially with cold PBS. Lymphoid tissues, spleen and mesenteric lymph nodes were harvested and smashed through 40 μm sterile strainers. ACK lysis (Buffer from Lonza) was performed with the spleen for 5 min at RT. Peyer’s patches were digested for 20 min at 37°C in a water bath in digestion buffer (5% fetal bovine serum, 50 μg/ml DNase I (Sigma Aldrich), 1 mg/ml Collagenase Type VIII (Sigma Aldrich), 2 mM CaCl2 (Sigma Aldrich), 5 mM MgCl2 (Sigma Aldrich) in RPMI 1640 medium (Thermo Fisher Scientific)) and smashed through a 40 μm sterile strainer. To isolate cells from the small intestine and the colon, intestinal tissues were harvested and cleaned. Epithelial cells and intraepithelial lymphocytes were removed by incubating the tissues for 20 min at 37°C with constant stirring at 350 rpm in extraction buffer (5% fetal bovine serum, 1 mM DTT (Thermo Fischer Scientific), 5 mM EDTA (Thermo Fisher Scientific) in RPMI 1640 medium (Thermo Fisher Scientific)). Tissues were minced and incubated in digestion buffer for 40 min at 37°C with constant stirring at 550 rpm and smashed through a 40 μm sterile strainer. To isolate CNS-infiltrating lymphocytes from EAE mice, brain and spinal cord were flushed out with PBS by hydrostatic pressure. The CNS was minced and digested in 2.5 mg/ml Collagenase D (Roche Diagnostics) in RPMI 1640 medium (Thermo Fisher Scientific) for 20 min at 37°C. Cells were smashed through a 40 μm sterile cell strainer and mononuclear cells were enriched on a discontinuous (37% and 70%) Percoll gradient (Cytiva). To analyze blood cells, mice were bled on the cheek and red blood cells were removed in two rounds of 5 min ACK lysis (Buffer from Lonza) at room temperature (RT). After tissue cell isolation, cells were stained for flow cytometry.
Flow cytometry analysis
Single-cell suspensions were stained in flow buffer (2% fetal bovine serum in PBS) with antibodies for surface proteins. Antibodies with specificity for the following cell surface proteins were purchased from Biolegend with different fluorochrome labels: CD45 (clone 3-F11), CD4 (clone RM4-5), TCRβ (clone H57-597), PD-L1 (clone 10F.9G2), CXCR6 (clone SA051D1), SLAMF6 (clone 330-AJ), CD8a (clone 53-6.7), PD-1 (clone 29F.1A12), NK1.1 (clone PK136), Ly-6C (clone HK1.4), I-A/E (clone M5/114.15.2), CD64 (clone X54-5/7.1), CD11b (clone M1/70), CD19 (clone 6D5), Gr1 (clone RB6-8C5), CD44 (clone IM7). The anti-mouse CD11c (clone N418) antibody was purchased from BD Biosciences. Cells were stained for viability using eF506 dye (eBioscience).
Surface antibody staining was performed for 30 min at 4°C in the dark. The following cell populations were identified in spleen and CNS based on cell marker expression: CD4+ T cells (CD45+ TCRβ+ CD4+), activated CD4+ T cells (CD45+ TCRβ+ CD4+ CD44+), CD8+ T cells (CD45+ TCRβ+ CD8+), activated CD8+ T cells (CD45+ TCRβ+ CD8+ CD44+), Th17 cells (CD45+ TCRβ+ CD4+ IL-17A-GFP+), B cells (CD45+ CD19+), natural killer (NK) cells (CD45+ NK1.1+), neutrophils (CD45+ CD11b+ Ly-6Cint Gr1+), dendritic cells (DCs) (CD45+ CD11c+ I-A/Ehigh), macrophages (MACS) (CD45+ CD11b+ Ly-6C− Gr1− CD64+), monocytes (CD45+ CD11b+ Gr1− CD64− Ly-6Clow or high), microglia (CD11b+ CD64+ CD44− CD45int).
For intracellular cytokine staining, cells were activated for 4 h at 37°C with the Cell Stimulation Cocktail (plus protein transport inhibitors) (eBioscience) and fixed and stained using the Fixation/Permeabilization Solution Kit (BD Biosciences) per manufacturer’s instructions. Antibodies with specificity for the following intracellular proteins were purchased from Biolegend with different fluorochrome labels: IFNγ (clone XMG1.2), GM-CSF (clone MP1-22E9), IL-17A (clone TC11-18H10.1), IL-2 (clone JES6-5H4). The anti-AREG antibody (clone 206220, R&D Systems) was conjugated to Pacific Blue according to the manufacturer’s instructions (Molecular Probes). Intracellular/intranuclear antibody staining was performed for 30 min at 4°C in the dark.
Flow cytometry analysis and Fluorescence Activated Cell Sorting (FACS) was performed on a BD LSRII (BD Biosciences) or BD LSRFortessa (BD Biosciences). FACS was performed on a BD FACS Aria IIIu flow cytometer (BD Biosciences). The data was analyzed with FlowJo software (TreeStar) and GraphPad Prism software (GraphPad).
T cell transfers
Active EAE was induced in 8- to 12-week-old Il17aCreGFP X Rosa26tdTomato littermates and spleens were isolated 14 days post immunization. Viable TCRβ+ CD4+ tdTomato+ SLAMF6+ or CXCR6+ populations were purified by FACS following MACS bead isolation of CD4+ cells (Miltenyi Biotec). 4×104 cells were subsequently intravenously transferred into CD45.1 recipient mice that have been immunized for active EAE 7 days prior to the transfer. 7 days post transfer tissues were isolated, and flow cytometry was performed as described above.
Recall experiments
8- to 12-week-old Il17aCreGFP X Rosa26tdTomato littermates were immunized for EAE induction. At 14 days post immunization, viable TCRβ+ CD4+ tdTomato+ SLAMF6+ or CXCR6+ populations were purified by MACS bead isolation of CD4+ cells followed by FACS. 2×104 SLAMF6+ or CXCR6+ cells were in vitro restimulated with 50 μg MOG35–55 peptide and 1×105 CD4− splenocytes for 3 days. Cells were harvested for intracellular cytokine staining.
For in vitro treatment of SLAMF6+ cells with IL-23, 2×104 SLAMF6+ cells were co-cultured with 2×105 CD4− splenocytes with or without IL-23 (20 ng/mL, R&D Systems, Cat# 1887-ML-010) for 3 days.
Bead-based immunoassay
Cytokine concentrations in the culture medium of the recall assay were determined by the LegendPlex Mouse Inflammation Panel (13-plex) (Biolegend, Cat# 740446) following the manufacturer’s instructions. Samples were acquired with a BD LSRII flow cytometer (BD Biosciences) and analyzed using the LegendPlex Data Analysis Software Suite.
Quantitative Polymerase Chain Reaction (qPCR)
Total RNA was extracted using the PicoPure RNA Isolation Kit (Applied Biosystems) according to the manufacturer’s instructions. Reverse transcription of mRNA was performed with the SuperScript VILO cDNA Synthesis Kit (Invitrogen) in a thermal cycler (Bio-Rad). qPCR was conducted using the TaqMan Fast Advanced Master Mix (Thermo Fisher Scientific) in the ViiA 7 Real-Time PCR system (Applied Biosystems). Gene expression was normalized to the expression of 18S rRNA and Actb. The following TaqMan probes (Thermo Fisher Scientific) were used: Ifit1 (Mm00515153_m1), Ifit3 (Mm01704846_s1), Irf7 (Mm00516793_g1), Isg15 (Mm01705338_s1), Isg20 (Mm00469585_m1), Zbp1 (Mm01247052_m1), Nkg7 (Cat# Mm01205900_g1), Ccl5 (Cat# Mm01302427_m1), Cd8a (Cat# Mm01182107_g1), Prf1 (Cat# Mm00812512_m1), Gzmk (Cat# Mm00492530_m1), Cxcl16 (Cat# Mm00469712_m1), Il12b (Cat# Mm01288989_m1), Il23a (Cat# Mm00518984_m1), 18S (Cat# 4352930E), and Actb (Cat# 4352341E). Relative expression levels are depicted as 2−ΔCT values, ΔCT = (gene of interest CT) - (geoMean Housekeeper CT).
Droplet-based scRNA-seq and scRNA/TCR-seq
For analysis at homeostasis, 8- to 12-week-old Il17aCreGFP X Rosa26tdTomato female littermates were used. For analysis during EAE disease, active EAE was induced in 8- to 12-week-old Il17aCreGFP X Rosa26tdTomato littermates as described above. Mice were harvested at disease onset (clinical score of 1). Viable CD4+ TCRβ+ tdTomato+ tissue Th17 cells were sorted by FACS for sequencing.
Cell hashing was performed with samples for scRNA/TCR-seq to process all tissues of one mouse together in one 10x channel. After the sort, cells were resuspended in flow buffer with the TotalSeq™-antibody pool (Biolegend) at 1:1000 final dilution for each antibody, followed by incubation for 30 min on ice. Next, cells were washed for a total of three washes with flow buffer.
Samples for scRNA/TCR-Seq with 5’ feature barcoding were then separated into droplet emulsions using the Chromium Single Cell 5’ V2 Solution, according to manufacturer’s instructions (10x Genomics). For samples that did not include scTCR-seq and 5’ feature barcoding, sorted cells were separated into droplet emulsions using the Chromium Single Cell 3′ Solution (v2 or v3) according to manufacturer’s instructions (10x Genomics). scRNA-seq, scTCR-seq and 5’ feature barcoding library preparations were performed according to manufacturer’s instructions (10x Genomics). scRNA-seq libraries and 5’ feature barcoding libraries were sequenced on the HiSeq X (Illumina) with the following read configuration: Read 1: 28 cycles, Read 2: 96 cycles, Index read 1: 8 cycles. scTCR-seq libraries were sequenced on the HiSeq X (Illumina) with the following read configuration: Read 1: 150 cycles, Read 2: 150 cycles, Index read 1: 8 cycles.
Bulk RNA-sequencing
1,000 living cells were sorted by FACS and immediately lysed in TCL buffer (QIAGEN) supplemented with 1% ß-mercaptoethanol (Sigma Aldrich). Full-length RNA-seq libraries were prepared following a modified SMART-Seq2 protocol (Picelli et al., 2014) as previously described (Satija et al., 2015). The cDNA concentration was measured using the Quant-iT PicoGreen dsDNA Assay Kit (Thermo Fisher Scientific) and normalized to 0.25ng/ul. The cDNA libraries were prepared using the Nextera XT DNA Library Preparation kit (Illumina). Final libraries were confirmed to have a size of 500 bp using a Bioanalyzer (Agilent). Prior to sequencing, the uniquely barcoded libraries were pooled, normalized to 2 nM, and denatured using 0.1 N NaOH. Flow cell cluster amplification and sequencing were performed according to the manufacturer’s protocols using the paired-end Illumina sequencing (38bp X 2) with 75 cycle Nextseq 500 high output V2 kit (Illumina).
Bulk ATAC-sequencing
6,000 viable TCRβ+ CD4+ tdTomato+ SLAMF6+ or CXCR6+cells were sorted from the spleen of Il17aCreGFP X Rosa26tdTomato mice and stored at −80°C in Bambanker™ cell freezing medium (LYMPHOTEC Inc.). For library preparation, cells were thawed at 37°C. Subsequently, cells were lysed and tagmented in 1X TD Buffer, 0.2 μl TDE1 (Illumina), 0.01% digitonin, and 0.3X PBS at 37°C for 30 min at 300 rpm, as previously described (Corces et al., 2016). DNA purification was performed with the MinElute PCR purification kit (QIAGEN). The complete eluate was pre-amplified with 5 PCR cycles using indexed primers with NEBNext High-Fidelity 2X PCR Master Mix (NEB) and the number of additional cycles was determined by SYBR Green quantitative PCR. After DNA purification, library quantification and quality control were performed with the Kapa Library Quantification Kit (Kapa Biosystems) and a Qubit dsDNA HS Assay kit (Invitrogen). Libraries were sequenced on an Illumina NextSeq 550 system with paired-end reads of 37 base pairs in length.
Preprocessing of scRNA-seq data
Paired reads were mapped to the reference mouse annotation mm10 and UMI counts were computed using CellRanger v3.0.2 (Zheng et al., 2017) with default parameters (except for chemistry=SC3Pv2). CellRanger filtered UMI count matrices were used for downstream analysis. Il17aGFP UMI counts were computed with a custom reference transcriptome built with the CellRanger mkref command that included the Il17aGFP transcript (Il17aGFPand mm10 chr19). Il17aGFP UMI counts were extracted from the CellRanger raw (unfiltered) count matrices and merged with whole transcriptome count matrix described above. For the scRNA/TCR-seq experiments with cell-hashing (batches 6–9), read and UMI counts were also mapped and computed using CellRanger v3.0.2 (chemistry=fiveprime), then DemuxEM v0.1.5 (Gaublomme et al., 2019) with default parameters was used to obtain tissue label assignment for every cell. Low quality cell profiles were excluded if they fulfilled one or more of the following criteria: (i) number of genes expressed <=500, (ii) >=5% of the total UMIs were mitochondrial RNA or (iii) <60% of a list of house-keeping genes (Table S6) were detected. We further excluded samples with less than 100 cells in non-cell-hashing batches or less than 50 cells in cell-hashing batches from further analysis (one scRNA-seq sample from PP, one scRNA/TCR-seq sample from MLN, COL, and SI).
Data from different batches were integrated using the Seurat v3 SCTransform integration (Stuart et al., 2019) workflow. Number of features in the SelectIntegrationFeatures function was set to infinity to allow as many variable features as possible (yielded 4,798 genes across all batches). Default parameters were used otherwise. Finally, we cleaned out non-Th17 cells and contaminations by iteratively over-cluster the cells in each tissue at each treatment condition using integrated batch-corrected expression data. Clusters that showed non-Th17 marker genes or were specific to a particular batch (the most abundant batch has significantly more cells than the second abundant batch in a cluster based on a Chi-square test (p < 0.05 and odds ratio >2)) were excluded. Following this process, 84,124 cells (37,773 naïve and 46,351 EAE) across 70 samples were used for downstream analysis.
Analyses of current Th17 cells during homeostasis (Figure 1 and Figure S1) used only non-cellhashing (batch 1–5) data, while other analyses of transcriptomics used both types of data unless otherwise specified. Batch corrected expression levels after integration were used for visualizations (e.g., heatmaps, dot plots, violin plots, etc.) unless otherwise specified.
Dimensionality reduction and clustering
Seurat v3 was used to preform dimensionality reduction and clustering. First, Principal Component Analysis (PCA) was performed on the batch corrected data with the RunPCA function. Next, a Shared Nearest Neighbor (SNN) graph was constructed using a set of highest-variance PCs using the FindNeighbors function, where a K-Nearest Neighbor (KNN, k.param = 20) graph was firstly built, the Jaccard index between every cell and its k nearest neighbors was calculated, then edges with low Jaccard index (prune.SNN <1/15) were removed. Clusters were constructed with the FindClusters function that maximizes the modularity of the SNN graph (Waltman and van Eck, 2013). The number of PCs (dims) to be used in downstream analysis was selected using an elbow plot, and the resolution parameter (resolution) in FindClusters changed slightly in the different data subsets due to differences in cell number and transcriptional variation and was selected such that the clusters obtained had biologically meaningful up-regulated genes. Uniform Manifold Approximation and Projection (UMAP) (McInnes et al., 2018) was performed using the RunUMAP function on the selected PCs and the UMAP coordinates were used for visualization. The parameters for the number of neighboring points used (n.neighbors), and the tightness of embedding (min.dist) were adjusted according to the elbow plot and the number of cells to visualize. The specific parameters used for the different analyses were as follows: dims = 1:20, resolution = 0.3, n.neighbors = 30, min.dist = 0.3 was used for GFP+ naïve cells inter-tissue clustering (Figure S1D); dims = 1:30, resolution = 0.15, n.neighbors = 30, min.dist = 0.3 was used for all naïve cells intra-tissue clustering (Figure 2A); dims = 1:30, resolution = 0.2, n.neighbors = 30, min.dist = 0.3 was used for all EAE cells intra-tissue clustering except SPL (Figure 3B, Figure S6B); dims = 1:30, resolution = 0.1, n.neighbors = 50, min.dist = 0.5 was used for all SPL EAE cells clustering (Figure 5A, Figure S6B).
Differential expression in scRNA-seq data
Differentially expressed genes were identified using edgeR (Robinson et al., 2010, McCarthy et al., 2012) with the input being raw UMI counts unless otherwise noted. Specifically, we used the quasi-likelihood method (glmQLFit and glmQLTest functions), adjusting for gene detection rate (z-normalized number of genes detected in each cell) and batch (a categorical variable with sum to zero contrasts). Genes were prefiltered such that only those detected in at least 10% of the cells in at least one of the levels in the categorical variable of interest (e.g. tissues, clusters, ex/current Th17 status) were considered in the model, unless otherwise noted (Soneson and Robinson, 2018).
Tissue signatures for current Th17 cells during homeostasis (Figure 1F) were generated by running edgeR with tissue as a categorical variable with SPL as reference, adjusting for above mentioned covariates. Batch-specific fold changes were computed for each gene based on edgeR fitted coefficients, and genes were grouped into modules if up- or down-regulated in the same set of tissues. If a gene was found to be up- and down-regulated (in different tissues), it was only included in the gene module where it was up-regulated. Tissue signatures for all Th17 cells (current and ex-Th17) during homeostasis (Figure S2E) were generated in the same way, except that all genes in tissue signatures for current Th17 cells were also included in the analyses in addition to those passed the prefiltering.
Cluster annotation of scRNA-seq data
Tissue-specific clusters were annotated to reflect the similarities and differences in subpopulation composition across tissues (Figure 2B and Figure S6C). First, cluster-specific genes were identified for each cluster within each tissue using Seurat FindAllMarkers function with default parameters. Second, an average expression profile was computed for each cluster for the union of all cluster-specific genes. Third, cosine similarities of average expression profiles were computed for each pair of clusters. Next, hierarchical clustering was conducted based on the cosine distances (1 – cosine similarities) using the Ward1 criterion (Murtagh and Legendre, 2014). Each group of clusters was assigned a function-related label according to the common cluster-specific genes.
Surface marker prediction in scRNA-seq data
COMET (Delaney et al., 2019) was applied to predict cell surface markers for clusters of interest. Batch corrected data were used, and the maximum number of genes in the combination (K) was set to 4. Other parameters were set to default.
Gene signature scoring in scRNA-seq data
Gene signature scores were computed using Seurat AddModuleScore function on integrated data with default parameters. Algorithm details were described in (Tirosh et al., 2016).
Analysis of DSS-induced colitis scRNA-seq data
Reads were mapped and count matrices were computed in the same way as in the preprocessing of scRNA-seq data section above. Low quality cell profiles were excluded if they fulfilled one or more of the following criteria: (i) number of genes expressed <=500 or >=2500, (ii) >=5% of the total UMIs were mitochondrial RNA or (iii) <=50% of a list of house-keeping genes (Table S6) were detected. Seurat v3 was then used to clean and analyze the data (default parameters were used unless otherwise specified). First, data were normalized with SCTransform function. Next, RunPCA function was applied to normalized data, followed by FindNeighbors and FindClusters function on top 30 PCs (selected according to the elbow plot) to identify 9 clusters. Treg-like, proliferating and contamination (CD74+) clusters were excluded to retain conventional Th17 cells (1,495 cells in total). RunPCA was applied again to the pruned dataset, and RunUMAP was applied to the top 30 PCs (selected according to the elbow plot) to obtain the UMAP coordinates for visualizations.
Analysis of public scRNA-seq data
Validation of Cd274 (PD-L1) in mouse and human data
Three publicly available datasets, Arazi et al. (Arazi et al., 2019), Hemmers et al. (Hemmers et al., 2019) and Tibbitt et al. (Tibbitt et al., 2019), were analyzed to validate if Cd274 is a possible marker gene for the ISG-high cells discovered in each of these studies. Standard Seurat clustering workflow was applied to each dataset to identify the ISG-high cells, and the odds ratio of Cd274 detection (non-zero reads) comparing the ISG-high cells vs. other CD4+ cells in the data was computed.
Validation of signatures in human scRNA-seq data
Public human scRNA-seq data for PBMC and CSF immune cells from healthy controls and MS patients were obtained from Schafflick et al. (Schafflick et al., 2020). PBMC and CSF data were preprocessed, normalized, integrated, and clustered respectively using the scRNA-seq analysis pipeline described above. Signature scores were computed with the AddModuleScore function in Seurat.
Detection of Il2b and Il23a in mouse CNS immune cells
scRNA-seq data for mouse CNS immune cells were obtained from Wheeler et al.(Wheeler et al., 2020). Data from each sample were cleaned up by excluding cells where <=400 genes were detected or >=5% of total UMIs were mitochondrial RNA, then processed by NormalizeData and FindVariableFeatures functions in Seurat v3. Samples were then integrated using the FindIntegrationAnchors and IntegrateData functions. Only samples from the priming EAE stage (n=6) were used to compute the proportion of cells with positive UMI counts. Cell type labels were provided by Wheeler et al.
Preprocessing of scTCR-seq data
Paired reads were mapped to GRCm38 V(D)J sequences (build from Ensembl Mus_musculus.GRCm38.90.chr_patch_hapl_scaff.gtf) and UMI counts were computed using CellRanger v3.0.2 with default parameters. Alpha and beta chains were filtered to keep only those that were from a detected cell, had high confidence and labeled as productive, as determined by CellRanger. Cell quality control was done based on the number of dominant alpha and beta chains. For each cell, 0, 1 or 2 dominant chains were identified for each of alpha and beta chains respectively. If only one chain was detected that chain was called dominant. If two chains were detected, call the more abundant (both if equal UMI counts) chain as dominant. If three or more chains were detected, the most abundant chain was determined dominant if it constituted more than 2 times the UMI counts than the second most abundant chain. Only cells with at least one dominant alpha (unless no valid alpha chain was detected in the first place) and at least one dominant beta chain were retained. Clonotypes were defined as sets of cells that have the same dominant alpha (if any) and dominant beta chain nucleotide sequences.
TCR repertoire similarity score (TRSS)
TCR repertoire similarity score (TRSS) reflects the similarity in clonal distribution across two sets of cells (e.g., two different clusters or tissue) and was measured with the Bhattacharyya coefficient (BC, Formula 1)
| (1) |
where and are the proportions of cells in group i and j that belongs to clonotype k, with a total of C clonotypes. This coefficient is between 0 and 1, and larger value indicates higher similarity in clonotype distributions. To increase power, cells from different mice were pooled for analyses unless otherwise noted. To identify cases of high TCR repertoire similarity, hierarchical clustering was performed on the Bhattacharyya distance (Formula 2) using the Ward2 criterion (Murtagh and Legendre, 2014). Distances that are infinitely large (when BC = 0) were capped at 10 times the maximum finite value in the distance matrix.
| (2) |
Calculation of clonal expansion score
To score clonal expansion we used the clonal expansion score previously proposed and used in STARTRAC (Zhang et al., 2018). The clonal expansion score depicts the diversity of clone sizes, defined as the complement of normalized Shannon entropy (Formula 3), where pk is the proportion of cells that belong to clonotype k, with a total of C clonotypes. The score ranges from 0 to 1. A score of 0 means all clones have equal sizes hence no interesting differences, whereas a score of 1 means there is only one expanded clonotype. Therefore, a large value between 0 and 1 indicates the existence of highly expanded clonotypes given the presence of non-expanded ones.
| (3) |
Identification of TCR specificity groups
GLIPH2 (Huang et al., 2020) was applied to all homeostasis cells with valid TCR information to identify clusters of TCRs that recognizes the same epitope based on CDR3β amino acid sequence similarities. GLIPH2 identified a total of 1,881 TCR specificity groups based on global CDR3β similarity. Among these, 552 groups had Fisher score <0.05, which are more likely induced by exposing to specific antigens than by chance. Specifically, GLIPH2 only allow one CDR3β sequence per cell, we split cells with 2 valid beta chains in two cells and collapsed the GLIPH2 clusters if they contain chains extracted from the same cell, resulting in 545 groups. Note that in Figure S6A, we further restricted to cells with a valid tissue label according in the demultiplexing step (see: Preprocessing of scRNA-seq data section above) and retained 322 groups.
Analysis of bulk RNA-seq data
Paired reads were mapped to mouse annotation mm10 using HISAT2 (Kim et al., 2019), and sequencing depth normalized TPM values were computed using RSEM (Li and Dewey, 2011) with default parameters. If a sample was sequenced in multiple lanes, normalized TPM values were summed up across these lanes to obtain sample-level data. Samples with low total number of reads, low alignment rate or low uniquely alignment rate compared to the rest of samples were excluded. Differentially expressed genes were identified using edgeR on TPMs with default parameters. Specifically, we used the generalized log-linear model and the likelihood ratio test (glmFit and glmLRT).
Analysis of bulk ATAC-seq data
A publicly available pipeline for ATAC-seq data analyses (Lee et al., 2016) was used to perform read alignment, filtering and quality control. Reads were aligned to reference mouse annotation mm10 using Bowtie2 (Langmead and Salzberg, 2012), and filtered for duplicated and mitochondrial reads. Peak-calling was performed using MACS2 (Zhang et al., 2008). Read counts per peak were computed using bedtools multicov tool, then analyzed for differential peak accessibility using DESeq2 (Love et al., 2014). PCA was applied to the top 50,000 highly variable accessible regions to examine the difference between SLAM6+ and CXCR6+ populations (Figure 5I). Peaks were associated to nearby genes based on cis-regulatory elements using Genomic Regions Enrichment of Annotations Tool (GREAT) (McLean et al., 2010) with the basal plus extension association rule and default parameters. If a gene is associated with more than one peak, it was assigned with the peak that has more significant differential peak accessibility (lower FDR). The ATAC signature score was computed as the average of log-normalized counts of the peaks associated with genes in the signature.
Gene set enrichment analysis
Gene set enrichment scores and p-values were computed with fGSEA (Korotkevich et al., 2019), a fast algorithm for Gene Set Enrichment Analysis (GSEA). For scRNA-seq data, genes were ranked according to the transformed F-test statistic in decreasing order, where the F values were obtained from the glmQLTest function in edgeR differential expression analysis, and the F values with a corresponding negative log-fold change were multiplied by (−1). For bulk RNA-seq data, genes were ranked according to the transformed likelihood ratio (LR) statistic in decreasing order, where the LR values were obtained from glmLRTest function in edgeR differential expression analysis and transformed in the same way as for scRNA-seq data.
Other statistical analysis of sequencing data
Proportions in Figure 2G, Figure S3I, Figure 7A, Figure 7C, and Figure 7D were computed for each sample respectively (each batch of batch 1–5 or each mouse in batch 6–9 was considered as one sample). A generalized linear mixed effects model with logistic link was fitted to test for a difference between the two groups, where a random intercept was used to model the variability across samples and a fixed slope for the group difference. P-values <0.05 are considered significant (NS, not significant; *, P <0.05; **, P <0.001; ***, P <0.001).
Spearman or Spearman correlations of scores were computed for pairs of signatures in Figure S5A–C. T-tests (cor.test function in the R stats package v3.6.2) were performed to assess if the correlations were significantly different from 0. P-values <0.05 are considered significant (NS, not significant; *, P <0.05; **, P <0.001; ***, P <0.001).
Two-tailed Wilcoxon rank-sum tests were used to assess the differences in Figure S4B, Figure S6H, Figure S7C, and Figure S7J. P-values <0.05 are considered significant (NS, not significant; *, P <0.05; **, P <0.001; ***, P <0.001).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis of non-sequencing data was performed with the GraphPad Prism software (GraphPad). Unless otherwise specified, data are presented as the mean with ± SEM and statistical significance was determined using unpaired two-tailed t-test. P-values <0.05 are considered significant (NS, not significant; *, P <0.05; **, P <0.01; ***, P <0.001; ****, P <0.0001). For the analysis of EAE disease curves, linear regression was performed for each condition followed by testing for the slopes to be significantly different.
Supplementary Material
Figure S1: Study design, Il17a scRNA-seq detection validation and clustering analysis of current tissue Th17 cell scRNA-seq, related to Figure 1 (A) Simplified representation of the Il17a and Rosa26 loci of Il17aCreGFP X Rosa26tdTomato mice.
(B) Gating strategy for the sorting of singlet viable CD45+ TCRβ+ CD4+ tdTomato+ cells for scRNA/TCR-seq.
(C) Comparison of frequencies of IL-17A-GFP+ of tdTomato+ cells in flow cytometry (left) vs. Il17aGFP transcript in scRNA-seq (right) (n=4–7).
(D) UMAP representation of unsupervised clustering of all current Th17 single cells (GFP+) (6,367 cells) at homeostasis.
(E) Heatmap representing cluster-specific up-regulated genes (FDR <0.05, fold change ≥1.5). Data were shown for a random sample of 1,000 cells.
(F) Bar plot depicting the tissue composition of each cluster.
(G) UMAP representation from (A) colored by batch for all tissues together (top left) and for each tissue respectively (other five subplots). Due to experimental complexity, not all tissues were present in all batches (Table S1). The Adjusted Mutual Information (AMI) between batch and clusters was close to 0 (and far from 1), suggesting the clusters were not driven by batch effects.
(H) Heatmap depicting the pairwise distance between tissues. Euclidean distance was computed for each pair of tissue centers, defined as the average of cell embeddings in the top 50 principal components. Cells from the proliferating and Treg clusters (Clusters 7 and 6, Figure S1D,E) were excluded from this analysis.
Figure S2: Joined single-cell profiling of ex- and current tissue Th17 cells, related to Figure 1 (A-C) UMAP of all current (GFP+) and ex-Th17 cells (GFP−) at homeostasis (37,773 cells). Color indicates the tissue origin (A), transcript expression of Foxp3 and Mki67 (B), and Th17 status (current or ex-Th17, C).
(D) Expression of selected Th17 effector genes (rows) in all Th17 cells in the tissues (columns) at homeostasis.
(E) Heatmap of up-regulated and down-regulated genes (FDR <0.05, fold change ≥1.5) in tissues vs. splenic current and ex-Th17 cells. Small multi-tissue gene sets (<20 genes) were not shown.
(F) Volcano plot for differentially expressed genes (FDR <0.05, fold change ≥1.2) comparing current vs. ex-Th17 cells in each tissue.
Figure S3: Validation of the ISG-high and the tdTomato+ Foxp3+ Treg subpopulations, related to Figure 2 (A) COMET (Delaney et al., 2019) prediction of PD-L1 (Cd274) as a favorable surface marker for the ISG-high Th17 subpopulation in the SPL (left) and MLN (right).
(B) Flow cytometry analysis of PD-L1 protein expression on tdTomato+ CD4+ T cells in the SPL (left) and MLN (right).
(C) Validation of PD-L1 as a marker for the ISG-high Th17 subpopulation by qPCR. Relative expression (RE) of differentially up-regulated genes in the ISG-high population in PD-L1− and PD-L1+ tdTomato+ CD4+ T cells by qPCR (n=3).
(D) Odds ratio of detecting Cd274 in the ISG-high cluster vs. other clusters in three published datasets (Arazi et al., 2019, Hemmers et al., 2019, Tibbitt et al., 2019). 95% confidence intervals were shown for each odds ratio. The dashed vertical line marks odds ratio = 1.
(E) TCR repertoire similarity analysis of the ISG-high clusters in the SPL (SPL5), MLN (MLN4), and PP (PP5) with all other intra-tissue clusters at homeostasis.
(F) Comparison of frequencies of Foxp3GFP+ cells of tdTomato+ cells in flow cytometry (left) vs. scRNA-seq (right) (n=3–6). The Foxp3 detection was based on Foxp3GFP protein in flow cytometry and based on the Foxp3 transcript in scRNA-seq.
(G) Representative flow cytometry plots of tdTomato+ Foxp3GFP+ cells in each tissue.
(H) TCR repertoire similarity analysis of intra-tissue clusters within the MLN, PP, and SI.
(I) Percentage of Treg-like and non-Treg colonic tdTomato+ cells that share their TCR sequence with splenic tdTomato+ cells (n=2–3).
(J)Presumptive model of the ancestry of tdTomato+ Foxp3+ Treg cells. The low TCR similarity of tdTomato+ Foxp3+ Treg cells with other tdTomato+ tissue Th17 cells suggests that tdTomato+ Foxp3+ Treg cells are not likely derived from Th17 cells but presumably from Treg cells.
Figure S4: Characterization of tissue Th17 cell subpopulations during EAE, related to Figure 3 and 4 (A) UMAP visualization of G1/S and G2/M signatures scores in CNS Th17 cells. G1/S and G2/M signatures were obtained from (Tirosh et al., 2016).
(B) Box plot depicting the total UMI counts of cells for each CNS Th17 cluster.
(C) Frequencies of current Th17 cells (GFP+) in each CNS Th17 cluster based on scRNA-seq.
(D) COMET (Delaney et al., 2019) prediction of PD-1 (Pdcd1) as a favorable surface marker for cluster 4.
(E,F) UMAP visualization of all current (GFP+) and ex-Th17 cells (GFP−) from naïve and EAE-diseased mice (84,124 cells). The cells are colored by batch (E) or by Th17 status (current or ex-Th17, F). Batches are not evenly distributed in (E) because not all tissues were present in all batches (Table S1).
(G) Bar plot depicting the distance between naïve and EAE cells in SPL, SI, and COL. For each tissue, Euclidean distance was computed between naïve and EAE cell centers, which were defined as the average of cell embeddings in the top 50 principal component spaces. Cells from proliferating and Treg clusters (Figure 4A) were excluded from this analysis.
(H) Bar plot comparing the number of differentially expressed genes between EAE and naïve cells in SPL, SI, and COL (FDR <0.05, fold change ≥1.5).
(I) UMAP visualization of all tdTomato+ single cells from the SPL (24,837 cells), SI (11,416 cells), and COL (15,801 cells) from naïve and EAE-diseased mice, colored by cluster assignment (left top row), Th17 status (left bottom row), and treatment (right top row). Bar plots (right bottom row) depicting the treatment composition of each cluster, and the Chi-square statistics for an association between treatment and cluster membership were shown for each tissue. All sub-figures other than the UMAPs colored by clusters were based on a sub-sample. Multiple subsamples were generated, and results were shown for one representative case.
Figure S5: Detection of the SLAMF6+ and CXCR6+ populations in human MS patients and mouse colitis, related to Figure 5 (A) Positive correlations of pathogenic Th17 pathway signatures (y-axis) (Gaublomme et al., 2015, Lee et al., 2012) and the CXCR6+ bulk RNA-seq signature (x-axis) (Figure 5F, Table S4) in cerebrospinal fluid (CSF) T cells in MS patients (Schafflick et al., 2020).
(B) Negative correlation of non-pathogenic Th17 pathway signature (Th17 self-renewing) (Gaublomme et al., 2015) and the CXCR6+ bulk RNA-seq signature in CSF T cells in MS patients.
(C) Dot plot comparing the correlations of the CXCR6+ bulk RNA-seq signature with the pathogenic and non-pathogenic Th17 pathway signatures (Gaublomme et al., 2015) in CSF T cells of healthy controls vs. MS patients.
(D) UMAP visualization of the SLAMF6+ (left) and CXCR6+ (right) bulk RNA-seq signatures in colonic tdTomato+ CD4+ single T cells of mice with DSS-induced colitis.
Figure S6: Intra-tissue heterogeneity of Th17 cells during EAE, related to Figure 6 (A) Heatmap depicting sharing of GLIPH2-identified (Huang et al., 2020) TCR specificity groups across tissue Th17 cells at homeostasis. All TCR specificity groups (rows) with GLIPH2 reported Fisher score <0.05 were included.
(B) UMAP visualization of all current (GFP+) and ex-Th17 cells (GFP−) single cells from the SPL, MLN, PP, SI, COL, CNS, and DLN during EAE, colored by cluster assignment (top row), cluster function annotation (middle row), and Th17 status (bottom row).
(C) Correlation of transcriptomic landscape for all pairs of intra-tissue clusters during EAE.
(D) Expression of selected genes used to define the cluster functional annotations.
(E) MA plot depicting the differentially expressed genes (FDR <0.05, fold change ≥1.5) in SI5 (Figure S6B) vs. all other cells in the SI.
(F) Gene set enrichment analysis results for selected GO terms and KEGG pathways enriched in SI5.
(G) Number and fraction of cells with valid clonotype data in each tissue that share TCR with CNS.
(H) SI5 signature score for cells that share the TCR with cells in the CNS (shared) vs. the cells that do not share the TCR with cells in the CNS (not shared) for each tissue. The SI5 signature was identified as described in Figure S6B.
Figure S7: Characterization of the conversion of the SLAMF6+ to the CXCR6+ population in vivo, related to Figure 6 and 7 (A) Frequencies of CXCR6+ tdTomato+ CD4+ T cells in the blood of naïve and EAE-diseased mice by flow cytometry (n= 3–6).
(B) Relative expression (RE) of Cxcl16 in different immune populations in the spleen and CNS of EAE mice, determined by qPCR (n=4).
(C) Violin plot depicting the proliferation signature score (Figure S1D,E) for the single cells in SPL0 and SPL1. Visualization was based on non-extreme expression values (between 0.1 and 99.9 percentiles) and jittered points were only shown for randomly selected 50% of the cells.
(D) EAE disease curve of immunized recipient mice that received CXCR6+ tdTomato+ cells 7 days after immunization (n=6–7).
(E) Frequencies of tdTomato+ CD4+ T cells in control vs. antibiotic-treated (Abx) mice in each tissue at homeostasis (n=4–5).
(F) Frequencies of the SLAMF6+ tdTomato+ CD4+ T cells in the spleen of control vs. Abx-treated mice at homeostasis (n=5).
(G) EAE disease curve of immunized Abx-treated or control mice (n=5).
(H) Numbers of SLAMF6+ (left) or CXCR6+ (right) tdTomato+ CD4+ T cells in the spleen of EAE-diseased Abx-treated or control mice harvested 9-days post immunization (n=3–4).
(I) GM-CSF and IFNγ concentration in the culture medium during recall assay, determined by LegendPlex (n=4).
(J) Violin plot depicting the IL-23 signature score (Wu et al., 2013) for the single cells in SPL0 and SPL1. Visualization was based on non-extreme expression values (between 0.1 and 99.9 percentiles) and jittered points were only shown for randomly selected 50% of the cells.
(K) Heatmap depicting the chromatin accessibility of published IL-23R signaling signatures in the ATAC-seq of the SLAMF6+ and CXCR6+ population. GSE39820 (Lee et al., 2012), GSE43955 (Yosef et al., 2013) (1- M9668; 2- M9672), GSE23505 (Ghoreschi et al., 2010), PID_IL23_Pathway (Schaefer et al., 2009), Langrish et al. (Langrish et al., 2005).
(L) Relative expression (RE) of Il12b and Il23a in splenic immune populations during EAE by qPCR (n=4). Act= Activated.
(M) Proportion of cells with non-zero expression of Il12b and Il23a in CNS immune cells from EAE (Wheeler et al., 2020) mice at EAE priming stage (n=6).
(N) Heatmap depicting the relative expression of differentially expressed genes (FDR <0.05) comparing CXCR6+ cells from Il17aCreIl23rfl/fl and Il17aCreIl23rWT mice.
(O) Bar plot of published Th17 pathway signatures (Lee et al., 2012, Gaublomme et al., 2015, Liberzon et al., 2015) from gene set enrichment analysis for CXCR6+ cells from Il17aCreIl23rfl/fl vs. Il17aCreIl23rWT mice.
Table S2: Tissue-specific Th17 signatures, related to Figure 1
Table S3: T-cell clonotype information and clonotype sharing across mice, related to Figure 2 and 4
(A) Summary, (B) Naïve shared, (C) EAE shared
Table S4: CXCR6+ vs. SLAMF6+ bulk RNA-seq signatures, related to Figure 5
Table S5: Bulk RNA-seq of CXCR6+ cells from Il17aCreIl23rfl/fl vs. Il17aCreIl23rWT mice, related to Figure 7
Table S6: List of housekeeping genes for QC, related to STAR Methods
Highlights.
Combined scRNA/TCR-seq reveals extensive heterogeneity of tissue Th17 cells
Th17 cells exhibit strong tissue-specific signatures
Stem-like SLAMF6+ and pathogenic CXCR6+ Th17 populations are induced in autoimmunity
IL-23 converts the stem-like IL-17+ to the pathogenic GM-CSF+ IFNγ+ population
Acknowledgments
We would like to thank all members of the Kuchroo laboratory for feedback. We thank Junrong Xia, Helene Stroh, Rajesh Kumar Krishnan, and Deneen Kozoriz for technical support and Leslie Gaffney for help with figures. Furthermore, we would like to thank Mary Collins for critical feedback on the manuscript and the Quintana laboratory for technical support for the photoconversion experiments. This work was supported by National Institute of Health grants (R01NS045937, R01NS30843, R01AI144166, P01AI073748, P01AI039671 and P01AI056299) to V.K.K. and by the Howard Hughes Medical Institute and Klarman Cell Observatory (A.R.). A.R. was an Investigator of the Howard Hughes Medical Institute when this study was conducted. A.S. is supported by a German Academic Scholarship Foundation (Studienstiftung des Deutschen Volkes) PhD fellowship. This manuscript was supported in part by a grant from the National Institute of Health P01 AI39671 (to V.K.K. and M.S.). G.M.z.H. was supported by the Deutsche Forschungsgemeinschaft (DFG; grants ME4050/1-1, ME4050/4-1) and a National Multiple Sclerosis Society postdoctoral fellowship (grant FG20118-A-1). R.A.I. was supported by the National Institute of Health grant R01HG005220.
Declaration of interests
A.R. was a SAB member of Thermo Fisher Scientific, Neogene Therapeutics, Asimov and Syros Pharmaceuticals. A.R. is a cofounder of and equity holder in Celsius Therapeutics and an equity holder in Immunitas. V.K.K. is a co-founder and has an ownership interest and is a member of SAB in Celsius Therapeutics, Tizona Therapeutics and Trishula Therapeutics. VKK is also on chair of the Board and has equity interests in Bicara Therpeutics. V.K.K. financial interests and conflicts are managed by Brigham and Women’s Hospital and Partners Health Care system. A.R. and O.R.R. are employees of Genentech (member of the Roche Group) since August and October 2020, respectively. None of these companies provide support for this work. A.R. was an HHMI Investigator while this study was conducted. A.S., M.S., A.R., and V.K.K. are co-inventors on US provisional patent application no. 17/063,617 and no. 62/968,981 filed by the Broad Institute relating to the subject matter of this manuscript. O.R.R. is co-inventor on patent applications filed by the Broad Institute relating to single-cell genomics. All other authors declare no competing interests.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ARAZI A, RAO DA, BERTHIER CC, DAVIDSON A, LIU Y, HOOVER PJ, CHICOINE A, EISENHAURE TM, JONSSON AH, LI S, LIEB DJ, ZHANG F, SLOWIKOWSKI K, BROWNE EP, NOMA A, SUTHERBY D, STEELMAN S, SMILEK DE, TOSTA P, APRUZZESE W, MASSAROTTI E, DALL’ERA M, PARK M, KAMEN DL, FURIE RA, PAYAN-SCHOBER F, PENDERGRAFT WF 3RD, MCINNIS EA, BUYON JP, PETRI MA, PUTTERMAN C, KALUNIAN KC, WOODLE ES, LEDERER JA, HILDEMAN DA, NUSBAUM C, RAYCHAUDHURI S, KRETZLER M, ANOLIK JH, BRENNER MB, WOFSY D, HACOHEN N, DIAMOND B & ACCELERATING MEDICINES PARTNERSHIP IN, S. L. E. N. 2019. The immune cell landscape in kidneys of patients with lupus nephritis. Nat Immunol, 20, 902–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEDOYA SK, LAM B, LAU K & LARKIN J 3RD 2013. Th17 cells in immunity and autoimmunity. Clin Dev Immunol, 2013, 986789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERER K, GERDES LA, CEKANAVICIUTE E, JIA X, XIAO L, XIA Z, LIU C, KLOTZ L, STAUFFER U, BARANZINI SE, KUMPFEL T, HOHLFELD R, KRISHNAMOORTHY G & WEKERLE H 2017. Gut microbiota from multiple sclerosis patients enables spontaneous autoimmune encephalomyelitis in mice. Proc Natl Acad Sci U S A, 114, 10719–10724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERER K, MUES M, KOUTROLOS M, RASBI ZA, BOZIKI M, JOHNER C, WEKERLE H & KRISHNAMOORTHY G 2011. Commensal microbiota and myelin autoantigen cooperate to trigger autoimmune demyelination. Nature, 479, 538–41. [DOI] [PubMed] [Google Scholar]
- BETTELLI E, CARRIER Y, GAO W, KORN T, STROM TB, OUKKA M, WEINER HL & KUCHROO VK 2006. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature, 441, 235–8. [DOI] [PubMed] [Google Scholar]
- BLASCHITZ C & RAFFATELLU M 2010. Th17 cytokines and the gut mucosal barrier. J Clin Immunol, 30, 196–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRADLEY CP, TENG F, FELIX KM, SANO T, NASKAR D, BLOCK KE, HUANG H, KNOX KS, LITTMAN DR & WU HJ 2017. Segmented Filamentous Bacteria Provoke Lung Autoimmunity by Inducing Gut-Lung Axis Th17 Cells Expressing Dual TCRs. Cell Host Microbe, 22, 697–704 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAO AT, YAO S, GONG B, ELSON CO & CONG Y 2012. Th17 cells upregulate polymeric Ig receptor and intestinal IgA and contribute to intestinal homeostasis. J Immunol, 189, 4666–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CODARRI L, GYULVESZI G, TOSEVSKI V, HESSKE L, FONTANA A, MAGNENAT L, SUTER T & BECHER B 2011. RORgammat drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat Immunol, 12, 560–7. [DOI] [PubMed] [Google Scholar]
- CORCES MR, BUENROSTRO JD, WU B, GREENSIDE PG, CHAN SM, KOENIG JL, SNYDER MP, PRITCHARD JK, KUNDAJE A, GREENLEAF WJ, MAJETI R & CHANG HY 2016. Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet, 48, 1193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSORICH I, DALLA-COSTA G, SORINI C, FERRARESE R, MESSINA MJ, DOLPADY J, RADICE E, MARIANI A, TESTONI PA, CANDUCCI F, COMI G, MARTINELLI V & FALCONE M 2017. High frequency of intestinal TH17 cells correlates with microbiota alterations and disease activity in multiple sclerosis. Sci Adv, 3, e1700492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUA DJ, SHERLOCK J, CHEN Y, MURPHY CA, JOYCE B, SEYMOUR B, LUCIAN L, TO W, KWAN S, CHURAKOVA T, ZURAWSKI S, WIEKOWSKI M, LIRA SA, GORMAN D, KASTELEIN RA & SEDGWICK JD 2003. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature, 421, 744–8. [DOI] [PubMed] [Google Scholar]
- DELANEY C, SCHNELL A, CAMMARATA LV, YAO-SMITH A, REGEV A, KUCHROO VK & SINGER M 2019. Combinatorial prediction of marker panels from single-cell transcriptomic data. Mol Syst Biol, 15, e9005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOWNS-CANNER S, BERKEY S, DELGOFFE GM, EDWARDS RP, CURIEL T, ODUNSI K, BARTLETT DL & OBERMAJER N 2017. Suppressive IL-17A(+)Foxp3(+) and ex-Th17 IL-17A(neg)Foxp3(+) Treg cells are a source of tumour-associated Treg cells. Nat Commun, 8, 14649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ESCOBAR G, MANGANI D & ANDERSON AC 2020. T cell factor 1: A master regulator of the T cell response in disease. Sci Immunol, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAGLIANI N, AMEZCUA VESELY MC, ISEPPON A, BROCKMANN L, XU H, PALM NW, DE ZOETE MR, LICONA-LIMON P, PAIVA RS, CHING T, WEAVER C, ZI X, PAN X, FAN R, GARMIRE LX, COTTON MJ, DRIER Y, BERNSTEIN B, GEGINAT J, STOCKINGER B, ESPLUGUES E, HUBER S & FLAVELL RA 2015. Th17 cells transdifferentiate into regulatory T cells during resolution of inflammation. Nature, 523, 221–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GALLI E, HARTMANN FJ, SCHREINER B, INGELFINGER F, ARVANITI E, DIEBOLD M, MRDJEN D, VAN DER MEER F, KRIEG C, NIMER FA, SANDERSON N, STADELMANN C, KHADEMI M, PIEHL F, CLAASSEN M, DERFUSS T, OLSSON T & BECHER B 2019. GM-CSF and CXCR4 define a T helper cell signature in multiple sclerosis. Nat Med, 25, 1290–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUBLOMME JT, LI B, MCCABE C, KNECHT A, YANG Y, DROKHLYANSKY E, VAN WITTENBERGHE N, WALDMAN J, DIONNE D, NGUYEN L, DE JAGER PL, YEUNG B, ZHAO X, HABIB N, ROZENBLATT-ROSEN O & REGEV A 2019. Nuclei multiplexing with barcoded antibodies for single-nucleus genomics. Nat Commun, 10, 2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUBLOMME JT, YOSEF N, LEE Y, GERTNER RS, YANG LV, WU C, PANDOLFI PP, MAK T, SATIJA R, SHALEK AK, KUCHROO VK, PARK H & REGEV A 2015. Single-Cell Genomics Unveils Critical Regulators of Th17 Cell Pathogenicity. Cell, 163, 1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GHORESCHI K, LAURENCE A, YANG XP, TATO CM, MCGEACHY MJ, KONKEL JE, RAMOS HL, WEI L, DAVIDSON TS, BOULADOUX N, GRAINGER JR, CHEN Q, KANNO Y, WATFORD WT, SUN HW, EBERL G, SHEVACH EM, BELKAID Y, CUA DJ, CHEN W & O’SHEA JJ 2010. Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature, 467, 967–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAAK S, CROXFORD AL, KREYMBORG K, HEPPNER FL, POULY S, BECHER B & WAISMAN A 2009. IL-17A and IL-17F do not contribute vitally to autoimmune neuroinflammation in mice. J Clin Invest, 119, 61–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAGHIKIA A, JORG S, DUSCHA A, BERG J, MANZEL A, WASCHBISCH A, HAMMER A, LEE DH, MAY C, WILCK N, BALOGH A, OSTERMANN AI, SCHEBB NH, AKKAD DA, GROHME DA, KLEINEWIETFELD M, KEMPA S, THONE J, DEMIR S, MULLER DN, GOLD R & LINKER RA 2015. Dietary Fatty Acids Directly Impact Central Nervous System Autoimmunity via the Small Intestine. Immunity, 43, 817–29. [DOI] [PubMed] [Google Scholar]
- HANG S, PAIK D, YAO L, KIM E, TRINATH J, LU J, HA S, NELSON BN, KELLY SP, WU L, ZHENG Y, LONGMAN RS, RASTINEJAD F, DEVLIN AS, KROUT MR, FISCHBACH MA, LITTMAN DR & HUH JR 2019. Bile acid metabolites control TH17 and Treg cell differentiation. Nature, 576, 143–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEMMERS S, SCHIZAS M, AZIZI E, DIKIY S, ZHONG Y, FENG Y, ALTAN-BONNET G & RUDENSKY AY 2019. IL-2 production by self-reactive CD4 thymocytes scales regulatory T cell generation in the thymus. J Exp Med, 216, 2466–2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERNANDEZ-SANTOS N & GAFFEN SL 2012. Th17 cells in immunity to Candida albicans. Cell Host Microbe, 11, 425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIROTA K, DUARTE JH, VELDHOEN M, HORNSBY E, LI Y, CUA DJ, AHLFORS H, WILHELM C, TOLAINI M, MENZEL U, GAREFALAKI A, POTOCNIK AJ & STOCKINGER B 2011. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat Immunol, 12, 255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIROTA K, TURNER JE, VILLA M, DUARTE JH, DEMENGEOT J, STEINMETZ OM & STOCKINGER B 2013. Plasticity of Th17 cells in Peyer’s patches is responsible for the induction of T cell-dependent IgA responses. Nat Immunol, 14, 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUANG H, WANG C, RUBELT F, SCRIBA TJ & DAVIS MM 2020. Analyzing the Mycobacterium tuberculosis immune response by T-cell receptor clustering with GLIPH2 and genome-wide antigen screening. Nat Biotechnol, 38, 1194–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUDSON WH, GENSHEIMER J, HASHIMOTO M, WIELAND A, VALANPARAMBIL RM, LI P, LIN JX, KONIECZNY BT, IM SJ, FREEMAN GJ, LEONARD WJ, KISSICK HT & AHMED R 2019. Proliferating Transitory T Cells with an Effector-like Transcriptional Signature Emerge from PD-1(+) Stem-like CD8(+) T Cells during Chronic Infection. Immunity, 51, 1043–1058 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HWANG SS, LIM J, YU Z, KONG P, SEFIK E, XU H, HARMAN CCD, KIM LK, LEE GR, LI HB & FLAVELL RA 2020. mRNA destabilization by BTG1 and BTG2 maintains T cell quiescence. Science, 367, 1255–1260. [DOI] [PubMed] [Google Scholar]
- IM SJ, HASHIMOTO M, GERNER MY, LEE J, KISSICK HT, BURGER MC, SHAN Q, HALE JS, LEE J, NASTI TH, SHARPE AH, FREEMAN GJ, GERMAIN RN, NAKAYA HI, XUE HH & AHMED R 2016. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature, 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IVANOV II, ATARASHI K, MANEL N, BRODIE EL, SHIMA T, KARAOZ U, WEI D, GOLDFARB KC, SANTEE CA, LYNCH SV, TANOUE T, IMAOKA A, ITOH K, TAKEDA K, UMESAKI Y, HONDA K & LITTMAN DR 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell, 139, 485–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KEBIR H, IFERGAN I, ALVAREZ JI, BERNARD M, POIRIER J, ARBOUR N, DUQUETTE P & PRAT A 2009. Preferential recruitment of interferon-gamma-expressing TH17 cells in multiple sclerosis. Ann Neurol, 66, 390–402. [DOI] [PubMed] [Google Scholar]
- KIM D, PAGGI JM, PARK C, BENNETT C & SALZBERG SL 2019. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol, 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINER E, WILLIE E, VIJAYKUMAR B, CHOWDHARY K, SCHMUTZ H, CHANDLER J, SCHNELL A, THAKORE PI, LEGROS G, MOSTAFAVI S, MATHIS D, BENOIST C & IMMUNOLOGICAL GENOME PROJECT C 2021. Gut CD4(+) T cell phenotypes are a continuum molded by microbes, not by TH archetypes. Nat Immunol, 22, 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KINUGASA T, SAKAGUCHI T, GU X & REINECKER HC 2000. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology, 118, 1001–11. [DOI] [PubMed] [Google Scholar]
- KLEINEWIETFELD M, MANZEL A, TITZE J, KVAKAN H, YOSEF N, LINKER RA, MULLER DN & HAFLER DA 2013. Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature, 496, 518–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMATSU N, OKAMOTO K, SAWA S, NAKASHIMA T, OH-HORA M, KODAMA T, TANAKA S, BLUESTONE JA & TAKAYANAGI H 2014. Pathogenic conversion of Foxp3+ T cells into TH17 cells in autoimmune arthritis. Nat Med, 20, 62–8. [DOI] [PubMed] [Google Scholar]
- KOMUCZKI J, TUZLAK S, FRIEBEL E, HARTWIG T, SPATH S, ROSENSTIEL P, WAISMAN A, OPITZ L, OUKKA M, SCHREINER B, PELCZAR P & BECHER B 2019. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1beta. Immunity, 50, 1289–1304 e6. [DOI] [PubMed] [Google Scholar]
- KORN T, BETTELLI E, OUKKA M & KUCHROO VK 2009. IL-17 and Th17 Cells. Annu Rev Immunol, 27, 485–517. [DOI] [PubMed] [Google Scholar]
- KOROTKEVICH G, SUKHOV V & SERGUSHICHEV A 2019. Fast gene set enrichment analysis. bioRxiv. [Google Scholar]
- KRANZ A, FU J, DUERSCHKE K, WEIDLICH S, NAUMANN R, STEWART AF & ANASTASSIADIS K 2010. An improved Flp deleter mouse in C57Bl/6 based on Flpo recombinase. Genesis, 48, 512–20. [DOI] [PubMed] [Google Scholar]
- KREBS CF, PAUST HJ, KROHN S, KOYRO T, BRIX SR, RIEDEL JH, BARTSCH P, WIECH T, MEYER-SCHWESINGER C, HUANG J, FISCHER N, BUSCH P, MITTRUCKER HW, STEINHOFF U, STOCKINGER B, PEREZ LG, WENZEL UO, JANNECK M, STEINMETZ OM, GAGLIANI N, STAHL RAK, HUBER S, TURNER JE & PANZER U 2016. Autoimmune Renal Disease Is Exacerbated by S1P-Receptor-1-Dependent Intestinal Th17 Cell Migration to the Kidney. Immunity, 45, 1078–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGMEAD B & SALZBERG SL 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods, 9, 357–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LANGRISH CL, CHEN Y, BLUMENSCHEIN WM, MATTSON J, BASHAM B, SEDGWICK JD, MCCLANAHAN T, KASTELEIN RA & CUA DJ 2005. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med, 201, 233–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAVIN Y, WINTER D, BLECHER-GONEN R, DAVID E, KEREN-SHAUL H, MERAD M, JUNG S & AMIT I 2014. Tissue-resident macrophage enhancer landscapes are shaped by the local microenvironment. Cell, 159, 1312–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE J, CHRISTOFORO G, CHRISTOFORO G, FOO CS, PROBERT C, KUNDAJE A, BOLEY N, KOHPANGWEI DACRE, M. & D. K 2016. kundajelab/atac_dnase_pipelines: 0.3.3. Zenodo. [Google Scholar]
- LEE Y, AWASTHI A, YOSEF N, QUINTANA FJ, XIAO S, PETERS A, WU C, KLEINEWIETFELD M, KUNDER S, HAFLER DA, SOBEL RA, REGEV A & KUCHROO VK 2012. Induction and molecular signature of pathogenic TH17 cells. Nat Immunol, 13, 991–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEE YK, MENEZES JS, UMESAKI Y & MAZMANIAN SK 2011. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proc Natl Acad Sci U S A, 108 Suppl 1, 4615–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LI B & DEWEY CN 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics, 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIBERZON A, BIRGER C, THORVALDSDOTTIR H, GHANDI M, MESIROV JP & TAMAYO P 2015. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst, 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LOVE MI, HUBER W & ANDERS S 2014. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARTHY DJ, CHEN Y & SMYTH GK 2012. Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res, 40, 4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCGINLEY AM, SUTTON CE, EDWARDS SC, LEANE CM, DECOURCEY J, TEIJEIRO A, HAMILTON JA, BOON L, DJOUDER N & MILLS KHG 2020. Interleukin-17A Serves a Priming Role in Autoimmunity by Recruiting IL-1beta-Producing Myeloid Cells that Promote Pathogenic T Cells. Immunity, 52, 342–356 e6. [DOI] [PubMed] [Google Scholar]
- MCINNES L, HEALY J & MELVILLE J 2018. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv preprint arXiv:1802.03426. [Google Scholar]
- MCLEAN CY, BRISTOR D, HILLER M, CLARKE SL, SCHAAR BT, LOWE CB, WENGER AM & BEJERANO G 2010. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol, 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIYAUCHI E, KIM SW, SUDA W, KAWASUMI M, ONAWA S, TAGUCHI-ATARASHI N, MORITA H, TAYLOR TD, HATTORI M & OHNO H 2020. Gut microorganisms act together to exacerbate inflammation in spinal cords. Nature, 585, 102–106. [DOI] [PubMed] [Google Scholar]
- MURPHY CA, LANGRISH CL, CHEN Y, BLUMENSCHEIN W, MCCLANAHAN T, KASTELEIN RA, SEDGWICK JD & CUA DJ 2003. Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med, 198, 1951–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURTAGH F & LEGENDRE P 2014. Ward’s Hierarchical Agglomerative Clustering Method: Which Algorithms Implement Ward’s Criterion? Journal of Classification, 31, 274–295. [Google Scholar]
- PANDURO M, BENOIST C & MATHIS D 2016. Tissue Tregs. Annu Rev Immunol, 34, 609–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PICELLI S, FARIDANI OR, BJORKLUND AK, WINBERG G, SAGASSER S & SANDBERG R 2014. Full-length RNA-seq from single cells using Smart-seq2. Nat Protoc, 9, 171–81. [DOI] [PubMed] [Google Scholar]
- PRATAMA A, SCHNELL A, MATHIS D & BENOIST C 2020. Developmental and cellular age direct conversion of CD4+ T cells into RORgamma+ or Helios+ colon Treg cells. J Exp Med, 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUINLAN AR & HALL IM 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RASOULI J, CASELLA G, YOSHIMURA S, ZHANG W, XIAO D, GARIFALLOU J, GONZALEZ MV, WIEDEMAN A, KUS A, MARI ER, FORTINA P, HAKONARSON H, LONG SA, ZHANG GX, CIRIC B & ROSTAMI A 2020. A distinct GM-CSF(+) T helper cell subset requires T-bet to adopt a TH1 phenotype and promote neuroinflammation. Sci Immunol, 5. [DOI] [PubMed] [Google Scholar]
- RILEY JL 2009. PD-1 signaling in primary T cells. Immunol Rev, 229, 114–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROBINSON MD, MCCARTHY DJ & SMYTH GK 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSENBLUM MD, REMEDIOS KA & ABBAS AK 2015. Mechanisms of human autoimmunity. J Clin Invest, 125, 2228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATIJA R, FARRELL JA, GENNERT D, SCHIER AF & REGEV A 2015. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol, 33, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAEFER CF, ANTHONY K, KRUPA S, BUCHOFF J, DAY M, HANNAY T & BUETOW KH 2009. PID: the Pathway Interaction Database. Nucleic Acids Res, 37, D674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHAFFLICK D, XU CA, HARTLEHNERT M, COLE M, SCHULTE-MECKLENBECK A, LAUTWEIN T, WOLBERT J, HEMING M, MEUTH SG, KUHLMANN T, GROSS CC, WIENDL H, YOSEF N & MEYER ZU HORSTE G 2020. Integrated single cell analysis of blood and cerebrospinal fluid leukocytes in multiple sclerosis. Nat Commun, 11, 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNEIDER WM, CHEVILLOTTE MD & RICE CM 2014. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol, 32, 513–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHNELL A, BOD L, MADI A & KUCHROO VK 2020. The yin and yang of co-inhibitory receptors: toward anti-tumor immunity without autoimmunity. Cell Res, 30, 285–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEBZDA E, ZOU Z, LEE JS, WANG T & KAHN ML 2008. Transcription factor KLF2 regulates the migration of naive T cells by restricting chemokine receptor expression patterns. Nat Immunol, 9, 292–300. [DOI] [PubMed] [Google Scholar]
- SEFIK E, GEVA-ZATORSKY N, OH S, KONNIKOVA L, ZEMMOUR D, MCGUIRE AM, BURZYN D, ORTIZ-LOPEZ A, LOBERA M, YANG J, GHOSH S, EARL A, SNAPPER SB, JUPP R, KASPER D, MATHIS D & BENOIST C 2015. MUCOSAL IMMUNOLOGY. Individual intestinal symbionts induce a distinct population of RORgamma(+) regulatory T cells. Science, 349, 993–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIDDIQUI I, SCHAEUBLE K, CHENNUPATI V, FUERTES MARRACO SA, CALDERON-COPETE S, PAIS FERREIRA D, CARMONA SJ, SCARPELLINO L, GFELLER D, PRADERVAND S, LUTHER SA, SPEISER DE & HELD W 2019. Intratumoral Tcf1(+)PD-1(+)CD8(+) T Cells with Stem-like Properties Promote Tumor Control in Response to Vaccination and Checkpoint Blockade Immunotherapy. Immunity, 50, 195–211 e10. [DOI] [PubMed] [Google Scholar]
- SKARNES WC, ROSEN B, WEST AP, KOUTSOURAKIS M, BUSHELL W, IYER V, MUJICA AO, THOMAS M, HARROW J, COX T, JACKSON D, SEVERIN J, BIGGS P, FU J, NEFEDOV M, DE JONG PJ, STEWART AF & BRADLEY A 2011. A conditional knockout resource for the genome-wide study of mouse gene function. Nature, 474, 337–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMILLIE CS, BITON M, ORDOVAS-MONTANES J, SULLIVAN KM, BURGIN G, GRAHAM DB, HERBST RH, ROGEL N, SLYPER M, WALDMAN J, SUD M, ANDREWS E, VELONIAS G, HABER AL, JAGADEESH K, VICKOVIC S, YAO J, STEVENS C, DIONNE D, NGUYEN LT, VILLANI AC, HOFREE M, CREASEY EA, HUANG H, ROZENBLATT-ROSEN O, GARBER JJ, KHALILI H, DESCH AN, DALY MJ, ANANTHAKRISHNAN AN, SHALEK AK, XAVIER RJ & REGEV A 2019. Intra- and Inter-cellular Rewiring of the Human Colon during Ulcerative Colitis. Cell, 178, 714–730 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SONESON C & ROBINSON MD 2018. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods, 15, 255–261. [DOI] [PubMed] [Google Scholar]
- STUART T, BUTLER A, HOFFMAN P, HAFEMEISTER C, PAPALEXI E, MAUCK WM 3RD, HAO Y, STOECKIUS M, SMIBERT P & SATIJA R 2019. Comprehensive Integration of Single-Cell Data. Cell, 177, 1888–1902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN JT, DUDL E, LEROY E, MURRAY R, SPRENT J, WEINBERG KI & SURH CD 2001. IL-7 is critical for homeostatic proliferation and survival of naive T cells. Proc Natl Acad Sci U S A, 98, 8732–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN TG, SEFIK E, GEVA-ZATORSKY N, KUA L, NASKAR D, TENG F, PASMAN L, ORTIZ-LOPEZ A, JUPP R, WU HJ, KASPER DL, BENOIST C & MATHIS D 2016. Identifying species of symbiont bacteria from the human gut that, alone, can induce intestinal Th17 cells in mice. Proc Natl Acad Sci U S A, 113, E8141–E8150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TIBBITT CA, STARK JM, MARTENS L, MA J, MOLD JE, DESWARTE K, OLIYNYK G, FENG X, LAMBRECHT BN, DE BLESER P, NYLEN S, HAMMAD H, ARSENIAN HENRIKSSON M, SAEYS Y & COQUET JM 2019. Single-Cell RNA Sequencing of the T Helper Cell Response to House Dust Mites Defines a Distinct Gene Expression Signature in Airway Th2 Cells. Immunity, 51, 169–184 e5. [DOI] [PubMed] [Google Scholar]
- TIROSH I, IZAR B, PRAKADAN SM, WADSWORTH MH 2ND, TREACY D, TROMBETTA JJ, ROTEM A, RODMAN C, LIAN C, MURPHY G, FALLAHI-SICHANI M, DUTTON-REGESTER K, LIN JR, COHEN O, SHAH P, LU D, GENSHAFT AS, HUGHES TK, ZIEGLER CG, KAZER SW, GAILLARD A, KOLB KE, VILLANI AC, JOHANNESSEN CM, ANDREEV AY, VAN ALLEN EM, BERTAGNOLLI M, SORGER PK, SULLIVAN RJ, FLAHERTY KT, FREDERICK DT, JANE-VALBUENA J, YOON CH, ROZENBLATT-ROSEN O, SHALEK AK, REGEV A & GARRAWAY LA 2016. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, 352, 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TOM MR, LI J, UENO A, FORT GASIA M, CHAN R, HUNG DY, CHENOO S, IACUCCI M, JIJON HB, KAPLAN GG, BECK PL, PANACCIONE R, BARKEMA HW, BURET AG, YAJNIK V & GHOSH S 2016. Novel CD8+ T-Cell Subsets Demonstrating Plasticity in Patients with Inflammatory Bowel Disease. Inflamm Bowel Dis, 22, 1596–608. [DOI] [PubMed] [Google Scholar]
- TOMURA M, YOSHIDA N, TANAKA J, KARASAWA S, MIWA Y, MIYAWAKI A & KANAGAWA O 2008. Monitoring cellular movement in vivo with photoconvertible fluorescence protein “Kaede” transgenic mice. Proc Natl Acad Sci U S A, 105, 10871–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WAGNER A, WANG C, FESSLER J, DETOMASO D, AVILA-PACHECO J, KAMINSKI J, ZAGHOUANI S, CHRISTIAN E, THAKORE P, SCHELLHAASS B, AKAMA-GARREN E, PIERCE K, SINGH V, RON-HAREL N, DOUGLAS VP, BOD L, SCHNELL A, PULESTON D, SOBEL RA, HAIGIS M, PEARCE EL, SOLEIMANI M, CLISH C, REGEV A, KUCHROO VK & YOSEF N 2021. Metabolic modeling of single Th17 cells reveals regulators of autoimmunity. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WALTMAN L & VAN ECK NJ 2013. A smart local moving algorithm for large-scale modularity-based community detection. The European physical journal B, 86, 1–14. [Google Scholar]
- WEAVER CT, ELSON CO, FOUSER LA & KOLLS JK 2013. The Th17 pathway and inflammatory diseases of the intestines, lungs, and skin. Annu Rev Pathol, 8, 477–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHEELER MA, CLARK IC, TJON EC, LI Z, ZANDEE SEJ, COUTURIER CP, WATSON BR, SCALISI G, ALKWAI S, ROTHHAMMER V, ROTEM A, HEYMAN JA, THAPLOO S, SANMARCO LM, RAGOUSSIS J, WEITZ DA, PETRECCA K, MOFFITT JR, BECHER B, ANTEL JP, PRAT A & QUINTANA FJ 2020. MAFG-driven astrocytes promote CNS inflammation. Nature, 578, 593–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU C, YOSEF N, THALHAMER T, ZHU C, XIAO S, KISHI Y, REGEV A & KUCHROO VK 2013. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature, 496, 513–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU HJ, IVANOV II, DARCE J, HATTORI K, SHIMA T, UMESAKI Y, LITTMAN DR, BENOIST C & MATHIS D 2010. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity, 32, 815–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YANG XO, NURIEVA R, MARTINEZ GJ, KANG HS, CHUNG Y, PAPPU BP, SHAH B, CHANG SH, SCHLUNS KS, WATOWICH SS, FENG XH, JETTEN AM & DONG C 2008. Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity, 29, 44–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YOSEF N, SHALEK AK, GAUBLOMME JT, JIN H, LEE Y, AWASTHI A, WU C, KARWACZ K, XIAO S, JORGOLLI M, GENNERT D, SATIJA R, SHAKYA A, LU DY, TROMBETTA JJ, PILLAI MR, RATCLIFFE PJ, COLEMAN ML, BIX M, TANTIN D, PARK H, KUCHROO VK & REGEV A 2013. Dynamic regulatory network controlling TH17 cell differentiation. Nature, 496, 461–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YU X, ROLLINS D, RUHN KA, STUBBLEFIELD JJ, GREEN CB, KASHIWADA M, ROTHMAN PB, TAKAHASHI JS & HOOPER LV 2013. TH17 cell differentiation is regulated by the circadian clock. Science, 342, 727–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG H, MADI A, YOSEF N, CHIHARA N, AWASTHI A, POT C, LAMBDEN C, SRIVASTAVA A, BURKETT PR, NYMAN J, CHRISTIAN E, ETMINAN Y, LEE A, STROH H, XIA J, KARWACZ K, THAKORE PI, ACHARYA N, SCHNELL A, WANG C, APETOH L, ROZENBLATT-ROSEN O, ANDERSON AC, REGEV A & KUCHROO VK 2020. An IL-27-Driven Transcriptional Network Identifies Regulators of IL-10 Expression across T Helper Cell Subsets. Cell Rep, 33, 108433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG L, YU X, ZHENG L, ZHANG Y, LI Y, FANG Q, GAO R, KANG B, ZHANG Q, HUANG JY, KONNO H, GUO X, YE Y, GAO S, WANG S, HU X, REN X, SHEN Z, OUYANG W & ZHANG Z 2018. Lineage tracking reveals dynamic relationships of T cells in colorectal cancer. Nature, 564, 268–272. [DOI] [PubMed] [Google Scholar]
- ZHANG Y, LIU T, MEYER CA, EECKHOUTE J, JOHNSON DS, BERNSTEIN BE, NUSBAUM C, MYERS RM, BROWN M, LI W & LIU XS 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol, 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHENG GX, TERRY JM, BELGRADER P, RYVKIN P, BENT ZW, WILSON R, ZIRALDO SB, WHEELER TD, MCDERMOTT GP, ZHU J, GREGORY MT, SHUGA J, MONTESCLAROS L, UNDERWOOD JG, MASQUELIER DA, NISHIMURA SY, SCHNALL-LEVIN M, WYATT PW, HINDSON CM, BHARADWAJ R, WONG A, NESS KD, BEPPU LW, DEEG HJ, MCFARLAND C, LOEB KR, VALENTE WJ, ERICSON NG, STEVENS EA, RADICH JP, MIKKELSEN TS, HINDSON BJ & BIELAS JH 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun, 8, 14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Study design, Il17a scRNA-seq detection validation and clustering analysis of current tissue Th17 cell scRNA-seq, related to Figure 1 (A) Simplified representation of the Il17a and Rosa26 loci of Il17aCreGFP X Rosa26tdTomato mice.
(B) Gating strategy for the sorting of singlet viable CD45+ TCRβ+ CD4+ tdTomato+ cells for scRNA/TCR-seq.
(C) Comparison of frequencies of IL-17A-GFP+ of tdTomato+ cells in flow cytometry (left) vs. Il17aGFP transcript in scRNA-seq (right) (n=4–7).
(D) UMAP representation of unsupervised clustering of all current Th17 single cells (GFP+) (6,367 cells) at homeostasis.
(E) Heatmap representing cluster-specific up-regulated genes (FDR <0.05, fold change ≥1.5). Data were shown for a random sample of 1,000 cells.
(F) Bar plot depicting the tissue composition of each cluster.
(G) UMAP representation from (A) colored by batch for all tissues together (top left) and for each tissue respectively (other five subplots). Due to experimental complexity, not all tissues were present in all batches (Table S1). The Adjusted Mutual Information (AMI) between batch and clusters was close to 0 (and far from 1), suggesting the clusters were not driven by batch effects.
(H) Heatmap depicting the pairwise distance between tissues. Euclidean distance was computed for each pair of tissue centers, defined as the average of cell embeddings in the top 50 principal components. Cells from the proliferating and Treg clusters (Clusters 7 and 6, Figure S1D,E) were excluded from this analysis.
Figure S2: Joined single-cell profiling of ex- and current tissue Th17 cells, related to Figure 1 (A-C) UMAP of all current (GFP+) and ex-Th17 cells (GFP−) at homeostasis (37,773 cells). Color indicates the tissue origin (A), transcript expression of Foxp3 and Mki67 (B), and Th17 status (current or ex-Th17, C).
(D) Expression of selected Th17 effector genes (rows) in all Th17 cells in the tissues (columns) at homeostasis.
(E) Heatmap of up-regulated and down-regulated genes (FDR <0.05, fold change ≥1.5) in tissues vs. splenic current and ex-Th17 cells. Small multi-tissue gene sets (<20 genes) were not shown.
(F) Volcano plot for differentially expressed genes (FDR <0.05, fold change ≥1.2) comparing current vs. ex-Th17 cells in each tissue.
Figure S3: Validation of the ISG-high and the tdTomato+ Foxp3+ Treg subpopulations, related to Figure 2 (A) COMET (Delaney et al., 2019) prediction of PD-L1 (Cd274) as a favorable surface marker for the ISG-high Th17 subpopulation in the SPL (left) and MLN (right).
(B) Flow cytometry analysis of PD-L1 protein expression on tdTomato+ CD4+ T cells in the SPL (left) and MLN (right).
(C) Validation of PD-L1 as a marker for the ISG-high Th17 subpopulation by qPCR. Relative expression (RE) of differentially up-regulated genes in the ISG-high population in PD-L1− and PD-L1+ tdTomato+ CD4+ T cells by qPCR (n=3).
(D) Odds ratio of detecting Cd274 in the ISG-high cluster vs. other clusters in three published datasets (Arazi et al., 2019, Hemmers et al., 2019, Tibbitt et al., 2019). 95% confidence intervals were shown for each odds ratio. The dashed vertical line marks odds ratio = 1.
(E) TCR repertoire similarity analysis of the ISG-high clusters in the SPL (SPL5), MLN (MLN4), and PP (PP5) with all other intra-tissue clusters at homeostasis.
(F) Comparison of frequencies of Foxp3GFP+ cells of tdTomato+ cells in flow cytometry (left) vs. scRNA-seq (right) (n=3–6). The Foxp3 detection was based on Foxp3GFP protein in flow cytometry and based on the Foxp3 transcript in scRNA-seq.
(G) Representative flow cytometry plots of tdTomato+ Foxp3GFP+ cells in each tissue.
(H) TCR repertoire similarity analysis of intra-tissue clusters within the MLN, PP, and SI.
(I) Percentage of Treg-like and non-Treg colonic tdTomato+ cells that share their TCR sequence with splenic tdTomato+ cells (n=2–3).
(J)Presumptive model of the ancestry of tdTomato+ Foxp3+ Treg cells. The low TCR similarity of tdTomato+ Foxp3+ Treg cells with other tdTomato+ tissue Th17 cells suggests that tdTomato+ Foxp3+ Treg cells are not likely derived from Th17 cells but presumably from Treg cells.
Figure S4: Characterization of tissue Th17 cell subpopulations during EAE, related to Figure 3 and 4 (A) UMAP visualization of G1/S and G2/M signatures scores in CNS Th17 cells. G1/S and G2/M signatures were obtained from (Tirosh et al., 2016).
(B) Box plot depicting the total UMI counts of cells for each CNS Th17 cluster.
(C) Frequencies of current Th17 cells (GFP+) in each CNS Th17 cluster based on scRNA-seq.
(D) COMET (Delaney et al., 2019) prediction of PD-1 (Pdcd1) as a favorable surface marker for cluster 4.
(E,F) UMAP visualization of all current (GFP+) and ex-Th17 cells (GFP−) from naïve and EAE-diseased mice (84,124 cells). The cells are colored by batch (E) or by Th17 status (current or ex-Th17, F). Batches are not evenly distributed in (E) because not all tissues were present in all batches (Table S1).
(G) Bar plot depicting the distance between naïve and EAE cells in SPL, SI, and COL. For each tissue, Euclidean distance was computed between naïve and EAE cell centers, which were defined as the average of cell embeddings in the top 50 principal component spaces. Cells from proliferating and Treg clusters (Figure 4A) were excluded from this analysis.
(H) Bar plot comparing the number of differentially expressed genes between EAE and naïve cells in SPL, SI, and COL (FDR <0.05, fold change ≥1.5).
(I) UMAP visualization of all tdTomato+ single cells from the SPL (24,837 cells), SI (11,416 cells), and COL (15,801 cells) from naïve and EAE-diseased mice, colored by cluster assignment (left top row), Th17 status (left bottom row), and treatment (right top row). Bar plots (right bottom row) depicting the treatment composition of each cluster, and the Chi-square statistics for an association between treatment and cluster membership were shown for each tissue. All sub-figures other than the UMAPs colored by clusters were based on a sub-sample. Multiple subsamples were generated, and results were shown for one representative case.
Figure S5: Detection of the SLAMF6+ and CXCR6+ populations in human MS patients and mouse colitis, related to Figure 5 (A) Positive correlations of pathogenic Th17 pathway signatures (y-axis) (Gaublomme et al., 2015, Lee et al., 2012) and the CXCR6+ bulk RNA-seq signature (x-axis) (Figure 5F, Table S4) in cerebrospinal fluid (CSF) T cells in MS patients (Schafflick et al., 2020).
(B) Negative correlation of non-pathogenic Th17 pathway signature (Th17 self-renewing) (Gaublomme et al., 2015) and the CXCR6+ bulk RNA-seq signature in CSF T cells in MS patients.
(C) Dot plot comparing the correlations of the CXCR6+ bulk RNA-seq signature with the pathogenic and non-pathogenic Th17 pathway signatures (Gaublomme et al., 2015) in CSF T cells of healthy controls vs. MS patients.
(D) UMAP visualization of the SLAMF6+ (left) and CXCR6+ (right) bulk RNA-seq signatures in colonic tdTomato+ CD4+ single T cells of mice with DSS-induced colitis.
Figure S6: Intra-tissue heterogeneity of Th17 cells during EAE, related to Figure 6 (A) Heatmap depicting sharing of GLIPH2-identified (Huang et al., 2020) TCR specificity groups across tissue Th17 cells at homeostasis. All TCR specificity groups (rows) with GLIPH2 reported Fisher score <0.05 were included.
(B) UMAP visualization of all current (GFP+) and ex-Th17 cells (GFP−) single cells from the SPL, MLN, PP, SI, COL, CNS, and DLN during EAE, colored by cluster assignment (top row), cluster function annotation (middle row), and Th17 status (bottom row).
(C) Correlation of transcriptomic landscape for all pairs of intra-tissue clusters during EAE.
(D) Expression of selected genes used to define the cluster functional annotations.
(E) MA plot depicting the differentially expressed genes (FDR <0.05, fold change ≥1.5) in SI5 (Figure S6B) vs. all other cells in the SI.
(F) Gene set enrichment analysis results for selected GO terms and KEGG pathways enriched in SI5.
(G) Number and fraction of cells with valid clonotype data in each tissue that share TCR with CNS.
(H) SI5 signature score for cells that share the TCR with cells in the CNS (shared) vs. the cells that do not share the TCR with cells in the CNS (not shared) for each tissue. The SI5 signature was identified as described in Figure S6B.
Figure S7: Characterization of the conversion of the SLAMF6+ to the CXCR6+ population in vivo, related to Figure 6 and 7 (A) Frequencies of CXCR6+ tdTomato+ CD4+ T cells in the blood of naïve and EAE-diseased mice by flow cytometry (n= 3–6).
(B) Relative expression (RE) of Cxcl16 in different immune populations in the spleen and CNS of EAE mice, determined by qPCR (n=4).
(C) Violin plot depicting the proliferation signature score (Figure S1D,E) for the single cells in SPL0 and SPL1. Visualization was based on non-extreme expression values (between 0.1 and 99.9 percentiles) and jittered points were only shown for randomly selected 50% of the cells.
(D) EAE disease curve of immunized recipient mice that received CXCR6+ tdTomato+ cells 7 days after immunization (n=6–7).
(E) Frequencies of tdTomato+ CD4+ T cells in control vs. antibiotic-treated (Abx) mice in each tissue at homeostasis (n=4–5).
(F) Frequencies of the SLAMF6+ tdTomato+ CD4+ T cells in the spleen of control vs. Abx-treated mice at homeostasis (n=5).
(G) EAE disease curve of immunized Abx-treated or control mice (n=5).
(H) Numbers of SLAMF6+ (left) or CXCR6+ (right) tdTomato+ CD4+ T cells in the spleen of EAE-diseased Abx-treated or control mice harvested 9-days post immunization (n=3–4).
(I) GM-CSF and IFNγ concentration in the culture medium during recall assay, determined by LegendPlex (n=4).
(J) Violin plot depicting the IL-23 signature score (Wu et al., 2013) for the single cells in SPL0 and SPL1. Visualization was based on non-extreme expression values (between 0.1 and 99.9 percentiles) and jittered points were only shown for randomly selected 50% of the cells.
(K) Heatmap depicting the chromatin accessibility of published IL-23R signaling signatures in the ATAC-seq of the SLAMF6+ and CXCR6+ population. GSE39820 (Lee et al., 2012), GSE43955 (Yosef et al., 2013) (1- M9668; 2- M9672), GSE23505 (Ghoreschi et al., 2010), PID_IL23_Pathway (Schaefer et al., 2009), Langrish et al. (Langrish et al., 2005).
(L) Relative expression (RE) of Il12b and Il23a in splenic immune populations during EAE by qPCR (n=4). Act= Activated.
(M) Proportion of cells with non-zero expression of Il12b and Il23a in CNS immune cells from EAE (Wheeler et al., 2020) mice at EAE priming stage (n=6).
(N) Heatmap depicting the relative expression of differentially expressed genes (FDR <0.05) comparing CXCR6+ cells from Il17aCreIl23rfl/fl and Il17aCreIl23rWT mice.
(O) Bar plot of published Th17 pathway signatures (Lee et al., 2012, Gaublomme et al., 2015, Liberzon et al., 2015) from gene set enrichment analysis for CXCR6+ cells from Il17aCreIl23rfl/fl vs. Il17aCreIl23rWT mice.
Table S2: Tissue-specific Th17 signatures, related to Figure 1
Table S3: T-cell clonotype information and clonotype sharing across mice, related to Figure 2 and 4
(A) Summary, (B) Naïve shared, (C) EAE shared
Table S4: CXCR6+ vs. SLAMF6+ bulk RNA-seq signatures, related to Figure 5
Table S5: Bulk RNA-seq of CXCR6+ cells from Il17aCreIl23rfl/fl vs. Il17aCreIl23rWT mice, related to Figure 7
Table S6: List of housekeeping genes for QC, related to STAR Methods
Data Availability Statement
scRNA/TCR-seq, bulk RNA-seq, and bulk ATAC-seq data have been deposited at GEO under accession number GSE188322 and are publicly available as of the date of publication. Detailed accession numbers are listed in the key resources table.
All original code has been deposited at GitHub (https://github.com/lhuang1/Th17_single_cell) and is publicly available as of the date of publication. The GitHub information is listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD45 APC-Cy7 (clone 3-F11) | Biolegend | Cat# 103116, RRID: AB_312981 |
| Anti-mouse CD4 APC (clone RM4-5) | Biolegend | Cat# 100516, RRID: AB_312719 |
| Anti-mouse CD4 APC-Cy7 (clone RM4-5) | Biolegend | Cat# 100526, RRID: AB_312727 |
| Anti-mouse TCRp PE-Cy7 (clone H57-597) | Biolegend | Cat# 109222, RRID: AB_893625 |
| Anti-mouse PD-L1 APC (clone 10F.9G2) | Biolegend | Cat# 124312, RRID: AB_10612741 |
| Anti-mouse CXCR6 APC (clone SA051D1) | Biolegend | Cat# 151106, RRID: AB_2572143 |
| Anti-mouse SLAMF6 Pacific Blue (clone 330-AJ) | Biolegend | Cat# 134608, RRID: AB_2188093 |
| Anti-mouse CD8a PE-Cy7 (clone 53-6.7) | Biolegend | Cat# 100722, RRID: AB_312761 |
| Anti-mouse PD-1 Brilliant Violet 711 (clone 29F.1A12) | Biolegend | Cat# 135231, RRID: AB_2566158 |
| Anti-mouse NK1.1 Brilliant Violet 421 (clone PK136) | Biolegend | Cat# 108732, RRID: AB_2562218 |
| Anti-mouse Ly-6C Brilliant Violet 605 (clone HK1.4) | Biolegend | Cat# 128036, RRID: AB_2562353 |
| Anti-mouse I-A/E Brilliant Violet 785 (clone M5/114.15.2) | Biolegend | Cat# 107645, RRID: AB_2565977 |
| Anti-mouse CD64 PE (clone X54-5/7.1) | Biolegend | Cat# 139304, RRID: AB_10612740 |
| Anti-mouse/human CD11b PE-Cy7 (clone M1/70) | Biolegend | Cat# 101216, RRID: AB_312799 |
| Anti-mouse CD19 APC-Cy7 (clone 6D5) | Biolegend | Cat# 115530, RRID: AB_830707 |
| Anti-mouse Gr1 APC (clone RB6-8C5) | Biolegend | Cat# 108412, RRID: AB_313377 |
| Anti-mouse/human CD44 PerCP/Cy5.5 (clone IM7) | Biolegend | Cat# 103032, RRID: AB_2076204 |
| Anti-mouse IFNγ APC-Cy7 (clone XMG1.2) | Biolegend | Cat# 505850, RRID: AB_2616698 |
| Anti-mouse GM-CSF PE-Cy7 (clone MP1-22E9) | Biolegend | Cat# 505412, RRID: AB_2721682 |
| Anti-mouse IL-17A FITC (clone TC11-18H10.1) | Biolegend | Cat# 506908, RRID: AB_536010 |
| Anti-mouse IL-2 Brilliant Violet 605 (clone JES6-5H4) | Biolegend | Cat# 503829, RRID: AB_11204084 |
| Anti-mouse CD11c PE-CF594 (clone N418) | BD Biosciences | Cat# 565591, RRID: AB_2869692 |
| Anti-mouse AREG antibody (clone 206220) | R&D Systems | Cat# MAB989, RRID: RRID: AB_2060661 |
| Chemicals, peptides, and recombinant proteins | ||
| MOG peptide | Genemed Synthesis | Cat# MOG3555-P2-5 |
| M. tuberculosis H37 Ra, desiccated | Becton Dickinson | Cat# 231141 |
| Adjuvant, Complete H37 Ra | Becton Dickinson | Cat# 231131 |
| Pertussis toxin | List Biological Laboratories | Cat# 181 |
| Ampicillin sodium salt | Sigma Aldrich | Cat# A9518 |
| Metronidazole | Acros Organics | Cat# AC210340050 |
| Vancomycin hydrochloride | Research products international | Cat# V06500-5.0 |
| Neomycin sulfate | Thermo Fischer Scientific | Cat# BP26695 |
| Fixable Viability Dye eFluor 506 | eBioscience | Cat# 65-0866-14 |
| DNase I | Sigma Aldrich | Cat# 11284932001 |
| Collagenase Type VIII | Sigma Aldrich | Cat# C2139 |
| CaCl2 | Sigma Aldrich | Cat# C5670 |
| MgCl2 | Sigma Aldrich | Cat# 63069 |
| DTT (dithiothreitol) | Thermo Fischer Scientific | Cat# R0861 |
| RPMI 1640 medium | Thermo Fischer Scientific | Cat# A1049101 |
| EDTA | Thermo Fischer Scientific | Cat# AM9261 |
| Collagenase D | Roche Diagnostics | Cat# 11088882001 |
| Percoll | Cytiva | Cat# 17-0891-01 |
| 2-mercaptoethanol | Sigma Aldrich | Cat# M6250 |
| Digitonin | Promega | Cat# G9441 |
| Dextran Sulfate Sodium Salt (DSS) | Affymetrix | Cat# 14489-100-GM |
| Critical commercial assays | ||
| Pacific Blue Antibody Labeling Kit | Molecular Probes | Cat# P30013 |
| ACK lysis buffer | Lonza | Cat# 10-548E |
| Cell Stimulation Cocktail (plus protein transport inhibitors) | eBioscience | Cat# 00-4975-93 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 554714 |
| CD4 (L3T4) MicroBeads, mouse | Miltenyi Biotec | Cat# 130117043 |
| Recombinant Mouse IL-23 Protein | R&D Systems | Cat# 1887-ML-010 |
| LegendPlex Mouse Inflammation Panel | Biolegend | Cat# 740446 |
| PicoPure™ RNA Isolation Kit | Applied Biosystems | Cat# KIT0204 |
| SuperScript™ VILO™ cDNA Synthesis Kit | Invitrogen | Cat# 11754050 |
| TaqMan™ Fast Advanced Master Mix | Thermo Fisher Scientific | Cat# 4444557 |
| HiSeq X Ten Reagent Kit v2.5 | Illumina | Cat# FC-501-2501 |
| TCL buffer | QIAGEN | Cat# 1031576 |
| Quant-iT PicoGreen dsDNA Assay Kit | Thermo Fischer Scientific | Cat# 1102-200 |
| Nextera XT DNA Library Preparation Kit | Illumina | Cat# FC-131-1024 |
| NextSeq 500 High Output v2 Kit (75 cycles) | Illumina | Cat# FC-404-2005 |
| BAMBANKER freeze medium | LYMPHOTEC Inc | Cat# NC9582225 |
| Illumina Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | Cat# 20034198 |
| MinElute PCR Purification Kit | QIAGEN | Cat# 28004 |
| NEBNext® High-Fidelity 2X PCR Master Mix | New England Biolabs | Cat# M0541L |
| SYBR™ Green I Nucleic Acid Gel Stain | Invitrogen | Cat# S7585 |
| KAPA Library Quantification Kit | KAPA Biosystems | Cat# KK4835 |
| Qubit™ dsDNA HS Assay Kit | Invitrogen | Cat# Q32851 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | Cat# 20024906 |
| Chromium Single Cell 3’ Reagent Kit (v2 Chemistry) | 10x Genomics | Cat# 120237 |
| Chromium Single Cell 5’ Library & Gel Bead Kit 16 rxn | 10x Genomics | Cat# 1000006 |
| Chromium Single Cell V(D)J Enrichment Kit, Mouse T Cell | 10x Genomics | Cat# 1000071 |
| Deposited data | ||
| Mouse reference genome NCBI build 38, GRCm38 (mm10) | Genome Reference Consortium | https://www.ncbi.nlm.nih.gov/assembly/GCF_000001635.20/ |
| Public human kidney immune cell scRNA-seq data | (Arazi et al., 2019) | https://www.immport.org/shared/study/SDY997 |
| Public mouse thymus immune cell scRNA-seq data | (Hemmers et al., 2019) | GEO: GSE134902 |
| Public mouse T helper cell scRNA-seq data | (Tibbitt et al., 2019) | GEO: GSE131935 |
| Public human PBMC and CSF immune cell scRNA-seq data | (Schafflick et al., 2020) | GEO: GSE138266 |
| Public mouse CNS immune cell scRNA-seq data | (Wheeler et al., 2020) | GEO: GSE130119 |
| Bulk RNA-seq and ATAC-seq of SLAMF6+ and CXCR6+ Th17 cells | This paper | GEO: GSE188319 |
| scRNA/TCR-seq of tissue Th17 cells in homeostasis and EAE | This paper | GEO: GSE188320 |
| scRNA-seq of colonic Th17 cells in colitis | This paper | GEO: GSE188321 |
| Experimental models: Mice | ||
| C57BL/6-Il17atmlBcgen/J (Il17aGFP) | Jackson Laboratory | JAX: 018472 |
| Il17atm1.1(icre)Stck/J (Il17aCre) | Jackson Laboratory | JAX: 016879 |
| B6;129S6-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (Rosa26tdTomato) | Jackson Laboratory | JAX: 007905 |
| B6.SJL-PtprcaPepcb/BoyJ (CD45.1) | Jackson Laboratory | JAX: 002014 |
| Foxp3tm1Kuch (Foxp3GFP) | (Bettelli et al., 2006) | N/A |
| Il23r fl/fl | This paper | |
| Tg(CAG-Kaede)15Kgwa (Kaede) | RIKEN BRC, Japan | N/A |
| Oligonucleotides | ||
| Eukaryotic 18S rRNA Endogenous Control | Thermo Fischer Scientific | Cat# 4352930E |
| Mouse ACTB (Actin, Beta) Endogenous Control | Thermo Fischer Scientific | Cat# 4352341E |
| Ifit1 (Mm00515153_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Ifit3 (Mm01704846_s1) | Thermo Fischer Scientific | Cat# 4448892 |
| Irf7 (Mm00516793_g1) | Thermo Fischer Scientific | Cat# 4448892 |
| Isg15 (Mm01705338_s1) | Thermo Fischer Scientific | Cat# 453320 |
| Isg20 (Mm00469585_m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Zbp1 (Mm01247052_m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Nkg7 (Cat# Mm01205900_g1) | Thermo Fischer Scientific | Cat# 4448892 |
| Ccl5 (Cat# Mm01302427_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Cd8a (Cat# Mm01182107_g1) | Thermo Fischer Scientific | Cat# 453320 |
| Prf1 (Cat# Mm00812512_m1) | Thermo Fischer Scientific | Cat# 453320 |
| Gzmk (Cat# Mm00492530 m1) | Thermo Fischer Scientific | Cat# 4448892 |
| Cxcl16 (Cat# Mm00469712_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Il12b (Cat# Mm01288989_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Il23a (Cat# Mm00518984_m1) | Thermo Fischer Scientific | Cat# 4331182 |
| Ad1 primer: AATGATACGGCGACCACCGAGATCTACACXXXXXXXXTCGTCGGCAGCGTCAGATGTGTAT | IDT | N/A |
| Ad2 primer: CAAGCAGAAGACGGCATACGAGATXXXXXXXXGTCTCGTGGGCTCGGAGATGTG | IDT | N/A |
| Software and algorithms | ||
| FlowJo | TreeStar | https://www.flowjo.com/solutions/flowjo |
| GraphPad Prism | GraphPad Software | https://www.graphpad.com/scientificsoftware/prism/ |
| R v3.6.2 | R core | https://www.r-project.org/ |
| CellRanger v3.0.2 | (Zheng et al., 2017) | https://support.10xgenomics.com/single-cell-gene-expression/software |
| DemuxEM v0.1.5 | (Gaublomme et al., 2019) | https://demuxem.readthedocs.io/ |
| Seurat v3.2.2 | (Stuart et al., 2019) | https://satijalab.org/seurat/ |
| edgeR | (Robinson et al., 2010, McCarthy et al., 2012) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| fGSEA | (Korotkevich et al., 2019) | https://bioconductor.org/packages/release/bioc/html/fgsea.html |
| COMET | (Delaney et al., 2019) | http://www.cometsc.com/index |
| GLIPH2 | (Huang et al., 2020) | http://50.255.35.37:8080/ |
| HISAT2 | (Kim et al., 2019) | http://daehwankimlab.github.io/hisat2/ |
| RSEM | (Li and Dewey, 2011) | https://deweylab.github.io/RSEM/ |
| ATAC-seq data analyses pipeline | (Lee et al., 2016) | https://github.com/kundajelab/atac_dnase_pipelines |
| Bowtie2 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 v2.1.2 | (Zhang et al., 2008) | https://pypi.org/project/MACS2/ |
| Bedtools v2.26.1 | (Quinlan and Hall, 2010) | https://github.com/arq5x/bedtools2 |
| GREAT v4.0.4 | (McLean et al., 2010) | http://great.stanford.edu/public/html/ |
| DESeq2 | (Love et al., 2014) | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Analysis code | This paper | https://github.com/lhuang1/Th17_single_cell |
| Other | ||
| 200 mW Electra Pro portable laser | Laserglow Technologies | GEL200XXX |