

The first structural characterization of long tandem repeats of the d(CGA) triplet motif in the parallel-stranded duplex form is provided and the results show that each strand within the duplex is conformationally distinct and plays a unique role in accommodating duplex asymmetry. The structural uniformity of tandem d(CGA) base-paired triplets obtained from different solution environments further demonstrates that the d(CGA)-based parallel-stranded duplex is a robust and highly predictable structure with applications in DNA nanotechnology.
Keywords: triplet-repeat DNA, parallel-stranded duplex, noncanonical, d(CGA) motif
Abstract
DNA can adopt noncanonical structures that have important biological functions while also providing structural diversity for applications in nanotechnology. Here, the crystal structures of two oligonucleotides composed of d(CGA) triplet repeats in the parallel-stranded duplex form are described. The structure determination of four unique d(CGA)-based parallel-stranded duplexes across two crystal structures has allowed the structural parameters of d(CGA) triplets in the parallel-stranded duplex form to be characterized and established. These results show that d(CGA) units are highly uniform, but that each strand in the duplex is structurally unique and has a distinct role in accommodating structural asymmetries induced by the C–CH+ base pair.
1. Introduction
DNA is a polymorphic biopolymer that can adopt an array of conformations beyond the traditional B-form double helix. Watson–Crick base-paired duplexes can access multiple helical forms (B-, A- or Z-form) depending on sequence and environmental conditions (Watson & Crick, 1953 ▸; Franklin & Gosling, 1953 ▸; Wang et al., 1979 ▸). The hydrogen-bonding and base-stacking interactions that stabilize antiparallel duplexes also allow DNA to access other noncanonical conformations, some of which have known biological implications, including G-quadruplexes (Spiegel et al., 2020 ▸), i-motifs (Abou Assi et al., 2018 ▸) and triplexes (Tateishi-Karimata & Sugimoto, 2021 ▸). Further, the formation of many noncanonical structures can be controlled by nucleotide-sequence composition and several environmental factors including pH, the presence and concentration of cations, and temperature (Saoji & Paukstelis, 2015 ▸; Benabou et al., 2019 ▸; Cristofari et al., 2019 ▸; Largy et al., 2016 ▸; Chen et al., 2017 ▸; Nguyen et al., 2017 ▸). The alternative structures formed by genomic DNA triplet-repeat sequences (Zheng et al., 1996 ▸; Paiva & Sheardy, 2004 ▸; Völker et al., 2002 ▸) have been implicated in their ability to expand and cause genetic instabilities (Wells, 1996 ▸; Lahue, 2020 ▸). Understanding these alternative structures and the conditions that may lead to their formation provides a fundamental basis for understanding the disease states. Additionally, the ability to control DNA conformations has utility in DNA nanotechnology applications, where noncanonical motifs expand the structural and functional diversity of nanostructures while retaining inherent programmability and predictability. Previous work has focused on incorporating functional noncanonical structures such as the G-quadruplex (Bourdoncle et al., 2006 ▸; Zhou et al., 2015 ▸; Sannohe & Sugiyama, 2012 ▸; Li & Mirkin, 2005 ▸), i-motif (Liedl & Simmel, 2005 ▸; Nesterova & Nesterov, 2014 ▸; Song et al., 2013 ▸; Shu et al., 2005 ▸), polyA motif (Chakraborty et al., 2009 ▸; Yu et al., 2018 ▸; Srivastava et al., 2018 ▸) and triple helix (Jung et al., 2006 ▸; Liu & Mao, 2014 ▸; Han et al., 2008 ▸) into DNA nanostructures, where changes in the local environment are used to tune the resulting structure.
The d(CGA) triplet-repeat motif is another such environmentally sensitive motif that can adopt different structural forms in a pH-dependent manner at near-physiological temperatures and salt concentrations (Luteran et al., 2020 ▸). Neutral pH favors a unimolecular antiparallel hairpin stabilized by canonical G–C base pairs (Zheng et al., 1996 ▸; Kejnovská et al., 2001 ▸), while acidic pH favors a noncanonical homo-base-paired parallel-stranded duplex (ps-duplex; Robinson et al., 1992 ▸; Robinson & Wang, 1993 ▸). Although the noncanonical d(CGA) motif can adopt distinct structural conformations, the ps-duplex is the predominantly studied form (Robinson et al., 1992 ▸; Sunami et al., 2002 ▸; Tripathi & Paukstelis, 2016 ▸; Tripathi et al., 2015 ▸; Wang & Patel, 1994 ▸). Originally described as Π-DNA, the d(CGA) n ps-duplex is stabilized by homo-base-pair interactions (C–CH+, G–G and A–A) and inter-strand base-stacking interactions (Robinson & Wang, 1993 ▸; Wang & Patel, 1994 ▸). The C–CH+ homo-base pair requires hemiprotonation at the N3 position to form three hydrogen bonds along the Watson–Crick face (Robinson & Wang, 1993 ▸). N2–N3 sugar-edge hydrogen bonds stabilize G–G homo-base pairs, while A–A homo-base pairs are formed through N6–N7 Hoogsteen face hydrogen bonds. Importantly, the GpA dinucleotide step provides significant stabilization to the ps-duplex by the formation of inter-strand G/A base-stacking interactions.
The structure and stability of the ps-duplex is highly influenced by the 5′-nucleotide of each triplet (Luteran et al., 2020 ▸). Similar G/A-stacking interactions have been observed in ps-duplex structures containing d(GGA) or d(TGA) triplets (Sunami et al., 2002 ▸; Tripathi et al., 2015 ▸; Robinson et al., 1994 ▸; Kettani et al., 1999 ▸; Rippe et al., 1992 ▸), although contiguous repeats of these sequences are unable to form ps-duplexes (Luteran et al., 2020 ▸). The ps-duplex region of an intercalation-locked tetraplex containing d(TGA) triplets forms a perfectly symmetrical duplex (Tripathi et al., 2015 ▸), while the same ps-duplex region containing d(CGA) triplets resulted in structural asymmetry and duplex bending (Tripathi & Paukstelis, 2016 ▸). The asymmetry is associated with a displacement from the helical axis at the C–CH+ base pair. Further, thermodynamic studies indicated that ps-duplex structures containing six tandem d(YGA) triplet repeats undergo a significant destabilization when the d(CGA) triplets are replaced with d(TGA) triplets (Luteran et al., 2020 ▸). Beyond the additional hydrogen-bond interaction within each C–CH+ base pair, the structural details as to why asymmetric d(CGA) duplexes are significantly more stable than symmetric d(TGA) triplets remain unclear.
The ability of d(CGA) to form distinct structural states appears to be a trait that is shared by several other triplet-repeat motifs, although d(CGA) is the only triplet that is known to form perfectly ps-duplex structures (Zheng et al., 1996 ▸; Paiva & Sheardy, 2004 ▸; Völker et al., 2002 ▸; Luteran et al., 2020 ▸). Genomic analyses of all possible triplet-repeat sequences identified to have ≥6 tandem repeat units have shown that such tracts of d(CAG) triplets are overrepresented in the human genome (1055 instances) and are indicated in disease pathologies, while tracts of d(CGA) triplets are the least frequently observed, occurring only 16 times (Kozlowski et al., 2010 ▸). A similarly low frequency and coverage of d(CGA) triplets was seen when a comparable genomic analysis was performed for other eukaryotic organisms (Astolfi et al., 2003 ▸). The formation of alternative structures and the relative stabilities of such structures are thought to be important factors contributing to the expansion of repeat sequences (Paiva & Sheardy, 2004 ▸; Poggi & Richard, 2021 ▸; Wells, 2007 ▸). Due to the challenges that they present to the replication machinery, repeat sequences that form alternative structures could influence pathological or evolutionary outcomes (Kejnovská et al., 2001 ▸; Khristich & Mirkin, 2020 ▸). Therefore, it is important to characterize the structural diversity of triplet-repeat sequences that have the ability to form such noncanonical structures.
In this work, we have determined the crystal structures of two oligonucleotides containing multiple tandem d(CGA) triplet repeats in the ps-duplex form. These structures are the longest ps-duplexes to be solved that are solely comprised of such triplets. The crystals grew from different solution conditions and resulted in distinct crystal-packing arrangements. The structure determination of four ps-duplexes across these two different crystal structures has allowed us to thoroughly characterize and define the structural features of d(CGA) triplets and the ps-duplexes that they form. Despite differences in crystallization and molecular packing, the resulting ps-duplex structures have strikingly low r.m.s.d. values, demonstrating the robust structural uniformity of the d(CGA) triplet-repeat motif in the ps-duplex form. Additionally, each ps-duplex contains two conformationally distinct d(CGA) triplets based on hydrogen-bonding and base-stacking interactions. Surprisingly, each strand contains only one triplet conformation. Thus, ps-duplexes containing d(CGA) repeats are not structurally symmetrical and the apparent structural asymmetry is propagated discretely throughout each strand.
2. Materials and methods
2.1. Oligonucleotide synthesis and purification
DNA oligonucleotides were synthesized on the 1 µmol scale using standard phosphoramidite chemistry on an Expedite 8909 Nucleic Acid Synthesizer (PerSeptive Biosystems, Framingham, Massachusetts, USA) with reagents from Glen Research (Sterling, Virginia, USA). Following deprotection with 30% ammonium hydroxide, the oligonucleotides were purified by 20% (19:1) acrylamide:bisacrylamide, 7 M urea gel electrophoresis, electro-eluted and dialyzed against deionized water.
2.2. Oligonucleotide crystallization
(CGA)5TGA was crystallized by mixing 2 µl 200 µM DNA solution with 2 µl crystallization solution [20% 2-methyl-2,4-pentanediol (MPD), 120 mM barium chloride, 30 mM sodium cacodylate pH 5.5]. GA(CGA)5 was crystallized by mixing 1 µl 125 µM DNA solution with 2 µl crystallization solution [8% PEG 400, 96 mM strontium chloride, 32 mM lithium chloride, 8 mM hexamminecobalt(III) chloride, 24 mM sodium cacodylate pH 7.4]. Crystallization was performed in sitting drops that were equilibrated against 300 µl 30% MPD or PEG 400 [for (CGA)5TGA and GA(CGA)5, respectively] in the well reservoir and incubated at 22°C. Crystals were observed within seven days of plating.
2.3. Data collection, processing, structure determination and refinement
Crystals were removed from drops with nylon cryo-loops, immediately dipped in the respective crystallization condition supplemented with 30% MPD or PEG 400 and plunged into liquid nitrogen. Diffraction data were collected at the Advanced Photon Source (APS), Argonne National Laboratory. Data for (CGA)5TGA were collected on the 24-ID-E beamline and data for GA(CGA)5 were collected on the 24-ID-C beamline.
Data processing for (CGA)5TGA was carried out with iMosflm (Leslie & Powell, 2007 ▸) and that for GA(CGA)5 was carried out with XDS (Kabsch, 2010 ▸) and AIMLESS (Evans & Murshudov, 2013 ▸). Initial phases were obtained by molecular replacement using Phaser (McCoy et al., 2007 ▸). The parallel-stranded homoduplex d(CGA) triplet region from PDB entry 1ixj (Sunami et al., 2002 ▸) was used as the search model for (CGA)5TGA, and two tandem d(CGA) units from the refined (CGA)5TGA structure were used as the search model for GA(CGA)5. Model building and refinement was carried out in Phenix (Liebschner et al., 2019 ▸) and Coot (Emsley et al., 2010 ▸), respectively, for both data sets. Data-collection and refinement statistics are given in Table 1 ▸.
Table 1. Data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| GA(CGA)5 | (CGA)5TGA | |
|---|---|---|
| PDB code | 7sb8 | 7t6y |
| Sequence | d(GACGACGACGACGACGA) | d(CGACGACGACGACGATGA) |
| Data collection | ||
| Wavelength (Å) | 0.979 | 0.979 |
| Space group | P1211 | C121 |
| a, b, c (Å) | 19.68, 30.42, 180.82 | 84.50, 32.35, 32.26 |
| α, β, γ (°) | 90, 90.4, 90 | 90, 91.03, 90 |
| Resolution range (Å) | 90.41–1.32 (1.36–1.32) | 32.25–2.30 (2.38–2.30) |
| Multiplicity | 4.5 (4.4) | 1.8 (1.8) |
| Completeness (%) | 94.7 (84.3) | 87.2 (88.6) |
| 〈I/σ(I)〉 | 9.9 (2.0) | 5.7 (5.0) |
| R p.i.m. † | 0.049 (0.350) | 0.109 (0.288) |
| CC1/2 ‡ | 0.995 (0.820) | 0.940 (0.395) |
| Refinement | ||
| No. of reflections | 48419 (4340) | 3487 (350) |
| R work | 0.171 (0.191) | 0.204 (0.225) |
| R free | 0.214 (0.292) | 0.242 (0.271) |
| No. of atoms | ||
| DNA | 2100 | 740 |
| Ligands | 101 | 10 |
| Solvent | 480 | 119 |
| R.m.s.d., bond lengths (Å) | 0.008 | 0.009 |
| R.m.s.d., bond angles (°) | 0.94 | 1.00 |
| Average B factor (Å2) | ||
| Overall | 18.69 | 13.22 |
| DNA | 16.06 | 12.47 |
| Ligands | 33.81 | 14.80 |
| Solvent | 27.01 | 17.97 |
Precision-indicating merging R factor (Weiss & Hilgenfeld, 1997 ▸). R
p.i.m. =
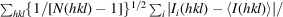
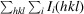 , where Ii
(hkl) is the ith observation of reflection hkl.
, where Ii
(hkl) is the ith observation of reflection hkl.
Correlation coefficient between reflection intensities from the data set randomly split into two halves.
2.4. Circular dichroism (CD)
CD spectra were obtained using a Jasco J-810 spectropolarimeter fitted with a thermostatted cell holder (Jasco, Easton, Maryland, USA). Samples were prepared using 10 µM DNA in 20 mM MES, 100 mM sodium chloride pH 5.5 or 20 mM sodium cacodylate, 100 mM sodium chloride pH 7.0. Samples were incubated at 4°C overnight prior to data collection. Data were collected at room temperature at wavelengths from 220 to 300 nm. Spectra are represented as the average of three scans.
3. Results and discussion
3.1. Overview
We determined the crystal structures of (CGA)5TGA and GA(CGA)5 in the ps-duplex form at 2.30 and 1.32 Å resolution, respectively (Table 1 ▸). (CGA)5TGA was crystallized at pH 5.5 to preferentially stabilize the ps-duplex form, while GA(CGA)5 was crystallized at pH 7.4 to characterize the antiparallel hairpin form. Despite being at a pH that strongly favors the hairpin form (Figs. 1 ▸ a and 1 ▸ b), GA(CGA)5 also crystallized as a ps-duplex. Several other structures that rely on C–CH+ hemiprotonation also crystallized as ps-duplexes at above-neutral pH, suggesting that factors beyond pH influence this structural preference (Tripathi et al., 2015 ▸; Kobuna et al., 2002 ▸). The high local concentration of DNA and the presence of crowding agents have been demonstrated to increase the observed pH of the structural transition in C–CH+-mediated structures (Kejnovská et al., 2001 ▸; Rajendran et al., 2010 ▸; Cui et al., 2013 ▸). CD measurements of d(CGA)-repeat sequences are consistent with these observations; the presence of crowding agents shifts the favorability range of the ps-duplex to higher pH (Fig. 1 ▸ c). Specifically, the addition of 30% PEG 2000 increased the pH of the structural transition by 0.33 ± 0.06 and 0.32 ± 0.13 pH units for (CGA)5TGA and (CGA)5, respectively (Fig. 1 ▸ d). Also, previous thermodynamic measurements have demonstrated a significantly greater stability of the ps-duplex over the antiparallel hairpin form (Luteran et al., 2020 ▸). Therefore, it is not surprising that the significantly more stable ps-duplex form is dominant in crowded crystallization conditions where structural stability is advantageous. It may also thus be possible for d(CGA) ps-duplexes to form in crowded cellular environments, similar to other C–CH+-mediated DNA structures (Tang et al., 2020 ▸; Zeraati et al., 2018 ▸; Dzatko et al., 2018 ▸). Despite testing multiple constructs of d(CGA)-derived oligonucleotides, we were unable to determine a structure in the hairpin form.
Figure 1.

CD spectra of d(CGA)-containing oligonucleotides. The positive band at 265 nm and negative band at 245 nm are characteristic of the ps-duplex form (Luteran et al., 2020 ▸; Kejnovská et al., 2001 ▸; Robinson & Wang, 1993 ▸). The antiparallel form has a weak positive band at 280 nm and a weak negative band at 260 nm (Luteran et al., 2020 ▸; Kejnovská et al., 2001 ▸; Robinson & Wang, 1993 ▸). (a) CD spectrum of GA(CGA)5 at pH 5.5 (blue) or pH 7.0 (black). (b) CD spectrum of (CGA)5TGA at pH 5.5 (blue) or pH 7.0 (black). (c) (CGA)5 forms a ps-duplex at pH 5.0 (light blue) and an antiparallel hairpin at pH 6.6 (dark blue) in the same buffer conditions as in (a) and (b). The formation of the ps-duplex form at pH 6.6 is favored in the presence of 30% PEG 400 (dark gray), PEG 2000 (medium gray) and PEG 4000 (light gray). (d) Crowding agents increase the pH of the structural transition from ps-duplex to antiparallel hairpin form. The transition was measured as the loss of the characteristic ps-duplex signal at 265 nm in native conditions (solid lines) or in the presence of 30% PEG 2000 (dashed lines) for (CGA)5 (gray) and (CGA)5TGA (pink).
3.2. Crystal packing
In the GA(CGA)5 crystal structure, six strands form three parallel-stranded homoduplexes (duplexes 1, 2 and 3) in the asymmetric unit (Fig. 2 ▸ a). Duplex 2 is coaxially stacked between duplexes 1 and 3 though 3′–5′ end stacking of the terminal G1–G1 and A17–A17 base pairs. This arrangement results in a junction of three tandem sets of inter-strand G/A stacking interactions at each duplex intersection to stabilize the crystal lattice (Fig. 2 ▸ b). This packing arrangement forms columns of alternating ps-duplexes propagating throughout the crystal along the c axis. This is the first instance of 3′–5′ end stacking in this class of ps-duplexes; other ps-duplexes containing the d(CGA) motif stack in the 3′–3′ or 5′–5′ orientation (Tripathi & Paukstelis, 2016 ▸). This difference is likely to be due to the lack of 5′-C. The exposed 5′-G allows the preferential formation of inter-duplex G/A stacking interactions with the 3′-A of another duplex that directly mimics the internal inter-strand G/A stacking interactions. Similar symmetry-related duplexes can be used to extend the ps-duplex structure beyond 17 nucleotides, but the absence of the 5′-C in this sequence disrupts the internal consistency of the d(CGA) repeating unit. In the (CGA)5TGA structure, two strands form one homoduplex (duplex 4) in the asymmetric unit (Fig. 2 ▸ c). The duplex is stacked with crystallographically identical duplexes via 5′–5′ stacking of C1–C1 base pairs and 3′–3′ stacking of A18–A18 base pairs.
Figure 2.

Overview of the d(CGA)-based parallel-stranded homoduplexes. The PyMOL graphics software was used for all figures (DeLano, 2002 ▸). (a) The asymmetric unit for GA(CGA)5. The individual chains within each duplex (1–3) are labeled and colored accordingly. Duplex 1: chain A, cyan; chain B, blue. Duplex 2: chain C, green; chain D, yellow. Duplex 3: chain E, magenta; chain F, light pink. (b) 3′ to 5′ end stacking of duplex 1 and 2. The 3′ A17–A17 base pair of duplex 1 forms stacking interactions with the 5′ G1–G1 base pair of duplex 2 to form three tandem G/A stacking interactions. (c) The (CGA)5TGA asymmetric unit. Each chain within duplex 4 is labeled and colored accordingly: chain A (salmon), chain B (gray).
Both crystals grew in the presence of divalent cations, which primarily mediate inter-duplex crystal-packing interactions (Supplementary Fig. S1). When possible, anomalous difference maps and coordination distances were used to verify the cation identity and placement (Supplementary Fig. S2). GA(CGA)5 (duplexes 1–3) crystallized in the presence of hexamminecobalt(III) (NCO) and Sr2+. Specifically, NCO is positioned in multiple conformations between the Hoogsteen faces of guanosines from two duplexes, with GN7–GN7 and GO6–GO6 distances of 8.7 ± 0.3 and 7.6 ± 0.1 Å, respectively (Supplementary Fig. S1a ). Sr2+ mediates the remaining inter-duplex guanosine positions in two distinct modes. The first set of Sr2+-mediated interactions are similar to those of NCO but have shorter GN7–GN7 and GO6–GO6 distances (7.9 ± 0.1 and 6.8 ± 0.1 Å, respectively; Supplementary Fig. S1b ). The remaining Sr2+ cations are similarly positioned between two guanosines from separate ps-duplexes, but the major-groove faces are positioned such that GN7 and GO6 are oriented together (9.31 ± 0.03 Å; Supplementary Fig. S1c ). In the (CGA)5TGA structure Ba2+ mediates inter-duplex packing in two distinct environments. One mode is almost identical to the first set of Sr2+-mediated interactions, where the GN7–GN7 and GO6–GO6 distances are 7.8 ± 0.1 and 6.9 ± 0.0 Å, respectively (Supplementary Fig. S1d ). The remaining Ba2+ cations are positioned between the major-groove face of one guanosine and the phosphate O atom of the opposing duplex guanosine, where the GN7–PO2 and GN6–PO2 distances are 9.4 ± 1.3 and 8.9 ± 0.9 Å, respectively (Supplementary Fig. S1e ). Despite the different cations and unique packing interactions, the resulting ps-duplex structures were highly uniform.
3.3. d(CGA) ps-duplexes are structurally isomorphous and highly uniform
Although these structures were solved from individual crystals with different DNA sequences, solution conditions and crystal-packing arrangements, the resulting ps-duplex structures are nearly identical over the length of the tandem d(CGA) repeats (Fig. 3 ▸ a). The three duplexes from the GA(CGA)5 structure have r.m.s.d. values between 0.421 and 0.451 Å for 700 atoms (Fig. 3 ▸ b). Most of the structural deviation arises from subtle differences in the phosphate backbones, which is likely to result from solvent interactions that influence crystal packing (Fig. 3 ▸ c). Despite being crystallized in different conditions and containing the C16T substitution, duplex 4 is also highly similar to duplexes 1–3 (r.m.s.d. values of 0.846, 0.855 and 0.877 Å, respectively, for 698 atoms; Figs. 3 ▸ a and 3 ▸ b). The structural deviations associated with duplex 4 are primarily observed near the substitution site. The weaker electron density and correspondingly higher B factors observed from A12 to A18 in duplex 4 may also contribute to increased r.m.s.d. values, although the overall ps-duplex structure is maintained.
Figure 3.

d(CGA) triplets in the ps-duplex form are structurally isomorphous. (a) Overlay of duplexes 1–4 illustrating the robust structural uniformity of the ps-duplex form across different sequences, solution conditions and crystal-packing arrangements. Structural deviations are primarily observed surrounding the C16T substitution position in (CGA)5TGA. Nucleotides are colored as follows: C, purple; G, light purple; A, gray; T, green. The 5′ C–CH+ homo-base pair was omitted from (CGA)5TGA in this overlay for simplicity. (b) R.m.s.d. values from pairwise alignment of duplexes 1–4. All ps-duplexes are highly similar, with r.m.s.d. values below 1.0. (c) Overlay of all d(CGA) triplets from duplexes 1–4 rotated 180° to show the difference in deviation along the phosphate backbone from each strand (colored teal or gray). Minimal overall deviations demonstrate the high structural predictability of the ps-duplex form of the d(CGA) triplet.
We also compared the structures of isolated d(CGA) base-paired triplets from all three duplexes with triplets within each duplex (intra-duplex) or from other duplexes (inter-duplex). Not surprisingly, comparison of all individual d(CGA) triplets results in high similarity, as evident from the low r.m.s.d. of the full duplexes. Individual d(CGA) triplets at different positions in the same duplex [intra-duplex d(CGA) triplets] are almost identical (0.122–0.557 Å for 124 atoms; Supplementary Fig. S3), indicating that there are no position-specific structural features along the helical length. The 3′-end d(CGA) triplet of duplexes 1–3 and the 3′-most d(CGA) triplet of duplex 4 are the sources of the largest deviations among intra-duplex triplets (r.m.s.d. ranging from 0.634 to 0.881 Å for 124 atoms). This position in duplexes 1–3 is likely to be associated with greater deviations due to duplex end flexibility or crystal contact interactions, while deviations in duplex 4 are likely to be influenced by the structural changes induced by the adjacent d(TGA) triplet. Similarly low r.m.s.d. values are observed when comparing individual d(CGA) triplets from different duplexes (inter-duplex; Supplementary Fig. S4). In the inter-duplex comparison, d(CGA) triplets from within duplex 3 exhibited the largest range of r.m.s.d. values when compared with triplets from other duplexes (0.459–1.043 Å for 124 atoms). Overall, the low r.m.s.d. values in the comparison of tandem and individual d(CGA) triplets illustrates that the ps-duplex forms of d(CGA) repeat-containing sequences are structurally isomorphous, even in different environmental contexts.
3.4. d(CGA) helical and base-pair parameters
The high degree of similarity of the four unique ps-duplexes obtained from our crystal structures has allowed us to establish helical and base-pair parameters for this motif (Supplementary Figs. S5 and S6). Like other d(CGA) and d(TGA) homoduplex structures (Tripathi & Paukstelis, 2016 ▸; Tripathi et al., 2015 ▸), the ps-duplexes form right-handed helices that lack distinct major and minor grooves. The d(CGA) ps-duplex form requires 9.0 ± 0.1 base pairs to complete one helical turn, resulting in an average helical pitch of 32.2 ± 0.5 Å. The decreased helical pitch of the ps-duplex form, compared with B-DNA, is primarily a result of the large helical rise (5.0 ± 0.1 Å) and twist (84 ± 1°) associated with the inter-strand G/A base step. As previously observed (Tripathi et al., 2015 ▸), there is a notable difference in base-pair parameters between purine and pyrimidines in the ps-duplex. Purine nucleotides adopt larger shear, propeller, stretch and buckle angles to accommodate the hydrogen bonds while maintaining the duplex-stabilizing inter-strand G/A base-stacking interactions. We quantified the range of helical and base-pair parameters using 3DNA version 2.4 (Lu & Olson, 2003 ▸) along each nucleotide position to highlight the periodic fluctuation of each parameter along individual d(CGA) triplets throughout the entire duplex (Supplementary Figs. S5 and S6).
We observed distinct ranges of phosphate backbone torsion angles (α, β, γ) among individual strands within each duplex. Notably, one strand in each duplex, referred to as the ‘loose’ strand, adopts a wide range of α, β and γ torsion angles (246 ± 66°, 180 ± 36° and 91 ± 66°, respectively), while the opposing strand of the duplex, referred to as the ‘rigid’ strand, has a much narrower range (292 ± 5°, 164 ± 14° and 59 ± 32°, respectively; Supplementary Fig. S7). The ability to adopt a wide range of torsion angles suggests that one strand is generally more flexible than its partner strand and led us to adopt the nomenclature of loose and rigid, respectively. This also indicates that each strand within the ps-duplex is conformationally unique.
3.5. Structural asymmetry is induced by the C–CH+ base pair
We assessed the overall symmetry of the ps-duplex form to determine how structural asymmetries are propagated through the ps-duplex. The linearity of each duplex along the base-pair units was measured by connecting the midpoint of the hydrogen-bonding partners of each homo-base pair (Fig. 4 ▸ a). Each d(CGA) triplet exhibits a similar bending pattern centered around the largest deviation from linearity (25.0 ± 3.9°) at the C–CH+ base pair (Fig. 4 ▸ b). The G–G centric angle does not propagate significant deviations from linearity, while the magnitude of the A–A centric deviation is highly dependent upon the identity of the following nucleotide (C or T). When a C–CH+ base pair is present, the adjacent 5′-A–A centric angle adopts a deviation (20.8 ± 4.3°) similar to the C–CH+ centric angle. Alternatively, the A–A centric deviation is smaller (9.0°) when followed by a T–T base pair (Fig. 4 ▸ b). Structural overlays of the A–(C/T) step indicate that this deviation coincides with the extension of one cytosine from the helical axis to align the Watson–Crick faces for the formation of the hemiprotonated C–CH+ base pair (Fig. 4 ▸ c), as previously noted (Tripathi & Paukstelis, 2016 ▸). There is also a slight displacement of the adjacent adenosine on the same strand which could be required to accommodate the cytosine deviation. This contrasts with the T–T base pair, which makes interactions in a perfectly symmetrical manner; therefore, the adjacent adenosine also remains unbent. We conclude that the A–A base pair provides structural flexibility to accommodate deviations from linearity induced by the C–CH+ base pair.
Figure 4.

Parallel-stranded homoduplex asymmetry. (a) Deviations from linearity along base pairs of ps-duplex 1. Left: the light gray cylinder represents the helical axis. Right: the deviation from linearity of a base pair is measured as the angle between the two cylinders adjacent to the base pair of interest. Individual cylinders were created by connecting points placed at the midpoint of the hydrogen-bonding partners of each base pair. The resulting cylinders are colored based on the identity of the base pairs they connect: G–A, gray; A–C, black; C–G, blue. (b) Deviation from linearity of each base pair along the (CGA)5TGA (purple) or GA(CGA)5 (red) sequence. Colored bars along the sequence correspond to the same cylinders connecting base pairs from (a). The angles measured for GA(CGA)5 are represented as the average of duplexes 1–3 and (CGA)5TGA is from duplex 4. (c) Overlay of d(YGA) triplets within (CGA)5TGA, rotated 180° to highlight the A–A and C–C base pairs. The loose (gray) and rigid (teal) strands of five A/C steps overlaid with one A/T step (black). Compared with the black strand, the teal strand does not show significant structural deviation, while the nucleotides within the gray strand are extended out of the helical axis.
3.6. Each strand within the ps-duplex has unique structural character
Backbone torsion-angle analysis and the duplex asymmetry suggested that the two strands of the ps-duplex have unique structural characteristics. These differences are correlated with two distinct hydrogen-bond interactions that form within the A/C step between d(CGA) triplets (Fig. 5 ▸ a). The first hydrogen bond is between cytosine N4 (C-N4) and a nonbridging phosphate oxygen (O2P) of the previous adenosine within the same strand. There is no bond equivalent to the C-N4 to O2P bond in the T–T base pair, further suggesting that this bond could be influential in controlling the relative position of the C–CH+ and A–A base pairs. The second hydrogen bond is between the same nonbridging phosphate O atom and the adenosine N6 (A-N6) of the opposing strand. Interestingly, depending on the strand within each duplex, there are unique differences in the A-N6 to O2P and C-N4 to O2P bond lengths (Fig. 5 ▸ b). In the rigid strand, all A-N6 to O2P and C-N4 to O2P bonds distances remain within 2.8–3.1 Å. However, the same bond lengths within the loose strand increase beyond a hydrogen-bond distance. The average A-N6 to O2P and C-N4 to O2P distances within the loose strand are 4.1 ± 0.4 and 3.5 ± 0.1 Å, respectively. The cytosine that is displaced from the helical axis is always on the loose strand, where the increased bond lengths and wider range of torsion angles within the loose strand coincide with this displacement.
Figure 5.

Hydrogen-bond distances and base-overlap areas are used to distinguish loose and rigid strands within the d(CGA) ps-duplex. (a) The A/C step highlighting the A-N6 to O2P and C-N4 to O2P interactions within loose (gray) and rigid (teal) strands. Chain A (duplexes 1 and 4), chain C (duplex 2) and chain E (duplex 3) have been characterized as loose strands. Chain B (duplexes 1 and 4), chain D (duplex 2) and chain F (duplex 3) have been characterized as rigid strands. (b) Loose (gray) and rigid (teal) strand bond distances represented along the GA(CGA)5 sequence. A-N6 to O2P distances are plotted as circles and C-N4 to O2P distances are plotted as diamonds. Each data point represents the average distance measured from duplexes 1 to 3. Loose-strand bond distances cycle between 3.5 ± 0.1 and 4.1 ± 0.1 Å depending on the identity of the nucleotide involved in the interaction, while rigid-strand bond distances remain between 2.8 and 3.1 Å regardless of the interaction. (c) Base-overlap areas are different for loose and rigid strands. View of all unique base-stacking interactions (inter-strand G/A, intra-strand A/C and intra-strand C/G) that contribute to d(CGA) triplet stabilization. The 90° rotation illustrates the difference in stacking-overlap area between strands. The rigid strand (teal) maintains consistent stacking-overlap areas, while the loose strand (gray) is highly variable. The star denotes the cytosine that is extended from the helical axis. (d) Base-stack overlap areas are represented as averages of overlap areas from d(CGA) triplets from duplexes 1 to 4 and are shown for the respective base steps.
Accompanying the differences in hydrogen bonding are distinct differences in base-stacking interactions between the loose and rigid strands (Fig. 5 ▸ c). The base-pair overlap areas (excluding exocyclic groups) calculated for each duplex using 3DNA version 2.4 (Lu & Olson, 2003 ▸) indicate that intra-rigid-strand A/C and C/G steps maintain similar overlap areas of 2.4 ± 0.5 and 2.3 ± 0.4 Å2, respectively (Fig. 5 ▸ d). The inter-strand G/A stacking interaction adjacent to the A/C step on the rigid strand also has a similar overlap area of 2.8 ± 0.9 Å2 (Fig. 5 ▸ d). However, the stacking areas of the loose strand are more variable. The A/C step on the loose strand has the lowest base-overlap area (0.4 ± 0.1 Å2), while the G/A (inter-strand) and C/G (intra-strand) stacking interactions surrounding the A/C step have the highest stacking overlap areas (4.6 ± 0.4 and 4.0 ± 0.2 Å2, respectively; Fig. 5 ▸ d). The large stacking interactions surrounding the bent A/C step within the loose strand contribute additional stabilization that may compensate for the increased base-to-phosphate hydrogen-bond distances.
The overall structural asymmetry and accompanying differences in hydrogen-bonding and base-stacking interactions among strands are observed throughout each ps-duplex studied. Although it would be conceivable to expect the structural asymmetry to be propagated on a per-triplet basis, we observed propagation on a per-strand basis over the entire length of the d(CGA) repeats. Thus, each ps-duplex is composed of two structurally unique strands where all triplets within a strand adopt either a loose or rigid character. The structural homogeneity of triplets within strands implies that duplexation of tandem d(CGA) triplets could occur in a cooperative manner. Further, the distinct conformations of each strand could play separate roles in accommodating the structural asymmetry. The rigid strand is the structural scaffold strand that maintains consistent hydrogen-bonding and stacking interactions, while the loose strand provides structural flexibility to stabilize and accommodate deviations from linearity induced by the C–CH+ base pair.
3.7. d(YGA) triplets are structurally compatible but not identical
It has been hypothesized that d(TGA) triplets could be useful discriminators in the programmable pairing of long stretches of d(CGA) triplets based on the slight structural and thermodynamic deviations incurred by the 5′-nucleotide (Luteran et al., 2020 ▸). Previous crystal structures have reported differences in d(CGA) (Tripathi & Paukstelis, 2016 ▸) and d(TGA) (Tripathi et al., 2015 ▸) triplets from separate sequence contexts, but have not yet examined the structural compatibility when d(YGA) triplets are present within the same sequence. The crystal structure of (CGA)5TGA has allowed us to evaluate the structural compatibility of d(CGA) and d(TGA) triplets within the same DNA sequence. We observed that the incorporation of a 3′-d(TGA) triplet significantly alters the bond distances of loose and rigid strands in upstream d(CGA) triplets. Within the d(CGA) triplet directly adjacent to the d(TGA) triplet, the rigid strand C-N4 to OP2 and A-N6 to OP2 bond distances increase from an average of 3.0 Å to 4.4 and 4.9 Å, respectively, while the loose-strand A-N6 to OP2 distance decreases from 4.0 to 3.5 Å (Supplementary Fig. S8a ). The C1′–C1′ distance for the T–T homo-base pair is 1.4 Å larger than the C–CH+ homo-base pair; therefore, upstream swelling of the rigid strand could be required to accommodate the wider T–T homo-base pair (Supplementary Fig. S8b ). Increased base-overlap areas of the G/A steps adjacent to the d(TGA) triplet could also contribute additional stabilization to compensate for the extended rigid-strand bond distances (Supplementary Figs. S8c and S8d ). The enthalpic destabilization observed in sequences containing d(TGA) triplets was previously attributed to the loss of one hydrogen bond on replacing the hemiprotonated C–CH+ with a T–T base pair (Luteran et al., 2020 ▸). The structure described here further suggests that this destabilization could also be due to the loss of the C-N4 to O2P hydrogen bond and swelling of adjacent d(CGA) triplets that coincides with the addition of a T–T base pair. The incorporation of a d(TGA) triplet at the 3′-end of a long stretch of d(CGA) triplets does not disrupt the overall ps-duplex structure, but induces slight structural changes in the adjacent d(CGA) triplet.
Although d(CGA) and d(TGA) triplets are not structurally identical within the ps-duplex, they could be used to control rigid and loose strands. 5-Br-UGA triplets have been shown to offer increased stability to the ps-duplex via the formation of a halogen bond with the phosphate O atom of an adjacent adenosine (Tripathi et al., 2015 ▸). To fully evaluate their potential use as discriminator triplets, structural analysis of d(CGA)-based repeat sequences containing internal d(TGA) triplets (and 5-Br-UGA triplets) at different positions are needed to understand their impact on adjacent d(CGA) triplets and the ps-duplex structure as a whole.
4. Concluding remarks
The crystal structures described here have allowed us to characterize the d(CGA) triplet-repeat motif in the ps-duplex form and establish structural features for its use as a building block in DNA nanotechnology applications. The generalized helical and base parameters established by these structures will serve as constraints for the incorporation of d(CGA)-based triplets into rational structure design. Particularly, the requirement of an integer number of base pairs per turn (9.0 ± 0.1 base pairs) simplifies its use from a design perspective, as the incorporation of three d(CGA) repeats completes exactly one helical turn. Consistent with previous d(CGA) base-paired triplets, we observed a structural asymmetry that is propagated throughout each duplex. These crystal structures containing multiple tandem d(CGA) triplets have demonstrated that this asymmetry is propagated on a per-strand basis, where all triplets within each strand adopt a specific conformation and play a unique role in accommodating the asymmetry. The rigid strand serves as the structural scaffold that maintains hydrogen-bonding and stacking interactions, while the loose strand provides structural flexibility to stabilize and accommodate deviations from linearity induced by the C–CH+ base pair. The distinct structural character between triplets within rigid and loose strands could be a useful tool in the structural programming of DNA-based architectures, where specific control of each strand within the duplex may be desirable. The structural similarity of tandem and individual d(CGA) base-paired triplets obtained from different solution environments demonstrates that the ps-duplex form is a robust and highly predictable structure. This strongly suggests that the d(CGA) motif can be used to reliably integrate the ps-duplex form into nanostructures. This motif has the added benefit of allowing conditional control of the ps-duplex form in solution through mild fluctuations in pH or the addition of crowding agents.
Although the formation of stable alternative DNA structures may be desirable for the rational design of DNA-based architectures, they may be unfavorable or selected against in biological systems. Repeat sequences that form alternative structures may undergo greater instability due to the challenges that they present to the replication machinery (Khristich & Mirkin, 2020 ▸). Additionally, there is also the possibility that readily formed, thermodynamically stable structures would be selected against evolutionarily, as they may have an even greater impact on endogenous replication systems. The mechanism by which ps-duplex structures such as those described here might form in a biological context is not immediately clear. Intramolecular ps-duplexes would require some form of looping from single-stranded regions to obtain the parallel-stranded orientation, while intermolecular contacts would likely require association from two single-stranded regions (Tchurikov et al., 1989 ▸). The formation of ps-duplex structures is possible during DNA replication, in which long strands of ssDNA are formed during lagging-strand synthesis. This is a commonly recognized mechanism for the expansion of trinucleotide-repeat sequences associated with hereditary diseases, where the formation of alternative structures (i.e. hairpins) results in template–strand misalignment (Wang & Vasquez, 2017 ▸; Mirkin & Smirnova, 2002 ▸). The subsequent resumption of DNA synthesis from the misaligned template results in repeat expansion. Alternatively, one intriguing possibility could be the formation of RNA/DNA hybrid ps-duplexes from transcriptionally active regions, in which nascent RNA triplet product and DNA triplet sense strands are nominally parallel and single-stranded. Such a hybrid structure would likely not be identical to the ps-duplex structures presented here, as the 2′-OH of the cytosines would make close contacts with the adjacent guanosine sugars.
To this point, d(CGA) triplet-repeat sequences have not been implicated in human disease and are found least frequently in eukaryotic genomes (Kozlowski et al., 2010 ▸; Astolfi et al., 2003 ▸). This raises the interesting possibility that their propensity to form highly stable ps-duplex structures could be a significant factor contributing to the underrepresentation of the d(CGA) triplet repeat in eukaryotic genomes. Although questions have arisen about the likelihood of C–CH+-dependent structures forming in vivo, mounting evidence, including the data presented here, suggests that such structures can form at near-neutral pH under crowding conditions.
5. Data availability
Atomic coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank under accession codes 7sb8 and 7t6y.
Supplementary Material
PDB reference: d[GA(CGA)5], 7sb8
PDB reference: d[(CGA)5TGA], 7t6y
Supplementary Figures. DOI: 10.1107/S2059798322000304/rr5217sup1.pdf
Acknowledgments
This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The EIGER 16M detector on 24-ID-E is funded by an NIH–ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a US Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
References
- Abou Assi, H., Garavís, M., González, C. & Damha, M. J. (2018). Nucleic Acids Res. 46, 8038–8056. [DOI] [PMC free article] [PubMed]
- Astolfi, P., Bellizzi, D. & Sgaramella, V. (2003). Gene, 317, 117–125. [DOI] [PubMed]
- Benabou, S., Mazzini, S., Aviñó, A., Eritja, R. & Gargallo, R. (2019). Sci. Rep. 9, 15807. [DOI] [PMC free article] [PubMed]
- Bourdoncle, A., Estévez Torres, A., Gosse, C., Lacroix, L., Vekhoff, P., Le Saux, T., Jullien, L. & Mergny, J.-L. (2006). J. Am. Chem. Soc. 128, 11094–11105. [DOI] [PubMed]
- Chakraborty, S., Sharma, S., Maiti, P. K. & Krishnan, Y. (2009). Nucleic Acids Res. 37, 2810–2817. [DOI] [PMC free article] [PubMed]
- Chen, X., Karpenko, A. & Lopez-Acevedo, O. (2017). ACS Omega, 2, 7343–7348. [DOI] [PMC free article] [PubMed]
- Cristofari, C., Rigo, R., Greco, M. L., Ghezzo, M. & Sissi, C. (2019). Sci. Rep. 9, 1210. [DOI] [PMC free article] [PubMed]
- Cui, J., Waltman, P., Le, V. H. & Lewis, E. A. (2013). Molecules, 18, 12751–12767. [DOI] [PMC free article] [PubMed]
- Delano, W. L. (2002). PyMOL. http://www.pymol.org.
- Dzatko, S., Krafcikova, M., Hänsel-Hertsch, R., Fessl, T., Fiala, R., Loja, T., Krafcik, D., Mergny, J.-L., Foldynova-Trantirkova, S. & Trantirek, L. (2018). Angew. Chem. Int. Ed. 57, 2165–2169. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Evans, P. R. & Murshudov, G. N. (2013). Acta Cryst. D69, 1204–1214. [DOI] [PMC free article] [PubMed]
- Franklin, R. E. & Gosling, R. G. (1953). Nature, 172, 156–157. [DOI] [PubMed]
- Han, X., Zhou, Z., Yang, F. & Deng, Z. (2008). J. Am. Chem. Soc. 130, 14414–14415. [DOI] [PubMed]
- Jung, Y. H., Lee, K.-B., Kim, Y.-G. & Choi, I. S. (2006). Angew. Chem. 118, 6106–6109.
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kejnovská, I., Tůmová, M. & Vorlíčková, M. (2001). Biochim. Biophys. Acta, 1527, 73–80. [DOI] [PubMed]
- Kettani, A., Bouaziz, S., Skripkin, E., Majumdar, A., Wang, W., Jones, R. A. & Patel, D. J. (1999). Structure, 7, 803–815. [DOI] [PubMed]
- Khristich, A. N. & Mirkin, S. M. (2020). J. Biol. Chem. 295, 4134–4170. [DOI] [PMC free article] [PubMed]
- Kobuna, T., Sunami, T., Kondo, J. & Takenaka, A. (2002). Nucleic Acids Symp. Ser. 2, 179–180. [DOI] [PubMed]
- Kozlowski, P., de Mezer, M. & Krzyzosiak, W. J. (2010). Nucleic Acids Res. 38, 4027–4039. [DOI] [PMC free article] [PubMed]
- Lahue, R. S. (2020). Neuronal Signal. 4, NS20200010. [DOI] [PMC free article] [PubMed]
- Largy, E., Marchand, A., Amrane, S., Gabelica, V. & Mergny, J.-L. (2016). J. Am. Chem. Soc. 138, 2780–2792. [DOI] [PubMed]
- Leslie, A. G. W. & Powell, H. R. (2007). Evolving Methods for Macromolecular Crystallography, edited by R. J. Read & J. L. Sussman, pp. 41–51. Dordrecht: Springer.
- Li, Z. & Mirkin, C. A. (2005). J. Am. Chem. Soc. 127, 11568–11569. [DOI] [PubMed]
- Liebschner, D., Afonine, P. V., Baker, M. L., Bunkóczi, G., Chen, V. B., Croll, T. I., Hintze, B., Hung, L.-W., Jain, S., McCoy, A. J., Moriarty, N. W., Oeffner, R. D., Poon, B. K., Prisant, M. G., Read, R. J., Richardson, J. S., Richardson, D. C., Sammito, M. D., Sobolev, O. V., Stockwell, D. H., Terwilliger, T. C., Urzhumtsev, A. G., Videau, L. L., Williams, C. J. & Adams, P. D. (2019). Acta Cryst. D75, 861–877.
- Liedl, T. & Simmel, F. C. (2005). Nano Lett. 5, 1894–1898. [DOI] [PubMed]
- Liu, Z. & Mao, C. (2014). Chem. Commun. 50, 8239–8241. [DOI] [PubMed]
- Lu, X.-J. & Olson, W. K. (2003). Nucleic Acids Res. 31, 5108–5121. [DOI] [PMC free article] [PubMed]
- Luteran, E. M., Kahn, J. D. & Paukstelis, P. J. (2020). Biophys. J. 119, 1580–1589. [DOI] [PMC free article] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Mirkin, S. M. & Smirnova, E. V. (2002). Nat. Genet. 31, 5–6. [DOI] [PubMed]
- Nesterova, I. V. & Nesterov, E. E. (2014). J. Am. Chem. Soc. 136, 8843–8846. [DOI] [PubMed]
- Nguyen, T., Fraire, C. & Sheardy, R. D. (2017). J. Phys. Chem. B, 121, 7872–7877. [DOI] [PubMed]
- Paiva, A. M. & Sheardy, R. D. (2004). Biochemistry, 43, 14218–14227. [DOI] [PubMed]
- Poggi, L. & Richard, G.-F. (2021). Microbiol. Mol. Biol. Rev. 85, 1–24. [DOI] [PMC free article] [PubMed]
- Rajendran, A., Nakano, S. & Sugimoto, N. (2010). Chem. Commun. 46, 1299–1301. [DOI] [PubMed]
- Rippe, K., Fritsch, V., Westhof, E. & Jovin, T. M. (1992). EMBO J. 11, 3777–3786. [DOI] [PMC free article] [PubMed]
- Robinson, H., van Boom, J. H. & Wang, A. H.-J. (1994). J. Am. Chem. Soc. 116, 1565–1566.
- Robinson, H., van der Marel, G. A., van Boom, J. H. & Wang, A. H.-J. (1992). Biochemistry, 31, 10510–10517. [DOI] [PubMed]
- Robinson, H. & Wang, A. H.-J. (1993). Proc. Natl Acad. Sci. USA, 90, 5224–5228. [DOI] [PMC free article] [PubMed]
- Sannohe, Y. & Sugiyama, H. (2012). Bioorg. Med. Chem. 20, 2030–2034. [DOI] [PubMed]
- Saoji, M. & Paukstelis, P. J. (2015). Acta Cryst. D71, 2471–2478. [DOI] [PMC free article] [PubMed]
- Shu, W., Liu, D., Watari, M., Riener, C. K., Strunz, T., Welland, M. E., Balasubramanian, S. & McKendry, R. A. (2005). J. Am. Chem. Soc. 127, 17054–17060. [DOI] [PubMed]
- Song, L., Ho, V. H. B., Chen, C., Yang, Z., Liu, D., Chen, R. & Zhou, D. (2013). Adv. Healthc. Mater. 2, 275–280. [DOI] [PubMed]
- Spiegel, J., Adhikari, S. & Balasubramanian, S. (2020). Trends Chem. 2, 123–136. [DOI] [PMC free article] [PubMed]
- Srivastava, S., Fukuto, M. & Gang, O. (2018). Soft Matter, 14, 3929–3934. [DOI] [PubMed]
- Sunami, T., Kondo, J., Kobuna, T., Hirao, I., Watanabe, K., Miura, K. & Takénaka, A. (2002). Nucleic Acids Res. 30, 5253–5260. [DOI] [PMC free article] [PubMed]
- Tang, W., Niu, K., Yu, G., Jin, Y., Zhang, X., Peng, Y., Chen, S., Deng, H., Li, S., Wang, J., Song, Q. & Feng, Q. (2020). Epigenetics Chromatin, 13, 12. [DOI] [PMC free article] [PubMed]
- Tateishi-Karimata, H. & Sugimoto, N. (2021). Nucleic Acids Res. 49, 7839–7855. [DOI] [PMC free article] [PubMed]
- Tchurikov, N. A., Chernov, B. K., Golova, Y. B. & Nechipurenko, Y. D. (1989). FEBS Lett. 257, 415–418. [DOI] [PubMed]
- Tripathi, S. & Paukstelis, P. J. (2016). ChemBioChem, 17, 1177–1183. [DOI] [PubMed]
- Tripathi, S., Zhang, D. & Paukstelis, P. J. (2015). Nucleic Acids Res. 43, 1937–1944. [DOI] [PMC free article] [PubMed]
- Völker, J., Makube, N., Plum, G. E., Klump, H. H. & Breslauer, K. J. (2002). Proc. Natl Acad. Sci. USA, 99, 14700–14705. [DOI] [PMC free article] [PubMed]
- Wang, A. H.-J., Quigley, G. J., Kolpak, F. J., Crawford, J. L., van Boom, J. H., van der Marel, G. & Rich, A. (1979). Nature, 282, 680–686. [DOI] [PubMed]
- Wang, G. & Vasquez, K. (2017). Genes, 8, 17. [DOI] [PMC free article] [PubMed]
- Wang, Y. & Patel, D. J. (1994). J. Mol. Biol. 242, 508–526. [PubMed]
- Watson, J. D. & Crick, F. H. C. (1953). Nature, 171, 737–738. [DOI] [PubMed]
- Weiss, M. S. & Hilgenfeld, R. (1997). J. Appl. Cryst. 30, 203–205.
- Wells, R. D. (1996). J. Biol. Chem. 271, 2875–2878. [DOI] [PubMed]
- Wells, R. D. (2007). Trends Biochem. Sci. 32, 271–278. [DOI] [PubMed]
- Yu, K.-K., Tseng, W.-B., Wu, M.-J., Alagarsamy, A. S. K. K., Tseng, W.-L. & Lin, P.-C. (2018). Sens. Actuators B Chem. 273, 681–688.
- Zeraati, M., Langley, D. B., Schofield, P., Moye, A. L., Rouet, R., Hughes, W. E., Bryan, T. M., Dinger, M. E. & Christ, D. (2018). Nat. Chem. 10, 631–637. [DOI] [PubMed]
- Zheng, M., Huang, X., Smith, G. K., Yang, X. & Gao, X. (1996). J. Mol. Biol. 264, 323–336. [DOI] [PubMed]
- Zhou, W., Liang, W., Li, X., Chai, Y., Yuan, R. & Xiang, Y. (2015). Nanoscale, 7, 9055–9061. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: d[GA(CGA)5], 7sb8
PDB reference: d[(CGA)5TGA], 7t6y
Supplementary Figures. DOI: 10.1107/S2059798322000304/rr5217sup1.pdf
Data Availability Statement
Atomic coordinates and structure factors for the reported crystal structures have been deposited in the Protein Data Bank under accession codes 7sb8 and 7t6y.