

Abstract

Based on the Interacting Quantum Atoms approach, we present herein a conceptual and theoretical framework of short-range electrostatic interactions, whose accurate description is still a challenging problem in molecular modeling. For all the noncovalent complexes in the S66 database, the fragment-based and atomic decomposition of the electrostatic binding energies is performed using both the charge density of the dimers and the unrelaxed densities of the monomers. This energy decomposition together with dispersion corrections gives rise to a pairwise approximation to the total binding energy. It also provides energetic descriptors at varying distance that directly address the atomic and molecular electrostatic interactions as described by point-charge or multipole-based potentials. Additionally, we propose a consistent definition of the charge penetration energy within quantum chemical topology, which is mainly characterized in terms of the intramolecular electrostatic energy. Finally, we discuss some practical implications of our results for the design and validation of electrostatic potentials.
1. Introduction
Electrostatic interactions are central to molecular modeling because of their slow decay and strength. Especially when polar atoms or charged species are involved, they largely determine the stability and activity of biomolecules such as proteins, nucleic acids, or lipids, among others.1,2 As such, a reliable description in molecular mechanics (MM) potentials is essential both in the short- and in the long-range.
Within the framework of MM methods, interactions comprising nonbonded atoms are usually represented by pairwise potentials such as the Lennard-Jones and the Coulomb ones. In the latter case, the use of point charges or higher order multipoles to avoid the integration of interacting charge densities has resulted in accurate electrostatics at long-range, with significant improvements to speed up and facilitate convergence such as the Ewald summation and its variants to perform, for example, molecular simulations in solution under periodic boundary conditions.3−8 At short-range, however, the approximations taken for long distances become less accurate or invalid,9 and a correct electrostatic description in this regime stills poses a challenge. Hence, there is a growing interest in improving short-range electrostatics (e.g., for troublesome hydrogen bonds), mainly focused on capturing the effects associated with the non-negligible interpenetration of densities, leading to the so-called charge penetration (CP) energy, which is typically defined as the difference between the electrostatic energy computed from continuous charge density distributions and that from multipolar approximations.10 Thus, several investigations have been devoted in the last years to incorporate the charge penetration energy into the MM electrostatic potentials.10−14
The separation of various energy terms as implemented in the MM potentials is somehow paralleled by the energy decomposition analysis (EDA) methods.15 A major goal of any EDA approach is to ascertain the nature and type of the interactions among molecules as well as to rationalize their stabilizing or destabilizing roles, which may have implications for the design, parametrization, and validation of MM potentials such as the electrostatic ones. However, there is no unique recipe to decompose the energy, and thus many EDAs have been developed rooted in different approaches. Hence, symmetry-adapted perturbation theory (SAPT) makes use of a perturbative approach to differentiate the distinct nature of the intermolecular interactions,16,17 while orbital-based EDAs exploit a stepped scheme to calculate the different energies according to some reference electronic states,18−20 and the interacting quantum atoms (IQA) method relies on a real space partition of the quantum mechanical (QM) density matrices,21,22 being thus classified as a quantum chemical topology (QCT) method.
According to recent studies, in spite of their crude approximations, it may be feasible to improve the classical MM potentials by utilizing the information provided by EDAs.23 More specifically, it has been shown that the SAPT energy components (electrostatics, induction, exchange-repulsion, and dispersion) can be modeled with relatively simple MM functions.24,25 In particular, it has been demonstrated that the combination of empirical damping functions accounting for the CP energy with point multipoles results in electrostatic energies at short-range that are quite close to the SAPT ones. Actually, the SAPT electrostatic energy provides the required reference to parametrize and validate the CP-augmented potentials. However, different interpretations of short-range energetic effects involving the overlap of the electron densities of two or more fragments may be possible depending on the particular EDA of choice.15 As such, other schemes such as the absolutely localized molecular orbital (ALMO) EDA, that relies on a different nonperturbative decomposition of energy terms, have also been proposed.26 In this work, we reexamine the nature of electrostatic interactions under the prism of an orbital-invariant, reference-free technique. The IQA approach fulfills these requirements as it is a QCT, real-space energy decomposition resorting to the partition of the reduced density matrices (RDMs). IQA distinguishes not only between electrostatic or exchange-correlation components of the interaction energy but also between intra- or interatomic (or fragment) contributions. Moreover, since IQA splits the total energy of a system and not only the interaction between selected fragments, it is capable of reconstructing (or dissecting) the energy ascribed to both covalent and noncovalent binding, allowing thus covalent bond energies to be characterized27 as well as the accuracy of the energy components handled by QM fragment methods to be investigated.28
Herein, we study in detail the electrostatic interactions involved in noncovalent complexes with a twofold goal. On the one hand, we aim to compare in a consistent and systematic manner the atomic and fragment contributions to the electrostatic energy as evaluated throughout a hierarchy of QM and MM approximations and at varying intermolecular distances. In this way, we seek to identify the best correspondence between the IQA and the MM electrostatic terms. On the other hand, we critically examine the CP concept and propose a novel definition relying on a joint orbital and real-space decomposition scheme, which can give new insight into the CP energy. To help fulfill these goals, the rest of the manuscript is structured as follows. First, we present and describe the theoretical scaffold that holds our work, paying particular attention to the IQA—and its IQF variant—energy decomposition, followed by subsections concerning the zeroth-order approximation, the electrostatic MM potentials, and a final assessment of the CP energy and the alternative definition proposed in this work. Subsequently, we describe some computational settings and the results of our test calculations, which were carried out on the S66 and S66x8 data sets.29,30 The various levels of description of the electrostatic interactions are then discussed based on their statistical correlation with benchmark data, their dependence with the intermolecular separation, etc. The QM and IQA calculations yield further information, not only about the magnitude of the CP energy, but more importantly, about its different role in the IQA descriptors. Finally, we conclude that the aim of improving the electrostatic description is essentially fulfilled at the expense of accounting for intramolecular effects.
2. Theory and Methods
2.1. IQA Decomposition of QM Energies
The interacting quantum atoms method is a robust and physically sound approach to decompose the total QM energy of a system into chemically meaningful components.21,22 It is based on partitioning the first- and second-order RDMs, as can be done with the real space partition proposed by Bader and co-workers within their Quantum Theory of Atoms in Molecules (QTAIM).31 Thus, the three-dimensional space is decomposed into atomic regions (ΩI) as the attraction basins of the gradient field of the electron density.
Given a global energy E of a system, IQA permits its decomposition into atomic components and pair interaction energies according to
| 1 |
where  is called the net atomic energy
and, under
the Born–Oppenheimer approximation, represents the energy due
to the kinetic energy of electrons plus all the interactions involved
(i.e., electron–electron and electron–nucleus) inside
the atomic basin of each atom I. Similarly, each
is called the net atomic energy
and, under
the Born–Oppenheimer approximation, represents the energy due
to the kinetic energy of electrons plus all the interactions involved
(i.e., electron–electron and electron–nucleus) inside
the atomic basin of each atom I. Similarly, each 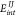 term comprises the
interaction energy between
the electrons (e) and nucleus (n) located in atom I with those ascribed to other atoms J, which can
be separated into n–e, e–e, and n–n contributions.
term comprises the
interaction energy between
the electrons (e) and nucleus (n) located in atom I with those ascribed to other atoms J, which can
be separated into n–e, e–e, and n–n contributions.
In order to compute the potential energy, the pair density ρ2(r1, r2) is
required. This object can be split according to ρ2(r1, r2) = ρ(r1)ρ(r2) + ρxc(r1, r2) in two contributions. On the one hand, ρ(r1)ρ(r2) represents
a noncorrelated product of densities, whereas electron correlation
is accounted for by the exchange-correlation (xc) density ρxc(r1, r2). Accordingly, the total interaction energy between
two atoms can be decomposed into a Coulomb or electrostatic term 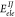 and a quantum mechanical
exchange-correlation
one
and a quantum mechanical
exchange-correlation
one 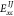 :
:
| 2 |
the latter term comprising
all the associated
QM effects that other (e.g., perturbative) approaches identify separately
as dispersion, charge-transfer, polarization, etc. However, such a
decomposition of 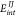 into two terms is
particularly relevant
when assessing the nature of a given bond or interaction, since the
electrostatic term is associated with ionicity and the exchange-correlation
contribution with covalency.22
into two terms is
particularly relevant
when assessing the nature of a given bond or interaction, since the
electrostatic term is associated with ionicity and the exchange-correlation
contribution with covalency.22
IQA admits the grouping of atomic terms into fragment contributions (e.g., functional groups, molecules). Thus, a fragment decomposition similar to eq 1 of a molecular aggregate constituted by two moieties A and B involves
| 3 |
| 4 |
where 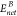 can be calculated analogously.
For practical
purposes, we use the IQA acronym to refer to the atomic analysis,
whereas for its fragment version the term interacting quantum fragments
(IQF) is preferred.
can be calculated analogously.
For practical
purposes, we use the IQA acronym to refer to the atomic analysis,
whereas for its fragment version the term interacting quantum fragments
(IQF) is preferred.
In a previous work,32 it was shown that IQF may be useful to dissect the formation energy of noncovalent complexes. Moreover, the IQA/IQF terms can be augmented with Grimme’s D3 dispersion correction33 as combined with the Becke-Johnson damping function34 to complement the DFT and HF descriptions. Using the IQF-D3 protocol, the formation energy of a two-fragment system AB given by the process A + B → A···B is split as
| 5 |
The deformation term 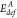 (
(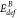 ) in the above equation corresponds
to the
net energy variation
) in the above equation corresponds
to the
net energy variation 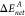 (
(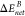 ) of fragment A (B), whereas the interfragment interaction energy
) of fragment A (B), whereas the interfragment interaction energy 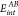 comprises the electrostatic
(
comprises the electrostatic
(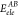 ), exchange-correlation
), exchange-correlation 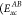 ), and dispersion (
), and dispersion (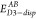 ) energies between
the two fragments, the
latter being thus separated from the whole exchange-correlation one.
Overall, the contribution of electrostatics and exchange-correlation
to ΔEform is split
between the intrafragment deformation and the interfragment interactions.
) energies between
the two fragments, the
latter being thus separated from the whole exchange-correlation one.
Overall, the contribution of electrostatics and exchange-correlation
to ΔEform is split
between the intrafragment deformation and the interfragment interactions.
2.2. Electrostatic Energy from Continuous Charge Densities
The purely electrostatic energy for a given system
with total charge density ρ(r) (ρ(r) ≡ ρtot(r)
= 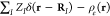 , including both the electron density
ρe(r) and the nuclear
charges ZI at positions RI) is readily computed using the Coulomb
law,
, including both the electron density
ρe(r) and the nuclear
charges ZI at positions RI) is readily computed using the Coulomb
law,
| 6 |
where, for the sake of simplicity, the electrostatic potential in this and the rest of the equations is expressed in atomic units. Interestingly, the QTAIM real space partition derived from ρe(r) allows us to decompose the electrostatic energy at the atomic level,
 |
7 |
Similarly, the fragment-based decomposition can be readily accomplished in an analogous way, allowing thus the specific assessment of the electrostatic component of the formation energy ΔEele of a two-fragment system AB as
| 8 |
where ΔEele is expressed in terms of two contributions, namely,
the intrafragment variations of electrostatic energy in the formation
process,  and
and 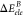 , and the interfragment electrostatic interaction,
, and the interfragment electrostatic interaction, 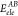 . At this point, we
note that although ΔEele is commonly termed as a
classical electrostatic interaction energy, we will refer to it as
the electrostatic contribution to the formation energy of the A···B
complex in order to help avoid confusions with the IQA/IQF interaction
energy terms. When the charge density is constructed from the unrelaxed
fragment densities as
. At this point, we
note that although ΔEele is commonly termed as a
classical electrostatic interaction energy, we will refer to it as
the electrostatic contribution to the formation energy of the A···B
complex in order to help avoid confusions with the IQA/IQF interaction
energy terms. When the charge density is constructed from the unrelaxed
fragment densities as 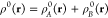 , the electrostatic contribution
to the
formation energy, which is named here as the zeroth-order energy
, the electrostatic contribution
to the
formation energy, which is named here as the zeroth-order energy 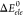 , equals the Coulomb interaction between
the unrelaxed densities:
, equals the Coulomb interaction between
the unrelaxed densities:
| 9 |
This energetic term corresponds to the so-called first-order polarization energy (or simply electrostatic energy) defined in SAPT,16 which has been adopted as a benchmark electrostatic energy for the validation of recently developed short-range electrostatic potentials.
2.3. Electrostatic Potentials in Molecular Mechanics
To
avoid the usage of continuous charge distributions, the MM methods
typically invoke the multipolar expansion as detailed in the Supporting Information (SI), which approximates
the zeroth-order energy defined in eq 9. Formally, the multipolar electrostatic energy 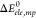 is affected by two
different error sources.
On the one hand, the underlying expansion must be truncated at some
order (lmax = 0, 1, 2,
...), resulting thus in a certain truncation error. On the other,
when
is affected by two
different error sources.
On the one hand, the underlying expansion must be truncated at some
order (lmax = 0, 1, 2,
...), resulting thus in a certain truncation error. On the other,
when 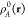 and
and 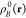 overlap to a significant extent, the rigorous
application of the multipole expansion is impeded so that its usage
at short distances implies some charge penetration error, which is
normally assumed to be dominant. Nevertheless, the multipole-based
potentials are still largely useful in many cases, and they enhance
convergence by distributing multipoles throughout the molecule at
the atomic sites and/or bond centers.9,35,36
overlap to a significant extent, the rigorous
application of the multipole expansion is impeded so that its usage
at short distances implies some charge penetration error, which is
normally assumed to be dominant. Nevertheless, the multipole-based
potentials are still largely useful in many cases, and they enhance
convergence by distributing multipoles throughout the molecule at
the atomic sites and/or bond centers.9,35,36
The MM electrostatic potentials can be classified into two groups. On one side, MM methods such as AMBER,37 CHARMM,38 GROMOS,39 and OPLS40 adopt simple electrostatic formulas with point charges (i.e., monopoles, with lmax = 0) that are ultimately the result of a fitting procedure against the molecular electrostatic potential (ESP). On the other side, more sophisticated methods, such as NEMO,41 AMOEBA,42 or the QTAIM-based FFLUX,43,44 include higher order multipoles (frequently up to the quadrupoles, lmax = 2) in order to capture the anisotropy of the distribution of electrons in space. These potentials are generally built from the QM density matrix of the molecule of interest by means of the distributed multipole analysis (DMA)36 or similar procedures. In addition, some methods (e.g., AMOEBA or NEMO) also refine the DMA multipoles to better reproduce the ESP values. In this way, the resulting charges/multipoles may include in an effective way both high-order multipolar contributions and CP effects. Actually, the performance of the MM potentials is examined statistically as a whole (i.e., using the full MM potential including all bonded and nonbonded terms) by various energetic and structural validation tests. A quite different approach is followed by the FFLUX force field. It makes use of QTAIM multipoles in contrast to the more widespread DMA methodologies and estimates them by means of a machine learning technique depending on each atom’s environment.
In comparison with the atomic/multipolar methods that are massively
employed in current simulation packages, the electrostatic MM potentials
that go beyond the multipolar approximation are much less consolidated.
In this category, we find different methodologies such as SIBFA,45 EFP,46 and AMOEBA+24 that complement the multipolar formulas with
other (so-called damping) functions to capture very-short-range electrostatics
and to remove the CP error. In this way, these potentials (whose general
form is shown in the SI) seek to reproduce 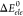 as evaluated by SAPT or similar methodologies.24
as evaluated by SAPT or similar methodologies.24
2.4. Charge Penetration Energy
The CP
energy Epen between two
molecules has been defined47 as the difference
between the exact zeroth-order electrostatic energy 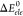 and its multipolar analogue
and its multipolar analogue 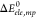 ,
,
| 10 |
Conceptually, this straightforward
definition of Epen is
satisfactory. It also shows that Epen is not only an interfragment quantity but rather an energy
that presents also intramolecular contributions according to the real
space partitioning of the whole 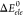 . In this respect, the energetic definition
suggests that the CP energy is not limited to the change in
the electrostatic interaction between two atoms due to their electron
cloud overlap and the associated loss of nuclear screening.48
. In this respect, the energetic definition
suggests that the CP energy is not limited to the change in
the electrostatic interaction between two atoms due to their electron
cloud overlap and the associated loss of nuclear screening.48
The rigorous evaluation of Epen for different systems at varying intermolecular separations would allow a deeper analysis of electrostatics and, eventually, the development of more accurate potentials. However, as noticed by Bojarowski et al.,47different methods of obtaining multipole moments lead to different radii of (pseudo)convergence, different levels of multipole expansions at which (pseudo)convergence is achieved, and different values of penetration energy. Therefore, the value of the CP energy as evaluated with eq 10 may depend on the particular method used to derive the multipoles. Moreover, the usage of truncated expansions introduces some additional truncation error so that both truncation and penetration effects become somewhat mixed in the resulting Epen values.49
An alternative to evaluate Epen has been proposed by Kairys and Jensen.50 Having noticed the relationship between the CP energy and the magnitude of the orbital overlap, they attempt to recover such an effect from scratch, with a derivation of Epen independently from the multipolar model used to estimate electrostatics at first stage. However, the authors find that the inherent dependence on the set of molecular orbitals used may lead to different CP values.
2.4.1. A Novel IQF Definition of the Charge Penetration Energy
By combining both the Bader partitioning scheme 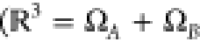 ) with a total
zeroth-order density decomposition
(
) with a total
zeroth-order density decomposition
( ), the following energy terms are obtained:
), the following energy terms are obtained:
-
(i)
the intramolecular interaction due to
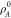 or
or 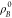 inside a given molecular basin ΩA or ΩB,
leading to
inside a given molecular basin ΩA or ΩB,
leading to 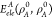 ,
, 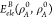 ,
, 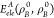 , and
, and 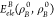 .
. -
(ii)
the intramolecular interaction between the two monomeric densities inside a given basin:
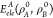 and
and 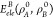 .
. -
(iii)
the intermolecular electrostatic energy between the same density pieces:
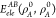 and
and 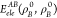 .
. -
(iv)
the intermolecular interaction between
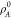 and
and  in opposite molecular basins:
in opposite molecular basins: 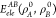 and
and 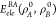 .
.
Hence, the total electrostatic energy of a complex AB can be written as
 |
11 |
In the notation used above the two interacting
densities are encompassed by parentheses, while the basins they are
integrated in are identified by the corresponding superscripts in
the given order (only one if both are the same). Hence, for instance,
the term 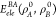 stands for
stands for 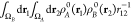 and
and 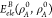 corresponds to
corresponds to 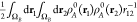 .
.
When the above double decomposition is applied to the electrostatic energies of the separate fragments, such as A, in the final complex, the electrostatic energy of the original species becomes
| 12 |
Note that this partitioning is derived from the AB zeroth-order (i.e., Hartree product) wave function and that geometry relaxation effects are not considered. By subtracting from eq 11 the previous fragment energies, the corresponding electrostatic contribution to the formation energy of the complex is obtained,
| 13 |
Among the
surviving terms in eq 13, 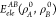 reveals itself
as the ordinary interaction
term between the two monomers A and B. It matches
reveals itself
as the ordinary interaction
term between the two monomers A and B. It matches 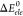 at long distances, while the other three
terms would present a similar behavior of increasing in magnitude
when shortening the intermolecular distances RAB and canceling out in the opposite situation.
Thus, those three terms can be directly related with the interpenetration
of molecular densities and grouped in the IQF-like electrostatic charge
penetration energy
at long distances, while the other three
terms would present a similar behavior of increasing in magnitude
when shortening the intermolecular distances RAB and canceling out in the opposite situation.
Thus, those three terms can be directly related with the interpenetration
of molecular densities and grouped in the IQF-like electrostatic charge
penetration energy
| 14 |
This term
fulfills 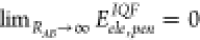 (and so its three components), while
(and so its three components), while 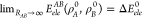 . Figure 1 represents the previous four
terms between the partitioned
. Figure 1 represents the previous four
terms between the partitioned 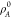 and
and 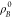 adding up to
adding up to 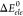 and compares them to the
and compares them to the 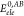 term between the total densities in each
basin.
term between the total densities in each
basin.
Figure 1.

Graphical scheme of the four contributions giving rise to 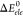 , where three of them (in dark blue) comprise
the IQF electrostatic penetration energy and the remaining one (dark
green) accounts for the interaction of
, where three of them (in dark blue) comprise
the IQF electrostatic penetration energy and the remaining one (dark
green) accounts for the interaction of 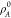 and
and 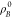 lying in the molecular basins ΩA and ΩB, respectively. The zeroth-order
IQF pairwise term
lying in the molecular basins ΩA and ΩB, respectively. The zeroth-order
IQF pairwise term 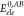 has been also included to highlight its
difference with the previous
has been also included to highlight its
difference with the previous 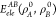 , as it accounts for an
interaction between
total densities inside each basin (the original
, as it accounts for an
interaction between
total densities inside each basin (the original 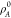 or
or 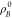 and the tail from the other that has penetrated
into another domain).
and the tail from the other that has penetrated
into another domain).
3. Computational Details
3.1. Molecular Geometries and Reference Interaction Energies
All the QM and classical electrostatic calculations were performed on the molecular geometries retrieved from the S66 database,29 which contains a set of 66 complexes featuring the most common noncovalent interactions in biomolecules. These can be classified depending on the atoms involved into polar, nonpolar, and mixed. Analogously, the different complexes have been grouped into H-bond, dispersion, and mixed according to the main interactions they experience (see Table S1). For representing both the atomic interactions and the subsets of complexes, a color code has been utilized: magenta for H-bond/polar, yellow for mixed, and blue for dispersion/nonpolar. In addition to the S66 set, a selection of 12 representative complexes from the S66x8 database,30 which is an extension of the former to eight different fractions of the equilibrium intermolecular distances, were also considered. The benchmark CCSD(T)/CBS interaction energies collected in S66 were employed as the reference values for comparative purposes.
3.2. HF-D3 Calculations
HF/cc-pVTZ calculations were carried out on the S66 and the S66x8 geometries using the GAMESS-US package.51 Grimme’s D3 dispersion potential as implemented in the DFT-D3 code52 was employed to incorporate the dispersion energy. Additionally, in order to correctly reproduce the asymptotic behavior of the dispersion energy at small distances, the Becke–Johnson damping function was chosen.53
We selected HF because it lacks entirely dispersion energy and thereby yields a straight physical partitioning of energy in combination with the D3 potential. We also note in passing that HF-D3 has been shown to describe correctly and efficiently the structure and energetics of biomolecules54 and that a variant of DFT-SAPT has been also developed in which the costly ab initio dispersion calculations are replaced by a reparametrized D3 potential.55 In addition, the HF-D3/cc-pVTZ energies reproduce quite well the reference CCSD(T)/CBS energies of the S66 structures (see Figure S1).
3.3. IQA Energy Decomposition Analysis
The decomposition of the QM and the
electrostatic energies derived
from continuous charge densities were performed with the PROMOLDEN
code.56 The integration settings comprised
β-spheres with radii of 60% of the distance between each nucleus
and its closest critical point. Within them, Lebedev angular grids
with 974 points were used, along with Euler–McLaurin radial
quadratures with 382 radial points. A bipolar expansion of 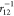 was selected with an lmax of 6. On the other hand, the outer
part of the basins (i.e., outside the β-spheres) employed the
same angular and radial quadratures, albeit increasing their respective
points up to 5810 and 512, with a maximum radius of 15 au. In this
case,
was selected with an lmax of 6. On the other hand, the outer
part of the basins (i.e., outside the β-spheres) employed the
same angular and radial quadratures, albeit increasing their respective
points up to 5810 and 512, with a maximum radius of 15 au. In this
case, 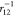 was expanded by means of a Laplace expansion
with lmax = 10.
was expanded by means of a Laplace expansion
with lmax = 10.
3.4. Point-Charge and Multipolar Calculations
Atomic charges were computed for the separate monomers in the S66 structures by means of the restrained electrostatic potential (RESP) method following the General Amber Force Field (GAFF)57 prescriptions with a HF/6-31G* level of theory. In the case of the atomic multipoles, two different sets were employed. On the one hand, AMOEBA multipoles were derived up to the quadrupoles (lmax = 2) following its corresponding parametrization protocol.42,58 On the other, QTAIM multipoles were obtained by means of the PROMOLDEN program with an lmax = 2. Both the AMOEBA and the QTAIM multipolar energies were obtained with the MPOLINT code.59
Additionally, a set of 12 S66x8 complexes was tested under the AMOEBA+ CP-corrected potentials.24 For this, TINKER was used to calculate the respective CP energies as the difference between the CP-corrected multipoles and the multipolar energies previously derived. The parameters of the damping functions were directly taken from the literature.24
3.5. Graphs and Statistical Analyses
Octave60 and GNUplot61 were, in turn, used to perform the statistical analyses and the correlation plots, while Python’s Matplotlib62 was chosen for the rest of the representations.
4. Results and Discussion
4.1. IQF-D3 Partitioning and Pairwise Approximation
The IQF-D3 decomposition of the HF/cc-pVTZ
binding energies for
the S66 complexes has been discussed at length in previous work.32 Herein, we focus on the decomposition of the
electrostatic descriptors into intra- and interfragment components.
Interestingly, we found that the combination of the interfragment
electrostatic interaction energy 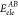 with the D3 dispersion
potential yields
pairwise energies that are quite well correlated with the S66 benchmark
values, the coefficient of determination being R2 = 0.990 with RMS errors of 5.7 kcal mol–1 (see Figure 2 and Table S2). Thus, the IQF
with the D3 dispersion
potential yields
pairwise energies that are quite well correlated with the S66 benchmark
values, the coefficient of determination being R2 = 0.990 with RMS errors of 5.7 kcal mol–1 (see Figure 2 and Table S2). Thus, the IQF 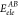 descriptors in conjunction
with the D3
potential capture the essential electrostatic and dispersion interactions
that determine the relative stability of the noncovalent complexes.
When addressing both terms independently (Figure S2), we find that the pure electrostatic
descriptors in conjunction
with the D3
potential capture the essential electrostatic and dispersion interactions
that determine the relative stability of the noncovalent complexes.
When addressing both terms independently (Figure S2), we find that the pure electrostatic 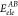 term exhibits a satisfactory
overall correlation
(R2 = 0.943) due to the fundamental role
of electrostatics in H-bond complexes. On the other hand, the D3 descriptor
has a null global correlation with the S66 reference energies, although
it is reasonable (R2 = 0.820) for the
dispersion complexes as expected. However, the mixed complexes are
not well-described by either the electrostatic or the dispersion energies
separately, and their combination becomes critical.
term exhibits a satisfactory
overall correlation
(R2 = 0.943) due to the fundamental role
of electrostatics in H-bond complexes. On the other hand, the D3 descriptor
has a null global correlation with the S66 reference energies, although
it is reasonable (R2 = 0.820) for the
dispersion complexes as expected. However, the mixed complexes are
not well-described by either the electrostatic or the dispersion energies
separately, and their combination becomes critical.
Figure 2.

Left: correlation between
the dispersion-augmented IQF intermolecular
electrostatic energy 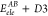 and the reference binding energies
and the reference binding energies 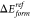 . Right: anticorrelation
featured by the
intrafragment electrostatic contribution to formation
. Right: anticorrelation
featured by the
intrafragment electrostatic contribution to formation 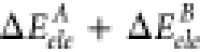 and the total kinetic plus exchange-correlation
contributions ΔT + ΔExc. The statistical analysis comprises
the coefficient of determination R2, Spearman’s
rank correlation coefficient ρ, and the root-mean-square error RMS. Data corresponding to the whole set of complexes is
depicted in black and that ascribed to the H-bond group is in magenta,
while mixed and dispersion complexes are in yellow and blue, respectively.
All the energies are in kcal mol–1.
and the total kinetic plus exchange-correlation
contributions ΔT + ΔExc. The statistical analysis comprises
the coefficient of determination R2, Spearman’s
rank correlation coefficient ρ, and the root-mean-square error RMS. Data corresponding to the whole set of complexes is
depicted in black and that ascribed to the H-bond group is in magenta,
while mixed and dispersion complexes are in yellow and blue, respectively.
All the energies are in kcal mol–1.
In contrast to the ability of the 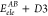 descriptors to capture the main features
of noncovalent binding, the combination of ΔEele, which includes both the intra- and
the intermolecular electrostatic effects, with the D3 potential deteriorates
the global correlation (R2 = 0.888) and
results in larger RMS errors (17.3 kcal mol–1). The full IQF decomposition (eq 8) explains this unbalanced description because the
intrafragment electrostatic energies, which contribute to the deformation
energies, tend to cancel out with the QM energy terms (electronic
kinetic energy and exchange-correlation) that are not required in
the simple electrostatic + dispersion picture (see Figure 2 right). Therefore, the pairwise
descriptors to capture the main features
of noncovalent binding, the combination of ΔEele, which includes both the intra- and
the intermolecular electrostatic effects, with the D3 potential deteriorates
the global correlation (R2 = 0.888) and
results in larger RMS errors (17.3 kcal mol–1). The full IQF decomposition (eq 8) explains this unbalanced description because the
intrafragment electrostatic energies, which contribute to the deformation
energies, tend to cancel out with the QM energy terms (electronic
kinetic energy and exchange-correlation) that are not required in
the simple electrostatic + dispersion picture (see Figure 2 right). Therefore, the pairwise 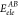 terms arise as the
most relevant IQF electrostatic
descriptors of noncovalent binding.
terms arise as the
most relevant IQF electrostatic
descriptors of noncovalent binding.
4.2. Validating and Analyzing the Zeroth-Order Approximation
The electrostatic IQF terms can be readily
evaluated under the zeroth-order approximation (i.e., 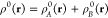 ). Thus, it turns out
that the interaction
energies
). Thus, it turns out
that the interaction
energies 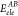 can be replaced effectively
by their zeroth-order
counterparts. Indeed, the pairwise
can be replaced effectively
by their zeroth-order
counterparts. Indeed, the pairwise 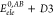 energies have low RMS errors
(3.1 kcal mol–1) and maintain a good correlation
(R2 = 0.971) with respect to the benchmark
data (Table S3). This behavior is also
satisfactory within the S66 subsets: R2 = 0.989 and 0.988 for the polar H-bonded systems and the dispersion-dominated
complexes, respectively, albeit the correlation is somewhat reduced
in the case of the mixed complexes (R2 = 0.755). Further support for the use of the zeroth-order energies
comes from the atomic level, where a high degree of coincidence between
the diatomic zeroth-order
energies have low RMS errors
(3.1 kcal mol–1) and maintain a good correlation
(R2 = 0.971) with respect to the benchmark
data (Table S3). This behavior is also
satisfactory within the S66 subsets: R2 = 0.989 and 0.988 for the polar H-bonded systems and the dispersion-dominated
complexes, respectively, albeit the correlation is somewhat reduced
in the case of the mixed complexes (R2 = 0.755). Further support for the use of the zeroth-order energies
comes from the atomic level, where a high degree of coincidence between
the diatomic zeroth-order 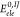 and fully relaxed
and fully relaxed 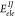 energies is also found
at the equilibrium
geometries (R2 = 0.995, see the SI).
energies is also found
at the equilibrium
geometries (R2 = 0.995, see the SI).
When addressing the distance dependence
of the previous term (see Figure 3), both 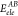 and
and 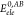 follow the same trends at varying intermolecular
separations RAB (given
as relative to the equilibrium distances Req). As expected, they start diverging at short distances
due to the strengthening of charge polarization, charge-penetration,
and charge-transfer effects that attenuate the pairwise electrostatic
forces. The magnitude of these effects is clearly system-dependent,
as well as the shape and slope of the
follow the same trends at varying intermolecular
separations RAB (given
as relative to the equilibrium distances Req). As expected, they start diverging at short distances
due to the strengthening of charge polarization, charge-penetration,
and charge-transfer effects that attenuate the pairwise electrostatic
forces. The magnitude of these effects is clearly system-dependent,
as well as the shape and slope of the 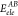 and
and 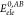 curves, revealing thus further details
about the role of electrostatics in these complexes. Thus, the electrostatic
stabilization of the four H-bond complexes and others (e.g., the π-complex
of the uracil dimer) is continuously reinforced upon shortening the
monomer–monomer distance, reflecting the major electrostatic
control of these systems. In contrast, the T-shaped benzene complexes
with methanol or N-methylacetamide reach an electrostatic
minimum at a distance longer than the equilibrium one while the small
electrostatic energies of the dispersion dimers (i.e., +1, −1
kcal/mol) change very little along the curves (some small leaps are
due to residual errors arising in the numerical integration over the
atomic basins).
curves, revealing thus further details
about the role of electrostatics in these complexes. Thus, the electrostatic
stabilization of the four H-bond complexes and others (e.g., the π-complex
of the uracil dimer) is continuously reinforced upon shortening the
monomer–monomer distance, reflecting the major electrostatic
control of these systems. In contrast, the T-shaped benzene complexes
with methanol or N-methylacetamide reach an electrostatic
minimum at a distance longer than the equilibrium one while the small
electrostatic energies of the dispersion dimers (i.e., +1, −1
kcal/mol) change very little along the curves (some small leaps are
due to residual errors arising in the numerical integration over the
atomic basins).
Figure 3.

Intermolecular electrostatic interactions for a subset
of the S66x8
complexes as provided by IQF (either exactly 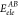 or under the zeroth-order
approximation
or under the zeroth-order
approximation 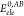 ), zeroth-order QTAIM multipoles
), zeroth-order QTAIM multipoles 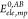 , AMOEBA multipolar
energies
, AMOEBA multipolar
energies 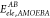 , and RESP atomic charges
, and RESP atomic charges 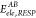 . Additionally, the
zeroth-order electrostatic
contribution to formation
. Additionally, the
zeroth-order electrostatic
contribution to formation 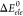 is also included. The complexes are colored
and displayed in columns according to the group they belong to, namely,
H-bond, mixed, or dispersion, respectively. The energies (Y-axis) are given in kcal mol–1, and the
abscissas represent the intermolecular distances relative to the equilibrium
ones (RAB/Req).
is also included. The complexes are colored
and displayed in columns according to the group they belong to, namely,
H-bond, mixed, or dispersion, respectively. The energies (Y-axis) are given in kcal mol–1, and the
abscissas represent the intermolecular distances relative to the equilibrium
ones (RAB/Req).
In Figure 3 the
deviation between the global 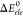 energies and the interfragment
energies and the interfragment 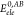 anticipates the underlying CP effects associated
with the density overlap. For the H-bond and some of the mixed complexes,
the two curves decrease with lowering separation, but they split gradually
for RAB/Req < 1.6. The global
anticipates the underlying CP effects associated
with the density overlap. For the H-bond and some of the mixed complexes,
the two curves decrease with lowering separation, but they split gradually
for RAB/Req < 1.6. The global 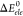 stabilization nearly doubles
stabilization nearly doubles 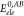 at Req, showing thus
the large impact of intramolecular electrostatics
as defined in the IQF framework. For the π-complexes (benzene–dimer,
benzene–methanol, ...) or the weakly interacting neopentane
dimer, the inter- vs intramolecular balance is differently modulated
because the deviation between the global and the interfragment electrostatics
becomes significant only at very close distances (e.g., RAB/Req < 1.1), which are indicative of mutual overlap. In these
systems,
at Req, showing thus
the large impact of intramolecular electrostatics
as defined in the IQF framework. For the π-complexes (benzene–dimer,
benzene–methanol, ...) or the weakly interacting neopentane
dimer, the inter- vs intramolecular balance is differently modulated
because the deviation between the global and the interfragment electrostatics
becomes significant only at very close distances (e.g., RAB/Req < 1.1), which are indicative of mutual overlap. In these
systems, 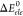 is thus reinforced by several kcal mol–1, which
are ascribed to the intrafragment electrostatic
stabilization achieved by the fragment-overlap (i.e., CP) effects.
Such effects have a minor influence on the small
is thus reinforced by several kcal mol–1, which
are ascribed to the intrafragment electrostatic
stabilization achieved by the fragment-overlap (i.e., CP) effects.
Such effects have a minor influence on the small 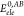 energies (<1–2 kcal mol–1), which tend
to remain nearly constant or become slightly attenuated.
As shown below (Section 4.5), the IQF analysis of the CP energy gives further insight
about the behavior of
energies (<1–2 kcal mol–1), which tend
to remain nearly constant or become slightly attenuated.
As shown below (Section 4.5), the IQF analysis of the CP energy gives further insight
about the behavior of 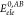 and
and 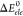 with RAB/Req.
with RAB/Req.
4.3. Comparison
between 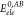 and Pairwise MM Energies
and Pairwise MM Energies
The pairwise
approximation that emerges from the IQF-D3 decomposition and the validity
of the zeroth-order approximation for the electrostatic interactions
provide an insightful theoretical support for the construction of
noncovalent MM potentials. In this scenario, 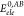 can be seen as the most suitable IQF descriptor
to assess the approximate electrostatic potentials. Hence, we calculated
the interfragment electrostatic energies using the RESP atomic charges
and the AMOEBA multipoles, as well as the QTAIM multipoles up to the
quadrupoles.
can be seen as the most suitable IQF descriptor
to assess the approximate electrostatic potentials. Hence, we calculated
the interfragment electrostatic energies using the RESP atomic charges
and the AMOEBA multipoles, as well as the QTAIM multipoles up to the
quadrupoles.
According to the statistical data in Table 1, either the RESP atomic charges
or the QTAIM/AMOEBA multipoles give interfragment electrostatic energies
that correlate considerably well with 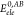 (R2 > 0.9 and RMS errors ∼ 1 kcal mol–1) for
the full S66 set and also for the H-bond/dispersion subsets. These
point-charge/multipolar electrostatic energies are less satisfactory
for the less abundant mixed complexes, although the multipolar potentials
yield a more accurate description (R2 ≃
0.6–0.8) than the RESP charges (R2 ≃ 0.5). In addition to
(R2 > 0.9 and RMS errors ∼ 1 kcal mol–1) for
the full S66 set and also for the H-bond/dispersion subsets. These
point-charge/multipolar electrostatic energies are less satisfactory
for the less abundant mixed complexes, although the multipolar potentials
yield a more accurate description (R2 ≃
0.6–0.8) than the RESP charges (R2 ≃ 0.5). In addition to 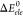 and the fully relaxed and zeroth-order
IQF pairwise terms, Figure 3 also displays the distance dependence of the QTAIM/AMOEBA/RESP
energies, that results quite close to that of the interfragment
and the fully relaxed and zeroth-order
IQF pairwise terms, Figure 3 also displays the distance dependence of the QTAIM/AMOEBA/RESP
energies, that results quite close to that of the interfragment 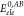 energies. Nevertheless, a closer inspection
reveals that the QTAIM/AMOEBA/RESP energies tend to overestimate the
stabilizing/destabilizing character of
energies. Nevertheless, a closer inspection
reveals that the QTAIM/AMOEBA/RESP energies tend to overestimate the
stabilizing/destabilizing character of 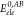 for the H-bond/dispersion dimers, respectively.
for the H-bond/dispersion dimers, respectively.
Table 1. Statistical Measurements Comprising
the Coefficient of Determination R2, Spearman’s
Rank Correlation Coefficient ρ, and the Root Mean Square Error RMS for the Correlation between 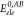 and either the QTAIM or AMOEBA Multipoles
(lmax = 2) or the RESP
Point Charges (lmax =
0).
and either the QTAIM or AMOEBA Multipoles
(lmax = 2) or the RESP
Point Charges (lmax =
0).
| multipolar approximation | complex type | R2 | ρ | RMS |
|---|---|---|---|---|
| QTAIM | global | 0.970 | 0.958 | 1.0 |
| H-bond | 0.956 | 0.904 | 1.4 | |
| mixed | 0.644 | 0.768 | 0.9 | |
| dispersion | 0.955 | 0.795 | 0.5 | |
| AOMEBA | global | 0.953 | 0.972 | 1.3 |
| H-bond | 0.904 | 0.841 | 2.0 | |
| mixed | 0.800 | 0.845 | 0.7 | |
| dispersion | 0.939 | 0.893 | 0.4 | |
| RESP | global | 0.974 | 0.962 | 0.8 |
| H-bond | 0.981 | 0.918 | 0.7 | |
| mixed | 0.456 | 0.687 | 1.1 | |
| dispersion | 0.948 | 0.831 | 0.3 |
The good agreement between the multipolar and the RESP energies in Table 1 and in Figure 3 suggests that the RESP fitting procedure may incorporate in an effective way higher order effects even at short distances. In addition, our results point out that the pure QTAIM multipoles can be employed in the construction of accurate electrostatic potentials, free from the inclusion of other effects that may be present when the DMA multipoles are fitted against the molecular ESP. In fact, the QTAIM multipoles, which are already considered in the FFLUX force field, readily reproduce the ESP without the need of any constraint.63
4.4. Comparing Diatomic Electrostatic Interactions
IQA permits an unambiguous decomposition of the continuous-density intermolecular interaction energy into a sum of atomic and diatomic terms that enables a thorough analysis of the global molecular properties based on their atomic origins, and a close comparison with the various MM descriptions at this atomic level.
As expected, the IQA diatomic
terms correlate almost perfectly with the QTAIM multipolar ones 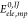 (see Figure 4). On the contrary,
the AMOEBA and RESP energies are
significantly less correlated (R2 of 0.7
and 0.4, respectively) and have large RMS errors.
For example, the largest discrepancies between
(see Figure 4). On the contrary,
the AMOEBA and RESP energies are
significantly less correlated (R2 of 0.7
and 0.4, respectively) and have large RMS errors.
For example, the largest discrepancies between 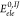 and the QTAIM-multipolar
and the QTAIM-multipolar  in the acetic
acid dimer (about 6 kcal
mol–1) arise from the atoms involved in the OH·O
H-bonds, the rest of pair interactions having much lower differences
(<0.5 kcal mol–1; see Tables S7–S9). When comparing
in the acetic
acid dimer (about 6 kcal
mol–1) arise from the atoms involved in the OH·O
H-bonds, the rest of pair interactions having much lower differences
(<0.5 kcal mol–1; see Tables S7–S9). When comparing 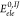 and
and 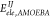 (or
(or 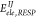 ), the largest discrepancies
amount to hundreds
of kcal mol–1 and involve not only short polar contacts
but methyl C atoms too (see Tables S10–S15).
), the largest discrepancies
amount to hundreds
of kcal mol–1 and involve not only short polar contacts
but methyl C atoms too (see Tables S10–S15).
Figure 4.

Comparison of the 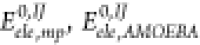 and
and 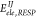 energies with the
energies with the 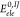 term (kcal mol–1). On
the left are the correlation plots and, on the right, each difference
as a function of the interatomic distance (Å).
term (kcal mol–1). On
the left are the correlation plots and, on the right, each difference
as a function of the interatomic distance (Å).
The dissimilarity between the 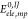 energies
and the
energies
and the 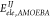 /
/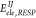 values was
not entirely unexpected given
that the RESP charges are derived from the molecular ESP and the AMOEBA
multipoles are obtained by the DMA protocol. In fact, a difference
of 1 order of magnitude between the atom–atom electrostatic
interactions from IQA and MM potentials has also been noticed previously.64 The present results show in further detail the
actual discrepancies between the various atomic representations and
suggest that, although the diverse atomic multipoles employed in classical
potentials yield similar molecular electrostatic energies, the atomic
decomposition is more questionable, which, in turn, can negatively
affect the interpretation of localized electrostatic interactions
and/or result in artifacts while dealing with QM and MM short-range
electrostatics in hybrid QM/MM methodologies.
values was
not entirely unexpected given
that the RESP charges are derived from the molecular ESP and the AMOEBA
multipoles are obtained by the DMA protocol. In fact, a difference
of 1 order of magnitude between the atom–atom electrostatic
interactions from IQA and MM potentials has also been noticed previously.64 The present results show in further detail the
actual discrepancies between the various atomic representations and
suggest that, although the diverse atomic multipoles employed in classical
potentials yield similar molecular electrostatic energies, the atomic
decomposition is more questionable, which, in turn, can negatively
affect the interpretation of localized electrostatic interactions
and/or result in artifacts while dealing with QM and MM short-range
electrostatics in hybrid QM/MM methodologies.
4.5. Charge Penetration under the QTAIM Scrutiny
Following the prescriptions
introduced in Theory
and Methods, the zeroth-order electrostatic formation energy 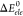 of each S66 complex was decomposed by combining
its real space partition into nonoverlapping atomic basins with the
zeroth-order density approximation (
of each S66 complex was decomposed by combining
its real space partition into nonoverlapping atomic basins with the
zeroth-order density approximation (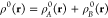 ). This
strategy leads to the IQF-based
charge penetration energies,
). This
strategy leads to the IQF-based
charge penetration energies, 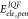 , resulting from the
sum of the intramolecular
terms
, resulting from the
sum of the intramolecular
terms 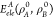 and
and 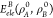 , as those accounting for the interaction
of both densities inside the same basin, and the intermolecular energy
, as those accounting for the interaction
of both densities inside the same basin, and the intermolecular energy  between the tails of
each molecular density
that penetrate into the opposite basin, as described in eq 14. This constitutes an effective penetration energy in the sense that the molecular identity between
two overlapping fragments becomes necessarily blurred so that fragment
properties are dependent upon the scheme followed to dissect the global
charge density into its constituents. Nevertheless, the topological
analysis of ρ0 yields a consistent identification
of molecular fragments so that we believe that the associated charge-penetration
analysis can give useful insight into the electrostatics of noncovalent
complexes.
between the tails of
each molecular density
that penetrate into the opposite basin, as described in eq 14. This constitutes an effective penetration energy in the sense that the molecular identity between
two overlapping fragments becomes necessarily blurred so that fragment
properties are dependent upon the scheme followed to dissect the global
charge density into its constituents. Nevertheless, the topological
analysis of ρ0 yields a consistent identification
of molecular fragments so that we believe that the associated charge-penetration
analysis can give useful insight into the electrostatics of noncovalent
complexes.
The application of eq 14 to 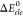 results in the energy contributions shown
in Figure 5. On the
one hand, the interfragment energy
results in the energy contributions shown
in Figure 5. On the
one hand, the interfragment energy 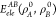 is formally not affected
by charge penetration
and plays a stabilizing role in all the H-bond complexes (slightly
repulsive in the dispersion complexes). On the other hand, the IQF
penetration term
is formally not affected
by charge penetration
and plays a stabilizing role in all the H-bond complexes (slightly
repulsive in the dispersion complexes). On the other hand, the IQF
penetration term 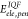 turns out to be of
equal importance in
the H-bond complexes or even more relevant in the dispersion subset
for which penetration energy describes the major part of
turns out to be of
equal importance in
the H-bond complexes or even more relevant in the dispersion subset
for which penetration energy describes the major part of 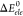 .
.
Figure 5.

Decomposition of 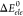 into
into  and the three IQF penetration
terms
and the three IQF penetration
terms 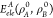 ,
, 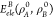 , and
, and 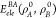 . Energies are given in
kcal mol–1.
. Energies are given in
kcal mol–1.
The decomposition of the penetration energy shows that it arises
mainly from the stabilizing interactions between 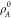 and
and 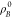 inside the same basin. This is an intramolecular effect as reflected by the magnitude of the
inside the same basin. This is an intramolecular effect as reflected by the magnitude of the 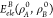 and
and 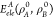 energies. As shown by the integration of
energies. As shown by the integration of 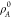 or
or 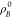 in the corresponding basins, the mutual
CP values range, for instance, from 0.035 e in the neopentane dimer
to 0.099 e in the case of the acetic acid dimer. These fractional
charges involve the e–e repulsion between the fragment electron
densities occupying the same space, such as
in the corresponding basins, the mutual
CP values range, for instance, from 0.035 e in the neopentane dimer
to 0.099 e in the case of the acetic acid dimer. These fractional
charges involve the e–e repulsion between the fragment electron
densities occupying the same space, such as 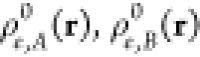 such that r ∈ ΩA (or equivalently in region ΩB), and the attraction experienced by the nuclei of
one fragment
such that r ∈ ΩA (or equivalently in region ΩB), and the attraction experienced by the nuclei of
one fragment  (or
(or 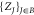 ) and
the fraction of electrons from the
other that has penetrated into the former
) and
the fraction of electrons from the
other that has penetrated into the former  (or similarly
(or similarly 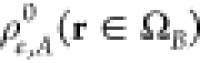 ). In light of these results, e–n
attraction greatly overcomes e–e repulsion between different
zeroth-order densities inside the same basin and gives rise to the
significant stabilizing energies observed. There is also a minor repulsive
contribution owing to the purely electronic repulsion between the
penetrating
). In light of these results, e–n
attraction greatly overcomes e–e repulsion between different
zeroth-order densities inside the same basin and gives rise to the
significant stabilizing energies observed. There is also a minor repulsive
contribution owing to the purely electronic repulsion between the
penetrating 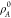 into ΩB and the
into ΩB and the  tail in ΩA, which is measured
by
tail in ΩA, which is measured
by 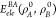 .
.
Further
insight can be gained by analyzing the distance dependence
of the various energy terms as shown in Figure 6. The plots confirm that the three components
of 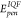 tend to zero when RAB/Req > 1.5 and further
highlight the role of the intrafragment
terms. Interestingly, the
tend to zero when RAB/Req > 1.5 and further
highlight the role of the intrafragment
terms. Interestingly, the 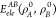 energy, formally lacking
penetration effects,
is modulated by the degree of the interfragment overlap so that the
decreasing trend in
energy, formally lacking
penetration effects,
is modulated by the degree of the interfragment overlap so that the
decreasing trend in 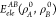 is damped out or inverted
at the shortest
distances. This is not entirely unexpected given that, as two initially
separated atomic basins (e.g, ΩI∈A and ΩJ∈B) approach one another, their volume, shape, and
electron population evolve along the RAB/Req curve in response to the density overlap. We note, however, that
the deviation of
is damped out or inverted
at the shortest
distances. This is not entirely unexpected given that, as two initially
separated atomic basins (e.g, ΩI∈A and ΩJ∈B) approach one another, their volume, shape, and
electron population evolve along the RAB/Req curve in response to the density overlap. We note, however, that
the deviation of 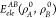 with respect to the interfragment
electrostatic
energy
with respect to the interfragment
electrostatic
energy 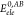 may constitute a useful index about the
specific impact of penetration effects on the pairwise electrostatics.
At this point, an important caveat should be noted. Within the QTAIM
framework, the
may constitute a useful index about the
specific impact of penetration effects on the pairwise electrostatics.
At this point, an important caveat should be noted. Within the QTAIM
framework, the 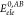 energy includes a fraction of stabilizing
penetration energy for RAB/Req < 1.2 given
that the loss of some electronic
energy includes a fraction of stabilizing
penetration energy for RAB/Req < 1.2 given
that the loss of some electronic  density from the basins of the monomer A is partially
compensated by the penetration of
density from the basins of the monomer A is partially
compensated by the penetration of 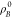 into the same basin. The fixed multipoles/charges
in the classical potentials somehow mimic this behavior so that they
remain closer to the
into the same basin. The fixed multipoles/charges
in the classical potentials somehow mimic this behavior so that they
remain closer to the 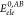 descriptors than to
descriptors than to 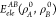 around the equilibrium
distance.
around the equilibrium
distance.
Figure 6.

Evolution of the energy terms from eq 13, along with the 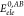 pair term as a function of the distance
for the set of S66x8 systems chosen. The complexes are grouped in
three columns as belonging to the H-bond, mixed, or dispersion subsets,
respectively.
pair term as a function of the distance
for the set of S66x8 systems chosen. The complexes are grouped in
three columns as belonging to the H-bond, mixed, or dispersion subsets,
respectively.
Finally, Figure 7 compares the IQF penetration term and other
relevant energetic quantities
with the analogue term derived from the AMOEBA+ model as a function
of the intermolecular distance. Thus, the combination of the multipolar 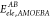 energies with the CP correction24
energies with the CP correction24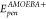 results in the
results in the 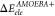 energies that approach to the reference
energies that approach to the reference 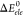 , which is equivalent to the SAPT electrostatic
energy. In effect, Figure 7 shows that
, which is equivalent to the SAPT electrostatic
energy. In effect, Figure 7 shows that 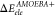 nearly matches
nearly matches 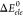 . Concerning the CP energies, it is important
to note again that the AMOEBA+ reference for measuring the CP energy
is different from that provided by the IQF-QTAIM approach. Nevertheless,
the two penetration energies exhibit a similar behavior with RAB, particularly for the more
stable H-bond complexes, which resemble also the variations experienced
by the intramolecular CP terms,
. Concerning the CP energies, it is important
to note again that the AMOEBA+ reference for measuring the CP energy
is different from that provided by the IQF-QTAIM approach. Nevertheless,
the two penetration energies exhibit a similar behavior with RAB, particularly for the more
stable H-bond complexes, which resemble also the variations experienced
by the intramolecular CP terms, 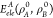 and
and 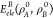 . Therefore, we conclude that the AMOEBA+
CP and similar corrections account mainly for intramolecular electrostatics.
. Therefore, we conclude that the AMOEBA+
CP and similar corrections account mainly for intramolecular electrostatics.
Figure 7.

Comparison between the AMOEBA+ model and the zeroth-order IQF energies for our model S66x8 complexes. The complexes have been displayed according to the group they belong to (either H-bond, mixed, or dispersion). Distances (X-axis) are relative to the equilibrium ones (RAB/Req) and energies (Y-axis) are in kcal mol–1.
5. Concluding Remarks
In this work we have analyzed
the short-range electrostatic interactions
in the S66 and S66x8 data sets through a hierarchy of approximations
at both the molecular and the atomic levels. We have shown first that
the IQA/IQF decomposition augmented with the D3 dispersion terms gives
support to the pairwise approach adopted by many MM potentials. In
this respect, the interfragment energies 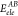 derived from the IQF
partitioning suffice
to capture the essential electrostatic effects explaining the binding
of the weakly interacting complexes. Moreover, the same role can be
played by the equivalent
derived from the IQF
partitioning suffice
to capture the essential electrostatic effects explaining the binding
of the weakly interacting complexes. Moreover, the same role can be
played by the equivalent 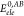 values, which are obtained from the unrelaxed
densities of the isolated monomers (i.e., the zeroth-order approximation).
values, which are obtained from the unrelaxed
densities of the isolated monomers (i.e., the zeroth-order approximation).
According to our results, the intermolecular 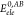 energy turns out to be the most appropriate
IQF descriptor to analyze and/or compare with electrostatic MM potentials.
In particular, we have considered two widely used potentials relying
on the RESP atomic charges or the AMOEBA distributed atomic multipoles,
respectively, as well as the multipolar potential up to the quadrupoles
derived directly from the QTAIM basins. The three MM pairwise approximations
correlate satisfactorily with the zeroth-order IQF term at varying
intermolecular distances and exhibit small RMS errors.
However, when the
energy turns out to be the most appropriate
IQF descriptor to analyze and/or compare with electrostatic MM potentials.
In particular, we have considered two widely used potentials relying
on the RESP atomic charges or the AMOEBA distributed atomic multipoles,
respectively, as well as the multipolar potential up to the quadrupoles
derived directly from the QTAIM basins. The three MM pairwise approximations
correlate satisfactorily with the zeroth-order IQF term at varying
intermolecular distances and exhibit small RMS errors.
However, when the 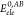 values are further decomposed into diatomic
contributions, large discrepancies between the RESP or the AMOEBA
atom–atom interactions and their zeroth-order IQA counterparts
are unveiled. Although this is understandable in terms of the specific
details of the RESP/AMOEBA charge/multipole derivations, it contrasts
sharply with the nearly perfect match between the QTAIM atomic multipolar
energies and the IQA reference values. Hence, MM potentials based
on the QTAIM multipoles—such as the QCT-based FFLUX—may
provide a more consistent description of electrostatic interactions
at both the molecular and the atomic levels.
values are further decomposed into diatomic
contributions, large discrepancies between the RESP or the AMOEBA
atom–atom interactions and their zeroth-order IQA counterparts
are unveiled. Although this is understandable in terms of the specific
details of the RESP/AMOEBA charge/multipole derivations, it contrasts
sharply with the nearly perfect match between the QTAIM atomic multipolar
energies and the IQA reference values. Hence, MM potentials based
on the QTAIM multipoles—such as the QCT-based FFLUX—may
provide a more consistent description of electrostatic interactions
at both the molecular and the atomic levels.
Besides forging
links between the IQF/IQA quantities and the MM
electrostatic potentials, we have studied the charge penetration effects
that arise from the mutual interpenetration of the zeroth-order molecular
densities in their opposite QTAIM basins as built from the final ρ0 of the complex. This QTAIM perspective allows us to dissect
the CP energy into different contributions that emphasize its intramolecular
character, which, in turn, is dominated by the attraction between
the nuclei of fragment A (B) and
the penetrating tail of density B (A). In this way we may clarify some practical issues related with
the CP corrections for MM potentials. For example, adding CP corrections
to MM potentials like RESP/AMOEBA, which target the zeroth-order interfragment
electrostatic energy, results, necessarily, in an unbalanced description.
This aspect, which has been overlooked in previous works,10,48,65 implies also that the electrostatic
energy employed in popular MM force fields (AMBER, CHARMM, ...) cannot
be compared with the global 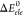 energy derived from continuous charge distributions,
but with its interfragment component. On the other hand, CP corrections
have been derived to improve the description of the QM–MM electrostatic
interactions in hybrid QM/MM methodologies.12 In this case, such corrections should mitigate short-range electrostatic
artifacts, particularly those associated with the QM–MM covalent
linkages. However, considering the highly dissimilar interatomic electrostatic
energies produced by the QM densities and the RESP/AMOEBA potentials,
the usage of electrostatic parameters more akin to the QM densities
at the atomic level may have a larger impact in improving the QM–MM
electrostatics.
energy derived from continuous charge distributions,
but with its interfragment component. On the other hand, CP corrections
have been derived to improve the description of the QM–MM electrostatic
interactions in hybrid QM/MM methodologies.12 In this case, such corrections should mitigate short-range electrostatic
artifacts, particularly those associated with the QM–MM covalent
linkages. However, considering the highly dissimilar interatomic electrostatic
energies produced by the QM densities and the RESP/AMOEBA potentials,
the usage of electrostatic parameters more akin to the QM densities
at the atomic level may have a larger impact in improving the QM–MM
electrostatics.
Finally, concerning the novel MM potentials
inspired by the QM
SAPT methodology, it is clear that the multipolar electrostatics (interfragment)
must be augmented by the CP potentials (intrafragment) if one seeks
to reproduce the global electrostatics 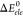 . Nevertheless, the IQF/IQA approach (and
other EDAs) points out that the intramolecular electrostatic energy
is closely related with other energy changes induced by fragment overlap
(e.g., deformation and interfragment exchange-correlation energy),
suggesting thus that the separate treatment of these effects by means
of independent potential terms might be inefficient and hamper parameter
development and transferability.
. Nevertheless, the IQF/IQA approach (and
other EDAs) points out that the intramolecular electrostatic energy
is closely related with other energy changes induced by fragment overlap
(e.g., deformation and interfragment exchange-correlation energy),
suggesting thus that the separate treatment of these effects by means
of independent potential terms might be inefficient and hamper parameter
development and transferability.
Acknowledgments
F.J.-G. and D.S. acknowledge the Spanish MICINN (Grant PGC2018-095953-B-I00), the FICyT (Grant FC-GRUPIN-IDI/2018/000177), and the European Union FEDER for financial support. F.J.-G. specially acknowledges the Spanish MICINN for a predoctoral grant (BES-2016-076986). Both authors are greatly grateful to Evelio Francisco for his contribution with the development of the MPOLINT code and to Ángel Martín Pendás for his valuable suggestions and careful reading of the manuscript.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jctc.1c00263.
(1) Multipole expansion formulation, (2) a brief description of the CP corrections, (3) classification of the S66 complexes, (4) HF-D3–CCSD(T) comparison, (5) statistical analysis of the fully relaxed and zeroth-order electrostatic descriptions, (6) correlation study between the interatomic
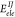 and
and 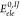 terms, and (7) atomic interactions within
the acetic acid dimer (PDF)
terms, and (7) atomic interactions within
the acetic acid dimer (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ren P.; Chun J.; Thomas D. G.; Schnieders M. J.; Marucho M.; Zhang J.; Baker N. A. Biomolecular electrostatics and solvation: a computational perspective. Q. Rev. Biophys. 2012, 45, 427–491. 10.1017/S003358351200011X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cisneros G. A.; Karttunen M.; Ren P.; Sagui C. Classical Electrostatics for Biomolecular Simulations. Chem. Rev. 2014, 114, 779–814. 10.1021/cr300461d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewald P. P. Die Berechnung optischer und elektrostatischer Gitterpotentiale. Ann. Phys. 1921, 369, 253–287. 10.1002/andp.19213690304. [DOI] [Google Scholar]
- Hockney R. W.; Eastwood J. W.. Computer Simulation Using Particles; IOP Publishing Ltd.: 1988. [Google Scholar]
- Pollock E.; Glosli J. Comments on P3M, FMM, and the Ewald method for large periodic Coulombic systems. Comput. Phys. Commun. 1996, 95, 93–110. 10.1016/0010-4655(96)00043-4. [DOI] [Google Scholar]
- Darden T.; York D.; Pedersen L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. 10.1063/1.464397. [DOI] [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T.; Lee H.; Pedersen L. G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. 10.1063/1.470117. [DOI] [Google Scholar]
- Herce D. H.; Perera L.; Darden T. A.; Sagui C. Surface solvation for an ion in a water cluster. J. Chem. Phys. 2005, 122, 024513. 10.1063/1.1829635. [DOI] [PubMed] [Google Scholar]
- Stone A.The Theory of Intermolecular Forces; Oxford University Press: 2013. [Google Scholar]
- Bojarowski S. A.; Kumar P.; Dominiak P. M. A Universal and Straightforward Approach to Include Penetration Effects in Electrostatic Interaction Energy Estimation. ChemPhysChem 2016, 17, 2455–2460. 10.1002/cphc.201600390. [DOI] [PubMed] [Google Scholar]
- Freitag M. A.; Gordon M. S.; Jensen J. H.; Stevens W. J. Evaluation of charge penetration between distributed multipolar expansions. J. Chem. Phys. 2000, 112, 7300–7306. 10.1063/1.481370. [DOI] [Google Scholar]
- Wang B.; Truhlar D. G. Including Charge Penetration Effects in Molecular Modeling. J. Chem. Theory Comput. 2010, 6, 3330–3342. 10.1021/ct1003862. [DOI] [PubMed] [Google Scholar]
- Wang B.; Truhlar D. G. Screened Electrostatic Interactions in Molecular Mechanics. J. Chem. Theory Comput. 2014, 10, 4480–4487. 10.1021/ct5005142. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Rackers J. A.; He C.; Qi R.; Narth C.; Lagardere L.; Gresh N.; Ponder J. W.; Piquemal J.-P.; Ren P. General Model for Treating Short-Range Electrostatic Penetration in a Molecular Mechanics Force Field. J. Chem. Theory Comput. 2015, 11, 2609–2618. 10.1021/acs.jctc.5b00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco E.; Martín Pendás A.. Energy Partition Analyses: Symmetry-Adapted Perturbation Theory and Other Techniques. In Non-Covalent Interactions in Quantum Chemistry and Physics; Otero de la Roza A., DiLabio G. A., Eds.; Elsevier: 2017; Chapter 2, pp 27–64. [Google Scholar]
- Jeziorski B.; Moszynski R.; Szalewicz K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. 10.1021/cr00031a008. [DOI] [Google Scholar]
- Patkowski K. Recent developments in symmetry-adapted perturbation theory. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2020, 10, e1452. 10.1002/wcms.1452. [DOI] [Google Scholar]
- Kitaura K.; Morokuma K. A new energy decomposition scheme for molecular interactions within the Hartree-Fock approximation. Int. J. Quantum Chem. 1976, 10, 325–340. 10.1002/qua.560100211. [DOI] [Google Scholar]
- Bickelhaupt F. M.; Baerends E. J.. Kohn-Sham Density Functional Theory: Predicting and Understanding Chemistry. Reviews in Computational Chemistry; John Wiley Sons, Ltd.: 2000; pp 1–86. [Google Scholar]
- Zhao L.; von Hopffgarten M.; Andrada D. M.; Frenking G. Energy decomposition analysis. Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2018, 8, e1345. 10.1002/wcms.1345. [DOI] [Google Scholar]
- Blanco M. A.; Martin Pendas A.; Francisco E. Interacting Quantum Atoms: A Correlated Energy Decomposition Scheme Based on the Quantum Theory of Atoms in Molecules. J. Chem. Theory Comput. 2005, 1, 1096–1109. 10.1021/ct0501093. [DOI] [PubMed] [Google Scholar]
- Francisco E.; Martin Pendas A.; Blanco M. A. A Molecular Energy Decomposition Scheme for Atoms in Molecules. J. Chem. Theory Comput. 2006, 2, 90–102. 10.1021/ct0502209. [DOI] [PubMed] [Google Scholar]
- Naseem-Khan S.; Gresh N.; Misquitta A. J.; Piquemal J.-P. Assessment of SAPT and Supermolecular EDA Approaches for the Development of Separable and Polarizable Force Fields. J. Chem. Theory Comput. 2021, 17, 2759–2774. 10.1021/acs.jctc.0c01337. [DOI] [PubMed] [Google Scholar]
- Liu C.; Piquemal J.-P.; Ren P. AMOEBA+ Classical Potential for Modeling Molecular Interactions. J. Chem. Theory Comput. 2019, 15, 4122–4139. 10.1021/acs.jctc.9b00261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandenbrande S.; Waroquier M.; Speybroeck V. V.; Verstraelen T. The Monomer Electron Density Force Field (MEDFF): A Physically Inspired Model for Noncovalent Interactions. J. Chem. Theory Comput. 2017, 13, 161–179. 10.1021/acs.jctc.6b00969. [DOI] [PubMed] [Google Scholar]
- Mao Y.; Demerdash O.; Head-Gordon M.; Head-Gordon T. Assessing Ion–Water Interactions in the AMOEBA Force Field Using Energy Decomposition Analysis of Electronic Structure Calculations. J. Chem. Theory Comput. 2016, 12, 5422–5437. 10.1021/acs.jctc.6b00764. [DOI] [PubMed] [Google Scholar]
- Pendás A. M.; Casals-Sainz J. L.; Francisco E. On Electrostatics, Covalency, and Chemical Dashes: Physical Interactions versus Chemical Bonds. Chem. - Eur. J. 2019, 25, 309–314. 10.1002/chem.201804160. [DOI] [PubMed] [Google Scholar]
- Massa L.; Keith T.; Cheng Y.; Matta C. F. The kernel energy method applied to quantum theory of atoms in molecules – energies of interacting quantum atoms. Chem. Phys. Lett. 2019, 734, 136650. 10.1016/j.cplett.2019.136650. [DOI] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. S66: A Well-balanced Database of Benchmark Interaction Energies Relevant to Biomolecular Structures. J. Chem. Theory Comput. 2011, 7, 2427–2438. 10.1021/ct2002946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Řezáč J.; Riley K. E.; Hobza P. Extensions of the S66 Data Set: More Accurate Interaction Energies and Angular-Displaced Nonequilibrium Geometries. J. Chem. Theory Comput. 2011, 7, 3466–3470. 10.1021/ct200523a. [DOI] [Google Scholar]
- Bader R.Atoms in molecules: a quantum theory; Clarendon Press: Oxford; New York, 1990. [Google Scholar]
- Suárez D.; Díaz N.; Francisco E.; Martín Pendás A. Application of the Interacting Quantum Atoms Approach to the S66 and Ionic-Hydrogen-Bond Datasets for Noncovalent Interactions. ChemPhysChem 2018, 19, 973–987. 10.1002/cphc.201701021. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Becke A. D.; Johnson E. R. A density-functional model of the dispersion interaction. J. Chem. Phys. 2005, 123, 154101. 10.1063/1.2065267. [DOI] [PubMed] [Google Scholar]
- Stone A. Distributed multipole analysis, or how to describe a molecular charge distribution. Chem. Phys. Lett. 1981, 83, 233–239. 10.1016/0009-2614(81)85452-8. [DOI] [Google Scholar]
- Stone A.; Alderton M. Distributed multipole analysis. Mol. Phys. 1985, 56, 1047–1064. 10.1080/00268978500102891. [DOI] [Google Scholar]
- Salomon-Ferrer R.; Case D. A.; Walker R. C. An overview of the Amber biomolecular simulation package. WIREs: Comput. Mol. Sci. 2013, 3, 198–210. 10.1002/wcms.1121. [DOI] [Google Scholar]
- Vanommeslaeghe K.; Hatcher E.; Acharya C.; Kundu S.; Zhong S.; Shim J.; Darian E.; Guvench O.; Lopes P.; Vorobyov I.; Mackerell A. D. Jr. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2009, 31, 671–690. 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oostenbrink C.; Soares T. A.; van der Vegt N. F. A.; van Gunsteren W. F. Validation of the 53A6 GROMOS force field. Eur. Biophys. J. 2005, 34, 273–284. 10.1007/s00249-004-0448-6. [DOI] [PubMed] [Google Scholar]
- Jorgensen W. L.; Maxwell D. S.; Tirado-Rives J. Development and Testing of the OPLS All-Atom Force Field on Conformational Energetics and Properties of Organic Liquids. J. Am. Chem. Soc. 1996, 118, 11225–11236. 10.1021/ja9621760. [DOI] [Google Scholar]
- Engkvist O.; Åstrand P.-O.; Karlström G. Accurate Intermolecular Potentials Obtained from Molecular Wave Functions: Bridging the Gap between Quantum Chemistry and Molecular Simulations. Chem. Rev. 2000, 100, 4087–4108. 10.1021/cr9900477. [DOI] [PubMed] [Google Scholar]
- Ponder J. W.; Wu C.; Ren P.; Pande V. S.; Chodera J. D.; Schnieders M. J.; Haque I.; Mobley D. L.; Lambrecht D. S.; DiStasio R. A.; Head-Gordon M.; Clark G. N. I.; Johnson M. E.; Head-Gordon T. Current Status of the AMOEBA Polarizable Force Field. J. Phys. Chem. B 2010, 114, 2549–2564. 10.1021/jp910674d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popelier P. L. A. QCTFF: On the construction of a novel protein force field. Int. J. Quantum Chem. 2015, 115, 1005–1011. 10.1002/qua.24900. [DOI] [Google Scholar]
- Hughes Z. E.; Thacker J. C. R.; Wilson A. L.; Popelier P. L. A. Description of Potential Energy Surfaces of Molecules Using FFLUX Machine Learning Models. J. Chem. Theory Comput. 2019, 15, 116–126. 10.1021/acs.jctc.8b00806. [DOI] [PubMed] [Google Scholar]
- Gresh N.; Cisneros G. A.; Darden T. A.; Piquemal J.-P. Anisotropic, Polarizable Molecular Mechanics Studies of Inter- and Intramolecular Interactions and Ligand-Macromolecule Complexes. A Bottom-Up Strategy. J. Chem. Theory Comput. 2007, 3, 1960–1986. 10.1021/ct700134r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon M. S.; Freitag M. A.; Bandyopadhyay P.; Jensen J. H.; Kairys V.; Stevens W. J. The Effective Fragment Potential Method: A QM-Based MM Approach to Modeling Environmental Effects in Chemistry. J. Phys. Chem. A 2001, 105, 293–307. 10.1021/jp002747h. [DOI] [Google Scholar]
- Bojarowski S. A.; Kumar P.; Dominiak P. M. Interplay of point multipole moments and charge penetration for intermolecular electrostatic interaction energies from the University at Buffalo pseudoatom databank model of electron density. Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater. 2017, 73, 598–609. 10.1107/S2052520617005510. [DOI] [PubMed] [Google Scholar]
- Rackers J. A.; Wang Q.; Liu C.; Piquemal J.-P.; Ren P.; Ponder J. W. An optimized charge penetration model for use with the AMOEBA force field. Phys. Chem. Chem. Phys. 2017, 19, 276–291. 10.1039/C6CP06017J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar P.; Bojarowski S. A.; Jarzembska K. N.; Domagała S.; Vanommeslaeghe K.; MacKerell A. D.; Dominiak P. M. A Comparative Study of Transferable Aspherical Pseudoatom Databank and Classical Force Fields for Predicting Electrostatic Interactions in Molecular Dimers. J. Chem. Theory Comput. 2014, 10, 1652–1664. 10.1021/ct4011129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kairys V.; Jensen J. H. Evaluation of the charge penetration energy between non-orthogonal molecular orbitals using the Spherical Gaussian Overlap approximation. Chem. Phys. Lett. 1999, 315, 140–144. 10.1016/S0009-2614(99)01187-2. [DOI] [Google Scholar]
- Schmidt M. W.; Baldridge K. K.; Boatz J. A.; Elbert S. T.; Gordon M. S.; Jensen J. H.; Koseki S.; Matsunaga N.; Nguyen K. A.; Su S.; Windus T. L.; Dupuis M.; Montgomery J. A. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. 10.1002/jcc.540141112. [DOI] [Google Scholar]
- Grimme S.; Antony J.; Ehrlich S.; Krieg H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- Grimme S.; Ehrlich S.; Goerigk L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. 10.1002/jcc.21759. [DOI] [PubMed] [Google Scholar]
- Goerigk L.; Collyer C. A.; Reimers J. R. Recommending Hartree–Fock Theory with London-Dispersion and Basis-Set-Superposition Corrections for the Optimization or Quantum Refinement of Protein Structures. J. Phys. Chem. B 2014, 118, 14612–14626. 10.1021/jp510148h. [DOI] [PubMed] [Google Scholar]
- Sedlak R.; Řezáč J. Empirical D3 Dispersion as a Replacement for ab Initio Dispersion Terms in Density Functional Theory-Based Symmetry-Adapted Perturbation Theory. J. Chem. Theory Comput. 2017, 13, 1638–1646. 10.1021/acs.jctc.6b01198. [DOI] [PubMed] [Google Scholar]
- Martín Pendás Á.; Francisco E. PROMOLDEN. A QTAIM/IQA code (unpublished). [Google Scholar]
- Wang J.; Wolf R. M.; Caldwell J. W.; Kollman P. A.; Case D. A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Ren P.; Wu C.; Ponder J. W. Polarizable Atomic Multipole-Based Molecular Mechanics for Organic Molecules. J. Chem. Theory Comput. 2011, 7, 3143–3161. 10.1021/ct200304d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francisco E. MPOLINT. A program to calculate Coulombic interactions by means of the multipole approximation (unpublished). [Google Scholar]
- Eaton J. W.; Bateman D.; Hauberg S.; Wehbring R.. GNU Octave version 4.4.0 manual: a high-level interactive language for numerical computations; 2018. [Google Scholar]
- Williams T.; Kelley C.; et al. Gnuplot 5.2: an Interactive Plotting Program; 2019. http://gnuplot.sourceforge.net/.
- Hunter J. D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. 10.1109/MCSE.2007.55. [DOI] [Google Scholar]
- Popelier P. L. A.; Kosov D. S. Atom–atom partitioning of intramolecular and intermolecular Coulomb energy. J. Chem. Phys. 2001, 114, 6539–6547. 10.1063/1.1356013. [DOI] [Google Scholar]
- Yuan Y.; Mills M. J. L.; Popelier P. L. A. Multipolar electrostatics for proteins: Atom-atom electrostatic energies in crambin. J. Comput. Chem. 2014, 35, 343–359. 10.1002/jcc.23469. [DOI] [PubMed] [Google Scholar]
- Zgarbová M.; Otyepka M.; Šponer J.; Hobza P.; Jurečka P. Large-scale compensation of errors in pairwise-additive empirical force fields: comparison of AMBER intermolecular terms with rigorous DFT-SAPT calculations. Phys. Chem. Chem. Phys. 2010, 12, 10476–10493. 10.1039/c002656e. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.