

Abstract

A general and efficient method for the deconjugative α-alkylation of α,β-unsaturated aldehydes promoted by a synergistic effect between tBuOK and NaH, which considerably increases the reaction rate under mild conditions, is reported. The β,γ-unsaturated aldehyde, resulting from the α-alkylation, is transformed in high yield into the corresponding allyl acetate via a lead(IV) acetate-mediated oxidative fragmentation. This strategy could be used for the construction of the carbon skeleton of a wide variety of alkyl or arylterpenoids.
Introduction
Terpenoids are a broad group of natural products, characterized by their structural diversity, that have found extensive use in medicine and other industries.1−3 However, their scarcity in their natural, marine, or terrestrial sources often forces the development of synthetic processes for their availability. Within the enormous variety of synthetic processes described to access this type of compounds, those that use simple terpenoids as a starting product to prepare more complex molecules deserve to be highlighted.4 This has the obvious advantage of reducing the number of synthetic sequence steps. In addition, if the terpene precursor is easily accessible, and therefore cheap, the process may have commercial interest.
β-Cyclocitral (1) is a cyclic monoterpene, commercially available at a low price, which has been used as a precursor of different terpenoids (Scheme 1). In this way, the reaction of aldehyde 1 with the suitable benzyllithium derivative 2 afforded the corresponding allyl homobenzyl alcohol, the precursor of the abietane quinone taxodione (3)5 and the totarane quinone maytenoquinone (4),6 both with antitumor properties, or the antifungal tricyclic diterpenoid 5.7 When β-cyclocitral (1) was treated with the suitable aryllithium derivative 6, taiwaniaquinoids such as dichroanone (7),8 taiwaniaquinone H (8),8 and dichroanal B (9)8b were obtained. These processes involve the condensation of a terpene synthon having electrophilic character with an aromatic derivative of nucleophile nature.
Scheme 1. Synthesis of Terpenoids from β-Cyclocitral (1) and Aryllithium Derivatives.

However, in certain cases, preparation of nucleophilic aromatic synthons became difficult because of either the substitution pattern or the presence of electrophilic groups (e.g., CN, COOR, COR, and so forth) in the aromatic ring, resulting in the consequent lengthening sequence. It is therefore of interest to investigate new approaches to avoid this inconvenience.
The construction of the carbon skeleton of compounds such as 3–5 and 7–9 could be achieved in an alternative way by performing the α-alkylation of the α,β-unsaturated terpenic aldehyde 1 with a suitable benzyl halide to afford the corresponding arylterpenylaldehydes, followed by cyclization. Despite its synthetic potential, the deconjugative α-alkylation of aldehydes has been studied very little (Scheme 2). In a pioneer work, De Graaf et al. described the direct alkylation of 1-cyclohexene-1-carbaldehyde with different agents in liquid ammonia at −60 °C in the presence of potassium amide, affording a mixture of products.9 After that, a procedure to achieve the direct α-alkylation of acyclic aldehydes utilizing NaOH and a phase-transfer agent in an inert solvent has been patented.10 Additionally, the palladium- and nickel-catalyzed deconjugative α-allylation with allyl alcohols of aldehydes, including a few α,β-unsaturated aldehydes, has also been described (Scheme 2).11 Other indirect methods, such as the alkylation of the corresponding dimethylhydrazones, in the presence of lead lithium diisopropylamide (LDA), have been described.12 Recently, the copper-catalyzed deconjugative α-alkylation of cyclic α,β-unsaturated nitriles has been reported.13
Scheme 2. Previous Direct α-Alkylation of α,β-Unsaturated Aldehydes.

In this paper, we describe the deconjugative α-alkylation of cyclohexene-1-carboxaldehydes and its synthetic application as a platform to access challenging terpene frameworks. In comparison to other existing direct α-alkylation of α,β-unsaturated aldehydes (Scheme 2), this methodology occurs under milder metal-free reaction conditions with a wider scope and compatible with electrophilic groups (Scheme 3).
Scheme 3. Synthetic Approaches to Terpenoids; (a) Conventional Methods for the Synthesis of Terpenoids from β-Cyclocitral (1) as an Electrophile; (b) Deconjugative α-Alkylation of Cyclohexene-1-carboxaldehydes for the Synthesis of Terpenoids.

Results and Discussion
Considering our working hypothesis, we initiated the study of the α-alkylation of β-cyclocitral (1) using allyl bromide (10a) as the electrophile. The reaction was performed in different solvents such as tetrahydrofuran (THF), acetonitrile, and toluene in the presence of tBuOK as a base, and the desired α-alkylated product (±)-11a, formed selectively from a trisubstituted dienolate, was obtained in poor to moderate yields (Table 1, entries 1–3). A higher amount of tBuOK did not improve the yield either (Table 1, entry 4). The addition of 18-crown-6-ether led to a mixture of (±)-11a and the corresponding O-alkylated derivative 12a in THF, acetonitrile, and toluene (Table 1, entries 5–7). Then, the effect of other bases was also considered in the model reaction. LiHMDS increased the C-/O-alkylation ratio [(±)-11a/12a ratio] up to 5:1 (Table 1, entry 8). On the contrary, the deconjugative α-alkylation of 1 with LDA did not work (Table 1, entry 9). In a similar fashion, NaH led to unreacted starting material in toluene, whereas a 1:1 mixture of (±)-11a/12a was obtained in THF at 60 °C (Table 1, entries 10–11). Gratifyingly, C-alkylation was selective over O-alkylation by using NaH and tBuOK in toluene, affording (±)-11a in 83% yield (Table 1, entry 12). This result pointed out that NaH could facilitate a fast deprotonation of tBuOH formed from the enolization and shift the equilibrium toward its conjugate base. In order to rule out that the possible formation in situ of tBuONa in the reaction mixture from tBuOK and NaH could be responsible for the improvement of the reaction yield, a control experiment was carried out with just tBuONa. A low conversion was observed for the reaction, which was increased after addition of NaH (Table 1, entries 13–14). The use of tBuONa/KH gave rise to a similar yield for (±)-11a to that observed with the pair tBuOK/NaH (Table 1, entry 15). These results support that tBuOH generated in the reaction is quenched by a metal hydride. Finally, we also tested a decrease of the amount of tBuOK to 0.5 equiv, giving rise to the lowering of the reaction conversion in comparison with using a stoichiometric amount of the base. However, a similar yield was obtained after heating at 60 °C for 1 h (Table 1, entries 16–17 vs entry 12).
Table 1. Optimization of Deconjugative α-Alkylation of 1 with Allyl Bromide (10a)a.

| entry | base | solvent | time (h) | yield 11a (%)b | yield 12a (%)b |
|---|---|---|---|---|---|
| 1 | tBuOK | THF | 16 | 8 | 0 |
| 2 | tBuOK | CH3CN | 16 | 33 | 0 |
| 3 | tBuOK | toluene | 16 | 43 | 0 |
| 4c | tBuOK | toluene | 16 | 48 | 0 |
| 5d | tBuOK | THF | 6 | 20 | 56 |
| 6d | tBuOK | CH3CN | 6 | 30 | 58 |
| 7d | tBuOK | toluene | 6 | 69 | 26 |
| 8 | LiHMDS | THF | 4 | 75 | 15 |
| 9e | LDA | THF | 4 | 0 | 0 |
| 10f | NaH | toluene | 16 | 0 | 0 |
| 11f,g | NaH | THF | 6 | 25 | 25 |
| 12f | NaH–tBuOK | toluene | 1 | 83 | 0 |
| 13 | tBuONa | toluene | 15 | 10 | 0 |
| 14h | NaH–tBuONa | toluene | 2 | 80 | 0 |
| 15i | KH–tBuONa | toluene | 3 | 73 | 0 |
| 16f,j | NaH–tBuOK | toluene | 13 | 30 | 0 |
| 17f,j,k | NaH–tBuOK | toluene | 5 | 78 | 0 |
The reaction was carried out with 1 (1.0 mmol), the base (1.1 mmol), and the solvent (20 mL). After 45 min, allyl bromide (1.5 mmol) was added.
Isolated yields.
3 equiv of tBuOK was used.
1 equiv of 18-crown-6-ether was used.
The reaction was carried out at −78 °C and allowed to warm to room temperature.
2 equiv of NaH 60% in the oil mineral was used.
The reaction was carried out at 60 °C.
Isolated yield after addition of 2 equiv of NaH 60% in the oil mineral.
2 equiv of KH 30% in the oil mineral was used.
0.5 equiv of tBuOK was used.
Isolated yield after heating at 60 °C.
The experiments shown in Table 1 confirm that the synergistic effect between tBuOK/tBuONa and metal hydrides plays a crucial role in the conversion and selectivity of the deconjugative α-alkylation, influencing the kinetic and increasing the reaction rate. A cooperative interaction between tBuOK and strong bases has been previously reported for the synthesis of 1-indanones from β-alkynyl ketones.14
With the optimized reaction conditions in hand, the scope and limitations of the deconjugative α-alkylation of β-cyclocitral (1) were evaluated with a series of activated alkyl and benzyl halides. In most cases, it undergoes α-alkylation in high to moderate yields under smooth conditions and short reaction times (Table 2). Methyl iodide (10b) and benzyl bromide (10e) promoted the deconjugative α-alkylation reaction in higher reaction yields (Table 2, entries 1 and 4) in comparison to the yields obtained when allylic or propargylic halides 10c–d were used (Table 2, entries 2–3). However, the effect of the electron-withdrawing or electron-releasing character of the benzyl group as well as the position of the substituents in the aromatic ring appears to affect the reaction course significantly. Strongly electron-releasing methoxy groups at meta- or para-positions on the aromatic ring and 1,3-benzodioxole groups afforded the corresponding deconjugated aldehydes (±)-11f and (±)-11h–i in moderate to good yields (Table 2, entries 5–8), whereas ortho-methoxy groups furnished the corresponding products (±)-11j–l in moderate yields (Table 2, entries 9–11). Furthermore, the use of benzyl bromides 10g and 10k gave slightly higher reaction yields than the corresponding benzyl chlorides 10f and 10j (Table 2, entry 5 vs entry 6 and entry 9 vs entry 10). Benzyl bromides bearing a bromo substituent at the ortho-position of the aromatic ring afforded the desired deconjugated aldehydes (±)-11m–n in good yields (Table 2, entries 12–13). Finally, the reaction was also compatible with other electrophilic groups, such as NO2 or CN, on the aromatic ring. Thus, compounds (±)-11o–p were obtained in the range of 77–80% yields (Table 2, entries 14–15).
Table 2. Reaction of β-Cyclocitral (1) with Activated Alkyl and Benzyl Halidesa.


Unless specified, the reaction was carried out with 1 (1.0 mmol), tBuOK (1.1 mmol), NaH (2 mmol), and toluene (20 mL). After 45 min, alkyl halide (1.5 mmol) was added.
Isolated yields.
Similar yields were obtained using 1 equiv of 18-crown-6 in the absence of NaH.
In order to demonstrate the general applicability of the deconjugative α-alkylation and to explore the further use of this reaction for getting access to other types of terpenoids, the alkylation of other α,β-unsaturated aldehydes with benzyl bromides has also been investigated (Table 3).
Table 3. Reaction of α,β-Unsaturated Aldehydes with Benzyl Bromidesa.


Unless specified, the reaction was carried out with 1.0 mmol of aldehyde, tBuOK (1.1 mmol), NaH (2 mmol), and toluene (20 mL). After 45 min, alkyl halide (1.5 mmol) was added.
Isolated yields.
Similar yields were obtained using 1 equiv of 18-crown-6 in the absence of NaH.
An approximate 1:1 epimeric mixture of 14b was deduced from the 13C NMR spectrum.
Cyclohex-1-ene-1-carboxaldehydes 13a and 13b, which lack substituents on carbon β, underwent benzylation in high yield in a short reaction time (Table 3, entries 1 and 2). The bicyclic sesquiterpene aldehydes 13c and 13d, whose absolute configuration is well known, also underwent this reaction in high yield, affording benzyl derivatives 14c and 14d, with complete diastereoselectivity (Table 3, entries 3 and 4). The disposition of the aldehyde group in both products has been confirmed by NOE experiments. The behavior of Δ8-drimenals 13c and 13d indicates the possibility of using this type of sesquiterpene α,β-unsaturated aldehydes to synthesize a large group of terpenoids such as benzofluorene derivatives by reaction with the appropriate benzyl halide.21 Finally, the α,β,γ,δ-unsaturated aldehyde safranal (13e) gave the corresponding α-benzylated β,γ,δ,ε-unsaturated aldehyde 14e in moderate yield (Table 3, entry 5).
The presence of the aldehyde group in these intermediates notably increases the synthetic potential of this new strategy. The formyl group can be removed and allow to introduce functionality in the final compounds. An interesting example of the latter would be the direct transformation of the β,γ-unsaturated aldehyde type 11 into the allyl acetates 15 via a lead(IV) acetate-mediated oxidative fragmentation. LTA has been previously used for the oxidative transformation of homoallyl alcohols to afford allyl acetate derivatives. The reaction proceeded with complete stereoselectivity; the acetyloxy group of the rearranged product and the hydroxymethyl group in the starting material are located at the same face of the molecule.22
To expand the scope of Preite’s reaction and illustrate the usefulness of this transformation, some of the synthesized β,γ-unsaturated aldehydes 11 were tested, and the results are shown in Table 4. The treatment of β,γ-unsaturated aldehydes (±)-11a, (±)-11e, (±)-11i, (±)-11m, and (±)-11o with Pb(OAc)4 in refluxing benzene afforded the corresponding allyl acetates (±)-15a, (±)-15e, (±)-15i, (±)-15m, and (±)-15o, respectively, in high yields and short reaction times (Table 4). With the aim of confirming the stereoselectivity previously described for the similar oxidative cleavage of homoallyl alcohols, we carried out the reaction with compound 14c. Unfortunately, it failed and produced a complex mixture. However, our results are in agreement with the concerted mechanistic pathway proposed by Preite et al. where a syn stereoselectivity was observed, with the hydroxymethyl group of the starting material and the acetyloxy group of rearranged compounds located at the same side of the molecule.22
Table 4. Transformation of β,γ-Unsaturated Aldehydes 11 into Allyl Acetates 15a.


Unless specified, the reaction was carried out with β,γ-unsaturated aldehyde (1 mmol), Pb(OAc)4 (1.1 mmol), and benzene (7 mL) at 80 °C.
Isolated yields.
Based on our experimental results and the literature precsedents,22,23Scheme 4 shows two tentative mechanistic pathways for the direct oxidative fragmentation of β,γ-unsaturated aldehydes. In a first step, one of the acetate groups of Pb(OAc)4 would be exchanged by the oxygen atom of the β,γ-unsaturated aldehyde to give intermediate I. Then, the released acetate ion could attack again the more accessible Pb(IV), with the simultaneous intramolecular nucleophilic addition of another Pb(IV) acetate group to the complexed aldehyde to afford intermediate II. The attack of one of the acetate groups to the olefin through a nine-membered cyclic transition state would generate the allyl acetate with the simultaneous removal of Pb(OAc)2 and mixed anhydride (pathway 1). However, a seven-membered cyclic transition state cannot be ruled out to give rise to intermediate III, which would be hydrolyzed to generate the allyl acetate (pathway 2).
Scheme 4. Tentative Mechanisms for the LTA-Mediated Transformation of β,γ-Unsaturated Aldehyde into Allyl Acetates.

Conclusions
In summary, a new alternative strategy for synthesizing a wide variety of terpenoid precursors is reported. This approach is based on the reaction of an electrophilic activated alkyl or benzyl halide with a nucleophilic terpenic α,β-unsaturated aldehyde. tBuOK and NaH interact synergistically, enhancing notably the kinetics and the selectivity for the C-alkylated derivatives. The β,γ-unsaturated aldehydes resulting from this alkylation undergo a lead(IV) acetate-mediated oxidative fragmentation, affording in high yield the corresponding allyl acetates. The deconjugative α-alkylation of α,β-unsaturated aldehydes and deformylation reaction described in this paper could serve as a platform to access in an efficient and simple manner to a wide variety of terpenoid skeletons.
Experimental Section
General Procedures
Aldehydes 1, 13a, 13b, and 13e were obtained from commercial suppliers and used without further purification. Unless stated otherwise, reactions were performed in oven-dried glassware under an argon atmosphere using dry solvents. Solvents were dried as follows: THF, diethyl ether (Et2O), and toluene over Na-benzophenone; dichloromethane (DCM) over CaH2; and acetonitrile over molecular sieves 4 Å. An oil bath was used as the heating source for the reactions that require heating. Thin-layer chromatography (TLC) was performed using F254 precoated plates (0.25 mm) and visualized by UV fluorescence quenching and phosphomolybdic acid solution in ethanol staining. Flash chromatography was performed on silica gel (230–400 mesh). Chromatography separations were carried out using a conventional column on silica gel 60 (230–400 mesh) using hexanes–AcOEt (AcOEt–hexane) or diethyl ether–hexane (ether–hexane) mixtures of increasing polarity. 1H and 13C{1H} NMR spectra were recorded at 600, 500, and 400 MHz and at 150, 125, and 100 MHz, respectively. CDCl3 was treated with K2CO3. Chemical shifts (δ H) are quoted in parts per million (ppm) referenced to the appropriate residual solvent peak and tetramethylsilane. Data for 1H NMR spectra are reported as follows: chemical shift (δ ppm) [multiplicity, coupling constant (Hz), and integration], with the abbreviations s, br s, d, br d, t, dt, dq, sept, and m denoting the singlet, broad singlet, doublet, broad doublet, triplet, doublet triplet, doublet quartet, septet, and multiplet, respectively. J = coupling constant in hertz (Hz). Data for 13C{1H} NMR spectra are reported in terms of chemical shift relative to Me4Si (δ 0.0), and the signals were assigned utilizing DEPT experiments and on the basis of heteronuclear correlations. Infrared (IR) spectra were recorded as thin films or as solids on an FTIR spectrophotometer with samples between sodium chloride plates or as potassium bromide pellets and are reported in frequency of absorption (cm–1). Only selected absorbances (νmax) are reported. ([α]D) measurements were carried out in a polarimeter utilizing a 1 dm length cell and CHCl3 as a solvent. Concentration is expressed in mg/mL. High-resolution mass spectra were recorded on a spectrometer utilizing a Q-TOF analyzer and ESI+ ionization.
General Procedure for the Preparation of Alkyl Bromides 10g–i, 10n, and 10p
To a solution of the corresponding commercially available alcohol (1 mmol) in diethyl ether (10 mL) was added PBr3 (1 mmol) at 0 °C, and the mixture was stirred at this temperature, being monitorized by TLC. Then, it was quenched with water (3 mL) and diluted with Et2O (20 mL). The organic layer was washed with water (3 × 10 mL), dried with anhydrous Na2SO4, and filtered, and the solvent was evaporated under reduced pressure to give the corresponding alkyl bromides 10g–i, 10n, and 10p.
1-(Bromomethyl)-4-methoxybenzene (10g)
Yellow oil [yield 537 mg (89%) from 414.6 mg (3 mmol) of (4-methoxyphenyl)methanol]. The spectroscopic data were in agreement with those described in the literature.15
1-Bromo-5-(bromomethyl)-2,3-dimethoxybenzene (10h)
White solid [yield 790 mg (85%) from 740.8 mg (3 mmol) of (3-bromo-4,5-dimethoxyphenyl)methanol]. The spectroscopic data were in agreement with those described in the literature.16
5-(Bromomethyl)benzo[d][1,3]dioxole (10i)
White solid [yield 619 mg (96%) from 456.2 mg (3 mmol) of benzo[d][1,3]dioxol-5-ylmethanol]. The spectroscopic data were in agreement with those described in the literature.17
5-Bromo-6-(bromomethyl)benzo[d][1,3]dioxole (10n)
White solid [yield 776 mg (88%) from 693.1 mg (3 mmol) of (6-bromobenzo[d][1,3]dioxol-5-yl)methanol]. The spectroscopic data were in agreement with those described in the literature.18
4-(Bromomethyl)benzonitrile (10p)
White solid [yield 531 mg (82%) from 439.8 mg (3 mmol) of 4-(hydroxymethyl)benzonitrile]. The spectroscopic data were in agreement with those described in the literature.19
Preparation of 1-(Chloromethyl)-3-isopropyl-2-methoxybenzene (10j)
A round-bottom flask charged with 2-isopropylphenol (10 g, 73 mmol) and p-formaldehyde (4.4 g, 88.8 mmol) in dry DCM (150 mL) under an argon atmosphere was cooled at −30 °C. Then, Et2AlCl (25% wt solution in toluene, 47.6 mL, 87.6 mmol) was added dropwise, and the reaction mixture was allowed to warm to room temperature. The reaction mixture was stirred for 3 h and was quenched with water (50 mL). DCM was evaporated under reduced pressure, and the aqueous layer was extracted with ethyl acetate (3 × 100 mL). The combined organic solution was dried over anhydrous sodium sulfate and filtered. The solvent was evaporated under reduced pressure to give a crude product which was purified by silica gel column chromatography (20% AcOEt/hexane), affording 2-(hydroxymethyl)-6-isopropylphenol. Colorless oil (10.97 g, 90%). 1H NMR (400 MHz, CD3COCD3): δ 8.46 (br s, OH), 7.14 (dd, J = 7.6, 1.7 Hz, 1H), 6.91 (m, 1H), 6.80 (t, J = 7.6 Hz, 1H), 5.23 (br s, OH), 4.87 (s, 2H), 3.40 (sept, J = 6.9 Hz, 1H), 1.26 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (100 MHz, CD3COCD3): δ 154.5 (C), 135.9 (C), 125.9 (CH), 125.8 (C), 125.5 (CH), 120.0 (CH), 64.4 (CH2), 27.1 (CH), 22.9 (CH3). IR (film): 3053, 2965, 1456, 1421, 1264, 733, 704 cm–1. HRMS (ESI/TOF) m/z: [M + Na]+ calcd for C10H14O2Na, 189.0891; found, 189.0887.
K2CO3 (13.65 g, 99 mmol) was added at 0 °C to a stirred solution of 2-(hydroxymethyl)-6-isopropylphenol (10.97 g, 66 mmol) in acetone (120 mL). After 15 min, Me2SO4 (6.3 mL, 66 mmol) was added and refluxed overnight. Then, water was added (50 mL) and the solvent was evaporated under reduced pressure. The aqueous layer was extracted with two portions of diethyl ether (2 × 80 mL), dried over anhydrous sodium sulfate, and filtered. The solvent was evaporated under reduced pressure to give a crude product which was purified by silica gel column chromatography (40% Et2O/hexane), affording (3-isopropyl-2-methoxyphenyl)methanol. Colorless oil (8.18 g, 68.9%). 1H NMR (500 MHz, CD3COCD3): δ 7.32 (m, 1H), 7.22 (dd, J = 7.6, 1.7 Hz, 1H), 7.10 (t, J = 7.6 Hz, 1H), 4.71 (d, J = 5.7 Hz, 2H), 4.12 (t, J = 5.7 Hz, OH), 3.76 (s, 3H), 3.36 (sept, J = 7.0 Hz, 1H), 1.23 (d, J = 7.0 Hz, 6H). 13C{1H} NMR (125 MHz, CD3COCD3): δ 155.7 (C), 142.0 (C), 135.9 (C), 126.9 (CH), 126.1 (CH), 125.0 (CH), 62.2 (CH2), 59.9 (CH3), 26.7 (CH), 24.1 (CH3). IR (film): 3053, 2966, 1451, 1428, 1264, 1206, 1096, 733, 703 cm–1. HRMS (ESI/TOF) m/z: [M + Na]+ calcd for C11H16O2Na, 203.1048; found, 203.1048.
Thionyl chloride (1.94 mL, 27 mmol) was slowly added to a solution of (3-isopropyl-2-methoxyphenyl)methanol (3.2 g, 18 mmol) and pyridine (1 drop) in dry CH2Cl2 (75 mL) at 0 °C. The reaction was allowed to warm to room temperature for 5 h and quenched with water (20 mL). The aqueous phase was extracted with DCM (3 × 50 mL), and the combined organic solutions were washed with brine (80 mL), dried over anhydrous sodium sulfate, and filtered. Evaporation of the solvent under reduced pressure yielded 1-(chloromethyl)-3-isopropyl-2-methoxybenzene (10j), which was used without chromatography purification. Colorless oil (2.3 g, 65.0%). 1H NMR (400 MHz, CDCl3): δ 7.33 (m, 2H), 7.19 (t, J = 7.6 Hz, 1H), 4.75 (s, 2H), 3.93 (s, 3H), 3.43 (sept, J = 6.9 Hz, 1H), 1.32 (d, J = 6.9 Hz, 6H). 13C{1H} NMR (125 MHz, CDCl3): δ 155.5 (C), 142.2 (C), 130.7 (C), 128.4 (CH), 127.4 (CH), 124.7 (CH), 62.5 (CH3), 41.3 (CH2), 26.2 (CH), 23.8 (CH3). IR (film): 2972, 1467, 1429, 1264, 1208, 1094, 1049, 1006, 799, 734, 703 cm–1. HRMS (ESI/TOF) m/z: [M–Cl]+ calcd for C11H15O, 163.1123; found, 163.1127.
Preparation of 1-(Bromomethyl)-3-isopropyl-2-methoxybenzene (10k)
The general procedure for the preparation of alkyl bromides was followed using (3-isopropyl-2-methoxyphenyl)methanol (3.8 g, 21.1 mmol) to afford 1-(bromomethyl)-3-isopropyl-2-methoxybenzene (10k), which was further used without chromatography purification. Yellow oil (4.3 g, 83.7%). 1H NMR (400 MHz, CD3COCD3): δ 7.29 (m, 2H), 7.10 (t, J = 7.7 Hz, 1H), 4.66 (s, 2H), 3.86 (s, 3H), 3.34 (sept, J = 6.7 Hz, 1H), 1.22 (d, J = 6.7 Hz, 6H). 13C{1H} NMR (125 MHz, CD3COCD3): δ 156.6 (C), 143.0 (C), 132.1 (C), 129.9 (CH), 128.3 (CH), 125.5 (CH), 62.5 (CH3), 29.4 (CH2), 26.9 (CH), 24.1 (CH3). IR (film): 2961, 2861, 2830, 1463, 1428, 1383, 1256, 1222, 1203, 1168, 1004, 795, 763 cm–1. HRMS (ESI/TOF) m/z: [M + H–HBr]+ calcd for C11H15O, 163.1123; found, 163.1126.
Preparation of 5-(Bromomethyl)-6-methoxybenzo[d][1,3]dioxole (10l)
To sesamol (5 g, 36 mmol) in water (110 mL) at 0 °C were added formaldehyde (37% wt in water, 5.5 mL, 72 mmol) and calcium oxide (1.02 g, 18 mmol). After 1 h, saturated aqueous ammonium chloride was added and the aqueous layer was extracted with ether (3 × 150 mL), dried over anhydrous Na2SO4, concentrated, and purified by column chromatography on silica gel (30% AcOEt/hexane) to give 6-(hydroxymethyl)benzo[d][1,3]dioxol-5-ol. Red solid (5.3 g, 82%), mp 183–185 °C. 1H NMR (500 MHz, CD3COCD3): δ 6.74 (s, 1H), 6.39 (s, 1H), 5.86 (s, 2H), 4.62 (s, 2H). 13C{1H} NMR (125 MHz, CD3COCD3): δ 151.2 (C), 148.2 (C), 141.6 (C), 120.5 (C), 108.5 (CH), 102.0 (CH), 98.9 (CH2), 61.9 (CH2). IR (film): 3500 (br s), 2922, 2853, 1503, 1484, 1190, 1157, 1039, 937, 852, 821 cm–1. HRMS (ESI/TOF) m/z: [M + H–H2O]+ calcd for C8H7O3, 151.0395; found, 151.0399.
6-(Hydroxymethyl)benzo[d][1,3]dioxol-5-ol (5.7 g, 34 mmol) was dissolved in acetone (60 mL), and K2CO3 (7.02 g, 50.9 mmol) was added and stirred at 0 °C for 15 min. Then, (CH3)2SO4 (3.2 mL, 34 mmol) was added and refluxed overnight. The solvent was evaporated, and water (40 mL) was added and extracted with diethyl ether (3 × 50 mL). The organic layer was dried over anhydrous Na2SO4, concentrated, and purified by flash chromatography on silica gel (30% Et2O/hexane) to give (6-methoxybenzo[d][1,3]dioxol-5-yl)methanol (4.3 g, 64.5%), which was used immediately. Spectroscopic data were consistent with those described in the literature.24
Finally, the general procedure for the preparation of alkyl bromides was followed to give 5-(bromomethyl)-6-methoxybenzo[d][1,3]dioxole (10l). Amorphous yellow solid (4.1 g, 76.8%). 1H NMR (500 MHz, CD3COCD3): δ 6.90 (s, 1H), 6.69 (s, 1H), 5.96 (s, 2H), 4.60 (s, 2H), 3.85 (s, 3H). 13C{1H} NMR (125 MHz, CD3COCD3): δ 154.8 (C), 150.4 (C), 142.2 (C), 119.3 (C), 111.1 (CH), 102.8 (CH), 96.1 (CH2), 57.3 (CH2), 31.2 (CH2). IR (film): 3018, 1504, 1466, 1215, 1040, 745 cm–1. HRMS (ESI/TOF) m/z: [M + H–HBr]+ calcd for C9H9O3, 165.0552; found, 165.0547.
General Procedure for the Deconjugative α-Alkylation of α,β-Unsaturated Aldehydes with Alkyl Halides
To a solution of unsaturated aldehydes 1 or 13a–e (1.0 mmol) in anhydrous toluene (15 mL) were added successively
sodium hydride (2 mmol, 60% dispersion in mineral oil) and potassium tert-butoxide (1.1 mmol), and the mixture was stirred for
45 min at room temperature. Then, a solution of the corresponding
alkyl halide (1.5 mmol) in toluene (5 mL) was added; the mixture was
stirred under an inert atmosphere for the specified time, and the
course of the reaction was monitored by TLC. When the starting material
was consumed, water (10 mL) was carefully added and the aqueous layer
was extracted with two portions of ethyl acetate (2 × 20 mL).
The combined organic solution was dried over anhydrous Na2SO4 and filtered, and the solvent was evaporated under
reduced pressure to give a crude, which was purified by silica gel
column chromatography. Elution with petroleum ether/ethyl acetate
mixtures yielded compounds (±)-11a–f, (±)-11h–j, (±)-11l–p, 12a, and (±)-14a–e in the yields indicated
in Tables 1–3.
(±)-1-Allyl-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11a) and (Z)-6-((Allyloxy)methylene)-1,5,5-trimethylcyclohex-1-ene (12a)
To a solution of β-cyclocitral (1) (200 mg, 1.31 mmol) in toluene (25 mL), NaH (105 mg, 2.62
mmol), tBuOK (162 mg, 1.45 mmol), and
allyl bromide (10a) (236 mg, 1.96 mmol) were added and
stirred for 1 h. Following the same workup used in the general procedure
and after column chromatography, using 2% EtOAc/hexane, compound 11a was obtained as a colorless oil (209 mg, 83%). ((±)-11a): 1H NMR (500 MHz, CDCl3): δ
9.64 (s, 1H), 5.85 (br s, 1H), 5.80 (m, 1H), 5.09 (d, J = 17.2 Hz, 1H), 4.98 (d, J = 9.9 Hz, 1H), 2.54
(m, 2H), 2.11 (br s, 2H), 1.61 (s, 3H), 1.56 (dt, J = 13.5, 6.7 Hz, 1H), 1.46 (dt, J = 13.5, 6.7 Hz,
1H), 1.03 (s, 3H), 0.96 (s, 3H). 13C{1H} NMR
(125 MHz, CDCl3): δ 204.6 (CH), 136.9 (CH), 130.1
(C), 127.9 (CH), 115.8 (CH2), 59.2 (C), 35.7 (C), 33.8
(CH2), 33.3 (CH2), 25.3 (CH3), 25.1
(CH3), 22.7 (CH2), 20.7 (CH3). IR
(film): 2916, 1717, 1674, 1447, 1378, 1225, 1051, 1025, 809 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C13H21O, 193.1592;
found, 193.1595. (12a) (see Table 1, entries 4–7 and 10): 1H NMR (400 MHz, CDCl3): δ 6.07 (s, 1H), 5.93 (ddt, J = 17.2, 10.5, 5.2 Hz, 1H), 5.44 (m, 1H), 5.32 (dq, J = 17.2, 1.7 Hz, 1H), 5.20 (dq, J = 10.5,
1.7 Hz, 1H), 4.27 (dt, J = 5.2, 1.7 Hz, 2H), 2.05
(m, 2H), 1.72 (q, J = 1.7 Hz, 3H), 1.41 (t, J = 6.2 Hz, 2H), 1.21 (s, 6H). 13C{1H} NMR (100 MHz, CDCl3): δ 142.7 (CH), 134.1 (C),
130.6 (C), 124.0 (CH), 123.6 (CH), 116.9 (CH2), 73.2 (CH2), 39.2 (CH2), 33.2 (C), 27.3 (2× CH3), 22.8 (CH2), 20.6 (CH3). IR (film): 2927,
1676, 1455, 1127, 929 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C13H21O, 193.1592; found,193.1594.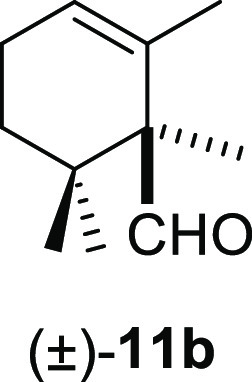
(±)-1,2,6,6-Tetramethylcyclohex-2-ene-1-carbaldehyde ((±)-11b)
To a solution of β-cyclocitral (1) (145 mg, 0.95 mmol) in toluene (15 mL), NaH (76 mg, 1.9
mmol), tBuOK (123 mg, 1.1 mmol), and iodomethane
(220 mg, 1.55 mmol) were added and stirred for 1 h. Following the
same workup used in the general procedure and after column chromatography,
using 5% EtOAc/hexane, compound 11b was obtained as a
colorless oil (147 mg, 93%). 1H NMR (500 MHz, CDCl3): δ 9.60 (s, 1H), 5.74 (br s, 1H), 2.10 (m, 2H), 1.81
(m, 1H), 1.51 (br s, 3H), 1.38 (dt, J = 12.8, 3.3
Hz, 1H), 1.07 (s, 3H), 0.91 (s, 3H), 0.85 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 203.6 (CH), 131.1
(C), 127.0 (CH), 56.5 (C), 34.4 (C), 33.1 (CH2), 24.9 (CH3), 24.2 (CH3), 22.7 (CH2), 19.9 (CH3), 12.6 (CH3). IR (film): 2963, 2874, 1703, 1634,
1366, 1309, 1233, 1180, 1122, 1079, 1032 cm–1. HRMS
(ESI/TOF) m/z: [M + H]+ calcd for C11H19O, 167.1436; found,167.1443.
(±)-2,6,6-Trimethyl-1-(3-methylbut-2-en-yl)cyclohex-2-ene-1-carbaldehyde ((±)-11c)
To a solution of β-cyclocitral
(1) (178 mg, 1.17 mmol) in toluene (20 mL), NaH (94 mg,
2.34 mmol), tBuOK (154 mg, 1.3 mmol),
and 10c (261 mg, 1.75 mmol) were added and stirred for
2 h. Following the same workup used in the general procedure and after
column chromatography, using 2% EtOAc/hexane, compound 11c was obtained as a colorless oil (239 mg, 64%). 1H NMR
(500 MHz, CDCl3): δ 9.61 (s, 1H), 5.82 (br s, 1H),
5.06 (m, 1H), 2.42 (m, 2H), 2.08 (m, 2H), 1.65 (d, J = 1.5 Hz, 3H), 1.62 (d, J = 1.5 Hz, 3H), 1.56 (q, J = 1.8 Hz, 3H), 1.48 (t, J = 6.8 Hz, 2H),
0.98 (s, 3H), 0.88 (s, 3H). 13C{1H} NMR (125
MHz, CDCl3): δ 205.0 (CH), 130.9 (C), 130.6 (C),
127.9 (CH), 122.2 (CH), 59.3 (C), 35.5 (C), 33.8 (CH2),
26.9 (CH2), 25.9 (CH3), 25.4 (CH3), 25.3 (CH3), 22.7 (CH2), 20.7 (CH3), 18.0 (CH3). IR (film): 2923, 1715, 1676, 1453, 1381,
1365, 1138, 1035, 754 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C15H25O, 221.1905; found, 221.1909.
(±)-2,6,6-Trimethyl-1-(prop-2-yn-1-yl)cyclohex-2-ene-1-carbaldehyde ((±)-11d)
To a solution of β-cyclocitral
(1) (230 mg, 1.51 mmol) in toluene (25 mL), NaH (121
mg, 3.02 mmol), tBuOK (186 mg, 1.66 mmol),
and propargyl chloride 10d (169 mg, 2.27 mmol) were added
and stirred for 2 h. Following the same workup used in the general
procedure and after column chromatography, using 5% EtOAc/hexane,
compound 11d was obtained as a colorless oil (192 mg,
67%). 1H NMR (500 MHz, CDCl3): δ 9.57
(s, 1H), 5.90 (br s, 1H), 2.75 (dd, J = 17.8, 2.7
Hz, 1H), 2.48 (dd, J = 17.8, 2.7 Hz, 1H), 2.12 (m,
2H), 1.95 (t, J = 2.7 Hz, 1H), 1.64 (s, 3H), 1.76–1.52
(m, 2H), 1.06 (s, 3H), 1.00 (s, 3H). 13C{1H}
NMR (125 MHz, CDCl3): δ 201.8 (CH), 129.0 (CH), 125.4
(C), 83.4 (C), 70.6 (CH), 63.7 (C), 59.2 (CH2), 35.4 (C),
33.8 (CH2), 25.4 (CH3), 22.6 (CH2), 20.1 (CH3), 17.4 (CH3). IR (film): 3324,
2919, 2126, 1715, 1673, 1448, 1374, 1224, 1053, 1032 cm–1. HRMS (ESI/TOF) m/z: [M + Na]+ calcd for C13H18ONa, 213.1255; found,
213.1249.
(±)-1-Benzyl-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11e)
To a solution of β-cyclocitral
(1) (210 mg, 1.38 mmol) in toluene (25 mL), NaH (110
mg, 2.76 mmol), tBuOK (170 mg, 1.52 mmol),
and benzyl bromide (10e) (354 mg, 2.07 mmol) were added
and stirred for 30 min. Following the same workup used in the general
procedure and after column chromatography, using 3% EtOAc/hexane,
compound 11e was obtained as a colorless oil (310 mg,
93%). 1H NMR (400 MHz, CDCl3): δ 9.79
(s, 1H), 7.25–7.20 (m, 5H), 5.80 (br s, 1H), 3.47 (d, J = 13.4 Hz, 1H), 2.75 (d, J = 13.4 Hz,
1H), 2.25–2.06 (m, 2H), 1.65 (ddd, J = 13.8,
10.5, 7.0 Hz, 1H), 1.24 (s, 3H), 1.21 (m, 1H), 1.09 (s, 3H), 1.04
(s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 206.4 (CH), 139.5 (C), 131.2 (C), 130.7 (2× CH),
127.9 (2× CH), 126.6 (CH), 126.0 (CH), 61.0 (C), 36.7 (C), 36.2
(CH2), 32.7 (CH2), 25.3 (CH3), 24.0
(CH3), 22.9 (CH2), 21.9 (CH3). IR
(film): 2930, 2860, 1727, 1529, 1470, 1369, 1347, 1241, 1017, 997,
962, 802, 755, 725, 672 cm–1. HRMS (ESI/TOF) m/z: [M–H2 + H]+ calcd for C17H21O, 241.1592; found, 241.1597.
(±)-1-(4-Methoxybenzyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11f)
To a solution of β-cyclocitral
(1) (250 mg, 1.64 mmol) in toluene (25 mL), NaH (131
mg, 3.28 mmol), tBuOK (362 mg, 1.80 mmol),
and 10g (385 mg, 1.91 mmol) were added and stirred for
30 min. Following the same workup used in the general procedure and
after column chromatography, using 5% EtOAc/hexane, compound 11f was obtained as a colorless syrup (326 mg, 73%). 1H NMR (500 MHz, CDCl3): δ 9.71 (s, 1H), 7.07
(d, J = 8.4 Hz, 2H), 6.73 (d, J =
8.4 Hz, 2H), 5.73 (br s, 1H), 3.75 (s, 3H), 3.31 (d, J = 13.1 Hz, 1H), 2.64 (d, J = 13.1 Hz, 1H), 2.06
(m, 2H), 1.56 (m, 1H), 1.20 (s, 3H), 1.12 (dd, J =
1.8, 6.6 Hz, 1H), 1.01 (s, 3H), 0.95 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 206.4 (C), 157.9
(C), 131.5 (2× CH), 131.4 (C), 131.3 (C), 126.4 (CH), 113.3 (2×
CH), 60.9 (C), 55.1 (CH3), 36.6 (C), 35.3 (CH2), 32.7 (CH2), 25.2 (CH2), 24.0 (CH3), 22.9 (CH3), 21.9 (CH3). IR (film): 2932,
1715, 1610, 1510, 1462, 1245, 1177, 1034, 821 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C18H25O2, 273.1855;
found, 273.1846.
(±)-1-(3-Bromo-4,5-dimethoxybenzyl)-2,6,6-trimethylcyclohex-2-enecarbaldehyde ((±)-11h)
To a solution of β-cyclocitral
(1) (177 mg, 1.16 mmol) in toluene (15 mL), NaH (93 mg,
2.32 mmol), tBuOK (143 mg, 1.28 mmol),
and 10h (540 mg, 1.74 mmol) were added and stirred for
1 h. Following the same workup used in the general procedure and after
column chromatography, using 7% EtOAc/hexane, compound 11h was obtained as a colorless syrup (389 mg, 88%). 1H NMR
(500 MHz, CDCl3): δ 9.67 (s, 1H), 6.92 (d, J = 1.9 Hz, 1H), 6.72 (d, J = 1.9 Hz, 1H),
5.76 (br s, 1H), 3.78 (s, 6H), 3.29 (d, J = 13.4
Hz, 1H), 2.53 (d, J = 13.4 Hz, 1H), 2.12 (m, 2H),
1.57 (ddd, J = 13.8, 10.4, 7.0 Hz, 1H), 1.20 (s,
3H), 1.19–1.10 (m, 1H), 1.02 (s, 3H), 0.92 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 205.9 (CH),
153.0 (C), 144.8 (C), 136.7 (C), 131.0 (C), 126.6 (2× CH), 116.8
(C), 114.3 (CH), 61.0 (CH3), 60.6 (CH3), 56.0
(C), 36.7 (C), 35.7 (CH2), 32.6 (CH2), 25.2
(CH3), 23.8 (CH3), 22.8 (CH2), 22.0
(CH3). IR (film): 2947, 1715, 1595, 1565, 1489, 1463, 1429,
1414, 1313, 1277, 1214, 1184, 1142, 1047, 1001, 879 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C19H26O3Br, 381.1065;
found, 381.1056.
(±)-1-(Benzo[d][1,3]dioxol-5-ylmethyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11i)
To a solution of β-cyclocitral
(1) (300 mg, 1.97 mmol) in toluene (25 mL), NaH (158
mg, 3.94 mmol), tBuOK (244 mg, 2.18 mmol),
and 10i (634 mg, 2.95 mmol) were added and stirred for
1 h. Following the same workup used in the general procedure and after
column chromatography, using 5% EtOAc/hexane, compound 11i was obtained as a white solid (501 mg, 89%). 1H NMR (500
MHz, CDCl3): δ 9.73 (s, 1H), 6.70 (br s, 1H), 6.67
(d, J = 8.0 Hz, 1H), 6.65 (d, J =
8.0 Hz, 1H), 5.89 (s, 2H), 5.76 (br s, 1H), 3.34 (d, J = 13.6 Hz, 1H), 2.60 (d, J = 13.6 Hz, 1H), 2.02–2.24
(m, 2H), 1.64 (m, 1H), 1.25 (s, 3H), 1.17 (m, 1H), 1.06 (s, 3H), 0.96
(s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 206.3 (CH), 147.2 (C), 145.8 (C), 133.1 (C), 131.3 (C),
126.5 (CH), 123.6 (CH), 111.0 (CH), 107.8 (CH), 100.7 (CH2), 61.0 (C), 36.7 (C), 36.0 (CH2), 32.7 (CH2), 25.2 (CH3), 23.9 (CH3), 22.9 (CH2), 22.06 (CH3). IR (film): 2947, 1718, 1530, 1350, 1261,
1082, 1028, 806, 723, 687 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C18H23O3, 287.1647; found, 287.1651.
(±)-1-(3-Isopropyl-2-methoxybenzyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11j)
To a solution of β-cyclocitral
(1) (112 mg, 0.74 mmol) in toluene (12 mL), NaH (59 mg,
1.48 mmol), tBuOK (244 mg, 0.81 mmol),
and 10k (270 mg, 1.11 mmol) were added and stirred for
45 min. Following the same workup used in the general procedure and
after column chromatography, using 5% EtOAc/hexane, compound 11j was obtained as a white solid (162 mg, 70%). 1H NMR (500 MHz, CDCl3): δ 9.75 (s, 1H), 7.07 (m,
1H), 7.02 (dd, J = 7.6, 1.7 Hz, 1H), 6.92 (t, J = 7.6 Hz, 1H), 5.63 (br s, 1H), 3.69 (s, 3H), 3.30 (d, J = 13.6 Hz, 1H), 3.26 (m, 1H), 2.94 (d, J = 13.6 Hz, 1H), 2.06 (m, 2H), 1.66 (m, 1H), 1.23 (m, 1H), 1.19 (d, J = 6.8 Hz, 3H), 1.16 (d, J = 6.8 Hz, 3H),
1.14 (br s, 3H), 0.99 (s, 3H), 0.98 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 206.5 (CH), 156.3 (C),
141.8 (C), 132.4 (C), 131.3 (C), 129.7 (CH), 126.7 (CH), 124.7 (CH),
123.8 (CH), 61.5 (CH3), 60.8 (C), 36.8 (C), 32.9 (CH2), 30.2 (CH2), 26.5 (CH), 25.3 (CH3),
24.1 (CH3), 23.9 (CH3), 23.6 (CH3), 22.9 (CH3), 21.6 (CH3). IR (film): 2960,
2864, 1719, 1689, 1458, 1427, 1384, 1254, 1201, 1165, 1060, 1010,
796, 765, 568 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C21H31O2, 315.2324; found, 315.2326.
(±)-1-((6-Methoxybenzo[d][1,3]dioxol-5-yl)methyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11l)
To a solution of β-cyclocitral
(1) (147 mg, 0.97 mmol) in toluene (15 mL), NaH (78 mg,
1.94 mmol), tBuOK (120 mg, 1.07 mmol),
and 10l (355 mg, 1.45 mmol) were added and stirred for
90 min. Following the same workup used in the general procedure and
after column chromatography, using 5% EtOAc/hexane, compound 11l was obtained as a colorless syrup (239 mg, 78%). 1H NMR (500 MHz, CDCl3): δ 9.72 (s, 1H), 6.67
(s, 1H), 6.40 (s, 1H), 5.83 (s, 2H), 5.59 (s, 1H), 3.64 (s, 3H), 3.22
(d, J = 13.5 Hz, 1H), 2.87 (d, J = 13.5 Hz, 1H), 2.05 (m, 2H), 1.82 (m, 1H), 1.15 (s, 3H), 1.14–1.10
(m, 1H), 0.97 (s, 6H). 13C{1H} NMR (125 MHz,
CDCl3): δ 206.6 (CH), 152.8 (C), 146.3 (C), 140.6
(C), 131.5 (C), 125.8 (CH), 120.0 (C), 111.6 (CH), 100.8 (CH), 94.3
(CH2), 60.5 (C), 56.0 (CH3), 36.7 (C), 32.7
(CH2), 30.6 (CH2), 25.3 (CH3), 23.7
(CH3), 22.9 (CH2), 21.9 (CH3). IR
(film): 2949, 2855, 1715, 1503, 1483, 1464, 1190, 1156, 1037, 1006,
935, 861, 824, 759 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C19H25O4, 317.1753; found, 317.1758.
(±)-1-(2-Bromobenzyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11m)
To a solution of β-cyclocitral
(1) (310 mg, 2.04 mmol) in toluene (25 mL), NaH (163
mg, 4.08 mmol), tBuOK (251 mg, 2.24 mmol),
and 10m (765 mg, 3.06 mmol) were added and stirred for
2 h. Following the same workup used in the general procedure and after
column chromatography, using 3% EtOAc/hexane, compound 11m was obtained as a colorless oil (602 mg, 92%). 1H NMR
(400 MHz, CDCl3): δ 9.70 (s, 1H), 7.45 (dd, J = 7.5, 1.4 Hz, 1H), 7.35 (dd, J = 7.7,
1.8 Hz, 1H), 7.12 (td, J = 7.5, 1.4 Hz, 1H), 6.98
(td, J = 7.7, 1.8 Hz, 1H), 5.67 (br s, 1H), 3.59
(d, J = 13.3 Hz, 1H), 3.06 (d, J = 13.3 Hz, 1H), 2.21–2.11 (m, 2H), 2.10–1.99 (m, 1H),
1.25–1.17 (m, 1H), 1.05 (s, 3H), 0.98 (s, 3H), 0.94 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ
206.6 (CH), 139.3 (C), 132.9 (CH), 132.8 (CH), 129.9 (C), 127.74 (CH),
127.70 (CH), 127.0 (CH), 126.1 (C), 60.9 (C), 37.4 (C), 35.7 (CH2), 32.6 (CH2), 25.3 (CH3), 23.3 (CH3), 23.2 (CH2), 21.9 (CH3). IR (film):
2950, 2875, 2834, 1716, 1470, 1438, 1387, 1367, 1025, 873, 765, 747,
659, 569 cm–1. HRMS (ESI/TOF) m/z: [M + Na]+ calcd for C17H22OBrNa, 321.0854; found, 321.0862.
(±)-1-((6-Bromobenzo[d][1,3]dioxol-5-yl)methyl)-2,6,6-trimethylcyclohex-2-ene-1-carbaldehyde ((±)-11n)
To a solution of β-cyclocitral
(1) (110 mg, 0.72 mmol) in toluene (12 mL), NaH (58 mg,
1.44 mmol), tBuOK (89 mg, 0.79 mmol),
and 10n (317 mg, 1.08 mmol) were added and stirred for
2 h. Following the same workup used in the general procedure and after
column chromatography, using 5% EtOAc/hexane, compound 11n was obtained as a white solid (223 mg, 85%). 1H NMR (400
MHz, CDCl3): δ 9.68 (s, 1H), 6.90 (s, 1H), 6.86 (s,
1H), 5.89 (s, 2H), 5.68 (br s, 1H), 3.45 (d, J =
13.4 Hz, 1H), 2.96 (d, J = 13.4 Hz, 1H), 2.14 (m,
2H), 2.10–1.96 (m, 1H), 1.28–1.15 (m, 1H), 1.10 (s,
3H), 1.04 (s, 3H), 0.91 (s, 3H). 13C{1H} NMR
(100 MHz, CDCl3): δ 206.7 (CH), 147.0 (C), 146.7
(C), 132.1 (C), 129.9 (C), 127.6 (CH), 116.0 (C), 112.6 (CH), 112.0
(CH), 101.5 (CH2), 60.8 (C), 37.3 (C), 35.7 (CH2), 32.6 (CH2), 25.3 (CH3), 23.3 (CH3), 23.1 (CH2), 22.2 (CH3). IR (film): 2921,
1715, 1502, 1474, 1407, 1388, 1367, 1267, 1227, 1167, 1113, 1037,
936, 873, 831, 756, 570 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C18H22O3Br, 365.0752; found, 365.0750.
(±)-2,6,6-Trimethyl-1-(3-nitrobenzyl)cyclohex-2-ene-1-carbaldehyde ((±)-11o)
To a solution of β-cyclocitral
(1) (192 mg, 1.26 mmol) in toluene (15 mL), NaH (101
mg, 2.52 mmol), tBuOK (156 mg, 1.39 mmol),
and 10o (408 mg, 1.89 mmol) were added and stirred for
10 min. Following the same workup used in the general procedure and
after column chromatography, using 8% EtOAc/hexane, compound 11o was obtained as a yellow syrup (289 mg, 80%). 1H NMR (600 MHz, CDCl3): δ 9.69 (s, 1H), 8.02 (m,
2H), 7.55 (d, J = 7.8 Hz, 1H), 7.36 (t, J = 7.8 Hz, 1H), 5.81 (s, 1H), 3.49 (d, J = 13.3
Hz, 1H), 2.73 (d, J = 13.3 Hz, 1H), 2.23–2.06
(m, 2H), 1.57 (ddd, J = 14.0, 10.6, 7.0 Hz, 1H),
1.20 (ddd, J = 14.0, 7.0, 2.3 Hz, 1H), 1.13 (s, 3H),
1.07 (s, 3H), 0.96 (s, 3H). 13C{1H} NMR (150
MHz, CDCl3): δ 205.6 (CH), 148.0 (C), 141.7 (C),
137.0 (CH), 130.0 (C), 128.7 (CH), 127.5 (CH), 125.4 (CH), 121.2 (CH),
61.1 (C), 36.8 (C), 35.8 (CH2), 32.6 (CH2),
25.3 (CH3), 23.9 (CH3), 22.8 (CH2), 22.0 (CH3). IR (film): 2926, 1716, 1527, 1348, 1261,
1222, 1061, 1029, 804, 756, 723, 687, 672 cm–1.
HRMS (ESI/TOF) m/z: [M + H]+ calcd for C17H22NO3, 288.1600;
found, 288.1608.
(±)-4-((1-Formyl-2,6,6-trimethylcyclohex-2-en-1-yl)methyl)benzonitrile ((±)-11p)
To a solution of β-cyclocitral
(1) (166 mg, 1.09 mmol) in toluene (15 mL), NaH (87 mg,
2.18 mmol), tBuOK (135 mg, 1.2 mmol),
and 10p (320 mg, 1.63 mmol) were added and stirred for
15 min. Following the same workup used in the general procedure and
after column chromatography, using 5% EtOAc/hexane, compound 11p was obtained as a yellow syrup (224 mg, 77%). 1H NMR (500 MHz, CDCl3): δ 9.67 (s, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.29 (d, J = 8.4 Hz, 2H),
5.76 (s, 1H), 3.43 (d, J = 13.2 Hz, 1H), 2.66 (d, J = 13.2 Hz, 1H), 2.20–2.03 (m, 2H), 1.64–1.51
(m, 1H), 1.19 (ddd, J = 13.9, 6.8, 2.2 Hz, 1H), 1.09
(s, 3H), 1.04 (s, 3H), 0.92 (s, 3H). 13C{1H}
NMR (125 MHz, CDCl3): δ 205.4 (CH), 145.5 (C), 131.6
(2× CH), 131.5 (2× CH), 130.2 (C), 127.3 (CH), 119.0 (C),
109.9 (C), 61.3 (C), 36.8 (C), 36.4 (CH2), 32.6 (CH2), 25.2 (CH3), 23.8 (CH3), 22.8 (CH2), 22.0 (CH3). IR (film): 2962, 2228, 1719, 1606,
1365, 1174, 1018, 816, 754, 816, 754 cm–1. HRMS
(ESI/TOF) m/z: [M-CH2+H]+ calcd for C17H20NO, 254.1545;
found, 254.1543.
(±)-1-Benzylcyclohex-2-enecarbaldehyde ((±)-14a)
To a solution of 13a (130 mg, 1.18
mmol) in toluene (15 mL), NaH (95 mg, 2.36 mmol), tBuOK (145 mg, 1.3 mmol), and benzyl bromide (10e) (303 mg, 1.77 mmol) were added and stirred for 20 min. Following
the same workup used in the general procedure and after column chromatography,
using 5% EtOAc/hexane, compound 14a was obtained as a
colorless oil (224 mg, 95%). 1H NMR (500 MHz, CDCl3): δ 9.55 (s, 1H), 7.30–7.10 (m, 5H), 6.01 (dt, J = 10.0, 3.8 Hz, 1H), 5.58 (d, J = 10.0
Hz, 1H), 2.90 (s, 2H), 2.05–1.88 (m, 3H), 1.64–1.52
(m, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 203.3 (CH), 136.5 (C), 132.0 (CH), 130.3 (2× CH),
128.1 (2× CH), 126.5 (CH), 126.3 (CH), 52.0 (C), 42.6 (CH2), 27.9 (CH2), 24.8 (CH2), 18.8 (CH2). IR (film): 3027, 2934, 2867, 2835, 1722, 1495, 1453, 1069,
762, 729, 701, 679 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C14H17O, 201.1279; found, 201.1282.
(1R,4S) and (1S,4S)-1-Benzyl-4-(prop-1-en-2-yl)cyclohex-2-enecarbaldehyde (14b)
To a solution of 13b (150
mg, 1 mmol) in toluene (15 mL), NaH (80 mg, 2 mmol), tBuOK (123 mg, 1.1 mmol), and benzyl bromide (10e) (256 mg, 1.5 mmol) were added and stirred for 20 min.
Following the same workup used in the general procedure and after
column chromatography, using 5% EtOAc/hexane, compound 14b was obtained as a colorless oil (223 mg, 93%, approx. 1:1 diasteroisomeric
ratio). 1H NMR (400 MHz, CDCl3): δ 9.56
(s, 2H), 7.39–7.10 (m, 10H), 5.91–5.86 (m, 2H), 5.68
(br s, 1H), 5.66 (br s, 1H), 4.77 (br s, 1H), 4.73 (br s, 1H), 4.71
(br s, 1H), 4.44 (br s, 1H), 2.91–2.87 (m, 4H), 2.68 (m, 2H),
2.01–1.98 (m, 2H), 1.81–1.61 (m, 4H), 1.69 (s, 6H),
1.55–1.42 (m, 2H). Major isomer: 13C{1H} NMR (125 MHz, CDCl3): δ 202.7 (CH), 147.8 (C),
136.3 (C), 135.1 (CH), 130.3 (2× CH), 128.1 (2× CH), 127.0
(CH), 126.6 (CH), 110.9 (CH2), 52.1 (C), 42.9 (CH), 42.6
(CH2), 27.1 (CH2), 24.7 (CH2), 20.6
(CH3). Minor isomer: 13C{1H} NMR
(125 MHz, CDCl3): δ 203.0 (CH), 147.1 (C), 136.3
(C), 134.3 (CH), 130.4 (2× CH), 128.1 (2× CH), 126.9 (CH),
126.6 (CH), 111.6 (CH2), 52.3 (C), 42.1 (CH2), 41.8 (CH), 24.7 (CH2), 23.3 (CH2), 21.4
(CH3). IR (film): 2936, 1720, 1644, 1496, 1453, 1374, 892,
831, 762, 735, 700 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C17H21O, 241.1592; found, 241.1601.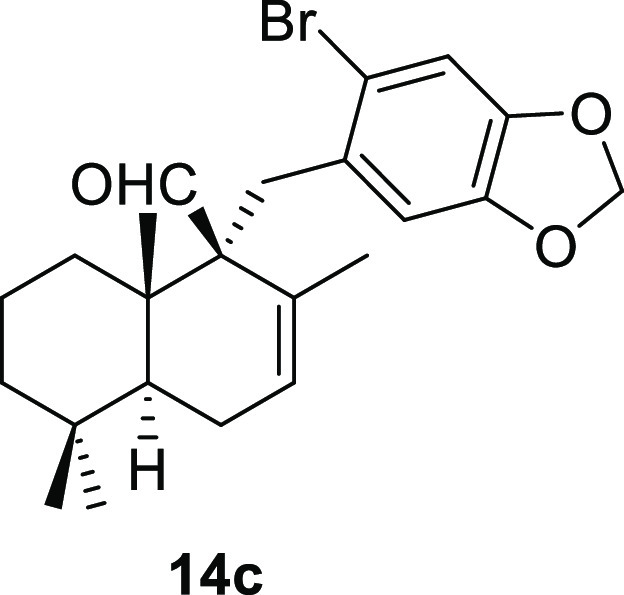
(1S,4aS,8aS)-1-((6-Bromobenzo[d][1,3]dioxol-5-yl)methyl)-2,5,5,8a-tetramethyl-1,4,4a,5,6,7,8,8a-octahydronaphthalene-1-carbaldehyde (14c)
To a solution of 13c (76
mg, 0.34 mmol) in toluene (10
mL), NaH (30 mg, 0.75 mmol), tBuOK (43
mg, 0.38 mmol), and 10n (153 mg, 0.52 mmol) were added
and stirred for 2 h. Following the same workup used in the general
procedure and after column chromatography, using 5% EtOAc/hexane,
compound 14c was obtained as a colorless syrup (128 mg,
86%). [α]D20 −3.0 (c 0.6, CHCl3). 1H NMR (400 MHz, CDCl3): δ 9.77 (s, 1H), 6.90 (s,
1H), 6.75 (s, 1H), 5.89 (s, 2H), 5.70 (br s, 1H), 3.52 (d, J = 13.3 Hz, 1H), 3.01 (d, J = 13.3 Hz,
1H), 2.19–2.12 (m, 1H), 2.01–1.91 (m, 1H), 1.59–1.42
(m, 5H), 1.26–1.16 (m, 2H), 1.10 (s, 3H), 1.07 (s, 3H), 0.96
(s, 3H), 0.93 (s, 3H). 13C{1H} NMR (100 MHz,
CDCl3): δ (ppm) 207.9 (CH), 147.0 (C), 146.7 (C),
132.2 (C), 129.5 (C), 128.4 (CH), 116.1 (C), 112.6 (CH), 112.1 (CH),
101.5 (CH2), 63.0 (C), 42.3 (C), 42.0 (CH2),
41.8 (CH), 36.0 (CH2), 33.9 (CH3), 33.4 (CH2), 33.3 (C), 24.7 (CH2), 22.3 (CH3),
22.0 (CH3), 18.2 (CH2), 17.5 (CH3). IR (film): 2949, 1716, 1672, 1503, 1478, 1367, 1228, 1114, 1039,
936, 882, 841, 655, 567 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C23H30O3Br, 433.1378; found, 433.1365.
(1S,4aS,5S,8aR)-Methyl 5-((6-Bromobenzo[d][1,3]dioxol-5-yl)methyl)-5-formyl-1,4a,6-trimethyl-1,2,3,4,4a,5,8,8a-octahydronaphthalene-1-carboxylate (14d)
To a solution of 13d (83
mg, 0.31 mmol) in toluene (10 mL), NaH (26 mg, 0.65 mmol), tBuOK (39 mg, 0.35 mmol), and 10n (138
mg, 0.47 mmol) were added and stirred for 2 h. Following the same
workup used in the general procedure and after column chromatography,
using 5% EtOAc/hexane, compound 14d was obtained as a
colorless syrup (133 mg, 90%). [α]D20 −26.0 (c 0.3,
CHCl3). 1H NMR (400 MHz, CDCl3):
δ 9.74 (s, 1H), 6.89 (s, 1H), 6.76 (s, 1H), 5.88 (s, 2H), 5.68
(s, 1H), 3.64 (s, 3H), 3.49 (d, J = 13.3 Hz, 1H),
2.94 (d, J = 13.3 Hz, 1H), 2.71–2.56 (m, 1H),
2.41–2.29 (m, 1H), 2.22–2.06 (m, 2H), 1.90–1.41
(m, 4H), 1.23 (s, 3H), 1.05 (s, 3H), 0.95 (s, 3H), 0.87–0.78
(m, 1H). 13C{1H} NMR (100 MHz, CDCl3): δ 207.2 (CH), 177.7 (C), 147.0 (C), 146.7 (C), 131.8 (C),
128.3 (C), 128.3 (CH), 116.1 (C), 112.6 (CH), 112.1 (CH), 101.5 (CH2), 62.3 (C), 51.4 (CH3), 44.3 (CH), 44.1 (C), 41.8
(C), 38.0 (CH2), 35.9 (CH2), 33.0 (CH2), 29.1 (CH3), 25.3 (CH2), 21.8 (CH3), 19.0 (CH2), 16.4 (CH3). IR (film): 2949,
1715, 1503, 1477, 1407, 1380, 1226, 1166, 1143, 1113, 1038, 984, 934,
910, 874, 841, 771, 730, 652, 566 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C24H30O5Br, 477.1277; found, 477.1276.
(±)-1-Benzyl-2,6,6-trimethylcyclohexa-2,4-diene-1-carbaldehyde ((±)-14e)
To a solution of 13e (163 mg, 1.08 mmol) in toluene (15 mL), NaH (84 mg, 2.1 mmol), tBuOK (134 mg, 1.19 mmol), and benzyl bromide (10e) (277 mg, 1.62 mmol) were added and stirred for 3 h. Following the same workup used in the general procedure and after column chromatography, using 5% EtOAc/hexane, compound 14e was obtained as a colorless syrup (188 mg, 72%). 1H NMR (500 MHz, CDCl3): δ 9.98 (s, 1H), 7.18–7.10 (m, 5H), 5.90 (m, 2H), 5.35 (m, 1H), 3.27 (d, J = 12.8 Hz, 1H), 2.87 (d, J = 12.8 Hz, 1H), 1.14 (s, 3H), 1.13 (s, 3H), 1.10 (s, 3H). 13C{1H} NMR (125 MHz, CDCl3): δ 206.8 (C), 138.8 (C), 135.8 (CH), 135.1 (C), 130.9 (CH), 127.6 (CH), 125.9 (CH), 121.9 (CH), 121.6 (CH), 61.8 (C), 40.1 (C), 30.8 (CH2), 22.5 (CH3), 22.4 (CH3), 22.0 (CH3). IR (film): 3028, 2961, 1718, 1494, 1453, 1362, 1076, 726, 700 cm–1. HRMS (ESI/TOF) m/z: [M–CH3]+ calcd for C16H19O, 227.1436; found, 227.1443.
Preparation of (4aS,8aS)-2,5,5,8a-Tetramethyl-3,4,4a,5,6,7,8,8a-octahydronaphthalene-1-carbaldehyde (13c)
Compound 13c was prepared following the procedure described in the literature. The spectroscopic data were in agreement with those described in the literature.20
Preparation of (1S,4aS,8aR)-Methyl 5-Formyl-1,4a,6-trimethyl-1,2,3,4,4a,7,8,8a-octahydronaphthalene-1-carboxylate (13d)
Compound 13d was prepared following the procedure described in the literature. The spectroscopic data were in agreement with those described in the literature.21
General Procedure for Deformylation of β,γ-Unsaturated Aldehydes (±)-11a, (±)-11e, (±)-11i, (±)-11m, and (±)-11o with Pb(OAc)4
To a solution of the corresponding
β,γ-unsaturated aldehydes (±)-11a, (±)-11e, (±)-11i, (±)-11m,
and (±)-11o (1 mmol) in dry benzene (7 mL) was added
Pb(OAc)4 (1.1 mmol), and the solution was heated at 80
°C for 10 min–2 h. Then, the reaction mixture
was quenched with 5% Na2SO3 (5 mL), and the
aqueous layer was extracted with two portions of ethyl acetate (2
× 15 mL). The combined organic solution was washed with brine
(10 mL), dried over anhydrous sodium sulfate, and filtered. The solvent
was evaporated under reduced pressure to give a crude product which
was purified by silica gel column chromatography (petroleum ether/ethyl
acetate), affording the corresponding allyl acetates (±)-15a, (±)-15e, (±)-15i,
(±)-15m, and (±)-15o in the yields
shown in Table 4.
(±)-3-Allyl-2,4,4-trimethylcyclohex-2-en-1-yl Acetate ((±)-15a)
To a solution of 11a (194 mg, 1.01
mmol) in benzene (7 mL) was added Pb(OAc)4 (492 mg, 1.11
mmol), and the mixture was heated at 80 °C for 7 min. Following
the same workup used in the general procedure, 15a (204
mg, 91%) was obtained as a colorless syrup after column chromatography
using 3% EtOAc/hexane. 1H NMR (500 MHz, CDCl3): δ 5.68 (m, 1H), 5.09 (t, J = 4.6 Hz, 1H),
4.95 (m, 1H), 4.81 (m, 1H), 2.76 (d, J = 5.9 Hz,
2H), 1.99 (s, 3H), 1.84 (m, 1H), 1.64 (m, 1H), 1.54 (m, 1H), 1.51
(s, 3H), 1.31 (ddd, J = 13.3, 7.0, 3.2 Hz, 1H), 0.97
(s, 3H), 0.91 (s, 3H). 13C{1H} NMR (100 MHz,
CDCl3): δ 171.2 (C), 141.7 (C), 136.5 (CH), 126.8
(C), 114.9 (CH2), 72.7 (CH), 35.3 (C), 34.8 (CH2), 32.8 (CH2), 28.2 (CH3), 26.8 (CH3), 25.4 (CH2), 21.4 (CH3), 16.6 (CH3). IR (film): 1719, 1634, 1469, 1370, 1244, 1174, 1145, 1017, 994,
961, 910, 865 cm–1. HRMS (ESI/TOF) m/z: [M–OAc]+ calcd for C12H19, 163.1487; found, 163.1483.
(±)-3-Benzyl-2,4,4-trimethylcyclohex-2-en-1-yl Acetate ((±)-15e)
To a solution of 11e (143 mg, 0.59 mmol) in benzene (4 mL) was added Pb(OAc)4 (288 mg, 0.65 mmol), and the mixture was heated at 80 °C for
2 h. Following the same workup used in the general procedure, 15e (114 mg, 71%) was obtained as a colorless syrup after
column chromatography using 5% EtOAc/hexane. 1H NMR (400
MHz, CDCl3): δ 7.29–7.23 (m, 2H), 7.19–7.10
(m, 3H), 5.27 (d, J = 4.8 Hz, 1H), 3.50 (s, 2H),
2.09 (s, 3H), 1.96 (dddd, J = 14.3, 11.4, 4.8, 3.2
Hz, 1H), 1.75 (dddd, J = 14.3, 7.2, 4.8, 3.2 Hz,
1H), 1.66 (ddd, J = 13.8, 11.4, 3.2 Hz, 1H), 1.54
(s, 3H), 1.40 (ddd, J = 13.8, 7.2, 3.2 Hz, 1H), 0.96
(s, 3H), 0.93 (s, 3H). 13C{1H} NMR (100 MHz,
CDCl3): δ 171.2 (C), 141.8 (C), 140.2 (C), 128.2
(2× CH), 128.0 (C), 127.8 (2× CH), 125.5 (CH), 72.6 (CH),
35.4 (C), 35.0 (CH2), 34.1 (CH2), 28.5 (CH3), 27.2 (CH3), 25.5 (CH2), 21.5 (CH3), 17.3 (CH3). IR (film): 2957, 2935, 2860, 1732,
1494, 1452, 1370, 1243, 1018, 961, 715 cm–1. HRMS
(ESI/TOF) m/z: [M + Na]+ calcd for C18H24O2Na, 295.1674;
found, 295.1679.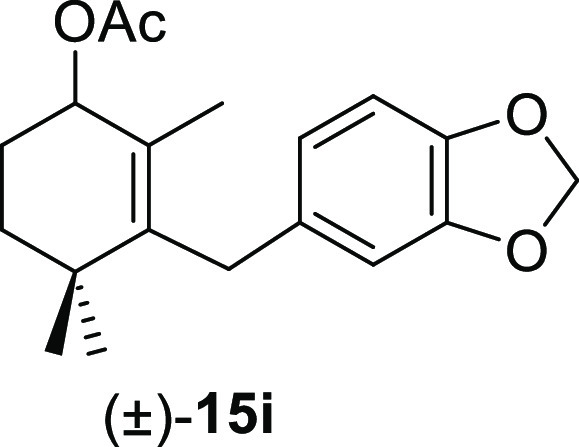
(±)-3-(Benzo[d][1,3]dioxol-5-ylmethyl)-2,4,4-trimethylcyclohex-2-en-1-yl Acetate ((±)-15i)
To a solution of 11i (204 mg, 0.71 mmol) in benzene (5 mL) was added Pb(OAc)4 (348 mg, 0.78 mmol), and the mixture was heated at 80 °C
for 50 min. Following the same workup used in the general procedure, 15i (206 mg, 92%) was obtained as an amorphous solid after
column chromatography using 3% EtOAc/hexane. 1H NMR (400
MHz, CDCl3): δ 6.71 (d, J = 8.0
Hz, 1H), 6.63 (br s, 1H), 6.59 (br d, J = 8.0 Hz,
1H), 5.91 (s, 2H), 5.25 (t, J = 4.7 Hz, 1H), 3.43
(d, J = 16.3 Hz, 1H), 3.39 (d, J = 16.3 Hz, 1H), 2.09 (s, 3H), 1.94 (m, 1H), 1.76 (m, 1H), 1.65 (ddd, J = 12.3, 12.3, 3.1, 1H), 1.55 (s, 3H), 1.41 (ddd, J = 13.1, 7.1 3.2 Hz, 1H), 0.95 (s, 3H), 0.93 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ
171.2 (C), 147.6 (C), 145.4 (C), 142.0 (CH), 134.1 (C), 128.1 (C),
120.6 (CH), 108.3 (CH), 108.1 (CH), 100.7 (CH2), 72.5 (C),
35.4 (C), 34.9 (CH2), 33.7 (CH2), 28.5 (CH3), 27.2 (CH3), 25.5 (CH2), 21.4 (CH3), 17.3 (CH3). IR (film): 1723, 1605, 1495, 1483,
1434, 1365, 1226, 1344, 1227, 1175, 1146, 1121, 1091, 1037, 1017,
923, 875, 793 cm–1. HRMS (ESI/TOF) m/z: [M + H]+ calcd for C19H25O4, 317.1753; found, 317.1751.
(±)-3-(2-Bromobenzyl)-2,4,4-trimethylcyclohex-2-en-1-yl Acetate ((±)-15m)
To a solution of 11m (53 mg, 0.16 mmol) in benzene (1 mL) was added Pb(OAc)4 (80 mg, 0.18 mmol), and the mixture was heated at 80 °C
for 1 h. Following the same workup used in the general procedure, 15m (48 mg, 83%) was obtained as a colorless syrup after column
chromatography using 5% EtOAc/hexane. 1H NMR (500 MHz,
CDCl3): δ 7.54 (dd, J = 7.8, 1.3
Hz, 1H), 7.22 (td, J = 7.5, 1.3 Hz, 1H), 7.08 (d, J = 7.8 Hz, 1H), 7.04 (t, J = 7.5 Hz, 1H),
5.28 (m, 1H), 3.48 (d, J = 17.5 Hz, 1H), 3.44 (d, J = 17.5 Hz, 1H), 2.09 (s, 3H), 1.98 (dddd, J = 14.2, 11.3, 5.0, 3.2 Hz, 1H), 1.80 (dddd, J =
14.2, 7.5, 4.6, 3.2 Hz, 1H), 1.69 (ddd, J = 13.4,
11.3, 3.2 Hz, 1H), 1.48–1.44 (m, 1H), 1.46 (s, 3H), 0.95 (s,
6H). 13C{1H} NMR (125 MHz, CDCl3):
δ 171.1 (C), 141.1 (C), 138.7 (C), 132.5 (CH), 129.1 (C), 128.7
(CH), 127.3 (CH), 127.1 (CH), 125.1 (C), 72.4 (CH), 35.3 (C), 34.8
(CH2), 34.7 (CH2), 28.2 (CH3), 27.0
(CH3), 25.4 (CH2), 21.4 (CH3), 17.1
(CH3). IR (film): 1723, 1438, 1387, 1231, 1155, 1095, 1076,
1025, 877, 770 cm–1. HRMS (ESI/TOF) m/z: [M–OAc]+ calcd for C16H20Br, 291.0748; found, 291.0754.
(±)-2,4,4-Trimethyl-3-(3-nitrobenzyl)cyclohex-2-en-1-yl Acetate ((±)-15o)
To a solution of 11o (67 mg, 0.23 mmol) in benzene (2 mL) was added Pb(OAc)4 (115 mg, 0.26 mmol), and the mixture was heated at 80 °C for 90 min. Following the same workup used in the general procedure, 15o (62 mg, 85%) was obtained as a colorless syrup after column chromatography using 3% EtOAc/hexane. 1H NMR (400 MHz, CDCl3): δ 8.00 (m, 2H), 7.44 (m, 2H), 5.27 (t, J = 4.7 Hz, 1H), 3.60 (d, J = 17.2 Hz, 1H), 3.57 (d, J = 17.2 Hz, 1H), 2.10 (s, 3H), 1.96 (m, 1H), 1.79 (m, 1H), 1.69 (ddd, J = 14.4, 11.6, 3.1 Hz, 1H), 1.52 (s, 3H), 1.43 (m, 1H), 0.94 (s, 3H), 0.93 (s, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 171.1 (C), 148.5 (C), 142.4 (C), 140.6 (C), 134.0 (CH), 129.4 (C), 129.1 (CH), 122.8 (CH), 120.9 (CH), 72.1 (CH), 35.4 (C), 34.7 (CH2), 33.6 (CH2), 28.4 (CH3), 27.1 (CH3), 25.3 (CH2), 21.4 (CH3), 17.4 (CH3). IR (film): 1725, 1469, 1696, 1346, 1231, 1169, 1144, 1095, 1076, 1017, 996, 961, 865, 802 cm–1. HRMS (ESI/TOF) m/z: [M–OAc]+ calcd for C16H20NO2, 258.1494; found, 258.1488.
Acknowledgments
The authors thank the Spanish Ministry of Economy and Competitiveness (Project CTQ2014-56611-R/BQU) for financial support and for the predoctoral fellowship granted to F.J. and the Regional Government of Andalucia (Project P11-CTS-7651) for financial support and assistance provided to the FQM-348 group. J.M.B.A. and M.J.D.P. thank the University of Cádiz for financial support (Ayudas de Investigación Plan Propio).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c00560.
Copies of 1H and 13C{1H} NMR spectra for all new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- For an example of the use of Taxol, see:Kanda Y.; Ishihara Y.; Wilde N. C.; Baran P. S. Two-phase total synthesis of taxanes: Tactics and strategies. J. Org. Chem. 2020, 85, 10293–10320. 10.1021/acs.joc.0c01287. [DOI] [PubMed] [Google Scholar]
- For an example of the use of Artemisin, see:D’Alessandro S.; Scaccabarozzi D.; Signorini L.; Perego F.; Ilboudo D. P.; Ferrante P.; Delbue S. The use of antimalarial drugs against viral infection. Microorganisms 2020, 8, 85. 10.3390/microorganisms8010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For an example of the use of Cantharidin, see:Naz F.; Wu Y.; Zhang N.; Yang Z.; Yu C. Anticancer attributes of Cantharidin: Involved molecular mechanisms and pathways. Molecules 2020, 25, 3279. 10.3390/molecules25143279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a For a recent review concerning the total synthesis of complex terpene natural products, utilizing terpene building blocks, see:Brill Z. G.; Condakes M. L.; Ting C. P.; Maimone T. J. Navigating the chiral pool in the total synthesis of complex terpene natural products. Chem. Rev. 2017, 117, 11753–11795. 10.1021/acs.chemrev.6b00834. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zentar H.; Arias F.; Haidour A.; Alvarez-Manzaneda R.; Chahboun R.; Alvarez-Manzaneda E. Protecting-group-free synthesis of cassane-type furan diterpenes via a decarboxylative dienone-phenol rearrangement. Org. Lett. 2018, 20, 7007–7010. 10.1021/acs.orglett.8b02867. [DOI] [PubMed] [Google Scholar]; c Gutierrez P.; Altarejos J.; Linares-Palomino P. J.; Chahboun R.; Alvarez-Manzaneda E. Synthesis of cassane-type diterpenes from abietane compounds: the first synthesis of taepeenin F.. Org. Chem. Front. 2018, 5, 2537–2541. 10.1039/c8qo00603b. [DOI] [Google Scholar]; d Hung K.; Hu X.; Maimone T. J. Total synthesis of complex terpenoids employing radical cascade processes. Nat. Prod. Rep. 2018, 35, 174–202. 10.1039/c7np00065k. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Gil J. A.; Arias F.; Chahboun R.; Alvarez-Manzaneda E. Synthesis of cyclosiphonodictyol A and its bis(sulfato). J. Org. Chem. 2020, 85, 3799–3805. 10.1021/acs.joc.9b03434. [DOI] [PubMed] [Google Scholar]; f Leger P. R.; Kuroda Y.; Chang S.; Jurczyk J.; Sarpong R. C-C bond cleavage approach to complex terpenoids: Development of a unified total synthesis of the phomactins. J. Am. Chem. Soc. 2020, 142, 15536–15547. 10.1021/jacs.0c07316. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Liu W.; Hong B.; Wang J.; Lei X. New strategies in the efficient total syntheses of polycyclic natural products. Acc. Chem. Res. 2020, 53, 2569–2586. 10.1021/acs.accounts.0c00531. [DOI] [PubMed] [Google Scholar]; h Harmange Magnani C. S.; Thach D. Q.; Haelsig K. T.; Maimone T. J. Syntheses of complex terpenes from simple polyprenyl precursors. Acc. Chem. Res. 2020, 53, 949–961. 10.1021/acs.accounts.0c00055. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Shen Y.; Li L.; Xiao X.; Yang S.; Hua Y.; Wang Y.; Zhang Y.-W.; Zhang Y. Site-specific photochemical desaturation enables divergent synthesis of Illicium sesquiterpenes. J. Am. Chem. Soc. 2021, 143, 3256–3263. 10.1021/jacs.1c00525. [DOI] [PubMed] [Google Scholar]
- Matsumoto T.; Usui S.; Morimoto T. A convenient synthesis of (±)-taxodione, (±)-ferruginol, and (±)-sugiol. Bull. Chem. Soc. Jpn. 1977, 50, 1575–1579. 10.1246/bcsj.50.1575. [DOI] [Google Scholar]
- Matsumoto T.; Ohmura T.; Usui S. The revised structure of dispermol and total synthesis of maytenoquinone, dispermol, and dispermone. Bull. Chem. Soc. Jpn. 1979, 52, 1957–1963. 10.1246/bcsj.52.1957. [DOI] [Google Scholar]
- Huang J.; Foyle D.; Lin X.; Yang J. Total synthesis and biological evaluation of an antifungal tricyclic o-hydroxy-p-quinone methide diterpenoid. J. Org. Chem. 2013, 78, 9166–9173. 10.1021/jo4013964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alvarez-Manzaneda E.; Chahboun R.; Cabrera E.; Alvarez E.; Alvarez-Manzaneda R.; Meneses R.; Es-Samti H.; Fernández A. A very efficient route toward the 4a-methyltetrahydrofluorene skeleton: Short synthesis of (±)-dichroanone and (±)-taiwaniaquinone H. J. Org. Chem. 2009, 74, 3384–3388. 10.1021/jo900153y. [DOI] [PubMed] [Google Scholar]; b Node M.; Ozeki M.; Planas L.; Nakano M.; Takita H.; Mori D.; Tamatani S.; Kajimoto T. Efficient asymmetric synthesis of abeo-abietane-type diterpenoids by using the intramolecular heck reaction. J. Org. Chem. 2010, 75, 190–196. 10.1021/jo901972b. [DOI] [PubMed] [Google Scholar]
- De Graaf S. A. G.; Oosterhoff P. E. R.; van der Gen A. Direct alkylation of α,β-unsaturated aldehydes. Tetrahedron Lett. 1974, 15, 1653–1656. 10.1016/s0040-4039(01)82544-2. [DOI] [Google Scholar]
- Hall J. B.; Wiegers W. J.. Process for the alkylation of α, β-unsaturated aldehydes. U.S. Patent 4,010,207 A, 1977.
- a Kimura M.; Horino Y.; Mukai R.; Tanaka S.; Tamaru Y. Strikingly simple direct α-allylation of aldehydes with allylic alcohols: Remarkable advance in the Tsuji-Trost reaction. J. Am. Chem. Soc. 2001, 123, 10401–10402. 10.1021/ja011656a. [DOI] [PubMed] [Google Scholar]; b Bernhard Y.; Thomson B.; Ferey V.; Sauthier M. Nickel-catalyzed α-allylation of aldehydes and tandem aldol condensation/allylation reaction with allylic alcohols. Angew. Chem., Int. Ed. 2017, 56, 7460–7464. 10.1002/anie.201703486. [DOI] [PubMed] [Google Scholar]
- Yamashita M.; Matsumiya K.; Nakano K.-i. Organic synthesis via dialkylhydrazones. Part 9. α-Alkylation of α,β -unsaturated aldehyde dimethylhydrazones accompanied with the double bond migration to β,γ. Bull. Chem. Soc. Jpn. 1993, 66, 1759–1763. 10.1246/bcsj.66.1759. [DOI] [Google Scholar]
- Yang X.; Nath D.; Morse J.; Ogle C.; Yurtoglu E.; Altundas R.; Fleming F. Cyclic alkenenitriles: Copper-catalyzed deconjugative α -alkylation. J. Org. Chem. 2016, 81, 4098–4102. 10.1021/acs.joc.6b00367. [DOI] [PubMed] [Google Scholar]
- Shi H.-N.; Huang M.-H.; Hao W.-J.; Tu X.-C.; Tu S.-J.; Jiang B. Synthesis of diastereoenriched 1-indanones via double-base cooperatively promoted 1,4-oxo-migration/cyclization of β-alkynyl ketones. J. Org. Chem. 2019, 84, 16027–16035. 10.1021/acs.joc.9b02525. [DOI] [PubMed] [Google Scholar]
- Khartulyari A. S.; Kapur M.; Maier M. E. Concise strategy to the core structure of the macrolide Queenslandon. Org. Lett. 2006, 8, 5833–5836. 10.1021/ol062479r. [DOI] [PubMed] [Google Scholar]
- Akbaba Y.; Türker Balaydın H.; Göksu S.; Şahin E.; Menzek A. Total synthesis of the Biologically active, naturally occurring 3,4-dibromo-5-[2-bromo-3,4-dihydroxy-6-(methoxymethyl)benzyl]benzene-1,2-diol and regioselective O-demethylation of aryl methyl ethers. Helv. Chim. Acta 2010, 93, 1127–1135. 10.1002/hlca.200900300. [DOI] [Google Scholar]
- Drew S. L.; Lawrence A. L.; Sherburn M. S. Total synthesis of Kingianins A, D, and F. Angew. Chem., Int. Ed. 2013, 52, 4221–4224. 10.1002/anie.201210084. [DOI] [PubMed] [Google Scholar]
- Spring D. R.; Krishnan S.; Blackwell H. E.; Schreiber S. L. Diversity-oriented synthesis of biaryl-containing medium rings using a one bead/one stock solution platform. J. Am. Chem. Soc. 2002, 124, 1354–1363. 10.1021/ja017248o. [DOI] [PubMed] [Google Scholar]
- Cantillo D.; de Frutos O.; Rincon J. A.; Mateos C.; Kappe C. O. A scalable procedure for light-induced benzylic brominations in continuous flow. J. Org. Chem. 2014, 79, 223–229. 10.1021/jo402409k. [DOI] [PubMed] [Google Scholar]
- a Barrero A. F.; Alvarez-Manzaneda E. J.; Chahboun R. Synthesis of wiedendiol-A and wiedendiol-B from labdane diterpenes. Tetrahedron 1998, 54, 5635–5650. 10.1016/s0040-4020(98)00235-x. [DOI] [Google Scholar]; b George J. H.; Baldwin J. E.; Adlington R. M. Enantiospecific, biosynthetically inspired formal total synthesis of (+)-Liphagal. Org. Lett. 2010, 12, 2394–2397. 10.1021/ol100756z. [DOI] [PubMed] [Google Scholar]; c Dethe D. H.; Sau S. K.; Mahapatra S. Biomimetic enantioselective total synthesis of (-)-Mycoleptodiscin A. Org. Lett. 2016, 18, 6392–6395. 10.1021/acs.orglett.6b03292. [DOI] [PubMed] [Google Scholar]
- For the synthesis of the benzofluorene derivative Dasyscyphin E using Δ7-drimenal as electrophile see:Jiménez F.; Fernández A.; Boulifa E.; Mansour A. I.; Alvarez-Manzaneda R.; Chahboun R.; Alvarez-Manzaneda E. Diastereoselective intramolecular Heck reaction assisted by an acetate group: Synthesis of the decahydrobenzofluorene derivative Dasyscyphin E. J. Org. Chem. 2017, 82, 9550–9559. 10.1021/acs.joc.7b01551. [DOI] [PubMed] [Google Scholar]
- Preite M. D.; Cuellar M. A. A new reaction: lead(IV) acetate-mediated oxidative fragmentation of homoallylic alcohols. Chem. Commun. 2004, 1970–1971. 10.1039/b405986g. [DOI] [PubMed] [Google Scholar]
- a Bertrand M. P.; Surzur J. M.; Boyer M.; Mihailović M. L. Mechanisms of oxidation of ethylenic alcohols by lead tetraacetate. ESR evidence for the influence of experimental conditions on the homolytic or heterolytic course of the reaction. Tetrahedron 1979, 35, 1365–1372. 10.1016/0040-4020(79)85030-9. [DOI] [Google Scholar]; b Paredes M. D.; Alonso R. Oxidation of α-hydroxysilanes by lead tetraacetate. Tetrahedron Lett. 1999, 40, 3973–3976. 10.1016/s0040-4039(99)00646-2. [DOI] [Google Scholar]
- Aslam S. N.; Stevenson P. C.; Phythian S. J.; Veitch N. C.; Hall D. R. Synthesis of cicerfuran, an antifungal benzofuran, and some related analogs. Tetrahedron 2006, 62, 4214–4226. 10.1016/j.tet.2006.02.015. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.