

Abstract
Hazard identification regarding adverse effects on the liver is a critical step in safety evaluations of drugs and other chemicals. Current testing paradigms for hepatotoxicity rely heavily on pre-clinical studies in animals and human data (epidemiology and clinical trials). Mechanistic understanding of the molecular and cellular pathways that may cause or exacerbate hepatotoxicity is well advanced, and holds promise for identification of hepatotoxicants. One of the challenges in translating mechanistic evidence into robust decisions about potential hepatotoxicity is the lack of a systematic approach to integrate these data to help identify liver toxicity hazards. Recently, marked improvements were achieved in the practice of hazard identification of carcinogens, female and male reproductive toxicants, and endocrine disrupting chemicals using the key characteristics approach. Here, we describe the methods by which key characteristics of human hepatotoxicants were identified and provide examples for how they could be used to systematically identify, organize and utilize mechanistic data when identifying hepatotoxicants.
Keywords: Risk factors, hazard, risk assessment, key characteristics, hepatotoxicity
The liver is a critical organ in the human body. Although it comprises only around 2% of an adult’s body weight, approximately 25% of the total cardiac output goes through the liver. Because the liver is the first organ that comes in contact with the blood from the gastro-intestinal tract, it is critically important for sensing and processing gut-derived molecular signals and xenobiotics that then may influence the function of other organs. It is responsible for a wide spectrum of essential functions including the uptake, metabolism, and excretion of various endogenous and exogenous substances. The liver is also a key organ for storage and metabolism of carbohydrates, vitamins, amino acids and ammonia, lipids and lipoproteins, and cholesterol, as well as synthesis and secretion of proteins, bile acids, hormones, and other mediators. The liver provides an immunological function through its role in phagocytosis and clearance of microorganisms and endotoxins from the portal blood.
In addition to these important physiologic functions, the liver is a key target of toxic response to xenobiotics and pathogens. It is the organ of first-pass metabolism for many chemicals, including drugs, occupational hazards, and environmental contaminants (pesticides, industrial chemicals and other pollutants). Chemicals and pathogens may be distributed to the liver via the circulation, given its high blood flow, or may be concentrated in the liver during enterohepatic circulation. As a result, the liver represents an important site for the generation and action of toxic metabolites and various chemicals are known to cause acute and chronic adverse effects on the liver. For instance, ethanol-associated liver disease is a common cause of chronic liver toxicity (1). Acute cases of hepatotoxicity are most often connected to drug-induced liver injury (DILI) (2) which accounts for more than 50% of acute liver failure cases (3). Hepatotoxicity is also a common finding for occupational as well as environmental chemicals, a number of which are associated with “toxicant-associated steatohepatitis” (4). Nearly a third (134 out of 417) of the chemicals examined by US EPA’s Integrated Risk Information System program have identified “critical effects” on the liver as basis for deriving of safe exposure levels. Among cancer hazard evaluations of the chemicals examined by the EPA, approximately half (53 out of 94) identified the liver as the tumor site in chronic studies in rats and mice. A recent review (5) of the 111 agents that are classified as carcinogenic to humans (Group 1) by the International Agency for Research on Cancer (IARC) revealed that liver is the second and fourth, respectively, most common chemical-associated target site for exogenous exposure-associated cancer in humans and experimental animals. Besides chemicals, other causes of chronic liver injury include nonalcoholic fatty liver disease, and pathogens.
Given the liver’s prominent roles in physiology and as a target of many injurious insults, hepatotoxicity is an important determinant of the body’s toxic responses. As a result, DILI is a leading cause for drug failure in pre-clinical development, clinical trials, and post-marketing. This underscores the critical need for robust methods to identify hepatotoxicants during drug development. One approach to find common elements among chemicals that may be hepatotoxic is to examine their chemical structure and/or metabolites through the development of structure-activity relationship models. However, while many such models have been developed, they remain imprecise. Indeed, it is widely accepted that drug attrition due to DILI is largely due to limited predictivity of the clinical outcomes by current preclinical models used for liver safety testing (6). Studies of liver toxicity by drugs and chemicals, and the mechanisms by which such effects may occur, are a very active area of research and discovery. Research into mechanisms of adverse effects holds promise for improving prediction of liver toxicity from drugs and other chemicals. An approach that can incorporate this new knowledge in identifying new hepatotoxicants is based on defining a finite set of properties of agents known to affect the liver, so called ‘key characteristics’ (7). Herein, we describe this approach, elaborate the key characteristics of hepatotoxicants, and describe how they can be applied.
The Key Characteristics Approach to Identifying Chemical Hazards to the Liver
The Key Characteristics (KCs) concept was first developed for, and based on, the known properties of established human carcinogens identified by IARC (7). This concept proved applicable for the systematic evaluation of the literature on mechanisms by which human carcinogens act, to identify gaps in knowledge, and to guide design of the cellular and molecular assays that may be used to replace long-term studies in experimental animals. The KCs of human carcinogens are now widely used by various regulatory agencies and form the basis for the evaluation and integration of mechanistic data. Others, in the pharmaceutical industry and elsewhere, also appreciated the value of KCs for the design of tests to evaluate the cancer hazards of novel chemicals to complement or replace existing approaches (8). A more broad application of this concept, beyond cancer hazard evaluations, was also advocated by the National Academies (9) “as a guide for evaluating the relationship between perturbations observed in assays, their potential to pose a hazard, and their contribution to risk.” Accordingly, the goal of the present work was to develop a consensus expert opinion on the KCs of known hepatotoxicants.
To consider how the KC concept might be applied to the etiology of liver disease, an inter-disciplinary working group of experts in hepatology, pathology, toxicology and risk assessment was convened. Representatives across various sectors (academia, clinical medicine, pharmaceutical safety evaluation and regulatory toxicology) were included to ensure that the outcomes of this expert working group’s efforts are broadly applicable. Twelve KCs of major relevance were identified for chemicals which cause liver injury. These KCs of hepatotoxicants (Table 1) are described in the paragraphs that follow.
Table 1.
Key Characteristics of Hepatotoxicants.
| Key Characteristic (KC) |
|---|
| 1. Is reactive and/or is metabolized (bioactivated) to reactive moieties |
| 2. Causes death (apoptosis and/or necrosis) of liver cells |
| 3. Affects liver cell proliferation and/or tissue regeneration |
| 4. Disrupts transport function |
| 5. Induces oxidative stress |
| 6. Triggers immune-mediated responses in liver |
| 7. Causes mitochondrial dysfunction |
| 8. Activates stress signaling pathways |
| 9. Causes cholestasis |
| 10. Disrupts cellular cytoskeleton |
| 11. Causes liver fibrosis |
| 12. Disrupts liver metabolism, including of lipids and proteins |
The Key Characteristics of Hepatotoxicants
KC1. Is reactive and/or is metabolized (bioactivated) to reactive moieties.
The liver is the primary site of biotransformation of endogenous molecules and xenobiotics; hepatocytes contain multiple families of enzymes essential for bioactivation, detoxification and transport of parent compounds and their metabolites. The process of biotransformation is generally directed at elimination of xenobiotics by changing their physical properties to those favoring excretion, but reactive intermediates can be formed during metabolic transformation. Many compounds that are known to be toxic to the liver are bioactivated in situ to reactive electrophiles, that form addition products with cellular macromolecules. Induction of xenobiotic metabolism by a variety of endogenous and exogenous compounds plays an important role in causing liver toxicity (10). Tissue-, sex-, age- and species-specific differences in expression and inducibility of metabolizing enzymes are critical determinants of sensitivity to toxic effects caused by xenobiotics in the liver and other organs (11). Classic examples of agents that require bioactivation in the liver to become hepatotoxic include acetaminophen, vinyl chloride, carbon tetrachloride, polycyclic aromatic hydrocarbons, and aflatoxin B1. Direct-acting electrophiles, such as sulfur mustards and other alkylating agents, phalloidin, and microcystins are also hepatotoxic albeit their toxic effects may not be liver-specific.
KC2. Causes death (apoptosis and/or necrosis) of liver cells.
Cell death governs outcomes and long-term consequences in response to many liver toxicants. Acute liver failure is characterized by cell death (12) and impairment of liver function is dependent on the cell type affected. Injury to hepatocytes results in impairment of drug metabolism and other liver functions, whereas cholangiocyte damage triggers cholestasis. Cell death in chronic liver injury is often more extensive and leads to long-term alterations in organ architecture and function, contributing to chronic hepatocyte turnover, the recruitment of immune cells, activation of hepatic stellate cells, and development of liver fibrosis, cirrhosis and cancer (12). Cell death in the liver occurs through a variety of pathways that lead to apoptosis or necrosis. Apoptosis is the physiological route to eliminate damaged or infected cells and to maintain tissue homeostasis and accompanies almost all types of liver injury. Death receptor-induced apoptosis in hepatocytes and cholangiocytes is mediated by mitochondrial and/or lysosomal permeabilization (13). Signaling between the endoplasmic reticulum and mitochondria can promote hepatocyte apoptosis in response to excesses in free fatty acids associated with metabolic syndrome. By contrast, necrosis may result from cellular damage or infiltration by pathogens (14). Other contributing forms of cell death including necroptosis and pyroptosis are under intense investigation.
KC3. Affects liver cell proliferation and/or tissue regeneration.
Increased cell proliferation is an important factor in cancer development and progression but also in response to liver damage. In liver, cell proliferation is typically driven by two main mechanisms: direct induction of cell cycle mechanisms by compounds such as peroxisome proliferators, barbiturates and dioxins, and compensatory cell proliferation in response to hepatocyte death. The latter can be caused by chemicals such as carbon tetrachloride, or by pathogens. Although mitotic division of mature hepatocytes is a normal part of liver cell turnover and repair, extensive or chronic injury can overwhelm the regenerative capacity of hepatocytes and prompt the activation of liver progenitor cells (15). Mediators of inflammation (e.g., cytokines) can prompt proliferation in response to injury and during development and growth of tumors (16).
KC4. Disrupts transport function.
Hepatic transporters are involved in movement of nutrients, waste products, and xenobiotics between hepatocytes and blood, and between hepatocytes and bile (17). As such, transport proteins serve a central role in liver homeostasis, and genetic or environmentally-induced deficits in their function can contribute to liver disease. As drug development shifted towards slower/non-metabolized molecules, disruption of hepatic transport pathways became a prominent component of drug safety research (18). Inhibition and altered expression/subcellular localization are the primary modes of transporter-mediated disruption, giving rise to altered endobiotic homeostasis and/or accumulation of hepatotoxic concentrations of xenobiotics. Studies of metabolism and transporter function are critical to unravel the dynamics of molecular hepatotoxicity (19). Transporter interactions have also been linked to hepatic accumulation of drugs and chemicals in liver.
KC5. Induces oxidative stress.
Oxidative stress is the result of an imbalance between production of reactive oxygen species (ROS) and cellular antioxidants. ROS are necessary for essential liver function such as fatty acid metabolism and immune responses, and play important roles in intra- and inter-cellular signaling. However, excess production of ROS can overwhelm cellular antioxidants, disrupt mitochondrial functions, cause damage to cellular macromolecules, promote endoplasmic reticulum stress, and activate pro-inflammatory responses (20). ROS in the liver are commonly produced in hepatocytes and by immune cells. Examples of substances that cause adverse responses in the hepatobiliary system through excessive ROS production and downstream cellular and tissue damage include chlorinated solvents (e.g., carbon tetrachloride), acetaminophen and other drugs, metals, and ethanol.
KC6. Triggers immune-mediated responses in liver.
Liver cell damage can initiate an inflammatory response driven by recruitment and activation of innate immune cells, including Kupffer cells, neutrophils and natural killer cells, that produce cytokines and other potentially damaging mediators (21). This inflammatory response can magnify injury initiated by hepatotoxicants and their reactive metabolites. Conversely, an innate immune response is necessary for liver repair. Recruited neutrophils phagocytose damaged cells, making way for tissue repair that involves proliferation of hepatocytes, which is initiated by growth factors produced by recruited macrophages. The inflammatory response is thought to play a role in both the pathogenesis of hepatocellular injury from acetaminophen and in replicative repair of the liver (22). The adaptive immune system is widely held to be involved in idiosyncratic DILI based on the strong evidence of the role of the human leukocyte antigen (HLA) alleles with genetic predisposition to liver injury. Many drugs, including nevirapine and flucloxacillin, are hypothesized to cause liver injury through an adaptive immune mechanism (23). Bioactivation of drugs to reactive metabolites that bind to proteins, which then act as haptens, can initiate an adaptive immune response (24). The healthy liver exists in a “tolerized” state that prevents damaging adaptive immune responses, so that immune tolerance likely must be overcome in order to initiate hepatocellular damage by this mechanism. A damaging adaptive immune response involves activation of lymphocytes that can effect killing of hepatocytes directly or indirectly through release of damaging mediators (23). The latter can involve secondary activation of innate immune cells that produce cytokines which bind to receptors and activate cell death pathways within hepatocytes.
KC7. Causes mitochondrial dysfunction.
Mitochondria are critical organelles in eukaryotic cells as they are responsible for aerobic metabolism. Oxidative phosphorylation is the primary physiological function of mitochondria and is dependent on electro-chemical and pH gradients across the inner membrane. Maintenance of electrochemical proton gradient and adenosine triphosphate (ATP) synthesis by mitochondria are critical to overall cell viability. Changes in permeability can lead to a collapse of ion gradients across the mitochondrial membrane, causing mitochondrial dysfunction (25). High cytosolic ATP/ADP ratio is important for oxidative metabolism and decreased mitochondrial function shifts cell metabolism to glycolysis. Because depletion of ATP is a salient feature of both necrotic and apoptotic cell death, induction of ATP-requiring metabolic pathways by drugs and chemicals can prime hepatocytes for cell death. In addition, through control of mitochondrial electron transport chain-generated oxidants, mitochondrial glutathione can modulate cell death (26). Drugs that cause liver injury often impact the mitochondria and activate signal transduction pathways important in determining cell survival or death. Classic hepatotoxic compounds that exert their effects in large part through the mitochondria are ethanol, acetaminophen, and components of herbal supplements including pyrrolizidine alkaloids. Mitochondrial disruption can also play a role in non-alcoholic fatty liver disease.
KC8. Activates stress signaling pathways.
Cell death signaling is often initiated by various forms of cell stress that involve a cascade of kinase activation which prompts mitochondrial dysfunction and a consequent reduction in ATP production leading to cell death (23). In hepatocytes, endoplasmic reticulum stress can also activate kinases that trigger cell death (27). For example, toxic doses of acetaminophen result in overproduction of ROS that initiates a kinase cascade, ultimately resulting in activation of c-Jun-N-terminal kinase (JNK). JNK interacts with mitochondrial membrane components to cause a permeability transition and loss of mitochondrial function and phosphorylates many targets to mediate cell death and inflammation (28). Chemical- and pathogen-induced stress to liver can also be mediated through stimulation of inflammatory cells and the production of cytokines that can initiate receptor-mediated apoptosis of hepatocytes. Importantly, the same signaling elements (e.g., kinases) that participate in cell death can also activate cell survival pathways (29). Chronic injury from hepatotoxicants such as ethanol, carbon tetrachloride, or vinyl chloride leads to fibrosis, the pathogenesis of which involves additional stress signaling pathways.
KC9. Causes cholestasis.
Cholestasis refers to the accumulation of bile salts or bile acids in the liver and in blood, as a consequence of alterations in hepatocellular transporter function or damage to the extrahepatic bile duct network (30). Drugs or xenobiotics can induce intrahepatic cholestasis by multiple mechanisms, for example by alterations in bile salt/acid transporters (expression, activity or intracellular localization) and disruption of hepatocellular physiology (e.g., disturbance of cell cytoskeleton or mitochondrial stress). Adaptive homeostasis of bile acids with the farnesoid X receptor (FXR) are also known to regulate the expression of bile acid metabolism and transporter pathways, and antagonism of FXR has emerged as a key contributor to hepatic cholestasis. Extrahepatic cholestasis can result from compound-induced intrinsic cytotoxicity in large bile ducts, as observed with 5-fluorouracyl. However, the most common mechanism involves an immune response to drugs, as hypothesized for example with amoxicillin-clavulanate (31).
KC10. Disrupts cellular cytoskeleton.
Differentiated hepatocytes and bile duct epithelial cells express a number of keratin molecules that form a filamentous network, or cytoskeleton, in the cytoplasm. Cellular keratins not only provide mechanical stability to cells, but also play important roles as targets and modulators of toxic stress and apoptosis (32). Keratins are abundant in hepatocytes and are common targets of xenobiotics or their metabolites; therefore, hepatotoxicants that affect keratin stability and assembly can have profound effects on cell proliferation, apoptosis and stress responses (e.g., unfolded protein response) (32). Examples of direct-acting agents that are known to disrupt hepatocyte cytoskeleton are phalloidin and microcystins. Reactive metabolites of acetaminophen, halothane and other liver toxicants also target keratins (33). Alterations of the hepatocyte keratin network are manifested as ballooning of hepatocytes and cytoplasmic inclusions, so called Mallory-Denk bodies. Ballooned hepatocytes exhibit a reduced density or loss of the cytoplasmic keratin network (32). Mallory-Denk bodies consist of misfolded and aggregated keratins and other proteins with altered molecular structure and chemical modifications.
KC11. Causes liver fibrosis.
Liver fibrosis is a product of the intrahepatic formation and persistence of extracellular matrix. It results from activation of perisinusoidal stellate cells (and possibly portal fibroblasts) and matrix production by these cells (34). In response to sustained injury of hepatocytes, biliary epithelial cells, or endothelial cells, pro-fibrotic cytokines produced by resident liver cells and infiltrating leukocytes activate the otherwise quiescent stellate cells (35). Activated stellate cells become myofibroblasts, which acquire contractile capacity and produce collagen and other extracellular matrix proteins. Deposition of collagenous matrix in the perisinusoidal space and other perivascular locations disrupts the normal exchange of plasma solutes between the blood and the hepatocytes, resulting in diminished liver function and derangements in hepatic blood flow. Liver fibrosis may result from a variety of causes of sustained injury to hepatocytes (ethanol, carbon tetrachloride, aflatoxin, iron, and pathogens), biliary epithelial cells (immune injury, bile acids, dihydrocollidine), or sinusoidal endothelial cells (pyrrolizidine alkaloids).
KC12. Disrupts liver metabolism, including of lipids and proteins.
The liver produces plasma proteins, lipoproteins, cholesterol, and phospholipids, metabolizes lipids, proteins, and carbohydrates, and removes ammonia via conversion to urea. Xenobiotics can disrupt these functions and cause adverse effects in liver and other tissues. Disruptions of lipid metabolism in liver (36) can occur via (i) inhibition of phospholipid degradation in the lysosome or enhanced phospholipid biosynthesis and subsequent accumulation of intracellular phospholipids (e.g. chloroquine, azithromycin); (ii) impairment of mitochondrial fatty acid oxidation through inhibition of beta-oxidation (e.g. valproate, amiodarone, perhexiline), inhibition of mitochondrial respiratory chain (e.g. tamoxifen, amiodarone, troglitazone), or depletion of mitochondrial DNA (e.g. fialuridine, zalcitabine, tacrine); and (iii) enhanced de novo lipogenesis (e.g. amiodarone, methotrexate), and impaired secretion of very low-density lipoproteins (e.g. tamoxifen, nucleoside reverse transcriptase inhibitors). Inhibition of transcription and protein synthesis in liver can occur with cycloheximide, fumonisin, and carbon tetrachloride. Finally, valproic acid can cause hyperammonemia and encephalopathy via inhibition of urea cycle enzymes.
Characterization of Drugs and Chemicals that Cause Liver Toxicity Using the KCs of Hepatotoxicants
Acetaminophen (Figure 1).
Figure 1.
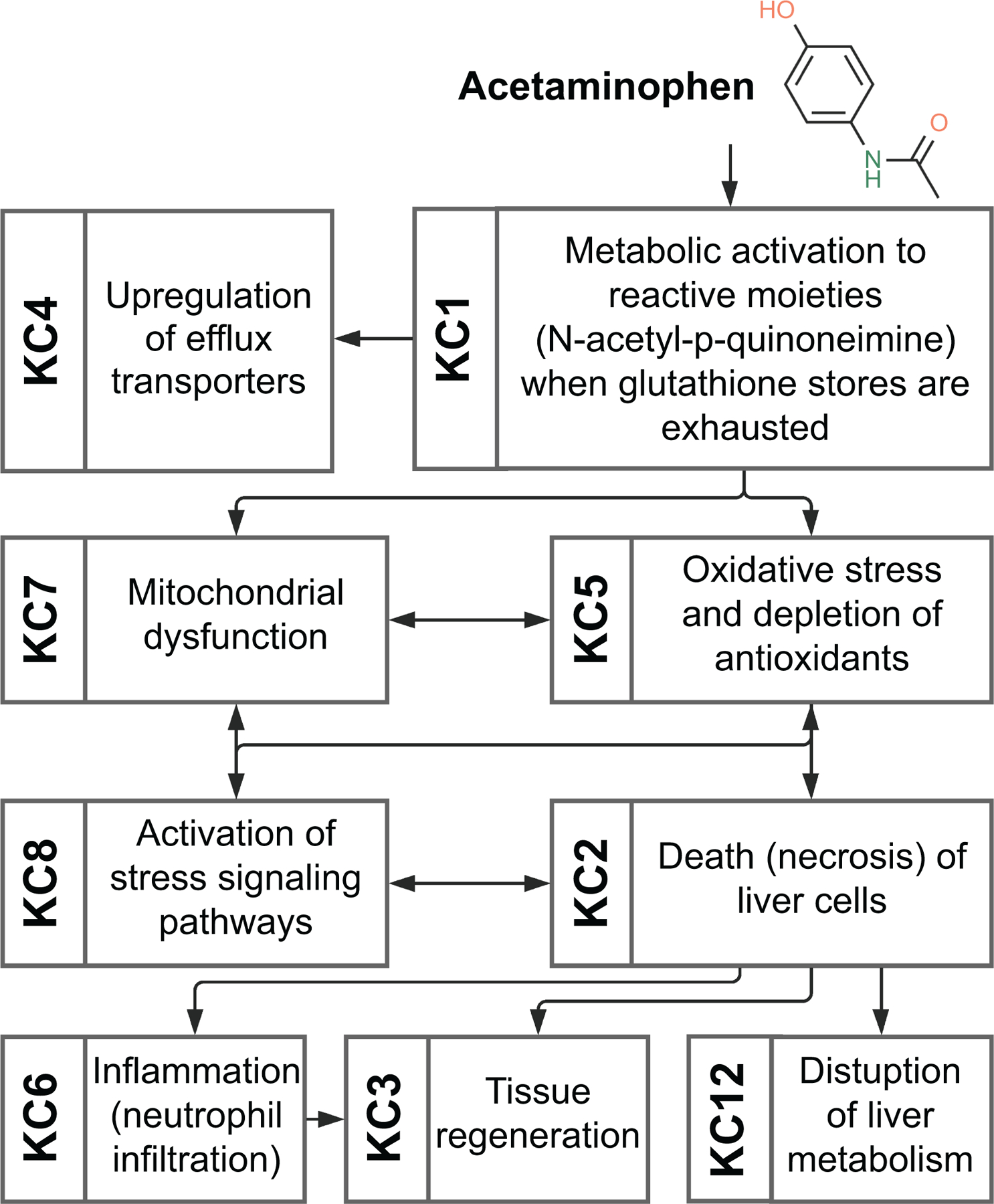
Key characteristics of hepatotoxicity associated with exposure to acetaminophen.
Acetaminophen consumed in overdose is responsible for about half of human cases of acute liver failure in the US and most European countries. It has become the most widely studied drug that causes hepatotoxicity. Acetaminophen exhibits many KCs of hepatotoxicants (KCs 1–8 and 12). At therapeutic doses, acetaminophen is conjugated and also metabolized to reactive N-acetyl-p-quinoneimine, which is inactivated by conjugation to glutathione; in overdose, the conjugation mechanisms are overwhelmed, leaving N-acetyl-p-quinoneimine (KC1) to bind to intracellular proteins, an event that initiates intracellular signaling that culminates in hepatocellular death (KC2) (23). Mitochondrial dysfunction is a key factor in the mechanism of injury (KC7), leading to enhanced production of reactive oxygen and nitrogen species and oxidative stress (KC5) (37). This precipitates a cascade of signaling events involving oxidation of thioredoxin, activation of apoptosis signaling and downstream activation of JNK (KC8) (29). Neutrophils are activated during acetaminophen overdose (KC6); they migrate to the injured parenchyma and appear to be important in converting pro-inflammatory macrophages into pro-resolving macrophages (38). This is followed by proliferation of hepatocytes (KC3) driven in part by inflammatory cytokines and growth factors; bile acids and phosphatidic acid also appear to play roles in liver regeneration (14). Acetaminophen-associated liver injury is accompanied by increased appearance of serum enzymes, elevated blood ammonia and decreases in export proteins such as coagulation factors and lipoproteins, which are synthesized by hepatocytes (KC12). In addition, several efflux transporters are upregulated in the livers of patients with acetaminophen-induced hepatic failure (KC4).
Amoxicillin-clavulanate (Figure 2).
Figure 2.
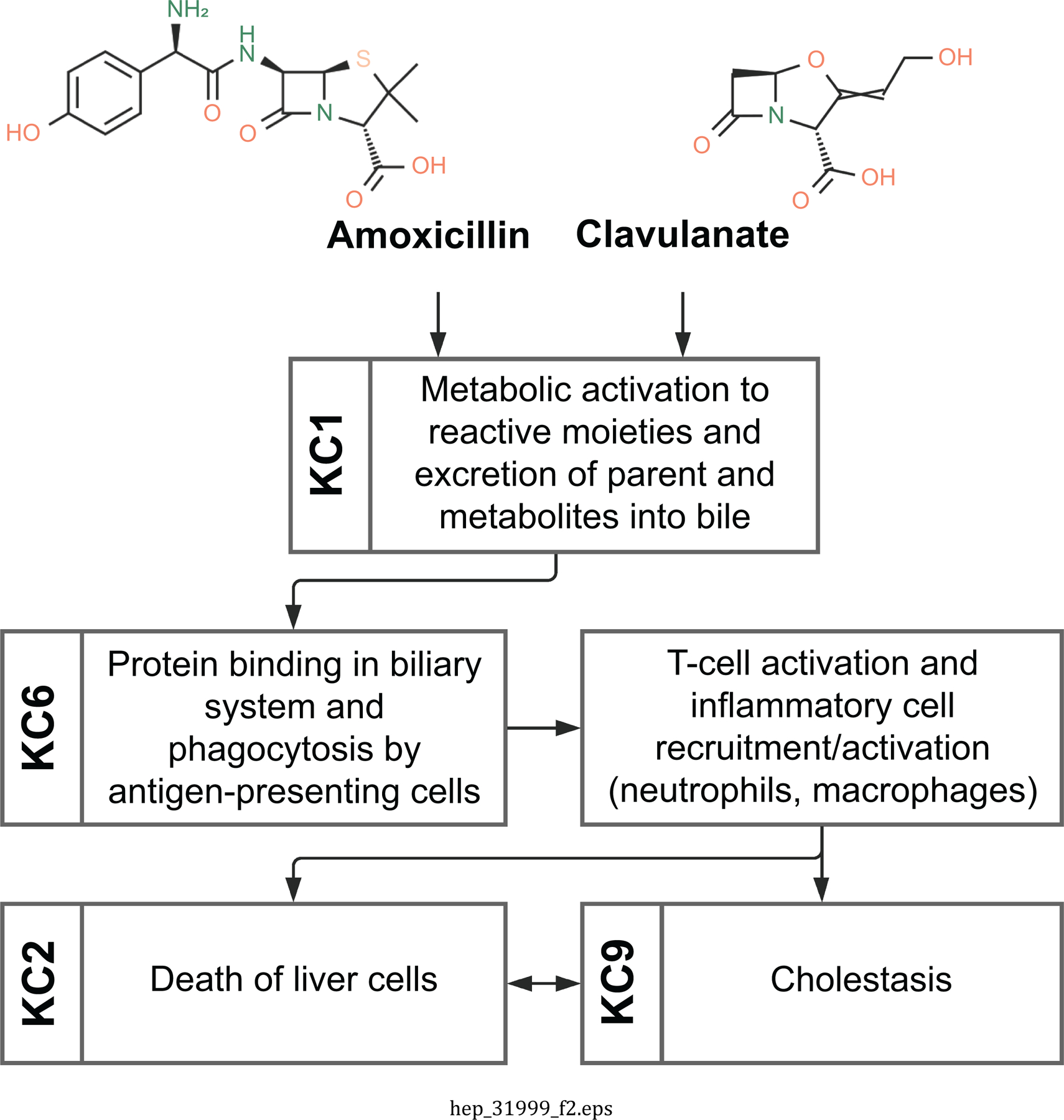
Key characteristics of hepatotoxicity associated with exposure to amoxycillin-clavulanate.
Amoxicillin-clavulanate is a combination antibiotic used to treat a variety of gram-positive bacterial infections. Amoxicillin-clavulanate administration has been associated with liver injury, typically delayed following exposure, in a small fraction of patients (39). Amoxicillin-clavulanate exhibits several KCs of hepatotoxicants (KCs 1, 2, 6, 9). Amoxicillin-clavulanate-related DILI is typically recognized as a cholestatic form of injury, in which there is injury to the small interlobular bile ducts, or as mixed cholestatic (KC9) and hepatocellular (KC2) form of injury (31). In early stages, this injury is histologically characterized by periportal inflammatory cell accumulation, swelling and vacuolation of biliary epithelial cytoplasm, increased mitotic figures, lymphocytic infiltration, and hepatocellular and canalicular accumulation of bile. In later stages, the injury may be histologically characterized by loss of interlobular ducts, irregular proliferations of bile ducts, and fibrosis. Clinical laboratory findings typically include increased serum enzymes and bilirubinemia; however, serum enzyme elevations are minor in many cases. The pathogenesis of amoxicillin-clavulanate-related DILI appears to result from an immune-mediated injury to the interlobular bile ducts (KC6). Both amoxicillin and clavulanate can form unique antigens (KC1) that trigger a T-lymphocyte mediated immune response, and this initiates injury to bile duct epithelial cells and inflammation in the liver (40). This immune-mediated pathogenesis is consistent with the prolonged latency, the idiosyncratic nature of the DILI, and susceptibility associated with certain HLA polymorphisms (41).
Vinyl chloride (Figure 3).
Figure 3.
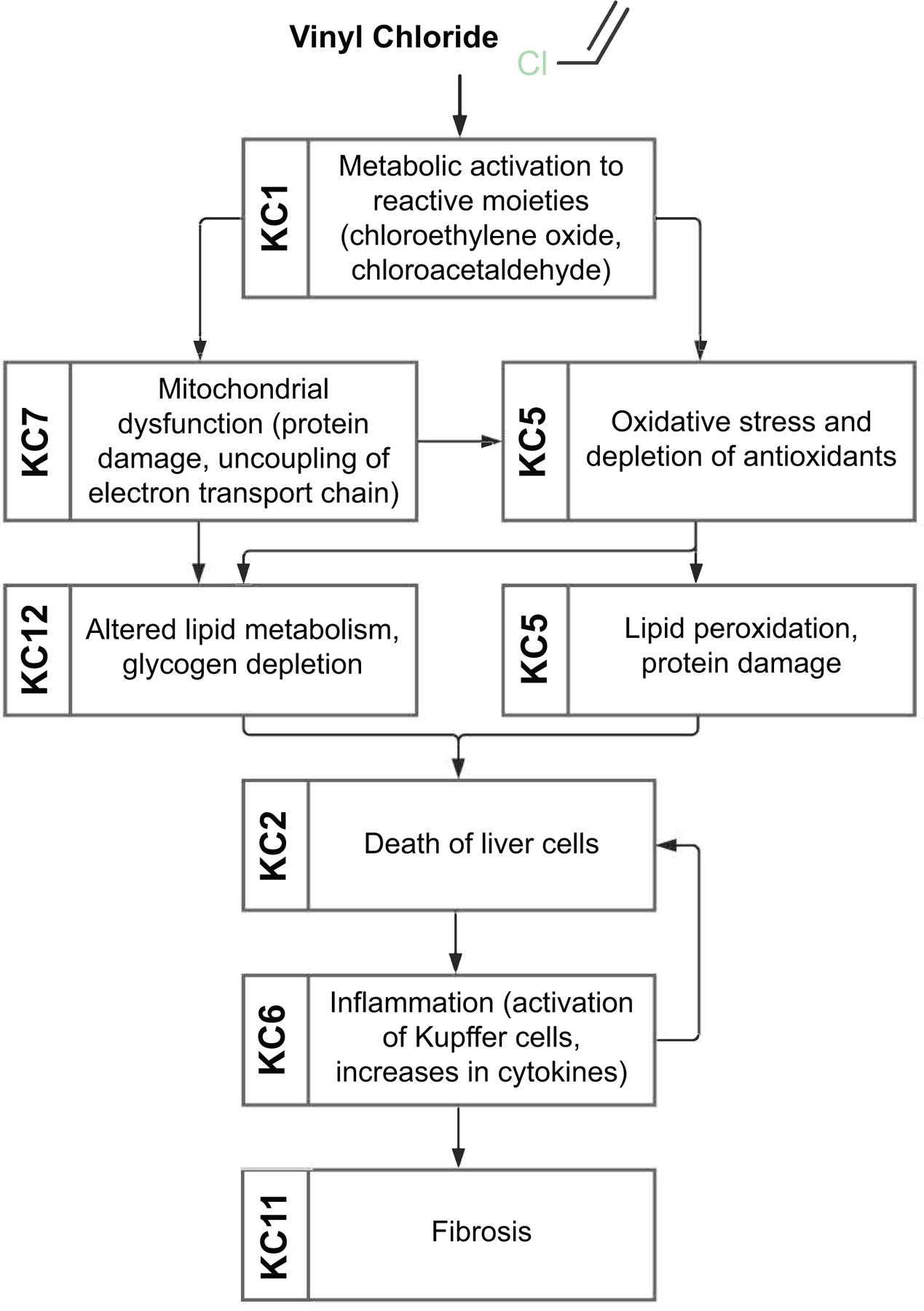
Key characteristics of hepatotoxicity associated with exposure to vinyl chloride.
Vinyl chloride is an industrial chemical used primarily as an intermediate in the production of polyvinyl chloride which is a material for plastic piping, flooring and many other consumer and industrial products. Human exposure to vinyl chloride is primarily by inhalation in the occupational setting (42). Epidemiological and experimental animal studies indicate that exposure to vinyl chloride causes both cancer (e.g. hepatocellular carcinoma, hemangiosarcoma) and non-cancer effects (e.g. fatty liver, steatohepatitis, peliosis hepatitis, and fibrosis) in liver. Vinyl chloride exhibits a range of KCs of hepatotoxicants (KCs 1, 2, 5–7, 11, 12). It is easily absorbed upon inhalation or oral exposure and is partitioned primarily to lipid-rich tissues such as liver. Liver metabolism of vinyl chloride yields highly reactive 2-chloroethylene oxide and 2-chloroacetaldehyde (KC1). These reactive metabolites are thought to induce mitochondrial damage by uncoupling of the electron transport chain (KC7), alter hepatocyte metabolic functions (KC12), and increase production of ROS (KC5), ultimately resulting in “toxicant-associated steatohepatitis” (43). Increased oxidative stress and fatty liver promote formation of cytotoxic lipid peroxidation products that have been associated with hepatocellular cell death (KC2) and immune-mediated signaling (KC6). Combined, these cellular responses activate pro-inflammatory signals which in turn promote remodeling of the extracellular matrix and fibrosis (KC11).
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD, Figure 4).
Figure 4.
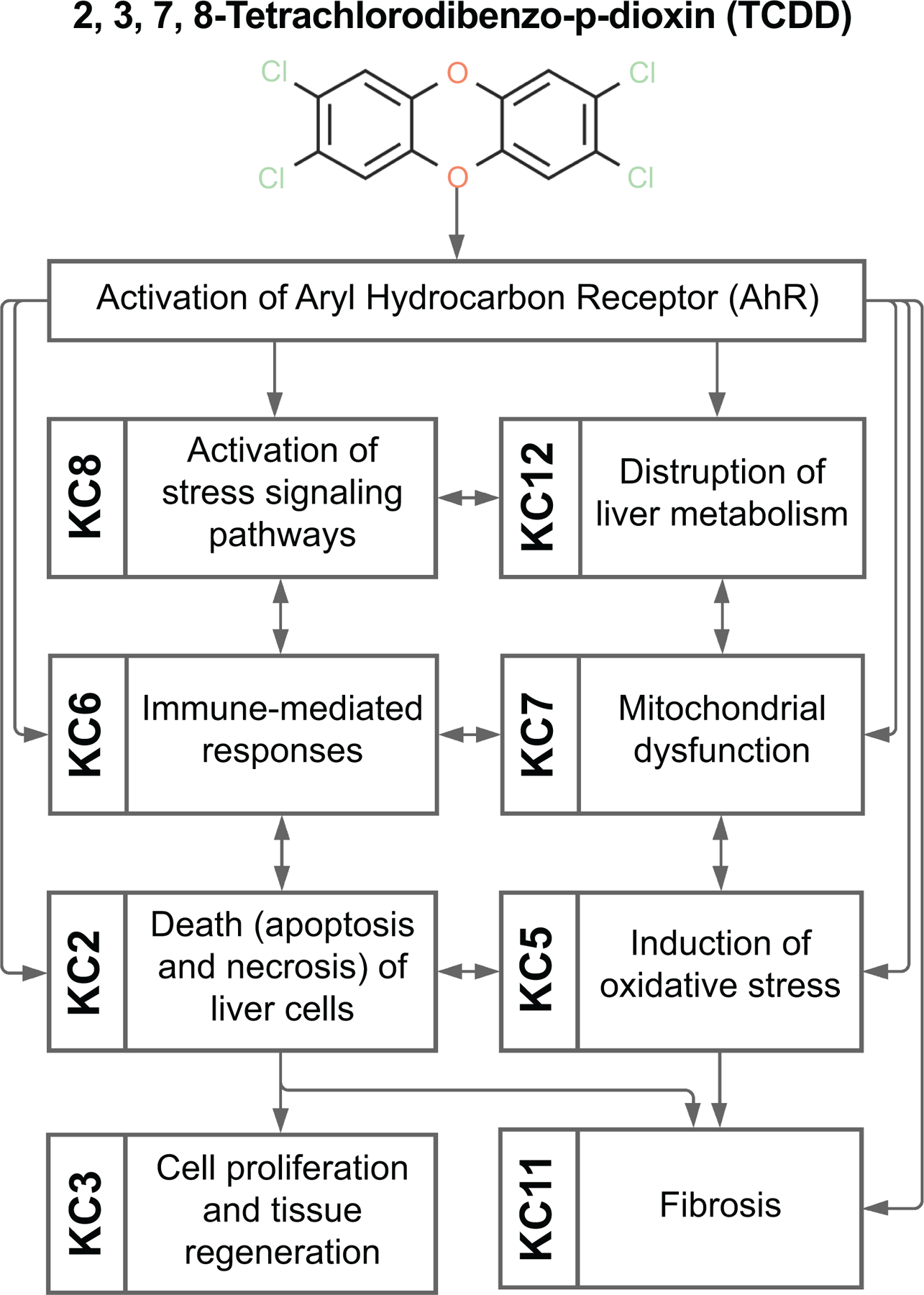
Key characteristics of hepatotoxicity associated with exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD).
A number of persistent environmental contaminants are high-affinity ligands for aryl hydrocarbon receptor (AhR), a central mediator of many pathological states in liver and other tissues (44). TCDD is a prototypical AhR activator that has been studied extensively as a representative for a larger class of “dioxins”. It exhibits many KCs of hepatotoxicants (KCs 2, 3, 5, 6, 7, 8, 11 and 12). In contrast to most hepatotoxicants, TCDD and other structurally-related AhR ligands do not require metabolism to exert liver toxicity, they are slowly metabolized into compounds with limited activity. TCDD-mediated induction of xenobiotic-metabolizing enzymes is AhR-dependent (45) and results in profound dysregulation of gene transcription in multiple pathways (KCs 8 and 12). These effects are thought to play critical roles in the adverse effects of TCDD and related compounds (46). Induction of cell proliferation (KC 3) occurs as a consequence of AhR-dependent gene transcription, and in response to liver cell death (KC 2) from oxidative stress (KC 5) and mitochondrial dysfunction (KC 7) (45). Effects on immune-mediated pathways (KC 6) and cell-specific (hepatocyte and/or stellate cell) responses to AhR activation determine the extent of liver fibrosis (KC 11) (46, 47).
Applications of the KCs of Hepatotoxicants for Identification and Characterization of the Causes of Liver Toxicity
Hazard evaluation as part of a human health risk assessment includes the identification and integration of mechanistic, toxicological, and epidemiological evidence (9). Recent developments in the field of systematic literature review have improved transparency of the evidence evaluation and integration steps in a hazard evaluation (48). Mechanistic studies (including those that involve the use of in vivo, in vitro, or in silico models) describe biological alterations caused by exposure to a drug or chemical which lead to an adverse response. Integration of mechanistic studies into a hazard evaluation can be a challenging process because of the complexity of experimental designs, diversity of research models, and volume of available mechanistic studies. The KC approach has been shown to facilitate the search, screening, organization, and evaluation of mechanistic evidence in a systematic and transparent manner (49). Furthermore, the KC approach evades potential biases introduced into an evidence analysis caused by adherence to a priori mechanistic hypotheses (7). With respect to studies of the liver disease, the presented set of twelve KCs of hepatotoxicants (Table 1) provides a framework to identify, organize, and evaluate relevant mechanistic and observational information as part of a systematic review and evaluation of chemical-induced hepatotoxicity.
In addition, these KCs can be used as a guide to develop compendia of assays that probe most salient pathways and mechanisms. For example, chemical bioactivation (KC1) is often probed with liver microsomes and cultured hepatocytes, and inclusion of metabolic enzymes is possible in microplate-based assays of other molecular phenotypes. Cell death (KC2) and proliferation (KC3) can be investigated with a number of cellular and molecular assays in hepatocytes and more complex models. Transporter function (KC4) is routinely examined with fluorescent probes, inhibitors and secretion of bile acids. ROS production (KC5) can be monitored with fluorescent probes and through formation of damaged DNA, proteins and lipids. Assays for mitochondrial dysfunction (KC7), activation of stress signaling pathways (KC8), and disruption of cellular cytoskeleton (KC10) range from cellular imaging to molecular phenotypes such as proteins and mRNA. Even though immune-mediated responses (KC6), liver fibrosis (KC11) and disruptions of liver metabolism (KC12) require more complex in vitro models that include multiple cells and extended culture periods, many assay systems are available (50). Hence, a systematic and comprehensive evaluation of the potential liver toxicity hazard can be performed to screen numerous compounds for liver toxicity.
CONCLUSIONS
Several important general considerations emerged from the exercise of defining key characteristics of hepatotoxicants. First, the focus was to define the features of agents that induce both acute and chronic liver injury, but not cancer. The rationale for this was that key characteristics of human carcinogens have already been established and are widely used for cancer hazard identification (7, 49); these already broadly cover the agents that may cause tumors in the liver. Second, the group debated at length the challenge of considering the dose in deriving a list of key characteristics. It was recognized that different mechanisms may operate at different dose levels, or that additional KCs might be evident at high doses than at lower doses. However, it was considered that the 12 KCs are inclusive of mechanisms operating at both low and high doses and in acute and chronic forms of liver toxicity. Third, the group further evaluated which assays and biomarkers might provide the strongest evidence for each of KCs in vitro, in experimental animals, and in humans and which agents were representative examples of hepatotoxicants that possessed many KCs; some of these are detailed by the examples above. Importantly, a consensus of the working group members was that the KCs of hepatotoxicants best illustrate how some agents can elicit liver injury. Although some hepatotoxicants might possess many of the KCs, others may possess few and there is no minimum or maximum number necessary to consider an agent as an hepatotoxicant.
It is noteworthy that toxicants can act by the same mechanism in several different organs. Indeed, many KCs that apply to liver will apply to other organs; the only liver-specific one is KC9, causes cholestasis. As a result, association of one or more KCs with a chemical should not be construed as indicating that exposure to this chemical will necessarily cause toxicity per se, or that the liver should be regarded as a target organ. Such overlap was considered a strength rather than a weakness of the KC approach because it supports the notion that various other factors, including the exposure dose, other aspects of toxicant exposure (e.g. dosing duration, regimen, and route), as well as the underlying genetic and environmental sensitivity of an individual might determine the target organ and the toxic response. This underscores the need for consideration of the sensitivity factors causing one organ to be a more sensitive “target” than another organ for a given common mechanism (mitochondrial dysfunction, oxidative stress as examples).
Acknowledgments
Financial support: This work was supported, in part, by the grants from the National Institute of Environmental Health Sciences (P42 ES 027704 and P42 ES004705).
Footnotes
Conflict of Interest Statement: The views expressed in this article are those of the author (s) and do not necessarily represent the views or policies of the U.S Environmental Protection Agency and other affiliations listed herein. Where authors are identified as personnel of the International Agency for Research on Cancer/World Health Organization, the authors alone are responsible for the views expressed in this article and they do not necessarily represent the decisions, policy or views of the International Agency for Research on Cancer/World Health Organization.
REFERENCES
- 1.Paik JM, Golabi P, Younossi Y, Mishra A, Younossi ZM. Changes in the Global Burden of Chronic Liver Diseases From 2012 to 2017: The Growing Impact of NAFLD. Hepatology 2020;72:1605–1616. [DOI] [PubMed] [Google Scholar]
- 2.Asrani SK, Devarbhavi H, Eaton J, Kamath PS. Burden of liver diseases in the world. J Hepatol 2019;70:151–171. [DOI] [PubMed] [Google Scholar]
- 3.Peters TS. Do preclinical testing strategies help predict human hepatotoxic potentials? Toxicol Pathol 2005;33:146–154. [DOI] [PubMed] [Google Scholar]
- 4.Wahlang B, Jin J, Beier JI, Hardesty JE, Daly EF, Schnegelberger RD, Falkner KC, et al. Mechanisms of Environmental Contributions to Fatty Liver Disease. Curr Environ Health Rep 2019;6:80–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krewski D, Rice JM, Bird M, Milton B, Collins B, Lajoie P, Billard M, et al. Concordance between sites of tumor development in humans and in experimental animals for 111 agents that are carcinogenic to humans. J Toxicol Environ Health B Crit Rev 2019;22:203–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monticello TM, Jones TW, Dambach DM, Potter DM, Bolt MW, Liu M, Keller DA, et al. Current nonclinical testing paradigm enables safe entry to First-In-Human clinical trials: The IQ consortium nonclinical to clinical translational database. Toxicol Appl Pharmacol 2017;334:100–109. [DOI] [PubMed] [Google Scholar]
- 7.Smith MT, Guyton KZ, Gibbons CF, Fritz JM, Portier CJ, Rusyn I, DeMarini DM, et al. Key Characteristics of Carcinogens as a Basis for Organizing Data on Mechanisms of Carcinogenesis. Environ Health Perspect 2016;124:713–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fielden MR, Ward LD, Minocherhomji S, Nioi P, Lebrec H, Jacobson-Kram D. Modernizing Human Cancer Risk Assessment of Therapeutics. Trends Pharmacol Sci 2018;39:232–247. [DOI] [PubMed] [Google Scholar]
- 9.National Academies of Sciences Engineering and Medicine. Using 21st Century Science to Improve Risk-Related Evaluations Washington, DC: The National Academies Press, 2017. [PubMed] [Google Scholar]
- 10.Parke DV. Mechanisms and consequences of the induction of microsomal enzymes of mammalian liver. Biochem J 1972;130:53P–55P. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maronpot RR, Yoshizawa K, Nyska A, Harada T, Flake G, Mueller G, Singh B, et al. Hepatic enzyme induction: histopathology. Toxicol Pathol 2010;38:776–795. [DOI] [PubMed] [Google Scholar]
- 12.Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Guicciardi ME, Malhi H, Mott JL, Gores GJ. Apoptosis and necrosis in the liver. Compr Physiol 2013;3:977–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2014;147:765–783 e764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kung JW, Currie IS, Forbes SJ, Ross JA. Liver development, regeneration, and carcinogenesis. J Biomed Biotechnol 2010;2010:984248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Roberts RA, James NH, Cosulich S, Hasmall SC, Orphanides G. Role of cytokines in non-genotoxic hepatocarcinogenesis: cause or effect? Toxicology Letters 2001;120:301–306. [DOI] [PubMed] [Google Scholar]
- 17.Gu X, Manautou JE. Molecular mechanisms underlying chemical liver injury. Expert Rev Mol Med 2012;14:e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schuetz JD, Swaan PW, Tweedie DJ. The role of transporters in toxicity and disease. Drug Metab Dispos 2014;42:541–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Smith MT. Mechanisms of troglitazone hepatotoxicity. Chem Res Toxicol 2003;16:679–687. [DOI] [PubMed] [Google Scholar]
- 20.Jaeschke H, Gores GJ, Cederbaum AI, Hinson JA, Pessayre D, Lemasters JJ. Mechanisms of hepatotoxicity. Toxicol Sci 2002;65:166–176. [DOI] [PubMed] [Google Scholar]
- 21.Roberts RA, Ganey PE, Ju C, Kamendulis LM, Rusyn I, Klaunig JE. Role of the Kupffer cell in mediating hepatic toxicity and carcinogenesis. Toxicol Sci 2007;96:2–15. [DOI] [PubMed] [Google Scholar]
- 22.Yang R, Tonnesseen TI. DAMPs and sterile inflammation in drug hepatotoxicity. Hepatol Int 2019;13:42–50. [DOI] [PubMed] [Google Scholar]
- 23.Iorga A, Dara L, Kaplowitz N. Drug-Induced Liver Injury: Cascade of Events Leading to Cell Death, Apoptosis or Necrosis. Int J Mol Sci 2017;18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adams DH, Ju C, Ramaiah SK, Uetrecht J, Jaeschke H. Mechanisms of immune-mediated liver injury. Toxicol Sci 2010;115:307–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rosser BG, Gores GJ. Liver cell necrosis: cellular mechanisms and clinical implications. Gastroenterology 1995;108:252–275. [DOI] [PubMed] [Google Scholar]
- 26.Fernandez-Checa JC, Kaplowitz N. Hepatic mitochondrial glutathione: transport and role in disease and toxicity. Toxicol Appl Pharmacol 2005;204:263–273. [DOI] [PubMed] [Google Scholar]
- 27.Ji C, Kaplowitz N. ER stress: can the liver cope? J Hepatol 2006;45:321–333. [DOI] [PubMed] [Google Scholar]
- 28.Win S, Than TA, Zhang J, Oo C, Min RWM, Kaplowitz N. New insights into the role and mechanism of c-Jun-N-terminal kinase signaling in the pathobiology of liver diseases. Hepatology 2018;67:2013–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Seki E, Brenner DA, Karin M. A liver full of JNK: signaling in regulation of cell function and disease pathogenesis, and clinical approaches. Gastroenterology 2012;143:307–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Padda MS, Sanchez M, Akhtar AJ, Boyer JL. Drug-induced cholestasis. Hepatology 2011;53:1377–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bonkovsky HL, Kleiner DE, Gu J, Odin JA, Russo MW, Navarro VM, Fontana RJ, et al. Clinical presentations and outcomes of bile duct loss caused by drugs and herbal and dietary supplements. Hepatology 2017;65:1267–1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fickert P, Fuchsbichler A, Wagner M, Silbert D, Zatloukal K, Denk H, Trauner M. The role of the hepatocyte cytokeratin network in bile formation and resistance to bile acid challenge and cholestasis in mice. Hepatology 2009;50:893–899. [DOI] [PubMed] [Google Scholar]
- 33.Cohen SD, Pumford NR, Khairallah EA, Boekelheide K, Pohl LR, Amouzadeh HR, Hinson JA. Selective protein covalent binding and target organ toxicity. Toxicology & Applied Pharmacology 1997;143:1–12. [DOI] [PubMed] [Google Scholar]
- 34.Kang N Mechanotransduction in Liver Diseases. Semin Liver Dis 2020;40:84–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Novo E, Bocca C, Foglia B, Protopapa F, Maggiora M, Parola M, Cannito S. Liver fibrogenesis: un update on established and emerging basic concepts. Arch Biochem Biophys 2020;689:108445. [DOI] [PubMed] [Google Scholar]
- 36.Begriche K, Massart J, Robin MA, Borgne-Sanchez A, Fromenty B. Drug-induced toxicity on mitochondria and lipid metabolism: mechanistic diversity and deleterious consequences for the liver. J Hepatol 2011;54:773–794. [DOI] [PubMed] [Google Scholar]
- 37.Jaeschke H, McGill MR, Ramachandran A. Oxidant stress, mitochondria, and cell death mechanisms in drug-induced liver injury: lessons learned from acetaminophen hepatotoxicity. Drug Metab Rev 2012;44:88–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Williams CD, Bajt ML, Sharpe MR, McGill MR, Farhood A, Jaeschke H. Neutrophil activation during acetaminophen hepatotoxicity and repair in mice and humans. Toxicol Appl Pharmacol 2014;275:122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bjornsson ES. Drug-induced liver injury due to antibiotics. Scand J Gastroenterol 2017;52:617–623. [DOI] [PubMed] [Google Scholar]
- 40.Meng X, Earnshaw CJ, Tailor A, Jenkins RE, Waddington JC, Whitaker P, French NS, et al. Amoxicillin and Clavulanate Form Chemically and Immunologically Distinct Multiple Haptenic Structures in Patients. Chem Res Toxicol 2016;29:1762–1772. [DOI] [PubMed] [Google Scholar]
- 41.Visentin M, Lenggenhager D, Gai Z, Kullak-Ublick GA. Drug-induced bile duct injury. Biochim Biophys Acta Mol Basis Dis 2018;1864:1498–1506. [DOI] [PubMed] [Google Scholar]
- 42.Wahlang B, Beier JI, Clair HB, Bellis-Jones HJ, Falkner KC, McClain CJ, Cave MC. Toxicant-associated steatohepatitis. Toxicol Pathol 2013;41:343–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Joshi-Barve S, Kirpich I, Cave MC, Marsano LS, McClain CJ. Alcoholic, Nonalcoholic, and Toxicant-Associated Steatohepatitis: Mechanistic Similarities and Differences. Cell Mol Gastroenterol Hepatol 2015;1:356–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wilson CL, Safe S. Mechanisms of ligand-induced aryl hydrocarbon receptor-mediated biochemical and toxic responses. Toxicol Pathol 1998;26:657–671. [DOI] [PubMed] [Google Scholar]
- 45.Budinsky RA, Schrenk D, Simon T, Van den Berg M, Reichard JF, Silkworth JB, Aylward LL, et al. Mode of action and dose-response framework analysis for receptor-mediated toxicity: The aryl hydrocarbon receptor as a case study. Crit Rev Toxicol 2014;44:83–119. [DOI] [PubMed] [Google Scholar]
- 46.Yan J, Tung HC, Li S, Niu Y, Garbacz WG, Lu P, Bi Y, et al. Aryl Hydrocarbon Receptor Signaling Prevents Activation of Hepatic Stellate Cells and Liver Fibrogenesis in Mice. Gastroenterology 2019;157:793–806 e714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li C, Liu Y, Dong Z, Xu M, Gao M, Cong M, Liu S. TCDD promotes liver fibrosis through disordering systemic and hepatic iron homeostasis. J Hazard Mater 2020;395:122588. [DOI] [PubMed] [Google Scholar]
- 48.Whaley P, Halsall C, Agerstrand M, Aiassa E, Benford D, Bilotta G, Coggon D, et al. Implementing systematic review techniques in chemical risk assessment: Challenges, opportunities and recommendations. Environ Int 2016;92–93:556–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guyton KZ, Rusyn I, Chiu WA, Corpet DE, van den Berg M, Ross MK, Christiani DC, et al. Application of the key characteristics of carcinogens in cancer hazard identification. Carcinogenesis 2018;39:614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soldatow VY, Lecluyse EL, Griffith LG, Rusyn I. In vitro models for liver toxicity testing. Toxicol Res (Camb) 2013;2:23–39. [DOI] [PMC free article] [PubMed] [Google Scholar]