

Abstract
In this review, we discuss the utility of quantitative electroencephalography (qEEG) parameters for detection of delayed cerebral ischemia (DCI) after aneurysmal subarachnoid hemorrhage in the context of the complex pathophysiology of DCI and the limitations of current diagnostic methods. Due to the multifactorial pathophysiology of DCI, methodologies solely assessing blood vessel narrowing (vasospasm), are insufficient to detect all DCI. qEEG has facilitated the exploration of EEG as a diagnostic modality of DCI. Multiple qEEG parameters such as alpha power, relative alpha variability, and alpha/delta ratio show reliable detection of DCI in multiple studies. Recent studies on epileptiform abnormalities suggest their potential for detection of DCI. qEEG is a promising continuous, non-invasive monitoring modality of DCI implementable in daily practice. Future work should validate these parameters in larger populations, facilitated by the development of automated detection algorithms and multimodal data integration.
Keywords: Quantitative electroencephalography, EEG, delayed cerebral ischemia, subarachnoid hemorrhage, Transcranial doppler, vasospasm
1. Introduction
Aneurysmal subarachnoid hemorrhage (SAH) accounts for only 3% of all stroke types but carries high morbidity and mortality (1). Delayed cerebral ischemia (DCI) occurs in 33–50% of SAH patients with the peak incidence between 3–14 days after ictus (2–4). DCI increases the risk of poor long-term outcomes including decreased instrumental activities of daily living (IADL), cognitive deficits, and reduced quality of life (4). DCI is preventable with timely intervention only if it is detected before clinical deterioration or radiographic infarction. While frequent clinical examinations are important for detecting new neurological deficits indicating ischemia, their utility is limited in high-grade SAH patients who have the highest risk of DCI. Furthermore, SAH is associated with multiple complications such as rebleeding, seizures and hydrocephalus, which can confound clinical examinations. Hence, much effort has focused on discovering accurate tests and parameters to predict impending DCI. Sonographic or radiographic modalities, which have varying degrees of accuracy, display changes in blood vessel caliber or blood flow and are the accepted standard of care measures for DCI surveillance. However, these studies can only be performed once or twice a day at best. Given the need for a diagnostic modality with the ability to provide real-time, noninvasive monitoring for DCI, continuous EEG has emerged as a valuable tool in assessing risk of impending DCI.
2. Definition of DCI
Historically, several terms have been used to describe DCI including delayed ischemic neurological deficits, symptomatic vasospasm, secondary cerebral ischemia, and cerebral infarction. To address confusion generated from these various terms and inconsistency in clinical study definitions, a multidisciplinary expert group proposed a unified definition of DCI as: cerebral infarction identified on CT or MRI or proven at autopsy after exclusion of procedure-related infarctions; or the new occurrence of focal neurological impairment, or a decrease of at least two points on the Glasgow Coma Scale, with or without radiological findings, that cannot be attributed to other causes by means of clinical assessment, CT, or MRI scanning of the brain and appropriate lab studies (5). This definition combines both clinical and radiographic criteria for DCI, thereby capturing the full breadth of patients who suffer from this complication. To provide clarity in our terminology for this review, we will be using the term “DCI” as defined here, whereas any reference to observations limited to blood vessel narrowing assessed by radiographic or sonographic imaging will be referred to as “vasospasm.”
3. Pathophysiology of DCI
The exact pathophysiology of DCI remains incompletely understood, contributing to the varied descriptions of DCI above. Conventionally, vasospasm, was believed to be the sole cause of DCI, but accruing evidence suggests DCI is secondary to other phenomena such as cortical spreading depolarization (CSD), impaired cerebral autoregulation, microcirculatory dysfunction, microthrombosis, and neuroinflammation (6–9).
3.1. Vasospasm
In 1949, Robertson first suggested temporary vasospasm as the cause of infarction in an area of the brain remote from the ruptured aneurysm (10), an observation which has since been supported by both radiographic and sonographic evidence of vasospasm after SAH (11–15). Current diagnostics and therapeutic strategies, such as endovascular treatment, are largely targeted at reducing vasospasm to reduce DCI and poor functional outcome (16)
However, the causal link between vasospasm and DCI has been questioned due to observations that DCI can occur without vasospasm and not all patients with vasospasm develop DCI (17–19). Failed treatment trials also call into question this relationship, such as recent trial failures of clazosentan, an endothelin-receptor antagonist, which induced significant reduction of moderate to severe vasospasm from 66% to 23% without changing the incidence of DCI (p=0.315) (20,21). This conflicting data alludes to more complex mechanisms of DCI not ascribed to vasospasm alone.
3.2. Cortical spreading depolarization (CSD)
CSD is a wave of depolarization spreading across the cortex propagating over minutes (22). This phenomenon has received more recent attention as a potential contributory mechanism of DCI as its presence has been observed in cases of DCI without vasospasm (23). Drier et al. used electrocorticography (ECoG) to assess the presence of CSD in 18 SAH patients and noted that DCI occurred in a time-locked fashion to CSD in 13 patients (24). CSD is proposed to cause DCI via increased metabolic demand during the CSD at an unsustainable level in the injured brain state of SAH, thereby directly causing neuronal injury and death (25,26). Simultaneously, persistent CSD triggered by insults to the brain such as SAH can induce vasoconstriction in contradiction to the normal response of vasodilation upon increased metabolism, which precipitates ischemia and accelerates the vicious cycle of prolonged CSD causing flow-metabolism uncoupling and energy failure, making it difficult to recover from CSD to the normal state (27,28). Moreover, the persistent excitation results in increased extracellular glutamate and the subsequent activation of NMDA-receptors which can lead to excitotoxic injury in energy-compromised neurons (Aiba and Shuttleworth, 2012). The trigger for CSD after SAH remains unclear. However, theories about relative ischemia and decreased extracellular nitric oxide levels suggest that in some cases there is a potential interplay by which vasospasm may induce CSD and increase the risk of DCI (29,30). While CSD is typically detected using ECoG, continuous EEG could prove useful. One study of CSD in TBI patients with simultaneous ECoG and cEEG recording found that spreading depolarizations induced a spread of depression on the order of minutes to hours (31). However, scalp EEG is limited as it is generally only sensitive to clustered spreading depolarizations (32). Future research is needed to better characterize scalp EEG signatures of CSD after SAH.
3.3. Impaired cerebral autoregulation and Microcirculatory dysfunction
Cerebral autoregulation (CA) is a mechanism to regulate regional cerebral blood flow (rCBF) with cerebral vasoconstriction and vasodilation to maintain constant cerebral perfusion pressure (CPP) under different physiologic conditions. Impaired cerebral autoregulation has been observed in SAH and may contribute to the development of DCI (33,34). Patients who deviated from autoregulation-based blood pressure targets had worse functional outcomes at 90 days compared to those who did not, suggesting that the disruption of CA contributes to post-bleed complications (35). Previous studies showed that vasospasm itself does not always result in reduced rCBF (36). It was postulated that impaired CA in microvasculature distal to the restricted segments of vasospasm plays a crucial role in failure to preserve rCBF, leading to the development of DCI (7,37). Some studies proposed that an independent process of microvascular spasm and dysfunction which is perpetuated by CSD in SAH with or without proximal vasospasm may cause cerebral infarction (38,39). One study found that patients who develop DCI and vasospasm have distinct autoregulation patterns from those who develop only DCI, implying that impaired autoregulation of cerebral blood flow might be an independent pathological process that can lead to DCI (40).
Impaired regional CBF was investigated in relation to microcirculatory dysfunction. While transcranial doppler (TCD) or conventional angiogram can visualize large cerebral vessels, they are poor at detecting changes in microcirculation. Abnormal measures of microcirculatory function were observed in DCI irrespective of vasospasm (41). This impaired microcirculation in DCI is believed to be related to endothelial dysfunction in SAH causing microthrombi, which can lead to infarction (41–44).
3.4. Neuroinflammation
Recently, inflammation-based mechanisms of DCI have gained attention. One study implicated the role of matricellular proteins (MCP) periostin, osteopontin, and galectin-3 in DCI; machine learning models incorporating plasma levels of these MCPs achieved approximately 90% prediction accuracy for DCI (45). As components of the extracellular matrix, MCPs affect vascular permeability and cell death, thereby contributing to other dysfunctional processes such as microcirculatory dysfunction and neuroinflammation (45). Overall, these results indicate that MCPs may comprise part of DCI pathology.
Several inflammatory biomarkers are elevated after SAH in patients who develop DCI (29,46,47). For example, the sulfonylurea receptor 1–transient receptor potential melastatin 4 (Sur1-Trpm4) channel is upregulated after SAH, and inhibition of this receptor in rats reduces neuroinflammation and cognitive impairment after SAH (48,49). Some inflammatory biomarkers, such as soluble growth stimulation expressed gene 2 (ST2), can independently predict DCI (50,51). Notably, ST2 is linked to new epileptiform abnormalities after SAH, making continuous EEG a useful tool to detect inflammation and predict DCI (52). Inflammation can also interact with thrombosis to create a positive feedback loop wherein a pro-inflammatory state can induce the formation of microthrombi and vice versa (53). Altogether, these results suggest an inflammatory pathology underlying DCI although they do little to elucidate the precise mechanism.
The above evidence supports that DCI has multifactorial pathophysiology (Figure 1). Therefore, diagnostic tools designed to detect vasospasm alone are insufficient to capture all cases of DCI, necessitating the development of novel diagnostic methods that directly assess known DCI contributors.
Figure 1.
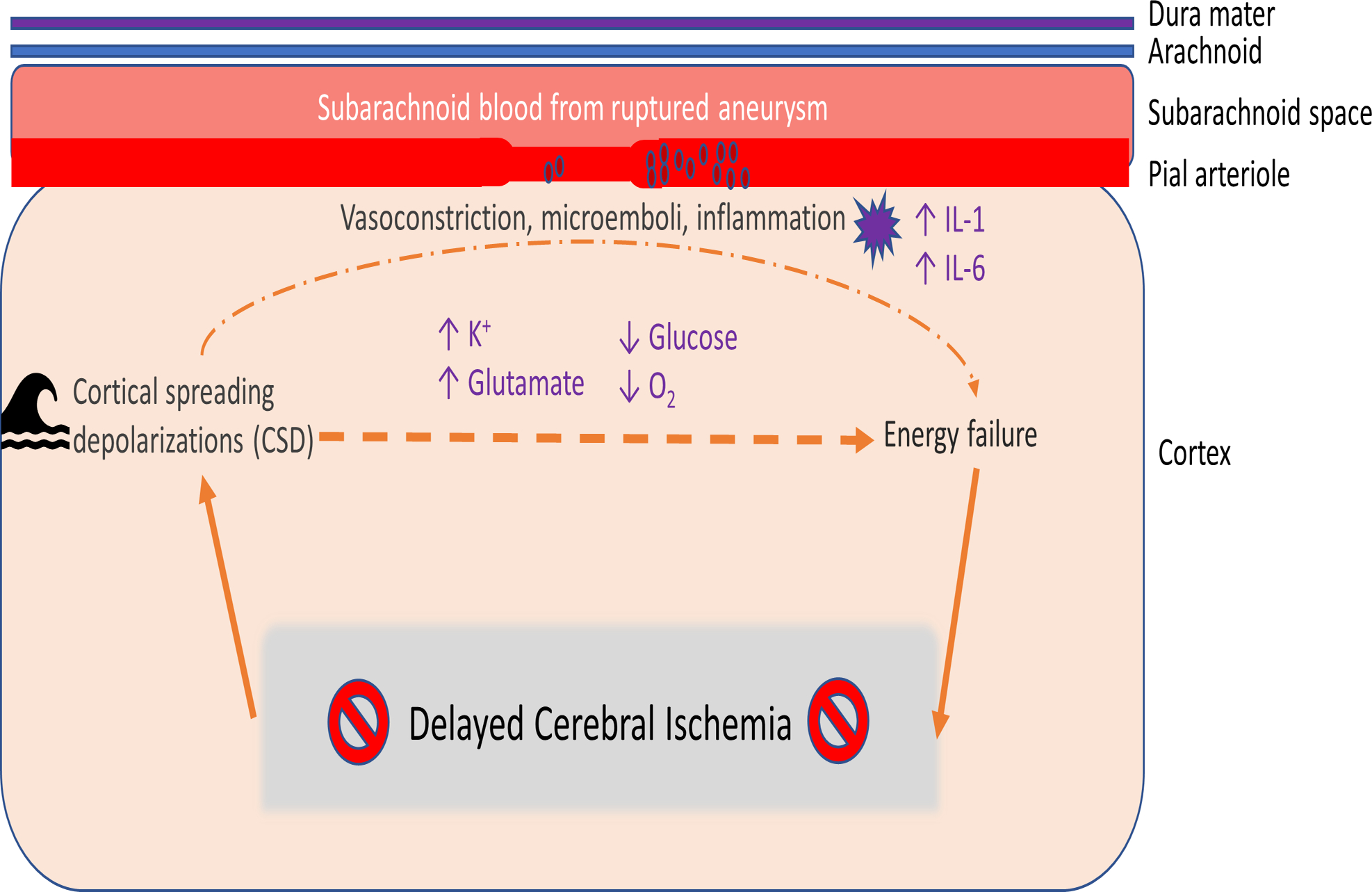
Multifactorial Pathophysiology of DCI. After aneurysmal subarachnoid hemorrhage, delayed cerebral ischemia can occur due to multifactorial pathophysiology. Vasospasm and microemboli, which can form as a result of a proinflammatory, hypercoagulable state after aneurysm rupture, produce regional hypoxia that can progress to infarction. Cortical spreading depressions, or waves of excitation propagating across the injured brain, can induce an inverse hemodynamic response wherein there is diminished blood flow to areas with increased metabolic demand. Persistent excitation leads to an increase in extracellular potassium and glutamate and an increase in intracellular calcium, which can lead to excitotoxic injury. When tissue at risk for infarction does not receive sufficient oxygen, DCI can result. Adapted from Leng, Fink, and Iadecola (2011).
4. Transcranial doppler (TCD) for monitoring vasospasm
Despite the emergence of other mechanisms of DCI, vasospasm is still the best understood mechanism contributing to DCI, and its detection and treatment is critical in reducing the devastating consequences of DCI. Thus, multiple modalities are used to screen for vasospasm including: TCD, CT or MR angiography, CT perfusion, and catheter-based digital subtraction angiography (DSA). While DSA remains the gold standard for detecting vasospasm (54), TCD is the most commonly used diagnostic tool for monitoring vasospasm as recommended in 2012 by the American Heart Association and American Stroke Association (AHA/ASA). Support for using TCDs is based on class IIa/level B evidence as it provides a rapid, noninvasive and portable modality without radiation exposure (55).
4.1. TCD parameters
Introduced in 1982, TCD detects differences in frequency between emitted ultrasonic waves and the reflected echoes which are proportional to the velocity of a moving object (56). Assuming that CBF is laminar and constant, according to the continuity equation, the velocity of blood flow and the area of the blood vessel are in inverse relationship, and thus, the increased speed of blood flow can reflect a narrowed blood vessel diameter such as seen in vasospasm (57,58). Several different parameters of TCD are used for monitoring vasospasm including mean flow velocity (MFV), pulsatility index (PI), and the Lindegaard Ratio (LR). MFV is measured in the bilateral middle (MCA), anterior (ACA), and posterior cerebral arteries (PCA), internal carotid arteries (ICA) as well as the basilar artery (BA); its increased value can be a surrogate marker of vascular narrowing (59). PI is calculated with peak systolic velocity (PSV) and end-diastolic velocity (EDV) to assess distal vascular resistance, and a low value can reflect dilated arteries distal to the narrowed segment of vasospasm (60). The caveat in assessing the diameter of the blood vessel with velocity is the assumption of constant CBF; in case of hyperemia, MFV can be elevated without changes of the diameter. LR, a ratio of MFVs in MCA and ICA, helps to differentiate between hemodynamic changes and narrowed blood vessels in the setting of high velocity; LR < 3 indicates hyperdynamic status while LR > 3 is more likely to represent vasospasm (59,61).
4.2. Limitations of TCD
TCD does not detect vasospasm equally for all blood vessels and is known for low sensitivity in detecting ACA or PCA vasospasm (59). Even for MCA vasospasm, accuracy fluctuates depending on the parameter threshold; only for the range of MFV <120 or >200 cm/s did it show reliable predictive values (62,63). Although LR is used to differentiate hyperemia and vasospasm, it is common that both states coexist and its interpretation can be affected by various clinical factors such as blood pressure (64). Additionally, TCD is highly dependent on the manual skills of the operator to insonate the maximal velocity signals. Anatomical variations of patients’ bony anatomy can limit insonation due to poor acoustic windows (65). Most importantly, even at the most resource-rich hospitals, TCDs are often done only once or twice a day (with guidelines recommending only every other day monitoring), thereby limiting assessment of vasospasm to the time of day performed. With these limitations and the increased recognition that vasospasm is only one of the contributors to DCI, there is broad interest in exploring other tools which circumvent these limitations.
5. EEG and Cerebral ischemia
Since the advent of scalp EEG, multiple studies have tried to identify unique patterns that reflect cerebral ischemia. Normal human CBF is estimated to be >50–54ml per 100g of brain tissue per minute (66). Early research observed the correlation between EEG changes and cerebral ischemia upon clamping the carotid artery during carotid endarterectomy (67,68). These studies noted abnormal EEG activity at CBF <17–18ml/100g/min and isoelectricity with a further decrease of CBF to <10–12ml/100g/min. As CBF progresses from normal to the range of anoxia, EEG showed a spectrum of gradual changes including emergence of delta activity and suppression of fast alpha and beta activities depending on the duration of the insult (68,69) (Figure 2). Subsequent studies using positron emission topography (PET) and MRI revealed that the CBF threshold for permanent infarction was around 8ml/100g/min albeit dependent on the duration of decreased CBF (70,71). Therefore, based on the observed EEG alterations during carotid endarterectomy, gradual predominance of slow activities along with disappearance of fast activities reliably indicates early cerebral ischemia.
Figure 2.
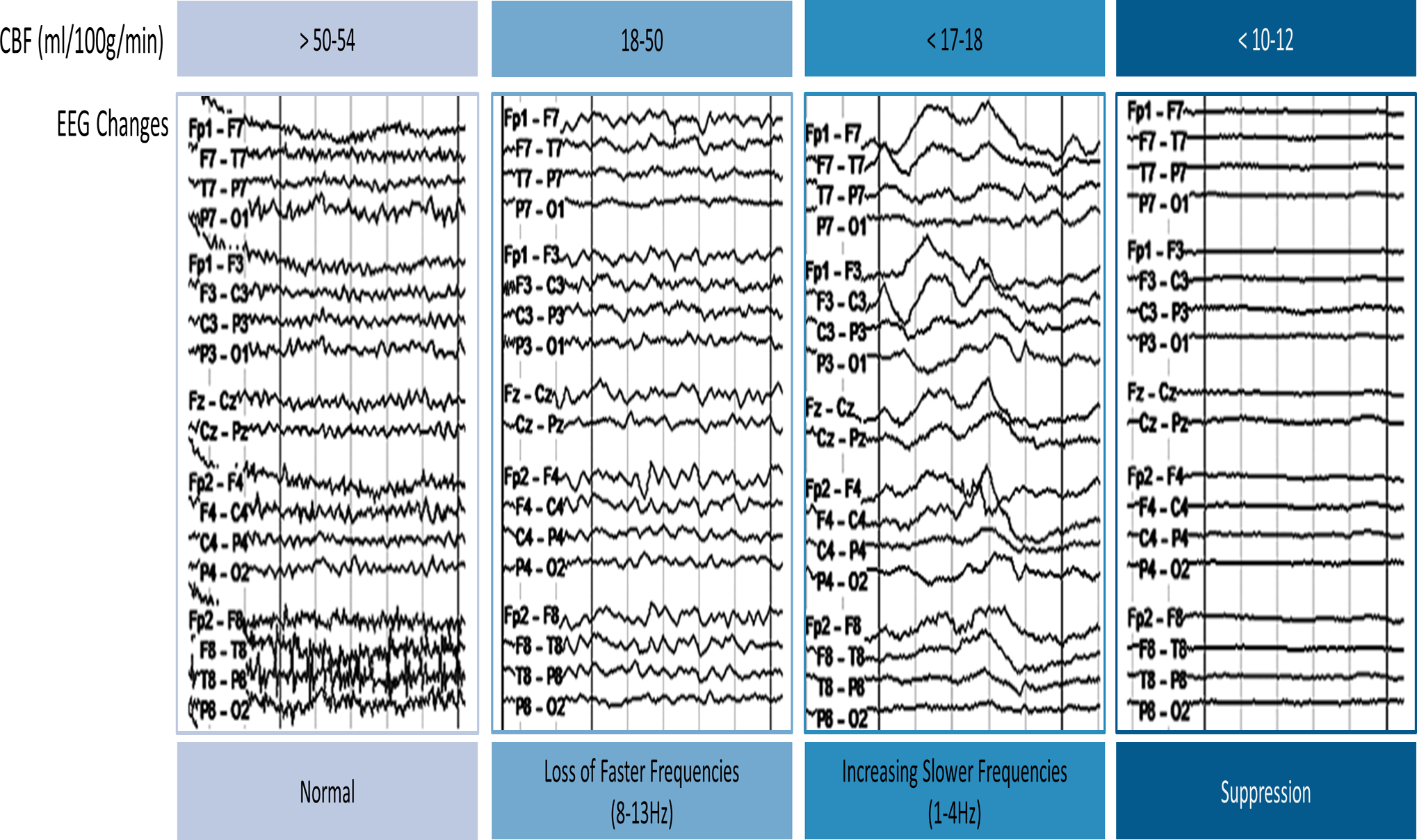
The changes of EEG activities in relation to the cerebral blood flow (CBF, ml/100g/min). As CBF decreases from the normal threshold of 50–54 ml/100g/min, fast activities are lost while slow activities become more prominent. If CBF is reduced below 10–12 ml/100g/min, EEG becomes nearly isoelectric.
5.1. Emergence of quantitative EEG (qEEG)
Although several studies reliably show notable EEG patterns associated with cerebral ischemia, it is challenging to utilize EEG for prolonged periods in real time. Furthermore, interpretation requires specific training and subtle changes can be missed with visual inspection alone (72). Therefore, qEEG has been pursued as a time-saving method by which to process and display various trends calculated from the EEG signal. Dietsch first attempted to perform qEEG with Fourier analysis in 1932, but it was not until the widespread use of digital EEG and the invention of the Fast Fourier Transformation (FFT) in 1965 that raw EEG data could be compressed into a density spectral array (73). Since then, multiple qEEG parameters have been developed such as spectrograms, asymmetry, and power band assessments.
5.2. EEG and DCI
Reports of EEG changes in relation to DCI after SAH were first published in the 1970s (74,75). The emergence of qEEG and the advances of commercial software facilitate our ability to define features of EEG associated with DCI (Table 1).
Table 1.
Summary of original studies on the features of raw EEG or qEEG. RAV, relative alpha variability; ADR, alpha/delta ratio; AT/D, alpha-theta/delta ratio; IIC, ictal-interictal continuum; GRDA, generalized rhythmic delta activity; mFS, modified Fisher scale; WFNS, World Federation of Neurosurgical Societies grading.
| Study type | N. of vasospasm or DCI / total N | SAH grade | EEG methods | EEG/qEEG features | |
|---|---|---|---|---|---|
| Parkes et al., 1971 | Prospective | 6/13 angiographic or clinical | All | 8-channel raw EEG | Increases in the percent time of theta and delta activity |
| Rivierez et al., 1991 | Retrospective | 104/151 angiographic | Fisher 1–3 | 8-channel raw EEG | Prognostic values of localized delta wave, delta diffuse wave, “axial bursts” |
| Labar et al., 1991 | Prospective | 8/11 clinical or radiographic | H-H 1–4 | 2-channel qEEG | Decreases in the total power |
| Vespa et al., 1997 | Prospective | 19/32 angiographic or sonographic | All | 8 electrodes, 10–20 system, qEEG | Decreases in RAV |
| Claassen et al, 2004 | Prospective | 9/34 angiographic, clinical, or radiographic | H-H 4–5 | F3–C3 and F4–C4, or Fp1–F3 and Fp2–F4, or F7–T3 and F8–T4 for the anterior circulation, P3–O1 and P4–O2, or T5–O1 and T6–O2 for the posterior circulation | Decreases in ADR, increases in the delta/total power ratio |
| Rathkrishnan et al., 2011 | Prospective | 8/12 clinical or radiographic | All H-H, mFS 3–4 | 20 electrodes, 10–20 System, qEEG. Analysis restricted to anterior quadrants | Composite alpha index (CAI) |
| Gollwitzer et al., 2015 | Prospective | 6/12 clinical or radiographic | All | 10 electrodes, 10–20 System, qEEG | Decreases in the alpha and theta powers |
| Rots et al., 2016 | Prospective | 11/21 clinical or radiographic | All | 8 electrodes, 10–20 system, qEEG | Decreases in either ADR, alpha power, alpha variability, combination of ADR and alpha variability |
| Kim, et al., 2017 | Retrospective | 53/124 clinical or radiographic | H-H 4–5, Fisher 3 | 10–20 system, qEEG | Late onset of any type of IICAs except GRDA |
| Rosenthal et al., 2018 | Prospective | 52/103 clinical or radiographic | H-H 4–5, mFS 3–4 | 10–20 System, qEEG | Worsening focal slowing, ADR, or RAV |
| Balança et al., 2018 | Retrospective | 9/15 radiographic | H-H or WFNS 4–5 | 10–20 system, qEEG | Decreases in AT/D |
6. The EEG/qEEG parameters related to DCI
6.1. qEEG absolute power parameters
6.1.1. Total Power (TP)
Innovative work in the early 1990s showed that TP, a summation of all frequency band powers (1–30 Hz in this study), was well-correlated (100% sensitivity) with clinical and radiographic changes in all grades of SAH; a decrease in TP confirmed cerebral ischemia (76). Subsequent studies have not replicated these findings, although differing research methodologies including varying number of electrodes and SAH severity may have impacted the results (77,78).
6.1.2. Alpha Power
In addition to TP, power of the alpha frequency band (variably defined as 8–12, 8–12.5, or 8–13 Hz) has been explored in multiple studies (77,79,80). In one study, a decrease in alpha power was the strongest predictor of vasospasm (Optimal cut-off 40%, AUC=0.66, Se=89%, Sp=77%) (80). Similarly, Rots and colleagues showed a decrease in alpha power as a distinct EEG feature in DCI (p=0.008) (78). Others have confirmed these associations and have gone on to show that a decrease in daily alpha power can precede clinical deterioration by more than 24 hours among assessed parameters including focal slowing, decreasing alpha/delta ratio, and epileptiform abnormalities (81).
6.1.3. Delta and Theta Power/Focal slowing
Early observations of increased slowing associated with progressive cerebral ischemia increased attention to these parameters in DCI. Parkes and James evaluated slowing on raw EEG and noted a significant increase in theta power with vasospasm (p<0.02) (82). However, a later study reported a reduction in theta power similar to alpha power decreases observed in DCI patients (80). While the utility of theta power in DCI prediction is unclear, delta power, and especially alpha/delta ratio as discussed below, show a strong correlation with DCI (p=0.003) (77). A recent study combining delta and theta powers in the definition of focal slowing, showed a positive trend of focal slowing on DCI prediction, but this did not reach statistical significance (Rosenthal et al., 2018). Thus, while there is some suggestion that absolute measures of low frequency bands are associated with DCI, their utility is mixed emphasizing the potential importance of assessing relative changes in the power of each frequency band.
6.2. qEEG relative power parameters
6.2.1. Relative Alpha Variability (RAV)
RAV, defined as the percentage of 6–14 Hz over the total 1–20 Hz frequency, is another qEEG feature found to change preceding or concomitant to vasospasm after SAH (Vespa et al., 1997). In this first study, all patients who were found to develop vasospasm on TCD or angiogram (19 out of 32) showed decreased RAV around the time of vasospasm. Moreover, for 10 of 19 patients, it was detected more than two days prior to vasospasm. Similarly, Rots et al. showed that decrease in RAV (8–12.5/1–30 Hz) strongly predicted DCI up to 7 hours before clinical or radiographic changes occurred especially involving all eight studied patients with the cut-off value of 38% (Rots et al., 2016). This close association was upheld in a study performed exclusively for high grade SAH patients (p=0.004) (Claassen et al., 2004). In a recent study assessing the diagnostic accuracy of EEG for detection of DCI, a decrease in RAV was found to have the lowest false positive rate of 2% and highest odds ratio (36.7 [4.70, 286.11], p<0.01) among assessed parameters including focal slowing, decreasing alpha/delta ratio, and epileptiform abnormalities (Rosenthal et al., 2018).
6.2.2. Alpha/delta ratio (ADR)
ADR is one of the most common parameters assessed for early detection of DCI combining the observations of a decrease in alpha and an increase in delta power as a reflection of DCI. A prior study shows the strongest association of ADR both globally and in affected territories with DCI (AUC=0.83, p<0.0001) for multiple EEG measures including those mentioned in the preceding sections (77). Subsequent studies with different methodologies support the correlation between a decrease in ADR and DCI but with varying sensitivity (78,80,81). A recent meta-analysis of five studies shows moderate correlation of ADR with DCI (AUC=0.84, Se=0.83, Sp=0.74) (83). An example of ADR reduction during DCI is illustrated in Figure 3.
Figure 3.
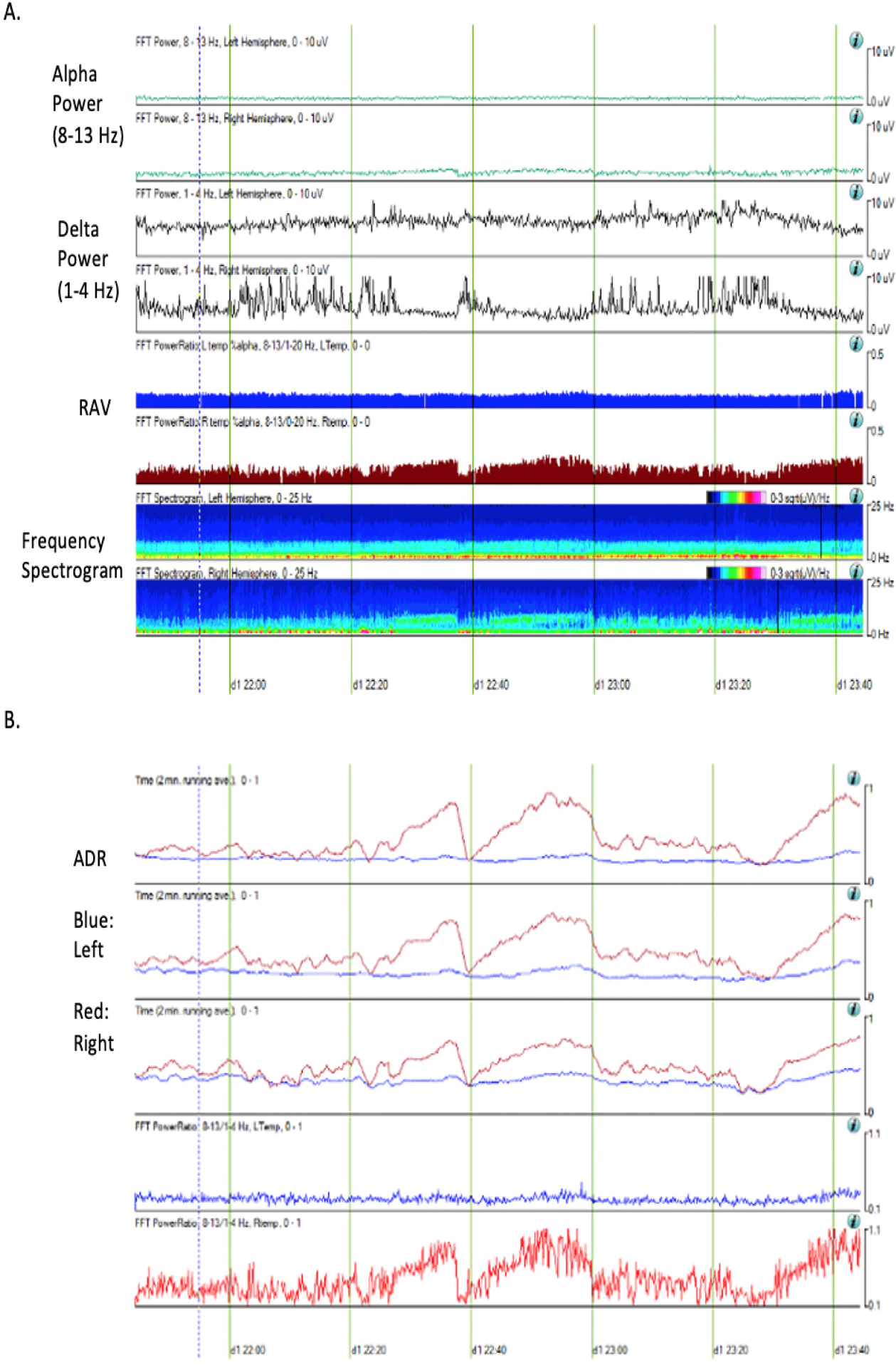
qEEG parameters of a 57-year old woman at the time of DCI who presented with SAH of Hunt Hess grade 3 and modified Fisher scale 3. She developed vasospasm in the left middle meningeal artery on post-bleed day 3. The trends displayed above were generated via Persyst software version 13 (Persyst Development Corporation, Prescott, AZ). Alpha power is decreased in the left hemisphere compared to the right hemisphere while delta power is increased in the left hemisphere. The left temporal lobe shows poor relative alpha variability (RAV) but the right temporal lobe maintains good RAV (A). Alpha/delta ratio (ADR) is decreased in the left temporal lobe compared to the right temporal lobe (B).
6.2.3. Alpha-theta/delta ratio (AT/D)
Most recently, AT/D was evaluated as an indicator of DCI in high-grade SAH (84). The potential relation of AT/D with DCI was postulated from the previous reports on the association of decreases in ADR and theta power with DCI (77,80). In a small study by Balança and colleagues, a transient decrease in AT/D was noted even in patients without DCI. However, prolonged decrease for the minimum 3.7 hours and 1.4 hours with cut-off values of 30% and 50%, respectively, showed the best prediction for DCI (AUC=0.94, Se=1, Sp=0.83). Moreover, in 6 of 9 patients with confirmed cerebral infarction, a decrease in AT/D was noted in the in the same region of infarction at least one day earlier.
6.3. Other
6.3.1. Epileptiform abnormalities (EA)
Most studies to date have focused on changes in background activity, but more recently investigators have begun to explore the relationship between DCI and epileptiform abnormalities including seizure, sporadic epileptiform discharge (ED), and periodic/rhythmic activity (85). All abnormal activities except seizure and generalized rhythmic delta activity (GRDA) showed statistically higher cumulative incidence in the DCI group compared to the non-DCI group for each abnormal activity. Seizure was also found to occur more frequently in patients with DCI, but presumably it did not reach statistical significance due to the small number, 7 out of 124 combining both groups. The study further investigated the temporal association of EAs with DCI. Most abnormalities occurred prior to the development of DCI, thereby suggesting the potential of EAs to predict DCI. In exploring the predictive value of all types of EAs, except seizure, new or increasing EAs were a strong predictor of impending DCI (63.5%) (81). The presence of either EA or abnormal background activity were even more predictive of DCI (96.2% vs 19.6%, DCI vs no DCI, OR = 102.5, 95% CI = 21.3–494, p<0.001) (81).
6.3.2. Combination of multiple parameters
Some groups assessed the composite measures of the suggested parameters as a marker for DCI. Rots and colleagues confirmed the findings of the prior studies using the ADR and RAV to predict DCI (AUC=0.90 and 0.73, respectively) (78). The receiver operating characteristic (ROC) curve of these two parameters combined resulted in better performance (AUC 0.92). Another study assessed the product of mean alpha power and its standard deviation (Composite Alpha Index, CAI) (79). This study showed changes in CAI were responsive to intravenous milrinone, a phosphodiesterase 3 inhibitor. CAI increased with milrinone initiation and maintenance, while a decrease was observed during weaning and discontinuation. Rosenthal and colleagues reported that combining new focal slowing, decreased ADR, and decreased RAV predicted DCI in 63.5% of cases. These studies suggest multiparameter qEEG may improve predictive power of DCI (81).
7. Discussion
qEEG is a valuable modality for monitoring DCI as it addresses the limitations of conventional tools such as TCD or DSA. It provides non-invasive, real-time continuous assessment of brain activity and requires a relatively shortened time for review compared to raw EEG assessment. With increasing availability of software capable of calculating these qEEG features in real time, there is great clinical potential for the timely treatment of DCI using this technology. It may even be used as a screening tool to specifically enroll patients with high-risk qEEG features in clinical trials testing novel therapies to mitigate and prevent DCI (32).
However, there are still gaps limiting its widespread utilization. While the above studies show the value of various qEEG parameters in predicting DCI, many of these studies use a limited number of patients with various definitions of DCI. Thus, these features must be validated as reliable markers on a larger scale under the unified definition of DCI. Moreover, the accuracy of the parameters has yet to be studied based on the location of ischemia or the involved vascular territories.
Constructing a DCI management algorithm with qEEG for daily clinical practice renders significant logistical challenges even if the qEEG parameters indicating DCI are validated. The interpretation of those parameters still necessitates real-time individual inspection in the context of the changes in raw EEG by neurophysiologists specially trained for reviewing qEEG and the recorded parameters. Therefore, an automated detection alarm system using these qEEG parameters should be developed for the widespread use of qEEG, sparing a continuous review of qEEG by an expert but still enabling real-time detection of DCI. However, these measures can be confounded by other concurrent pathologic states such as high intracranial pressure, hydrocephalus, surgery, or medications and thus must be contextualized. Furthermore, due to the prevalence of artifacts in the ICU, automated calculation of these qEEG features without a priori selection of artifact-free segments of data or manual review by trained neurophysiologists, is challenging for fully automated DCI detection algorithms (86). This limitation was demonstrated in a study attempting to automate an algorithm for DCI detection using ADR and RAV qEEG parameters (87). They found suboptimal sensitivity and specificity with significant drop offs in either parameter depending on the chosen threshold of ADR or RAV changes. Thus, further work needs to be done to improve automated calculations in combination with artifact reduction algorithms. Designing a multi-feature algorithm based on all qEEG features mentioned above including the incorporation of epileptiform activity is the next step in optimizing a DCI prediction algorithm that can be clinically implemented.
A multi-modal approach, incorporating TCDs along with qEEG features may improve upon the performance of either diagnostic tool alone. Although TCD was used as a modality to confirm vasospasm in a few previous studies, the predictive value of combining TCD and qEEG for DCI detection has not been studied. Newer technologies such as near-infrared spectroscopy (NIRS) may serve as another modality by which to improve DCI prediction. NIRS is another non-invasive real-time bedside modality for monitoring regional cerebral ischemia. It uses a characteristic of oxyhemoglobin and deoxyhemoglobin absorbing a specific spectrum of light wavelengths for detection of tissue oxygenation and has been used mainly in vascular and cardiac surgeries (88). Its use is technically limited to the frontal areas of the brain and is a technology not yet widely available at many institutions with controversy on its utility and unestablished protocols. However, a few small studies have shown that NIRS may serve a useful tool in DCI detection (89,90). Thus, incorporating TCD and newer monitoring modalities, such as NIRS, with qEEG may improve DCI prediction.
Ultimately, qEEG is a promising modality implementable in daily practice allowing real-time, non-invasive monitoring with global coverage across the brain. It simultaneously has the potential benefit of cost- and time-efficiency, especially with the future development of automated systems, which could facilitate prompt detection and early intervention on DCI. Clinical trials testing novel interventions specifically targeted at those with high-risk EEG signatures, can then be developed to improve patient outcomes in SAH.
Footnotes
The authors have no conflicts of interest or funding sources for this work.
References
- 1.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Vol. 139, Circulation. 2019. 56–66 p. [DOI] [PubMed] [Google Scholar]
- 2.Roos YBWEM, Haan RJ De, Beenen LFM, Groen RJM, Albrecht KW, Vermeulen M. Complications and outcome in patients with aneurysmal subarachnoid haemorrhage : a prospective hospital based cohort study in The Netherlands. 2000;337–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macdonald RL. History and Definition of Delayed Cerebral Ischemia. In: Acta neurochirurgica Supplement. 2013. p. 1–5. [DOI] [PubMed] [Google Scholar]
- 4.Frontera JA, Fernandez A, Schmidt JM, Claassen J, Wartenberg KE, Badjatia N, et al. Defining vasospasm after subarachnoid hemorrhage: What is the most clinically relevant definition? Stroke. 2009;40(6):1963–8. [DOI] [PubMed] [Google Scholar]
- 5.Vergouwen MDI, Vermeulen M, van Gijn J, Rinkel GJE, Wijdicks EF, Muizelaar JP, et al. Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke. 2010;41(10):2391–5. [DOI] [PubMed] [Google Scholar]
- 6.Dreier JP, Major S, Manning A, Woitzik J, Drenckhahn C, Steinbrink J, et al. Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain [Internet]. 2009. Jul [cited 2016 Jan 21];132(Pt 7):1866–81. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=2702835&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Budohoski KP, Czosnyka M, Smielewski P, Kasprowicz M, Helmy A, Bulters D, et al. Impairment of cerebral autoregulation predicts delayed cerebral ischemia after subarachnoid hemorrhage: A prospective observational study. Stroke. 2012;43(12):3230–7. [DOI] [PubMed] [Google Scholar]
- 8.Vergouwen MDI, Vermeulen M, Coert BA, Stroes ESG, Roos YBWEM. Microthrombosis after aneurysmal subarachnoid hemorrhage: An additional explanation for delayed cerebral ischemia. Vol. 28, Journal of Cerebral Blood Flow and Metabolism. 2008. p. 1761–70. [DOI] [PubMed] [Google Scholar]
- 9.Carr KR, Zuckerman SL, Mocco J. Inflammation, cerebral vasospasm, and evolving theories of delayed cerebral ischemia. Vol. 2013, Neurology Research International. 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Robertson EG. Cerebral lesions due to intracranial aneurysms. Brain. 1949. Jun;72(Pt. 2):150–85. [DOI] [PubMed] [Google Scholar]
- 11.Ecker A, Riemenschneider PA. Arteriographic demonstration of spasm of the intracranial arteries, with special reference to saccular arterial aneurysms. J Neurosurg. 1951;8(6):660–7. [DOI] [PubMed] [Google Scholar]
- 12.Fisher CM, Roberson GH, Ojemann RG. Cerebral vasospasm with ruptured saccular aneurysm--the clinical manifestations. Neurosurgery. 1977;1(3):245–8. [DOI] [PubMed] [Google Scholar]
- 13.Fisher CM, Kistler JP, Davis JM. Relation of cerebral vasospasm to subarachnoid hemorrhage visualized by computerized tomographic scanning. Neurosurgery. 1980;6(1):1–9. [DOI] [PubMed] [Google Scholar]
- 14.Crowley RW, Medel R, Dumont AS, Ilodigwe D, Kassell NF, Mayer SA, et al. Angiographic vasospasm is strongly correlated with cerebral infarction after subarachnoid hemorrhage. Stroke. 2011. Apr;42(4):919–23. [DOI] [PubMed] [Google Scholar]
- 15.Kumar G, Shahripour RB, Harrigan MR. Vasospasm on transcranial Doppler is predictive of delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage: a systematic review and meta-analysis. 2016;124(May):1257–64. [DOI] [PubMed] [Google Scholar]
- 16.Jabbarli R, Pierscianek D, Rölz R, Darkwah Oppong M, Kaier K, Shah M, et al. Endovascular treatment of cerebral vasospasm after subarachnoid hemorrhage: More is more. Neurology. 2019. Jul;93(5):e458–66. [DOI] [PubMed] [Google Scholar]
- 17.Dankbaar JW, Rijsdijk M, Van Der Schaaf IC, Velthuis BK, Wermer MJH, Rinkel GJE. Relationship between vasospasm, cerebral perfusion, and delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage. Neuroradiology. 2009. Dec;51(12):813–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lefournier V, Krainik A, Gory B, Derderian F, Bessou P, Fauvage B, et al. Quantification du vasospasme cérébral après hémorragie sous-arachnoïdienne par scanner de perfusion. J Neuroradiol. 2010. Dec;37(5):284–91. [DOI] [PubMed] [Google Scholar]
- 19.Brown RJ, Kumar A, Dhar R, Sampson TR, Diringer MN. The relationship between delayed infarcts and angiographic vasospasm after aneurysmal subarachnoid hemorrhage. Neurosurgery. 2013. May;72(5):702–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacDonald RL, Kassell NF, Mayer S, Ruefenacht D, Schmiedek P, Weidauer S, et al. Clazosentan to overcome neurological ischemia and infarction occurring after subarachnoid hemorrhage (CONSCIOUS-1): Randomized, double-blind, placebo-controlled phase 2 dose-finding trial. Stroke. 2008. Nov;39(11):3015–21. [DOI] [PubMed] [Google Scholar]
- 21.Macdonald RL, Higashida RT, Keller E, Mayer SA, Molyneux A, Raabe A, et al. Clazosentan, an endothelin receptor antagonist, in patients with aneurysmal subarachnoid haemorrhage undergoing surgical clipping: A randomised, double-blind, placebo-controlled phase 3 trial (CONSCIOUS-2). Lancet Neurol. 2011. Jul;10(7):618–25. [DOI] [PubMed] [Google Scholar]
- 22.Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, Strong AJ. Clinical relevance of cortical spreading depression in neurological disorders: Migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. Vol. 31, Journal of Cerebral Blood Flow and Metabolism. 2011. p. 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woitzik J, Dreier JP, Hecht N, Fiss I, Sandow N, Major S, et al. Delayed cerebral ischemia and spreading depolarization in absence of angiographic vasospasm after subarachnoid hemorrhage. J Cereb Blood Flow Metab [Internet]. 2012. Feb [cited 2016 Jan 27];32(2):203–12. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3272613&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dreier JP, Woitzik J, Fabricius M, Bhatia R, Major S, Drenckhahn C, et al. Delayed ischaemic neurological deficits after subarachnoid haemorrhage are associated with clusters of spreading depolarizations. Brain. 2006;129:3224–37. [DOI] [PubMed] [Google Scholar]
- 25.von Bornstädt D, Houben T, Seidel JL, Zheng Y, Dilekoz E, Qin T, et al. Supply-demand mismatch transients in susceptible peri-infarct hot zones explain the origins of spreading injury depolarizations. Neuron [Internet]. 2015. Mar 4 [cited 2016 Jan 27];85(5):1117–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25741731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung DY, Oka F, Ayata C. Spreading Depolarizations: A Therapeutic Target Against Delayed Cerebral Ischemia After Subarachnoid Hemorrhage. J Clin Neurophysiol [Internet]. 2016. Jun [cited 2017 Jan 6];33(3):196–202. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00004691-201606000-00004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bosche B, Graf R, Ernestus RI, Dohmen C, Reithmeier T, Brinker G, et al. Recurrent spreading depolarizations after subarachnoid hemorrhage decreases oxygen availability in human cerebral cortex. Ann Neurol. 2010. May;67(5):607–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dreier JP. The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease. Vol. 17, Nature Medicine. Nat Med; 2011. p. 439–47. [DOI] [PubMed] [Google Scholar]
- 29.Oka F, Hoffmann U, Lee JH, Shin HK, Chung DY, Yuzawa I, et al. Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents. J Cereb Blood Flow Metab. 2017. May;37(5):1829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menyhárt Á, Zölei-Szénási D, Puskás T, Makra P, Orsolya MT, Szepes BE, et al. Spreading depolarization remarkably exacerbates ischemia-induced tissue acidosis in the young and aged rat brain. Sci Rep. 2017. Dec;7(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartings JA, Wilson JA, Hinzman JM, Pollandt S, Dreier JP, DiNapoli V, et al. Spreading depression in continuous electroencephalography of brain trauma. Ann Neurol [Internet]. 2014. Nov [cited 2016 Jan 28];76(5):681–94. Available from: http://www.ncbi.nlm.nih.gov/pubmed/25154587 [DOI] [PubMed] [Google Scholar]
- 32.Drenckhahn C, Winkler MKL, Major S, Scheel M, Kang E-J, Pinczolits A, et al. Correlates of spreading depolarization in human scalp electroencephalography. A J Neurol. 2012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishii R Regional cerebral blood flow in patients with ruptured intracranial aneurysms. J Neurosurg. 1979;50(5):587–94. [DOI] [PubMed] [Google Scholar]
- 34.Jaeger M, Soehle M, Schuhmann MU, Meixensberger J. Clinical significance of impaired cerebrovascular autoregulation after severe aneurysmal subarachnoid hemorrhage. Stroke. 2012. Aug;43(8):2097–101. [DOI] [PubMed] [Google Scholar]
- 35.Silverman A, Kodali S, Strander S, Gilmore EJ, Kimmel A, Wang A, et al. Deviation From Personalized Blood Pressure Targets Is Associated With Worse Outcome After Subarachnoid Hemorrhage. Stroke. 2019. Oct;50(10):2729–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Geraud G, Tremoulet M, Guell A, Bes A. The prognostic value of noninvasive CBF measurement in subarachnoid hemorrhage. Stroke. 1984;15(2):301–5. [DOI] [PubMed] [Google Scholar]
- 37.Hattingen E, Blasel S, Dettmann E, Vatter H, Pilatus U, Seifert V, et al. Perfusion-weighted MRI to evaluate cerebral autoregulation in aneurysmal subarachnoid haemorrhage. Neuroradiology. 2008. Nov;50(11):929–38. [DOI] [PubMed] [Google Scholar]
- 38.Shin HK, Dunn AK, Jones PB, Boas DA, Moskowitz MA, Ayata C. Vasoconstrictive neurovascular coupling during focal ischemic depolarizations. J Cereb Blood Flow Metab [Internet]. 2006. Aug [cited 2016 Feb 8];26(8):1018–30. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16340958 [DOI] [PubMed] [Google Scholar]
- 39.Tso MK, Loch Macdonald R. Acute Microvascular Changes after Subarachnoid Hemorrhage and Transient Global Cerebral Ischemia. Stroke Res Treat. 2013;2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santos GA, Petersen N, Zamani AA, Larose S, Monk A, Sorond FA. Pathophysiologic differences in cerebral autoregulation after subarachnoid hemorrhage. 2016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rowland MJ, Hadjipavlou G, Kelly M, Westbrook J, Pattinson KTS. Delayed cerebral ischaemia after subarachnoid haemorrhage: looking beyond vasospasm. Br J Anaesth [Internet]. 2012;109(3):315–29. Available from: http://bja.oxfordjournals.org/lookup/doi/10.1093/bja/aes264 [DOI] [PubMed] [Google Scholar]
- 42.Hirashima Y, Nakamura S, Suzuki M, Kurimoto M, Endo S, Ogawa A, et al. Cerebrospinal fluid tissue factor and thrombin-antithrombin III complex as indicators of tissue injury after subarachnoid hemorrhage. Stroke. 1997;28(9):1666–70. [DOI] [PubMed] [Google Scholar]
- 43.Frijns CJM, Fijnheer R, Algra A, Van Mourik JA, Van Gijn J, Rinkel GJE. Early circulating levels of endothelial cell activation markers in aneurysmal subarachnoid haemorrhage: Associations with cerebral ischaemic events and outcome. J Neurol Neurosurg Psychiatry. 2006. Jan;77(1):77–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lad SP, Hegen H, Gupta G, Deisenhammer F, Steinberg GK. Proteomic biomarker discovery in cerebrospinal fluid for cerebral vasospasm following subarachnoid hemorrhage. J Stroke Cerebrovasc Dis. 2012. Jan;21(1):30–41. [DOI] [PubMed] [Google Scholar]
- 45.Tanioka S, Ishida F, Nakano F, Kawakita F, Kanamaru H, Nakatsuka Y, et al. Machine Learning Analysis of Matricellular Proteins and Clinical Variables for Early Prediction of Delayed Cerebral Ischemia After Aneurysmal Subarachnoid Hemorrhage. Mol Neurobiol. 2019. Oct;56(10):7128–35. [DOI] [PubMed] [Google Scholar]
- 46.Hendryk S, Jarzab B, Josko J. Increase of the IL-1β and IL-6 levels in CSF in patients with vasospasm following aneurysmal SAH. Vol. 2, Neuroendocrinology Letters Nos.1. 2004. [PubMed] [Google Scholar]
- 47.Al-Mufti F, Amuluru K, Damodara N, Dodson V, Roh D, Agarwal S, et al. Admission neutrophil-lymphocyte ratio predicts delayed cerebral ischemia following aneurysmal subarachnoid hemorrhage. J Neurointerv Surg. 2019. Nov;11(11):1135–40. [DOI] [PubMed] [Google Scholar]
- 48.Marc Simard J, Geng Z, Kyoon Woo S, Ivanova S, Tosun C, Melnichenko L, et al. Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J Cereb Blood Flow Metab. 2009;29(2):317–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tosun C, Kurland DB, Mehta R, Castellani RJ, Dejong JL, Kwon MS, et al. Inhibition of the sur1-trpm4 channel reduces neuroinflammation and cognitive impairment in subarachnoid hemorrhage. Stroke. 2013. Dec;44(12):3522–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ma C, Zhou W, Yan Z, Qu M, Bu X. Toll-like receptor 4 (TLR4) is correlated with delayed cerebral ischemia (DCI) and poor prognosis in aneurysmal subarachnoid hemorrhage. J Neurol Sci. 2015. Dec;359(1–2):67–71. [DOI] [PubMed] [Google Scholar]
- 51.Bevers MB, Wolcott Z, Bache S, Hansen C, Sastre C, Mylvaganam R, et al. Soluble ST2 links inflammation to outcome after subarachnoid hemorrhage. Ann Neurol. 2019;86(3):384–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lissak IA, Zafar SF, Westover MB, Schleicher RL, Kim JA, Leslie-Mazwi T, et al. Soluble ST2 Is Associated With New Epileptiform Abnormalities Following Nontraumatic Subarachnoid Hemorrhage. Stroke. 2020. Apr;51(4):1128–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.McBride DW, Blackburn SL, Peeyush Kumar T, Matsumura K, Zhang JH. The role of thromboinflammation in delayed cerebral ischemia after subarachnoid hemorrhage. Vol. 8, Frontiers in Neurology. Frontiers Media S.A.; 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shankar JJS, Tan IYL, Krings T, Terbrugge K, Agid R. CT angiography for evaluation of cerebral vasospasm following acute subarachnoid haemorrhage. Neuroradiology. 2012. Mar;54(3):197–203. [DOI] [PubMed] [Google Scholar]
- 55.Connolly ES, Rabinstein AA, Carhuapoma JR, Derdeyn CP, Dion J, Higashida RT, et al. Guidelines for the Management of Aneurysmal Subarachnoid Hemorrhage. Stroke. 2012. Jun;43(6):1711–37. [DOI] [PubMed] [Google Scholar]
- 56.Sarkar S, Ghosh S, Ghosh SK, Collier A. Role of transcranial Doppler ultrasonography in stroke. Vol. 83, Postgraduate Medical Journal. BMJ Publishing Group; 2007. p. 683–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cipolla MJ. Control of Cerebral Blood Flow. 2009; [Google Scholar]
- 58.Purkayastha S, Sorond F. Transcranial doppler ultrasound: Technique and application. Semin Neurol. 2012;32(4):411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Naqvi J, Yap KH, Ahmad G, Ghosh J. Transcranial Doppler ultrasound: a review of the physical principles and major applications in critical care. Int J Vasc Med [Internet]. 2013. Jan [cited 2016 Jan 19];2013:629378. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3876587&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rajajee V, Fletcher JJ, Pandey AS, Gemmete JJ, Chaudhary N, Jacobs TL, et al. Low pulsatility index on transcranial doppler predicts symptomatic large-vessel vasospasm after aneurysmal subarachnoid hemorrhage. Vol. 70, Neurosurgery. Neurosurgery; 2012. p. 1195–206. [DOI] [PubMed] [Google Scholar]
- 61.Lindegaard KF, Nornes H, Bakke SJ, Sorteberg W, Nakstad P. Cerebral vasospasm diagnosis by means of angiography and blood velocity measurements. Acta Neurochir (Wien). 1989. Mar;100(1–2):12–24. [DOI] [PubMed] [Google Scholar]
- 62.Vora YY, Suarez-Almazor M, Steinke DE, Martin ML, Findlay JM. Role of transcranial Doppler monitoring in the diagnosis of cerebral vasospasm after subarachnoid hemorrhage. Neurosurgery. 1999. Jun;44(6):1237–48. [PubMed] [Google Scholar]
- 63.Carrera E, Schmidt JM, Oddo M, Fernandez L, Claassen J, Seder D, et al. Transcranial doppler for predicting delayed cerebral ischemia after Subarachnoid hemorrhage. Neurosurgery. 2009;65(2):316–23. [DOI] [PubMed] [Google Scholar]
- 64.Tsivgoulis G, Alexandrov AV, Sloan MA, Usf T/, Program S. Advances in Transcranial Doppler Ultrasonography 2009; [DOI] [PubMed] [Google Scholar]
- 65.Bonow RH, Young CC, Bass DI, Moore A, Levitt MR. Transcranial Doppler ultrasonography in neurological surgery and neurocritical care. Neurosurg Focus. 2019;47(6):E2. [DOI] [PubMed] [Google Scholar]
- 66.Van Putten MJAM, Hofmeijer J. EEG monitoring in cerebral ischemia: Basic concepts and clinical applications. Vol. 33, Journal of Clinical Neurophysiology. Lippincott Williams and Wilkins; 2016. p. 203–10. [DOI] [PubMed] [Google Scholar]
- 67.Sharbrough FW, Messick JM, Sundt TM. Correlation of continuous electroencephalograms with cerebral blood flow measurements during carotid endarterectomy. Stroke. 1973;4(4):674–83. [DOI] [PubMed] [Google Scholar]
- 68.Jordan KG. Emergency EEG and continuous EEG monitoring in acute ischemic stroke. Vol. 21, Journal of Clinical Neurophysiology. 2004. p. 341–52. [PubMed] [Google Scholar]
- 69.Hossmann K‐A. Viability thresholds and the penumbra of focal ischemia. Ann Neurol. 1994;36(4):557–65. [DOI] [PubMed] [Google Scholar]
- 70.Baron JC. Perfusion thresholds in human cerebral ischemia: Historical perspective and therapeutic implications. In: Cerebrovascular Diseases. S. Karger AG; 2001. p. 2–8. [DOI] [PubMed] [Google Scholar]
- 71.Bandera E, Botteri M, Minelli C, Sutton A, Abrams KR, Latronico N. Cerebral blood flow threshold of ischemic penumbra and infarct core in acute ischemic stroke: a systematic review. Stroke. 2006. May;37(5):1334–9. [DOI] [PubMed] [Google Scholar]
- 72.Foreman B, Claassen J. Quantitative EEG for the detection of brain ischemia. Crit Care [Internet]. 2012. Jan [cited 2015 Aug 21];16(2):216. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3681361&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kaiser DA. Basic principles of quantitative EEG. Vol. 12, Journal of Adult Development. Springer; 2005. p. 99–104. [Google Scholar]
- 74.Kiloh L Clinical electroencephalography. Butterworths; 1972. [Google Scholar]
- 75.Van der Drift J, Kok N. The EEG in cerebrovascular disorders in relation to pathology. Handbook of Electroencephalography and Clinical Neurophysiology. 1972. [Google Scholar]
- 76.Labar DR, Fisch BJ, Pedley T a., Fink ME, Solomon R a. Quantitative EEG monitoring for patients with subarachnoid hemorrhage. Electroencephalogr Clin Neurophysiol [Internet]. 1991. May;78(5):325–32. Available from: http://linkinghub.elsevier.com/retrieve/pii/001346949190094K [DOI] [PubMed] [Google Scholar]
- 77.Claassen J, Hirsch LJ, Kreiter KT, Du EY, Sander Connolly E, Emerson RG, et al. Quantitative continuous EEG for detecting delayed cerebral ischemia in patients with poor-grade subarachnoid hemorrhage. Clin Neurophysiol [Internet]. 2004;115(12):2699–710. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1388245704002585 [DOI] [PubMed] [Google Scholar]
- 78.Rots ML, van Putten MJAM, Hoedemaekers CWE, Horn J. Continuous EEG Monitoring for Early Detection of Delayed Cerebral Ischemia in Subarachnoid Hemorrhage: A Pilot Study. Neurocrit Care. 2016. Apr;24(2):207–16. [DOI] [PubMed] [Google Scholar]
- 79.Rathakrishnan R, Gotman J, Dubeau F, Angle M. Using continuous electroencephalography in the management of delayed cerebral ischemia following subarachnoid hemorrhage. Neurocrit Care. 2011;14(2):152–61. [DOI] [PubMed] [Google Scholar]
- 80.Gollwitzer S, Groemer T, Rampp S, Hagge M, Olmes D, Huttner HB, et al. Early prediction of delayed cerebral ischemia in subarachnoid hemorrhage based on quantitative EEG: A prospective study in adults. Clin Neurophysiol [Internet]. 2015;126(8):1514–23. Available from: http://linkinghub.elsevier.com/retrieve/pii/S1388245714007895 [DOI] [PubMed] [Google Scholar]
- 81.Rosenthal ES, Biswal S, Zafar SF, O’Connor KL, Bechek S, Shenoy AV., et al. Continuous electroencephalography predicts delayed cerebral ischemia after subarachnoid hemorrhage: A prospective study of diagnostic accuracy. Ann Neurol. 2018. May;83(5):958–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Parkes JD, James IM. Electroencephalographic and cerebral blood flow changes following spontaneous subarachnoid hæmorrhage. Brain. 1971;94(1):69–76. [DOI] [PubMed] [Google Scholar]
- 83.Yu Z, Wen D, Zheng J, Guo R, Li H, You C, et al. Predictive Accuracy of Alpha-Delta Ratio on Quantitative Electroencephalography for Delayed Cerebral Ischemia in Patients with Aneurysmal Subarachnoid Hemorrhage: Meta-Analysis. World Neurosurg. 2019. Jun;126:e510–6. [DOI] [PubMed] [Google Scholar]
- 84.Balança B, Dailler F, Boulogne S, Ritzenthaler T, Gobert F, Rheims S, et al. Diagnostic accuracy of quantitative EEG to detect delayed cerebral ischemia after subarachnoid hemorrhage: A preliminary study. Clin Neurophysiol. 2018;129(9):1926–36. [DOI] [PubMed] [Google Scholar]
- 85.Kim JA, Rosenthal ES, Biswal S, Zafar S, Shenoy AV., O’Connor KL, et al. Epileptiform abnormalities predict delayed cerebral ischemia in subarachnoid hemorrhage. Clin Neurophysiol. 2017;128(6):1091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Muniz CF, Shenoy AV., O’Connor KL, Bechek SC, Boyle EJ, Guanci MM, et al. Clinical development and implementation of an institutional guideline for prospective EEG monitoring and reporting of delayed cerebral ischemia. J Clin Neurophysiol. 2016;33(3):217–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Wickering E, Gaspard N, Zafar S, Moura VJ, Biswal S, Bechek S, et al. Automation of Classical QEEG Trending Methods for Early Detection of Delayed Cerebral Ischemia: More Work to Do. J Clin Neurophysiol [Internet]. 2016. Jun [cited 2018 May 15];33(3):227–34. Available from: http://content.wkhealth.com/linkback/openurl?sid=WKPTLP:landingpage&an=00004691-201606000-00008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Scheeren TWL, Schober P, Schwarte LA. Monitoring tissue oxygenation by near infrared spectroscopy (NIRS): Background and current applications. In: Journal of Clinical Monitoring and Computing. J Clin Monit Comput; 2012. p. 279–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yokose N, Sakatani K, Murata Y, Awano T, Igarashi T, Nakamura S, et al. Bedside assessment of cerebral vasospasms after subarachnoid hemorrhage by near infrared time-resolved spectroscopy. In: Advances in Experimental Medicine and Biology. Springer, Boston, MA; 2010. p. 505–11. [DOI] [PubMed] [Google Scholar]
- 90.Maslehaty H, U K-T, AK P, H B, HM M. Continuous Measurement of Cerebral Oxygenation With Near-Infrared Spectroscopy After Spontaneous Subarachnoid Hemorrhage. ISRN Neurol. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]