

Abstract
A number of systemic autoinflammatory diseases (SAIDs) arise from gain-of-function mutations in genes encoding IL-1 activating inflammasomes and cytoplasmic nucleic acid sensors including the receptor and sensor STING and result in increased IL-1 and Type-I interferon (IFN) production respectively. Blocking these pathways in human diseases has provided proof-of-concept confirming the prominent roles of these cytokines in disease pathogenesis. Recent insights into the multilayered regulation of these sensor pathways and insights into their role in amplifying the disease pathogenesis of monogenic and complex genetic diseases spurred new drug development. This review provides insights into the pathogenesis and genetic causes of these “prototypic” diseases caused by gain-of function mutations in IL-1 activating inflammasomes (inflammasomopathies) and in interferon-activating pathways (interferonopathies) including SAVI (STING-associated vasculopathy with onset in infancy), AGS (Aicardi-Goutières Syndrome) and proteasome associated autoinflammatory syndromes (PRAAS) that link activation of the viral sensors STING, “self” nucleic acid metabolism and the ubiquitin-proteasome-system to “Type-I IFN production” and human diseases. Clinical responses and biomarker changes to JAK inhibitors confirm a role of IFNs and a growing number of diseases with “interferon signatures” unveil extensive crosstalk between major inflammatory pathways. Understanding these interactions promises new tools in tackling the significant clinical challenges in treating patients with these conditions.
Keywords: interferonopathies, inflammasomopathies, Aicardi-Goutières Syndrome, STING, JAK inhibitor, interferon, inflammasome, proteasome-associated autoinflammatory syndrome (PRAAS)
This review summarizes the genetic causes, pathomechanisms and clinical disease manifestations of systemic autoinflammatory diseases (SAIDs) caused by inflammasome and viral sensing pathway dysregulation. The discovery of the genetic causes provided key insights into disease pathogenesis and treatment and linked intracellular germline-encoded pattern recognition receptor pathways with the activation of major proinflammatory cytokines that lead to the identification of novel treatment targets.
Inflammasomes are cytosolic multiprotein oligomers that sense microbial or danger signals and induce IL-1 and IL-18 activation and/or cell death. Gain-of-function (GOF) mutations in four inflammasome-encoding genes cause inflammasomopathies that all enhance the assembly of IL-1 activating inflammasomes1, 2. Most of those conditions respond to treatment with IL-1 inhibitors including specific IL-1β blocking antibodies, thus confirming their predominant pathogenic link to inflammasome mediated IL-1β activation3, 4. However, clinical heterogeneity points to disease or inflammasome-specific differences in organ susceptibility to inflammation and organ damage that have not been fully explored or resolved.
On the other hand, autoinflammatory Type I interferonopathies comprise an expanding group of autoinflammatory diseases caused by mutations in the Type I interferon (IFN-I) axis5–8, that present with an enhanced interferon response gene signature (IRGS) in blood cells and a characteristic set of clinical and laboratory features9. The response to treatment with JAK inhibitors that can reduce interferon (IFN) signaling10–12 has been used to “validate” a role for IFN-I in these diseases. However, the inflammatory targets of JAK inhibitors are diverse and clinical validation exclusively targeting IFN-I signaling have not been conducted. As the downstream inflammatory pathways in the diseases termed interferonopathies can be complex, a systematic evaluation of the contribution of IFN to disease pathogenesis is critical.
Although the genetic causes and targeted treatment for the two disease groups differ, all patients present with systemic inflammation (elevation of acute phase reactants and in many instances rashes) and making a clinical diagnosis particularly early in the course of the disease challenging. However, hematologic changes during disease flares differ in IL-1 mediated compared to IFN mediated diseases, with increase in leukocyte and thrombocyte counts in IL-1 mediated disease flares and with decreases in absolute lymphocyte count and a rise in the IRGS in the interferonopathies (Table 1). In fact, the overlapping and differing hematologic and clinical features can aid in making a clinical diagnosis before genetic work up is completed and may allow for swift initiation of treatment.
Table 1.
Differences in flare characteristics in patients with IL-1 mediated vs. IFN-I mediated inflammatory flares
| Inflammatory markers | IL-1 mediated disease flares* | IFN mediated disease flares |
|---|---|---|
| Clinical parameters: | ||
| CRP | Increases with disease flares | Increases with disease flares |
| ESR | Increases with disease flares | Increases with disease flares |
| Hgb | Decreases with disease flares | Decreases with disease flares |
| WBC | Increases with disease flares due to granulocytosis | Remains the same or decreases with disease flares |
| ALC | Remains the same or increases with disease flares | Decreases with disease flares |
| Plt count | Increases with disease flares | Decreases with disease flares |
| Research parameter/biomarker | ||
| IFN-score | Remains within normal range with disease flares | Increases significantly with disease flares |
CRP=C-reactive protein, ESR=erythrocyte sedimentation rate, Hgb=hemoglobin, WBC=white blood cell, ALC=absolute lymphocyte count, Plt=platelet, N=No, Y=Yes
For clinical decisions age related normal values apply, however the table indicates that the direction of change for WBC, ALC and PLT count and IFN-score differ in IL-1 and IFN mediated changes
The values below are derived from patients with the IL-1 mediated diseases CAPS and DIRA Disease related flares seen in patients with CANDLE/PRAAS and other interferonopathies differ from the disease flares seen in patients with IL-1 mediated diseases. Although patients in both disease groups develop fever and rashes and elevated acute phase reactants, the laboratory parameters drawn at the time of a disease flare illustrate characteristic laboratory features of the disease flares that distinguish IL-1 and IFN mediated diseases.
However, differences in clinical disease characteristics, complex dysregulation of multiple inflammatory pathways recent data on upregulation of IL-1 mediated pathways by IFN-I, point to extensive crosstalk in responding to intracellular stress and in coordinating inflammatory signaling pathways.
INFLAMMASOMOPATHIES AND INFLAMMASOME MEDIATED AUTOINFLAMMATORY DISEASES
Familial Mediterranean fever (FMF) was the first autoinflammatory disease genetically described. Its discovery led to the concept of innate immune dysregulation as pathogenic mechanism for “periodic fever syndromes” and resulted in the separation of autoinflammatory diseases caused by predominantly innate immune dysregulation and autoimmune diseases with predominantly adaptive immune dysregulation. In the pre-WES (whole exome sequencing) era, disease-causing mutations in the gene MEFV, which encodes the protein pyrin, were first identified from a 115-kb familial Mediterranean fever candidate interval on chromosome 16p by positional cloning, in 199713, 14. Similarly, autosomal dominant missense mutations in NLRP3 were identified in families with familial cold autoinflammatory syndrome (FCAS) in 200115. These discoveries preceded the description of the paradigm shifting concept of the IL-1 activating NLRP1 inflammasome by Jürg Tschopp and colleagues in 200216. Within 20 years using data mining of homologous proteins, a total of 6 inflammasomes were molecularly characterized. Autoactivating mutations in 4 of these inflammasomes, NLRP1, NLRP3, NLRC4 and MEFV/Pyrin cause monogenic human autoinflammatory diseases. A list of different pathogen-associated molecular patterns (PAMPs) and/or danger associated molecular patterns (DAMPs) that are known to activate these various inflammasomes are provided in (Fig 1). Molecular mechanisms leading to AIM2 and to CASP4/5 as well as to the non-canonical inflammasome activation have been characterized, but yet monogenic diseases caused by mutations in these genes have not been identified. 1, 2, 4
FIG 1. Characteristics of various inflammasomes.
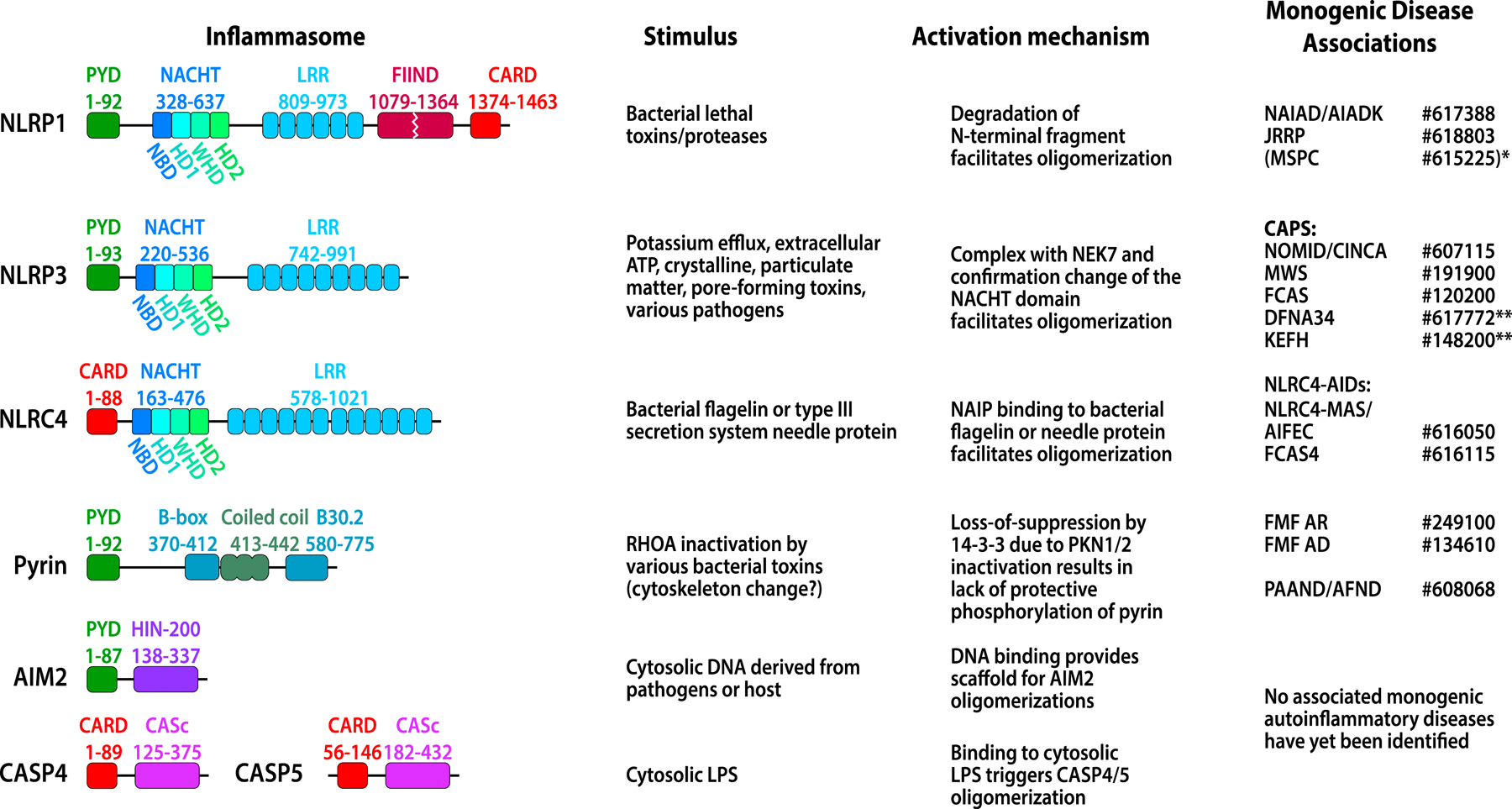
CARD domain or PYD domain is responsible for recruiting ASC and for forming ASC specks. Other domains function as regulatory domains to prevent inflammasome autoactivation (detailed in Fig 2). Diseases related to the inflammasomes are shown in the last column with the MIM numbers (www.oimm.org). Diseases in bracket typically do not present with systemic inflammation. See text for full name of the diseases. NLRP1-associated: NAIAD or AIADK; JRRP. *MSPC is caused by mutations in the pyrin domain of NLRP1 and leads to a precancerous skin condition without systemic inflammation. NLRP3-associated: The disease spectrum of CAPS includes the 3 severity phenotypes: NOMID, MWS, and FCAS. ** DFNA34, (Deafness, autosomal dominant 34) and **KEFH, (Keratoendothelitis fugax hereditaria) present with inflammation restricted to the inner ear and the cornea respectively with absent or minimal systemic inflammation. NLRC4-associated AIDs include NLRC4-MAS/AIFEC presenting with recurrent MAS and FCAS4 presenting with more systemic inflammation and rash. MEFV/pyrin-associated AIDs include additive gain-of function, AR (autosomal recessive) and AD (autosomal dominant) FMF, and PAAND/AFND caused by mutations in two 14–3-3-binding serine residues. Mutations in AIM2 and CASP4 have so far not been associated with human monogenic diseases.
The mechanisms of inflammasome activation and regulation are subject to ongoing research but share common principles and were refined by recent cryo-EM studies of the NLRP3 and NLRC4 inflammasomes, which have provided novel insights into inflammasome activation17–19. Recognition of an inflammasome activating PAMP/DAMP triggers a conformational change and oligomerization of an inflammasome receptor1, 2, 17–19 (Fig 2). The oligomerized pyrin domains (PYDs) or caspase activation and recruitment domains (CARDs) then recruit apoptosis-associated speck-like protein containing a card (ASC), which polymerizes to form large protein aggregates termed ASC “specks”20. Pro-caspase-1 is recruited to the ASC specks via CARD-CARD interaction and is activated21. Active caspase-1 cleaves pro-IL1, pro-IL18 and gasdermin D into active moieties22. These studies revealed a sensitive system where small amounts of PAMP/DAMP propagate a rapidly escalating response via the formation of filaments and specks. Most inflammasomes with the exception of AIM2 (Fig 1), have regulatory autoinhibitory domains, and ligand binding relieves an autoinhibitory conformation to allow activation (for example NLRP3, NLRC4 activation shown in Fig 2A&B)17–19. In the case of NLRP1, the autoinhibitory domain needs to be degraded to trigger inflammasome activation, and most of the disease-causing mutations are located in the autoinhibitory domains23, 24 (Fig 2C).
FIG 2. Mechanism of NLRP3 (A), NLRC4 (B), and NLRP1 (C) inflammasome activation.
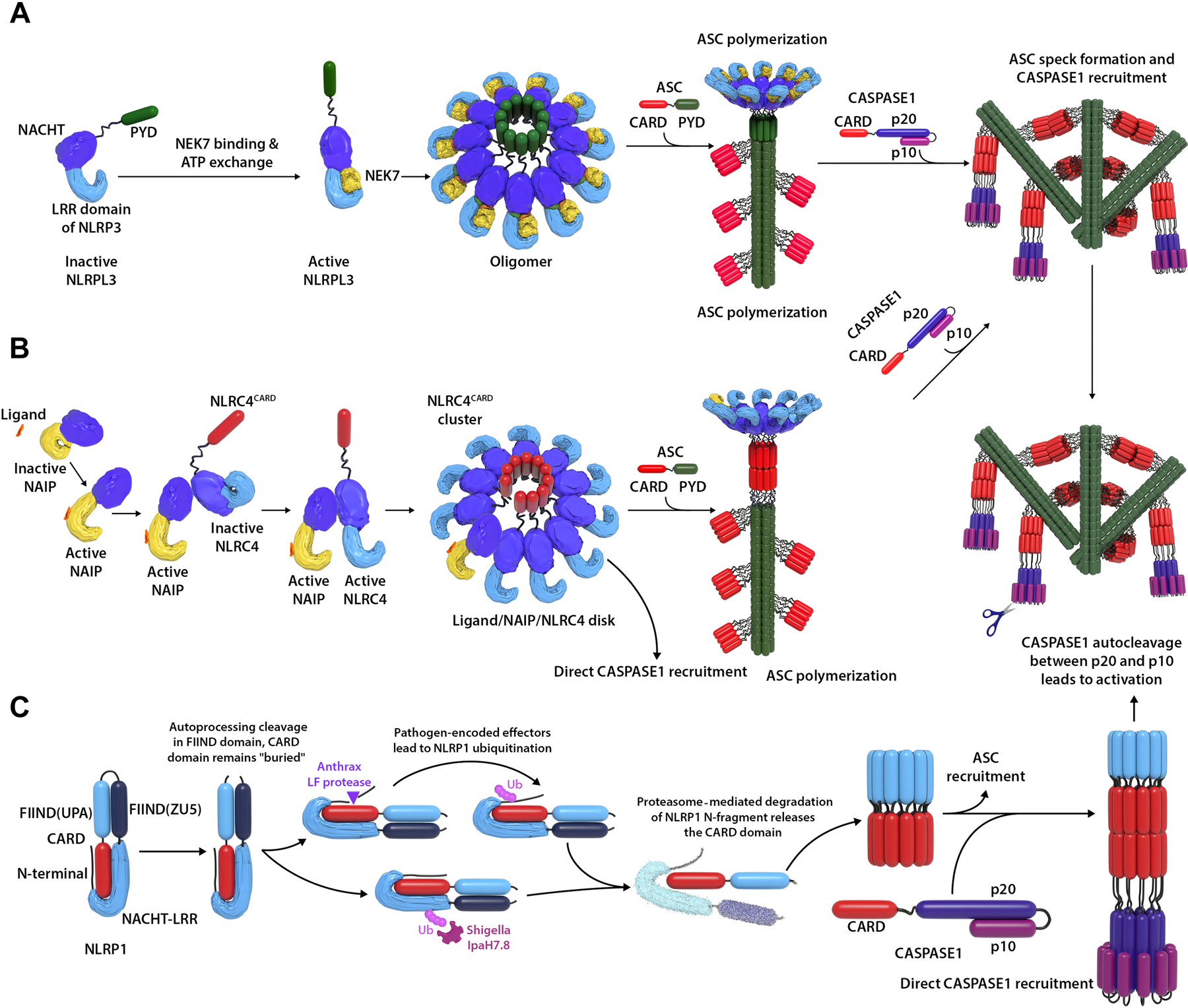
(A) NEK7 binding and ATP exchange in NLRP3 cause a confirmation change of the NACHT domain, which allows NLRP3 oligomerization and formation of an NLRP3 disc. The PYD domains in the “disc” then recruit ASC via PYD-PYD interaction, which triggers polymer assembly of ASC. The ASC polymers can further form specks via CARD-CARD interaction between the ASC polymers. This creates a platform to recruit CASPASE1 via CARD-CARD interaction between ASC and CASPASE1. CASPASE1 is then activated by autocleavage between p20 and p10 domain, which leads to NLRP3 inflammasome activation. NLRC4 is activated in a similar way (B), except initiated by ligand binding to NAIP and the ligand/NAIP/NLRC4 disc recruits ASC via CARD-CARD interaction. (C) NLRP1 is a “booby trap” for pathogens. In an attempt to deactivate the host immune system, some pathogen lethal factors try to degrade NLRP1. However, degradation of the NLRP1 N-terminal fragment leads to release of the CARD domain which triggers NLRP1 activation. CARD domain in NLRC4 or NLRP1 can recruit ASC to form ASC specks; they can also directly recruit CASPASE1 as shown in (C), right panel, direct CASPASE1 recruitment.
NLRP1 inflammasome – a “booby trap” for pathogen lethal factors
NLRP1 was the first molecularly characterized inflammasome. It is the only inflammasome receptor with a FIIND (function-to-find) domain and is activated by bacterial lethal factors. Posttranslational autoproteolysis within the FIIND domain (between residue p.F1212 and p.S1213 for humans) cleaves the protein into N- and C-terminal fragments that remain noncovalently attached in an autoinhibited state (Fig 2). Degradation of the N-terminal fragment triggered by bacterial lethal factors (including cleavage by Bacillus anthracis lethal factor protease, or ubiquitination by Shigella flexneri effector IpaH7.8) frees the C-terminal fragment and allows oligomerization and activation23, 24. These studies revealed an elegant mechanism of host defense. While many pathogens target degradation of host defense proteins to improve their survival, degradation of the “NLRP1 N-terminal fragment” triggers NLRP1 inflammasome activation and decreases pathogen survival, which has aptly been characterized as “booby trap”25. Based on this model, the N-terminal fragment might interact with the C-terminal fragment to suppress the activation of the latter (Fig 2). Indeed, almost all current NLRP1 autoinflammatory mutations lie in the N-terminus before the cleavage site at p.F1212-p.S121326–29. The only exception is the mutation at residue p.P1214, which is located right after the cleavage site and might in fact also be crucial for the interaction with the N-terminal fragment for autoinhibition.
Autosomal dominant GOF mutations in the PYD domain of NLRP1, located in chromosome 17p13.2, cause the pre-cancerous conditions, multiple self-healing palmoplantar carcinoma (MSPC)26 and familial keratosis lichenoides chronica (FKLC)27; and a missense mutation, p.M77T, causes inherited corneal intraepithelial dyskeratosis without systemic inflammation (Mendelian Inheritance in Man number (MIM#) 615225, www.omim.org)27, 30. Furthermore, single nucleotide polymorphisms (SNPs) in NLRP1 have been associated with diseases including vitiligo and have been termed vitiligo-associated multiple autoimmune disease susceptibility 1 (VAMAS1)31. Similarly, an autosomal recessive mutation between the NACHT and leucine-rich repeat (LRR) domain, p.T755N, causes congenital juvenile recurrent respiratory papillomatosis (JRRP, MIM# 618803), which is thought to be caused by infections with human papilloma virus HPV-6 or −11 and presents without systemic inflammation29. These diseases point to a potential role of NLRP1 not only in inflammation but also in keratinocyte, melanocyte and epithelial cell differentiation32,33 and may provide insights in how viral infections affect and modify keratinocyte and epithelial cell differentiation34. In contrast, recessive GOF mutations between the NACHT and the FIIND domain or an autosomal dominant mutation at the autolytic cleavage domain of FIIND, cause NLRP1-associated autoinflammation with arthritis and dyskeratosis (NAIAD/AIADK, MIM# 617388) that clinically presents with systemic inflammation that responds to IL-1 blockade28.
NLRP3 inflammasome – the frontrunner of inflammasome biology
The NLRP3 inflammasome was the first cytoplasmic sensor discovered to be linked to IL-1 production in human disease and to establish a role for disease15, 35–41. Familial cold-induced inflammatory syndrome-1 (FCAS1, MIM# 120200) caused by autosomal GOF mutations in NLRP3 was first identified in 2000/2001 and now includes the disease spectrum of Muckle-Wells syndrome (MWS; MIM# 191900), and the most severe phenotype of neonatal-onset multisystem inflammatory disease (NOMID), also known as Chronic infantile neurologic cutaneous and articular (CINCA) syndrome, (MIM# 607115)15, 35–41. Clinical features that are unique to the cryopyrinopathies include a neutrophilic urticaria presented with the systemic inflammatory attacks (induced in FCAS and chronic in MWS and NOMID) and the early development of sensorineural hearing loss and neutrophilic aseptic meningitis in the more severe disease spectrum. These pathologies provide early clues to a diagnosis. Interestingly, DFNA34, Deafness, autosomal dominant 34 is caused by a heterozygous mutation, p.R920Q, in NLRP3 (canonical transcript NM_00107982)42, and KEFH, Keratoendothelitis fugax hereditaria is caused by a heterozygous mutation, p.D21H, in NLRP3, present with organ specific, localized inflammation suggestive of local interactive factors that activate the inflammasome43, 44.
NLRP3 has been the center of inflammasome studies, as it is activated by a surprisingly diverse set of stimuli. These include ATP, crystalline, pore-forming toxins, and various types of pathogens45–49, which have been extensively reviewed50–54. These stimuli all induce “cell stress/damage” and lead to potassium efflux55, 56, but the exact nature of the NLRP3 activator that cause increased inflammasome activation in hematopoietic and non-hematopoietic cells remain elusive. Recently, the Cryo-EM structure of the NLRP3-NEK7 complex revealed that both NEK7 binding and the NACHT conformational change are required for NLRP3 activation19 (Fig. 2). Consistent with this model, almost all disease-causing cryopyrin-associated periodic syndrome (CAPS) mutations lie in the NACHT domain and surround the ADP-binding site. These pathogenic mutations affect the stability of the autoinhibitory domain and lead to autoactivation. Disease-causing variants were also found in the NEK7 binding site within the LRR domain at residues p.G755 and p.R918. In fact, the missense mutations, p.G755A and p.G755R increase the interaction with NEK7, which causes autoactivation19. Furthermore, some NLRP3 mutations are located inside or close to sumoylation motifs and impair NLRP3 sumoylation which leads to activation of the NLRP3 inflammasome57 and Table 2. These studies suggest mechanisms of how disease-causing mutations cause NLRP3 inflammasome autoactivation, which implies more broadly that disease-causing mutations at other post translational modification sites may similarly lower the autoinhibition of the NLRP3 inflammasome53, 54.
Table 2.
NLRP3 mutation in close proximity with potential sumoylation sites
| Sumoylation Lysin Position | Sumoylation Motif* | Position of closest human mutation | Pathogenicity prediction and clinical phenotype |
|---|---|---|---|
| K88 | (AKRD) | D90Y | novel, VUS, (Undefined-AID) |
| K133 | (MKKD) YR | R137H | MAF 0.0005, VUS, possibly pathogenic (MWS) |
| K204 | (IKME) | E206G | novel, possibly pathogenic, (Undefined-AID) |
| K552 | (LKLP) SR | R556X | MAF 0.0000039, likely pathogenic (atypical FMF) |
| K652 | (PKIE) | P651S | novel, likely pathogenic (MWS) |
| K689 | (PKEE) EEEEK | E690K, E692K | Both novel, likely pathogenic (NOMID/CINCA) |
closest variant/mutation reported in Infevers is indicated in red in the respective (sumoylation motif) or closest to the sumoylated lysin (K).
Pyrin inflammasome – battlefield of host and pathogen
FMF (MIM# 249100) is a periodic fever syndrome that classically presents with 1–3-day episodes of recurrent systemic inflammation and serositis that is most prevalent in Mediterranean countries13, 14. Mechanisms triggering activation of the pyrin inflammasome were characterized in 2014, when Xu et al. showed that pyrin senses Rho GTPase inactivation by bacterial toxins58. Pyrin does not directly sense pathogen molecular patterns but senses a physiological change caused by the infectious agent, which resembles plant “resistant” genes59. Rho GTPase inactivation prevents the phosphorylation of PKN1/2 and subsequently of 2 pyrin residues, that when phosphorylated, bind a protective guard molecule, 14–3-3, which keeps the pyrin inflammasome inactive60. This mechanism further explains how mevalonate kinase deficiency (MKD) or HIDS (MIM# 260920), a periodic fever syndrome caused by LOF mutations in MVK, shares clinical similarities including periodic (typically) 3-day fever and rash flares, with FMF61–63. MVK encodes mevalonate kinase, deficiency of which creates a shortage of geranylgeranyl-pyrophosphate, the substrate for PTM by geranylgeranylation of the key molecular switches that control distinct signaling pathways, including GTPases, Kras and RhoA. Thus RhoA inactivation through lack of PTM prevents the protective phosphorylation of pyrin and leads to pyrin inflammasome activation and inflammation60, 64.
Furthermore, autosomal-dominant missense mutations of the binding sites of 14–3-3 to pyrin at the serine residues, p.S208 or p.S242, cause chronic autoactivation and a clinical syndrome presenting with recurrent episodes of neutrophilic dermatosis, fever, elevated acute-phase reactants, termed pyrin-associated auto-inflammation with neutrophilic dermatosis (PAAND) also termed acute febrile neutrophilic dermatosis (AFND) (MIM# 608068)65.
A role for pyrin in pathogen sensing was established as Yersinia pestis, the organism causing the plague, deactivates pyrin66. YopM, a Yersinia pestis lethal factor, binds to the autoinhibitory domain of pyrin, B30.2 domain, and connects host ribosome S6 kinases (RSKs) with pyrin that enable its phosphorylation and suppress pyrin inflammasome activation. Interestingly, FMF mutations in the B30.2 domain autoactivate pyrin but reduce binding affinity to YopM, which prevents YopM mediated pyrin inactivation66. This selective advantage of FMF causing pyrin mutations has been proposed to confer protection from Yersinia pestis infections and may have led to positive haplotype selection of some frequent FMF mutations that explain the high FMF carrier frequencies in the Mediterranean and Middle East66.
NLRC4 inflammasome – ultra high IL18 and the risk of developing macrophage activation syndrome
The NAIP-NLRC4 inflammasome recognizes flagellin and bacterial type-3-secretion-system needle protein. Ligands that bind NAIPs trigger a conformational change of NAIP and the recruitment of 10–12 NLRC4 monomers to oligomerize (Fig 2). GOF mutations in NLRC4 cause a human autoinflammatory disease (MIM# 616050, 616115)67–69. Different from the other inflammasomes, these patients have constitutively highly elevated serum IL-18 levels that predisposes to disease flares and the development of macrophage activation syndrome (MAS) in the context of infections. Preliminary results and limited treatment experience with recombinant IL-18BP suggest a role for IL-18 in triggering MAS in these patients70. Although IL18 was also induced by other inflammasomes, the level is much lower compared to NLRC4 patients. The constitutively high serum IL18 levels may predispose to the development of MAS by also enhancing IFNγ production. IFNγ blockade in murine models was as effective as treatment of MAS with IL-18 inhibition 71, 72. The presence of high IL-18 levels in NLRC4 GOF mutant mouse chimera who received wild type bone marrow suggests that NLRC4 inflammasome activation can lead to production of high IL-18 levels in non-hematopoietic cells and points to the need to improve insights into the respective roles of inflammasomes in regulating immune and non-immune functions in non-hematopoietic cells. Serum IL-18 has since become a clinical marker in screening for the risk for genetic predisposition of MAS and in monitoring disease progression71, 73.
TYPE I INTERFERONOPATHIES – DYSREGULATION OF INTERFERON INDUCTION AND SIGNALING
The autoinflammatory Type I interferonopathies present with systemic and organ specific inflammation and a chronically elevated IRGS in blood cells that correlates with disease flares. Insights into the disease pathogenesis come from discoveries that link the type I interferon induction to activation of viral sensors.
Accumulation of cytosolic nucleic acids is a signal of viral infection, which triggers IFN-I production through activation of cytosolic viral sensors including the DNA sensor cGAS (belonging to the nucleotidyltransferase family, encoded by MB21D1), or RNA sensors MDA5 or RIG-I (encoded by IFIH1 or DDX58 respectively). All viral sensors induce transcription of IFN-I when activated, which signals in an autocrine and paracrine fashion through the IFN-I receptor and leads to the induction of interferon response genes74 (Fig 3). Recognition of cytosolic dsDNAs by cGAS trigger the synthesis of a second messenger 2’3’cGAMP75–77, which binds to the adaptor STING78–81 and activates the protein kinase, TBK1, that phosphorylates the transcription factor, IRF3, which then translocates into the nucleus and initiates the transcription of IFN-I. Similarly, cytosolic RNAs induce IFN-I by binding to the signaling adaptor MAVS, which leads to the activation of NF-κB and the induction of a TBK1 and IRF3-mediated type I interferon response82–85. The interferons stimulate through the IFN-I receptors IFNAR1 and/or IFNAR2, which leads to activation of the JAK-STAT pathway, formation of the ISRE (interferon-stimulated response element) signaling complex (STAT1, STAT2 and IFR9), and transcription of genes with ISRE binding promotor sites, namely the interferon response genes (IRG)86. Expanding functions of STING were discovered recently not only in pathogen recognition but in maintaining cell homeostasis and in regulating and coordinating interferon-independent pathways87–90.
FIG 3. Interferon mediated/amplified diseases are caused by dysregulation of interferon production and signaling.
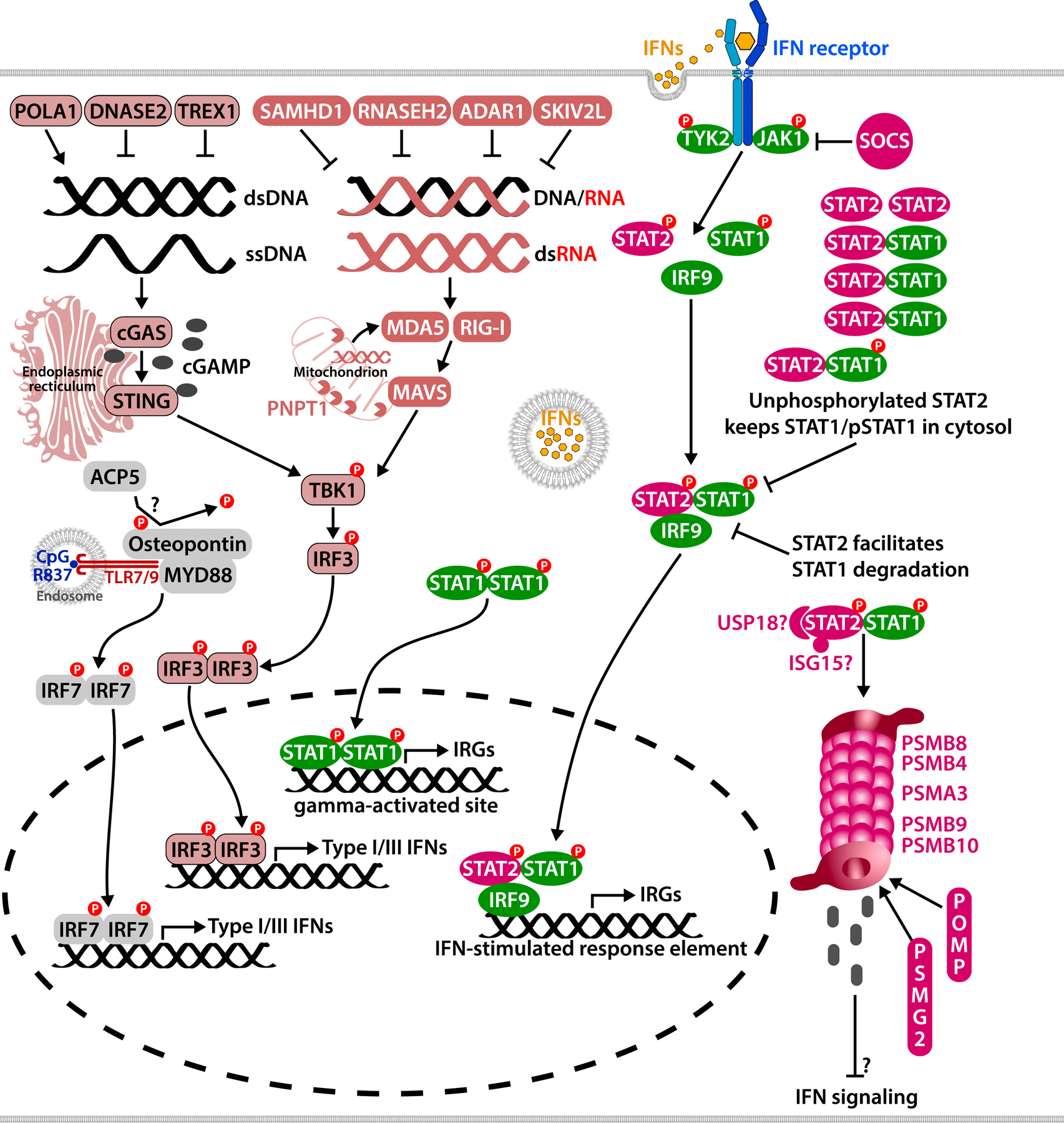
Viral replication in the cell leads to aberrant accumulation of cytosolic nucleic acids, which are sensed by the host and trigger type I/III interferon production. Cytosolic DNAs are sensed by cGAS, which produces 2’3’cGAMP, a ligand for the adaptor STING. Cytosolic RNAs are sensed by RIG-I or MDA5, which activates the adaptor MAVS. Adaptor activation leads to TBK1 phosphorylation, which phosphorylates the transcription factor IRF3 and triggers the Type I/III interferon production. Cytosolic nucleic acids are also generated in cell homeostatic processes such as DNA replication, and are degraded/reduced by various enzymes including nucleases. Mitochondrial RNAs are degraded by RNases including PNPT1. Type I and II interferons bind to their respective receptors and mediate the transcription of interferon response genes. Interferon signaling is tightly controlled by signaling suppressors including USP18, ISG15, SOCS family proteins, and STAT2, which bind to the signaling complexes for proteasome degradation. Mutations in proteasome components also lead to IFN production by yet unknown mechanisms. Disease-causing mutations in ACP5 are associated with enhanced interferon signaling, possibly via Osteopontin and IRF7. Due to space limitations, two AGS associated genes, LSM11, RNU7–1, are not shown in the figure.
Cytosolic nucleic acids are also generated in physiologic conditions, such as during DNA replication and RNA transcription. These self-nucleic acids are tightly controlled to avoid their accumulation in the cytosol and the subsequent induction of an interferon response. In fact, loss-of-function mutations (LOF) in a number of genes that are involved in nucleic acid metabolism, which includes the DNase DNASE2, TREX1, the Rnases RNASEH2A, RNASEH2B, RNASEH2C, the DNA replication regulators SAMHD1 and POLA191, and the RNA editing enzyme ADAR74, 92–94 (Figure 3) cause “interferonopathies”. The nucleic acid sensing pathways that lead to IFN production in these diseases have been extensively reviewed10–12, 74 and will only be summarized below.
Aicardi-Goutières Syndrome (AGS) and AGS-like diseases – dysregulation in cytosolic DNA/RNA clearance and sensing by the intracellular sensors
Genetic discoveries have linked mutations in self DNA/RNA sensing pathways to clinically relevant IFN dysregulation and provided insights into pathogenesis and novel targets of treatment. LOF mutations in the nucleic acid regulators TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR (MIM# 225750, 610333, 610181, 610329, 612952, 615010), and GOF mutations in the cytosolic RNA sensor MDA5 encoded by IFIH1 (MIM# 615846) cause severe leukoencephalopathy syndromes with systemic IFN signatures/IRGS and vasculopathy with variable disease severity called, Aicardi-Goutières syndrome (AGS).92–100 Recently, recessive biallelic LOF mutations in LSM11 and RNU7–1 (MIM# 619486, 619487) which encode components of the replication-dependent histone pre-mRNA–processing complex were identified to cause AGS100. Heterozygous GOF mutations in p.R822Q of IFIH1 or another cytosolic RNA sensor, RIG-I encoded by DDX58, cause Singleton-Merten syndromes 1 and 2 (MIM # 182250 and 616298 respectively), which is characterized by variable expression of glaucoma, aortic calcification, and skeletal abnormalities, without dental anomalies101.
Autosomal recessive LOF mutations in TTC37, SKIV2L affect processing of RNA and cause a syndrome of intractable diarrhea and mild neurologic symptoms (MIM# 222470 and 614602). These two genes encode the 2 components of the Ski2‐Ski3‐Ski8 (SKI) complex, a multi-protein complex that is involved in the 3’-end degradation of messenger RNAs. Patients also presents with liver disease leading to liver cirrhosis, immunodeficiency (69%), hair abnormalities, facial dysmorphism, and mild mental retardation called Trichohepato-enteric syndromes 1 and 2 (THES1 and THES2)102–105.
Mitochondrial double-stranded (ds)RNA can also accumulate and leak into the cytosol and activate the MDA5 RNA sensing pathway106. This is prevented by the mitochondrial RNA helicase SUV3 and the polynucleotide phosphorylases PNPase PNPT1107, 108, and RNASET2109, which degrade the mitochondrial dsRNA. LOF mutations in PNPT1 lead to enhanced IRGS that resulted in an early-onset mitochondrial disorder with severe encephalomyopathy, choreoathetotic movements, hypotonia, variable deafness, and combined oxidative phosphorylation deficiency 13 (COXPD1) (MIM# 614932)107; and recessive LOF mutations in RNASET2 (MIM# 612951) lead to infantile-onset cystic leukoencephalopathy and upregulation of ISGs109.
SAVI (STING-associated vasculopathy with onset in infancy) and SAVI-like diseases – constitutive activation of STING, an intracellular sensor and adaptor molecule
GOF mutations in the cytosolic DNA sensing pathway adaptor and di-nucleic acid sensor, STING (encoded by STING1) cause SAVI (STING-associated vasculopathy with onset in infancy, MIM# 615934), which presents with early-onset systemic inflammation, cold-induced vasculopathy and/or interstitial lung disease. So far, SAVI-causing mutations were found in a total of 11 different residues110–117, including 3 recently identified novel sites118. SAVI presents with variable and often severe and progressive interstitial lung disease and with peripheral vasculitis and patients develop a vasculopathy that can be mild or severe and lead to gangrene and progressive loss of tissue.
Recently other monogenic diseases with SAVI-like features of vasculopathy and/or interstitial lung disease demonstrated a role of STING in their disease pathogenesis. LOF mutations in ADA2 cause the autoinflammatory disease, DADA2 (MIM# 615688) 119–125; and COPA (COPI coat complex subunit alpha) deficiency causes the autoinflammatory disease, COPA syndrome (MIM# 616414) 126. Some DADA2 patients present with peripheral vasculitis similar to SAVI patients that involves cold-exposed, predominantly acral areas; and some COPA patients present with lung disease reminiscent of the interstitial lung disease in SAVI. Indeed, adenosine deaminase 2 encoded by ADA2, was shown to have DNase activity127; ADA2 deficiency in the THP-1 human monocyte cell line leads to cytosolic DNA accumulation and subsequent enhanced interferon induction in a STING-dependent manner127. COPA was recently found to mediate STING transportation from Golgi to endoplasmic reticulum128–130, and deficiency in COPA retains STING in the Golgi, which leads to constitutive activation of STING and enhanced production of IFN-I117, 131, 132. Furthermore, a role of the innate immune sensors on shaping adaptive immune function has been explored in COPA patients. It was proposed that the enhanced IRGS in thymic epithelia cells may impair the thymic selection of T cells and result in an increase in autoreactive T cells and a decrease in regulatory T cells thus driving autoimmunity in COPA syndrome128, 133.
Besides COPA, the calcium sensor STIM1 also regulates STING localization by retaining STING at the endoplasmic reticulum134; furthermore, STIM1 deficiency (MIM# 612783) also leads to enhanced IRGS134–137. However, the clinical phenotype of STIM1 deficiency differs from SAVI in that patients present with defective enamel and nail development and recurrent infections due to defective T-cell function138, which indicates that STIM1 has additional functions other than regulating STING localization.
Spondyloenchondrodysplasia (SPENCD; MIM# 607944) is caused by LOF mutations in ACP5 and is another disease with clinical similarities to SAVI139, 140. The mechanism remains unclear. Some reports indicate that ACP5 is involved in dephosphorylating Osteopontin (OPN), an important regulator in TLR7/8 mediated interferon-alpha induction and development of adaptive immune response141, 142. ACP5 deficiency was thought to lead to accumulation of phosphorylated OPN, which causes autoinflammation139, 143. However, direct evidence of ACP5 inactivating OPN has not been reported, and another report on SPENCD didn’t reveal a role of OPN140. Moreover, patients with GOF mutations in SPP1, the gene encoding OPN, develop autoantibodies, and showed predominately adaptive immune dysregulation144.
Regulation of the IFN response gene signature through other mechanisms
Novel Pseudo-TORCH syndromes with skin necrosis reveal dysregulation in interferon signaling through prevention of binding of the negative regulator USP18
Interferon signaling is also tightly regulated (Fig 3) through regulation of induction/transcription of IFN-Is and through regulation of signaling though the IFN receptor. SOCS1 can directly suppress the JAK activity, and the JAK-STAT signaling complex is degraded by the proteasome system to turn off the signaling145–147. It has been hypothesized that SOCS1, USP18 and ISG15 may mediate the ubiquitination and/or ubiquitin-like modifications of signaling proteins, which mark them for proteasome degradation148. Haploinsufficiency in SOCS1 has recently been associated with early-onset autoimmunity 149 and multisystem inflammatory syndrome in children (MIS-C) 150. Discoveries of LOF mutations in USP18 or ISG15 and GOF mutations in STAT2 have confirmed the critical role of USP18 as negative regulator in IFN signaling.151–154 Interestingly, all three conditions lead to a defined clinical phenotype presenting with intracranial bleeds and/or calcifications and necrotizing skin lesions in the context of enhanced interferon signaling responses. USP18 deficiency has been termed pseudo-TORCH syndrome 2 (MIM# 617397), ISG15 deficiency leads to immunodeficiency 38 with basal ganglia calcification (MIM# 616126), and STAT2 mutations at position p.R148 cause pseudo-TORCH syndrome-3 (MIM# 618886).
The molecular mechanisms remain incompletely understood, but ISG15 seems to stabilize binding of USP18 to JAK1 which inhibits activation of STATs. STAT2 may function as a negative regulator of interferon signaling either by sequestering or degrading STAT1155, 156. STAT2 also functions as transcription factor in interferon signaling8, 157, and the mutation at the p.R148 residue specifically affects the suppressor function of STAT2 while maintaining its function as transcription factor153, 154.
CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome)/PRAAS (proteasome-associated autoinflammatory syndrome), an Interferonopathy caused by proteasome deficiencies
Besides the interferonopathies caused by defects in the viral sensor/interferon signaling axis, there is an increasing number of “immune dysregulatory diseases” that present with IFN signatures that are mediated independent of the viral sensors and may or may not be the major mediators of the inflammatory response. These conditions present with clinical features of neutrophilic or monocytic panniculitis and progressive lipodystrophy and include CANDLE (chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome)/PRAAS (proteasome-associated autoinflammatory syndrome, MIM# 256040, 618048, 617591) caused by LOF mutations in genes encoding for components of the proteasome system, including the proteasome subunits PSMB8, PSMB9, PSMB4, PSMA3, PSMB10, and proteasome assembling proteins POMP and PSMG2, which all lead to an elevated IRGS. 158–161 In some patients, elements of an elevated integrated stress response (ISR) have been described162, 163. The response to JAK inhibitors that correlates with the reduction in IRGS and clinical improvement164, 165 suggests a prominent role of IFN signaling in CANDLE, which suggested that CANDLE is an interferonopathy with IFN driving the abnormal immune response. The immune dysregulation in CANDLE is however complex, and the pathomechanisms that link the IFN signature to proteasome defects remain incompletely understood. Deficiencies in the proteasome system affect the degradation of essential proteins in IFN signaling, and link impaired protein metabolism to the triggers of an elevated interferon response gene signature. A role for the UPR in inducing IFN is being explored166.
MORE THAN AN “INTERFERONOPATHY”? – COMPLEX SIGNALING PATHWAYS REVEALED BY THE MONOGENIC “AUTOINFLAMMTORY INTERFERONOPATHIES” PROPOSE NOVEL TREATMENT TARGETS BEYOND INHIBITION OF IFN SIGNALING
Recently CANDLE-like diseases that present with panniculitis were identified by genetic analysis of patients with an elevated IFN signature in peripheral blood and include NEMO-NDAS (NEMO exon5 deletion associated autoinflammatory syndrome)167. Four patients, 3 male and one female, who presented with moderately elevated interferon response gene signatures harbored de novo splicing site variants in the X-linked gene IKBKG 167. These splicing variants all lead to an in-frame deletion of exon 5 (exon 5 skipping) in IKBKG which encodes NF-kB essential modulator (NEMO). Like CANDLE, patients with NEMO-NDAS present with panniculitis, but progressive B cell lymphopenia and hypogammaglobulinemia, progressive liver disease and CNS bleeds early in life distinguish NEMO-NDAS from CANDLE. Different from previously reported patients with NEMO deficiency168–171, infections are not a prominent feature in most NEMO-NDAS patients; some patients with NEMO-NDAS develop conical teeth. Incomplete responses to JAK inhibitors and superior clinical responses to TNF inhibition167 suggested that increased IFN signaling may not be the critical pathomechanisms that drives the systemic inflammatory response.
An elevated IRGS during active disease is a hallmark of AGS, SAVI and CANDLE, but variable responses to treatment with JAK inhibitors suggest more complex immune dysregulation. While CANDLE patients respond well to JAK inhibitors as outlined above, responses are only partial for patients with SAVI164, 172–174 and AGS175. Recent discoveries of interferon-independent functions of STING, which include activation of the NFκB signaling pathway, dysregulation of autophagy and cell death88, 89, 176–178 support the involvement of interferon and JAK-independent functions in disease pathogenesis.
Moreover, we do not understand the variable disease manifestations and severity in patients with SAVI110–117. Recent data point to heterogeneous mechanisms of STING autoactivation, including the 180° rotation model for connector region mutations131, the C-terminal tail blocking model for polymer interface mutations132, and a reconciled model based on novel disease-causing mutations118. The role of these variants in differentially activating interferon-independent pathways is currently not understood but may explain some of the phenotypic heterogeneity in SAVI patients with various disease-causing mutations. Furthermore, murine models of STING point to IRF3 independent mechanisms that cause lung disease and to the heterogeneity in clinical presentation and disease severity based on allele-specific mutation and level of STING expression in various hematopoietic and non-hematopoietic cell types and provide tools for further dissecting the effect of STING signaling and the development of STING inhibitors as target for treatment179, 180
Current disease models fail to explain the clinical heterogeneity of disease manifestations between SAVI and AGS. STING has a well-documented role in AGS (Figure 3). In murine models of Trex1 deficiency, the phenotype can be rescued by knocking out Sting192. However, patients with AGS develop severe CNS disease, that can include calcifying leukoencephalopathy and microcephaly. These clinical features are not reproduced in the murine model and are not seen in patients with SAVI who have constitutive STING activation. In contrast, the severe vasculitis and interstitial lung disease that are hallmark presentations in SAVI, are absent in patients with AGS. These observations suggest that STING-independent pathomechanisms may contribute to CNS disease in patients with AGS. In fact, overexpression of IFNα in the CNS of mice mimics the human encephalopathy and suggests a role for IFN signaling in causing neuronal damage and death181 and generation of iPS cell derived TREX1-deficient neuronal progenitor cells differentiated into neurons, and astrocytes demonstrated a significant increase in intracellular DNA species including L1 retroelements, which correlated with neuronal toxicity. Inhibition of L1 reverse transcription IFN signaling inhibition in TREX1-deficient astrocytes improved cell death suggesting tissue specific effects of the mutations182. Understanding the pathogenesis of CNS disease in AGS may advance the discovery of neuroprotective strategies that may ultimately be beneficial in neurogenerative diseases beyond AGS.
Constitutive STING activation is seminal in the pathogenesis of interstitial lung disease in SAVI patients and recent data that disease-causing mutations in COPA, the gene encoding COPI Coat Complex Subunit Alpha, fails to constrain STING activation in the Golgi by preventing its transport from the Golgi to the ER, links STING activation to the pulmonary disease in COPA patients128–130. Moreover, data on a role for STING in other potentially “interferon-amplified” diseases including SLE183 make STING an attractive molecular target for therapeutic intervention not only in SAVI and AGS which may address the control of IFN independent immune dysregulation of STING.
Cosstalk between IFN, inflammasome pathways and other pathways add additional layers of complexity to these diseases. NLRP3 inflammasome can be activated by intracellularly activated complement components, which is fundamental to human TH1 induction and regulation.184 NLRC4 germline GOF mutation, in the NBD domain, the HD1 domain, (p.T337S p.V341A) or in the LRR domain, (p.W665C)185 can all cause MAS and early-onset enterocolitis (NLRC4-MAS), which are IL-1 and IL-18 driven and result in the induction of IFN-γ 71. The presence of an IFN signature in a subset of patients with high IL-18 levels and pulmonary alveolar proteinosis167 remains poorly understood; but is reminiscent of a role of IFN-I (described in a mucosal infection model) in upregulating IL-18 as demonstrated in a murine model of mucosal infection with HSV2. Infection induces CCL2 production from mucosal cells that recruit inflammatory monocytes. IFN-I then binds IFNAR on inflammatory monocytes, signals through IRF9 to induce the release of IL-18, which then binds to IL-18R on NK cells to induce IFN-γ186. It was suggested that caspase-1 activation downstream of IFN-I is mediated by the absent in melanoma 2 (AIM2) inflammasome complex187; a mechanism that has not been explored in these patients with high IL-18 levels and pulmonary alveolar proteinosis. Moreover, heterozygous RELA mutations cause early-onset systemic lupus erythematosus by skewing the NF-κB pathway towards transcriptional activation of IFN-I genes.188
Lastly, crosstalk between viral sensing pathways and inflammasome and cell death pathways have been described189, 190. RIG-I can trigger a MAVS-independent pathway that involves the signaling adaptor ASC that in an NLRP3-independent fashion leads to the production of IL-1β by caspase-1 activation.190 Furthermore, sensing of cytosolic DNA through activation of the cGAS-STING pathway and RNA ligands through TLR3 or RIG-I sensors lead to necroptotic cell death in primary cells, which requires synergy of the IFN-I and TNF signaling pathway; and administration of a STING agonist leads to a fatal, shock-like inflammatory disease in mice189. These pathways have not systematically been explored in human diseases but raise questions whether similar mechanisms may contribute to the often-sudden death in patients with interferon signatures who develop severe infections167. The presence of an IFN signature in patients with CANDLE-like diseases such as NEMO-NDAS and the better clinical responses to TNF inhibitors compared to JAK inhibitors further raises important questions regarding the impact and involvement of inflammatory cell stress and TNF mediated cell death pathways in the disease pathogenesis. In fact, necroptosis has recently been implicated in the disease pathomechanism of a patient with NEMO mediated disease presenting with immunodeficiency and inflammatory disease manifestations191.
CONCLUDING REMARKS
Basic research and disease-based discovery of monogenic defects in inflammasome and viral sensors provide insights into the clinical impact of these sensor mechanisms on human disease. The respective key cytokine mediators, IL-1 and IFN-I significantly influence the spectrum of the systemic and organ-specific disease manifestations in patients with the various inflammasomopathies and autoinflammatory interferonopathies. The clinical improvement with the use of targeted treatments that block IL-1, and Type-I interferon signaling respectively validate the role of these potent cytokines in causing and amplifying the inflammatory disease manifestations, but partial responses to IL-1 inhibitor and JAK inhibitor therapy reveal the unresolved complexity of disease-causing pathways in some diseases. For example, the discovery of constitutively high serum IL-18 levels in patients with GOF mutations in NLRC4 link the NLRC4 inflammasome to the development of macrophage activation syndrome and Type-II interferon mediated pathology. New pathomechanistic insights link activation of the viral adaptor and sensor STING, to the development of pulmonary fibrosis in SAVI and COPA syndrome, but in murine models lung disease is independent of IRF3, thus suggesting a complex role of STING in pulmonary disease and in regulating adaptive immune dysfunction; and point to STING as therapeutic target in a growing number of immunedysregulatory diseases. Lastly, new diseases with complex immune dysregulation including NEMO-NDAS reveal crosstalk between IFN pathways, NFκB signaling pathways and cell death pathways and pose new challenges to diagnosis and treatment, but also bear prospect that understanding these interactions may reveals novel targets for better treatments.
Funding:
This work is supported by the Intramural Research Program of NIAID, NIH.
ABBREVIATIONS
- SAVI
STING-associated vasculopathy with onset in infancy
- AGS
Aicardi-Goutières Syndrome
- CANDLE
chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome
- PRAAS
proteasome-associated autoinflammatory syndrome
- IFN
interferon
- IFN-I
Type I interferon
- IRGS
interferon response gene signature
- FMF
Familial Mediterranean fever
- MIM
Mendelian Inheritance in Man
- ASC
apoptosis-associated speck-like protein containing a CARD
- CARD
caspase activation and recruitment domain (CARD)
- PYD
pyrin domain
- PAMPs
pathogen-associated molecular patterns
- DAMPs
danger-associated molecular patterns
- GOF
gain-of-function
- LOF
loss-of function
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
Dr. Goldbach-Mansky has received study support under government CRADAs from SOBI, Regeneron, Novartis and Eli Lilly. All other authors have nothing to disclose.
References:
- 1.Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016; 16:407–20. [DOI] [PubMed] [Google Scholar]
- 2.Shen C, Sharif H, Xia S, Wu H. Structural and mechanistic elucidation of inflammasome signaling by cryo-EM. Curr Opin Struct Biol 2019; 58:18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harapas CR, Steiner A, Davidson S, Masters SL. An Update on Autoinflammatory Diseases: Inflammasomopathies. Curr Rheumatol Rep 2018; 20:40. [DOI] [PubMed] [Google Scholar]
- 4.Schnappauf O, Chae JJ, Kastner DL, Aksentijevich I. The Pyrin Inflammasome in Health and Disease. Front Immunol 2019; 10:1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Villarino AV, Kanno Y, O’Shea JJ. Mechanisms and consequences of Jak-STAT signaling in the immune system. Nat Immunol 2017; 18:374–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin FC, Young HA. Interferons: Success in anti-viral immunotherapy. Cytokine Growth Factor Rev 2014; 25:369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Floyd K, Hick VE, Morrison JF. The influence of visceral mechanoreceptors on sympathetic efferent discharge in the cat. J Physiol 1982; 323:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fensterl V, Chattopadhyay S, Sen GC. No Love Lost Between Viruses and Interferons. Annu Rev Virol 2015; 2:549–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Canna SW, Goldbach-Mansky R. New monogenic autoinflammatory diseases--a clinical overview. Semin Immunopathol 2015; 37:387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Crow YJ, Manel N. Aicardi-Goutieres syndrome and the type I interferonopathies. Nat Rev Immunol 2015; 15:429–40. [DOI] [PubMed] [Google Scholar]
- 11.Davidson S, Steiner A, Harapas CR, Masters SL. An Update on Autoinflammatory Diseases: Interferonopathies. Curr Rheumatol Rep 2018; 20:38. [DOI] [PubMed] [Google Scholar]
- 12.Lee-Kirsch MA. The Type I Interferonopathies. Annu Rev Med 2017; 68:297–315. [DOI] [PubMed] [Google Scholar]
- 13.Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. The International FMF Consortium. Cell 1997; 90:797–807. [DOI] [PubMed] [Google Scholar]
- 14.French FMFC. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31. [DOI] [PubMed] [Google Scholar]
- 15.Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–26. [DOI] [PubMed] [Google Scholar]
- 17.Hu Z, Zhou Q, Zhang C, Fan S, Cheng W, Zhao Y, et al. Structural and biochemical basis for induced self-propagation of NLRC4. Science 2015; 350:399–404. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science 2015; 350:404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sharif H, Wang L, Wang WL, Magupalli VG, Andreeva L, Qiao Q, et al. Structural mechanism for NEK7-licensed activation of NLRP3 inflammasome. Nature 2019; 570:338–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014; 156:1193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lu A, Li Y, Schmidt FI, Yin Q, Chen S, Fu TM, et al. Molecular basis of caspase-1 polymerization and its inhibition by a new capping mechanism. Nat Struct Mol Biol 2016; 23:416–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015; 526:660–5. [DOI] [PubMed] [Google Scholar]
- 23.Chui AJ, Okondo MC, Rao SD, Gai K, Griswold AR, Johnson DC, et al. N-terminal degradation activates the NLRP1B inflammasome. Science 2019; 364:82–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sandstrom A, Mitchell PS, Goers L, Mu EW, Lesser CF, Vance RE. Functional degradation: A mechanism of NLRP1 inflammasome activation by diverse pathogen enzymes. Science 2019; 364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Flemming A Functional degradation ignites the inflammasome. Nat Rev Immunol 2019; 19:349. [DOI] [PubMed] [Google Scholar]
- 26.Mamai O, Boussofara L, Denguezli M, Escande-Beillard N, Kraeim W, Merriman B, et al. Multiple self-healing palmoplantar carcinoma: a familial predisposition to skin cancer with primary palmoplantar and conjunctival lesions. J Invest Dermatol 2015; 135:304–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhong FL, Mamai O, Sborgi L, Boussofara L, Hopkins R, Robinson K, et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 2016; 167:187–202 e17. [DOI] [PubMed] [Google Scholar]
- 28.Grandemange S, Sanchez E, Louis-Plence P, Tran Mau-Them F, Bessis D, Coubes C, et al. A new autoinflammatory and autoimmune syndrome associated with NLRP1 mutations: NAIAD (NLRP1-associated autoinflammation with arthritis and dyskeratosis). Ann Rheum Dis 2017; 76:1191–8. [DOI] [PubMed] [Google Scholar]
- 29.Drutman SB, Haerynck F, Zhong FL, Hum D, Hernandez NJ, Belkaya S, et al. Homozygous NLRP1 gain-of-function mutation in siblings with a syndromic form of recurrent respiratory papillomatosis. Proc Natl Acad Sci U S A 2019; 116:19055–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Soler VJ, Tran-Viet KN, Galiacy SD, Limviphuvadh V, Klemm TP, St Germain E, et al. Whole exome sequencing identifies a mutation for a novel form of corneal intraepithelial dyskeratosis. J Med Genet 2013; 50:246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin Y, Birlea SA, Fain PR, Gowan K, Riccardi SL, Holland PJ, et al. Variant of TYR and autoimmunity susceptibility loci in generalized vitiligo. N Engl J Med 2010; 362:1686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhai Z, Vaddi PK, Samson JM, Takegami T, Fujita M. NLRP1 Functions Downstream of the MAPK/ERK Signaling via ATF4 and Contributes to Acquired Targeted Therapy Resistance in Human Metastatic Melanoma. Pharmaceuticals (Basel) 2020; 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Burian M, Yazdi AS. NLRP1 Is the Key Inflammasome in Primary Human Keratinocytes. J Invest Dermatol 2018; 138:2507–10. [DOI] [PubMed] [Google Scholar]
- 34.Eller MS, Yaar M, Ostrom K, Harkness DD, Gilchrest BA. A role for interleukin-1 in epidermal differentiation: regulation by expression of functional versus decoy receptors. J Cell Sci 1995; 108 ( Pt 8):2741–6. [DOI] [PubMed] [Google Scholar]
- 35.Dode C, Le Du N, Cuisset L, Letourneur F, Berthelot JM, Vaudour G, et al. New mutations of CIAS1 that are responsible for Muckle-Wells syndrome and familial cold urticaria: a novel mutation underlies both syndromes. Am J Hum Genet 2002; 70:1498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feldmann J, Prieur AM, Quartier P, Berquin P, Certain S, Cortis E, et al. Chronic infantile neurological cutaneous and articular syndrome is caused by mutations in CIAS1, a gene highly expressed in polymorphonuclear cells and chondrocytes. Am J Hum Genet 2002; 71:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Aksentijevich I, Nowak M, Mallah M, Chae JJ, Watford WT, Hofmann SR, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Goldbach-Mansky R, Dailey NJ, Canna SW, Gelabert A, Jones J, Rubin BI, et al. Neonatal-onset multisystem inflammatory disease responsive to interleukin-1beta inhibition. N Engl J Med 2006; 355:581–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hoffman HM, Gregory SG, Mueller JL, Tresierras M, Broide DH, Wanderer AA, et al. Fine structure mapping of CIAS1: identification of an ancestral haplotype and a common FCAS mutation, L353P. Hum Genet 2003; 112:209–16. [DOI] [PubMed] [Google Scholar]
- 40.Hoffman HM, Rosengren S, Boyle DL, Cho JY, Nayar J, Mueller JL, et al. Prevention of cold-associated acute inflammation in familial cold autoinflammatory syndrome by interleukin-1 receptor antagonist. Lancet 2004; 364:1779–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Neven B, Callebaut I, Prieur AM, Feldmann J, Bodemer C, Lepore L, et al. Molecular basis of the spectral expression of CIAS1 mutations associated with phagocytic cell-mediated autoinflammatory disorders CINCA/NOMID, MWS, and FCU. Blood 2004; 103:2809–15. [DOI] [PubMed] [Google Scholar]
- 42.Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, et al. NLRP3 mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc Natl Acad Sci U S A 2017; 114:E7766–E75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turunen JA, Wedenoja J, Repo P, Jarvinen RS, Jantti JE, Mortenhumer S, et al. Keratoendotheliitis Fugax Hereditaria: A Novel Cryopyrin-Associated Periodic Syndrome Caused by a Mutation in the Nucleotide-Binding Domain, Leucine-Rich Repeat Family, Pyrin Domain-Containing 3 (NLRP3) Gene. Am J Ophthalmol 2018; 188:41–50. [DOI] [PubMed] [Google Scholar]
- 44.Ruusuvaara P, Setala K. Keratoendotheliitis fugax hereditaria. A clinical and specular microscopic study of a family with dominant inflammatory corneal disease. Acta Ophthalmol (Copenh) 1987; 65:159–69. [DOI] [PubMed] [Google Scholar]
- 45.Zhong Z, Liang S, Sanchez-Lopez E, He F, Shalapour S, Lin XJ, et al. New mitochondrial DNA synthesis enables NLRP3 inflammasome activation. Nature 2018; 560:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhong Z, Zhai Y, Liang S, Mori Y, Han R, Sutterwala FS, et al. TRPM2 links oxidative stress to NLRP3 inflammasome activation. Nat Commun 2013; 4:1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hornung V, Bauernfeind F, Halle A, Samstad EO, Kono H, Rock KL, et al. Silica crystals and aluminum salts activate the NALP3 inflammasome through phagosomal destabilization. Nat Immunol 2008; 9:847–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dostert C, Petrilli V, Van Bruggen R, Steele C, Mossman BT, Tschopp J. Innate immune activation through Nalp3 inflammasome sensing of asbestos and silica. Science 2008; 320:674–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440:228–32. [DOI] [PubMed] [Google Scholar]
- 50.He Y, Hara H, Nunez G. Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends Biochem Sci 2016; 41:1012–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol 2010; 10:210–5. [DOI] [PubMed] [Google Scholar]
- 52.Rathinam VA, Fitzgerald KA. Inflammasome Complexes: Emerging Mechanisms and Effector Functions. Cell 2016; 165:792–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Christgen S, Place DE, Kanneganti TD. Toward targeting inflammasomes: insights into their regulation and activation. Cell Res 2020; 30:315–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019; 19:477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Munoz-Planillo R, Kuffa P, Martinez-Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013; 38:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019; 573:590–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barry R, John SW, Liccardi G, Tenev T, Jaco I, Chen CH, et al. SUMO-mediated regulation of NLRP3 modulates inflammasome activity. Nat Commun 2018; 9:3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Xu H, Yang J, Gao W, Li L, Li P, Zhang L, et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 2014; 513:237–41. [DOI] [PubMed] [Google Scholar]
- 59.Arora S, Steuernagel B, Gaurav K, Chandramohan S, Long Y, Matny O, et al. Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat Biotechnol 2019; 37:139–43. [DOI] [PubMed] [Google Scholar]
- 60.Park YH, Wood G, Kastner DL, Chae JJ. Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 2016; 17:914–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Houten SM, Kuis W, Duran M, de Koning TJ, van Royen-Kerkhof A, Romeijn GJ, et al. Mutations in MVK, encoding mevalonate kinase, cause hyperimmunoglobulinaemia D and periodic fever syndrome. Nat Genet 1999; 22:175–7. [DOI] [PubMed] [Google Scholar]
- 62.Cuisset L, Drenth JP, Simon A, Vincent MF, van der Velde Visser S, van der Meer JW, et al. Molecular analysis of MVK mutations and enzymatic activity in hyper-IgD and periodic fever syndrome. Eur J Hum Genet 2001; 9:260–6. [DOI] [PubMed] [Google Scholar]
- 63.Drenth JP, Cuisset L, Grateau G, Vasseur C, van de Velde-Visser SD, de Jong JG, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–81. [DOI] [PubMed] [Google Scholar]
- 64.Akula MK, Shi M, Jiang Z, Foster CE, Miao D, Li AS, et al. Control of the innate immune response by the mevalonate pathway. Nat Immunol 2016; 17:922–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Masters SL, Lagou V, Jeru I, Baker PJ, Van Eyck L, Parry DA, et al. Familial autoinflammation with neutrophilic dermatosis reveals a regulatory mechanism of pyrin activation. Sci Transl Med 2016; 8:332ra45. [DOI] [PubMed] [Google Scholar]
- 66.Park YH, Remmers EF, Lee W, Ombrello AK, Chung LK, Shilei Z, et al. Ancient familial Mediterranean fever mutations in human pyrin and resistance to Yersinia pestis. Nat Immunol 2020; 21:857–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Canna SW, de Jesus AA, Gouni S, Brooks SR, Marrero B, Liu Y, et al. An activating NLRC4 inflammasome mutation causes autoinflammation with recurrent macrophage activation syndrome. Nat Genet 2014; 46:1140–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Romberg N, Al Moussawi K, Nelson-Williams C, Stiegler AL, Loring E, Choi M, et al. Mutation of NLRC4 causes a syndrome of enterocolitis and autoinflammation. Nat Genet 2014; 46:1135–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kitamura A, Sasaki Y, Abe T, Kano H, Yasutomo K. An inherited mutation in NLRC4 causes autoinflammation in human and mice. J Exp Med 2014; 211:2385–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Canna SW, Girard C, Malle L, de Jesus A, Romberg N, Kelsen J, et al. Life-threatening NLRC4-associated hyperinflammation successfully treated with IL-18 inhibition. J Allergy Clin Immunol 2017; 139:1698–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Weiss ES, Girard-Guyonvarc’h C, Holzinger D, de Jesus AA, Tariq Z, Picarsic J, et al. Interleukin-18 diagnostically distinguishes and pathogenically promotes human and murine macrophage activation syndrome. Blood 2018; 131:1442–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Girard-Guyonvarc’h C, Palomo J, Martin P, Rodriguez E, Troccaz S, Palmer G, et al. Unopposed IL-18 signaling leads to severe TLR9-induced macrophage activation syndrome in mice. Blood 2018; 131:1430–41. [DOI] [PubMed] [Google Scholar]
- 73.Krei JM, Moller HJ, Larsen JB. The role of interleukin-18 in the diagnosis and monitoring of hemophagocytic lymphohistiocytosis/macrophage activation syndrome - a systematic review. Clin Exp Immunol 2021; 203:174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Crowl JT, Gray EE, Pestal K, Volkman HE, Stetson DB. Intracellular Nucleic Acid Detection in Autoimmunity. Annu Rev Immunol 2017; 35:313–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Sun L, Wu J, Du F, Chen X, Chen ZJ. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013; 339:786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wu J, Sun L, Chen X, Du F, Shi H, Chen C, et al. Cyclic GMP-AMP is an endogenous second messenger in innate immune signaling by cytosolic DNA. Science 2013; 339:826–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhang X, Shi H, Wu J, Zhang X, Sun L, Chen C, et al. Cyclic GMP-AMP containing mixed phosphodiester linkages is an endogenous high-affinity ligand for STING. Mol Cell 2013; 51:226–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Burdette DL. STING is a direct innate immune sensor of cyclic di-GMP. Nature 2011; 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008; 455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin L MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol. Cell. Biol 2008; 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhong B The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity 2008; 29. [DOI] [PubMed] [Google Scholar]
- 82.Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006; 441:101–5. [DOI] [PubMed] [Google Scholar]
- 83.Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med 2008; 205:1601–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity 2006; 24:633–42. [DOI] [PubMed] [Google Scholar]
- 85.Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med 2006; 203:1795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schneider WM, Chevillotte MD, Rice CM. Interferon-stimulated genes: a complex web of host defenses. Annu Rev Immunol 2014; 32:513–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hopfner KP, Hornung V. Molecular mechanisms and cellular functions of cGAS-STING signalling. Nat Rev Mol Cell Biol 2020; 21:501–21. [DOI] [PubMed] [Google Scholar]
- 88.Wu J, Dobbs N, Yang K, Yan N. Interferon-Independent Activities of Mammalian STING Mediate Antiviral Response and Tumor Immune Evasion. Immunity 2020; 53:115–26 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yamashiro LH, Wilson SC, Morrison HM, Karalis V, Chung JJ, Chen KJ, et al. Interferon-independent STING signaling promotes resistance to HSV-1 in vivo. Nat Commun 2020; 11:3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Motwani M, Pesiridis S, Fitzgerald KA. DNA sensing by the cGAS-STING pathway in health and disease. Nat Rev Genet 2019; 20:657–74. [DOI] [PubMed] [Google Scholar]
- 91.Starokadomskyy P, Gemelli T, Rios JJ, Xing C, Wang RC, Li H, et al. DNA polymerase-alpha regulates the activation of type I interferons through cytosolic RNA:DNA synthesis. Nat Immunol 2016; 17:495–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gall A, Treuting P, Elkon KB, Loo YM, Gale M Jr., Barber GN, et al. Autoimmunity initiates in nonhematopoietic cells and progresses via lymphocytes in an interferon-dependent autoimmune disease. Immunity 2012; 36:120–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Mackenzie KJ, Carroll P, Lettice L, Tarnauskaite Z, Reddy K, Dix F, et al. Ribonuclease H2 mutations induce a cGAS/STING-dependent innate immune response. EMBO J 2016; 35:831–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Pokatayev V, Hasin N, Chon H, Cerritelli SM, Sakhuja K, Ward JM, et al. RNase H2 catalytic core Aicardi-Goutieres syndrome-related mutant invokes cGAS-STING innate immune-sensing pathway in mice. J Exp Med 2016; 213:329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, et al. Mutations in the gene encoding the 3’−5’ DNA exonuclease TREX1 cause Aicardi-Goutieres syndrome at the AGS1 locus. Nat Genet 2006; 38:917–20. [DOI] [PubMed] [Google Scholar]
- 96.Yang YG, Lindahl T, Barnes DE. Trex1 exonuclease degrades ssDNA to prevent chronic checkpoint activation and autoimmune disease. Cell 2007; 131:873–86. [DOI] [PubMed] [Google Scholar]
- 97.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, et al. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutieres syndrome and mimic congenital viral brain infection. Nat Genet 2006; 38:910–6. [DOI] [PubMed] [Google Scholar]
- 98.Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GM, Gornall HL, et al. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 2015; 167A:296–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rice GI, Bond J, Asipu A, Brunette RL, Manfield IW, Carr IM, et al. Mutations involved in Aicardi-Goutieres syndrome implicate SAMHD1 as regulator of the innate immune response. Nat Genet 2009; 41:829–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Uggenti C, Lepelley A, Depp M, Badrock AP, Rodero MP, El-Daher MT, et al. cGAS-mediated induction of type I interferon due to inborn errors of histone pre-mRNA processing. Nat Genet 2020; 52:1364–72. [DOI] [PubMed] [Google Scholar]
- 101.Jang MA, Kim EK, Now H, Nguyen NT, Kim WJ, Yoo JY, et al. Mutations in DDX58, which encodes RIG-I, cause atypical Singleton-Merten syndrome. Am J Hum Genet 2015; 96:266–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hartley JL, Zachos NC, Dawood B, Donowitz M, Forman J, Pollitt RJ, et al. Mutations in TTC37 cause trichohepatoenteric syndrome (phenotypic diarrhea of infancy). Gastroenterology 2010; 138:2388–98, 98 e1–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Morgan NV, Hartley JL, Setchell KD, Simpson MA, Brown R, Tee L, et al. A combination of mutations in AKR1D1 and SKIV2L in a family with severe infantile liver disease. Orphanet J Rare Dis 2013; 8:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Fabre A, Bourgeois P, Coste ME, Roman C, Barlogis V, Badens C. Management of syndromic diarrhea/tricho-hepato-enteric syndrome: A review of the literature. Intractable Rare Dis Res 2017; 6:152–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hiejima E, Yasumi T, Nakase H, Matsuura M, Honzawa Y, Higuchi H, et al. Tricho-hepato-enteric syndrome with novel SKIV2L gene mutations: A case report. Medicine (Baltimore) 2017; 96:e8601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Borowski LS, Dziembowski A, Hejnowicz MS, Stepien PP, Szczesny RJ. Human mitochondrial RNA decay mediated by PNPase-hSuv3 complex takes place in distinct foci. Nucleic Acids Res 2013; 41:1223–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018; 560:238–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Liu X, Fu R, Pan Y, Meza-Sosa KF, Zhang Z, Lieberman J. PNPT1 Release from Mitochondria during Apoptosis Triggers Decay of Poly(A) RNAs. Cell 2018; 174:187–201 e12. [DOI] [PubMed] [Google Scholar]
- 109.Henneke M, Diekmann S, Ohlenbusch A, Kaiser J, Engelbrecht V, Kohlschutter A, et al. RNASET2-deficient cystic leukoencephalopathy resembles congenital cytomegalovirus brain infection. Nat Genet 2009; 41:773–5. [DOI] [PubMed] [Google Scholar]
- 110.Liu Y, Jesus AA, Marrero B, Yang D, Ramsey SE, Sanchez GAM, et al. Activated STING in a vascular and pulmonary syndrome. N Engl J Med 2014; 371:507–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jeremiah N, Neven B, Gentili M, Callebaut I, Maschalidi S, Stolzenberg MC, et al. Inherited STING-activating mutation underlies a familial inflammatory syndrome with lupus-like manifestations. J Clin Invest 2014; 124:5516–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Konig N, Fiehn C, Wolf C, Schuster M, Cura Costa E, Tungler V, et al. Familial chilblain lupus due to a gain-of-function mutation in STING. Ann Rheum Dis 2017; 76:468–72. [DOI] [PubMed] [Google Scholar]
- 113.Melki I, Rose Y, Uggenti C, Van Eyck L, Fremond ML, Kitabayashi N, et al. Disease-associated mutations identify a novel region in human STING necessary for the control of type I interferon signaling. J Allergy Clin Immunol 2017; 140:543–52 e5. [DOI] [PubMed] [Google Scholar]
- 114.Konno H, Chinn IK, Hong D, Orange JS, Lupski JR, Mendoza A, et al. Pro-inflammation Associated with a Gain-of-Function Mutation (R284S) in the Innate Immune Sensor STING. Cell Rep 2018; 23:1112–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Saldanha RG, Balka KR, Davidson S, Wainstein BK, Wong M, Macintosh R, et al. A Mutation Outside the Dimerization Domain Causing Atypical STING-Associated Vasculopathy With Onset in Infancy. Front Immunol 2018; 9:1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Keskitalo S, Haapaniemi E, Einarsdottir E, Rajamaki K, Heikkila H, Ilander M, et al. Novel TMEM173 Mutation and the Role of Disease Modifying Alleles. Front Immunol 2019; 10:2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Lin B, Berard R, Al Rasheed A, Aladba B, Kranzusch PJ, Henderlight M, et al. A novel STING1 variant causes a recessive form of STING-associated vasculopathy with onset in infancy (SAVI). J Allergy Clin Immunol 2020; 146:1204–8 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lin B, Torreggiani S, Kahle D, Rumsey DG, Wright BL, Montes-Cano MA, et al. Case Report: Novel SAVI-Causing Variants in STING1 Expand the Clinical Disease Spectrum and Suggest a Refined Model of STING Activation. Front Immunol 2021; 12:636225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee PY, Kellner ES, Huang Y, Furutani E, Huang Z, Bainter W, et al. Genotype and functional correlates of disease phenotype in deficiency of adenosine deaminase 2 (DADA2). J Allergy Clin Immunol 2020; 145:1664–72 e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhou Q, Yang D, Ombrello AK, Zavialov AV, Toro C, Zavialov AV, et al. Early-onset stroke and vasculopathy associated with mutations in ADA2. N Engl J Med 2014; 370:911–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Navon Elkan P, Pierce SB, Segel R, Walsh T, Barash J, Padeh S, et al. Mutant adenosine deaminase 2 in a polyarteritis nodosa vasculopathy. N Engl J Med 2014; 370:921–31. [DOI] [PubMed] [Google Scholar]
- 122.van Montfrans J, Zavialov A, Zhou Q. Mutant ADA2 in vasculopathies. N Engl J Med 2014; 371:478. [DOI] [PubMed] [Google Scholar]
- 123.Van Eyck L, Liston A, Wouters C. Mutant ADA2 in vasculopathies. N Engl J Med 2014; 371:480. [DOI] [PubMed] [Google Scholar]
- 124.Van Eyck L, Liston A, Meyts I. Mutant ADA2 in vasculopathies. N Engl J Med 2014; 371:478–9. [DOI] [PubMed] [Google Scholar]
- 125.Van Montfrans JM, Hartman EA, Braun KP, Hennekam EA, Hak EA, Nederkoorn PJ, et al. Phenotypic variability in patients with ADA2 deficiency due to identical homozygous R169Q mutations. Rheumatology (Oxford) 2016; 55:902–10. [DOI] [PubMed] [Google Scholar]
- 126.Watkin LB, Jessen B, Wiszniewski W, Vece TJ, Jan M, Sha Y, et al. COPA mutations impair ER-Golgi transport and cause hereditary autoimmune-mediated lung disease and arthritis. Nat Genet 2015; 47:654–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Greiner-Tollersrud OK, Krausz M, Bartok E, Boehler V, Ebersbach H, Baash S, et al. ADA2 is a lysosomal DNase regulating the type-I interferon response. bioRxiv 2020. [Google Scholar]
- 128.Deng Z, Chong Z, Law CS, Mukai K, Ho FO, Martinu T, et al. A defect in COPI-mediated transport of STING causes immune dysregulation in COPA syndrome. Journal of Experimental Medicine 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Lepelley A, Martin-Niclós MJ, Le Bihan M, Marsh JA, Uggenti C, Rice GI, et al. Mutations in COPA lead to abnormal trafficking of STING to the Golgi and interferon signaling. Journal of Experimental Medicine 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Steiner A, Schaale KH, Prigione I, De Nardo D, Dagley LF, Yu C-H, et al. Activation of STING due to COPI-deficiency. BioRxiv 2020. [Google Scholar]
- 131.Shang G, Zhang C, Chen ZJ, Bai XC, Zhang X. Cryo-EM structures of STING reveal its mechanism of activation by cyclic GMP-AMP. Nature 2019; 567:389–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Ergun SL, Fernandez D, Weiss TM, Li L. STING Polymer Structure Reveals Mechanisms for Activation, Hyperactivation, and Inhibition. Cell 2019; 178:290–301 e10. [DOI] [PubMed] [Google Scholar]
- 133.Deng Z, Law CS, Ho FO, Wang KM, Jones KD, Shin JS, et al. A Defect in Thymic Tolerance Causes T Cell-Mediated Autoimmunity in a Murine Model of COPA Syndrome. J Immunol 2020; 204:2360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Srikanth S, Woo JS, Wu B, El-Sherbiny YM, Leung J, Chupradit K, et al. The Ca(2+) sensor STIM1 regulates the type I interferon response by retaining the signaling adaptor STING at the endoplasmic reticulum. Nat Immunol 2019; 20:152–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Li Y, James SJ, Wyllie DH, Wynne C, Czibula A, Bukhari A, et al. TMEM203 is a binding partner and regulator of STING-mediated inflammatory signaling in macrophages. Proc Natl Acad Sci U S A 2019; 116:16479–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Parry DA, Holmes TD, Gamper N, El-Sayed W, Hettiarachchi NT, Ahmed M, et al. A homozygous STIM1 mutation impairs store-operated calcium entry and natural killer cell effector function without clinical immunodeficiency. J Allergy Clin Immunol 2016; 137:955–7 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Rice L, Stockdale C, Berry I, O’Riordan S, Pysden K, Anwar R, et al. A Report of Novel STIM1 Deficiency and 6-Year Follow-Up of Two Previous Cases Associated with Mild Immunological Phenotype. J Clin Immunol 2019; 39:249–56. [DOI] [PubMed] [Google Scholar]
- 138.Byun M, Abhyankar A, Lelarge V, Plancoulaine S, Palanduz A, Telhan L, et al. Whole-exome sequencing-based discovery of STIM1 deficiency in a child with fatal classic Kaposi sarcoma. J Exp Med 2010; 207:2307–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Lausch E, Janecke A, Bros M, Trojandt S, Alanay Y, De Laet C, et al. Genetic deficiency of tartrate-resistant acid phosphatase associated with skeletal dysplasia, cerebral calcifications and autoimmunity. Nat Genet 2011; 43:132–7. [DOI] [PubMed] [Google Scholar]
- 140.Briggs TA, Rice GI, Daly S, Urquhart J, Gornall H, Bader-Meunier B, et al. Tartrate-resistant acid phosphatase deficiency causes a bone dysplasia with autoimmunity and a type I interferon expression signature. Nat Genet 2011; 43:127–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Leavenworth JW, Verbinnen B, Yin J, Huang H, Cantor H. A p85alpha-osteopontin axis couples the receptor ICOS to sustained Bcl-6 expression by follicular helper and regulatory T cells. Nat Immunol 2015; 16:96–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shinohara ML, Lu L, Bu J, Werneck MB, Kobayashi KS, Glimcher LH, et al. Osteopontin expression is essential for interferon-alpha production by plasmacytoid dendritic cells. Nat Immunol 2006; 7:498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.An J, Briggs TA, Dumax-Vorzet A, Alarcon-Riquelme ME, Belot A, Beresford M, et al. Tartrate-Resistant Acid Phosphatase Deficiency in the Predisposition to Systemic Lupus Erythematosus. Arthritis Rheumatol 2017; 69:131–42. [DOI] [PubMed] [Google Scholar]
- 144.Rittling SR, Singh R. Osteopontin in Immune-mediated Diseases. J Dent Res 2015; 94:1638–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Durham GA, Williams JJL, Nasim MT, Palmer TM. Targeting SOCS Proteins to Control JAK-STAT Signalling in Disease. Trends Pharmacol Sci 2019; 40:298–308. [DOI] [PubMed] [Google Scholar]
- 146.Linossi EM, Nicholson SE. Kinase inhibition, competitive binding and proteasomal degradation: resolving the molecular function of the suppressor of cytokine signaling (SOCS) proteins. Immunol Rev 2015; 266:123–33. [DOI] [PubMed] [Google Scholar]
- 147.Yoshimura A, Naka T, Kubo M. SOCS proteins, cytokine signalling and immune regulation. Nat Rev Immunol 2007; 7:454–65. [DOI] [PubMed] [Google Scholar]
- 148.Hermann M, Bogunovic D. ISG15: In Sickness and in Health. Trends Immunol 2017; 38:79–93. [DOI] [PubMed] [Google Scholar]
- 149.Hadjadj J, Castro CN, Tusseau M, Stolzenberg MC, Mazerolles F, Aladjidi N, et al. Early-onset autoimmunity associated with SOCS1 haploinsufficiency. Nat Commun 2020; 11:5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee PY, Platt CD, Weeks S, Grace RF, Maher G, Gauthier K, et al. Immune dysregulation and multisystem inflammatory syndrome in children (MIS-C) in individuals with haploinsufficiency of SOCS1. J Allergy Clin Immunol 2020; 146:1194–200 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Meuwissen ME, Schot R, Buta S, Oudesluijs G, Tinschert S, Speer SD, et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J Exp Med 2016; 213:1163–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhang X, Bogunovic D, Payelle-Brogard B, Francois-Newton V, Speer SD, Yuan C, et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 2015; 517:89–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Duncan CJA, Thompson BJ, Chen R, Rice GI, Gothe F, Young DF, et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci Immunol 2019; 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gruber C, Martin-Fernandez M, Ailal F, Qiu X, Taft J, Altman J, et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J Exp Med 2020; 217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ho J, Pelzel C, Begitt A, Mee M, Elsheikha HM, Scott DJ, et al. STAT2 Is a Pervasive Cytokine Regulator due to Its Inhibition of STAT1 in Multiple Signaling Pathways. PLoS Biol 2016; 14:e2000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Arimoto KI, Lochte S, Stoner SA, Burkart C, Zhang Y, Miyauchi S, et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat Struct Mol Biol 2017; 24:279–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity 2000; 13:795–804. [DOI] [PubMed] [Google Scholar]
- 158.Agarwal AK, Xing C, DeMartino GN, Mizrachi D, Hernandez MD, Sousa AB, et al. PSMB8 encoding the beta5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome. Am J Hum Genet 2010; 87:866–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Brehm A, Liu Y, Sheikh A, Marrero B, Omoyinmi E, Zhou Q, et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest 2015; 125:4196–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Liu Y, Ramot Y, Torrelo A, Paller AS, Si N, Babay S, et al. Mutations in proteasome subunit beta type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum 2012; 64:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.de Jesus AA, Brehm A, VanTries R, Pillet P, Parentelli AS, Montealegre Sanchez GA, et al. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J Allergy Clin Immunol 2019; 143:1939–43 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Cetin G, Klafack S, Studencka-Turski M, Kruger E, Ebstein F. The Ubiquitin-Proteasome System in Immune Cells. Biomolecules 2021; 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Poli MC, Ebstein F, Nicholas SK, de Guzman MM, Forbes LR, Chinn IK, et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am J Hum Genet 2018; 102:1126–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Sanchez GAM, Reinhardt A, Ramsey S, Wittkowski H, Hashkes PJ, Berkun Y, et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J Clin Invest 2018; 128:3041–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Boyadzhiev M, Marinov L, Boyadzhiev V, Iotova V, Aksentijevich I, Hambleton S. Disease course and treatment effects of a JAK inhibitor in a patient with CANDLE syndrome. Pediatr Rheumatol Online J 2019; 17:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ebstein F, Poli Harlowe MC, Studencka-Turski M, Kruger E. Contribution of the Unfolded Protein Response (UPR) to the Pathogenesis of Proteasome-Associated Autoinflammatory Syndromes (PRAAS). Front Immunol 2019; 10:2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.de Jesus AA, Hou Y, Brooks S, Malle L, Biancotto A, Huang Y, et al. Distinct interferon signatures and cytokine patterns define additional systemic autoinflammatory diseases. J Clin Invest 2020; 130:1669–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Smahi A, Courtois G, Vabres P, Yamaoka S, Heuertz S, Munnich A, et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000; 405:466–72. [DOI] [PubMed] [Google Scholar]
- 169.Zonana J, Elder ME, Schneider LC, Orlow SJ, Moss C, Golabi M, et al. A novel X-linked disorder of immune deficiency and hypohidrotic ectodermal dysplasia is allelic to incontinentia pigmenti and due to mutations in IKK-gamma (NEMO). Am J Hum Genet 2000; 67:1555–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Jain A, Ma CA, Liu S, Brown M, Cohen J, Strober W. Specific missense mutations in NEMO result in hyper-IgM syndrome with hypohydrotic ectodermal dysplasia. Nat Immunol 2001; 2:223–8. [DOI] [PubMed] [Google Scholar]
- 171.Maubach G, Schmadicke AC, Naumann M. NEMO Links Nuclear Factor-kappaB to Human Diseases. Trends Mol Med 2017; 23:1138–55. [DOI] [PubMed] [Google Scholar]
- 172.Fremond ML, Rodero MP, Jeremiah N, Belot A, Jeziorski E, Duffy D, et al. Efficacy of the Janus kinase 1/2 inhibitor ruxolitinib in the treatment of vasculopathy associated with TMEM173-activating mutations in 3 children. J Allergy Clin Immunol 2016; 138:1752–5. [DOI] [PubMed] [Google Scholar]
- 173.Volpi S, Insalaco A, Caorsi R, Santori E, Messia V, Sacco O, et al. Efficacy and Adverse Events During Janus Kinase Inhibitor Treatment of SAVI Syndrome. J Clin Immunol 2019; 39:476–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Raffaele CGL, Messia V, Moneta G, Caiello I, Federici S, Pardeo M, et al. A patient with stimulator of interferon genes-associated vasculopathy with onset in infancy without skin vasculopathy. Rheumatology (Oxford) 2020; 59:905–7. [DOI] [PubMed] [Google Scholar]
- 175.Vanderver A, Adang L, Gavazzi F, McDonald K, Helman G, Frank DB, et al. Janus Kinase Inhibition in the Aicardi-Goutieres Syndrome. N Engl J Med 2020; 383:986–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Gui X, Yang H, Li T, Tan X, Shi P, Li M, et al. Autophagy induction via STING trafficking is a primordial function of the cGAS pathway. Nature 2019; 567:262–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Wu J, Chen YJ, Dobbs N, Sakai T, Liou J, Miner JJ, et al. STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell death. J Exp Med 2019; 216:867–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gaidt MM, Ebert TS, Chauhan D, Ramshorn K, Pinci F, Zuber S, et al. The DNA Inflammasome in Human Myeloid Cells Is Initiated by a STING-Cell Death Program Upstream of NLRP3. Cell 2017; 171:1110–24 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Motwani M, Pawaria S, Bernier J, Moses S, Henry K, Fang T, et al. Hierarchy of clinical manifestations in SAVI N153S and V154M mouse models. Proc Natl Acad Sci U S A 2019; 116:7941–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Luksch H, Stinson WA, Platt DJ, Qian W, Kalugotla G, Miner CA, et al. STING-associated lung disease in mice relies on T cells but not type I interferon. J Allergy Clin Immunol 2019; 144:254–66 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, et al. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol 1998; 161:5016–26. [PubMed] [Google Scholar]
- 182.Thomas CA, Tejwani L, Trujillo CA, Negraes PD, Herai RH, Mesci P, et al. Modeling of TREX1-Dependent Autoimmune Disease using Human Stem Cells Highlights L1 Accumulation as a Source of Neuroinflammation. Cell Stem Cell 2017; 21:319–31 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Kato Y, Park J, Takamatsu H, Konaka H, Aoki W, Aburaya S, et al. Apoptosis-derived membrane vesicles drive the cGAS-STING pathway and enhance type I IFN production in systemic lupus erythematosus. Ann Rheum Dis 2018; 77:1507–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Arbore G, West EE, Spolski R, Robertson AAB, Klos A, Rheinheimer C, et al. T helper 1 immunity requires complement-driven NLRP3 inflammasome activity in CD4(+) T cells. Science 2016; 352:aad1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Alehashemi S, Goldbach-Mansky R. Human Autoinflammatory Diseases Mediated by NLRP3-, Pyrin-, NLRP1-, and NLRC4-Inflammasome Dysregulation Updates on Diagnosis, Treatment, and the Respective Roles of IL-1 and IL-18. Front Immunol 2020; 11:1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Lee AJ, Chen B, Chew MV, Barra NG, Shenouda MM, Nham T, et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J Exp Med 2017; 214:1153–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Fernandes-Alnemri T, Yu JW, Juliana C, Solorzano L, Kang S, Wu J, et al. The AIM2 inflammasome is critical for innate immunity to Francisella tularensis. Nat Immunol 2010; 11:385–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Barnabei L, Lamrini H, Castela M, Jeremiah N, Stolzenberg M-C, Chentout L, et al. Heterozygous RELA mutations cause early-onset systemic lupus erythematosus by hijacking the NF-κB pathway towards transcriptional activation of type-I Interferon genes. bioRxiv 2020:2020.04.27.046102. [Google Scholar]
- 189.Brault M, Olsen TM, Martinez J, Stetson DB, Oberst A. Intracellular Nucleic Acid Sensing Triggers Necroptosis through Synergistic Type I IFN and TNF Signaling. J Immunol 2018; 200:2748–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Poeck H, Bscheider M, Gross O, Finger K, Roth S, Rebsamen M, et al. Recognition of RNA virus by RIG-I results in activation of CARD9 and inflammasome signaling for interleukin 1 beta production. Nat Immunol 2010; 11:63–9. [DOI] [PubMed] [Google Scholar]
- 191.Pescatore A, Esposito E, Draber P, Walczak H, Ursini MV. NEMO regulates a cell death switch in TNF signaling by inhibiting recruitment of RIPK3 to the cell death-inducing complex II. Cell Death Dis 2016; 7:e2346. [DOI] [PMC free article] [PubMed] [Google Scholar]