

Abstract
Background and purpose:
The faster rates of cognitive decline and predominance of atypical forms in early-onset Alzheimer’s disease (EOAD) suggest that neuropsychiatric symptoms could be different in EOAD compared to late-onset AD (LOAD); however, prior studies based on non-biomarker-diagnosed cohorts show discordant results. Our goal was to determine the profile of neuropsychiatric symptoms in EOAD and LOAD, in a cohort with biomarker/postmortem-confirmed diagnoses. Additionally, the contribution of co-pathologies was explored.
Methods:
In all, 219 participants (135 EOAD, 84 LOAD) meeting National Institute on Aging and Alzheimer’s Association criteria for AD (115 amyloid positron emission tomography/cerebrospinal fluid biomarkers, 104 postmortem diagnosis) at the University of California San Francisco were evaluated. The Neuropsychiatric Inventory—Questionnaire (NPI-Q) was assessed at baseline and during follow-up. The NPI-Q mean comparisons and regression models adjusted by cognitive (Mini-Mental State Examination) and functional status (Clinical Dementia Rating Sum of Boxes) were performed to determine the effect of EOAD/LOAD and amnestic/non-amnestic diagnosis on NPI-Q. Regression models assessing the effect of co-pathologies on NPI-Q were performed.
Results:
At baseline, the NPI-Q scores were higher in EOAD compared to LOAD (p < 0.05). Longitudinally, regression models showed a significant effect of diagnosis, where EOAD had higher NPI-Q total, anxiety, motor disturbances and night-time behavior scores (p < 0.05). No differences between amnestics/non-amnestics were found. Argyrophilic grain disease co-pathology predicted a higher severity of NPI-Q scores in LOAD.
Conclusions:
Anxiety, night-time behaviors and motor disturbances are more severe in EOAD than LOAD across the disease course. The differential patterns of neuropsychiatric symptoms observed between EOAD/LOAD could suggest a pattern of selective vulnerability extending to the brain’s subcortical structures. Further, co-pathologies such as argyrophilic grain disease in LOAD may also play a role in increasing neuropsychiatric symptoms.
Keywords: Alzheimer’s disease, behavioral symptoms, locus coeruleus, phenotype, sleep
INTRODUCTION
Neuropsychiatric symptoms such as irritability, agitation and sleep disturbances are common in Alzheimer’s disease (AD) [1–3]. Previous studies suggest that these symptoms increase in severity across the disease course, although neuropsychiatric symptoms are present from the early stages of the disease [4–6]. Behavioral symptoms negatively impact both patients and families, decreasing quality of life, increasing caregiver burden and increasing the likelihood of institutionalization [7–9]. Neuropsychiatric symptoms are still underdiagnosed and undertreated in clinical environments despite being an essential and determining factor for disease progression and patient well-being [2,10].
Alzheimer’s disease is a heterogeneous disorder, presenting with different ages of onset and clinical phenotypes that feature unique patterns of cognitive impairment, cortical atrophy, neuropathological burden and disease progression [11,12]. This heterogeneity may influence neuropsychiatric symptoms. For instance, the faster rates of cognitive decline and the predominance of atypical phenotypes associated with early-onset AD (EOAD) suggest that neuropsychiatric symptoms could be more common in EOAD than in late-onset AD (LOAD). Yet, prior studies investigating the differences between AD variants showed discordant results [13–17]. The inconsistent results could be due to the methodology across the studies, such as the use of non-biomarker-diagnosed cohorts and the inclusion of patients with a wide range of functional or cognitive impairment. Neuropsychiatric symptoms have been mostly evaluated in cross-sectional studies; therefore how they evolve across time in different AD variants is not well understood in well-characterized cohorts [18]. Better characterization of neuropsychiatric profiles across the AD spectrum is crucial to understanding its underlying brain changes, promoting early diagnosis and developing more tailored and effective treatment strategies.
Therefore, our goals were to study the differing profiles of the neuropsychiatric symptoms in biomarker/postmortem-confirmed EOAD and LOAD (i) cross-sectionally, (ii) longitudinally and (iii) differentially between amnestic and non-amnestic phenotypes within EOAD and LOAD. It is hypothesized that EOAD would manifest more severe neuropsychiatric symptoms than LOAD both at baseline and longitudinally over time, and that the group EOAD versus LOAD difference would be driven by the non-amnestic phenotype.
METHODS
Participants
In all, 219 participants were recruited from the Memory and Aging Center, University of California San Francisco (UCSF), from 2008 to 2020. The study was approved by the UCSF Institutional Review Board, and all participants gave their written, informed consent. All participants were assessed with a comprehensive neuropsychological battery. Global cognition and functional status were assessed by the Mini-Mental State Examination (MMSE) and Clinical Dementia Rating (CDR) [19,20]. All patients and their informants were systematically asked about the age of symptom onset on the first visit. The inclusion criteria required the patient to be fluent in English, have adequate visual and auditory acuity, be able to undergo extensive psychometric testing and have an MMSE ≥ 15 or a CDR less than 2 at baseline. The study also required the presence of a willing and able informant who had daily contact with the subject. Of the total, 115 patients met the National Institute on Aging and Alzheimer’s Association (NIA-AA) diagnostic criteria for mild cognitive impairment (MCI) or dementia due to AD [21,22] and 104 had a confirmed postmortem diagnosis [23]. Based on the age of onset of the symptoms, the participants were classified as follows:
EOAD group (n = 135; age of onset of symptoms ≤65 years): patients with positive amyloid positron emission tomography (amyloid-PET) (n = 60) or typical AD cerebrospinal fluid (CSF) biomarker profile (n = 9) meeting the NIA-AA criteria for MCI due to AD (n = 48) or AD dementia (n = 21) [24,25], and 66 with autopsy-proven diagnosis.
LOAD group (n = 84; age of onset of symptoms >65 years): patients with typical AD positive amyloid-PET (n = 43) or typical AD CSF biomarker profile (n = 3) meeting the NIA-AA criteria for MCI due to AD (n = 34) or AD dementia (n = 12), and 38 with an autopsy-proven diagnosis.
Furthermore, based on the clinical presentation and the neuropsychological profile, participants were classified as an amnestic or non-amnestic variant, considering the predominant impairment of memory or non-memory cognitive domains, respectively. Non-memory phenotypes included visuospatial (posterior cortical atrophy), language (logopenic variant of primary progressive aphasia), motor (cortico-basal syndrome), executive or cognitive behavioral domains (dysexecutive-behavioral variant) [26].
Neuropathological evaluation
Neuropathological diagnoses (n = 104) were based on an extensive dementia-oriented postmortem assessment at the UCSF/Neurodegenerative Disease Brain Bank. The neuropathological evaluation was performed as previously described by Spina et al., 2021 [27]. Twenty-four tissue blocks covering dementia-related regions of interest were dissected from the fixed slabs, and hematoxylin and eosin and immunohistochemical stains were applied following standard diagnostic procedures developed for patients with dementia. Neuropathological diagnosis followed currently accepted guidelines. The overall severity of AD neuropathological change was assigned using the NIA-Reagan criteria and NIA-AA criteria for AD [23]. The presence of Lewy body disease (LBD), TDP-43, argyrophilic grain disease (AGD),cerebral amyloid angiopathy, hippocampal sclerosis and vascular brain injury co-pathologies was assessed according to published criteria as described by Spina et al. [27].
Neuropsychiatric inventory
All informants completed the self-administered Neuropsychiatric Inventory—Questionnaire (NPI-Q) about the patients they care for [28]. The NPI-Q rates the existence and severity of neuropsychiatric alterations within the last month including the following 12 domains: delusions, hallucinations, agitation/aggression, depression/dysphoria, anxiety, elation/euphoria, apathy/indifference, disinhibition, irritability/lability, motor disturbance, night-time behaviors (awakenings, early risings, excessive naps) and appetite/eating. The severity of the symptoms in each domain are evaluated on a three-point scale. The total NPI-Q severity scores reflect the sum of individual domain scores.
Neuropsychiatric treatment
The prescription of neuropsychiatric treatments (present/absent) was collected for the baseline and follow-up visits considering the prescription of at least one of the following categories: selective serotonin reuptake inhibitors, serotonin antagonist and reuptake inhibitors (trazodone), noradrenergic and specific serotonergic antidepressants (mirtazapine), benzodiazepines and atypical antipsychotic (quetiapine).
Follow-up
A subsample of 114 patients underwent longitudinal NPI-Q evaluations during annual follow-up visits (EOAD, n = 64; LOAD, n = 50). The number of longitudinal assessments varied amongst patients from one to four.
Statistical analyses
Statistical analyses were conducted using Stata/IC 16.1. Differences between EOAD and LOAD in demographics were analyzed by Student’s t test for continuous data and the χ2 test for categorical data. Differences in NPI-Q outcomes (NPI-Q total score and NPI-Q domains) at baseline were analyzed by Student’s t test. Linear regression models were also performed at baseline controlling for MMSE and CDR Sum of Boxes (SoB) to avoid potential biases due to cognitive or functional status. Linear mixed effects models were used to analyze longitudinal changes in NPI-Q scores from baseline, using diagnosis (EOAD vs. LOAD) as the main predictor and adjusting by MMSE and CDR SoB score and time to follow-up. Time to follow-up was defined as the time elapsed from the first NPI-Q evaluation (baseline) to each of the consecutive assessment time-points. The interaction between EOAD/LOAD diagnosis and time to follow-up was also included in the model to evaluate slope differences. Additionally sub-analyses were performed evaluating the differences in NPI-Q scores between amnestic and non-amnestic phenotypes in EOAD and LOAD groups, separately. Moreover, as an exploratory analysis, clinicopathological correlations between NPI-Q total score and different co-pathologies within the EOAD and LOAD autopsy-proven cohorts were analyzed. Linear mixed effects models in NPI-Q total score were performed using the presence of co-pathology as a main predictor and adjusting by MMSE and CDR SoB score and time to follow-up. The NPI-Q domain scores were further analyzed only for those co-pathologies showing a significant effect on the NPI-Q total score.
RESULTS
Demographics and clinical data in EOAD and LOAD
Demographic and clinical data for EOAD and LOAD groups are provided in Table 1. The age of onset was significantly different between EOAD and LOAD, as expected. Conversely, no differences were found between groups regarding sex and time from symptom onset to diagnosis. EOAD showed lower MMSE and higher CDR SoB at first visit. All participants had high school/secondary level education (means 16.1 EOAD vs. 17.1 LOAD years).
TABLE 1.
Demographics and clinical data at baseline in EOAD and LOAD considering amnestic and non-amnestic subgroups
| EOAD |
LOAD |
Significance |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| Total sample n = 135 |
Amnestic n = 93 |
Non-amnestic n = 42 |
Total sample n = 84 |
Amnestic n = 71 |
Non-amnestic n = 13 |
EOAD versus LOAD | EOAD amnestic versus non-amnestic | LOAD amnestic versus non-amnestic | |
| Age (years) | 55.1 ± 5.7 | 55.3 ± 5.9 | 54.9 ± 5.3 | 73.4 ± 5.7 | 73.6 ± 5.4 | 73.4 ± 4.9 | p = 0.000 | ns | ns |
| Gender (men) | 54% | 56% | 52% | 63% | 63% | 62% | ns | ns | ns |
| Years of education | 16.1 ± 2.8 | 16.1 ± 2.5 | 15.9 ± 2.3 | 17.3 ± 2.1 | 17.1 ± 2.7 | 16.5 ± 2 | p = 0.005 | ns | ns |
| Disease duration | 4.5 ± 2.5 | 4.4 ± 2.4 | 4.6 ± 2.8 | 3.8 ± 3.1 | 4.1 ± 3.2 | 1.9 ± 0.5 | ns | ns | p = 0.027 |
| CDR total | 0.9 ± 0.5 | 0.8 ± 0.7 | 0.9 ± 0.6 | 0.8 ± 0.4 | 0.8 ± 0.7 | 0.6 ± 0.5 | ns | ns | ns |
| Mild cognitive impairment (CDR 0.5) | 50% | 48% | 55% | 57% | 53% | 77% | |||
| Mild dementia (CDR 1) | 39% | 43% | 29% | 37% | 40% | 23% | |||
| Moderate dementia (CDR 2) | 11% | 9% | 16% | 6% | 7% | 0% | |||
| CDR SoB | 4.6 ± 2.9 | 4.5 ± 2.6 | 4.9 ± 3.4 | 3.9 ± 2.8 | 4.1 ± 3.3 | 3 ± 1.9 | p = 0.035 | ns | ns |
| MMSE | 22.1 ± 7 | 22.6 ± 6 | 19.9 ± 8 | 23.9 ± 5 | 23.9 ± 4 | 23.7 ± 5 | p = 0.001 | ns | ns |
| Anticholinesterase inhibitors | 72% | 71% | 75% | 64% | 63% | 69% | ns | ns | ns |
| Neuropsychiatric treatment | 45% | 46% | 44% | 29% | 34% | 8% | p = 0.011 | ns | p = 0.029 |
| SSRIs | 90% | 38% | 29% | 92% | 27% | 8% | |||
| SARI (trazodone) | 9% | 3% | 5% | 8% | 3% | 0% | |||
| NaSSA (mirtazapine) | 5% | 5% | 0% | 0% | 0% | 0% | |||
| Benzodiazepine | 13% | 5% | 8% | 12% | 4% | 0% | |||
| Atypical antipsychotic (quetiapine) | 4% | 2% | 0% | 8% | 3% | 0% | |||
Note: Data are presented as mean ± standard deviation.
Abbreviations: CDR, Clinical Dementia Rating scale; CDR SoB, CDR Sum of Boxes; EOAD, early-onset Alzheimer’s disease; LOAD, late-onset Alzheimer’s disease; MMSE, Mini-Mental State Examination; NaSSA, noradrenergic and specific serotonergic antidepressant; SARI, serotonin receptor antagonists and reuptake inhibitors; SSRIs, selective serotonin reuptake inhibitors.
Regarding pharmacological treatments, no differences in the prescription of anticholinesterase inhibitors were found between EOAD and LOAD, either at baseline or longitudinally. Conversely, the prescription of neuropsychiatric medications was higher in EOAD both at baseline (Table 1) and at follow-up (p < 0.01).
Demographics and clinical data in amnestic and non-amnestic groups
Detailed demographic and clinical data for amnestic and non-amnestic phenotypes within the EOAD and LOAD groups are provided in Table 1. No differences were found in age of onset, sex, cognitive (MMSE) or functional status (CDR SoB). Within the LOAD group, amnestic patients showed greater disease duration (defined as time from the first symptom to the diagnosis) and higher prescription of neuropsychiatric treatments. Conversely, these were similar between amnestic and non-amnestic patients in the EOAD group.
Baseline neuropsychiatric symptoms between EOAD and LOAD
Mean differences between EOAD and LOAD diagnostic groups
Differences of NPI-Q scores between diagnostic groups are shown in Figure 1. Mean comparisons (t test) showed that NPI-Q total scores were higher in EOAD compared to LOAD. Regarding NPI-Q specific domains, EOAD showed higher scores in most domains (p < 0.05): agitation, anxiety, elation, apathy, disinhibition, irritability, motor disturbances, night-time behaviors and appetite.
FIGURE 1.
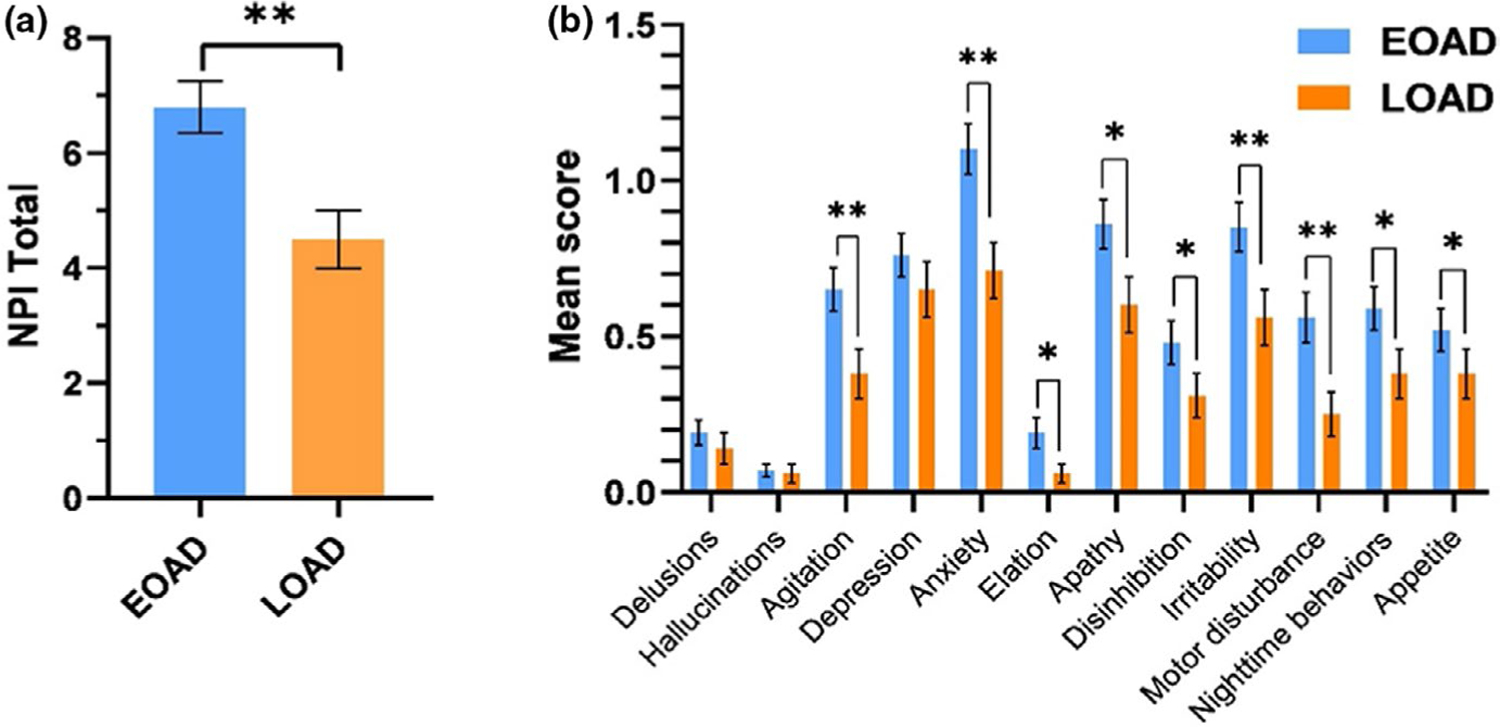
Baseline NPI-Q scores in EOAD and LOAD groups. (a) Mean NPI-Q total scores in EOAD and LOAD. (b) Mean scores across the different NPI-Q domains in EOAD and LOAD. Significant difference *p < 0.05, **p < 0.01
Linear regression models on NPI-Q baseline scores between EOAD and LOAD diagnostic groups
Since EOAD patients had lower cognitive and functional performances, a linear regression model was performed controlling for MMSE and CDR SoB at baseline. The results showed the significant effect of EOAD diagnosis on higher NPI total (coef. 1.59, p < 0.01), anxiety scores (coef. 0.18, p < 0.05), motor disturbances (coef. 0.15, p < 0.05) and night-time events (coef. 0.19, p < 0.05). When controlling for prescription of neuropsychiatric treatment, the results did not change.
Longitudinal neuropsychiatric symptoms between EOAD and LOAD
EOAD versus LOAD as a predictor for longitudinal trajectories of neuropsychiatric symptoms
Detailed longitudinal trajectories of neuropsychiatric symptoms in EOAD and LOAD diagnostic groups are shown in Figure 2. Mixed effects models adjusted for MMSE, CDR SoB score and time to follow-up highlighted EOAD/LOAD diagnosis as a predictor for NPI-Q total score (coef. 1.52, p < 0.01), night-time behaviors (coef. 0.26, p < 0.01), anxiety (coef. 0.32, p < 0.01) and motor disturbances (coef. 0.27, p < 0.01) with higher scores in each for EOAD compared to LOAD. EOAD showed higher NPI-Q scores than LOAD along the disease course, although the slopes (rates of change) did not differ between groups.
FIGURE 2.
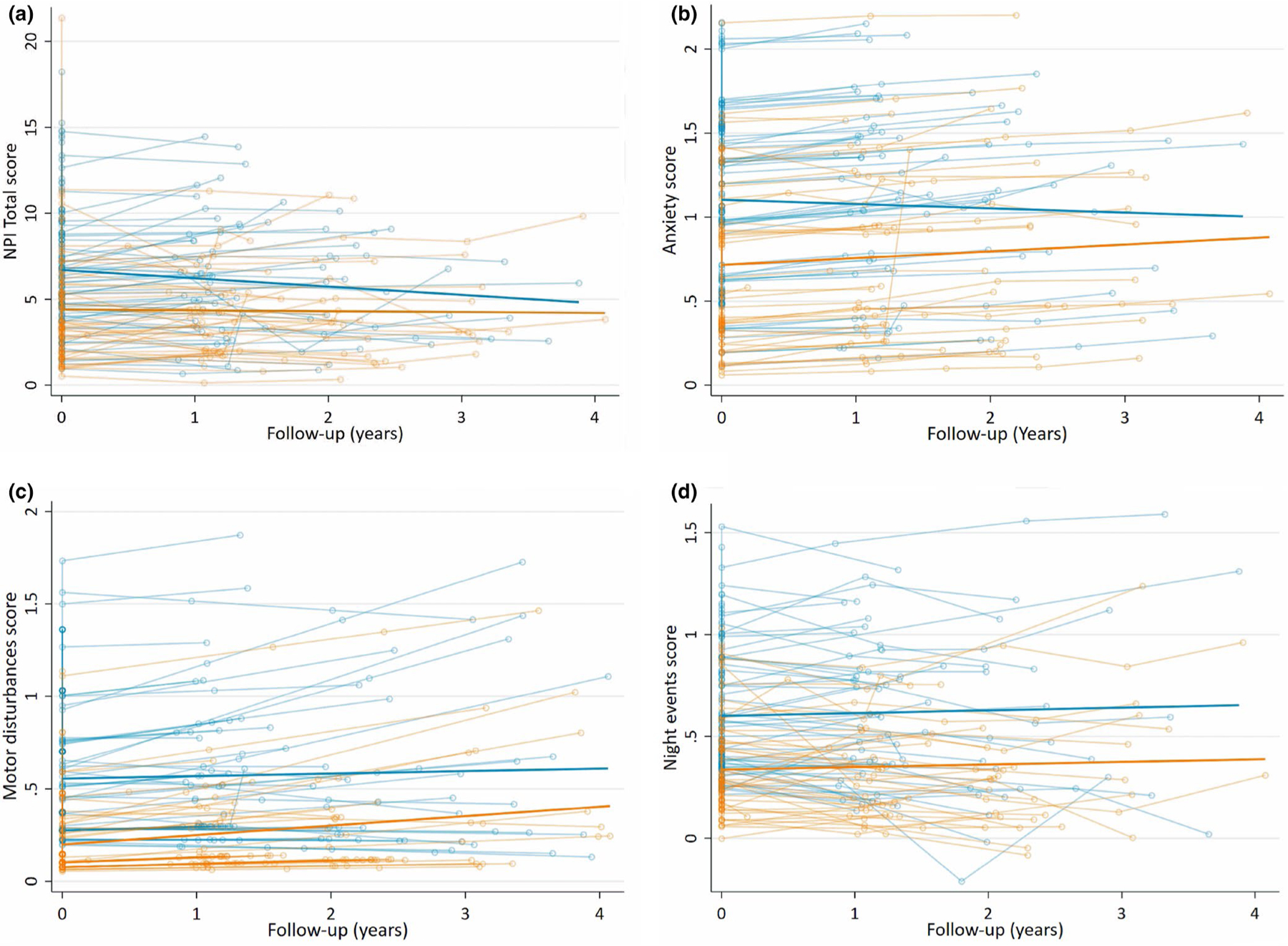
Longitudinal trajectories of NPI-Q scores in EOAD and LOAD. The individual and mean differential group trajectories between EOAD and LOAD (p < 0.05) adjusted by MMSE and CDR SoB in (a) NPI-Q total score, (b) anxiety score, (c) motor disturbances score and (d) night-time behaviors score
In addition, the effect of EOAD and LOAD diagnosis was analyzed separately within the amnestic and non-amnestic phenotypes. The results mostly resembled the whole group EOAD and LOAD comparison: amnestic EOAD (n = 93) predicted higher NPI total (p < 0.01), night events, motor disturbances, anxiety and apathy (p < 0.05) than amnestic LOAD (n = 71). No statistically significant effect of non-amnestic EOAD (n = 42) over non-amnestic LOAD (n = 13) was found because of the small sample size.
Sub-analyses comparing amnestic and non-amnestic phenotypes in EOAD
Results from the amnestic–non-amnestic sub-analyses in EOAD can be found in Figure 3a,b. Amnestic (n = 93) and non-amnestic (n = 42) phenotypes showed similar NPI-Q total scores at baseline. Amnestic/non-amnestic phenotypes did not predict NPI-Q total scores or any of the subdomains either at baseline or longitudinally.
FIGURE 3.
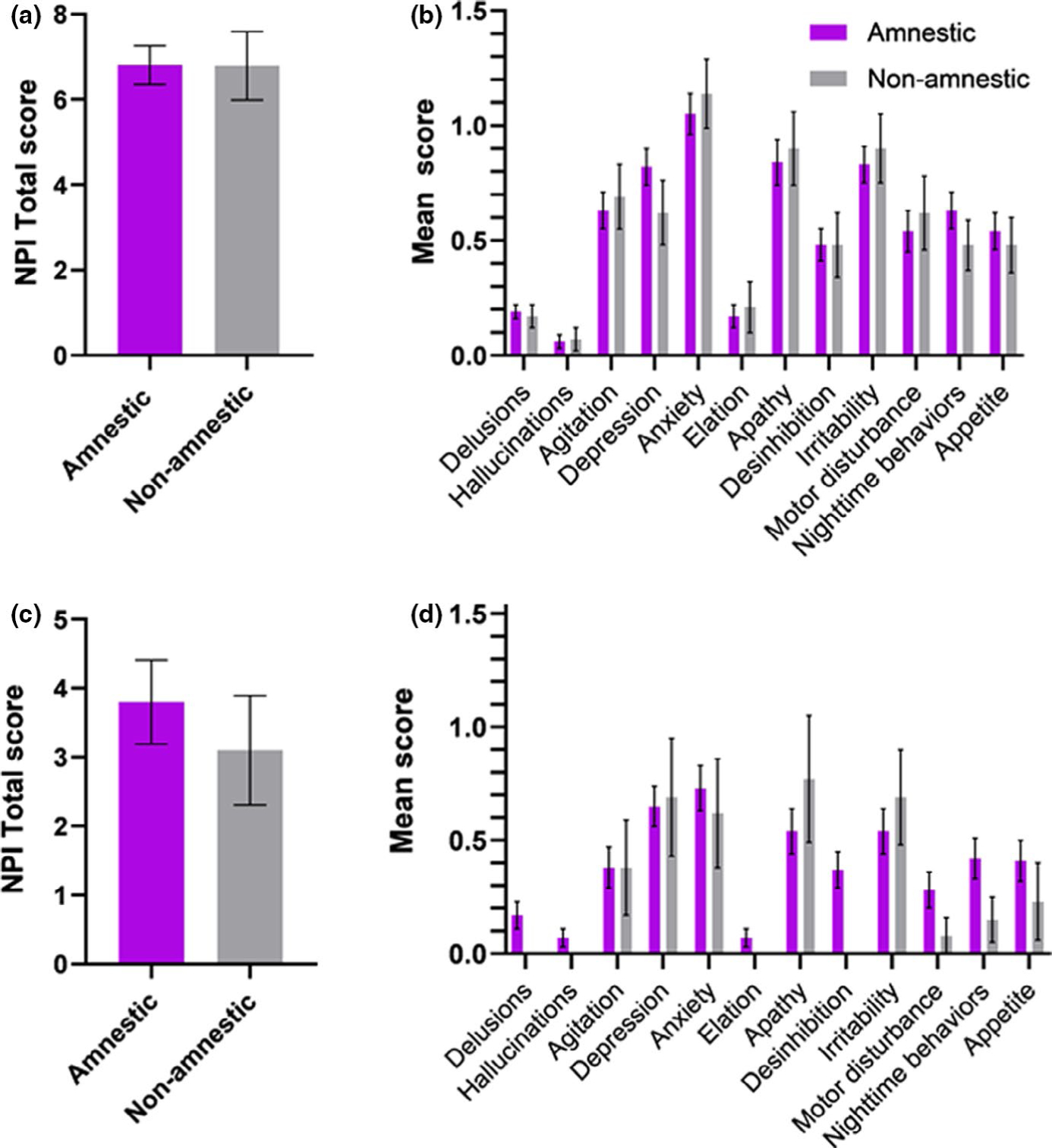
Baseline NPI-Q scores in amnestic and non-amnestic groups in EOAD and LOAD. (a) Mean NPI-Q total scores in amnestic and non-amnestic phenotypes in EOAD patients. (b) Means of the different NPI-Q domains in amnestic and non-amnestic phenotypes in EOAD. (c) Mean NPI-Q total scores in amnestic and non-amnestic phenotypes in LOAD patients. (d) Means of the different NPI-Q domains in amnestic and non-amnestic phenotypes in LOAD. No statistically significant difference was found between groups
Sub-analyses comparing amnestic and non-amnestic phenotypes in LOAD
Amnestic (n = 71) and non-amnestic (n = 13) phenotypes showed similar NPI-Q total and domain scores at baseline (Figure 3c,d). Amnestic/non-amnestic phenotypes did not predict differential NPI-Q total or domain scores longitudinally.
Clinical-pathological correlations
The prevalence of co-pathologies within EOAD and LOAD cohorts is shown in Table 2 and has been previously described as part of a larger cohort of patients from the same brain bank [27]. Mixed effects models showed the presence of AGD as the only co-pathology, being a significant predictor for higher NPI-Q total scores in LOAD (coef. 0.41, p < 0.05). Further analyses evaluating its effect on NPI domain scores showed AGD pathology as a significant predictor for higher delusions (coef. 0.41, p < 0.05), hallucinations (coef. 0.34, p < 0.05) and appetite (coef. 0.59, p < 0.05). Although not significant, there were trends for higher agitation (coef. 0.37, p = 0.052) and depression scores (coef. 0.47, p = 0.052).With regard to EOAD, neither AGD nor other co-pathologies showed a significant effect on NPI total score.
TABLE 2.
Prevalence of co-pathologies within the autopsy-proven EOAD and LOAD cohorts
| EOAD (n = 66) | LOAD (n = 38) | |
|---|---|---|
| LBD (%) | 47 | 39 |
| CAA (%) | 91 | 84 |
| TDP (%) | 5 | 34 |
| AGD (%) | 44 | 42 |
| HS (%) | 2 | 26 |
| VBI (%) | 32 | 63 |
| ARTAG (%) | 27 | 45 |
| CTE (%) | 0 | 3 |
| FTLD tau (%) | 0 | 0 |
Abbreviations: AGD, argyrophilic grain disease; ARTAG, CAA, cerebral amyloid angiopathy; CTE, FTLD, HS, hippocampal sclerosis; LBD, Lewy body disease; TDP, TDP-43; VBI, Vascular Brain Injury.
DISCUSSION
A cross-sectional and longitudinal study was performed evaluating the severity of neuropsychiatric symptoms in EOAD and LOAD in a biomarker-diagnosed cohort. Our findings highlight that the neuropsychiatric symptoms are more severe in EOAD than LOAD across the disease course, independent of the disease stage (MMSE, CDR SoB) and despite the prescription of neuropsychiatric treatments. These differences are strongly driven by higher levels of anxiety, night-time behaviors and motor disturbances. Conversely, the severity of neuropsychiatric symptoms was similar between amnestic and non-amnestic phenotypes within the EOAD and LOAD cohorts.
Our findings suggest that, at baseline, EOAD patients have more severe neuropsychiatric symptoms compared to LOAD. Although this result is in alignment with the general perception within AD clinics, the difference observed between EOAD and LOAD in the overall severity of the neuropsychiatric symptoms during the early clinical stages of the disease was unclear in previous literature. Prior cross-sectional studies investigating the potential differences in neuropsychiatric symptoms using the NPI-Q total score showed contradictory outcomes, indicating that the severity of neuropsychiatric alterations might be similar or even higher in individuals presenting with LOAD [13–16]. It is believed that these discordant results are best explained by several methodological differences such as the use of non-biomarker-based AD cohorts or the inclusion of a varied range of disease stages.
For example, a 2-year longitudinal study comparing neuropsychiatric symptoms in EOAD and LOAD (non-biomarker-based diagnoses) found that the incidence and prevalence of neuropsychiatric symptoms were lower in EOAD than in LOAD, suggesting that EOAD patients were more resilient to behavioral symptoms [29]. Conversely, our longitudinal results show that there are worse neuropsychiatric symptoms in EOAD than in LOAD along the entire disease course and independent of disease stage. Furthermore, the severity of these symptoms is higher in EOAD, even though these patients are more frequently prescribed with neuropsychiatric medications. EOAD tends to have a more aggressive disease course at the clinical and neuropathological levels than LOAD, featuring faster onset of cognitive decline and higher cortical burden of amyloid/tau pathology [12,30–34]. Our study supports the idea that EOAD has a more severe progression at the behavioral level as well.
More specifically, anxiety, motor disturbances and night-time behaviors are factors that drive the higher severity of neuropsychiatric symptoms in EOAD. Interestingly, this is in concordance with a prior cross-sectional study which, despite not detecting differences in the total NPI-Q score, showed higher anxiety, irritability and sleep subscale scores in EOAD than in LOAD [14]. All together this indicates that EOAD and LOAD have different profiles of neuropsychiatric symptoms.
Growing evidence suggests that the development of behavioral symptoms such as mood changes or sleep disorders in AD is related to the degeneration of the neuromodulatory subcortical systems in the brain, involving the locus coeruleus in the pons (noradrenergic system), dorsal raphe nucleus (serotoninergic system), substantia nigra (dopaminergic system) in the midbrain, and nucleus basalis of Meynert in the basal forebrain (cholinergic system) [35–37]. Through their ascending projections to the cortex, these nuclei regulate mood and behavior by synthesizing aminergic and cholinergic neurotransmitters. The imbalance of these neurotransmitters in AD could contribute not only to the cognitive impairment but also to its neuropsychiatric symptoms in the early stages of the disease [6,35]. The existence of different patterns of neuropsychiatric symptoms (i.e., sleep, anxiety, irritability, abnormal motor behaviors) in AD variants suggests that these subcortical regions might be affected differently within the AD spectrum, thus indicating the presence of a pattern of selective vulnerability extending to subcortical structures.
For instance, sleep disorders, that are common in AD, might vary between AD variants. Sleep alterations appear early in the disease course of AD, even before cognitive impairment, due to the early degeneration of the arousal system [4,6,38,39]. The AD-tau degeneration falls mainly on the locus coeruleus, causing noradrenergic dysfunction as the main determinant for dysregulation of sleep–wake patterns in AD [35,38,40]. Our findings indicate that sleep alterations are more severe in EOAD than LOAD, suggesting that the arousal system may be more compromised in early-onset presentations. Interestingly, prior neuropathological studies support this hypothesis, by demonstrating that the magnitude of degeneration within the locus coeruleus is higher in EOAD compared to LOAD [41,42]. Altogether, this reinforces the existence of different sleep profiles within AD variants, probably due to different degenerative patterns within the arousal system; it is believed that this hypothesis warrants further research. The clinical-pathological correlates already established between sleep alterations and brainstem nuclei degeneration in AD (i.e., locus coeruleus), together with the availability of objective measurements for this neuropsychiatric symptom (i.e., polysomnography, actigraphy), make sleep alterations a perfect model to investigate mechanisms of subcortical selective vulnerability in AD [43]. Another possible explanation is that with EOAD psychosocial factors such as early retirement, loss of financial independence and familial responsibilities play a role in these symptoms [44–46].
Anxiety and motor disturbances are prevalent in AD patients and prior literature suggests that they are more severe in EOAD [14,47,48]. The anxiety/motor disturbance effect could be explained in part by possible EOAD-related psychological aspects associated with the impact on EOAD starting during earlier life stages, such as early retirement, offspring responsibilities, economic consequences, social implications, stigma and emotional suffering [49]. As with sleep, the anxiety level differences between AD variants could be related to the disease burden on the subcortical structures regulating mood and anxiety (i.e., dorsal raphe). The dorsal raphe nucleus is severely affected in the disease course, similarly to the locus coeruleus, showing the earliest and most severe AD-tau lesions in AD, the neurofibrillary tangles [50]. Degeneration of the dorsal raphe leads to the dysregulation of the serotonergic transmitter system, producing anxiety and mood changes early in the disease course.
Conversely, it was found that amnestic and non-amnestic phenotypes manifest similarly with regard to neuropsychiatric symptoms. Some findings suggest that non-amnestic phenotypes are more aware of their cognitive deficits, which could drive higher levels of anxiety or depression amongst non-amnestic AD patients compared to amnestic ones. Our results challenge this idea by showing similar severity of neuropsychiatric symptoms between amnestic and non-amnestic phenotypes, which is in line with a prior study suggesting that patient’s awareness of cognitive deficits is not higher in non-amnestic AD [51]. Furthermore, the use of the term dysexecutive-behavioral variant has been questioned by recent research which considers the dysexecutive and behavioral syndromes as distinct [52]. These recent findings are in alignment with the results that non-amnestic (including dysexecutive) and amnestic do not differ in terms of neuropsychiatric symptoms.
Overall, our results could suggest that the selective vulnerability of subcortical structures in AD may be more related to the age of onset (EOAD vs. LOAD) than to the AD clinical phenotype (amnestic vs. non-amnestic). However, our findings could be driven by the relatively small sample sizes of the variant subgroups and warrant future study.
In addition, our results highlight the potential role that the co-pathologies have in the development of neuropsychiatric symptoms amongst AD patients. The results show AGD as a predictor for several NPI-Q scores in LOAD (i.e., NPI-Q total, delusions, hallucinations and appetite) which suggests that AGD may be a relevant driver of the severity of many behavioral disturbances in LOAD. These results are in line with prior studies that demonstrate that AGD is a highly frequent sporadic tauopathy, manifesting with slowly progressive amnestic mild cognitive impairment and accompanied by a high prevalence of neuropsychiatric symptoms [50,53,54]. Despite the high overlap between AD and AGD in neuropathological studies, they are defined as two clearly different entities with differing patterns of spread. For instance, within the hypothalamus, AGD pathology shows an early and prominent involvement of the lateral tuberal nuclei and relative resistance of the tuberomammillary nuclei, which is opposite to AD pathology [54]. Interestingly, the lateral tuberal nuclei are involved in feeding regulation, which could explain appetite change in the context of LOAD associated with AGD pathology [53].
On the other hand, recent evidence suggests that EOAD is associated with more non-AD neuropathological changes than previously thought [27]. LBD (synucleinopathy), particularly in the amygdala, is the most common co-pathology in EOAD, occurring even more frequently than in LOAD. It is believed that LBD co-pathology might have an important role in the expression of AD clinical phenotypes in EOAD, particularly involving neuropsychiatric symptoms. Along this line, a recent study demonstrated the relevance of neuropathological changes in the presence or even the subtype of psychotic symptoms, since LBD/AD pathology is more likely to have hallucinations such as misperception and hallucinations of people/animals/objects [55]. Conversely, the results from our cohort did not support this contribution of LBD as co-pathology to the severity of behavioral symptoms in EOAD, which may be due to the limited sample size of the autopsy-proven cohort. Therefore, further studies directly evaluating how LBD pathology shapes the neuropsychiatric symptoms in a treatment-free EOAD cohort are needed.
It is believed that better characterization of the neuropsychiatric symptoms featuring AD variants would help clinicians identify those patients who are especially at risk for developing certain behavioral disorders. It would also promote research to further understand their underpinning brain changes on the subcortical structures. Given that the neuropsychiatric symptoms on AD have such a negative impact on patients’ and families’ quality of life, and that there is a need for effective treatments, a better understanding of the underlying mechanisms within the AD spectrum is an opportunity to develop tailored treatment strategies [43].
This study’s main strength is that all of the patients included had an autopsy-proven or biomarker-based diagnosis with at least one biological biomarker (i.e., PET amyloid, CSF biomarkers), improving diagnostic clarity for understanding the specificity of the NPI-Q findings for EOAD and LOAD. It is also the first study to include not only the NPI-Q evaluation at baseline but also up to 4 years of follow-up. The main limitation of this study is that the neuropsychiatric evaluation was performed by an informant-based subjective assessment which could hamper the interpretation of the results. However, it has been shown to be sensitive and is the primary tool used in clinical settings [28]. Although the cohort is well characterized, the sample size may not be enough to detect more subtle differences between the amnestic and non-amnestic subgroups or between the comorbidity pathological subtypes beyond AGD in LOAD. Moreover, the results of clinical-pathological correlations may not reflect real-time pathological changes because of the postmortem nature of the evaluation itself. An additional limitation is the difference in MMSE and CDR SoB between EOAD and LOAD. Whilst analyses were adjusted for these two scores, there may have been some masking or distortion in our findings as a result. Therefore, further research across a larger spectrum of MMSE and CDR SoB represented in each group across time is warranted. A number of quantitative analyses were performed; however, our hypotheses were specific and the directions and magnitudes of the outcomes fit a biologically coherent pattern; thus formal multiple comparisons and adjustments were not required [56].
CONCLUSION
Neuropsychiatric symptoms present differently across AD variants, suggesting a selective vulnerability in subcortical structures. Further understanding of the specific subcortical changes underlying these clinical features between AD variants is needed in order to develop more tailored diagnostic and treatment strategies across the AD spectrum.
ACKNOWLEDGEMENTS
The authors thank the patients and their relatives for their participation in this research. They would also like to thank all of the research coordinators and nurses who helped to collect these data over the years, in particular Robin Ketelle and Ken Edwards.
Funding information
This work was supported by the Global Brain Health Institute, Tau Consortium/Rainwater Charity Foundation, National Institute on Aging grants NIA R01 AG060477, NIA R01 AG064314, K24AG053435, K23-AG031861, K08 AG052648, R01-AG027859, P01-AG1972403 and P50-AG023501, State of California Department of Health Services Alzheimer’s Disease Research Center of California grant 04–33516
Footnotes
CONFLICT OF INTEREST
Authors declare no conflict of interest.
DATA AVALIABILITY STATEMENT
The data that support the findings of this study are available on request from the corresponding author. The data are not publicly available due to privacy or ethical restrictions.
REFERENCES
- 1.Lanctôt KL, Amatniek J, Ancoli-Israel S, et al. Neuropsychiatric signs and symptoms of Alzheimer’s disease: new treatment paradigms. Alzheimers Dement N Y N 2017;3(3):440–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Clement A, Wiborg O, Asuni AA. Steps towards developing effective treatments for neuropsychiatric disturbances in Alzheimer’s disease: insights from preclinical models, clinical data, and future directions. Front Aging Neurosci 2020;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gatchel JR, Donovan NJ, Locascio JJ, et al. Depressive symptoms and tau accumulation in the inferior temporal lobe and entorhinal cortex in cognitively normal older adults: a pilot study. J Alzheimers Dis 2017;59(3):975–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liguori C, Placidi F, Izzi F, Spanetta M, Mercuri NB, Di Pucchio A. Sleep dysregulation, memory impairment, and CSF biomarkers during different levels of neurocognitive functioning in Alzheimer’s disease course. Alzheimers Res Ther 2020;12(1):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tanaka H, Hashimoto M, Fukuhara R, et al. Relationship between dementia severity and behavioural and psychological symptoms in early-onset Alzheimer’s disease. Psychogeriatrics 2015;15(4):242–247. [DOI] [PubMed] [Google Scholar]
- 6.Ehrenberg AJ, Suemoto CK, França Resende EDP, et al. Neuropathologic correlates of psychiatric symptoms in Alzheimer’s disease. J Alzheimers Dis 2018;66(1):115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Okuda S, Tetsuka J, Takahashi K, Toda Y, Kubo T, Tokita S. Association between sleep disturbance in Alzheimer’s disease patients and burden on and health status of their caregivers. J Neurol 2019;266(6):1490–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lyketsos CG, Carrillo MC, Ryan JM, et al. Neuropsychiatric symptoms in Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2011;7(5):532–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Germain S, Adam S, Olivier C, et al. Does cognitive impairment influence burden in caregivers of patients with Alzheimer’s disease? J Alzheimers Dis JAD 2009;17(1):105–114. [DOI] [PubMed] [Google Scholar]
- 10.McCleery J, Sharpley AL. Pharmacotherapies for sleep disturbances in dementia. Cochrane Database Syst Rev 2020;11:CD009178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murray ME, Graff-Radford NR, Ross OA, Petersen RC, Duara R, Dickson DW. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: a retrospective study. Lancet Neurol 2011;10(9):785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petersen C, Nolan AL, de Paula França Resende E, et al. Alzheimer’s disease clinical variants show distinct regional patterns of neurofibrillary tangle accumulation. Acta Neuropathol (Berl) 2019;138(4):597–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ferreira MDC, Abreu MJ, Machado C, Santos B, Machado Á, Costa AS. Neuropsychiatric profile in early versus late onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen 2018;33(2):93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Baillon S, Gasper A, Wilson-Morkeh F, Pritchard M, Jesu A, Velayudhan L. Prevalence and severity of neuropsychiatric symptoms in early-versus late-onset Alzheimer’s disease. Am J Alzheimers Dis Other Demen 2019;34(7–8):433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mushtaq R, Pinto C, Tarfarosh SFA, et al. A comparison of the behavioral and psychological symptoms of dementia (BPSD) in early-onset and late-onset Alzheimer’s disease—a study from south east Asia (Kashmir, India). Cureus 2016;8(5):e625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park HK, Choi SH, Park SA, et al. Cognitive profiles and neuropsychiatric symptoms in Korean early-onset Alzheimer’s disease patients: a CREDOS study. J Alzheimers Dis 2015;44(2):661–673. [DOI] [PubMed] [Google Scholar]
- 17.Hori K, Oda T, Asaoka T, et al. First episodes of behavioral symptoms in Alzheimer’s disease patients at age 90 and over, and early-onset Alzheimer’s disease: comparison with senile dementia of Alzheimer’s type. Psychiatry Clin Neurosci 2005;59(6):730–735. [DOI] [PubMed] [Google Scholar]
- 18.Singleton EH, Rijkers C, Groot C, et al. The evolution of neuropsychiatric symptoms in atypical variants of Alzheimer’s disease. Alzheimers Dement 2020;16(S6):1–3. 10.1002/alz.045236 [DOI] [Google Scholar]
- 19.Folstein MF, Folstein SE, McHugh PR. “Mini-mental state”. A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12(3):189–198. [DOI] [PubMed] [Google Scholar]
- 20.Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology 1993;43(11):2412–2414. [DOI] [PubMed] [Google Scholar]
- 21.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2011;7(3):263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hyman BT, Phelps CH, Beach TG, et al. National Institute on Aging–Alzheimer’s Association guidelines for the neuropathologic assessment of Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2012;8(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7(3):270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging—Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011;7:263–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ossenkoppele R, Pijnenburg YAL, Perry DC, et al. The behavioural/dysexecutive variant of Alzheimer’s disease: clinical, neuroimaging and pathological features. Brain 2015;138(9):2732–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Spina S, La Joie R, Petersen C, et al. Comorbid neuropathological diagnoses in early versus late-onset Alzheimer’s disease. Brain J Neurol 2021;144(7):2186–2198. 10.1093/brain/awab099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kaufer DI, Cummings JL, Ketchel P, et al. Validation of the NPI-Q, a brief clinical form of the neuropsychiatric inventory. J Neuropsychiatry Clin Neurosci 2000;12(2):233–239. [DOI] [PubMed] [Google Scholar]
- 29.van Vliet D, de Vugt ME, Aalten P, et al. Prevalence of neuropsychiatric symptoms in young-onset compared to late-onset Alzheimer’s disease—part 1: findings of the two-year longitudinal NeedYD-study. Dement Geriatr Cogn Disord 2012;34(5–6):319–327. [DOI] [PubMed] [Google Scholar]
- 30.Wattmo C, Wallin ÅK. Early-versus late-onset Alzheimer’s disease in clinical practice: cognitive and global outcomes over 3 years. Alzheimers Res Ther 2017;9(1):70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smits LL, Pijnenburg YAL, van der Vlies AE, et al. Early onset APOE E4-negative Alzheimer’s disease patients show faster cognitive decline on non-memory domains. Eur Neuropsychopharmacol 2015;25(7):1010–1017. [DOI] [PubMed] [Google Scholar]
- 32.Marshall GA, Fairbanks LA, Tekin S, Vinters HV, Cummings JL. Early-onset Alzheimer’s disease is associated with greater pathologic burden. J Geriatr Psychiatry Neurol 2007;20(1):29–33. [DOI] [PubMed] [Google Scholar]
- 33.Middleton LE, Grinberg LT, Miller B, Kawas C, Yaffe K. Neuropathologic features associated with Alzheimer disease diagnosis: age matters. Neurology 2011;77(19):1737–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.La Joie R, Visani AV, Baker SL, et al. Prospective longitudinal atrophy in Alzheimer’s disease correlates with the intensity and topography of baseline tau-PET. Sci Transl Med 2020;12(524):eaau5732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Theofilas P, Dunlop S, Heinsen H, Grinberg LT. Turning on the light within: subcortical nuclei of the isodentritic core and their role in Alzheimer’s disease pathogenesis. J Alzheimers Dis 2015;46(1):17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years. J Neuropathol Exp Neurol 2011;70(11):960–969. [DOI] [PubMed] [Google Scholar]
- 37.Ishii T. Distribution of Alzheimer’s neurofibrillary changes in the brain stem and hypothalamus of senile dementia. Acta Neuropathol (Berl) 1966;6(2):181–187. [DOI] [PubMed] [Google Scholar]
- 38.Oh J, Eser RA, Ehrenberg AJ, et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement J Alzheimers Assoc 2019;15(10):1253–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ehrenberg AJ, Nguy AK, Theofilas P, et al. Quantifying the accretion of hyperphosphorylated tau in the locus coeruleus and dorsal raphe nucleus: the pathological building blocks of early Alzheimer’s disease. Neuropathol Appl Neurobiol 2017;43(5):393–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jacobs HIL, Riphagen JM, Ramakers IHGB, Verhey FRJ. Alzheimer’s disease pathology: pathways between central norepinephrine activity, memory, and neuropsychiatric symptoms. Mol Psychiatry 2021;26(3):897–906. [DOI] [PubMed] [Google Scholar]
- 41.Theofilas P, Ehrenberg AJ, Nguy A, et al. Probing the correlation of neuronal loss, neurofibrillary tangles, and cell death markers across the Alzheimer’s disease Braak stages: a quantitative study in humans. Neurobiol Aging 2018;61:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bolton CJ, Tam JW. Differential involvement of the locus coeruleus in early- and late-onset Alzheimer’s disease: a potential mechanism of clinical differences? J Geriatr Psychiatry Neurol 2021. doi: 10.1177/08919887211044755 [DOI] [PubMed] [Google Scholar]
- 43.Falgàs N, Walsh CM, Neylan TC, Grinberg LT. Deepen into sleep and wake patterns across Alzheimer’s disease phenotypes. Alzheimers Dement J Alzheimers Assoc 2021;17(8):1403–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nagata T, Shinagawa S, Nakajima S, Mimura M, Shigeta M. Association between neuropsychiatric improvement and neurocognitive change in Alzheimer’s disease: analysis of the CATIE-AD study. J Alzheimers Dis 2018;66(1):139–148. [DOI] [PubMed] [Google Scholar]
- 45.Nagata T, Nakajima S, Shinagawa S, et al. Psychosocial or clinico-demographic factors related to neuropsychiatric symptoms in patients with Alzheimer’s disease needing interventional treatment: analysis of the CATIE-AD study. Int J Geriatr Psychiatry 2017;32(12):1264–1271. [DOI] [PubMed] [Google Scholar]
- 46.Rouch I, Pongan E, Trombert B, et al. One-year evolution of behavioral and psychological symptoms of dementia in patients initially hospitalized in cognitive behavioral units: the EVITAL prospective cohort. J Alzheimers Dis 2017;57(1):147–155. [DOI] [PubMed] [Google Scholar]
- 47.Kaiser NC, Liang L-J, Melrose RJ, Wilkins SS, Sultzer DL, Mendez MF. Differences in anxiety among patients with early-versus late-onset Alzheimer’s disease. J Neuropsychiatry Clin Neurosci 2014;26(1):73–80. [DOI] [PubMed] [Google Scholar]
- 48.Panegyres PK, Chen HY. Coalition against major diseases (CAMD). Early-onset Alzheimer’s disease: a global cross-sectional analysis. Eur J Neurol 2014;21(9):1149–1154, e64–65. [DOI] [PubMed] [Google Scholar]
- 49.Mendez MF. The relationship between anxiety and Alzheimer’s disease. J Alzheimers Dis Rep 2021;5(1):171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Grinberg LT, Rüb U, Ferretti REL, et al. The dorsal raphe nucleus shows phospho-tau neurofibrillary changes before the transentorhinal region in Alzheimer’s disease. A precocious onset? Neuropathol Appl Neurobiol 2009;35(4):406–416. [DOI] [PubMed] [Google Scholar]
- 51.Lehrner J, Kogler S, Lamm C, et al. Awareness of memory deficits in subjective cognitive decline, mild cognitive impairment, Alzheimer’s disease and Parkinson’s disease. Int Psychogeriatr 2015;27(3):357–366. [DOI] [PubMed] [Google Scholar]
- 52.Townley RA, Graff-Radford J, Mantyh WG, et al. Progressive dysexecutive syndrome due to Alzheimer’s disease: a description of 55 cases and comparison to other phenotypes. Brain Commun 2020;2(1):fcaa068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rodriguez RD, Suemoto CK, Molina M, et al. Argyrophilic grain disease: demographics, clinical, and neuropathological features from a large autopsy study. J Neuropathol Exp Neurol 2016;75(7):628–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez RD, Grinberg LT. Argyrophilic grain disease: an underestimated tauopathy. Dement Neuropsychol 2015;9(1):2–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Naasan G, Shdo SM, Rodriguez EM, et al. Psychosis in neurodegenerative disease: differential patterns of hallucination and delusion symptoms. Brain J Neurol 2021;144(3):999–1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gelman A, Hill J, Yajima M. Why we (usually) don’t have to worry about multiple comparisons. J Res Educ Eff 2012;5(2):189–211. [Google Scholar]