

Abstract
Recently, there have been impressive advancements in understanding of the immune mechanisms underlying cutaneous inflammatory diseases. To understand these diseases on a deeper level and clarify the therapeutic targets more precisely, numerous studies including in vitro experiments, animal models, and clinical trials have been conducted. This has resulted in a paradigm shift from non-specific suppression of the immune system to selective, targeted immunotherapies. These approaches target the molecular pathways and cytokines responsible for generating inflammatory conditions and reinforcing feedback mechanisms to aggravate inflammation. Among the numerous types of skin inflammation, psoriasis and atopic dermatitis (AD) are common chronic cutaneous inflammatory diseases. Psoriasis is a IL-17–mediated disease driven by IL-23, while AD is predominantly mediated by Th2 immunity. Autoimmune bullous diseases are autoantibody-mediated blistering disorders, including pemphigus and bullous pemphigoid. Alopecia areata is an organ-specific autoimmune disease mediated by CD8+ T-cells that targets hair follicles. This review will give an updated, comprehensive summary of the pathophysiology and immune mechanisms of inflammatory skin diseases. Moreover, the therapeutic potential of current and upcoming immunotherapies will be discussed.
Keywords: Atopic dermatitis; Psoriasis; Pemphigus; Pemphigoid, bullous; Alopecia areata; Biologics; Immunopathology
INTRODUCTION
Inflammatory skin diseases, including psoriasis, atopic dermatitis (AD), autoimmune bullous diseases (AIBDs) and alopecia areata (AA), cause major health burdens, deterioration of quality of life, and are associated with various comorbidities. Conventionally, treatment of such skin conditions has focused on controlling symptoms with topical and systemic corticosteroids and other systemic immunosuppressants, which can not only cause dissatisfactory effects, but also occasionally cause serious adverse events.
Dysregulation of the cutaneous immune system leads to various pathogenic outcomes involving inflammatory immune cells and structural tissue cells. Further understanding of the molecular mechanisms and immune biology of cutaneous inflammatory conditions has led to identification of the pathways and cytokines involved. These then become molecular targets for the development of immunotherapies including biologic agents that target specific cell-surface molecules or extracellular molecules. Furthermore, small molecule inhibitors can affect intracellular signaling by targeting receptor-associated kinases.
Herein, we present an overview of the molecular immune pathogenesis of inflammatory skin diseases, followed by discussion of approved immunotherapeutic agents and treatments currently under development.
PSORIASIS
Psoriasis is a chronic inflammatory skin disease that appears as demarcated erythematous plaques and silvery-scaled patches. The prevalence of psoriasis ranges between 0.1% and 1.5% across countries (1). Skin lesions vary in size and can occur anywhere on the body, although they are usually located on buttocks, scalp, and extensor areas of knees and elbows. While these clinical features are observed in plaque psoriasis, the most common clinical type, other subtypes also exist, including guttate, erythrodermic, flexural, palmoplantar, nail, and pustular forms. Psoriasis can be associated with other diseases such as psoriatic arthritis, cardiovascular diseases, metabolic syndrome, inflammatory bowel diseases, and psychological disorders. Psoriatic arthritis occurs in approximately 20% of patients with psoriasis and presents as seronegative inflammatory arthritis in distal interphalangeal joints, dactylitis, and enthesitis (2).
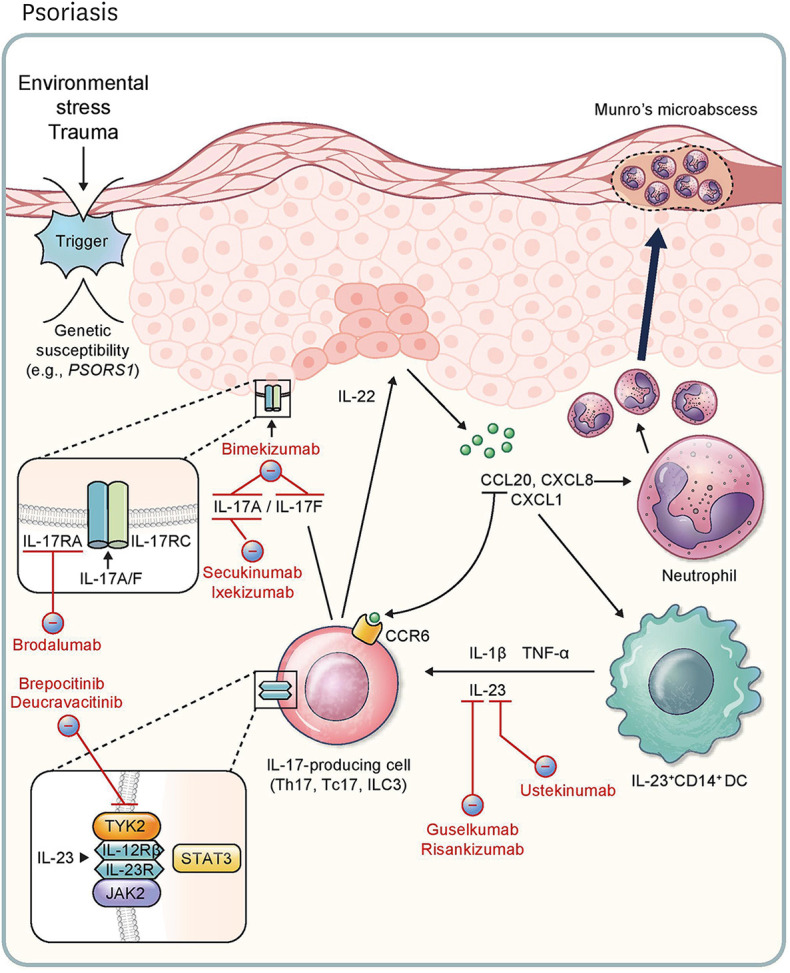
Histologic findings of psoriasis typically include dermal infiltration of immune cells, including T cells and myeloid cells, under epidermal hyperplasia with neutrophil condensation. In addition, skin lesions in psoriasis patients improve after treatment with immunosuppressive drugs such as cyclosporine and methotrexate. These histologic and clinical findings provide insight into the pathogenic role of immune cells in this disease (3). Numerous immunologic, genetic, and clinical studies have highlighted the involvement of the IL-23/IL-17 axis and IL-17+ T cells, such as Th17 and Tc17 cells, in the central mechanisms of psoriasis (Fig. 1) (4,5,6). The importance of Ag recognition in function of the adaptive immune system is supported by the genetic susceptibility of HLA-C*06:02 in PSORS1 locus (7) and the presence of autoreactive and PD-1+ T cells in psoriatic skin (8,9,10). However, IL-23, a p19 and p40 heterodimer, is the most important stimulator of IL-17+ T cells in psoriasis. After the stimulation of IL-17+ T cells with IL-23, STAT3 is phosphorylated by JAK2 and Tyrosine kinase 2 (TYK2) and then induces the transcription factor RORγt. pSTAT3 and RORγt bind to IL17A and IL17F promoters and induce the expression of IL-17 (6). A recent study revealed that CD1c+CD14+ classical dendritic cell type 3 (cDC3) is the major source of IL-23 in psoriatic skin (11). Homodimers or heterodimers of IL-17A and IL-17F secreted by T cells primarily target keratinocytes in psoriatic skin, signaling through the IL-17RA-IL-17RC complex (12). IL-17-activated keratinocytes secrete chemokines (e.g., CCL20, CXCL1, and CXCL8) that attract CCR6+IL-17+ T cells, DCs, and neutrophils (13). This feedforward mechanism amplifies disease activity in psoriasis.
Figure 1. Pathophysiology of and immunotherapeutic agents for psoriasis. External environmental stress and trauma is a possible trigger of psoriasis, especially in individuals with genetic susceptibilities (e.g., PSORS1). This leads to the release of CCL20, CXCL8, and CXCL1, stimulating CCR6+IL-17+ lymphocytes, neutrophils, and IL-23+CD14+ DCs, respectively. Release of IL-17A and IL-17F via IL-17-producing cells (Th17, Tc17, and ILC3) results in the activation of the IL-17RA/IL-17RC complex in keratinocytes, further feeding the inflammatory response. Production of IL-22 by ILC3 accelerates hyperproliferation of keratinocytes. DCs induce cytokines like IL-1β, TNF-α, and IL-23. IL-17-producing cells are stimulated by IL-23 through the JAK2/TYK2-STAT3 pathway. Ustekinumab targets the p40 subunit shared by IL-12 and IL-23, and guselkumab and risankizumab inhibit the p19 subunit of IL-23. Secukinumab and ixekizumab are inhibitors of IL-17A. Bimekizumab is a monoclonal antibody targeting IL-17A and IL-17F. Brodalumab is an inhibitor of IL-17RA. With respect to JAK signaling, deucravacitinib acts to inhibit TYK2, and brepocitinib inhibits TYK2 and JAK1 simultaneously.
Current immunotherapies for psoriasis
For the evaluation of treatment efficacy in psoriasis, disease severity is measured using the Psoriasis Area and Severity Index (PASI), with 75%, 90%, and 100% reductions in PASI scores are referred to as PASI-75, PASI-90, and PASI-100, respectively.
Although TNF inhibitors were initially approved as biologics for psoriasis (14), they show better therapeutic efficacy in psoriatic arthritis than psoriasis. Ustekinumab, an inhibitor of the p40 subunit shared by IL-23 and IL-12, was approved for the treatment of moderate to severe psoriasis (15). Since ustekinumab targets both Th1 and Th17 immunity, specific IL-23 inhibitors targeting the p19 subunit have subsequently been developed. Among p19 inhibitors, guselkumab (70%–73% of PASI-90 at week 16) and risankizumab (75% of PASI-90 at week 16) are superior to ustekinumab and are well tolerated (16,17,18). In parallel, IL-17 inhibitors have also been developed for the treatment of psoriasis. Secukinumab (54%–59% of PASI-90 at week 12) and ixekizumab (68%–71% of PASI-90 at week 12) target IL-17A and have shown higher therapeutic efficacy than ustekinumab (18,19,20). Further, brodalumab, an anti-IL-17RA monoclonal Ab, showed 69%–70% PASI-90 at week 12, which is also superior to ustekinumab (18,21). IL-23 inhibitors are commonly used in severe psoriasis with or without mild psoriatic arthritis because of the convenient dosing interval, whereas IL-17 inhibitors are generally selected when patients have pronounced psoriatic arthritis or require rapid improvement (6).
New immunotherapies for psoriasis
Bimekizumab, a monoclonal Ab targeting IL-17A and IL-17F, is a promising candidate for the treatment of psoriasis. In a phase 3 trial, bimekizumab was superior to secukinumab, showing 86% PASI-90 at week 16 (22). In addition, several oral drug candidates target intracellular signal pathways in IL-17+ T cells. Binding of IL-23 to the IL-23 receptor in T cells induces IL-17 through the JAK-STAT signaling pathway and thus, JAK inhibitors have been developed for clinical use. Deucravacitinib, a TYK2 inhibitor, showed 75% PASI-75 and 43% PASI-90 at week 12 in a phase 2 trial (23). Brepocitinib, a JAK1 and TYK2 inhibitor, was found to provide clinical efficacy in a phase 2 trial with 86% PASI-75 and 52% PASI-90 at week 12 (24). Although the clinical efficacy of JAK inhibitors is well established, the US Food and Drug Administration (FDA) recently expressed concern about an increased risk of serious heart-related events, thromboembolism, cancer, and death with these therapeutics (25). Thus, more clinical studies are warranted.
ATOPIC DERMATITIS
AD is a chronic, recurrent inflammatory skin disease characterized by recurrent eczema and severe itching (26). The prevalence of AD is increasing worldwide and in developed countries, it affects up to 20% of children and 2.1%–4.9% of adults (26). AD can arise at any time in a person’s life, but frequently occurs during early childhood. In recent years, its increasing incidence in the adult population has become a serious problem (27,28). Although genetic predisposition is significant in AD, environmental factors are also increasingly known to be of importance (29). AD patients have dry and sensitive skin, and suffer from severe itching arising from eczematous lesions in localized or diffuse areas of the body (30). In cases during infancy, edematous erythema and excoriations are widespread on the face and trunk, and the lesions become localized to flexural areas of dry skin and chronic lichenification in childhood. Adolescents and adult patients often present with focal eczema on eyelids, hands, and flexural areas (26). AD is also considered part of the atopic march that includes food allergy, asthma, and allergic rhinoconjunctivitis (31).
Although the exact pathogenesis of AD remains to be elucidated, it is thought to involve the interaction of three major mechanisms (Fig. 2): skin barrier defects, alterations in the skin microbiome, and Th2-biased immune dysregulation (26). Genetic predispositions such as filaggrin mutations and environmental factors disturb physiologic epidermal barrier function, and these dysfunctions contribute to the changes in the skin microbiome and immune system (26). In healthy individuals, Propionibacterium acnes and Staphylococcus epidermidis are abundant in the skin microbiome (32), and the skin commensal Staphylococcus spp. inhibits the growth of Staphylococcus aureus (33). In conditions of AD, however, colonization of S. aureus increases in the skin microbiome, promoting dysregulation of the skin barrier and immunity (34). In this review, we mainly focus on the skin immune system in the pathophysiology of AD.
Figure 2. Pathophysiology of and immunotherapeutic agents for atopic dermatitis. Genetic predisposition and environmental factors induce barrier dysfunction. Further, change of surface microbiome diversity, especially decrease of S. epidermidis and increase of S. aureus, enhances barrier dysfunction and increases vulnerability of skin epidermis to external allergens. TSLP, IL-25, and IL-33 released from keratinocytes promote a type 2 response through ILC2, Th2 cells, and Tfh2 cells, which are induced from activated skin LCs and IDECs. IL-4, IL-13, and IL-5 are released from these lymphocytes. IL-4 and IL-13 activate type I (IL-4Rα/CD132) and type II (IL-4Rα/IL-13Rα1) receptors on B cells, keratinocytes, and sensory neurons, resulting in activation of JAK-STAT pathways. Tfh2 cells induce IgE+ B cells, stimulating mast cells and basophils via FcεRI. AMPs (e.g., human β-defensin 3) are decreased in keratinocytes in response to IL-4 and IL-13. Neuronal itch is induced by IL-4, IL-13, IL-31, IL-33 and TSLP, and the itch-scratch cycle is intensified through the process. Type 2 cytokines also aggravate the imbalance of skin surface microbiota, and IL-5 recruits eosinophils, continuing the vicious circle. Dupilumab inhibits type I and II receptors by blocking IL-4Rα. Tralokinumab neutralizes IL-13 and nemolizumab inhibits IL-31R. Upadacitinib and abrocitinib are oral, selective JAK1 inhibitors, and baricitinib is an oral JAK1 and JAK2 inhibitor. Topical agents ruxolitinib (a JAK1 and JAK2 inhibitor) and delgocitinib (a pan-JAK inhibitor) are approved treatments for atopic dermatitis.

Th2-mediated skin inflammation is considered a central pathway in AD. Ag uptake by Ag-presenting cells is increased through loose tight junctions in the AD epidermis (35). These Ags are derived from a variety of sources, including air allergens (e.g., house dust mite), food, and microorganisms (26). Keratinocytes also secrete TSLP, IL-25, and IL-33 due to disruption of the epidermal barrier (36). Increased Ag exposure and signaling from keratinocytes activates Th2 cells to release IL-4, IL-5, and IL-13 and induce IgE production in B cells. IL-4 signals through the type I receptor IL-4Rα/CD132 and type II receptor IL-4Rα/IL-13Rα1. IL-13 shares the type II receptor with IL-4 and can bind to the IL-13Rα2 decoy receptor (37). Binding of cytokines to type I and type II receptors activates the JAK1/STAT6 pathway in hematopoietic and non-hematopoietic cells (37). These type 2 cytokines also cause skin barrier damage and increase the colonization of S. aureus (38). Barrier defects also induce keratinocyte production of IL-23, which leads to the activation of IL-23R expressing DCs triggering the Th22 immune response. In addition, CCR6+ Th22 cells promote epidermal hyperplasia and lichenification via the IL-22/IL-22R axis in chronic atopy (39).
Neuronal itch can be induced by the receptors for TSLP, IL-4, IL-13, IL-31, and IL-33 expressed on sensory neurons (40,41). Allergen-induced crosslinking of IgE activates granulocytes, including mast cells and basophils, via Fcε receptor I (FcεRI). Basophils recognize allergen-specific IgE through FcεRI and then release leukotriene C4, which activates CysLTR2 in sensory neurons causing acute atopic pruritus (42,43).
Current immunotherapies for AD
Conventional systemic management of AD consists of corticosteroids and immunosuppressants such as cyclosporine A and methotrexate (44). Topical treatment of AD includes corticosteroids and calcineurin inhibitors (26). However, patients with refractory AD respond poorly to conventional treatments, and long-term usage of systemic drugs carries a risk of side effects.
Recent discoveries in the pathogenesis of AD have enabled the development of biologics and small molecule therapies to target refractory cases. The Eczema Area and Severity Index (EASI) is used to measure disease severity in AD. EASI-75, EASI-90 and EASI-100 indicate reductions of 75%, 90%, and 100% in EASI scores, respectively (45). Currently, dupilumab, a human IgG4 monoclonal Ab blocking IL-4Rα, is the only systemic biologic approved by both the FDA and European Medicines Agency (EMA) (46). Various studies have shown that dupilumab (300 mg every 2 weeks, with a loading dose of 600 mg) is effective in relieving both pruritus and inflammation (47). There are no life-threatening safety concerns with dupilumab, while mild conjunctivitis occurred in 6.5% of patients (48). More than half of AD patients reached EASI-75 at week 16, and 36% of patients achieved EASI-90 at week 16 with dupilumab (45). Tralokinumab, an EMA-approved human IL-13 neutralizing Ab, exhibited 33.2% of EASI-75 in a phase 3 study (11.4% of EASI-75 in the placebo group) (49). Upadacitinib is an EMA-approved oral JAK1 selective inhibitor. A phase 3 study showed that 30 mg/d of upadacitinib is a safe and effective treatment option, with 80% of AD patients reaching PASI-75 at week 16 (50,51). In a randomized clinical trial comparing the efficacy and safety of upadacitinib and dupilumab, 71% of patients achieved EASI-75 with upadacitinib at week 16, whereas 61.1% of patients achieved EASI-75 with dupilumab at week 16 (52). Baricitinib, an oral JAK inhibitor that blocks JAK1/JAK2, is also approved by the EMA for patients with severe adult AD. EASI-75 was achieved in 24.8% of patients taking 4 mg/d of baricitinib for 16 weeks compared to 8.8% of patients in placebo group (53). The topical JAK1/JAK2 inhibitor ruxolitinib is approved by the FDA for patients with mild to moderate AD, and 62% of patients (1.5% ruxolitinib cream, twice daily) achieved EASI-75 at week 8 (54). Delgocitinib is a topical pan-JAK inhibitor approved in Japan. In a phase 3 study, the change in EASI score was −44.3% in the delgocitinib group (0.5% delgocitinib ointment, twice daily) after 4 weeks of treatment, compared with a 1.7% increase in the vehicle group (55).
New immunotherapies for AD
Various biologics and small molecule therapies have shown promise for modulating clinical symptoms of AD. Nemolizumab is a monoclonal Ab that blocks IL-31RA and is known to alleviate itching and the severity of eczema. In a phase 3 study, the visual analogue scale score decreased by 42.8% in the nemolizumab group (60 mg every 4 weeks) compared to a decrease of 21.4% in the placebo group, and EASI score dropped by 45.9% in the nemolizumab group and 33.2% in the placebo group (56). Treatment with abrocitinib, a selective JAK1 inhibitor, resulted in 63% of patients reaching EASI-75 at week 12 with a daily dose of 200 mg (57), and is currently awaiting FDA approval.
AUTOIMMUNE BULLOUS DISEASES
AIBDs are a group of rare, life-threatening blistering diseases mediated by autoantibodies targeting proteins in desmosomes or hemidesmosomes of keratinocytes in epidermis. According to the location of blisters, AIBDs are divided into intraepidermal and subepidermal types. Intraepidermal AIBD is called pemphigus, and bullous pemphigoid is the most common subepidermal AIBD.
Pemphigus
Pemphigus is characterized by suprabasal acantholytic blisters on skin and/or mucosa. Autoantibodies in pemphigus are directed against desmoglein 1 (Dsg1) and/or Dsg3, the cell-cell adhesion proteins of desmosomes (58). Pemphigus usually occurs between the ages of 50 and 60 years and is mainly classified as pemphigus vulgaris and pemphigus foliaceus in accordance with the major target Ags and clinical phenotypes. Pemphigus vulgaris caused by anti-Dsg3 autoantibody predominantly affects the oral mucosa, whereas pemphigus foliaceus induced by anti-Dsg1 autoantibody presents with superficial blisters on the skin but not the mucosa. These clinical differences between the two types of pemphigus are thought to be caused by the different expression patterns of Dsg1 and 3 in the oral mucosa and the skin (59). Moreover, the clinical features are similar to those of Dsg1- and 3-deficient patients (60,61).
Titers of anti-Dsg IgG autoantibodies positively correlate with disease activity (62) and pathogenic monoclonal antibodies from pemphigus patients are necessary and sufficient to cause acantholytic blisters (63). These autoantibodies mechanically interfere with the adhesion of Dsg and internalize Dsgs from the cell surface through various cellular signaling mechanisms (Fig. 3A) (64). Pathogenic autoreactive B cells are known to undergo somatic hypermutation in patients with pemphigus vulgaris despite the ability of some germline-reverted monoclonal antibodies to bind Dsg3 (65,66). Affinity maturation and isotype switching in B cells requires help from T follicular helper (Tfh) cells. In the mouse model of pemphigus vulgaris, inducible costimulator-positive (ICOS+) Tfh cells are required for disease induction, and Dsg3-specific ICOS+ Tfh cells are associated with anti-Dsg3 Ab production (67). Furthermore, specific subtypes of circulating Tfh cells are associated with anti-Dsg3 autoantibody production in peripheral blood of pemphigus vulgaris patients (67,68).
Figure 3. Pathophysiology and immunotherapeutic agents of autoimmune bullous diseases. (A) In pemphigus, Tfh cells activate autoreactive B cells, which differentiate into antibody-producing cells generating pathogenic anti-Dsg1/3 autoantibodies. Autoantibodies circulate through vessels and relocate to the epidermal intercellular space. FcRn expressed on endothelial cells lengthens the half-life of IgG autoantibodies. Binding of autoantibodies to Dsg1/3 induces steric hindrance and enhances endocytosis, leading to loss of cell-to-cell adhesion of keratinocytes. Rituximab depletes autoreactive B cells by targeting cell surface antigen CD20. Rilzabrutinib and tirabrutinib are small molecule drugs that bind BTK in B cells and inhibit aberrant B-cell receptor signaling. Dsg3 CAAR T cells specifically target and destroy Dsg3-specific B cells. ALXN1830 and efgartigimod shorten the half-life of antibodies by blocking FcRn. Intravenous immunoglobulin (IVIG) inhibits antibody-producing cells by activating the FcγRIIb inhibitory receptor and enhances the degradation of autoantibodies by saturating FcRn. (B) In BP, autoreactive B cells switch to become antibody-producing cells resulting in production of pathogenic anti-BP180/BP230 autoantibodies which migrate to dermo-epidermal junction. IgG and IgE autoantibodies against BP180 promote complement activation leading to infiltration of granulocytes such as neutrophils and eosinophils into the dermis. Release of various proteases plays an important role in creating clefts and in blister formation. Along with pemphigus, rituximab depletes CD20+ B cells. IVIG saturates FcRn reducing the life span of autoantibodies, and suppresses antibody-producing cells by activating FcγRIIb. Omalizumab, a monoclonal antibody against IgE, can decrease the level of pathogenic IgE autoantibodies. Sutimlimab decreases complement activation at the dermo-epidermal junction.

Current immunotherapies for pemphigus
Systemic corticosteroids and steroid-sparing immunosuppressive drugs such as mycophenolate mofetil are the main treatments for pemphigus; however, achieving clinical remission with these drugs alone takes a long time (69). Intravenous immunoglobulin can reduce pathogenic IgG autoantibody by various mechanisms, including activation of Fcγ receptor IIb (FcγRIIb) inhibitory receptor in B cells and saturation of the neonatal Fc receptor (FcRn), which prolongs the half-life of IgG. Intravenous immunoglobulin was found to significantly, but not dramatically, reduce disease activity and autoantibody titers in a randomized trial for pemphigus (70). Rituximab, a monoclonal Ab against the CD20 Ag of B cells, depletes B cells and has been used to treat pemphigus since the 2000s (71,72). Rituximab was found to strikingly shorten the time to clinical remission and reduce the use of systemic corticosteroid significantly compared to mycophenolate mofetil (71,73). In a meta-analysis, clinical remission was found to be achieved 3 to 6 months after rituximab therapy in 75% of pemphigus patients (74). First-line treatment of rituximab (1,000 mg on days 0 and 14 and 500 mg at months 12 and 18) also showed favorable results (89% of complete remission and 24% of relapse) in a randomized trial (75).
New immunotherapies for pemphigus
Despite the fact that rituximab notably improves clinical outcome in terms of time to remission and use of systemic corticosteroids, the depletion of non-pathogenic B-cells increases the risk of serious infections (75,76). To overcome this problem, T cells expressing a chimeric autoantibody receptor (CAAR) composed of Dsg3 combined to CD137-CD3ζ signaling domains were developed (77). Preclinical data showed Dsg3 CAAR T cells specifically target and lyse Dsg3-specific B cells, thus preserving non-pathogenic B cells (78). Rilzabrutinib and tirabrutinib, oral Bruton’s tyrosine kinase inhibitors that inhibit B-cell activation, were found to decrease the dose of systemic corticosteroid needed and reduce disease activity in pemphigus vulgaris in a phase 2 trial (79,80). In addition, efgartigimod and ALXN1830, FcRn blocking antibodies, showed an early onset of clinical improvement in pemphigus patients in phase 2 trials (81,82).
Bullous pemphigoid
Bullous pemphigoid is the most common subepidermal AIBD clinically featuring pruritic tense bullae with erosions on the skin and/or mucosa. Target Ags of bullous pemphigoid are BP180 (bullous pemphigoid Ag 2; BPAG2) and BP230 (bullous pemphigoid Ag 1; BPAG1), which are the components of hemidesmosomes connecting basal keratinocytes to dermal structures. Several drugs such as dipeptidyl dipeptidase-4 inhibitors and immune checkpoint inhibitors (e.g., PD-1 and PD-L1 inhibitors) may be triggers for bullous pemphigoid (83,84). Bullous pemphigoid is thought to be associated with neurologic disorders including dementia, multiple sclerosis, and Parkinson’s disease (85). Because it preferentially occurs in older adults with comorbidities and requires long-term use of systemic corticosteroids and immunosuppressive drugs, patients with bullous pemphigoid have a higher mortality rate (approximately 20%) than healthy older adults (86).
IgG and IgE autoantibodies targeting the extracellular NC16A domain of BP180 (Fig. 3B) are associated with disease activity (87,88), and their pathogenicity was confirmed by the passive transfer of human autoantibodies to humanized or human skin-grafted mouse models (89,90). Complement activation and granulocyte infiltration are followed by the autoantibody deposition on the dermo-epidermal junction (91). These factors are known to contribute to blister formation (89,90,91). In addition to the presence of autoreactive B cells (92), dysfunction of regulatory B cells is observed in peripheral blood of patients with bullous pemphigoid (93). Additionally, a decrease in regulatory T cells might lead to disease since evidence of bullous pemphigoid has been found in Foxp3-deficient mice and humans (94,95,96).
Current and new immunotherapies for bullous pemphigoid
Bullous pemphigoid is mainly treated with systemic and topical corticosteroids and immunosuppressive agents (97,98). Dapsone and doxycycline can be applied as steroid-sparing agents (99,100,101). Rituximab treatment showed remission at 5 months after treatment and increased survival rates over a long-term period (102). Intravenous immunoglobulin has also proven beneficial, but with limited therapeutic effects (103). Based on the existence of pathogenic IgE autoantibodies in bullous pemphigoid, omalizumab, a monoclonal Ab against IgE, has been reported to be effective for the treatment of bullous pemphigoid (104). However, clinical results are variable among patients because it cannot reduce pathogenic IgG autoantibodies (104). Sutimlimab, a C1s inhibitor, decreased complement deposition at the dermo-epidermal junctions in patients with bullous pemphigoid in a phase I trial (105).
ALOPECIA AREATA (AA)
AA, a chronic autoimmune inflammatory disease causing sudden hair loss, has a lifetime prevalence of 2% (106). It is characterized by patch-like distribution without scarring, often in sharply defined areas, with dystrophic hairs called exclamation point hairs (107). There is a genetic predisposition for AA, and several studies have shown up to a 10-fold increased risk for first-degree relatives of AA patients (108). In the early stage of AA, the proportion of telogen hairs and dystrophic hair shafts increase before hair loss (109). Along with the disease progression, peribulbar inflammation takes place, and the hair follicles miniaturize leading to developmental arrest in the anagen phase (110). Affected hair follicles prematurely enter the telogen phase and undergo shortened cycles.
AA is known to be a T-cell-mediated inflammatory disease (Fig. 4). Normal anagen hair follicles are considered immune privileged sites with low expression of the MHC (111). This immune privilege is broken in AA lesions, where MHC I and II molecules in the hair follicles are increased with recruitment of CD8+ and CD4+ T cells and APCs (112). These features first gave rise to the concept that hair follicle autoantigens play a primary pathogenic role in AA. Several autoantigens in AA have been proposed so far, but the exact autoantigens remain elusive (113). In addition to TCR-mediated activation in T cells, a genome-wide association study suggests that a TCR-independent, natural killer group 2D (NKG2D)-mediated pathway is involved in the pathogenesis of AA (114). NKG2D+CD8+ T cells exert their cytotoxic functions on hair follicles through NKG2D, contributing to pathogenesis in the C3H/HeJ AA mouse model (115). In human AA, ligands of NKG2D, such as MICA and ULBP3, were found to be upregulated in the hair follicle (114,116), and NKG2D+CD8+ T cells were infiltrated into and activated in perifollicular areas (114,117).
Figure 4. Pathophysiology and immunotherapeutic agents of alopecia areata. In alopecia areata, NKG2D+CD8+ T cells infiltrate into the dermis and relocate to the hair follicle bulb. IL-15 is an important cytokine for pathogenesis of this disease through activation and proliferation of NKG2D+CD8+ T cells. The IL-15 receptor complex is composed of IL-15Rα expressed on follicular epithelial cells and CD122 and Fcγ on T cells, and trans-activates CD8+ T cells through multiple pathways including Ras-Raf-MEK-MAPK, PI3K-Akt-mTOR, and JAK1/3-STAT5 signaling. These pathways upregulate expression of KLRK1, encoding NKG2D, and IFNG. IFN-γ binds to the IFN-γ receptor on follicular epithelial cells and triggers JAK1/2-STAT1 signaling, thereby upregulating expression of MICA, ULBP3, IL15, and CXCR3. Ruxolitinib and baricitinib inhibit JAK1 and JAK2, and brepocitinib inhibits JAK1. Tofacitinib is a JAK1 and JAK3 inhibitor, and ritlecitinib is a selective JAK3 inhibitor.

IFN-γ and IL-15 are important cytokines involved in the pathogenesis of AA. IL-15 is known to upregulate NKG2D and drive TCR-independent activation in CD8+ T cells (118). In AA lesions, IL-15Rα and IL-15Rβ are upregulated in the hair follicle and CD8+ T cells, respectively, indicating that trans-presentation of IL-15 activates CD8+ T cells (115). After CD8+ T cells recognize IL-15, downstream signaling pathways, including JAK1/3-STAT5, MAP kinase, and mTOR pathways, are activated (118). Activated CD8+ T cells secrete IFN-γ which upregulates NKG2D ligands, MHC I and II, IL-15, and chemokine ligands of CXCR3 (e.g., CXCL9, CXCL10, and CXCL11) through the JAK1/2-STAT1 pathway in the follicular epithelium (119,120). These subsequent responses induce the recruitment, activation, and effector function of T cells, and this vicious cycle exacerbates inflammation in AA.
Current immunotherapies for AA
Currently, no treatment for AA has been approved by the FDA nor EMA, but various methods have been used for AA treatment (121). Severity of Alopecia Tool (SALT) score, indicating the rate of hair loss, is used to evaluate disease severity and treatment efficacy. Both systemic and local application of corticosteroids were found to be effective, but often resulted in limited curative rates of hair growth in moderate-to-severe AA. Cyclosporine, often used in combination with systemic corticosteroids, showed a hair regrowth rate of up to 76.6% (122). Methotrexate, alone or in combination with systemic corticosteroids, allowed full recovery in 64% of patients with alopecia totalis and universalis (123). Another treatment option for AA is topical immunotherapies such as diphenylcyclopropenone (DPCP) or squaric acid dibutylester, which induce delayed-type hypersensitivity (124). While the exact mechanism underlying the efficacy of this treatment is unknown, DPCP has shown a response rate of up to 72.2% for treatment of chronic and extensive AA (24).
New immunotherapies for AA
In treating AA, about 25% of patients are refractory to conventional therapy and 13.5-33% of patients recur (125). Recent studies suggest that blocking the JAK pathway could be an option for refractory AA (126). Currently, there are several oral JAK inhibitors for treatment of AA, including tofacitinib (JAK 1 and 3 inhibitor), ruxolitinib (JAK 1 and 2 inhibitor), baricitinib (JAK 1 and 2 inhibitor), ritlecitinib (JAK 3 inhibitor), and brepocitinib (Tyrosine kinase 2 and JAK 1 inhibitor). JAK inhibitors can affect both NKG2D+CD8+ T cells and follicular epithelial cells (115), interrupting the positive feedback loop involving IFN-γ and IL-15 (113,127,128). In a retrospective study of tofacitinib, 58% of patients with at least 40% scalp hair loss achieved greater than 50% change in SALT score over 4–18 months of treatment (129). In a comparative study, tofacitinib (20 mg twice daily) and ruxolitinib (5 mg twice daily) showed similar efficacy after 6 months of treatment (130). In a phase 2 trial, 51.9% of severe AA patients (SALT > 50) taking 4 mg/d of baricitinib achieved SALT ≤ 20 at week 36, while 3.6% of the placebo group achieved SALT ≤ 20 (131). In a phase 2a study of severe AA (SALT > 50), 50% of patients receiving ritlecitinib (200 mg/d for 4 weeks, then 50 mg/d for 20 weeks) and 64% of patients receiving brepocitinib (60 mg/d for 4 weeks, then 30 mg/d for 20 weeks) achieved a 30% improvement in SALT score (SALT30), while 2% of the placebo group achieved SALT30 (132).
CONCLUSION
Advancements in understanding of skin immunity and molecular pathogenesis have resulted in promising therapeutic approaches for refractory inflammatory skin disorders. Although glucocorticoids and traditional immunosuppressants persist as the most prevalent treatment methods, monoclonal antibodies targeting pathogenic cytokines and their receptors and small molecule inhibitors of cytokine signaling have recently arisen as potential solutions. Still, many inquiries remain unanswered and possible safety issues exist, especially in regard to the FDA’s latest drug safety communication on JAK inhibitors. With continuous research in the fields of dermatology and immunology, further understanding of the pathological mechanisms underlying distinct inflammatory disorders will be gained. Along with this, ongoing and future clinical trials will identify novel, more effective targeted immunotherapy approaches for cutaneous inflammatory conditions.
ACKNOWLEDGEMENTS
This study was supported by the National Research Foundation of South Korea (grant NRF-2021R1C1C1007179).
The authors thank Medical Illustration & Design, part of the Medical Research Support Services of Yonsei University College of Medicine, for all artistic support related to this work.
Abbreviations
- AA
alopecia areata
- AD
atopic dermatitis
- AIBD
autoimmune bullous disease
- Akt
protein kinase B
- BP180
bullous pemphigoid antigen 2 (BPAG2)
- BP230
bullous pemphigoid antigen 1 (BPAG1)
- BTK
Bruton's tyrosine kinase
- CAAR
chimeric autoantibody receptor
- DC
dendritic cells
- DPCP
diphenylcyclopropenone
- Dsg
desmoglein
- EASI
Eczema Area and Severity Index
- EMA
European Medicines Agency
- FcRn
neonatal Fc receptor
- FcγRIIb
Fcγ receptor IIb
- FcεRI
Fcε receptor I
- FDA
US Food and Drug Administration
- ICOS
inducible costimulator
- IDEC
Inflammatory dendritic epidermal cells
- ILC2
Innate lymphoid cell group 2
- ILC3
Innate lymphoid cell group 3
- IVIG
Intravenous immunoglobulin
- LC
Langerhans cell
- NKG
natural killer group
- PASI
Psoriasis Area and Severity Index
- SALT
Severity of Alopecia Tool
- Tc17
IL-17-secreting CD8+ T cells
- Tfh
T follicular helper
- TYK
tyrosine kinase
Footnotes
Conflicts of Interest: The authors declare no potential conflicts of interest.
- Conceptualization: Kim JH.
- Supervision: Kim JH.
- Visualization: Song A.
- Writing - original draft: Song A, Kim JH.
- Writing - review & editing: Lee SE, Kim JH.
References
- 1.Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, Mehta NN, Finlay AY, Gottlieb AB. Psoriasis. Nat Rev Dis Primers. 2016;2:16082. doi: 10.1038/nrdp.2016.82. [DOI] [PubMed] [Google Scholar]
- 2.Alinaghi F, Calov M, Kristensen LE, Gladman DD, Coates LC, Jullien D, Gottlieb AB, Gisondi P, Wu JJ, Thyssen JP, et al. Prevalence of psoriatic arthritis in patients with psoriasis: a systematic review and meta-analysis of observational and clinical studies. J Am Acad Dermatol. 2019;80:251–265.e219. doi: 10.1016/j.jaad.2018.06.027. [DOI] [PubMed] [Google Scholar]
- 3.Heydendael VM, Spuls PI, Opmeer BC, de Borgie CA, Reitsma JB, Goldschmidt WF, Bossuyt PM, Bos JD, de Rie MA. Methotrexate versus cyclosporine in moderate-to-severe chronic plaque psoriasis. N Engl J Med. 2003;349:658–665. doi: 10.1056/NEJMoa021359. [DOI] [PubMed] [Google Scholar]
- 4.Lee E, Trepicchio WL, Oestreicher JL, Pittman D, Wang F, Chamian F, Dhodapkar M, Krueger JG. Increased expression of interleukin 23 p19 and p40 in lesional skin of patients with psoriasis vulgaris. J Exp Med. 2004;199:125–130. doi: 10.1084/jem.20030451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, et al. Genome-wide scan reveals association of psoriasis with IL-23 and NF-kappaB pathways. Nat Genet. 2009;41:199–204. doi: 10.1038/ng.311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ghoreschi K, Balato A, Enerbäck C, Sabat R. Therapeutics targeting the IL-23 and IL-17 pathway in psoriasis. Lancet. 2021;397:754–766. doi: 10.1016/S0140-6736(21)00184-7. [DOI] [PubMed] [Google Scholar]
- 7.Nair RP, Stuart PE, Nistor I, Hiremagalore R, Chia NV, Jenisch S, Weichenthal M, Abecasis GR, Lim HW, Christophers E, et al. Sequence and haplotype analysis supports HLA-C as the psoriasis susceptibility 1 gene. Am J Hum Genet. 2006;78:827–851. doi: 10.1086/503821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Arakawa A, Siewert K, Stöhr J, Besgen P, Kim SM, Rühl G, Nickel J, Vollmer S, Thomas P, Krebs S, et al. Melanocyte antigen triggers autoimmunity in human psoriasis. J Exp Med. 2015;212:2203–2212. doi: 10.1084/jem.20151093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lande R, Botti E, Jandus C, Dojcinovic D, Fanelli G, Conrad C, Chamilos G, Feldmeyer L, Marinari B, Chon S, et al. The antimicrobial peptide LL37 is a T-cell autoantigen in psoriasis. Nat Commun. 2014;5:5621. doi: 10.1038/ncomms6621. [DOI] [PubMed] [Google Scholar]
- 10.Kim JH, Choi YJ, Lee BH, Song MY, Ban CY, Kim J, Park J, Kim SE, Kim TG, Park SH, et al. Programmed cell death ligand 1 alleviates psoriatic inflammation by suppressing IL-17a production from programmed cell death 1-high T cells. J Allergy Clin Immunol. 2016;137:1466–1476.e1463. doi: 10.1016/j.jaci.2015.11.021. [DOI] [PubMed] [Google Scholar]
- 11.Nakamizo S, Dutertre CA, Khalilnezhad A, Zhang XM, Lim S, Lum J, Koh G, Foong C, Yong PJ, Tan KJ, et al. Single-cell analysis of human skin identifies CD14+ type 3 dendritic cells co-producing IL1B and IL23A in psoriasis. J Exp Med. 2021;218:218. doi: 10.1084/jem.20202345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moos S, Mohebiany AN, Waisman A, Kurschus FC. Imiquimod-induced psoriasis in mice depends on the IL-17 signaling of keratinocytes. J Invest Dermatol. 2019;139:1110–1117. doi: 10.1016/j.jid.2019.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Kim TG, Jee H, Fuentes-Duculan J, Wu WH, Byamba D, Kim DS, Kim DY, Lew DH, Yang WI, Krueger JG, et al. Dermal clusters of mature dendritic cells and T cells are associated with the CCL20/CCR6 chemokine system in chronic psoriasis. J Invest Dermatol. 2014;134:1462–1465. doi: 10.1038/jid.2013.534. [DOI] [PubMed] [Google Scholar]
- 14.Leonardi CL, Powers JL, Matheson RT, Goffe BS, Zitnik R, Wang A, Gottlieb AB Etanercept Psoriasis Study Group. Etanercept as monotherapy in patients with psoriasis. N Engl J Med. 2003;349:2014–2022. doi: 10.1056/NEJMoa030409. [DOI] [PubMed] [Google Scholar]
- 15.Griffiths CE, Strober BE, van de Kerkhof P, Ho V, Fidelus-Gort R, Yeilding N, Guzzo C, Xia Y, Zhou B, Li S, et al. Comparison of ustekinumab and etanercept for moderate-to-severe psoriasis. N Engl J Med. 2010;362:118–128. doi: 10.1056/NEJMoa0810652. [DOI] [PubMed] [Google Scholar]
- 16.Gordon KB, Strober B, Lebwohl M, Augustin M, Blauvelt A, Poulin Y, Papp KA, Sofen H, Puig L, Foley P, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392:650–661. doi: 10.1016/S0140-6736(18)31713-6. [DOI] [PubMed] [Google Scholar]
- 17.Diels J, Thilakarathne P, Cameron C, McElligott S, Schubert A, Puig L. Adjusted treatment COMPArisons between guSelkumab and uStekinumab for treatment of moderate-to-severe plaque psoriasis: the COMPASS analysis. Br J Dermatol. 2020;183:276–284. doi: 10.1111/bjd.18634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sbidian E, Chaimani A, Garcia-Doval I, Doney L, Dressler C, Hua C, Hughes C, Naldi L, Afach S, Le Cleach L. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. 2021;4:CD011535. doi: 10.1002/14651858.CD011535.pub4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thaçi D, Blauvelt A, Reich K, Tsai TF, Vanaclocha F, Kingo K, Ziv M, Pinter A, Hugot S, You R, et al. Secukinumab is superior to ustekinumab in clearing skin of subjects with moderate to severe plaque psoriasis: CLEAR, a randomized controlled trial. J Am Acad Dermatol. 2015;73:400–409. doi: 10.1016/j.jaad.2015.05.013. [DOI] [PubMed] [Google Scholar]
- 20.Reich K, Pinter A, Lacour JP, Ferrandiz C, Micali G, French LE, Lomaga M, Dutronc Y, Henneges C, Wilhelm S, et al. Comparison of ixekizumab with ustekinumab in moderate-to-severe psoriasis: 24-week results from IXORA-S, a phase III study. Br J Dermatol. 2017;177:1014–1023. doi: 10.1111/bjd.15666. [DOI] [PubMed] [Google Scholar]
- 21.Lebwohl M, Strober B, Menter A, Gordon K, Weglowska J, Puig L, Papp K, Spelman L, Toth D, Kerdel F, et al. Phase 3 studies comparing brodalumab with ustekinumab in psoriasis. N Engl J Med. 2015;373:1318–1328. doi: 10.1056/NEJMoa1503824. [DOI] [PubMed] [Google Scholar]
- 22.Reich K, Warren RB, Lebwohl M, Gooderham M, Strober B, Langley RG, Paul C, De Cuyper D, Vanvoorden V, Madden C, et al. Bimekizumab versus secukinumab in plaque psoriasis. N Engl J Med. 2021;385:142–152. doi: 10.1056/NEJMoa2102383. [DOI] [PubMed] [Google Scholar]
- 23.Papp K, Gordon K, Thaçi D, Morita A, Gooderham M, Foley P, Girgis IG, Kundu S, Banerjee S. Phase 2 trial of selective tyrosine kinase 2 inhibition in psoriasis. N Engl J Med. 2018;379:1313–1321. doi: 10.1056/NEJMoa1806382. [DOI] [PubMed] [Google Scholar]
- 24.Forman SB, Pariser DM, Poulin Y, Vincent MS, Gilbert SA, Kieras EM, Qiu R, Yu D, Papacharalambous J, Tehlirian C, et al. Tyk2/JAK1 inhibitor PF-06700841 in patients with plaque psoriasis: phase IIa, randomized, double-blind, placebo-controlled trial. J Invest Dermatol. 2020;140:2359–2370.e2355. doi: 10.1016/j.jid.2020.03.962. [DOI] [PubMed] [Google Scholar]
- 25.Murphrey M, Waldman RA, Druso T, Grant-Kels JM. Special editorial: when prescribing Janus kinase inhibitors for dermatologic conditions, be mindful of the fda’s 9/1/2021 data safety communication. J Am Acad Dermatol. 2022;86:42–43. doi: 10.1016/j.jaad.2021.09.051. [DOI] [PubMed] [Google Scholar]
- 26.Langan SM, Irvine AD, Weidinger S. Atopic dermatitis. Lancet. 2020;396:345–360. doi: 10.1016/S0140-6736(20)31286-1. [DOI] [PubMed] [Google Scholar]
- 27.Abuabara K, Hoffstad O, Troxel AB, Gelfand JM, McCulloch CE, Margolis DJ. Patterns and predictors of atopic dermatitis disease control past childhood: an observational cohort study. J Allergy Clin Immunol. 2018;141:778–780.e6. doi: 10.1016/j.jaci.2017.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abuabara K, Yu AM, Okhovat JP, Allen IE, Langan SM. The prevalence of atopic dermatitis beyond childhood: a systematic review and meta-analysis of longitudinal studies. Allergy. 2018;73:696–704. doi: 10.1111/all.13320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sandilands A, Terron-Kwiatkowski A, Hull PR, O’Regan GM, Clayton TH, Watson RM, Carrick T, Evans AT, Liao H, Zhao Y, et al. Comprehensive analysis of the gene encoding filaggrin uncovers prevalent and rare mutations in ichthyosis vulgaris and atopic eczema. Nat Genet. 2007;39:650–654. doi: 10.1038/ng2020. [DOI] [PubMed] [Google Scholar]
- 30.Bieber T. Atopic dermatitis: an expanding therapeutic pipeline for a complex disease. Nat Rev Drug Discov. 2021;21:21–40. doi: 10.1038/s41573-021-00266-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang L, Fu J, Zhou Y. Research progress in atopic march. Front Immunol. 2020;11:1907. doi: 10.3389/fimmu.2020.01907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Byrd AL, Belkaid Y, Segre JA. The human skin microbiome. Nat Rev Microbiol. 2018;16:143–155. doi: 10.1038/nrmicro.2017.157. [DOI] [PubMed] [Google Scholar]
- 33.Zipperer A, Konnerth MC, Laux C, Berscheid A, Janek D, Weidenmaier C, Burian M, Schilling NA, Slavetinsky C, Marschal M, et al. Human commensals producing a novel antibiotic impair pathogen colonization. Nature. 2016;535:511–516. doi: 10.1038/nature18634. [DOI] [PubMed] [Google Scholar]
- 34.Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, Kong HH, Amagai M, Nagao K. Dysbiosis and staphylococcus aureus colonization drives inflammation in atopic dermatitis. Immunity. 2015;42:756–766. doi: 10.1016/j.immuni.2015.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoshida K, Kubo A, Fujita H, Yokouchi M, Ishii K, Kawasaki H, Nomura T, Shimizu H, Kouyama K, Ebihara T, et al. Distinct behavior of human Langerhans cells and inflammatory dendritic epidermal cells at tight junctions in patients with atopic dermatitis. J Allergy Clin Immunol. 2014;134:856–864. doi: 10.1016/j.jaci.2014.08.001. [DOI] [PubMed] [Google Scholar]
- 36.Hammad H, Lambrecht BN. Barrier epithelial cells and the control of type 2 immunity. Immunity. 2015;43:29–40. doi: 10.1016/j.immuni.2015.07.007. [DOI] [PubMed] [Google Scholar]
- 37.Gandhi NA, Bennett BL, Graham NM, Pirozzi G, Stahl N, Yancopoulos GD. Targeting key proximal drivers of type 2 inflammation in disease. Nat Rev Drug Discov. 2016;15:35–50. doi: 10.1038/nrd4624. [DOI] [PubMed] [Google Scholar]
- 38.Lee SJ, Kim SE, Shin KO, Park K, Lee SE. Dupilumab therapy improves stratum corneum hydration and skin dysbiosis in patients with atopic dermatitis. Allergy Asthma Immunol Res. 2021;13:762–775. doi: 10.4168/aair.2021.13.5.762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jin M, Yoon J. From bench to clinic: the potential of therapeutic targeting of the il-22 signaling pathway in atopic dermatitis. Immune Netw. 2018;18:e42. doi: 10.4110/in.2018.18.e42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Oetjen LK, Mack MR, Feng J, Whelan TM, Niu H, Guo CJ, Chen S, Trier AM, Xu AZ, Tripathi SV, et al. Sensory neurons co-opt classical immune signaling pathways to mediate chronic itch. Cell. 2017;171:217–228.e13. doi: 10.1016/j.cell.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trier AM, Mack MR, Fredman A, Tamari M, Ver Heul AM, Zhao Y, Guo CJ, Avraham O, Ford ZK, Oetjen LK, et al. IL-33 signaling in sensory neurons promotes dry skin itch. J Allergy Clin Immunol. 2021 doi: 10.1016/j.jaci.2021.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon D, Braathen LR, Simon HU. Eosinophils and atopic dermatitis. Allergy. 2004;59:561–570. doi: 10.1111/j.1398-9995.2004.00476.x. [DOI] [PubMed] [Google Scholar]
- 43.Wang F, Trier AM, Li F, Kim S, Chen Z, Chai JN, Mack MR, Morrison SA, Hamilton JD, Baek J, et al. A basophil-neuronal axis promotes itch. Cell. 2021;184:422–440.e17. doi: 10.1016/j.cell.2020.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Kim JE, Park GH, Bae JM, Byun JY, Shin MK, Han TY, Hong SP, Jang YH, Kim HO, et al. Consensus update for systemic treatment of atopic dermatitis. Ann Dermatol. 2021;33:497–514. doi: 10.5021/ad.2021.33.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Simpson EL, Bieber T, Guttman-Yassky E, Beck LA, Blauvelt A, Cork MJ, Silverberg JI, Deleuran M, Kataoka Y, Lacour JP, et al. Two phase 3 trials of dupilumab versus placebo in atopic dermatitis. N Engl J Med. 2016;375:2335–2348. doi: 10.1056/NEJMoa1610020. [DOI] [PubMed] [Google Scholar]
- 46.Gooderham MJ, Hong HC, Eshtiaghi P, Papp KA. Dupilumab: a review of its use in the treatment of atopic dermatitis. J Am Acad Dermatol. 2018;78(Suppl 1):S28–S36. doi: 10.1016/j.jaad.2017.12.022. [DOI] [PubMed] [Google Scholar]
- 47.Beck LA, Thaçi D, Hamilton JD, Graham NM, Bieber T, Rocklin R, Ming JE, Ren H, Kao R, Simpson E, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis. N Engl J Med. 2014;371:130–139. doi: 10.1056/NEJMoa1314768. [DOI] [PubMed] [Google Scholar]
- 48.Schneeweiss MC, Kim SC, Wyss R, Schneeweiss S, Merola JF. Dupilumab and the risk of conjunctivitis and serious infection in patients with atopic dermatitis: a propensity score-matched cohort study. J Am Acad Dermatol. 2021;84:300–311. doi: 10.1016/j.jaad.2020.09.084. [DOI] [PubMed] [Google Scholar]
- 49.Wollenberg A, Blauvelt A, Guttman-Yassky E, Worm M, Lynde C, Lacour JP, Spelman L, Katoh N, Saeki H, Poulin Y, et al. Tralokinumab for moderate-to-severe atopic dermatitis: results from two 52-week, randomized, double-blind, multicentre, placebo-controlled phase III trials (ECZTRA 1 and ECZTRA 2) Br J Dermatol. 2021;184:437–449. doi: 10.1111/bjd.19574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Reich K, Teixeira HD, de Bruin-Weller M, Bieber T, Soong W, Kabashima K, Werfel T, Zeng J, Huang X, Hu X, et al. Safety and efficacy of upadacitinib in combination with topical corticosteroids in adolescents and adults with moderate-to-severe atopic dermatitis (AD Up): results from a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2021;397:2169–2181. doi: 10.1016/S0140-6736(21)00589-4. [DOI] [PubMed] [Google Scholar]
- 51.Guttman-Yassky E, Teixeira HD, Simpson EL, Papp KA, Pangan AL, Blauvelt A, Thaçi D, Chu CY, Hong HC, Katoh N, et al. Once-daily upadacitinib versus placebo in adolescents and adults with moderate-to-severe atopic dermatitis (Measure Up 1 and Measure Up 2): results from two replicate double-blind, randomised controlled phase 3 trials. Lancet. 2021;397:2151–2168. doi: 10.1016/S0140-6736(21)00588-2. [DOI] [PubMed] [Google Scholar]
- 52.Blauvelt A, Teixeira HD, Simpson EL, Costanzo A, De Bruin-Weller M, Barbarot S, Prajapati VH, Lio P, Hu X, Wu T, et al. Efficacy and safety of upadacitinib vs dupilumab in adults with moderate-to-severe atopic dermatitis: a randomized clinical trial. JAMA Dermatol. 2021;157:1047–1055. doi: 10.1001/jamadermatol.2021.3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simpson EL, Lacour JP, Spelman L, Galimberti R, Eichenfield LF, Bissonnette R, King BA, Thyssen JP, Silverberg JI, Bieber T, et al. Baricitinib in patients with moderate-to-severe atopic dermatitis and inadequate response to topical corticosteroids: results from two randomized monotherapy phase III trials. Br J Dermatol. 2020;183:242–255. doi: 10.1111/bjd.18898. [DOI] [PubMed] [Google Scholar]
- 54.Papp K, Szepietowski JC, Kircik L, Toth D, Eichenfield LF, Leung DY, Forman SB, Venturanza ME, Sun K, Kuligowski ME, et al. Efficacy and safety of ruxolitinib cream for the treatment of atopic dermatitis: results from 2 phase 3, randomized, double-blind studies. J Am Acad Dermatol. 2021;85:863–872. doi: 10.1016/j.jaad.2021.04.085. [DOI] [PubMed] [Google Scholar]
- 55.Nakagawa H, Nemoto O, Igarashi A, Saeki H, Kaino H, Nagata T. Delgocitinib ointment, a topical Janus kinase inhibitor, in adult patients with moderate to severe atopic dermatitis: a phase 3, randomized, double-blind, vehicle-controlled study and an open-label, long-term extension study. J Am Acad Dermatol. 2020;82:823–831. doi: 10.1016/j.jaad.2019.12.015. [DOI] [PubMed] [Google Scholar]
- 56.Kabashima K, Matsumura T, Komazaki H, Kawashima M Nemolizumab-JP01 Study Group. Trial of nemolizumab and topical agents for atopic dermatitis with pruritus. N Engl J Med. 2020;383:141–150. doi: 10.1056/NEJMoa1917006. [DOI] [PubMed] [Google Scholar]
- 57.Simpson EL, Sinclair R, Forman S, Wollenberg A, Aschoff R, Cork M, Bieber T, Thyssen JP, Yosipovitch G, Flohr C, et al. Efficacy and safety of abrocitinib in adults and adolescents with moderate-to-severe atopic dermatitis (JADE MONO-1): a multicentre, double-blind, randomised, placebo-controlled, phase 3 trial. Lancet. 2020;396:255–266. doi: 10.1016/S0140-6736(20)30732-7. [DOI] [PubMed] [Google Scholar]
- 58.Amagai M, Klaus-Kovtun V, Stanley JR. Autoantibodies against a novel epithelial cadherin in pemphigus vulgaris, a disease of cell adhesion. Cell. 1991;67:869–877. doi: 10.1016/0092-8674(91)90360-b. [DOI] [PubMed] [Google Scholar]
- 59.Mahoney MG, Wang Z, Rothenberger K, Koch PJ, Amagai M, Stanley JR. Explanations for the clinical and microscopic localization of lesions in pemphigus foliaceus and vulgaris. J Clin Invest. 1999;103:461–468. doi: 10.1172/JCI5252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Samuelov L, Sarig O, Harmon RM, Rapaport D, Ishida-Yamamoto A, Isakov O, Koetsier JL, Gat A, Goldberg I, Bergman R, et al. Desmoglein 1 deficiency results in severe dermatitis, multiple allergies and metabolic wasting. Nat Genet. 2013;45:1244–1248. doi: 10.1038/ng.2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim JH, Kim SE, Park HS, Lee SH, Lee SE, Kim SC. A homozygous nonsense mutation in the dsg3 gene causes acantholytic blisters in the oral and laryngeal mucosa. J Invest Dermatol. 2019;139:1187–1190. doi: 10.1016/j.jid.2018.09.038. [DOI] [PubMed] [Google Scholar]
- 62.Amagai M, Komai A, Hashimoto T, Shirakata Y, Hashimoto K, Yamada T, Kitajima Y, Ohya K, Iwanami H, Nishikawa T. Usefulness of enzyme-linked immunosorbent assay using recombinant desmogleins 1 and 3 for serodiagnosis of pemphigus. Br J Dermatol. 1999;140:351–357. doi: 10.1046/j.1365-2133.1999.02752.x. [DOI] [PubMed] [Google Scholar]
- 63.Payne AS, Ishii K, Kacir S, Lin C, Li H, Hanakawa Y, Tsunoda K, Amagai M, Stanley JR, Siegel DL. Genetic and functional characterization of human pemphigus vulgaris monoclonal autoantibodies isolated by phage display. J Clin Invest. 2005;115:888–899. doi: 10.1172/JCI24185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kasperkiewicz M, Ellebrecht CT, Takahashi H, Yamagami J, Zillikens D, Payne AS, Amagai M. Pemphigus. Nat Rev Dis Primers. 2017;3:17026. doi: 10.1038/nrdp.2017.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cho A, Caldara AL, Ran NA, Menne Z, Kauffman RC, Affer M, Llovet A, Norwood C, Scanlan A, Mantus G, et al. Single-cell analysis suggests that ongoing affinity maturation drives the emergence of pemphigus vulgaris autoimmune disease. Cell Rep. 2019;28:909–922.e906. doi: 10.1016/j.celrep.2019.06.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cho MJ, Lo AS, Mao X, Nagler AR, Ellebrecht CT, Mukherjee EM, Hammers CM, Choi EJ, Sharma PM, Uduman M, et al. Shared VH1-46 gene usage by pemphigus vulgaris autoantibodies indicates common humoral immune responses among patients. Nat Commun. 2014;5:4167. doi: 10.1038/ncomms5167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kim AR, Han D, Choi JY, Seok J, Kim SE, Seo SH, Takahashi H, Amagai M, Park SH, Kim SC, et al. Targeting inducible costimulator expressed on cxcr5(+)pd-1(+) th cells suppresses the progression of pemphigus vulgaris. J Allergy Clin Immunol. 2020;146:1070–1079.e1078. doi: 10.1016/j.jaci.2020.03.036. [DOI] [PubMed] [Google Scholar]
- 68.Holstein J, Solimani F, Baum C, Meier K, Pollmann R, Didona D, Tekath T, Dugas M, Casadei N, Hudemann C, et al. Immunophenotyping in pemphigus reveals a TH17/TFH17 cell-dominated immune response promoting desmoglein1/3-specific autoantibody production. J Allergy Clin Immunol. 2021;147:2358–2369. doi: 10.1016/j.jaci.2020.11.008. [DOI] [PubMed] [Google Scholar]
- 69.Kim MR, Kim HC, Kim SC. Long-term prognosis of pemphigus in Korea: retrospective analysis of 199 patients. Dermatology. 2011;223:182–188. doi: 10.1159/000332848. [DOI] [PubMed] [Google Scholar]
- 70.Amagai M, Ikeda S, Shimizu H, Iizuka H, Hanada K, Aiba S, Kaneko F, Izaki S, Tamaki K, Ikezawa Z, et al. A randomized double-blind trial of intravenous immunoglobulin for pemphigus. J Am Acad Dermatol. 2009;60:595–603. doi: 10.1016/j.jaad.2008.09.052. [DOI] [PubMed] [Google Scholar]
- 71.Joly P, Mouquet H, Roujeau JC, D’Incan M, Gilbert D, Jacquot S, Gougeon ML, Bedane C, Muller R, Dreno B, et al. A single cycle of rituximab for the treatment of severe pemphigus. N Engl J Med. 2007;357:545–552. doi: 10.1056/NEJMoa067752. [DOI] [PubMed] [Google Scholar]
- 72.Kim JH, Kim YH, Kim MR, Kim SC. Clinical efficacy of different doses of rituximab in the treatment of pemphigus: a retrospective study of 27 patients. Br J Dermatol. 2011;165:646–651. doi: 10.1111/j.1365-2133.2011.10411.x. [DOI] [PubMed] [Google Scholar]
- 73.Werth VP, Joly P, Mimouni D, Maverakis E, Caux F, Lehane P, Gearhart L, Kapre A, Pordeli P, Chen DM, et al. Rituximab versus mycophenolate mofetil in patients with pemphigus vulgaris. N Engl J Med. 2021;384:2295–2305. doi: 10.1056/NEJMoa2028564. [DOI] [PubMed] [Google Scholar]
- 74.Wang HH, Liu CW, Li YC, Huang YC. Efficacy of rituximab for pemphigus: a systematic review and meta-analysis of different regimens. Acta Derm Venereol. 2015;95:928–932. doi: 10.2340/00015555-2116. [DOI] [PubMed] [Google Scholar]
- 75.Joly P, Maho-Vaillant M, Prost-Squarcioni C, Hebert V, Houivet E, Calbo S, Caillot F, Golinski ML, Labeille B, Picard-Dahan C, et al. First-line rituximab combined with short-term prednisone versus prednisone alone for the treatment of pemphigus (Ritux 3): a prospective, multicentre, parallel-group, open-label randomised trial. Lancet. 2017;389:2031–2040. doi: 10.1016/S0140-6736(17)30070-3. [DOI] [PubMed] [Google Scholar]
- 76.Schmidt E, Kasperkiewicz M, Joly P. Pemphigus. Lancet. 2019;394:882–894. doi: 10.1016/S0140-6736(19)31778-7. [DOI] [PubMed] [Google Scholar]
- 77.Ellebrecht CT, Bhoj VG, Nace A, Choi EJ, Mao X, Cho MJ, Di Zenzo G, Lanzavecchia A, Seykora JT, Cotsarelis G, et al. Reengineering chimeric antigen receptor T cells for targeted therapy of autoimmune disease. Science. 2016;353:179–184. doi: 10.1126/science.aaf6756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee J, Lundgren DK, Mao X, Manfredo-Vieira S, Nunez-Cruz S, Williams EF, Assenmacher CA, Radaelli E, Oh S, Wang B, et al. Antigen-specific B cell depletion for precision therapy of mucosal pemphigus vulgaris. J Clin Invest. 2020;130:6317–6324. doi: 10.1172/JCI138416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Murrell DF, Patsatsi A, Stavropoulos P, Baum S, Zeeli T, Kern JS, Roussaki-Schulze AV, Sinclair R, Bassukas ID, Thomas D, et al. Proof of concept for the clinical effects of oral rilzabrutinib, the first Bruton tyrosine kinase inhibitor for pemphigus vulgaris: the phase II BELIEVE study. Br J Dermatol. 2021;185:745–755. doi: 10.1111/bjd.20431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Yamagami J, Ujiie H, Aoyama Y, Ishii N, Tateishi C, Ishiko A, Ichijima T, Hagihara S, Hashimoto K, Amagai M. A multicenter, open-label, uncontrolled, single-arm phase 2 study of tirabrutinib, an oral Bruton’s tyrosine kinase inhibitor, in pemphigus. J Dermatol Sci. 2021;103:135–142. doi: 10.1016/j.jdermsci.2021.07.002. [DOI] [PubMed] [Google Scholar]
- 81.Goebeler M, Bata-Csörgő Z, De Simone C, Didona B, Remenyik E, Reznichenko N, Stoevesandt J, Ward ES, Parys W, de Haard H, et al. Treatment of pemphigus vulgaris and foliaceus with efgartigimod, a neonatal Fc receptor inhibitor: a phase II multicentre, open-label feasibility trial. Br J Dermatol. 2021 doi: 10.1111/bjd.20782. [DOI] [PubMed] [Google Scholar]
- 82.Werth VP, Culton DA, Concha JS, Graydon JS, Blumberg LJ, Okawa J, Pyzik M, Blumberg RS, Hall RP., 3rd Safety, tolerability, and activity of alxn1830 targeting the neonatal fc receptor in chronic pemphigus. J Invest Dermatol. 2021;141:2858–2865.e4. doi: 10.1016/j.jid.2021.04.031. [DOI] [PubMed] [Google Scholar]
- 83.Lee H, Chung HJ, Pawar A, Patorno E, Kim DH. Evaluation of risk of bullous pemphigoid with initiation of dipeptidyl peptidase-4 inhibitor vs second-generation sulfonylurea. JAMA Dermatol. 2020;156:1107–1114. doi: 10.1001/jamadermatol.2020.2158. [DOI] [PubMed] [Google Scholar]
- 84.Wongvibulsin S, Pahalyants V, Kalinich M, Murphy W, Yu KH, Wang F, Chen ST, Reynolds K, Kwatra SG, Semenov YR. Epidemiology and risk factors for the development of cutaneous toxicities in patients treated with immune-checkpoint inhibitors: a United States population-level analysis. J Am Acad Dermatol. 2021 doi: 10.1016/j.jaad.2021.03.094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lai YC, Yew YW, Lambert WC. Bullous pemphigoid and its association with neurological diseases: a systematic review and meta-analysis. J Eur Acad Dermatol Venereol. 2016;30:2007–2015. doi: 10.1111/jdv.13660. [DOI] [PubMed] [Google Scholar]
- 86.Tedbirt B, Gillibert A, Andrieu E, Hébert V, Bastos S, Korman NJ, Tang MB, Li J, Borradori L, Cortés B, et al. Mixed individual-aggregate data on all-cause mortality in bullous pemphigoid: a meta-analysis. JAMA Dermatol. 2021;157:421–430. doi: 10.1001/jamadermatol.2020.5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kobayashi M, Amagai M, Kuroda-Kinoshita K, Hashimoto T, Shirakata Y, Hashimoto K, Nishikawa T. BP180 ELISA using bacterial recombinant NC16a protein as a diagnostic and monitoring tool for bullous pemphigoid. J Dermatol Sci. 2002;30:224–232. doi: 10.1016/s0923-1811(02)00109-3. [DOI] [PubMed] [Google Scholar]
- 88.van Beek N, Lüttmann N, Huebner F, Recke A, Karl I, Schulze FS, Zillikens D, Schmidt E. Correlation of serum levels of IgE autoantibodies against BP180 with bullous pemphigoid disease activity. JAMA Dermatol. 2017;153:30–38. doi: 10.1001/jamadermatol.2016.3357. [DOI] [PubMed] [Google Scholar]
- 89.Nishie W, Sawamura D, Goto M, Ito K, Shibaki A, McMillan JR, Sakai K, Nakamura H, Olasz E, Yancey KB, et al. Humanization of autoantigen. Nat Med. 2007;13:378–383. doi: 10.1038/nm1496. [DOI] [PubMed] [Google Scholar]
- 90.Fairley JA, Burnett CT, Fu CL, Larson DL, Fleming MG, Giudice GJ. A pathogenic role for IgE in autoimmunity: bullous pemphigoid IgE reproduces the early phase of lesion development in human skin grafted to nu/nu mice. J Invest Dermatol. 2007;127:2605–2611. doi: 10.1038/sj.jid.5700958. [DOI] [PubMed] [Google Scholar]
- 91.Nelson KC, Zhao M, Schroeder PR, Li N, Wetsel RA, Diaz LA, Liu Z. Role of different pathways of the complement cascade in experimental bullous pemphigoid. J Clin Invest. 2006;116:2892–2900. doi: 10.1172/JCI17891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Leyendeckers H, Tasanen K, Bruckner-Tuderman L, Zillikens D, Sitaru C, Schmitz J, Hunzelmann N. Memory B cells specific for the NC16A domain of the 180 kDa bullous pemphigoid autoantigen can be detected in peripheral blood of bullous pemphigoid patients and induced in vitro to synthesize autoantibodies. J Invest Dermatol. 2003;120:372–378. doi: 10.1046/j.1523-1747.2003.12071.x. [DOI] [PubMed] [Google Scholar]
- 93.Liu Z, Dang E, Li B, Qiao H, Jin L, Zhang J, Wang G. Dysfunction of CD19+CD24hiCD27+ B regulatory cells in patients with bullous pemphigoid. Sci Rep. 2018;8:703. doi: 10.1038/s41598-018-19226-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Muramatsu K, Ujiie H, Kobayashi I, Nishie W, Izumi K, Ito T, Yoshimoto N, Natsuga K, Iwata H, Shimizu H. Regulatory t-cell dysfunction induces autoantibodies to bullous pemphigoid antigens in mice and human subjects. J Allergy Clin Immunol. 2018;142:1818–1830.e1816. doi: 10.1016/j.jaci.2018.03.014. [DOI] [PubMed] [Google Scholar]
- 95.Haeberle S, Wei X, Bieber K, Goletz S, Ludwig RJ, Schmidt E, Enk AH, Hadaschik EN. Regulatory t-cell deficiency leads to pathogenic bullous pemphigoid antigen 230 autoantibody and autoimmune bullous disease. J Allergy Clin Immunol. 2018;142:1831–1842.e1837. doi: 10.1016/j.jaci.2018.04.006. [DOI] [PubMed] [Google Scholar]
- 96.McGinness JL, Bivens MM, Greer KE, Patterson JW, Saulsbury FT. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked syndrome (IPEX) associated with pemphigoid nodularis: a case report and review of the literature. J Am Acad Dermatol. 2006;55:143–148. doi: 10.1016/j.jaad.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 97.Joly P, Roujeau JC, Benichou J, Picard C, Dreno B, Delaporte E, Vaillant L, D’Incan M, Plantin P, Bedane C, et al. A comparison of oral and topical corticosteroids in patients with bullous pemphigoid. N Engl J Med. 2002;346:321–327. doi: 10.1056/NEJMoa011592. [DOI] [PubMed] [Google Scholar]
- 98.Feliciani C, Joly P, Jonkman MF, Zambruno G, Zillikens D, Ioannides D, Kowalewski C, Jedlickova H, Kárpáti S, Marinovic B, et al. Management of bullous pemphigoid: the European Dermatology Forum consensus in collaboration with the European Academy of Dermatology and Venereology. Br J Dermatol. 2015;172:867–877. doi: 10.1111/bjd.13717. [DOI] [PubMed] [Google Scholar]
- 99.Williams HC, Wojnarowska F, Kirtschig G, Mason J, Godec TR, Schmidt E, Chalmers JR, Childs M, Walton S, Harman K, et al. Doxycycline versus prednisolone as an initial treatment strategy for bullous pemphigoid: a pragmatic, non-inferiority, randomised controlled trial. Lancet. 2017;389:1630–1638. doi: 10.1016/S0140-6736(17)30560-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sticherling M, Franke A, Aberer E, Gläser R, Hertl M, Pfeiffer C, Rzany B, Schneider S, Shimanovich I, Werfel T, et al. An open, multicentre, randomized clinical study in patients with bullous pemphigoid comparing methylprednisolone and azathioprine with methylprednisolone and dapsone. Br J Dermatol. 2017;177:1299–1305. doi: 10.1111/bjd.15649. [DOI] [PubMed] [Google Scholar]
- 101.Park CS, Kim SH, Lee CK. Immunotherapy of autoimmune diseases with nonantibiotic properties of tetracyclines. Immune Netw. 2020;20:e47. doi: 10.4110/in.2020.20.e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yoo DS, Lee JH, Kim SC, Kim JH. Mortality and clinical response of patients with bullous pemphigoid treated with rituximab. Br J Dermatol. 2021;185:210–212. doi: 10.1111/bjd.19890. [DOI] [PubMed] [Google Scholar]
- 103.Amagai M, Ikeda S, Hashimoto T, Mizuashi M, Fujisawa A, Ihn H, Matsuzaki Y, Ohtsuka M, Fujiwara H, Furuta J, et al. A randomized double-blind trial of intravenous immunoglobulin for bullous pemphigoid. J Dermatol Sci. 2017;85:77–84. doi: 10.1016/j.jdermsci.2016.11.003. [DOI] [PubMed] [Google Scholar]
- 104.De D, Kaushik A, Handa S, Mahajan R, Schmidt E. Omalizumab: an underutilized treatment option in bullous pemphigoid patients with co-morbidities. J Eur Acad Dermatol Venereol. 2021;35:e469–e472. doi: 10.1111/jdv.17229. [DOI] [PubMed] [Google Scholar]
- 105.Freire PC, Munoz CH, Derhaschnig U, Schoergenhofer C, Firbas C, Parry GC, Panicker S, Gilbert JC, Stingl G, Jilma B, et al. Specific inhibition of the classical complement pathway prevents c3 deposition along the dermal-epidermal junction in bullous pemphigoid. J Invest Dermatol. 2019;139:2417–2424.e2412. doi: 10.1016/j.jid.2019.04.025. [DOI] [PubMed] [Google Scholar]
- 106.Mirzoyev SA, Schrum AG, Davis MD, Torgerson RR. Lifetime incidence risk of alopecia areata estimated at 2.1% by Rochester Epidemiology Project, 1990–2009. J Invest Dermatol. 2014;134:1141–1142. doi: 10.1038/jid.2013.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Trüeb RM, Dias MF. Alopecia areata: a comprehensive review of pathogenesis and management. Clin Rev Allergy Immunol. 2018;54:68–87. doi: 10.1007/s12016-017-8620-9. [DOI] [PubMed] [Google Scholar]
- 108.Martinez-Mir A, Zlotogorski A, Gordon D, Petukhova L, Mo J, Gilliam TC, Londono D, Haynes C, Ott J, Hordinsky M, et al. Genomewide scan for linkage reveals evidence of several susceptibility loci for alopecia areata. Am J Hum Genet. 2007;80:316–328. doi: 10.1086/511442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Eckert J, Church RE, Ebling FJ. The pathogenesis of alopecia areata. Br J Dermatol. 1968;80:203–210. doi: 10.1111/j.1365-2133.1968.tb11960.x. [DOI] [PubMed] [Google Scholar]
- 110.Whiting DA. Histopathologic features of alopecia areata: a new look. Arch Dermatol. 2003;139:1555–1559. doi: 10.1001/archderm.139.12.1555. [DOI] [PubMed] [Google Scholar]
- 111.Paus R, Ito N, Takigawa M, Ito T. The hair follicle and immune privilege. J Investig Dermatol Symp Proc. 2003;8:188–194. doi: 10.1046/j.1087-0024.2003.00807.x. [DOI] [PubMed] [Google Scholar]
- 112.Bodemer C, Peuchmaur M, Fraitaig S, Chatenoud L, Brousse N, De Prost Y. Role of cytotoxic T cells in chronic alopecia areata. J Invest Dermatol. 2000;114:112–116. doi: 10.1046/j.1523-1747.2000.00828.x. [DOI] [PubMed] [Google Scholar]
- 113.Gilhar A, Laufer-Britva R, Keren A, Paus R. Frontiers in alopecia areata pathobiology research. J Allergy Clin Immunol. 2019;144:1478–1489. doi: 10.1016/j.jaci.2019.08.035. [DOI] [PubMed] [Google Scholar]
- 114.Petukhova L, Duvic M, Hordinsky M, Norris D, Price V, Shimomura Y, Kim H, Singh P, Lee A, Chen WV, et al. Genome-wide association study in alopecia areata implicates both innate and adaptive immunity. Nature. 2010;466:113–117. doi: 10.1038/nature09114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Xing L, Dai Z, Jabbari A, Cerise JE, Higgins CA, Gong W, de Jong A, Harel S, DeStefano GM, Rothman L, et al. Alopecia areata is driven by cytotoxic T lymphocytes and is reversed by JAK inhibition. Nat Med. 2014;20:1043–1049. doi: 10.1038/nm.3645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ito T, Ito N, Saatoff M, Hashizume H, Fukamizu H, Nickoloff BJ, Takigawa M, Paus R. Maintenance of hair follicle immune privilege is linked to prevention of NK cell attack. J Invest Dermatol. 2008;128:1196–1206. doi: 10.1038/sj.jid.5701183. [DOI] [PubMed] [Google Scholar]
- 117.Borcherding N, Crotts SB, Ortolan LS, Henderson N, Bormann NL, Jabbari A. A transcriptomic map of murine and human alopecia areata. JCI Insight. 2020;5:e137424. doi: 10.1172/jci.insight.137424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee H, Jeong S, Shin EC. Significance of bystander t cell activation in microbial infection. Nat Immunol. 2021 doi: 10.1038/s41590-021-00985-3. [DOI] [PubMed] [Google Scholar]
- 119.Freyschmidt-Paul P, McElwee KJ, Hoffmann R, Sundberg JP, Vitacolonna M, Kissling S, Zöller M. Interferon-gamma-deficient mice are resistant to the development of alopecia areata. Br J Dermatol. 2006;155:515–521. doi: 10.1111/j.1365-2133.2006.07377.x. [DOI] [PubMed] [Google Scholar]
- 120.Rajabi F, Drake LA, Senna MM, Rezaei N. Alopecia areata: a review of disease pathogenesis. Br J Dermatol. 2018;179:1033–1048. doi: 10.1111/bjd.16808. [DOI] [PubMed] [Google Scholar]
- 121.Alkhalifah A, Alsantali A, Wang E, McElwee KJ, Shapiro J. Alopecia areata update: part II. Treatment. J Am Acad Dermatol. 2010;62:191–202. doi: 10.1016/j.jaad.2009.10.031. [DOI] [PubMed] [Google Scholar]
- 122.Kim BJ, Min SU, Park KY, Choi JW, Park SW, Youn SW, Park KC, Huh CH. Combination therapy of cyclosporine and methylprednisolone on severe alopecia areata. J Dermatolog Treat. 2008;19:216–220. doi: 10.1080/09546630701846095. [DOI] [PubMed] [Google Scholar]
- 123.Joly P. The use of methotrexate alone or in combination with low doses of oral corticosteroids in the treatment of alopecia totalis or universalis. J Am Acad Dermatol. 2006;55:632–636. doi: 10.1016/j.jaad.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 124.Gordon PM, Aldrige RD, McVittie E, Hunter JA. Topical diphencyprone for alopecia areata: evaluation of 48 cases after 30 months’ follow-up. Br J Dermatol. 1996;134:869–871. [PubMed] [Google Scholar]
- 125.Uchiyama M, Egusa C, Hobo A, Irisawa R, Yamazaki M, Tsuboi R. Multivariate analysis of prognostic factors in patients with rapidly progressive alopecia areata. J Am Acad Dermatol. 2012;67:1163–1173. doi: 10.1016/j.jaad.2012.06.006. [DOI] [PubMed] [Google Scholar]
- 126.Wang EH, Sallee BN, Tejeda CI, Christiano AM. Jak inhibitors for treatment of alopecia areata. J Invest Dermatol. 2018;138:1911–1916. doi: 10.1016/j.jid.2018.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Leonard WJ, Lin JX. Cytokine receptor signaling pathways. J Allergy Clin Immunol. 2000;105:877–888. doi: 10.1067/mai.2000.106899. [DOI] [PubMed] [Google Scholar]
- 128.Schwartz DM, Kanno Y, Villarino A, Ward M, Gadina M, O’Shea JJ. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat Rev Drug Discov. 2017;17:78. doi: 10.1038/nrd.2017.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Liu LY, Craiglow BG, Dai F, King BA. Tofacitinib for the treatment of severe alopecia areata and variants: a study of 90 patients. J Am Acad Dermatol. 2017;76:22–28. doi: 10.1016/j.jaad.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 130.Almutairi N, Nour TM, Hussain NH. Janus kinase inhibitors for the treatment of severe alopecia areata: An open-label comparative study. Dermatology. 2019;235:130–136. doi: 10.1159/000494613. [DOI] [PubMed] [Google Scholar]
- 131.King B, Ko J, Forman S, Ohyama M, Mesinkovska N, Yu G, McCollam J, Gamalo M, Janes J, Edson-Heredia E, et al. Efficacy and safety of the oral Janus kinase inhibitor baricitinib in the treatment of adults with alopecia areata: Phase 2 results from a randomized controlled study. J Am Acad Dermatol. 2021;85:847–853. doi: 10.1016/j.jaad.2021.05.050. [DOI] [PubMed] [Google Scholar]
- 132.King B, Guttman-Yassky E, Peeva E, Banerjee A, Sinclair R, Pavel AB, Zhu L, Cox LA, Craiglow B, Chen L, et al. A phase 2a randomized, placebo-controlled study to evaluate the efficacy and safety of the oral Janus kinase inhibitors ritlecitinib and brepocitinib in alopecia areata: 24-week results. J Am Acad Dermatol. 2021;85:379–387. doi: 10.1016/j.jaad.2021.03.050. [DOI] [PubMed] [Google Scholar]