

Abstract
Mammalian innate immune response to virus infection is meditated by many cell-intrinsic pathways. RNA viruses, such as Sendai virus, which replicate in the cytoplasm, trigger the RIG-I-like receptor pathway, which activates the transcription factor, IRF3. Activated IRF3 translocates to the nucleus and induces transcription of the genes which encode interferons, the major antiviral cytokines. Interestingly, IRF3 activates another interferon-independent antiviral pathway, called RIG-I induced pathway of apoptosis (RIPA). For activating RIPA, IRF3 translocates from the cytoplasm to mitochondria. RIPA requires linear polyubiquitination of IRF3 by the enzyme complex, LUBAC; ubiquitinated IRF3 binds to Bax and translocates it to mitochondria causing the release of Cytochrome C, activation of caspases and apoptosis of the infected cell. Here, we report that Otulin, the deubiquitinase that removes linear polyubiquitin chains, inhibits RIPA by deubiquitinating IRF3. Ablation of Otulin expression enhanced RIPA and its overexpression inhibited RIPA. In virus-infected cells, to overcome Otulin-mediated inhibition, RIPA actively degrades Otulin. This degradation required sequential actions of RIPA-activated Caspase 3 and proteasomes. Caspase 3 cleaved Otulin at D31; the D31A mutant was not cleaved at all. The caspase-cleaved fragment was totally degraded by proteasomes, which was preceded by its K48-linked ubiquitination. Mass spectrometric analysis of Otulin identified K64 and K197 as the ubiquitinated residues. Otulin interacted with LUBAC after virus infection and the E3-ubiquitin ligase, HOIP, a component of LUBAC, ubiquitinated Otulin to trigger its proteasome-mediated degradation. To assess the impact of Otulin degradation on RIPA-mediated antiviral action, we expressed, in Otulin-ablated cells, a non-degradable mutant of Otulin, in which D31, K64 and K197 had been mutated. The cells expressing the Otulin mutant were less susceptible to virus-induced apoptosis, because RIPA was less active, and consequently virus replication was more robust. Thus, our study has revealed an important positive feedback loop of RIPA.
Subject terms: Cell death and immune response
Introduction
Viral infection in host cells triggers antiviral response after recognition of the pathogen-associated molecular patterns (PAMPs) by different host-pathogen recognition receptors (PRRs), such as RIG-I-like receptors (RLRs), cGAS/STING, and Toll-like receptors (TLRs) [1–3]. RLRs are cytoplasmic sensors of viral RNA having a DExD/H box helicase domain and a carboxyl-terminal CARD domain [4, 5]. Many RNA viruses, such as Sendai virus, Dengue virus, and Influenza virus are recognized by the RLR pathway [6, 7]. After recognition of viral RNA, RLR signaling activates transcription factors, including IRF3 and NF-ΚB, which then induce the synthesis of different interferons (IFN) and the IFN-stimulated genes (ISGs) to inhibit viral infection [8, 9]. IRF3, the major transcription factor that induces IFN, is activated through its phosphorylation by RLR signaling. Activation of RIG-I by viral RNA leads to recruitment of RIG-I to MAVS to activate the cytoplasmic kinase TBK1. Activated TBK1 phosphorylates IRF3 at specific serine residues. Phosphorylated IRF3 dimerizes and translocates from the cytoplasm to the nucleus and binds to IFN-stimulated response elements (ISRE) of target genes to induce type-I IFN production [10, 11].
IRF3 can also trigger a non-transcriptional IFN-independent antiviral pathway, called RLR-induced IRF3 mediated Pathway of Apoptosis (RIPA) [12–14]. RIPA controls viral replication by triggering apoptosis of virus-infected cells and does not require IFN or ISGs [15]. RIPA activation requires several RLR-transcriptional pathway components, such as MAVS, TRAF3, and TBK1, in addition to TRAF2 and TRAF6 [12, 16]. In RIPA, IRF3 is recruited by the ubiquitinating LUBAC complex, consisting of HOIP, HOIL, and Sharpin. HOIP, an E3 ligase, attaches linear polyubiquitin chains on IRF3, which activates it to interact with the pro-apoptotic protein, Bax. The activated IRF3-Bax complex translocates to mitochondria leading to the release of Cytochrome C in the cytoplasm [12, 13, 16]. Cytoplasmic Cytochrome C activates the apoptosome complex, composed of APAF-1, pro-caspase 9, pro-caspase 3 and an inhibitor of apoptotic caspases, such as XIAP, which prevents pro-caspase 9 cleavage and activation of the apoptotic cascade [17, 18]. The apoptotic activity of RIPA has been shown to be temporally regulated. In the early phase of infection, RIPA is blocked by XIAP. Moreover, viruses activate the PI3K/AKT pathway in the early phase of infection to stabilize XIAP, thereby preventing early cell death. However, during the late phase of infection, XIAP is degraded to relieve the inhibition of RIPA. If the PI3K/AKT axis is inhibited, RIPA causes early apoptosis of virus-infected cells [19]. Using cells and mice expressing an IRF3 mutant, which cannot be activated as a transcription factor, it was firmly established that RIPA mediates significant antiviral effects [15, 16, 20].
RIPA has been also implicated in metabolic liver diseases, such as alcoholic hepatitis (AH) and alcohol-related liver disease (ALD), where IRF3 transcription-independent functions have been shown to differentially contribute to ethanol-induced liver injury and high-fat diet (HFD)-induced liver injury models. In Gao-binge liver injury model, RIPA- mediated apoptosis of restorative Ly6Clow monocytes shifts the immune environment to more pro-inflammatory state thereby exacerbating the alcoholic hepatitis [21]. In contrast in HFD mediated liver injury model, non-transcriptional IRF3 mice (IRF3S1/S1) were protected [22]. The differences have been shown to be associated with increased or decreased ubiquitination of IRF3.
RLR-signaling requires ubiquitination of RIG-I and other signaling proteins to promote activation of IRF3 as a transcription factor [23, 24]. This process is regulated by specific deubiquitinases (DUB), such as CYLD and USP15, which remove the polyubiquitin chains from the signaling proteins [25–27]. We inquired whether IRF3 activation in RIPA, through its linear polyubiquitination, was similarly regulated by a DUB. Otulin is a DUB that can remove linear polyubiquitin chains from signaling proteins, such as STAT1 and Nemo [28–32]. We report here that Otulin inhibits RIPA, by deubiquitinating IRF3. However, to evade this negative regulation, enzymes activated in RIPA actively degrade Otulin, thereby creating a positive feed-back loop to RIPA’s antiviral action.
Results
Otulin attenuates the RIPA pathway
In the RIPA pathway, linear polyubiquitin chains are added to IRF3 by the LUBAC complex and we were interested to learn whether RIPA can be negatively regulated by the removal of polyubiquitin chains from IRF3 by a deubiquitinase. Otulin is a deubiquitinase that can cleave linear polyubiquitin chains from its substrates and we tested its ability to reduce IRF3 linear polyubiquitination (polyUb). Sendai virus (SeV) infection, which activates RIPA, was performed in human HT1080 cells with or without ectopic expression of Otulin, followed by immunoprecipitation of IRF3 from the extracts of these cells. We observed that in Otulin over-expressing cells, IRF3 linear polyUb was decreased significantly, indicating that Otulin inhibited IRF3 linear polyUb (Fig. 1A). Total linear ubiquitination of the input was also assessed, with and without SeV infection, as shown in the right panel of Fig. 1A. We observed minor changes in total linear ubiquitination of the input as expected; however the corresponding lanes of IRF3 linear ubiquitination (Fig. 1A, left panel) showed significant changes indicating the effect of Otulin on IRF3 to be a specific effect. To further confirm this observation, we generated Otulin knockdown (OT-KD) cells (Supplementary Fig. S1A). We observed that IRF3 linear polyUb was significantly enhanced in OT-KD cells, compared to WT cells, thereby indicating a physiological role of Otulin in regulating IRF3 polyUb (Fig. 1B). We used high salt washes (1 M NaCl) to detect IRF3 linear poly-ubiquitination in HT0180 WT and OT-KD cells during RIPA activation to rule out the contribution of bait-interacting proteins and we found similar results as observed for Fig. 1B (data not shown). To demonstrate the dependence of IRF3 linear polyUb on the enzymatic activity of Otulin, OT-KD cells were reconstituted with either Otulin WT or the enzymatically inactive Otulin C129A mutant. SeV infection of cells expressing Otulin C129A showed an IRF3 linear polyUb level similar to that in OT-KD cells, whereas in cells expressing WT Otulin, IRF3 linear polyUb was much reduced (Fig. 1C). After confirming the role of Otulin on IRF3 linear ubiquitination, we performed a co-immunoprecipitation assay to investigate Otulin-IRF3 interaction during RIPA activation. The result showed strong interaction of IRF3 with Otulin (Fig. 1D). Because RIPA activation induces apoptosis, we analyzed, as a measure of apoptosis, cleaved PARP and cleaved Caspase 3 levels in HT1080 WT and OT-KD cells, post-SeV infection. We found that OT-KD cells had increased levels of cleaved PARP and cleaved Caspase 3, indicating enhanced apoptosis (Fig. 1E). We also analyzed apoptotic cell death, in HT080 WT and OT-KD cells post-SeV infection, by Annexin-V staining. OT-KD cells showed more cell death than HT080 WT (Supplementary Fig. S2E) whereas IFN production was similar in both cell lines (Supplementary Figs. S2F, S2G, and S2H). This enhanced apoptosis in OT-KD cells inhibited virus replication, as measured by SeV-P mRNA (Supplementary Fig. S1B) and protein expression (Fig. 1F). As expected, these cells produced less infectious virus (Fig. 1G). RIPA requires an interaction of IRF3 with Bax and its translocation to mitochondria. We observed that IRF3 interaction with Bax and its mitochondrial localization were more pronounced in infected OT-KD cells, compared to WT cells (Supplementary Fig. S1C, D). We also tested whether the anti-apoptotic effect of Otulin was dependent on IRF3. We observed that the absence of Otulin expression enhanced apoptosis and inhibited viral replication in HT1080 WT cells, but not in HT1080sh.IRF3 cells, in which IRF3 expression had been knocked down (Supplementary Fig. S1E). The above results indicate that Otulin specifically attenuates RIPA pathway to support viral replication.
Fig. 1. Otulin attenuates RIPA pathway.

A IRF3 linear ubiquitination was analyzed in HT1080 cell line transfected with either empty vector (lanes 1, 2, 3 and 4) or HA-Otulin (lane 5) followed by Sendai virus (SeV) infection (lanes 2, 4 and 5) for 24 h (MOI 10). Immunoprecipitation was performed with IRF3 antibody and immunoblotting was done with LUB9 antibody for detection of linear ubiquitination. Right panel shows the total linear ubiquitination of the input. B IRF3 linear ubiquitination was assessed in HT1080 WT and HT1080 Otulin-knockdown (OT-KD) cells post-SeV infection, using LUB9 antibody. C HT1080 Otulin knockdown cells (OT-KD) were reconstituted with either Otulin WT or enzymatically inactive mutant Otulin C129A and IRF3 linear ubiquitination was assessed by LUB9 antibody post SeV infection. D Otulin-IRF3 interaction was assayed by their co-immunoprecipitation from extracts of HT1080 cells that had been infected with SeV for 1 h and 3 h. E HT1080 WT and HT1080 OT-KD cells were either mock or SeV infected and C-PARP and C-caspase 3 levels were measured for assessing apoptotic cell death at the indicated time points. F SeV-C protein levels were assessed post-SeV infection in WT and OT-KD cells as a measure of viral protein synthesis at the indicated time points. G SeV yields were measured after infection of WT and OT-KD cells with SeV for 24 h (MOI 10). Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
RIPA causes degradation of Otulin
As Otulin appeared to be a crucial player in the regulation of the RIPA pathway, we inquired whether cellular Otulin levels were affected by RIPA activation, by measuring Otulin protein levels at different time points after SeV infection. Interestingly, we found that Otulin was degraded in infected cells (Fig. 2A). To test whether RLR activation causes this degradation, we used the synthetic dsRNA, poly I:C, to activate it. After poly I:C transfection in HT1080 cells, Otulin degradation was observed (Supplementary Fig. S2A), suggesting that Otulin degradation takes place upon RLR activation. Similarly in mouse L929 cells, both SeV infection and poly I:C transfection caused degradation of Otulin, confirming that RLR activation degrades Otulin in both human and mouse cell lines (Supplementary Fig. S2B, C). Next, we inquired whether Otulin degradation was caused by RIPA, by testing the needs of different components of RIPA for this process. HT1080 RIG-IC cells express a RIG-I mutant that lacks its CARD domains and acts as a dominant-negative inhibitor of RIG-I. We observed that Otulin degradation was absent in RIG-IC cells (Fig. 2B). As expected, PARP cleavage was inhibited in these cells as well. This result indicates that Otulin degradation is dependent on RIG-I activation and it correlates with RIPA activation, as shown by PARP cleavage. Similarly, two other essential components of RIPA, IRF3 and Bax, were required for Otulin degradation in infected cells (Fig. 2C, D). To further confirm RIPA-dependence of Otulin degradation, we used RIPA deficient HT1080 cell lines expressing the IRF3 KK313/315RR and IRF3 K193R mutants [16]. As expected, we observed that there was no Otulin degradation in RIPA deficient cell lines (Fig. 2E, F). The dependence of Otulin degradation on the transcriptional activity of IRF3 was ruled out by testing HT1080 and MEF cells expressing the transcriptionally inert IRF3 S1 mutant; in both these cell lines Otulin was degraded (Fig. 2G, Supplementary Fig. S2D). As expected, IFN was not induced in these cells (Supplementary Figs. S2F, S2G and S2H). Moreover, Otulin knockdown in IRF3 S1 cells and WT cells caused similar apoptosis as indicated by C-PARP levels and Annexin-V staining (Fig. 2G, Supplementary Figs. S2D and S2E) indicating that Otulin-mediated anti-apoptotic function does not require transcriptional activity of IRF3. Further, Otulin degradation appears to be a specific mechanism to regulate RIPA, because apoptosis activation by Staurosporine treatment did not trigger degradation of Otulin (Fig. 2H and Supplementary Fig. S2I). These data strongly suggest that Otulin degradation in SeV-infected cells is mediated by RIPA.
Fig. 2. RIPA causes degradation of Otulin.

A HT1080 cells were infected with SeV (MOI 10) for the indicated time points and analyzed for Otulin expression, using Otulin antibody. Viral protein synthesis was assessed with SeV C antibody. B Otulin degradation and PARP cleavage were analyzed in HT1080 WT or HT1080 cells expressing RIG-Ic (RIG-I dominant-negative mutant); cells were either mock or SeV infected for 24 h. Otulin degradation was assessed with Otulin antibody while Cleaved PARP levels were assessed as a measure for apoptosis. C Otulin degradation and PARP cleavage were analyzed in MEF WT or MEF IRF3-KO cells, which were either mock or SeV infected for 24 h. Otulin degradation was assessed with Otulin antibody while C-PARP levels were assessed as a measure for apoptosis. D Otulin degradation and PARP cleavage were analyzed in MEF WT or MEF Bax-KO cells, which were either mock or SeV infected for 24 h. Otulin degradation was assessed with Otulin antibody while C-PARP levels were assessed as a measure for apoptosis. E Otulin degradation and PARP cleavage were analyzed in HT1080 WT and RIPA deficient HT1080 IRF3-K1 cells, which were either mock or SeV infected for 24 h. F Analysis similar to E comparing WT and IRF3-K4 cells. G Otulin degradation and PARP cleavage were analyzed in HT1080 WT, HT1080-OT-KD, HT1080S1 and HT080S1-OT-KD cells after mock or SeV infection for 24 h. H Otulin degradation and PARP cleavage were analyzed in HT1080 cells which were either treated with Staurosporine (50 nM) or infected with SeV for 12 h.). Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
Both caspases and proteasome are needed for Otulin degradation
After confirming the role of RIPA, we examined the role of caspases on Otulin degradation, as RIPA leads to activation of caspases. HT1080 cells were pre-treated with the pan-caspase inhibitor, Z-VAD-FMK, for 1 h followed by SeV infection for 24 h with the inhibitor present throughout the infection. Z-VAD-FMK prevented Otulin degradation, suggesting a role of caspases (Fig. 3A, lane 3). As caspase-3 is a major player in RIPA pathway, we tested the role of this caspase in Otulin degradation by using caspase 3 knockdown cells and caspase-3 specific inhibitor Z-DEVD-FMK. Both caspase 3 knockdown and Z-DEVD-FMK prevented Otulin degradation during RIPA, indicating an essential role of caspase-3 (Fig. 3B, Supplementary Fig. S3A). Because caspase-3 is activated to mediate RIPA, we examined, by co-immunoprecipitation, the interaction of Otulin with caspase-3 during SeV infection in the presence or absence of Z-VAD-FMK. We found that caspase-3 and Otulin interacted during SeV infection (Fig. 3C, lane 3) and this interaction was significantly increased upon caspase inhibition (Fig. 3C, lane 4) indicating Z-VAD-FMK prevents pro-caspase-3 activation and hence Otulin degradation. C-caspase-3 which represents the activated form, also immunoprecipitated with Otulin during RIPA activation. These results indicate that caspase-3 interacts with Otulin during infection and causes its degradation.
Fig. 3. Both caspases and proteasome are needed for Otulin degradation.

A HT1080 cells were treated with the pan-caspase inhibitor Z-VAD-FMK (10 μM) and monitored for Otulin degradation post-SeV infection of 24 h (MOI 10); the treatment with the inhibitor started 1 h before and continued during virus infection. B Otulin degradation was analyzed in HT1080 WT and HT1080 Caspase 3 knockdown (KD) cells post-SeV infection of 24 h. C-caspase 3 was detected as a measure for caspase 3 activation. C Co-immunoprecipitation of Caspase 3 and Otulin in the presence and absence of Z-VAD-FMK during SeV infection for 6 h; the treatment with the inhibitor started 1 h before and continued during virus infection. D HT1080 cells were transfected with poly (I:C) (1 μg) for 24 h to activate RIPA. Z-VAD-FMK treatment started 1 h before transfection and continued throughout the transfection. MG132 (10 μM) treatment was given for 4 h before harvesting cells. Cell lysates were subjected to immunoblot analysis with Otulin antibody to determine full length and cleaved Otulin. Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
One of the major pathways for protein degradation is the proteasome pathway. To test if Otulin is degraded through proteasome pathway, we used the inhibitor, MG132, to block it. RIPA was activated in HT1080 cells by transfecting poly I:C. As expected, Otulin level was decreased (Fig. 3D, lane 2), but not if both caspases and proteasomes were inhibited (Fig. 3D, lane 4). Surprisingly, when only proteasomes were inhibited, a cleaved fragment of Otulin accumulated (Fig. 3D, lane 3). Similar results were obtained when RIPA was activated by SeV infection of cells treated with the inhibitors (Supplementary Fig. S3B), and the appearance of the cleaved fragment required both MG132 treatment and RIPA activation (Supplementary Fig. S3C). Proteasomal degradation of proteins is preceded by their K48-linked ubiquitination and as expected, Otulin was ubiquitinated in infected cells (Supplementary Fig. S3D). These results indicate that Otulin is cleaved by RIPA-activated caspase 3 first, to generate a cleaved fragment, which is ubiquitinated and further degraded by the proteasome.
Identification of the Caspase-3 target site in Otulin
From the previous results, it was clear that Otulin is cleaved by caspase 3 activated by RIPA and then degraded by proteasomes. To find out the caspase-3 target site in Otulin, we used N-terminal HA-tagged Otulin. After reconstitution of HA-Otulin in OT-KD cells, these cells were treated with MG132 and transfected with poly I:C for RIPA activation and assessed for Otulin cleavage using HA and Otulin antibody. Interestingly, we did not detect the cleaved fragment of Otulin using the HA antibody (Fig. 4A, top panel), whereas the Otulin antibody was able to detect it (Fig. 4A, middle panel), indicating that Otulin was cleaved by caspase 3 at a site near the N-terminus. A putative caspase target site, identified at residues 28–32 of human Otulin (Supplementary Fig. S4), was well conserved in Otulin of other species (Fig. 4B) indicating that D31 could be the cleavage site. Indeed, mutating it blocked Otulin cleavage by RIPA; the D31A Otulin mutant was not cleaved in SeV-infected cells in the presence of MG132 (Fig. 4C, lane 6) or its absence (Fig. 4C, lane 5). Therefore, D31 was identified as the target site of cleavage by caspase 3.
Fig. 4. Identification of the Caspase 3 target site in Otulin.

A HT1080-OT-KD cells expressing HA-Otulin were transfected with poly (I:C) (1 μg) for 24 h to activate RIPA and analyzed for Otulin cleavage using HA and Otulin antibody. MG132 treatment was given for 4 h before harvesting cells. B Multiple sequence alignments showing conservation of Caspase 3 site D31 in various organisms. C Otulin WT and Otulin D31A expressing OT-KD cells were infected with SeV (MOI 10) for 24 h followed by MG132 treatment of 4 h before harvesting the cells. Otulin cleavage was detected using Otulin antibody (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
Otulin is K48-ubiquitinated
The degradation of a protein through the proteasome pathway is usually preceded by its K48-linked ubiquitination. Therefore, we measured Otulin-K48 ubiquitination in SeV-infected cells in the presence or absence of MG132. Immunoblot analysis showed that Otulin was K48 ubiquitinated in the infected cells (Fig. 5A, lane 3); the K48 ubiquitination was more pronounced in infected cells that were treated with MG132 (Fig. 5A, lane 4), probably because ubiquitinated Otulin could not be degraded in these cells by proteasomes. We used high salt washes (1 M NaCl) to detect Otulin-K48 ubiquitination during RIPA activation to rule out the contribution of bait-interacting proteins and we found similar results as observed for Fig. 5A (data not shown). K48 poly-ubiquitination inputs did not reveal any difference between mock and SeV-infected cells (Supplementary Fig. S6A, lane 2 and 3), while MG132 treatment showed more K48 poly-ubiquitination as expected. These data indicate RIPA-mediated Otulin K48 poly-ubiquitination as a specific effect. We observed that Otulin K48 ubiquitination started early in the infection which then led to its degradation, while K48 poly-ubiquitination inputs remains unaffected in this process (Fig. 5B, Supplementary Fig. S6B). The nature of K48-ubiquitin linkages on Otulin was further confirmed by expressing His-Ub-WT or His-Ub-K48R in infected cells. Isolation of His-Ub, using Ni-NTA resin, revealed that Otulin was ubiquitinated by WT, but not K48R, Ub, thereby confirming the K48-nature of Otulin ubiquitination (Fig. 5C). To identify the E3 ligase that ubiquitinates Otulin, we considered HOIP, a component of LUBAC, as a candidate, because it can ligate K48Ub, as well as linear Ub, to its substrates [33–37]. Indeed, overexpression of HOIP, but not the other two components of LUBAC, Sharpin and HOIL, enhanced K48 ubiquitination of Otulin (Fig. 5D, lane 3). Here also, K48 poly-ubiquitination inputs did not show any significant change thereby suggesting HOIP as an E3-ligase to induce specifically Otulin K48 poly-ubiquitination during RIPA activation (Supplementary Fig. S6C). Moreover, as evidenced by their co-immunoprecipitation, endogenous HOIP was bound to endogenous Otulin in infected cells (Fig. 5E, lane 3), presumably along with the other two components of the LUBAC complex (Supplementary Fig. S5A, lane 3). The deubiquitinase activity of Otulin was not needed for binding to HOIP, as evidenced by HOIP interaction with the C129A mutant of Otulin (Supplementary Fig. S5B, lane 5). K48 ubiquitination of Otulin required RIPA activation, because it did not occur in infected cells expressing RIG-IC, the dominant-negative inhibitor of RIG-I (Supplementary Fig. S5C), or in cells devoid of IRF3 (Supplementary Fig. S5D). Moreover, prior cleavage of Otulin by caspase 3 was required for its K48-ubiquitination: blocking caspase 3 action by its inhibitor (Fig. 5F, lane 3) or by mutating its target site (D31) in Otulin (Fig. 5G, lane 3) eliminated K48 ubiquitination. These results indicated that the substrate for K48 ubiquitination was the caspase-cleaved Otulin, rather than Otulin itself. The ubiquitination inputs during RIPA activation for RIG-IC, shIRF3, Z-VAD-FMK and Otulin D31A showed similar K48 poly-ubiquitination levels indicating a role of RIPA and caspase-3 specifically in Otulin K48 poly-ubiquitination (Supplementary Fig. S6D, S6E, S6F, and S6G).
Fig. 5. Otulin is K48 ubiquitinated.

A Otulin K48 ubiquitination was analyzed in HT1080 cells after SeV infection (MOI 10) for 24 h followed by immunoprecipitation with Otulin antibody and immunoblotting with K-48 ubiquitin antibody. MG132 was added for 4 h before harvesting cells. B Otulin K48-ubiquitination was analyzed post-SeV infection for the indicated time points. Immunoprecipitation was performed with Otulin antibody and immunoblotting was done with K-48 ubiquitin antibody. C To show Otulin K48 ubiquitination after transfection of His-Ub WT and His-UbK48R in HT1080 cells overnight followed by SeV infection for 24 h. Cells were lysed in denaturing lysis buffer (6 M guanidine-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole; pH 8.0), sonicated, and centrifuged. His-Ub was pulled down with Ni-NTA resin and immunoblot analysis was done with Otulin antibody. D Otulin K48 ubiquitination was assessed in cells transfected with Flag-tagged HOIP, HOIL or Sharpin. HOIP/HOIL/Sharpin were transfected overnight followed by SeV infection for 24 h. E Immunoprecipitation assay to show an interaction of HOIP with Otulin post-SeV infection for 1 h. F Otulin K48 ubiquitination was analyzed in the presence or absence of Z-VAD-FMK post-SeV infection. Immunoprecipitation assay was performed with Otulin antibody and immunoblot analysis was done with K48-Ub antibody. G OT-KD cells reconstituted with Otulin WT and Otulin D31A were assessed for Otulin K48 ubiquitination post-SeV infection. Immunoprecipitation assay was performed with Otulin antibody and immunoblot analysis was done with K48-Ub antibody. Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
Identification of the K48 ubiquitination sites
To identify the lysine residues in Otulin that are K48-ubiquitination, we subjected Otulin, purified from SeV-infected cells, to LC-MS/MS analysis. Otulin-GFP was expressed in HT1080 cells followed by SeV infection. The purified Otulin was digested with trypsin and subjected to LC-MS/MS. Two ubiquitinated peptides corresponding to AADEIEKGlyGlyEKELLIHER and KGlyGlyKWAGLAEMR were detected by LC-MS/MS analysis. The lysine residues which showed ubiquitin linkages appeared to be K64 and K197 (Fig. 6A–C). To validate these sites, we generated the K64R and the K197R mutants of Otulin as well as a double mutant, K64R/K197R. These mutants were reconstituted in OT-KD cells followed by SeV infection. Immunoprecipitation of Otulin followed by immunoblot analysis with the K48 antibody showed that individual lysine mutation reduced K48 ubiquitination partially while the double mutations abolished it completely (Fig. 6D). These results suggest that K64 and K197 are the sites needed for Otulin K48 ubiquitination and both participate in Otulin degradation.
Fig. 6. Identification of the K48-ubiquitination sites.

A, B HT1080 cells were transfected with Otulin-GFP overnight followed by SeV infection for 12 h. MG132 was given for 4 h before harvesting cells. Otulin was purified with GFP-Trap beads, digested with trypsin and ubiquitinated peptides corresponding to (A) KGlyGlyKWAGLAEMR and (B) AADEIEKGlyGlyEKELLIHER and were detected by LC-MS/MS analysis. C Table showing peptides detected in LC-MS/MS analysis and the positions of lysine residues. D K48-Ubiquitination assay of Otulin WT and Otulin lysine mutants in reconstituted OT-KD cells to assess of the effect of lysine mutations on Otulin K48 ubiquitination post-SeV infection of 24 h. Immunoprecipitation assay was performed with Otulin antibody and immunoblot analysis was done with K48-Ub antibody. Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
Consequences of Otulin degradation on the RIPA pathway
Results, presented above, showed that Otulin degradation was dependent on caspase-3 mediated cleavage at D31 followed by its K48-ubiquitination at K64 and K197 by HOIP. We used this knowledge to design a triple mutant (TM) of Otulin, carrying the three mutations, D31A, K64R, and K197R, which was expected to be stable even in cells with activated RIPA. This mutant was expressed in OT-KD cells to generate a cell line expressing Otulin TM. Otulin-TM was stable for 24 h during viral infection, while degradation of Otulin WT was observed at as early as 6 h of infection (Fig. 7A). We observed that IRF3 linear polyubiquitination was significantly reduced in cells expressing Otulin-TM (Fig. 7B, lane 4), presumably because of a higher deubiquitinase activity of stable Otulin-TM. Consequently, RIPA-mediated apoptosis of the infected cells, as measured by C-PARP generation by PARP-cleavage and Annexin-V staining, was strongly inhibited in Otulin-TM expressing cells (Fig. 7C, Supplementary Fig. S7A). These cells expressed more SeV-P mRNA (Supplementary Fig. S6) and SeV C protein (Fig. 7D); more importantly, a higher level of infectious virus was produced by the cells expressing Otulin-TM (Fig. 7E). These results demonstrated that RIPA-mediated degradation of Otulin produces a strong positive feed-back loop to promote RIPA’s antiviral action.
Fig. 7. Consequences of Otulin degradation on the RIPA pathway.

A HT1080 OT-KD cells were reconstituted with Otulin WT or Otulin TM (TM represents the K64R, K197R, D31A mutant). Otulin WT and Otulin TM protein levels were assessed by Western blots, post-SeV infection (MOI 10) for the indicated time points. B IRF3 linear ubiquitination was analyzed in OT-KD cells reconstituted with either Otulin WT or Otulin TM mutant post-SeV infection of 24 h. Immunoprecipitation assay was performed by IRF3 antibody and immunoblotting was done by LUB9 antibody to detect IRF3 linear ubiquitination. C C-PARP levels were analyzed post-SeV infection of 24 h in HT1080 WT and HT1080 TM cells as a measure of apoptotic cell death. D SeV-C protein levels were assessed post-SeV infection in HT1080WT and HT1080 TM cells as a measure of viral protein synthesis. E HT1080 WT and HT1080 TM cells were infected with SeV for 24 h (MOI 10) and infectious viral yields were measured. Quantification of western blots and statistical analyses are provided in Supplementary Fig. 8 (mean ± SEM, n = 3; ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001).
Discussion
Mammalian cells respond to viral infections by triggering several cell-intrinsic antiviral pathways [38]. The most well-known response leads to the induction of type I IFN and the IFN-stimulated antiviral genes; all virus-activated pattern recognition receptors trigger this response [39]. But, in addition, there are PRR-specific antiviral pathways, such as RIPA, which is triggered selectively by RLRs, in response to cytoplasmic viral dsRNA, produced during virus replication or cellular dsRNA, produced in response to various types of stress [40]. A salient feature of RIPA is the linear poly-ubiquitination of IRF3, which causes mitochondrial apoptosis [16]. We have identified Otulin as the deubiquitinase, which negatively regulates RIPA. Overexpression of Otulin attenuated the action of RIPA, whereas ablation of its expression enhanced RIPA. We also discovered that RIPA counteracts the action of Otulin by causing its degradation.
The mechanism of Otulin degradation by RIPA has been analyzed (Fig. 8). Viral dsRNA-binding to RIG-I translocates it to the mitochondrial membrane-anchored protein, MAVS, which is the assembly platform of the components of the signaling complex, including IRF3 [12, 41]. A critical event for RIPA activation is the recruitment of LUBAC to this complex. LUBAC, consisting of the three proteins, Sharpin, HOIL and HOIP, adds linear poly-ubiquitin chains to IRF3 using the E3 ligase activity of HOIP. Ubiquitinated IRF3 binds Bax, a pro-apoptotic protein, and brings it to the mitochondrial membrane causing the release of Cytochrome C to the cytoplasm, which in turn activates the apoptasome complex leading to the activation of Caspase 3 and cellular apoptosis, as measured by PARP cleavage [16]. We presented results here showing that Otulin, which removes linear poly-ubiquitin chains, was also recruited by the LUBAC complex, but only after RIPA activation. Moreover, the deubiquitinase enzyme activity of Otulin was not required for binding to LUBAC, which was mediated through an interaction between Otulin and HOIP. Otulin can suppress auto-ubiquitination of LUBAC and thereby promote its enzyme activity [32]; however, if this effect was in play in RIPA, it would have augmented IRF3 ubiquitination. Instead, our observation that in the presence of Otulin there was a lower level of ubiquitination of IRF3, strongly suggests that it directly removed linear ubiquitin chains from IRF3.
Fig. 8. Otulin degradation boosts RIPA.
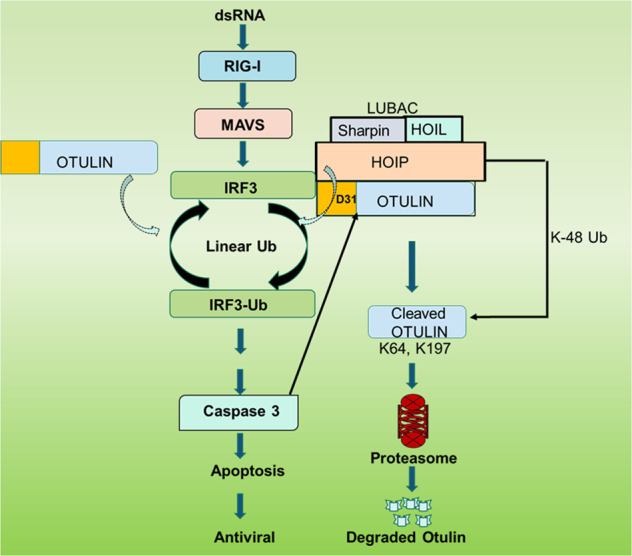
The model shows different steps of Otulin degradation triggered by RIPA. Otulin removes linear ubiquitin chains from IRF3 to inhibit the RIPA pathway and enhance viral replication. RIPA-activated caspase 3 leads to Otulin degradation by cleaving it at D31 position. The cleaved fragment is then K48-ubiquitinated by HOIP at K64 and K197, which leads to its proteasomal degradation.
The inhibitory activity of Otulin was evaded by the proteolytic action of RIPA-activated Caspase 3, which cleaved Otulin at D31, near its N-terminus. The N-terminally trimmed Otulin was rapidly degraded; but it accumulated in cells, if proteasome activity was impaired. Proteasomal degradation of target proteins is usually preceded by their K48-linked ubiquitination and we identified K64 and K197 of Otulin as the recipients of the K48 poly-ubiquitin chains. Interestingly, our results indicate that the N-terminal trimming of Otulin was required for its ability to be a target of K48 ubiquitination. Moreover, HOIP, which can modify its target protein by adding either linear or K48 poly-ubiquitin chains, was responsible for Otulin K48 ubiquitination. Therefore, HOIP promoted RIPA by both IRF3 linear ubiquitination and Otulin K48 ubiquitination.
The positive feedback loop to RIPA, triggered by Otulin degradation, is biologically significant. Using a degradation-resistant mutant of Otulin, we demonstrated that SeV replicated more efficiently in cells devoid of this loop; thus, confirming the importance of Otulin in regulation of RIPA’s antiviral effects. It is reasonable to anticipate that this feedback loop is equally important for boosting RIPA’s role in biological systems, other than viral infection, such as metabolic liver diseases.
Materials and methods
Reagents and antibodies
Proteasome pathway inhibitor MG132 (C2211) was obtained from Sigma Aldrich. Poly (I:C) was purchased from GE healthcare (27473201). Otulin antibody was purchased from Cell Signaling Technology (14127), PARP antibody (9542), Cleaved caspase 3 (9661), GFP antibody (2956), Bax antibody (2772), HOIP antibody (99633, CST), Sharpin antibody (12541, CST), Cleaved PARP (9546, CST), Actin antibody (3700, CST), IRF3 antibody (4302, CST), Ub antibody (SC8017), K48 antibody (ab140601), LUB9 (AB130, Life sensors) for linear ubiquitination, anti-HA antibody (ab18181, abcam), anti-HA antibody (H690, Sigma), Z-VAD-FMK (SC3067, Chem Cruz), Z-DEVD-FMK (264155,Sigma-Aldrich), Anti-Flag M2 affinity gel (A2220, Sigma), Anti-V5 antibody (R96025, Invitrogen), Protein A/G PLUS agarose (SC-2003), Actin antibody (3700, CST), IRF3 antibody (ab68481, abcam). WT and ubiquitin plasmids were obtained from Addgene. SHARPIN, HOIL and HOIP plasmids were provided by Jae Jung (Cleveland Clinic). Caspase-3-GFP (HG10050-ANG, Sino Biological Inc.), P-TRIP-GFP-IRF3 (127663, Addgene), Otulin-GFP plasmid (EX-H2249-M03, GeneCopoeia). GFP-TRAP beads (gta-10, Chromotek) were commercial products. Mito Tracker Red CMXRos (9082, CST) was used at 100 nM for 1 h before harvesting cells. Lipofectamine 2000 (11668019) was obtained from Invitrogen and used for transfection of different cell lines. Antibody against SeV C protein was raised at the Hybridoma Core, Lerner Research Institute.
Cell cultures
HT1080 WT (CCL-121), HT1080 V5-IRF3 (generated in the lab), HEK-293T (CRL-11268), L929 (CCL-1) were obtained from ATCC (Manassas, VA, USA), HT1080/RIG-Ic, and HT080/shIRF3 cells, MEF WT, MEF irf3−/−, MEF bax−/−, HT1080 S1, HT1080 IRF3K1, HT1080 IRF3-K4, MEF S1 as previously described [16]. HT0180/Otulin KD, HT1080S1-OT-KD, MEFS1-OT-KD cell lines were generated by lentivirus transduction. For this HEK-293T cells were transfected with shRNA Otulin Lentiviral Vector (Sigma, TRCN000013451), pCMVR8.74 (packaging plasmid), p-VSVG (psuedotyping plasmid) to generate lentiviral particles. The cell supernatant containing lentiviral particles was collected after 48 h and was used to infect HT1080 cells. These cells were then used to generate HT1080 Otulin-Knockdown (OT-KD) by picking clones following their selection in puromycin (1 μg/ml) rich medium. HT1080 Caspase 3 knockdown cells were generated by lentiviral transduction as described above using lentiviral vector TRCN0000003551 from Sigma. All the cell lines were maintained in DMEM supplemented with 10% FBS, 100 units/ml of penicillin and 100 μg/ml of streptomycin. LLCKM2 (ATCC, CCL-7) was maintained in Medium 199. Trypan blue staining was used to seed equal number of healthy live cells before performing experiments.
dsRNA stocks were prepared by re-suspending poly(I):poly(C) in the PBS and shearing RNA by passing through a 26-guage needle. Poly (I:C) was transfected using Lipofectamine 2000 Invitrogen in all the experiments. The transfections were performed according to the manufacturer’s protocol.
SeV infection and SeV plaque assay
SeV Cantell strain was obtained from Charles Rivers SPAFAS (Preston, CT). Viral infection was carried out in minimal amount of DMEM supplemented with 2% FBS. SeV was used at a concentration of 80 HAU/ml (MOI 10). Cells were incubated with virus for 1 h with gentle shaking after every 10 min. The infection medium was removed after 1 h and the cells were washed with twice with DMEM. These cells were then allowed to grow in complete media before harvesting.
SeV viral plaque assay was performed in LLCMK2 cells. HT1080 WT and HT1080 OT-KD cells were infected with SeV at an MOI of 10 for 24 h and the medium containing virus was then collected and used for plaque assay. Serial dilutions were made and 200 μl of it was used to infect the confluent monolayers of LLCMK2 cells for 90 min with gentle agitation after 30 min. The viral inoculum was removed and cells were overlaid with medium containing 0.5% agar. After 3 days the agar was removed and the cells were washed with PBS. Virus colonies were detected by incubating these cells with 0.1% suspension of guinea pig blood for 30 min. The monolayer was washed with PBS to score viral plaques as described earlier [42].
Site-directed mutagenesis and quantitative real-time PCR
Otulin mutants were generated using Q5-site directed mutagenesis kit (E0554S, NEB) as per the manufacturer’s protocol. The primers used are as follows:
Otulin D31A forward primer 5’cacggcgcgggccggcgggaagg 3’
Otulin D31A reverse primer 5’gccgccgcctcccgcgcc 3’
Otulin K64R forward primer 5’tgaaatagaacgggagaaagaattgcttatacatgaaagag 3’
Otulin K64R reverse primer 5’tctgcagcacggtacatg 3’
Otulin K197R forward primer 5’tctgctgaggcggaagtgggcag 3’
Otulin K197R reverse primer 5’gtaagggactctttaattttatcaac3’
For quantitative RT-PCR analyses, RNA was isolated from cells using Roche RNA extraction kit. cDNA was prepared using ImProm-II reverse transcription system (Promega) with random hexamers. 0.5 ng cDNA was used in 384 well plate for real-time PCR using power SYBR Green PCR Master mix (Applied Biosystems). The qPCR was carried out in a Roche Light Cycler 480 instrument II. The expression levels of mRNAs were normalized with18s rRNA.
Western blot
Cells were lysed in 50 mM Tris buffer, pH 7.4 containing 150 mM of NaCl, 0.1% Triton X-100, 1 mM sodium orthovanadate, 10 mM of NaF, 10 mM of β-glycerophosphate, 5 mM sodium pyrophosphate and protease/phosphatase inhibitors cocktail (Roche) 4 °C for 30 min and centrifuged at 21,130 × g at 4 °C for 15 min. The proteins were electrophoresed on SDS–PAGE gels and then transferred to PVDF membrane (Bio-Rad). Then, the membranes were kept in 5% skim milk in TBST buffer (150 mM NaCl; Tris, pH 7.4 and 0.1% Tween-20) for 60 min at room temperature and then incubated with the primary antibody at 4 °C overnight. Western blot experiments were performed with the indicated antibodies and visualized by Pierce ECL2 Western Blotting Substrate (Thermo Scientific). The Western blot image data were processed by Photoshop.
Immunoprecipitation
For immunoprecipitation assays, cells were lysed in RIPA buffer containing 20 mM HEPES (pH 7.5), 150 mM NaCl, 10 mM NaF, 1.5 mM MgCl2, 10 mM β-glycerophosphate, 2 mM EDTA, 5 mM Na-pyrophosphate, 12.5 mM β-glycerophosphate, 1 mM Na3VO4, 1% (v/v) Triton X-100, 0.2% NP-40, and protease inhibitors (Roche Applied Science, Indianapolis, IN, USA) by keeping on ice for 30 m. Whole-cell lysate was centrifuged for 15 min at 4 °C. The cell lysates were subjected to preclearing for 1 h with anti-mouse or anti-rabbit depending on the source of antibody used for IP assay. After preclearing, the supernatant was incubated overnight with IP antibodies at 4 °C followed by incubation with Protein A/G PLUS-Agarose beads (Santa Cruz) for 2 h with rotation. The related beads were washed 3x times with IP buffer for 10 m each wash and boiled in 2xLaemmli sample buffer for 10 min at 95 °C. For high salt washes, 1 M NaCl was used to remove the contribution of any non-specific binding proteins. The samples were run in 10% SDS–PAGE Gel and transferred onto PVDF membranes (Bio-Rad). Western blot experiments were performed via the indicated antibodies and visualized by Pierce ECL2 Western Blotting Substrate (Thermo Scientific).
For IRF3 Linear Ubiquitination and Otulin K48 ubiquitination assays, cells were lysed and processed as above. 10 mM N-ethylmaleimide (NEM, Sigma Aldrich) was added to lysis buffer if ubiquitination was analyzed. MG132 10 μM was added for 4 h in case of Otulin K48 poly-ubiquitination IP assays before harvesting cells. IRF3 linear ubiquitination assay and Otulin K48 poly-ubiquitination assays were performed by seeding the cells in 10 cm dishes followed by SeV infection at an MOI of 10 for 24 h. The cell density in both the cases was ~106. Mock infected cells were taken as control. After infection, cells were lysed and 1 mg of protein was used for performing ubiquitination assay. Preclearing was done for 1 h. For IRF3 Linear ubiquitination 2 μg of IRF3 antibody was used for overnight incubation while for Otulin K48 poly-ubiquitination Otulin antibody was used at 1:50 dilution with overnight incubation. After performing immunoprecipitation assay as described above; IRF3 linear ubiquitination was analyzed with LUB9 antibody while Otulin K48 ubiquitination was analyzed using K48 antibody.
Ubiquitination assay
In vivo ubiquitination assay was performed to detect Otulin ubiquitination. His-expressing plasmid was transfected for 12 h followed by Sendai virus infection for 24 h.10 µM of MG132 (Sigma-Aldrich) was added to the cells for 4 h before harvesting the cells. Thereafter, the cells were collected in 1xPBS and resuspended in 1 ml of lysis buffer (6 M guanidinium-HCl, 0.1 M Na2HPO4/NaH2PO4, 10 mM imidazole; pH 8.0), sonicated, and centrifuged. Ni-NTA beads (P-6611, Sigma) (50 µl) were added to the supernatant and the mixture was incubated at room temperature for 6 h while rotating. Subsequently, the beads were washed for 5 min at room temperature with 750 µl of each of the following buffers: buffer A (6 M guanidium-HCl, 0.1 M Na2HPO4/ NaH2P04, 10 mM imidazole; pH 8.0); buffer TI (25 mM Tris, 20 mM imidazole; pH 6.8). After the last wash, Ni-NTA beads were incubated in 50 µl of buffer containing 200 mM imidazole, 5% SDS, 0.15 M Tris, 30% glycerol and 0.72 M β-mercaptoethanol at pH 6.7 for 10 min at 95 °C. The eluates were mixed in a 1:1 ratio with 2xLaemmli buffer and resolved by SDS-PAGE followed by western blot analysis with the indicated antibodies.
Flow cytometry
Flow cytometric analysis was performed to assess the cell death by using Annexin V staining. SeV-infected cells were analyzed for apoptosis after a 24 h of post infection. Apoptosis was detected by PE Annexin V Apoptosis Detection Kit I (BD Pharmingen 559763) by staining cells with Annexin-V for 15 min at RT in the dark as per the manufacturer’s protocol.
Mass spectrometry
Otulin-GFP was transfected in HT1080 cells for overnight followed by infection of 12 h, MG132 treatment was given for 4 h before harvesting the cells. Otulin-GFP was isolated using GFP-Trap beads. A 10 µl (10 ng/µl) aliquot of trypsin in 100 mM ammonium bicarbonate was added directly onto the washed beads for digestion. The samples were vortexed for 15 s every 2–3 min for the first 15 min. Digestion was continued overnight at 37 °C in an incubator without agitation. After the overnight digest, a second 10 µl aliquot of protease was added to each sample and digested for an additional 4 h at 37 °C. The tubes were centrifuged and the supernatant was removed. The digested samples were diluted with 100% (vol/vol) formic acid to give a final concentration of 5% formic acid (vol/vol). The digests were cleaned using a C18 ultra micro spin column prior to LC-MS/MS analysis. Digested peptides were analyzed on a ThermoFisher Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer (Bremen, Germany). 5 μl of the peptide extract were injected and peptides eluted from the column by a 90-min acetonitrile/0.1% formic acid gradient at a flow rate of 0.30 μl/min and introduced to the source of the mass spectrometer on-line. The data were analyzed with the search engine Sequest. Two databases were used to search the MS/MS spectra including the Uniprot human protein database and database containing GFP-tagged Otulin. Oxidation of Methionine, acetylation of protein N-terminus, and lysine ubiquitination were set as dynamic modifications. The precursor mass tolerance for these searches was set to 10 ppm and the fragment ion mass tolerance was set to 0.6 Da. A false discovery rate (FDR) was set to 1% for both peptide and protein with a minimum length of seven amino acids, one unique and two total peptides were required for positive identification. Positively identified ubiquitinated Otulin peptides were manually inspected.
Confocal microscopy
Human HT1080 cells were transfected with indicated plasmids using Lipofecatmine 2000. The cells were fixed with 4% paraformaldehyde for 20 min and permeabilized with 0.2% TritonX-100 for 15 min. The cells were then washed with PBS three times and then blocked with 5% bovine serum for 1 h at room temperature. Then cells were incubated with related antibody overnight at 4 °C. Goat anti rabbit Alexa Flour 594 from Invitrogen (1 h) was used as secondary antibody. Objects were mounted using VECTASHIELD/DAPI, and images were acquired confocal laser scanning microscopy (TCS SP8; Leica). Images were processed with Leica LCS software. Fiji (Image J) software was used to determine colocalization of proteins as shown by white dots.
Statistical analysis
All statistical analysis were performed using Prism 7.0 (GraphPad software). The mean ± SEM of all biological replicates was used to make graphs. Statistical analysis was calculated by using two-tailed Student’s t test or one-way ANOVA test. P value ≤0.05 was considered significant. ns > 0.05; *P < 0.05; **P < 0.01, ***P < 0.001. For Western blot, co-IP, and confocal experiments, unless indicated otherwise, results are representative of at least three independent experiments.
Supplementary information
Acknowledgements
We thank all the Sen Laboratory members for technical support and inputs. We thank Akansha Jalota for her help with Flow Cytometry Assay. We thank Chenyao Wang for his help with confocal microscopy experiment. We also thank Belinda Willard for the mass spectrometric analysis and Judith A. Drazba for providing confocal microscopy assistance. This work was supported by the National Institutes of Health grants CA068782 and AA027456 to GCS. The Proteomics core is supported by 1S10OD023436‐01 and the Imaging core is supported by 1S10OD019972-01 from the NIH.
Author contributions
RR performed the experiments, interpreted the results, and wrote parts of the manuscript; GCS designed the experiments, interpreted the results, and wrote parts of the manuscript.
Data availability
Mass spectrometry data for Otulin ubiquitination was provided by Lerner Research Institute, Mass Spectrometry Laboratory for Protein Sequencing, The Cleveland Clinic Foundation with Report number:3800 Sen-Raja.
Competing interests
The authors declare no competing interests.
Footnotes
Edited by M. Piacentini
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-021-00870-4.
References
- 1.Gurtler C, Bowie AG. Innate immune detection of microbial nucleic acids. Trends Microbiol. 2013;21:413–20. doi: 10.1016/j.tim.2013.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koyama S, Ishii KJ, Coban C, Akira S. Innate immune response to viral infection. Cytokine. 2008;43:336–41. doi: 10.1016/j.cyto.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 3.Chow J, Franz KM, Kagan JC. PRRs are watching you: localization of innate sensing and signaling regulators. Virology. 2015;479–480:104–9. doi: 10.1016/j.virol.2015.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luo D, Ding SC, Vela A, Kohlway A, Lindenbach BD, Pyle AM. Structural insights into RNA recognition by RIG-I. Cell. 2011;147:409–22. doi: 10.1016/j.cell.2011.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bruns AM, Horvath CM. Antiviral RNA recognition and assembly by RLR family innate immune sensors. Cytokine Growth Factor Rev. 2014;25:507–12. doi: 10.1016/j.cytogfr.2014.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ramos HJ, Gale M., Jr RIG-I like receptors and their signaling crosstalk in the regulation of antiviral immunity. Curr Opin Virol. 2011;1:167–76. doi: 10.1016/j.coviro.2011.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen S, Thomsen AR. Sensing of RNA viruses: a review of innate immune receptors involved in recognizing RNA virus invasion. J Virol. 2012;86:2900–10. doi: 10.1128/JVI.05738-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rehwinkel J, Gack MU. RIG-I-like receptors: their regulation and roles in RNA sensing. Nat Rev Immunol. 2020;20:537–51. doi: 10.1038/s41577-020-0288-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loo YM, Gale M., Jr Immune signaling by RIG-I-like receptors. Immunity. 2011;34:680–92. doi: 10.1016/j.immuni.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ikushima H, Negishi H, Taniguchi T. The IRF family transcription factors at the interface of innate and adaptive immune responses. Cold Spring Harb Symp Quant Biol. 2013;78:105–16. doi: 10.1101/sqb.2013.78.020321. [DOI] [PubMed] [Google Scholar]
- 11.Hiscott J. Triggering the innate antiviral response through IRF-3 activation. J Biol Chem. 2007;282:15325–9. doi: 10.1074/jbc.R700002200. [DOI] [PubMed] [Google Scholar]
- 12.Chattopadhyay S, Marques JT, Yamashita M, Peters KL, Smith K, Desai A, et al. Viral apoptosis is induced by IRF-3-mediated activation of Bax. EMBO J. 2010;29:1762–73. doi: 10.1038/emboj.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chattopadhyay S, Yamashita M, Zhang Y, Sen GC. The IRF-3/Bax-mediated apoptotic pathway, activated by viral cytoplasmic RNA and DNA, inhibits virus replication. J Virol. 2011;85:3708–16. doi: 10.1128/JVI.02133-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chattopadhyay S, Sen GC. IRF-3 and Bax: a deadly affair. Cell Cycle. 2010;9:2479–80. doi: 10.4161/cc.9.13.12237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chattopadhyay S, Fensterl V, Zhang Y, Veleeparambil M, Yamashita M, Sen GC. Role of interferon regulatory factor 3-mediated apoptosis in the establishment and maintenance of persistent infection by Sendai virus. J Virol. 2013;87:16–24. doi: 10.1128/JVI.01853-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chattopadhyay S, Kuzmanovic T, Zhang Y, Wetzel JL, Sen GC. Ubiquitination of the transcription factor IRF-3 activates RIPA, the apoptotic pathway that protects mice from viral pathogenesis. Immunity. 2016;44:1151–61. doi: 10.1016/j.immuni.2016.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schafer ZT, Kornbluth S. The apoptosome: physiological, developmental, and pathological modes of regulation. Dev Cell. 2006;10:549–61. doi: 10.1016/j.devcel.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 18.Dorstyn L, Akey CW, Kumar S. New insights into apoptosome structure and function. Cell Death Differ. 2018;25:1194–208. doi: 10.1038/s41418-017-0025-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.White CL, Chattopadhyay S, Sen GC. Phosphatidylinositol 3-kinase signaling delays sendai virus-induced apoptosis by preventing XIAP degradation. J Virol. 2011;85:5224–7. doi: 10.1128/JVI.00053-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Peters K, Chattopadhyay S, Sen GC. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol. 2008;82:3500–8. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sanz-Garcia C, Poulsen KL, Bellos D, Wang H, McMullen MR, Li X, et al. The non-transcriptional activity of IRF3 modulates hepatic immune cell populations in acute-on-chronic ethanol administration in mice. J Hepatol. 2019;70:974–84. doi: 10.1016/j.jhep.2019.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanz-Garcia C, McMullen MR, Chattopadhyay S, Roychowdhury S, Sen G, Nagy LE. Nontranscriptional activity of interferon regulatory factor 3 protects mice from high-fat diet-induced liver injury. Hepatol Commun. 2019;3:1626–41. doi: 10.1002/hep4.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Maelfait J, Beyaert R. Emerging role of ubiquitination in antiviral RIG-I signaling. Microbiol Mol Biol Rev. 2012;76:33–45. doi: 10.1128/MMBR.05012-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chiang C, Liu G, Gack MU. Viral evasion of RIG-I-like receptor-mediated immunity through dysregulation of ubiquitination and ISGylation. Viruses. 2021;13:182. [DOI] [PMC free article] [PubMed]
- 25.Pauli EK, Chan YK, Davis ME, Gableske S, Wang MK, Feister KF, et al. The ubiquitin-specific protease USP15 promotes RIG-I-mediated antiviral signaling by deubiquitylating TRIM25. Sci Signal. 2014;7:ra3. doi: 10.1126/scisignal.2004577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9:930–6. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu Q, Wu Y, Qin Y, Hu J, Xie W, Qin FX, et al. Broad and diverse mechanisms used by deubiquitinase family members in regulating the type I interferon signaling pathway during antiviral responses. Sci Adv. 2018;4:eaar2824. doi: 10.1126/sciadv.aar2824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Belgnaoui SM, Paz S, Samuel S, Goulet ML, Sun Q, Kikkert M, et al. Linear ubiquitination of NEMO negatively regulates the interferon antiviral response through disruption of the MAVS-TRAF3 complex. Cell Host Microbe. 2012;12:211–22. doi: 10.1016/j.chom.2012.06.009. [DOI] [PubMed] [Google Scholar]
- 29.Zuo Y, Feng Q, Jin L, Huang F, Miao Y, Liu J, et al. Regulation of the linear ubiquitination of STAT1 controls antiviral interferon signaling. Nat Commun. 2020;11:1146. doi: 10.1038/s41467-020-14948-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivkin E, Almeida SM, Ceccarelli DF, Juang YC, MacLean TA, Srikumar T, et al. The linear ubiquitin-specific deubiquitinase gumby regulates angiogenesis. Nature. 2013;498:318–24. doi: 10.1038/nature12296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Damgaard RB, Walker JA, Marco-Casanova P, Morgan NV, Titheradge HL, Elliott PR, et al. The deubiquitinase OTULIN Is an essential negative regulator of inflammation and autoimmunity. Cell. 2016;166:1215–30 e20. doi: 10.1016/j.cell.2016.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heger K, Wickliffe KE, Ndoja A, Zhang J, Murthy A, Dugger DL, et al. OTULIN limits cell death and inflammation by deubiquitinating LUBAC. Nature. 2018;559:120–4. doi: 10.1038/s41586-018-0256-2. [DOI] [PubMed] [Google Scholar]
- 33.Inn KS, Gack MU, Tokunaga F, Shi M, Wong LY, Iwai K, et al. Linear ubiquitin assembly complex negatively regulates RIG-I- and TRIM25-mediated type I interferon induction. Mol Cell. 2011;41:354–65. doi: 10.1016/j.molcel.2010.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rittinger K, Ikeda F. Linear ubiquitin chains: enzymes, mechanisms and biology. Open Biol. 2017;7:170026. [DOI] [PMC free article] [PubMed]
- 35.Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, et al. Linear ubiquitination prevents inflammation and regulates immune signalling. Nature. 2011;471:591–6. doi: 10.1038/nature09816. [DOI] [PubMed] [Google Scholar]
- 36.Stieglitz B, Morris-Davies AC, Koliopoulos MG, Christodoulou E, Rittinger K. LUBAC synthesizes linear ubiquitin chains via a thioester intermediate. EMBO Rep. 2012;13:840–6. doi: 10.1038/embor.2012.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirisako T, Kamei K, Murata S, Kato M, Fukumoto H, Kanie M, et al. A ubiquitin ligase complex assembles linear polyubiquitin chains. EMBO J. 2006;25:4877–87. doi: 10.1038/sj.emboj.7601360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yan N, Chen ZJ. Intrinsic antiviral immunity. Nat Immunol. 2012;13:214–22. doi: 10.1038/ni.2229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goubau D, Deddouche S, Reis e Sousa C. Cytosolic sensing of viruses. Immunity. 2013;38:855–69. [DOI] [PMC free article] [PubMed]
- 40.Chattopadhyay S, Sen GC. RIG-I-like receptor-induced IRF3 mediated pathway of apoptosis (RIPA): a new antiviral pathway. Protein Cell. 2017;8:165–8. doi: 10.1007/s13238-016-0334-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ren Z, Ding T, Zuo Z, Xu Z, Deng J, Wei Z. Regulation of MAVS expression and signaling function in the antiviral innate immune response. Front Immunol. 2020;11:1030. doi: 10.3389/fimmu.2020.01030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tatsumoto N, Miyauchi T, Arditi M, Yamashita M. Quantification of infectious sendai virus using plaque assay. Bio Protoc. 2018;8:e3068. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry data for Otulin ubiquitination was provided by Lerner Research Institute, Mass Spectrometry Laboratory for Protein Sequencing, The Cleveland Clinic Foundation with Report number:3800 Sen-Raja.