

Abstract
Ferroptosis is an iron-dependent form of cell death driven by biochemical processes that promote oxidation within the lipid compartment. Calcium (Ca2+) is a signaling molecule in diverse cellular processes such as migration, neurotransmission, and cell death. Here, we uncover a crucial link between ferroptosis and Ca2+ through the identification of the novel tetraspanin MS4A15. MS4A15 localizes to the endoplasmic reticulum, where it blocks ferroptosis by depleting luminal Ca2+ stores and reprogramming membrane phospholipids to ferroptosis-resistant species. Specifically, prolonged Ca2+ depletion inhibits lipid elongation and desaturation, driving lipid droplet dispersion and formation of shorter, more saturated ether lipids that protect phospholipids from ferroptotic reactive species. We further demonstrate that increasing luminal Ca2+ levels can preferentially sensitize refractory cancer cell lines. In summary, MS4A15 regulation of anti-ferroptotic lipid reservoirs provides a key resistance mechanism that is distinct from antioxidant and lipid detoxification pathways. Manipulating Ca2+ homeostasis offers a compelling strategy to balance cellular lipids and cell survival in ferroptosis-associated diseases.
Subject terms: Lipidomics, Cancer genetics, Cell biology
Introduction
Ferroptosis is a type of oxidative cell death induced by glutathione (GSH) deprivation or uncontrolled reactive oxygen species (ROS). During ferroptosis, polyunsaturated phospholipid peroxides induced by reactive iron accumulate to lethal levels, resulting in membrane lapse [1]. The selenoenzyme glutathione peroxidase 4 (GPX4) is a central enzyme protecting lipids from oxidative species that uses GSH as an essential cofactor to convert lipid hydroperoxides to lipid alcohols [2, 3]. Loss of GPX4 activity and deprivation of GSH both lead to lipoxygenase activation in a process closely linked to inflammation [4, 5]. Lipoxygenases oxidize polyunsaturated fatty acids (PUFAs) to generate metabolites which additionally promote calcium (Ca2+) influx for the final, catastrophic phase of cell death [6].
Calcium is a store-operated signal transduction molecule controlling diverse cellular processes such as growth and migration. It is intricately linked to cancer and the pathogenesis of degenerative diseases, which feature imbalanced metabolism and excessive ROS [7–9]. The endoplasmic reticulum (ER) is the main intracellular Ca2+ storage site and plays a key role in the maintenance of Ca2+ homeostasis and regulation of protein, lipid, and glucose metabolism. In response to extrinsic stimuli, inositol 1,4,5-trisphosphate (IP3) and ryanodine receptors release Ca2+ from the ER to the cytosol, whereas the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pumps Ca2+ against the gradient to maintain a concentration difference between the ER lumen and the cytosol at rest.
Previous studies have shown that ER Ca2+ homeostasis is critical for adipogenesis and lipid storage [10, 11]. Altering Ca2+ balance can regulate activity of key enzymes in de novo lipogenesis, including fatty acid synthase (FAS) and stearoyl-CoA desaturase 1 (Scd1), or, induce lipolysis [12, 13]. IP3 receptor (IP3R) mutants have conserved pathways of energy metabolism, with higher serum triglycerides and free fatty acids in mice [14] and an obese phenotype with enlarged lipid droplets (LDs) and elevated fat storage in Drosophila [15]. As SERCA is solely responsible for transporting Ca2+ into the ER lumen, the SERCA inhibitor thapsigargin inhibits early adipogenesis in cultured cells [16, 17]. In Drosophila fat cells, inhibiting dSERCA promotes lipodystrophy, aberrant LD formation and ectopic lipid accumulation by regulating intracellular Ca2+ homeostasis [18].
Apart from late stage store-operated calcium (SOCE) induced death [19, 20], the intracellular role of Ca2+ in ferroptosis is obscure, in particular its role in regulating phospholipids. Phospholipid plasticity, dependent on diet and de novo lipogenesis, contributes to malignant transformation [21–25]. In particular, cancer cells with a higher degree of saturated membrane phospholipids are protected against ROS [26]. Thus ‘front-loading’ highly saturated membrane lipids may have the consequence of eliminating or quenching the primary biochemical substrates of ferroptosis.
In this report, we demonstrate that the novel tetraspanin MS4A15 interacts with ER-resident Ca2+ regulators to specifically block ferroptosis by altering the lipid profile of overexpressing cells. MS4A15 belongs to the membrane-spanning 4-domains subfamily A (MS4A) whose members function within oligomeric complexes. It is proposed that MS4A proteins act as ion channels through association with other subunits [27]. Specifically, MS4A1, MS4A2, and MS4A12 have been shown to possess Ca2+-regulating abilities [28–31].
We show here that MS4A15 drives lipid remodeling by depleting luminal Ca2+, favoring accumulation of protective monounsaturated fatty acid (MUFA)-containing phospholipids and plasmalogen ether lipids while limiting polyunsaturated alkyl chains. Strikingly, this effect is mimicked by constitutively inhibiting endoplasmic Ca2+ uptake with thapsigargin, or by stimulating phospholipase C (PLC), which both reduce ER Ca2+ levels. Restoration of luminal Ca2+ homeostasis re-sensitizes Ms4a15-overexpressing cells, and extraordinarily, ferroptosis-resistant cell lines. This phenomenon shows that persistent luminal Ca2+ depletion circumvents synthesis of ferroptosis-sensitive substrates in human cancer cell lines. This is the first report directly linking modulation of ER Ca2+ homeostasis to lipid remodeling and ferroptosis sensitivity.
Results
Ms4a15 expression specifically blocks ferroptosis
Ms4a15 was identified in a CRISPR activation screen protecting against ferroptosis [32]. To test if MS4A15 extensively inhibits ferroptosis, we generated pooled Ms4a15-overexpressing mouse immortalized fibroblasts (Ms4a15 OE) [32, 33] and characterized resistance to different ferroptosis inducers (1S,3R)-RSL3 (RSL3), imidazole ketone erastin (IKE), ferroptosis inducer derived from CIL56 (FIN56), and genetic ablation of Gpx4 (Fig. 1A, B) compared to empty vector-containing cells (control). In each case, elevated Ms4a15 mRNA expression (~20-fold increase) robustly increased viability similar to the level of control cells treated with α-tocopherol (αToc), an inhibitor of ferroptosis [34, 35]. In contrast, Ms4a15 knockout cells showed no viability change; however expression was detected only in trace quantities in parental MF cells (Supplementary Fig. 1A, I). We examined then if Ms4a15 OE leveraged general protection against cell death. Resistance to induced apoptosis, necroptosis and several chemotherapeutic agents was not observed, while partial protection was observed against staurosporine and paclitaxel (Supplementary Fig. 1B).
Fig. 1. MS4A15 specifically protects cells against ferroptosis.

A Dose-response curve of Ms4a15-overexpressing immortalized mouse fibroblasts (Ms4a15 OE) compared to empty vector control cells (control) against RSL3 treatment (16 h). Viability was detected by percent Resazurin conversion relative to respective untreated cells. Addition of 10 μM α-tocopherol (αToc) serves as rescue control for ferroptosis. Inset shows relative Ms4a15 expression by qPCR (rel. mRNA). cT values are 31.1 and 27.4 for control and Ms4a15 OE, respectively. B Survival of Ms4a15 OE cells compared to control against ferroptosis inducers: 2 μM IKE (16 h), Gpx4–/– (72 h) by 1 μM 4-hydroxy-tamoxifen induction and 10 μM FIN56 with 10 μM αToc rescue. Significance was evaluated by two-tailed t-test. C Lipid peroxidation induced by RSL3 (0.3 μM) treatment for 3 h in Ms4a15 OE and control cells measured by BODIPY 581/591 C11 stain (BODIPY-C11). A typical FACS histogram of three independent experiments is depicted. D Brightfield and propidium iodide images and quantification (PI %) of Ms4a15 OE cells compared to control following 16 h RSL3 (0.5 μM) challenge (left). PI values at this timepoint likely underestimate cell death due to cell detachment, as observed in phase contrast images. (Right) Clonogenic survival at 7 d following 16 h RSL3 (1.25 μM) treatment in a colony-forming assay. E 3D-spheroids of Ms4a15 OE and control cells grown for 4 d and treated with 2 µM RSL3 for 16 h before PI staining. Relative (rel.) PI intensity was calculated versus untreated spheroids (n = 4). F Viability of human HT1080 (D) and Calu-1 (E) MS4A15-overexpressing cells (±FLAG) challenged with IKE compared to empty control. Insets show MS4A15 expression by qPCR or western. G mRNA expression level of MS4A15 in 1375 CCLE cancer cell lines compared with GAPDH. Relative mRNA expression is shown as mean ± SD of n = 3 technical replicates of three independent experimental repetitions. Viability data are plotted as representative mean ± SD of n = 3 technical replicates for independent experiments repeated at least three times with similar outcomes. Curve statistics, p-values of two-way ANOVA, are shown for comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next examined glycerophospholipid (GP) oxidation using BODIPY 581/591 C11 (BODIPY-C11). Treatment for 3 h with RSL3-induced robust BODIPY-C11 oxidation in control cells, while Ms4a15 OE cells were unchanged (Fig. 1C). We validated corresponding cell survival under different conditions with propidium iodide (PI), colony-forming, and 3-dimensional spheroid assays (Fig. 1D, E), all which showed stable protection by Ms4a15 OE against ferroptosis.
Human MS4A15 protein is 87% identical with mouse (Supplementary Fig. 1C) and expressed in lung tissue [36]. Conserved protection was observed in human MS4A15-overexpressing (MS4A15 OE) HT-1080 fibrosarcoma and Calu-1 non-small-cell lung cancer cells treated with IKE (Fig. 1F). However, due to absent MS4A15 expression in cell lines (1345 of 1375 have ≤1 TPM; Fig. 1G) [37, 38], siRNA knockdown cells were not more sensitive to ferroptotic challenge (Supplementary Fig. 1D). We further noted that despite high expression in primary adenocarcinomas, MS4A15 is lost in cultured lung cancer cell lines in a direct relationship to cell adhesion markers (Supplementary Fig. 1E). A defective cell migration phenotype is thus consistent with decreased metastasis/increased survival of lung cancer patients with high MS4A15-expressing tumors (Supplementary Fig. 1F, G).
Together, these results show that MS4A15 is linked to cell migration and can robustly protect against ferroptosis. MS4A15 protein is increased following ferroptosis induction, suggesting its presence is instrumental to survival (Supplementary Fig. 1H). Notably, this resistance is accomplished without substantially affecting regulators of ferroptosis [1] (Supplementary Fig. 1I).
MS4A15 associates with ER-resident Ca2+ regulators
To further investigate its role we immunoprecipitated human FLAG-tagged MS4A15 from HEK293T cell lysates and quantified co-eluting proteins (Fig. 2A). Differentially identified proteins (fold change (FC) log2(MS4A15/GFP)) were compared to GFP-expressing control cells. A robust enrichment was seen for MS4A15 (p = 2.32E–05, two-tailed t-test; log2FC = 9.17) while an expected negative enrichment was seen for GFP (p = 0.012; log2FC = −3.98).
Fig. 2. MS4A15 regulates calcium-mediated ferroptosis.

A Enrichment of MS4A15-FLAG co-immunoprecipitated proteins in HEK293T cells as determined by label-free proteomic quantification. Mean abundance ratios were calculated compared to GFP-expressing cells incubated with anti-FLAG as a control. Dotted horizontal line indicates significance (paired t-test, p < 0.05). B Single sample Gene Set Enrichment Analysis (GSEA) correlation analysis in primary lung tumors between MS4A15 and Ca2+ transmembrane transporters (RSEM, RNA-Seq by Expectation-Maximization). Significance was evaluated by Pearson correlation. C Western blot of IP3R1 protein in Ms4a15 OE and control cells. Vinculin is given as loading control. D Schematic of calcium related processes in (E–G). Activation of G protein-coupled receptors (GPCRs) such as Bradykinin receptor stimulates phospholipase C (PLC) cleavage of phosphatidylinositol 4,5-bisphosphate (PIP2) to yield IP3, triggering Ca2+ release from the endoplasmic reticulum (ER). Thapsigargin (Tgn) blocks SERCA-mediated ER Ca2+ uptake, while ionophore catalyzes nonspecific store release in Ca2+ free medium. Membrane channels mediate uptake following re-addition of CaCl2-containing medium. E Calcium levels detected by cytosolic sensor GCaMP6s using flow cytometry (normalized fluorescence, ex488/em530 nm). Top panels: ER Ca2+ release mediated by 50 nM Bradykinin (∆) or 5 μM Ionophore (^) in Ms4a15 OE compared to control cells in Ca2+-free buffer. Bottom panels: control cells pretreated with 50 nM Tgn for 3 h. Addition of 2 mM CaCl2 (▲). Data shown are representative results of three independent repetitions performed in triplicate with similar outcomes. Fluorescent images were acquired 30 s following Bradykinin stimulation for respective genotypes. F Time-dependent (0 h–14 days) effect of Tgn pretreatment on lipid peroxidation detected by BODIPY-C11 induced by RSL3 (0.3 μM for 3 h) in control cells (left panels) compared to DMSO. A typical FACS histogram of three independent repetitions is depicted. Viability of control cells pretreated with 2.5 nM Tgn for 7 days or 14 days prior to RSL3 induction (untreated, 0 days). Fluorescent images were acquired 30 s following Bradykinin stimulation for 14 d treated cells. G Dose-dependent sensitization of Ms4a15 OE cells to RSL3 by overexpressing Serca2 (Ms4a15 OE + Serca2 OE) or empty virus control (Ms4a15 OE + control) in Ms4a15 OE cells (left panel). Restoration of Ca2+ dynamics is indicated by Bradykinin (right panels). Insets show SERCA2 expression by western and viability (PI%) measurements in respective cell lines. Viability data are representative mean ± SD of n = 4 (F) or n = 3 (G) replicates for experiments repeated independently at least three times. Curve p-values of two-way ANOVA comparisons are shown. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
The highest scorings proteins associate with IP3-receptors in the ER, including: TMEM33 (p = 4.33E–06; log2FC = 20.46), a Ca2+ regulator affecting acute kidney injury [39, 40], ERLINs, which regulate IP3 receptors, DNAJs regulating degradation, and ARFs controlling G-protein coupled receptors (GPCRs). Consistent with a proposed role in Ca2+ regulation, we observed MS4A15 localization to the ER (Supplementary Fig. 1J).
KEGG pathways from primary lung adenocarcinomas in The Cancer Genome Atlas (TCGA) [41] showed a strong association of MS4A15 with smooth muscle contraction triggered by Ca2+ release, PPAR signaling, arachidonic acid metabolism, and Ca2+ signaling (Supplementary Fig. 2A). We also observed a direct correlation between MS4A15 and Ca2+ transporter genes in primary lung tumors (Fig. 2B). Highly co-regulated genes include CLIC5, producing PIP2, a metabolic precursor of IP3; cardiac troponin (TNNC1), encoding a Ca2+ buffering protein and SUSD2 mediating adhesion (Supplementary Fig. 2B, C). CLIC5, TNNC1, and SUSD2 also showed strong z-score correlations in solid tumor regulation of Ca2+ transport (Supplementary Fig. 2D). Due to the enrichment of these Ca2+ modulators, we examined the hallmark Inositol trisphosphate receptor (IP3R1) expression (Fig. 2C). Marked IP3R1 downregulation in Ms4a15 OE cells indicated an inverse relationship but only partial co-localization was evident (Supplementary Fig. 2E). Together with the IP data, this suggests that reduced IP3R1 levels are a consequence of altered Ca2+ regulation rather than direct interaction.
MS4A15 regulates Ca2+-mediated ferroptosis
In light of these observations we examined Ca2+ signaling in Ms4a15 OE cells. Extracellular stimuli such as EGF can trigger phospholipase C to generate IP3, which stimulates cytosolic Ca2+ release or MAPK/PKC to mediate cellular response [30, 42, 43]. We observed in Ms4a15 OE cells that phospho-ERK levels show a slight concentration-dependent sensitization to EGF stimulation (Supplementary Fig. 3A). However, STAT3 and AKT were unchanged, arguing against parallel activation of signaling pathways.
We therefore directly measured Ca2+ response using the fluorescent sensor GCaMP6s. In Ca2+ free medium, bradykinin activates its GPCR, releasing Ca2+ from ER stores (Fig. 2D). In Ms4a15 OE cells stimulated with bradykinin, however, the Ca2+ response was strikingly reduced (Fig. 2E). Re-addition of CaCl2 induced robust transients in control cells but a limited response in Ms4a15 OE cells, suggesting the inactivation of SOCE. The permeant ionophore A23187 corroborated a potent decrease in total Ca2+ released from Ms4a15 OE internal stores (Fig. 2E).
This profile is similar to that of cells treated with thapsigargin (Tgn), a potent inhibitor of SERCAs that supply the lumen with Ca2+ (Fig. 2D, E). Remarkably, Tgn disruption of ER Ca2+ import in control cells showed diminished lipid peroxidation corresponding to treatment duration (Fig. 2F). Whereas simultaneous application of Tgn with RSL3 did not affect resistance, 7 and 14 days pretreatment comprehensively protected cells. Pretreatment with Tgn abolished bradykinin and ionophore-induced store release, but increased Ca2+ uptake from the extracellular milieu (Fig. 2E, F). This shows that while cytosolic Ca2+ levels in Tgn-treated cells may be partially rebalanced, Ms4a15 OE cells are refractory to uptake.
We next investigated if Ms4a15 OE resistance was due to ER-Ca2+ depletion or SOCE-related effects. Inhibition of SOCE Ca2+ import by CoCl2 as well as forced influx via ionophore did not markedly affect Ms4a15 OE cell sensitivity (Supplementary Fig. 3B, C). In addition, rapid uptake store-operated membrane channel (Orai) expression was virtually unchanged, consistent with unchanged ferroptosis sensitivity upon SOCE-inhibition with BAPTA-AM (Supplementary Fig. 3D, E). Together with Tgn-mediated survival, these outcomes indicate SOCE does not contribute to ferroptosis resistance in these cells.
From this, we reasoned that Tgn and Ms4a15 OE may limit lipid oxidation via persistent Ca2+ depletion. We therefore tested if restoration of ER Ca2+ levels could re-sensitize Ms4a15 OE cells. Strikingly, elevating SERCA2 in Ms4a15 OE and control cells recapitulated parental Bradykinin-mediated Ca2+ release and sensitized cells to RSL3-induced ferroptosis (Fig. 2G, Supplementary Fig. 3F), indicating that replenishing ER Ca2+ stores can re-sensitize cells.
Aberrant ER Ca2+ homeostasis is associated with stress and the unfolded protein response (UPR) [44], thus we examined hallmarks of UPR, Xbp1 splicing and Chop/Ddit3 and Gadd34/Ppp1r15a expression but could not discern UPR activation (Supplementary Fig. 3G, H). Moreover, short- and long-term tunicamycin treatments that trigger ER stress via UDP-HexNAc inhibition were ineffective against ferroptosis (Supplementary Fig. 3I). Taken together, we conclude that persistent disruption of ER Ca2+ homeostasis in Ms4a15 OE and Tgn-treated cells leads to ferroptosis resistance in a manner unrelated to ER stress.
MS4A15 regulates lipid saturation and length
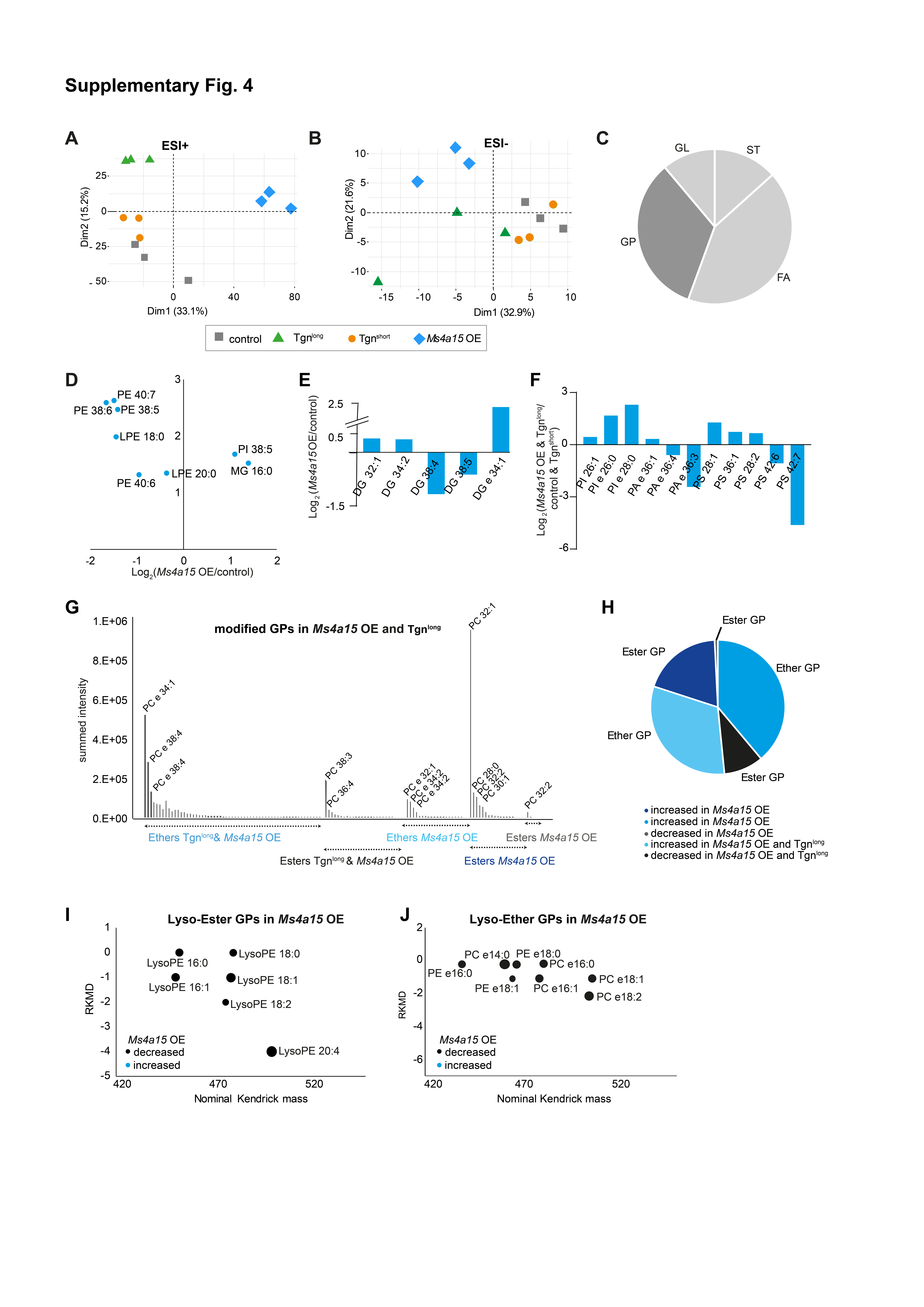
MS4A15 informatics revealed a role for Ca2+ in the biosynthesis of ER-synthesized lipids (Supplementary Fig. 2A). We investigated if Ca2+ dyshomeostasis in Ms4a15 OE cells and Tgn-treated cells impacts cellular lipid composition. We performed LC-MS² based lipidomics to broadly examine lipid types [45] and chose a 16 h treatment (Tgnlong) time point to minimize secondary effects. Unsupervised statistical analysis of >4600 extracted lipid species revealed a clear association of Ms4a15 OE with Tgnlong samples in both modes, whereas 3 h treatment (Tgnshort) delivered comparable lipid profiles to vehicle-treated controls (Fig. 3A, Supplementary Fig. 4A, B, Supplementary Table 1).
Fig. 3. Lipid dysregulation in Ms4a15 OE cells.

Global lipidomics analysis of Ms4a15 OE, control cells treated with Tgnshort (3 h), control cells treated with Tgnlong (16 h), and untreated control cells. A Dendrogram indicating separation of untreated Ms4a15 OE and Tgnlong treated control cells from untreated and Tgnshort treated control cells by hierarchical cluster analysis. Similarly regulated lipid species from Ms4a15 OE and Tgnlong were extracted and plotted in the heatmap. B Lipid abundance heatmap showing z-score profiles of species similarly downregulated in both Ms4a15 OE and Tgnlong (group I), exclusively upregulated in Ms4a15 OE (group II), and similarly upregulated in Ms4a15 OE and Tgnlong (group III). Sample colors correspond to A. (n = 3, Wilcoxon–Mann–Whitney-Test, BH corrected). C Modulated lipid classes in groups I–III by LIPID MAPS Structure Database. GP glycerophospholipid, FA fatty acid, ST sterol Lipid, SP sphingolipid, GL glycerolipid. Ether GPs and ester GPs are in dark colors. D Free fatty acid fold change in Ms4a15 OE and Tgnlong compared to untreated control. SFAs saturated fatty acids, MUFAs monounsaturated fatty acids, PUFAs polyunsaturated fatty acids. Significant p-values of two-way t-test comparisons versus control are shown. E Kendrick plot of significantly modulated diacylglycerophospho-ethanolamine (PE) and -choline (PC) ester phospholipids. All species have a referenced Kendrick mass-defect (RKMD) value of 0 (saturated chains) or a negative integer (number of unsaturated bonds). Dot sizes indicate absolute values of log2(mean Ms4a15 OE/mean control) (n = 3). p < 0.05, Wilcoxon–Mann–Whitney-Test, BH corrected). F Model structures of diacyl (esters), plasmanyl (ethers) and plasmenyl (vinyl-ethers). The latter are also termed plasmalogens. G Kendrick plot of significantly modulated ether GPs (PE and PC). Dot sizes indicate summed peak intensity. For given species isomeric plasmalogens are validated by acidic hydrolysis (see Supplementary Table 2) and (H). ‘PC e’ or ‘PE e’ represent the respective ether species of PC or PE, n = 3, p < 0.05, Wilcoxon–Mann–Whitney-Test, BH corrected). H Acidic hydrolysis abundance illustrated for one ester (top), one alkyl-ether (middle) and one vinyl-ether GP (bottom). I Summed intensities for all detected GP show a slight reduction of ester GPs as well as enrichment in ether GP for Ms4a15 OE and Tgnlong. Data shown represent mean ± SD of n = 3 technical replicates.
We focused on shared lipid modifications in Ms4a15 OE and Tgnlong as well as exclusively dysregulated lipids in Ms4a15 OE (Fig. 3B). Classes of significantly altered species are shown for lipids downregulated in Ms4a15 OE and Tgnlong (group I), those exclusively enriched in Ms4a15 OE (group II), and those enriched in both Ms4a15 OE and Tgnlong (group III) (ESI+, Fig. 3C; ESI−, Supplementary Fig. 4C). The data show the vast majority of modulated lipids are glycerophospholipids (GP), followed by several free fatty acid (FA) species (Fig. 3D).
Ms4a15 OE delivered a different free FA profile compared to control cells. Significant increases of the main saturated FAs, palmitic (C16:0) and stearic (C18:0) acid, were observed while PUFA fatty acids such as arachidonic (20:4, AA), andrenic (22:4), eicosapentaenoic acid (20:5, EPA), docasapentaenoic acid (22:5, DPA), and doxosahexaenoic acid (22:6, DHA) were decreased. Tgnlong cells shared a similar albeit less robust profile than Ms4a15 OE, possibly due to the abbreviated treatment (Fig. 3D).
A Kendrick plot (Fig. 3E) revealed a marked decrease in higher molecular weight PUFA-containing glycerophospho-ethanolamines (PEs) and -cholines (PCs) esters in Ms4a15 OE cells. In addition, we observed decreased esterfied PUFAs in all GP classes, glycerolipids (GL) as well as in lyso-species (Fig. 3E, Supplementary Fig. 4D–J). Notably, the decrease in PUFA-containing species was accompanied by an increase in MUFA- and saturated acyl-containing GPs (SFA). These lipids suggest increased dependence on de novo synthesis, as they are highly enriched in breast cancer tumors [24]. An elegant ferroptosis-protective mechanism of exogenous MUFA supplementation resulting in PUFA downregulation has recently been elucidated [46].
A distinct enrichment of ether lipids—specialized GPs with an sn-1 ether linkage—was seen for all fatty acids compositions (Fig. 3F, G; ‘e’ indicating ‘ether’). The total ether lipid pool was upregulated in Ms4a15 OE and Tgnlong conditions: 25% in controls versus 36% in Ms4a15 OE, and Tgnshort 25% versus Tgnlong 37% (Fig. 3I). In particular, MUFA-containing ethers were enriched (Fig. 3G).
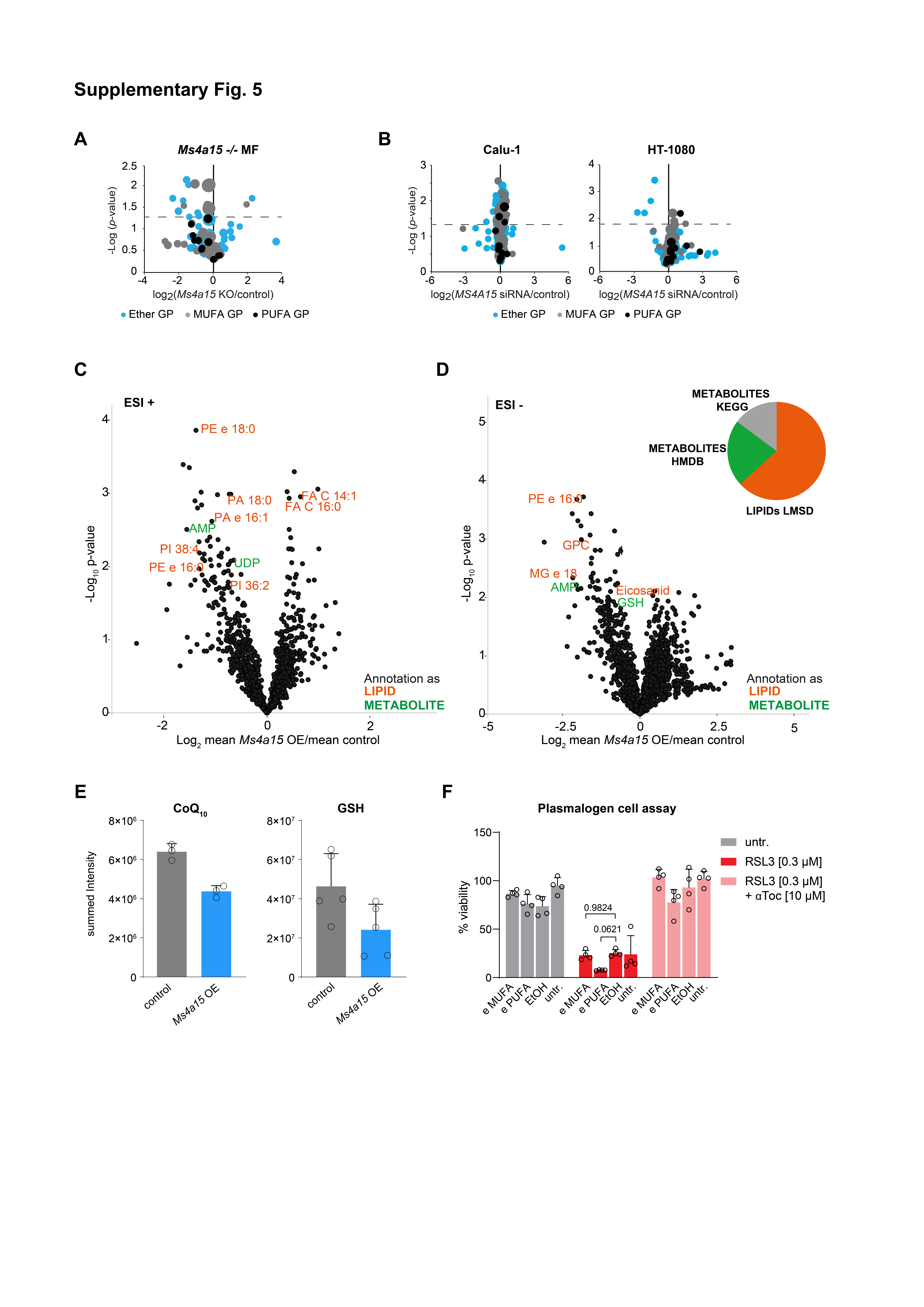
Ether lipids may consist of alkyl-ether or vinyl-ether moieties, with a double bond proximal to the oxygen, termed plasmalogens (Fig. 3F). MS2 cannot differentiate between isomeric alkyl-ether and vinyl-ether, thus we verified MUFA plasmalogens as the main species in Ms4a15 OE cells by acidic hydrolysis (Fig. 3H). Co-elution of a plasmalogen and an isomeric saturated ether was seen for several species, while many upregulated ethers were entirely plasmalogens (Supplementary Table 2). Consistently, Ms4a15 knockout MF cells show a decrease in the same ether species and MUFA-GPs, however, these lipids were mostly unaffected in knockdown Calu-1 and HT-1080, in agreement with unchanged viability for these cell lines (Supplementary Fig. 5A, B)
Finally, global analysis of non-targeted metabolomics of Ms4a15 OE showed the most highly dysregulated metabolites are GP/GL lipids found in LIPID MAPS (Supplementary Fig. 5C, D, Supplementary Table 3). GSH and ubiquinone (CoQ10) metabolites showed negligible change, further supporting a Ca2+-based effect on lipid structure and viability (Supplementary Fig. 5E).
Ms4a15 OE ether-MUFAs are anti-ferroptotic reservoirs
To clarify the mechanism of how Ms4a15 OE cells evade cell death we examined lipid behavior during ferroptosis. PUFA-containing GPs are characteristic targets for peroxidation and are consequently degraded [47, 48]. Upon ferroptosis initiation, depletion of PUFA PE was observed in controls as well as several Ms4a15 OE species (Fig. 4A). We therefore compared all affected lipid species by global non-supervised principal component analysis (PCA), resulting in group separation with minimal convergence (Fig. 4B). This suggests that ferroptosis is classically initiated in cells but peroxidation degrades additional lipid species in Ms4a15 OE cells. We therefore investigated their origin with respect to dysregulated lipids found in the Ms4a15 OE pool.
Fig. 4. Ms4a15 OE defines MUFA-lipids and -plasmalogens as ferroptosis targets.

A Targets of lipid peroxidation are affected by RSL3 treatment (3 h) in control and Ms4a15 OE cells. ‘LPE’ indicates lyso-form of PE. B PCA scores plot for positive (ESI+) and negative (ESI−) electrospray ionization mode indicating the global lipid profile in Ms4a15 OE and control cells under untreated or RSL3 treatment conditions. PCAs were based on 924 annotated PL, GL and FA primary affected lipid classes in ferroptosis. C Significantly increased lipids in Ms4a15 OE are affected by 3 h RSL3 treatment of Ms4a15 OE. Volcano plot of log2(fold change) following RSL3 treatment. Larger dots are significant (p < 0.05, n = 3, two-sided Welch test) for changes due to RSL3. D–F Significantly modulated lipids in Ms4a15 OE compared to control, ranked by abundance. Summed peak area of all samples (A) is shown in (D). E Fold change of these species in Ms4a15 OE compared to control cells, under untreated conditions. F Fold change of these species in Ms4a15 OE and control cells upon 3 h RSL3 treatment. G–I Significantly modulated lipids in Ms4a15 OE compared to control, ranked by RSL3 effect. Summed peak area of all samples (A) is shown (G). H Fold change of these species in Ms4a15 OE compared to control cells, under untreated conditions. I Fold change of these species in Ms4a15 OE and control cells upon 3 h RSL3 treatment. Data shown represent mean of n = 3 technical replicates.
We found that RSL3-treatment depleted the same lipids that are elevated in Ms4a15 OE cells (Figs. 3E, G, 4C). We therefore examined if significantly upregulated and highly abundant lipids are preferred targets of RSL3 (Fig. 4D), however, the pattern is independent of initial concentration. In Ms4a15 OE, RSL3 treatment extensively modifies most ether-lipids and MUFA-containing GPs, rather than single or highly concentrated species (Fig. 4C–F). However, highly abundant MUFA ester-PC 32:1 (log2 = 0.39 increase) and MUFA ether-PC 34:1 (log2 = 0.89) (Fig. 4E) are depleted by log2 = −0.27 and log2 = −0.42 in Ms4a15 OE cells treated with RSL3 (Fig. 4F), respectively, while these same lipids are unaffected in controls. Instead, degradation of highly abundant PUFA ether-PC 36:4 and PE 36:5 was observed in control cells.
We investigated which Ms4a15 OE lipids are most affected by RSL3 treatment and observed the largest changes in upregulated ether lipids, both MUFA and PUFA, suggesting that the plasmalogen vinyl ether bond is reactive with ferroptotic ROS (Fig. 4A, G–I). The largest change was seen for PE e 36:1 (log2 = −4.55), highly enriched in Ms4a15 OE (log2 = 2.91), indicating both properties (MUFA and vinyl ether) are adept at absorbing this reaction.
In summary, significantly elevated lipid species in Ms4a15 OE cells, 16- and 18-carbon plasmalogens and MUFA-containing GPs, comprise the primary targets of RSL3-induced degradation in Ms4a15 OE. This reveals that the ensuing lipid remodeling is important for ferroptosis protection.
MUFA-plasmalogens protect PEs against oxidation
We further examined the behavior of plasmalogens under oxidizing cell-free conditions with AAPH in the presence of PEs using BODIPY-C11 as a sensor. Consistent with Zou et al. [48] we observed increased oxidation in the presence of PUFA-plasmalogen PE (P-16:0/20:4). However, MUFA-plasmalogen PC (P-18:0/18:1) displayed protection of BODIPY-C11 oxidation, similar to ferrostatin-1 (Fig. 5A). We examined PE-ester phospholipid stability by MS2 and observed that MUFA-plasmalogens strongly protected against PE decay by AAPH (Fig. 5B). However, exogenous addition to control cells showed increased lethality for PUFA- but no change for MUFA-plasmalogens (Supplementary Fig. 5F). This may be due to sn-2 remodeling of MUFA-plasmalogens in cells producing high levels of PUFAs. Nevertheless, minor synergistic viability was observed only for MUFA-plasmalogen in the presence of αToc (Fig. 5C), suggesting (sensitizing) PUFA-lipids are more potent than (protective) MUFA-plasmalogens.
Fig. 5. Distinct activities of MUFA- and PUFA-containing plasmalogens and lipids.

A Antioxidant activity of plasmalogens (50 parts per million, ppm) “e MUFA” (P-18:0/18:1) PC or “e PUFA” (P-16:0/20:4) PE and 3 ppm ferrostatin-1 (Fer-1) on BODIPY-C11 oxidation in the presence of 50 ppm ester lipids (PE 18:0/22:6 and PE 16:0/20:4) in 2,2’-Azobis(2-amidinopropane) dihydrochloride (AAPH). Fer-1 is given as control. Significance was evaluated by two-tailed t-test. B Peak area stability (LC-MS²) of PUFA ester lipids (PE 16:0/20:4) in presence of plasmalogens in 2,2’-Azobis(2-amidinopropane) dihydrochloride (AAPH). C Cell viability of control cells incubated with 25 µM plasmalogens (e MUFA and e PUFA) or EtOH for 8 h then challenged with 0.3 µM RSL3 in the presence of αToc in a dose-dependent manner. D Viability of Ms4a15 OE cells pretreated with PUFAs eicosapentaenoic acid (C20:5, EPA), docasapentaenoic acid (C22:5, DPA), and doxosahexaenoic acid (C22:6, DHA) with ferroptosis induction by 2 μM RSL3 and αToc rescue. Significance was evaluated by two-tailed t-test. E RSL3 treatment of 72 h siRNA knockdown of Scd1, Fads2, or Hsd17b12 compared to siGFP in control cells as individual experiments (left panel) or all three siRNAs together (3x siRNA, right panel). Inset shows relative gene expression by qPCR (rel. mRNA). F Heatmap showing dysregulation of genes involved in lipid droplet formation. G BODIPY 493/503 staining of lipid droplets of Ms4a15 OE, control and 14 d Tgn-treated cells. High-content images (upper) showing lipid droplet dispersion. Quantification of lipid droplet number (LDs/cell) and area (μm2/cell) was performed by Harmony software (PerkinElmer). H Analysis of average lipid droplet number and area (left) and fluorescence intensity (right). Data were obtained from three independent experiments and a representative experiment shown with analysis by Harmony software. Lipid droplet intensity is depicted via a flow cytometry histogram of a representative experiment of three independent repetitions. Significance was evaluated by two-tailed t-test. Cell-free assay and viability assays are reported as mean ± SD of n = 3 (A, C, E) or n = 4 (D) technical replicates of three independent experiments with similar outcomes. Curve statistics, p-values of two-way ANOVA, shown above comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Lipid elongation and desaturation mediate resistance
Ms4a15 OE lipids are shorter but more saturated (Fig. 3E, G). Thus, these lipids may derive from de novo lipogenesis upon compromised ER-resident elongase and desaturase activities. Analogously, ML239 agonizes fatty acid Δ6 desaturase 2 (FADS2) activity to increase PUFA synthesis and ferroptosis sensitivity [49]. We considered that supplementation with free exogenous PUFA fatty acids may overcome protective lipids. We treated Ms4a15 OE cells for 48 h with 20:5n-3 (EPA), 22:5n-3 (DPA) and 22:6n-3 (DHA) and observed that longer, more unsaturated DPA and DHA potentiated ferroptosis more robustly than EPA (Fig. 5D).
These data are consistent with elongase and desaturase deficits. Their corresponding genes are so far absent from ferroptosis screens, possibly reflecting independent desaturation activities [50]. Accordingly, individual siRNA inhibition of stearoyl-CoA desaturase 1 (Scd1), Fads2, or very-long-chain 3-oxoacyl-CoA reductase (Hsd17b12) did not protect against ferroptosis, while pooling all three siRNAs partially protected (Fig. 5E).
Scd1 and Fads2 are counterregulated with Ms4a15 OE and act downstream of key lipid regulator Pparγ to promote lipid droplets (LDs), which are formed in the ER and act as reservoirs to control lipotoxicity and ER homeostasis under stress. RNAseq revealed Pparγ misregulation in Ms4a15 OE cells together with genes controlling LD dynamics (Fig. 5F, Supplementary Fig. 2A), while high-content analysis showed widespread LD dispersion in Ms4a15 OE and Tgn-treated cells (Fig. 5G). A significant mean decrease in number but unchanged area and fluorescent intensity indicated that LDs are redistributed to smaller droplets in the cytosol rather than lost (Fig. 5H). Collectively, these data show that depletion of ER calcium lead to qualitative changes in ferroptosis-sensitive lipids in concert with subcellular LD rearrangement.
Global Ca2+ genes define a signature for ferroptosis
We speculated that changes in Ca2+ homeostasis resulting in ferroptosis-resistant lipids may contribute to resistance in different cell lines. We tested this theory by cross-referencing sensitivity of the 100 most RSL3-resistant and -sensitive cancer cell lines from the CTRP database [49] to KEGG Ca2+ gene expression [37].
Using unsupervised clustering of Ca2+ genes, we observed segregation corresponding to sensitivity (Supplementary Fig. 6A, Supplementary Table 4). Several clusters dominated sensitive lines, in particular coordinated downregulation of EGFR, ERBB2/3 (HER2/3), ITPR3 (IP3R3), and GNAQ, coupled to activation of PLC-beta and Ca2+ release. Reduced GPCR and PLC subtypes was also prominent among sensitive hematopoietic/lymphoid-derived cell lines, which favor cadherin/integrin-based homing and are exquisitely sensitive to ferroptosis [32, 51, 52]. PCA also distinctly separated resistant and sensitive CTRP cell lines (Fig. 6A). Of these, ATP2A3 and PLCG2 were key drivers of the RSL3 sensitive group, while EGFR, ERBB2/3 and ADRB2 were in the RSL3 resistant group. Together, these results are consistent with re-sensitization of Ms4a15 OE cells by Atp2a2/Serca2 overexpression and suggest that signaling molecules can influence Ca2+ homeostasis and PUFA/MUFA/plasmalogen ratios.
Fig. 6. Global Ca2+ genes define a signature for ferroptosis sensitivity.

A PCA biplot of mRNA gene expression of RSL3 resistant/sensitive cell lines. The distance between points approximates gene expression differences among groupings. Arrows indicate driver genes with greater biplot scores. Arrow length indicates relative abundance differences in associated samples. B Dose response curves of control cells against RSL3 (0.4 μM) after 48 h pretreatment with PLC activator m-3M3FBS and inactive analog o-3M3FBS. Addition of 10 μM αToc serves as rescue control. Ca2+ store depletion was detected by ionophore. C Volcano plot of lipid changes in 3M3FBS samples for Ms4a15 OE modulated species. Dot size indicates abundance of single lipid species. D–F Dose response curve of cell lines pretreated with PLC inhibitors (2 µM U73122 + 1.5 µM edelfosine, EDEL) for 48 h. RSL3 was applied for ferroptosis induction and αToc serves as rescue control. E Ca2+ store accumulation was detected by ionophore. F Volcano plot of lipid changes for treated versus untreated (untr.) cells for Ms4a15 OE modulated species. Dot size indicates single species abundance. G Schematic overview of MS4A15-mediated ferroptosis resistance. Overexpression of MS4A15 leads to ER Ca2+ store depletion, resulting in decreased PUFA- and long chain acyl-lipids. Simultaneously, increased MUFAs and MUFA-plasmalogens act as ROS sinks to protect cells from lipid peroxidation. Viability data are plotted as representative mean ± SD of n = 3 technical replicates for independent experiments repeated at least three times with similar outcomes. Curve statistics, p-values of two-way ANOVA, shown above comparisons. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
EGFR and EGFR/ERBB3 dimers can activate Ca2+ release via PLCs. We mimicked this signal and associated ER Ca2+ depletion by constitutively activating PLC for 48 h with m-3M3FBS in control cells (Fig. 6B). Extensive ferroptosis protection was observed, while o-3M3FBS (a control for nonspecific antioxidant activity) showed no effect, consistent with observed changes in ether and MUFA-lipids (Fig. 6C). PLCs are classically coupled to GPCRs, therefore we tested “orphan” GPCR Olfr39 overexpressing cells identified in the same screen [32] and observed the prototypical pattern of Ms4a15 OE in viability, Ca2+, and lipid regulation (Supplementary Fig. 6B–D).
Given this result, we tested if augmenting ER Ca2+ could sensitize ferroptosis-resistant cell lines. We applied PLC inhibitors U73122 and edelfosine for 48 h to broadly inhibit Ca2+ release (Fig. 6D, E, Supplementary Fig. 6E). Testing of two resistant breast cancer cell lines revealed that MDA-MB-231 could be sensitized to RSL3, while MCF-7 cells were unaffected. MDA-MB-231 are EGFR-addicted cells, whereas MCF-7 are estrogen dependent. Similarly, lung cancer line NCIH1975 has a high dependency on EGFR [53] and could be sensitized while A549 could not. Finally, of two investigational cell lines, HEK293T and HeLa, only HEK293T responded to synthetic sensitization with relevant lipids plots showing degrees of ether and MUFA-lipid depletion (Fig. 6F). Together, these results demonstrate that elevating ER Ca2+ levels by blocking signals at the membrane can sensitize certain ferroptosis-resistant cell lines.
Discussion
In this report we define a unique mechanism for ferroptosis resistance based on the discovery of MS4A15, an uncharacterized four-pass membrane protein. MS4A proteins have previously been proposed to oligomerize into ion channels to facilitate Ca2+ movement [27], however recent work suggests the requirement for other channel proteins [54]. In contrast to other members at the plasma membrane, MS4A15 is localized to the ER where it constitutively depletes Ca2+ stores. Consistent with previous studies showing that MS4A proteins promote Ca2+ flux [30, 55, 56], overexpression of Ms4a15 profoundly altered Ca2+ homeostasis and depressed IP3R1 expression, resulting in extensive lipid remodeling (see graphical summary Fig. 6G). This effect is similar to treatment with thapsigargin, a specific inhibitor of ER Ca2+ uptake, and can be reversed by PLC inhibitors.
The primary consequence of decreased luminal Ca2+ levels is depletion of long PUFA-GPs in favor of shorter MUFA-GPs and -ether lipids, particularly plasmalogens. Long chain PUFA-GPs are targets of ferroptosis oxidation in control cells, while Ms4a15 OE demonstrate preferential degradation of MUFA-GPs and plasmalogens. Until now, exogenous MUFAs [46] and nonspecific ether lipids [57] have hinted to ferroptosis protection. Ms4a15 OE provides the first demonstration that endogenous MUFAs and specifically MUFA-plasmalogens are targets of ferroptotic ROS in the low luminal Ca2+ state.
Plasmalogens have been suspected to harbor antioxidant capacity [58, 59]. In vitro, plasmalogens delay degradation of sn-2 GPs in the presence of oxidants, suggesting the vinyl ether bond protects against radical-generated oxidation [60]. Importantly, the antioxidant capacity appears to be intramolecular [61]. Lipid peroxidation propagation is stopped by the absorption of ROS at delocalized electrons of the vinyl ether bond. During preparation of this manucript, PUFA-containing plasmalogens were shown to promote ferroptosis [48], which complements our findings that alkyl chains strongly dictate sensitivity. MUFA-plasmalogens, therefore, act as anti-ferroptotic reservoirs by absorbing ROS and limiting their propagation in the membrane [62].
Disruption of ER Ca2+ homeostasis has been linked to lipogenesis [18]. Similarly, in rats, Ca2+ deficiency leads to loss of long chain PUFAs [63]. Our results suggest that the activities of elongases and desaturases may require stable luminal Ca2+ to synthesize PUFA-containing lipids. Moreover, depletion of Ca2+ stores causes the dispersion of lipid droplets, which are tightly coupled to cellular metabolism and storage of diverse lipid species. In this respect, the lack of PUFAs may be compensated by de novo lipogenesis, driving increased MUFA-GPs and plasmalogens and changes in lipid droplet dynamics. As LDs sequester not only neutral lipids but also PUFA-containing phospholipids, these are not released into the fatty acid pool for re-esterification in membranes as observed in MS4A15 OE cells. LDs also provide physical separation from peroxidation at the membrane [64, 65]. Thus, qualitative remodeling of lipids to MUFA-GPs in MS4A15 OE cells also triggers a redistribution of LDs, producing smaller, dispersed lipid droplets that may additionally limit oxidation [64, 66]. However, the relationship between LD localization and ferroptosis sensitivity is still unexplored.
Ferroptosis has been widely linked to cancer, yet how precancerous cells limit ferroptosis-inducing PUFAs is enigmatic [67]. Our findings linking calcium and ferroptosis are relevant in this context as the number of oncogenes and tumor suppressors that control Ca2+ homeostasis and cell death is increasing [68–70]. For instance, the RAS oncogene limits IP3R activity and ER Ca2+ flux [71]. Analogously, Serca2 haploinsufficiency and thapsigargin cause tumors in mice [72, 73]. Thus, it is plausible that changes in the calcium/lipid axis disrupt an endogenous ferroptotic mechanism to abate neoplastic transformation. Ours and others recent work has demonstrated evidence for endogenously produced antioxidants to overcome ROS-induced lipid peroxides [32, 74, 75], or limit PUFA insertion into membranes [76]. Targeting these pathways provides an opportunity to limit therapy resistance in tumors. Hence, modulating Ca2+ homeostasis provides an additional lever to influence cell survival.
An overlap between ferroptosis and oxytosis has been suggested as the late lethal influx of Ca2+ is conserved in some cells. A conclusion of MS4A15 limiting acute Ca2+ flux is nevertheless unlikely. BODIPY-C11 analysis of Ms4a15 OE cells, in contrast to short Tgn-treatment, demonstrably lack early-forming oxidized lipids. Moreover, SOCE blockage did not markedly affect ferroptosis sensitivity while Tgn alters cell viability and lipid profiles, despite its highly active Ca2+ uptake. Thus, acute Ca2+ flux and persistent Ca2+ dyshomeostasis are distinct cell death phenomena with the latter primarily affecting biosynthesis of ferroptosis substrates.
In conclusion, MS4A15 unites several distinct ferroptosis phenomena. It coordinates lipid remodeling by regulating ER Ca2+ levels, while ER-synthesized MUFA-GPs and -plasmalogens abate ferroptosis-induced lipid peroxidation. Taken together, these data strongly support the conclusion that MS4A15 is an independent contributor to ferroptosis resistance.
Materials and methods
Cell lines and culture conditions
Cell lines used in the study: Immortalized conditional Gpx4 −/− mouse embryonic fibroblasts expressing Cre-ERt2 (MEF, male) [33] were previously generated [32] with the CRISPR activation system [77] and a mouse Ms4a15 CRISPR guide (Supplementary Table 5) for overexpression, Calu-1 (gift from Brent Stockwell), HEK293T (fetal, ATCC Cat# CRL-3216), H1975 (female, ATCC Cat# CRL-5908); MDA-MB-231 (female, ATCC Cat# HTB-26), MCF-7 (female, ATCC Cat# HTB-22), HeLa (female, ATCC Cat# CCL-2), A549 (male, ATCC Cat# CCL-185).
Calu-1 cells were maintained in RPMI Medium (Thermo Fisher Scientific) with 15% fetal bovine serum (FBS, Biochrom). Other cell lines were maintained in DMEM (Thermo Fisher Scientific) containing 10% FBS. All cells were grown in medium supplemented with 1% L-Glutamine (Thermo Fisher Scientific) and 1% Penicillin-Streptomycin (Thermo Fisher Scientific) at 37 °C in a humidified atmosphere of 5% CO2. Cell lines were regularly checked for mycoplasma and morphological conformity with ATCC’s specifications.
Generation of cell lines
To generate pooled OE cell lines, individual guides were cloned into lenti-sgRNA(MS2)_Neo (neomycin resistance substituted for zeomycin in Addgene plasmid # 61427) and packaged with lentiviral third generation ecotropic system. Control cells were infected with empty lentivirus. A guide for Serca2 activation (Supplementary Table 5) was cloned into lenti-sgRNA(MS2)_Zeo (Addgene plasmid # 61427) [77] to generate Ms4a15 + Serca2 OE cell line via stable infection of the MF Ms4a15 OE cell line. Cell pools were selected for 1 week with 1 mg/mL G418 Sulfate (Geneticin Selective Antibiotic, Thermo Fisher Scientific) and 200 µg/mL Zeo (Thermo Fisher Scientific), respectively. Viral production and infection were performed as previously reported [32]. Ms4a15 CRISPR homozygous mutations (30% efficiency) were generated in parental MF cells and validated by genotyping PCR and a 17-bp deletion in exon 2 by Tide (shinyapps.datacurators.nl/tide/). All guides and genotyping primers are listed in Supplementary Table 5.
Human MS4A15-overexpressing HT1080 and Calu-1 cell lines
To generate pooled MS4A15-overexpressing HT1080 cells, corresponding guides were cloned into lenti-sgRNA(MS2)_Neo and packaged with lentiviral third generation system (see above) and expressed with helper constructs [77]. To generate MS4A15-overexpressing Calu-1 cells, a human pLVTHM hMS4A15-FLAG-T2A-neo expression construct was cloned and lentivirus applied to parental Calu-1 cells and selected with G418 for 7 days before cell death experiments.
Generation of monoclonal anti-human MS4A15 antibody
For generation of monoclonal antibodies against MS4A15, a Lou/c rat was immunized with 40 µg ovalbumin-coupled peptide spanning aa50-62 (AQTPRATQPPDLR) of human MS4A15, 5 nmol CpG (TIB MOLBIOL), and an equal volume of Incomplete Freund’s adjuvant (IFA; Sigma). After 12 weeks, a boost injection without IFA was given 3 days before fusion of rat spleen cells with P3X63Ag8.653 myeloma cells. Hybridoma supernatants were screened in a bead-based flow cytometry assay (iQue, Intellicyte; Sartorius) on his-tagged, biotinylated peptide captured on streptavidin beads (PolyAN) and incubated for 90 min with hybridoma supernatant and Atto 488-coupled isotype-specific monoclonal mouse-anti-rat IgG secondary antibodies. Antibody binding was analyzed using ForeCyt software (Sartorius). Positive supernatants were validated by western blot of Ms4a15 OE and control cell lysates. Hybridoma cells were subcloned five limiting dilution rounds to obtain the stable monoclonal cell clone MS4A 5E6 (rat IgG2c/ƙ). Experiments in this work were performed with hybridoma supernatant.
Assessment of cell viability
Unless indicated otherwise, 2 × 103 MF or 4 × 103 human cells were seeded in 96-well plates and treated with the corresponding compounds as indicated in figures and figure legends. RSL3/IKE was added to the cells 1 day before Resazurin incubation. Resazurin (Sigma) was added to a final concentration of 50 µM, cell viability was assessed after 6–8 h incubation. The Envision 2104 Multilabel plate reader (PerkinElmer) was used for measuring the fluorescence at 540 nm excitation/590 nm emission. In general, at least 3 wells under each condition were averaged and all cell viability results are presented as percentage relative to the respective untreated or vehicle-treated control as mean ± SD. For propidium iodide (PI) stains, cells were treated with 0.5 μM RSL3 overnight and incubated with 3 μM PI for 15 min. Cell images were taken with an Operetta High-Content Screening System (PerkinElmer) with a ×20 objective. For colony-forming assays, cells were treated with 1.25 μM RSL3 overnight, then trypsinized single-cells, diluted 1:300 and seeded into six-well plates. After 7 d colonies were stained with cresyl violet and imaged.
Three-dimensional spheroids. MF control and Ms4a15 OE cells were seeded into the GravityTRAP ULA 96-well plates (InSphero/PerkinElmer) to form 3D spheroids. Interwell variations <10% were confirmed and spheres were grown for 4 days, treated with 2 µM RSL3 for additional 16 h and stained with PI. Spheroids were imaged directly with an Operetta High-content system. Images from a single plate were acquired using Brightfield and PI channels and ×20 High-NA objective in wide field mode. Ten planes of each sample were tracked and four replicates per cell condition were collected with the same parameters and PI intensity of different cell conditions were analyzed with Harmony software (PerkinElmer) using the same settings to optimize the results.
siRNA knockdown
Mission esiRNAs targeting human TMEM33 (EHU035611), EGFP (EHUEGFP), murine Tmem33 (Emu078331), murine Fads2 (EMU027741), murine Scd1 (EMU023031) and murine Hsd17b12 (EMU064031) were purchased from Sigma. 1.5 × 105 cells were typically seeded in six-well plates 1 day before. Prior to transfection, 200 ng of siRNA and 3 μl Lipofectamine RNAiMAX Transfection Reagent (Thermo Fisher Scientific) were mixed and incubated at room temperature for 15 min in serum-free media, then added dropwise on top of the cells. After 48 h transfection, cells were harvested for subsequent experiments.
Quantitative PCR
Total RNA was isolated with the InviTrap Spin Universal RNA Mini Kit (Stratec). Random hexamer primer and AMV Reverse Transcriptase (NEB) were used for reverse transcription. Quantitative PCR reactions were carried out using the LightCycler480 (Roche) with Power SYBR Green PCR Master Mix (Thermo Fisher Scientific). Using GAPDH or Actin as a reference gene, the relative expression levels compared to the control were calculated by the ΔΔCp method. Primer sequences are listed in Supplementary Table 5.
Lipid peroxidation analysis by flow cytometry
Cells were seeded in six-well plates to reach 70% confluency. The next day, 0.3 µM RSL3 was added for 3 h. Cells were loaded with 2 µM BODIPY 581/591 C11 (Thermo Fisher Scientific) for 30 min and harvested for analysis on an Attune acoustic flow cytometer (Applied Biosystems). At least 30,000 events per condition were collected from the BL-1 channel (excited by 488 nm laser). Each experiment was repeated at least three times independently and representative results are shown.
Intracellular calcium measurements
Cells containing the cytosolic calcium sensor GCaMP6s were seeded the day before in 10 cm dishes to reach 70% confluency. The following day, cells were treated with Accutase (Sigma) and resuspended in PBS, washed twice with Ca2+-free buffer (NaCl 116 mM, KCl 5.6 mM, MgCl2 1.2 mM, NaHCO3 5 mM, NaH2PO4 1 mM, HEPES 20 mM, Glucose 1 g/L). Cell pellets were resuspended in 2 mL of Ca2+-free buffer and were analyzed with a BD FACSCanto II (Becton Dickinson). Untreated cell suspensions were recorded for 2 min (approx. 2000 events/second) to establish a baseline signal. Ca2+ release mediated by Bradykinin (Sigma) and Ionophore (Sigma) was measured for 4 and 6 min, respectively. After Bradykinin stimulation, 2 mM CaCl2 was added to the cells and data for the uptake of Ca2+ was collected for additional 9 min. Kinetic data were created by FlowJo V10 of viable, GFP positive cells and exported for visualization to GraphPrad Prism 8. All experiments were repeated at least three times.
AAPH oxidation assay using BODIPY 581/591 C11
Ester lipids, plasmalogens and ferrostatin (fer-1) were added into 150 µL PBS as indicated to achieve 150, 150, and 9 ppm, respectively. Freshly dissolved 1.875 µM BODIPY 581/591 C11 in 150 µL PBS and 7.5 mM 2,2′-Azobis(2- amidinopropane) dihydrochloride (AAPH, VWR International) in 150 µL PBS were separately added to start the oxidation. PBS containing the same ratios of ethanol/methanol/DMSO served as control. After mixing thoroughly, reaction samples were incubated in the dark for 30 min at room temperature. 100 µL sample per well was measured using an Envision 2104 System (PerkinElmer) in black 96-well plates as triplicates. Fluorescence intensity at excitation 495 nm/emission 520 nm was evaluated and normalized to ethanol/methanol/DMSO control. Ferrostatin-1 was used as an antioxidant positive control.
Lipid cell assays
20 mM PUFA lipids were mixed with 2.5 mM BSA at a ratio of 1:4 and incubation at 37 °C for 45 min, pre-warmed media was subsequently added into the mixture. Ms4a15 OE cells were pre-seeded the day before, the PUFA/BSA mixture was added to the cells to achieve a final PUFA concentration of 25 µM. After 48 h incubation, cells were challenged with 2 µM RSL3.
For plasmalogen experiments, MF control cells were seeded the day before on 96-well plates. The following day, cells were washed with PBS and incubated with 25 µM plasmalogens in serum-free medium for 8 h. After serum starvation, 10% FBS was added back and the cells were treated with RSL3 and aToc to achieve final concentrations as indicated. Cell viability assay was performed as described above.
EGF signaling in cultured cells
MF cells were pre-seeded in six-well plates 1 day before for reaching 70% confluency. The culture medium was changed to serum-free medium and incubated at 37 °C for 4 h starvation. Subsequently, the serum-starved MF cells were stimulated with 0–5 ng/mL EGF for 10 min at 37 °C, washed with PBS and lysed for western blot analysis.
Western blotting
Cells were lysed for 20 min in lysis buffer (63 mM Tris-HCl, pH 6.8, 10% glycerol, 2% SDS, 2.5% DTT and 1x protease inhibitor tablet (Roche)) and DNA was shredded with a sonicator. After separation on a 6–12% SDS-PAGE gel according to the protein sizes, proteins were transferred to PVDF membranes. After blocking with 5% non-fat milk for 1 h at room temperature, the membranes were incubated in specific primary antibodies diluted in 2.5% BSA at 4 °C overnight. The next day, membranes were incubated with secondary antibodies for 2 h at room temperature. ECL prime western blotting detection reagents (Bio-Rad) were used at a ratio of 1:1 for chemiluminescence detection. Each experiment presented was repeated at least three times. Primary antibodies used in this study: MS4A15 (HMGU, N/A,1:10), ATP2A2 (Elabscience, E-AB-30196, 1:250), FLAG (Sigma, F7425, 1:2000), MYC (Abcam, ab206486, 1:2000), ERK1/2 (Cell Signaling, 4696, 1:1000), pERK1/2 (Cell Signaling, 9101, 1:1000), STAT3 (Cell Signaling, 9139, 1:1000), pSTAT3 (Cell Signaling, 4113,1:1000), AKT (Cell Signaling, 9272, 1:1000), pAKT (Cell Signaling, 9271,1:1000), ß-Actin (Cell Signaling, 3700, 1:2000), alpha-Tublin (Cell Signaling, 2125, 1:2000) and Vinculin (Abcam, ab130007, 1:500).
Confocal microscopy and immunofluorescence
Cells were plated at a density of 4 × 103 cells/well on 96-well plates (Perkin Elmer Cell Carrier Ultra Viewer). Cells were transfected with corresponding expression constructs for 24 h before 4% formaldehyde fixation. Images were taken with a laser scanning confocal microscope (Olympus FluoView 1200; Olympus Corporation). Nuclei were labeled with DAPI staining (blue). MS4A15 was visualized with Anti-FLAG antibody (Sigma F7425; 1:500) and a secondary goat anti rabbit antibody (Cy3 Jackson Immuno 111-165-003; 1:500). TMEM33 was visualized with Anti-MYC tag antibody (Abcam 9E10; 1:200) and a secondary donkey anti-mouse antibody (Alexa 647 Invitrogen A-32733; 1:500). IP3R1 was visualized with anti-IP3R1 antibody (Biozol BLD-817701; 1:500) and a secondary donkey anti-mouse antibody (Alexa 647 Invitrogen A-32733; 1:500). ER was tracked with ER marker Concanavalin A/Alexa fluor 488 conjugate (Invitrogen C11252; 100 μg/mL).
Lipid droplets analysis by high-content imaging
Cells were seeded in 96-well plates to reach 80% confluency. The next day, cells were loaded with 2 µM BODIPY 493/503 for 30 min and washed with PBS twice before fixation. The images were taken using an Operetta High-Content Screening System (PerkinElmer) with GFP filter (excitation 488 nm, emission 509 nm) with the same parameters.
High-resolution high-speed time-lapse live-cell imaging
High-throughput wound healing assay: culture-Inserts (ibidi 80209) were used to create a 500 µm gap, in two reservoirs for culturing cells. 8 × 103 MF cells were seeded in each reservoir and cultured for 24 h until they attached in monolayers. The cells were imaged at ×20 magnification after insers removal using an Operetta High-Content Screening System (PerkinElmer) equipped with digital phase contrast for live-cell imaging. Eight images per well were collected with the same parameters and analyzed with Harmony software (PerkinElmer) using the same settings to optimize the comparison results between different cell lines.
RNA-Seq
RNA-Seq was performed as described earlier [78]. Briefly, RNA was isolated from whole-cell lysates using InviTrap Spin Universal RNA Mini Kit (Stratec) according to the manufacturer’s instructions. For library preparation, 1 μg of RNA was poly(A) selected, fragmented, and reverse transcribed with the Elute, Prime, Fragment Mix (Illumina). End repair, A-tailing, adaptor ligation, and library enrichment were performed as described in the Low Throughput protocol of the TruSeq RNA Sample Prep Guide (Illumina). RNA libraries were assessed for quality and quantity with the Agilent 2100 BioAnalyzer and the Quant-iT PicoGreen dsDNA Assay Kit (Life Technologies). RNA libraries were sequenced as 100 bp paired-end runs on an Illumina HiSeq4000 platform.
Immunoprecipitation assay
HEK 293 T cells were seeded at 1 × 106 cells per well in 10 cm plates the day before. Transfection was performed in triplicates with 10 μg of each plasmid (GFP and MS4A15) using Lipofectamine 2000 following the manufacturer’s instructions. Cells were harvested after 24 h in PBS and crosslinked using 1% formaldehyde at room temperature for 7 min, followed by 3 min centrifugation at 1800 × g. Supernatant was removed and the reaction was quenched with 0.5 mL ice-cold 1.25 M glycine/PBS. Cells were washed once in 1.25 M glycine/PBS and lysed for 60 min on ice with homogenization in 1 mL RIPA buffer (50 mM Tris HCl, pH 8.0, 150 mM sodium chloride, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 1 mM EDTA, protease inhibitors (Complete mini, EDTA-free, Roche)). Spun for 30 min at 20,000 × g to remove insoluble debris, the lysates were precleared by incubation for 2 h with 20 μl protein G agarose beads (Protein A/G PLUS-Agarose, Santa Cruz). The precleared lysates were incubated with 2 μl FLAG (Sigma, F7425) antibody for 1 h, subsequently 20 μl of beads were added and immunoprecipitation was performed overnight. All steps were carried out with mild agitation at 4 °C. The beads were washed three times with RIPA buffer and incubated in 1 x Roti Loading Dye (Carl Roth) at 65 °C for 5 min. Samples were stored at −80 °C for mass spectrometric analysis.
Quantitative mass spectrometry in data‐dependent acquisition mode
Dried beads after pulldown of MS4A15 from formaldehyde-fixed samples were resuspended in 50 µL 1x Laemmli and de-crosslinked for 60 min at 99 °C. after reduction and alkylation using DTT and IAA, the proteins were centrifuged on a 30 kDa cutoff filter device (Sartorius), washed twice with UA buffer (8 M urea in 0.1 M Tris/HCl pH 8.5) and twice with 50 mM ammoniumbicarbonate. The proteins were digested for 2 h at room temperature using 0.5 µg Lys-C (Wako Chemicals) and for 16 h at 37 °C using 1 µg trypsin (Promega). After centrifugation (10 min at 14,000 g) the eluted peptides were acidified with 0.5% TFA and stored at −20 °C.
LC-MS/MS analysis was performed on a Q-Exactive HF mass spectrometer (Thermo Scientific) online coupled to an Ultimate 3000 nano-RSLC (Thermo Scientific). Tryptic peptides were automatically loaded on a C18 trap column (300 µm inner diameter (ID) x 5 mm, Acclaim PepMap100 C18, 5 µm, 100 Å, LC Packings) at 30 µL/min flow rate prior to C18 reversed phase chromatography on the analytical column (nanoEase MZ HSS T3 Column, 100 Å, 1.8 µm, 75 µm × 250 mm, Waters) at 250 nl/min flow rate in a 95 min nonlinear acetonitrile gradient from 3 to 40% in 0.1% formic acid. Profile precursor spectra from 300 to 1500 m/z were recorded at 60,000 resolution with an automatic gain control (AGC) target of 3e6 and a maximum injection time of 50 ms. TOP10 fragment spectra of charges 2–7 were recorded at 15,000 resolution with an AGC target of 1e5, a maximum injection time of 50 ms, an isolation window of 1.6 m/z, a normalized collision energy of 27 and a dynamic exclusion of 30 s.
Proteomics
Briefly, 1 × 107 Ms4a15 OE and parental MF cells per replicate (n = 5) were lysed and equal amounts were proteolyzed using a modified FASP procedure [79]. The proteins were digested for 2 h at room temperature using 0.5 µg Lys-C (Wako Chemicals) and for 16 h at 37 °C using 1 µg trypsin (Promega), eluted by centrifugation, acidified with TFA and stored at −20 °C. Peptides were measured on a Q-Exactive HF mass spectrometer online coupled to an Ultimate 3000 nano-RSLC (Thermo Scientific) in data-independent acquisition (DIA) mode as described previously (Lepper et al., [80]). Raw files were analyzed using the Spectronaut Pulsar software (Biognosys; [81]) with a false discovery rate setting of <1%, using an in-house mouse spectral meta library generated using Proteome Discoverer 2.1 (Thermo Scientific), the Byonic search engine (Protein Metrics) and the Swissprot Mouse database (release 2016_02). Quantification was based on MS2 area levels of all unique peptides per protein fulfilling the percentile 0.3 setting. Normalized protein quantifications were exported and used for calculations of fold-changes and significance values.
Metabolite extraction and global metabolomics
Ms4a15 OE and control were prepared as described [32]. For analysis, cells were resuspended in 800 µL methanol and transferred into beat tubes. Eppendorf cups were flushed additionally with 200 µL to transfer remaining cells. Cells were lysed using 2 × 15 s, below 4 °C (Precellys, Bertin) and centrifuged with 12,000 rpm for 15 min. The supernatant was immediately diluted 1:10 in methanol. Mass spectra were acquired on a 12 T solariX FT-ICR mass spectrometer (Bruker Daltonics) using an Apollo II electrospray source (Bruker Daltonics), in broad band detection mode with a time domain transient of 2 Megawords in positive and negative electrospray mode. The instrument was calibrated with a 1 ppm arginine solution. A mass error below 100 ppb was achieved. Injected velocity was set to 120 µL/h. Mass lists were generated with a signal-to-noise ratio (S/N) of four, exported, and combined to one data matrix by applying a 1 ppm window. Ions (m/z mass/charge) were annotated using MassTRIX allowing 1 ppm mass tolerance. Unidentified metabolites were annotated by elemental composition using mass-differences based network approach allowing 0.1 ppm mass tolerance [82].
Lipid extraction and global lipidomics
Procedures for lipid extraction and global lipidomics profiling using UPLC-MS were described previously [45]. In short, we used a two-step MTBE extraction in a cooled Precellys (Bertin). The organic content was analyzed using data-dependent auto LC-MS² (maXis, Bruker Daltonics) coupled to an UHPLC ACQUITY (Waters) using reverse phase chromatography (CORTECS UPLC C18 column, 150 mm × 2.1 mm ID 1.6 µm, Waters Corporation) in both positive and negative electrospray modes. The injection volume was set to 10 µL. Lipid elution was achived using 10 mM ammonium formate and 0.1% formic acid in 60% acetonitrile/water mixture (A) and in 90% isopropanol/acetonitrile mixture (B) as mobile phase. Quality control consisting of an aliquot of each sample and pure solvent blanks were used for column equilibration. The MS analysis alternated between MS and data-dependent MSn scans using dynamic exclusion. Alignment, peak picking and identification as well as quality control processing was done in Genedata software (Genedata Expressionist 13.5, Genedata). Retention time and detected m/z were used to annotate lipid species according to the Lipid Classification System guidelines of LIPID MAPS Structure Database (LMSD) [83] (max 0.005 Da error), while single lipid species identification was substantiated by MS2 fragmentation (see Supplementary Table 2). MS2 information was first annotated based on MoNA library with MSPepSearch [84] and with MetFrag [85], followed by a further validation by manual curation [86]. Furthermore, the existence of the vinyl ether linkage was verified via acidic hydrolysis following previously published protocol [87, 88]. Samples were evaporated and reconstituted in methanol prior MS analysis. Under the chosen conditions, only vinyl ether linkages in plasmenyl-compounds are cleaved. Ether and ester bindings stay intact.
Quantification and statistical analysis
Statistics summary
Unless otherwise stated, general statistical analyses and data visualization were performed in GraphPad Prism version 8.0 and R version 3.6.3. All of the statistical details can be found in the figures, figure legends, and results, including the statistical tests used, exact p-values, and dispersion and precision measures. Curve statistics were performed in GraphPad Prism using Two-way ANOVA and Tukey’s multiple comparisons test.
RNAseq analysis
The STAR aligner [89] (version 2.4.2a) with modified parameter settings (--twopassMode = Basic) is used for split-read alignment against the mouse genome assembly mm10 and UCSC knownGene annotation. To quantify the number of reads mapping to annotated genes we use HTseq-count [90] (v0.6.0). FPKM (Fragments Per Kilobase of transcript per Million fragments mapped) values are calculated using custom scripts and differential gene expression analysis was performed with the R Bioconductor package “DESeq2” [91].
Immunoprecipitation analysis
Generated raw files were analyzed using Progenesis QI for proteomics (version 4.1, Nonlinear Dynamics, part of Waters) for label-free quantification as described previously [92]. Resulting normalized protein abundances were used for calculation of fold-changes and statistical values.
The log2 of the normalized protein abundance ratios MS4A15/GFP and -log10 of corresponding p-values of all quantified proteins were visualized in a volcano plot. A very specific pulldown in the MS4A15-PD samples and very low protein abundances in the GFP controls lead to the appearance of mainly only one “arm” of the volcano plot.
Metabolomic analysis
Statistical analysis was performed in R studio (R 1.2.5019). To identify metabolites that show significant change a Mann–Whitney U test for non-parametric variables was performed, and BH corrected for multiple testing. Missing values were imputed by randomly generated minimum values and the data was TIC normalized. Unit variance scaling and mean centering was applied before statistical testing. PLS-DA models were built in SIMCA-P (Umetrics) and validated by performing 100 random permutations.
Heatmap proteomics representation
For heatmap of known ferroptosis genes from Stockwell [1], individual log2 samples were divided by the sum of each row and clustered by Euclidean distance using Gene Cluster 3 [93]. The results were mapped with Java Treeview [94].
KEGG Calcium clustering
To generate the clustered dataset shown in Fig. 6A and Supplementary Table 4, CTRP2.0 data were downloaded from CTD2 data-portal [95]. Top 100 resistant/sensitive cell lines are AUC v20.data.curves_post_qc.txt values. CCLE expression data were downloaded from https://depmap.org/portal/download/all/?release=DepMap+Public+20Q1&file=CCLE_expression_full.csv.
KEGG Calcium signaling pathway genes were downloaded from https://www.genome.jp/dbget-bin/get_linkdb?-t+orthology+path:ko04020. After normalization, Gene Cluster 3.0 with hierarchical clustering for cell lines was used according to Euclidean distance with complete linkage; clustering for genes used City Block clustering. Data were visualized using Java TreeView.
Principal component analysis
Gene expression data consists of 204 human cell lines (observations) from two different known groups (Resistant group (R) and Sensitive group (S)) described by 193 genes (variables).
PCA was performed in R (version 3.6.3) to visualize the clustering of the gene expression data using log-fold transcript abundance of gene arrays in each group. Variables were pretreated to eliminate redundant columns with more than 40 zero values by applying the function implemented in R/colSums (RS = 0). The following analysis was performed by variables with the highest 100 median absolute deviations (MAD). Multivariate biplot were performed to characterize the variability of the data in each group using “ggplot2” [96], “factoextra” [97], and “ade4” [98] packages.
ssGSEA implementation
The correlations between gene expression levels were calculated by Pearson’s test. The 50 genes with the most significant correlation coefficients were identified from whole transcriptome. The heatmap was plotted with R package “pheatmap” [99].
GO_CALCIUM_ION_TRANSMEMBRANE_TRANSPORT, KEGG_CELL_ADHESION, and KEGG_CALCIUM_SIGNALING_PATHWAY term lists were derived from GSEA. The correlation between each term and gene expression level was calculated by Pearson’s test and plotted with package “ggplot2” [96]. Briefly, all tumor samples were centered into 40 values by their expression level of MS4A15. Each dot represents the average MS4A15 expression level of 40 tumor samples. The most significant correlation between each GO terms and MS4A15 expression was identified and plotted with R package “ggplot2” [96].
Lung Adenocarcinoma (LUAD) and solid tumor transcriptome data were downloaded via the TCGA website. R (version: 3.5.3) was used for these analyses. The enrichment scores of the terms (GO or KEGG) were evaluated using single-sample gene set enrichment analysis (ssGSEA) (R package “GSVA” [100]).
Supplementary information
{kind=link}
Supplementary Fig. 1. MS4A15 specifically regulates ferroptosis.
{kind=link}
Supplementary Fig. 2. MS4A15 informatics defines intracellular Ca2+ role.
{kind=link}
Supplementary Fig. 3. MS4A15 upregulation regulates calcium homeostasis.
{kind=link}
Supplementary Fig. 4. Lipid metabolites in Ms4a15 OE cells.
{kind=link}
Supplementary Fig. 5. Metabolomics analysis and cell viability.
{kind=link}
Supplementary Fig. 6. Ca2+ genes in ferroptosis and cell viability.
Supplementary Table 1. Raw data of lipidomics analysis.
Supplementary Table 2. Based on MS² fragmentation pattern identified phospholipids.
Supplementary Table 3. Raw data for metabolomics analysis.
Supplementary Table 4. CCLE expression data and full heatmap.
Supplementary Table 5. Oligonucleotide sequences used in this study.
Acknowledgements
The results shown here are in whole or part based upon data generated by the TCGA Research Network: https://www.cancer.gov/tcga. Calu-1 cells, (1S, 3R)-RSL3 (RSL3) and imidazole ketone erastin (IKE) were kindly provided by Brent Stockwell. We also thank Brent Stockwell for critical reading of the manuscript. We thank Michelle Vincendeau and Stefan Schwarzmayr for supporting the RNAseq experiments. Lenti dCAS-VP64_Blast was a gift from Feng Zhang. Lenti MS2-P65-HSF1_Hygro was a gift from Feng Zhang. Lenti sgRNA(MS2)_zeo backbone was a gift from Feng Zhang. pLV hU6-sgRNA hUbC-dCas9-KRAB-T2a-Puro was a gift from Charles Gersbach. pHCMV-EcoEnv was a gift from Miguel Sena-Esteves. pRSV-Rev was a gift from Didier Trono. pMDLg/pRRE was a gift from Didier Trono. We thank Daniel Krappmann, Martin Göttlicher, and Kamyar Hadian for gifting cell lines and materials.
Funding
This work was supported by Helmholtz Zentrum Munich GmbH (JAS); this research was also supported by the Chinese Scholarship Council (SX, 201609505007) and the Innovation Platform for Academicians of Hainan Province. Open Access funding enabled and organized by Projekt DEAL.
Author contributions
Study initiation, SX, SP, and JAS; conceptualization, JAS; methodology, SX, CM, SP, JM-P, XB, RF, XJ, and JAS; Investigation, SX, CM, SP, JM-P, XB, RF, XJ, SMH, PS-K, and JAS; Data Analysis, SX, CM, SP, VANK, JM-P, XB, and JAS; writing—original draft, CM, JAS; writing— review and editing, SX, CM, SP, VANK, and JAS; funding acquisition, SX and JAS; resources: JAS; supervision, JAS.
Data availability
All data for this study are included. Transcriptomics data generated in this study are available via GEO: GSE160574. Scripts and additional data related to this work will be available upon request to the lead contact.
Competing interests
JAS holds patents related to ferroptosis. The other authors declare no competing interests.
Footnotes
Edited by K. Newton
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Shan Xin, Constanze Müller and Susanne Pfeiffer.
Supplementary information
The online version contains supplementary material available at 10.1038/s41418-021-00883-z.
References
- 1.Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent-and AIF-mediated cell death. Cell Metab. 2008;8:237–48. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 5.Li Y, Maher P, Schubert D. A role for 12-lipoxygenase in nerve cell death caused by glutathione depletion. Neuron. 1997;19:453–63. doi: 10.1016/s0896-6273(00)80953-8. [DOI] [PubMed] [Google Scholar]
- 6.Lewerenz J, Hewett SJ, Huang Y, Lambros M, Gout PW, Kalivas PW, et al. The cystine/glutamate antiporter system xc− in health and disease: from molecular mechanisms to novel therapeutic opportunities. Antioxid Redox Signal. 2013;18:522–55. doi: 10.1089/ars.2011.4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Emerit J, Edeas M, Bricaire F. Neurodegenerative diseases and oxidative stress. Biomedicine Pharmacother. 2004;58:39–46. doi: 10.1016/j.biopha.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 8.Orrenius S, Gogvadze V, Zhivotovsky B. Calcium and mitochondria in the regulation of cell death. Biochemical biophysical Res Commun. 2015;460:72–81. doi: 10.1016/j.bbrc.2015.01.137. [DOI] [PubMed] [Google Scholar]
- 9.Roderick HL, Cook SJ. Ca2+ signalling checkpoints in cancer: remodelling Ca2+ for cancer cell proliferation and survival. Nat Rev Cancer. 2008;8:361–75. doi: 10.1038/nrc2374. [DOI] [PubMed] [Google Scholar]
- 10.Zhai M, Yang D, Yi W, Sun W. Involvement of calcium channels in the regulation of adipogenesis. Adipocyte. 2020;9:132–41. doi: 10.1080/21623945.2020.1738792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Arruda AP, Hotamisligil GS. Calcium Homeostasis and Organelle Function in the Pathogenesis of Obesity and Diabetes. Cell Metab. 2015;22:381–97. doi: 10.1016/j.cmet.2015.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones BH, Kim JH, Zemel MB, Woychik RP, Michaud EJ, Wilkison WO, et al. Upregulation of adipocyte metabolism by agouti protein: possible paracrine actions in yellow mouse obesity. Am J Physiol-Endocrinol Metab. 1996;270:E192–E196. doi: 10.1152/ajpendo.1996.270.1.E192. [DOI] [PubMed] [Google Scholar]
- 13.Xue B, Moustaid N, Wilkison WO, Zemel MB. The agouti gene product inhibits lipolysis in human adipocytes via a Ca2+-dependent mechanism. Faseb j. 1998;12:1391–6. [PubMed] [Google Scholar]
- 14.Ye R, Ni M, Wang M, Luo S, Zhu G, Chow RH, et al. Inositol 1,4,5-trisphosphate receptor 1 mutation perturbs glucose homeostasis and enhances susceptibility to diet-induced diabetes. J Endocrinol. 2011;210:209–17. doi: 10.1530/JOE-11-0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Subramanian M, Metya SK, Sadaf S, Kumar S, Schwudke D, Hasan GJDM, et al. Altered lipid homeostasis in Drosophila InsP3 receptor mutants leads to obesity and hyperphagia. Dis Model Mech. 2013;6:734–44. doi: 10.1242/dmm.010017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ntambi JM, Takova TJD. Role of Ca2+ in the early stages of murine adipocyte differentiation as evidenced by calcium mobilizing agents. Differentiation. 1996;60:151–8. doi: 10.1046/j.1432-0436.1996.6030151.x. [DOI] [PubMed] [Google Scholar]
- 17.Shi H, Halvorsen Y-D, Ellis PN, Wilkison WO, Zemel MBJPG. Role of intracellular calcium in human adipocyte differentiation. Physiol Genomics. 2000;3:75–82. doi: 10.1152/physiolgenomics.2000.3.2.75. [DOI] [PubMed] [Google Scholar]
- 18.Bi J, Wang W, Liu Z, Huang X, Jiang Q, Liu G, et al. Seipin promotes adipose tissue fat storage through the ER Ca2+-ATPase SERCA. Cell Metab. 2014;19:861–71. doi: 10.1016/j.cmet.2014.03.028. [DOI] [PubMed] [Google Scholar]
- 19.Lewerenz J, Ates G, Methner A, Conrad M, Maher P. Oxytosis/Ferroptosis-(Re-) Emerging Roles for Oxidative Stress-Dependent Non-apoptotic Cell Death in Diseases of the Central Nervous System. Front Neurosci. 2018;12:214. doi: 10.3389/fnins.2018.00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Maher P, van Leyen K, Dey PN, Honrath B, Dolga A, Methner A. The role of Ca(2+) in cell death caused by oxidative glutamate toxicity and ferroptosis. Cell Calcium. 2018;70:47–55. doi: 10.1016/j.ceca.2017.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lane J, Mansel RE, Jiang WG. Expression of human delta-6-desaturase is associated with aggressiveness of human breast cancer. Int J Mol Med. 2003;12:253–7. [PubMed] [Google Scholar]
- 22.Azrad M, Turgeon C, Demark-Wahnefried W. Current evidence linking polyunsaturated Fatty acids with cancer risk and progression. Front Oncol. 2013;3:224–224. doi: 10.3389/fonc.2013.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yan G, Li L, Zhu B, Li Y. Lipidome in colorectal cancer. Oncotarget. 2016;7:33429–39. doi: 10.18632/oncotarget.7960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hilvo M, Denkert C, Lehtinen L, Müller B, Brockmöller S, Seppänen-Laakso T, et al. Novel theranostic opportunities offered by characterization of altered membrane lipid metabolism in breast cancer progression. Cancer Res. 2011;71:3236–45. doi: 10.1158/0008-5472.CAN-10-3894. [DOI] [PubMed] [Google Scholar]
- 25.Peck B, Schulze A. Lipid desaturation–the next step in targeting lipogenesis in cancer? FEBS J. 2016;283:2767–78. doi: 10.1111/febs.13681. [DOI] [PubMed] [Google Scholar]
- 26.Rysman E, Brusselmans K, Scheys K, Timmermans L, Derua R, Munck S, et al. De novo lipogenesis protects cancer cells from free radicals and chemotherapeutics by promoting membrane lipid saturation. Cancer Res. 2010;70:8117–26. doi: 10.1158/0008-5472.CAN-09-3871. [DOI] [PubMed] [Google Scholar]
- 27.Ishibashi K, Suzuki M, Sasaki S, Imai MJG. Identification of a new multigene four-transmembrane family (MS4A) related to CD20, HTm4 and β subunit of the high-affinity IgE receptor. Gene. 2001;264:87–93. doi: 10.1016/s0378-1119(00)00598-9. [DOI] [PubMed] [Google Scholar]
- 28.Cruse G, Kaur D, Leyland M, Bradding PJTFJ. A novel FcεRIβ‐chain truncation regulates human mast cell proliferation and survival. FASEB J. 2010;24:4047–57. doi: 10.1096/fj.10-158378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deans JP, Li H, Polyak MJ. CD20‐mediated apoptosis: signalling through lipid rafts. Immunology. 2002;107:176–82. doi: 10.1046/j.1365-2567.2002.01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koslowski M, Sahin U, Dhaene K, Huber C, Tureci O. MS4A12 is a colon-selective store-operated calcium channel promoting malignant cell processes. Cancer Res. 2008;68:3458–66. doi: 10.1158/0008-5472.CAN-07-5768. [DOI] [PubMed] [Google Scholar]
- 31.Shan D, Ledbetter JA, Press OW. Signaling events involved in anti-CD20-induced apoptosis of malignant human B cells. Cancer Immunol, Immunother. 2000;48:673–83. doi: 10.1007/s002620050016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kraft VA, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F, et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Central Sci. 2019;6:41–53. doi: 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Seiler A, Schneider M, Förster H, Roth S, Wirth EK, Culmsee C, et al. Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab. 2008;8:237–48. doi: 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 34.Bannai S, Tsukeda H, Okumura H. Effect of antioxidants on cultured human diploid fibroblasts exposed to cystine-free medium. Biochemical biophysical Res Commun. 1977;74:1582–8. doi: 10.1016/0006-291x(77)90623-4. [DOI] [PubMed] [Google Scholar]
- 35.Cao JY, Dixon SJ. Mechanisms of ferroptosis. Cell Mol Life Sci. 2016;73:2195–209. doi: 10.1007/s00018-016-2194-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, et al. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 37.Ghandi M, Huang FW, Jané-Valbuena J, Kryukov GV, Lo CC, McDonald ER, et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature. 2019;569:503–8. doi: 10.1038/s41586-019-1186-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.http://ds.biogps.org/?dataset=GSE1133&gene=219995 G-MA. June 21, 2021.
- 39.Arhatte M, Gunaratne GS, El Boustany C, Kuo IY, Moro C, Duprat F, et al. TMEM33 regulates intracellular calcium homeostasis in renal tubular epithelial cells. Nat Commun. 2019;10:2024. doi: 10.1038/s41467-019-10045-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Savage AM, Kurusamy S, Chen Y, Jiang Z, Chhabria K, MacDonald RB, et al. tmem33 is essential for VEGF-mediated endothelial calcium oscillations and angiogenesis. Nat Commun. 2019;10:732. doi: 10.1038/s41467-019-08590-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Collisson EA, Campbell JD, Brooks AN, Berger AH, Lee W, Chmielecki J, et al. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–50. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Poursaitidis I, Wang X, Crighton T, Labuschagne C, Mason D, Cramer SL, et al. Oncogene-selective sensitivity to synchronous cell death following modulation of the amino acid nutrient cystine. Cell Rep. 2017;18:2547–56. doi: 10.1016/j.celrep.2017.02.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, et al. RAS–RAF–MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447:865–9. doi: 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–29. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- 45.Witting M, Schmitt-Kopplin P. The Caenorhabditis elegans lipidome: A primer for lipid analysis in Caenorhabditis elegans. Arch Biochem Biophys. 2016;589:27–37. doi: 10.1016/j.abb.2015.06.003. [DOI] [PubMed] [Google Scholar]
- 46.Magtanong L, Ko P-J, To M, Cao JY, Forcina GC, Tarangelo A, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol. 2019;26:420–32. doi: 10.1016/j.chembiol.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13:81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–8. doi: 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rees MG, Seashore-Ludlow B, Cheah JH, Adams DJ, Price EV, Gill S, et al. Correlating chemical sensitivity and basal gene expression reveals mechanism of action. Nat Chem Biol. 2016;12:109–16. doi: 10.1038/nchembio.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vriens K, Christen S, Parik S, Broekaert D, Yoshinaga K, Talebi A, et al. Evidence for an alternative fatty acid desaturation pathway increasing cancer plasticity. Nature. 2019;566:403–6. doi: 10.1038/s41586-019-0904-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sahin AO, Buitenhuis M. Molecular mechanisms underlying adhesion and migration of hematopoietic stem cells. Cell Adh Migr. 2012;6:39–48. doi: 10.4161/cam.18975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, et al. Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature. 2017;547:453. doi: 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, et al. Computational correction of copy number effect improves specificity of CRISPR–Cas9 essentiality screens in cancer cells. Nat Genet. 2017;49:1779–84. doi: 10.1038/ng.3984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rougé L, Chiang N, Steffek M, Kugel C, et al. Structure of CD20 in complex with the therapeutic monoclonal antibody rituximab. Science. 2020;367:eaaz9356. doi: 10.1126/science.aaz9356. [DOI] [PubMed] [Google Scholar]
- 55.Eon Kuek L, Leffler M, Mackay GA, Hulett MD. The MS4A family: counting past 1, 2 and 3. Immunol Cell Biol. 2016;94:11–23. doi: 10.1038/icb.2015.48. [DOI] [PubMed] [Google Scholar]
- 56.Polyak MJ, Li H, Shariat N, Deans JP. CD20 homo-oligomers physically associate with the B cell antigen receptor. Dissociation upon receptor engagement and recruitment of phosphoproteins and calmodulin-binding proteins. J Biol Chem. 2008;283:18545–52. doi: 10.1074/jbc.M800784200. [DOI] [PubMed] [Google Scholar]
- 57.Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Developmental Cell. 2020;54:447–54. doi: 10.1016/j.devcel.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gorgas K, Teigler A, Komljenovic D, Just WW. The ether lipid-deficient mouse: Tracking down plasmalogen functions. Biochimica et Biophysica Acta (BBA) - Mol Cell Res. 2006;1763:1511–26. doi: 10.1016/j.bbamcr.2006.08.038. [DOI] [PubMed] [Google Scholar]
- 59.Brites P, Waterham HR, Wanders RJ. Functions and biosynthesis of plasmalogens in health and disease. Biochim et Biophys Acta. 2004;1636:219–31. doi: 10.1016/j.bbalip.2003.12.010. [DOI] [PubMed] [Google Scholar]
- 60.Reiss D, Beyer K, Engelmann B. Delayed oxidative degradation of polyunsaturated diacyl phospholipids in the presence of plasmalogen phospholipids in vitro. Biochem J. 1997;323:807–14. doi: 10.1042/bj3230807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Broniec A, Klosinski R, Pawlak A, Wrona-Krol M, Thompson D, Sarna T. Interactions of plasmalogens and their diacyl analogs with singlet oxygen in selected model systems. Free Radic Biol Med. 2011;50:892–8. doi: 10.1016/j.freeradbiomed.2011.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Messias MCF, Mecatti GC, Priolli DG, de Oliveira Carvalho P. Plasmalogen lipids: functional mechanism and their involvement in gastrointestinal cancer. Lipids Health Dis. 2018;17:41. doi: 10.1186/s12944-018-0685-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang YS, McAdoo KR, Mitchell J, Horrobin DF. Effects of calcium deprivation on n-6 fatty acid metabolism in growing rats. Biochemical Med Metab Biol. 1988;40:61–67. doi: 10.1016/0885-4505(88)90105-3. [DOI] [PubMed] [Google Scholar]
- 64.Bai Y, Meng L, Han L, Jia Y, Zhao Y, Gao H, et al. Lipid storage and lipophagy regulates ferroptosis. Biochemical biophysical Res Commun. 2019;508:997–1003. doi: 10.1016/j.bbrc.2018.12.039. [DOI] [PubMed] [Google Scholar]
- 65.Bailey AP, Koster G, Guillermier C, Hirst EM, MacRae JI, Lechene CP, et al. Antioxidant role for lipid droplets in a stem cell niche of drosophila. Cell. 2015;163:340–53. doi: 10.1016/j.cell.2015.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20:137–55. doi: 10.1038/s41580-018-0085-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther. 2020;5:108. doi: 10.1038/s41392-020-00216-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bittremieux M, Parys JB, Pinton P, Bultynck GER. functions of oncogenes and tumor suppressors: modulators of intracellular Ca(2+) signaling. Biochim et biophys Acta. 2016;1863:1364–78. doi: 10.1016/j.bbamcr.2016.01.002. [DOI] [PubMed] [Google Scholar]
- 69.Cui C, Merritt R, Fu L, Pan Z. Targeting calcium signaling in cancer therapy. Acta Pharmaceutica Sin B. 2017;7:3–17. doi: 10.1016/j.apsb.2016.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kania E, Roest G, Vervliet T, Parys JB, Bultynck G. IP3 Receptor-mediated calcium signaling and its role in autophagy in cancer. Front Oncol. 2017;7:140. doi: 10.3389/fonc.2017.00140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pierro C, Cook SJ, Foets TC, Bootman MD, Roderick HL. Oncogenic K-Ras suppresses IP3-dependent Ca2+ release through remodelling of the isoform composition of IP3Rs and ER luminal Ca2+ levels in colorectal cancer cell lines. J Cell Sci. 2014;127:1607–19. doi: 10.1242/jcs.141408. [DOI] [PubMed] [Google Scholar]
- 72.Hakii H, Fujiki H, Suganuma M, Nakayasu M, Tahira T, Sugimura T, et al. Thapsigargin, a histamine secretagogue, is a non-12-O-tetradecanolphorbol-13-acetate (TPA) type tumor promoter in two-stage mouse skin carcinogenesis. J Cancer Res Clin Oncol. 1986;111:177–81. doi: 10.1007/BF00389230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Liu LH, Boivin GP, Prasad V, Periasamy M, Shull GE. Squamous cell tumors in mice heterozygous for a null allele of Atp2a2, encoding the sarco(endo)plasmic reticulum Ca2+-ATPase isoform 2 Ca2+ pump. J Biol Chem. 2001;276:26737–40. doi: 10.1074/jbc.C100275200. [DOI] [PubMed] [Google Scholar]
- 74.Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8. doi: 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 75.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575:688–92. doi: 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Konermann S, Brigham MD, Trevino AE, Joung J, Abudayyeh OO, Barcena C, et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature. 2015;517:583–8. doi: 10.1038/nature14136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Haack TB, Kopajtich R, Freisinger P, Wieland T, Rorbach J, Nicholls TJ, et al. ELAC2 mutations cause a mitochondrial RNA processing defect associated with hypertrophic cardiomyopathy. Am J Hum Genet. 2013;93:211–23. doi: 10.1016/j.ajhg.2013.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wiśniewski JR, Zougman A, Nagaraj N, Mann M. Universal sample preparation method for proteome analysis. Nat Methods. 2009;6:359–62. doi: 10.1038/nmeth.1322. [DOI] [PubMed] [Google Scholar]
- 80.Lepper MF, Ohmayer U, von Toerne C, Maison N, Ziegler AG, Hauck SM. Proteomic Landscape of Patient-Derived CD4+ T Cells in Recent-Onset Type 1 Diabetes. J Proteome Res. 2018;17:618–34. doi: 10.1021/acs.jproteome.7b00712. [DOI] [PubMed] [Google Scholar]
- 81.Bruderer R, Bernhardt OM, Gandhi T, Miladinović SM, Cheng L-Y, Messner S, et al. Extending the limits of quantitative proteome profiling with data-independent acquisition and application to acetaminophen-treated three-dimensional liver microtissues. Mol Cell Proteom. 2015;14:1400–10. doi: 10.1074/mcp.M114.044305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Muller C, Dietz I, Tziotis D, Moritz F, Rupp J, Schmitt-Kopplin P. Molecular cartography in acute Chlamydia pneumoniae infections—a non-targeted metabolomics approach. Anal Bioanal Chem. 2013;405:5119–31. doi: 10.1007/s00216-013-6732-5. [DOI] [PubMed] [Google Scholar]
- 83.Sud M, Fahy E, Cotter D, Brown A, Dennis EA, Glass CK, et al. LMSD: LIPID MAPS structure database. Nucleic Acids Res. 2007;35:D527–532. doi: 10.1093/nar/gkl838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burke MC, Mirokhin YA, Tchekhovskoi DV, Markey SP, Heidbrink Thompson J, Larkin C, et al. The hybrid search: a mass spectral library search method for discovery of modifications in proteomics. J Proteome Res. 2017;16:1924–35. doi: 10.1021/acs.jproteome.6b00988. [DOI] [PubMed] [Google Scholar]
- 85.Ruttkies C, Schymanski EL, Wolf S, Hollender J, Neumann S. MetFrag relaunched: incorporating strategies beyond in silico fragmentation. J Cheminformatics. 2016;8:3. doi: 10.1186/s13321-016-0115-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kerwin JL, Tuininga AR, Ericsson LH. Identification of molecular species of glycerophospholipids and sphingomyelin using electrospray mass spectrometry. J Lipid Res. 1994;35:1102–14. [PubMed] [Google Scholar]
- 87.Murphy EJ, Stephens R, Jurkowitz-Alexander M, Horrocks LA. Acidic hydrolysis of plasmalogens followed by high-performance liquid chromatography. Lipids. 1993;28:565–8. doi: 10.1007/BF02536090. [DOI] [PubMed] [Google Scholar]
- 88.Boncompain G, Muller C, Meas-Yedid V, Schmitt-Kopplin P, Lazarow PB, Subtil A. The intracellular bacteria Chlamydia hijack peroxisomes and utilize their enzymatic capacity to produce bacteria-specific phospholipids. PLoS One. 2014;9:e86196. doi: 10.1371/journal.pone.0086196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hauck SM, Dietter J, Kramer RL, Hofmaier F, Zipplies JK, Amann B, et al. Deciphering membrane-associated molecular processes in target tissue of autoimmune uveitis by label-free quantitative mass spectrometry. Mol Cell Proteomics. 2010;9:2292–305. doi: 10.1074/mcp.M110.001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. doi: 10.1093/bioinformatics/bth078. [DOI] [PubMed] [Google Scholar]
- 94.Saldanha AJ. Java Treeview—extensible visualization of microarray data. Bioinformatics. 2004;20:3246–8. doi: 10.1093/bioinformatics/bth349. [DOI] [PubMed] [Google Scholar]
- 95.Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–61. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wickham H, ggplot2: elegant graphics for data analysis. (Springer, 2016).
- 97.Kassambara A, Mundt F. Package ‘factoextra’. Extract and visualize the results of multivariate data analyses 2017;76. http://www.sthda.com/english/rpkgs/factoextra.
- 98.Dray S, Dufour A-B. The ade4 package: implementing the duality diagram for ecologists. J Stat Softw. 2007;22:1–20. [Google Scholar]
- 99.Kolde R, Kolde MR. Package ‘pheatmap’. R Package. 2015;1. https://cran.r-project.org/package=pheatmap.
- 100.Hänzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinforma. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Fig. 1. MS4A15 specifically regulates ferroptosis.
Supplementary Fig. 2. MS4A15 informatics defines intracellular Ca2+ role.
Supplementary Fig. 3. MS4A15 upregulation regulates calcium homeostasis.
Supplementary Fig. 4. Lipid metabolites in Ms4a15 OE cells.
Supplementary Fig. 5. Metabolomics analysis and cell viability.
Supplementary Fig. 6. Ca2+ genes in ferroptosis and cell viability.
Supplementary Table 1. Raw data of lipidomics analysis.
Supplementary Table 2. Based on MS² fragmentation pattern identified phospholipids.
Supplementary Table 3. Raw data for metabolomics analysis.
Supplementary Table 4. CCLE expression data and full heatmap.
Supplementary Table 5. Oligonucleotide sequences used in this study.
Data Availability Statement
All data for this study are included. Transcriptomics data generated in this study are available via GEO: GSE160574. Scripts and additional data related to this work will be available upon request to the lead contact.