

Abstract
Neutrophils are critical for inflammation and innate immunity, and their adhesion to vascular endothelium is a crucial step in neutrophil recruitment. Mitofusin-2 (MFN2) is required for neutrophil adhesion, but molecular details are unclear. Here, we demonstrated that β2-integrin-mediated slow-rolling and arrest, but not PSGL-1-mediated cell rolling, are defective in MFN2-deficient neutrophil-like HL60 cells. This adhesion defect is associated with reduced expression of fMLP (N-formylmethionyl-leucyl-phenylalanine) receptor FPR1 as well as the inhibited β2 integrin activation, as assessed by conformation-specific monoclonal antibodies. MFN2 deficiency also leads to decreased actin polymerization, which is important for β2 integrin activation. Mn2+-induced cell spreading is also inhibited after MFN2 knockdown. MFN2 deficiency limited the maturation of β2 integrin activation during the neutrophil-directed differentiation of HL60 cells, which is indicated by CD35 and CD87 markers. MFN2 knockdown in β2-integrin activation-matured cells (CD87high population) also inhibits integrin activation, indicating that MFN2 directly affects β2 integrin activation. Our study illustrates the function of MFN2 in leukocyte adhesion and may provide new insights into the development and treatment of MFN2 deficiency-related diseases.
Keywords: actin polymerization, β2 integrin activation, CD87, HL60, Mitoifusin-2, Neutrophil adhesion, neutrophil maturation
1 ∣. INTRODUCTION
The most abundant leukocytes in humans are neutrophils, which serve as the first responders to acute infection and cell damage and are critical for inflammatory and immunological responses.1 An intrinsic neutrophil defect leads to pathologies, such as leukocyte adhesion deficiency syndromes.2,3 In response to inflammation or infection, circulating neutrophils undergo a recruitment cascade1,4-7: The interactions of neutrophil P-selectin glycoprotein ligand-1 (PSGL-1) and corresponding endothelial selectins recruit neutrophils from the circulation, tether them to the endothelial surface, and cause them to roll on the endothelial surface.8-13 Long tethers form slings, which help to stabilize rolling.12,14,15 During rolling, the interactions of PSGL-1 and selectins induce the extension (E+) but not high-affinity (H+) of β2 integrins, which allow neutrophils to briefly engage endothelial intercellular adhesion molecule 1 (ICAM-1) and cause the rolling to slow (slow-rolling).16-21 In turn, this slow-rolling facilitates neutrophil interactions with endothelial chemokines22,23 and induces both E+ and H+ of β2 integrins, leading to a firm arrest.18-20,24-29 Next, spherical neutrophils spread30,31 and intravascularly crawl.31-36 Finally, neutrophils will undergo transendothelial migration through the vascular wall31,37,38 and ultimately migrate38-45 to specific tissue sites.
In this cascade, neutrophil adhesion under flow, including rolling, slow-rolling, and arrest phases, is the incipient step in transendothelial migration. β2 integrins are critical for neutrophil slow-rolling and arrest. Resting β2 integrins assume a bent conformation with a low affinity (E−H−) for the ligand. The “switchblade” model of integrin activation46 holds that integrin E+ is followed by a rearrangement in the ligand-binding site leading to H+. E+ reveals a neoepitope in the knee of the β2 subunit recognized by the KIM127 antibody.47,48 The H+ conformation of the β2 I-like domain reveals a neoepitope detected by mAb 24 (mAb24).49-51 As mentioned before, the interaction between selectins and PSGL-1 gives rise to the E+ of β2 integrins during neutrophil rolling. E+H− β2 integrins interact with endothelial ICAMs with weak force and turn neutrophils into the slow-rolling phase. The chemokine-induced inside-out signaling triggers both H+ and E+ of β2 integrins that interact with endothelial ICAMs with a strong force and cause cell arrest.18-20,24-29
Mitofusin-2 (MFN2) is a conserved, dynamin-like GTPase located in the mitochondrial outer membrane, which tethers mitochondria to each other. Mitochondria are dynamic organelles with the ability to fuse and divide (fission), forming constantly changing tubular networks in most eukaryotic cells.52 MFN2 mediates mitochondrial fusion and plays a role in the maintenance of the mitochondrial network.53,54 MFN2 is also important for the tethering of endoplasmic reticulum to mitochondria as well as calcium uptake by mitochondria.55 Previous studies found that MFN2 is important for neutrophil migration43,56 and the development of the hematopoietic system.57 However, the specific role(s) of MFN2 in neutrophil trafficking or other functions are largely unknown. Increased understanding of MFN2 actions during inflammation could reveal novel therapeutic targets in neutrophil-mediated disease.
2 ∣. MATERIALS AND METHODS
2.1 ∣. Reagents
Recombinant human P-selectin-Fc, ICAM-1-Fc, recombinant mouse ICAM-1-Fc, and Alexa Fluor 647 (AF647)-conjugated mouse anti-human CD18 (pan-β2 integrins) antibody (Ab) were purchased from R&D Systems. Zombie Yellow Fixable Viability Kit, Alexa Fluor 488 (AF488)-conjugated, Alexa Fluor 700 (AF700)-conjugated, and allophycocyanin (APC)-conjugated conformation-specific Ab mAb24 reporting the H+ of β2 integrins, AF647-conjugated conformation-specific Ab CBRM1/5 reporting the H+ of αM integrins, APC-conjugated mouse anti-human PSGL-1 (CD162) Ab, PerCP-conjugated mouse anti-human CD11a Ab, Pacific Blue-conjugated mouse anti-human CD11b Ab, AF700-conjugated mouse anti-human CD11c Ab, AF700-conjugated, APC-conjugated, unconjugated CD18 (pan-β2 integrins, clone TS1/18) Ab, APC-conjugated mouse anti-human FPR1 Ab, PE-conjugated mouse anti-human CD87 Ab, Brilliant Violet 650 (BV650)-conjugated rat anti-mouse Ly6G Ab, rat anti-mouse CD11a Ab (M17/4), rat anti-mouse CD11b Ab (M1/70), APC-conjugated mouse anti-human IgG Fc Ab, all isotype control Abs, and intracellular staining perm wash buffer were purchased from Biolegend. Fluorescein isothiocyanate (FITC)-conjugated mouse anti-human FPR2 Ab was purchased from Santa Cruz Biotechnology. AF647-conjugated mouse anti-human CD35 Ab, Dylight 800-conjugated goat anti-rabbit IgG secondary Ab, Dylight 680-conjugated goat anti-mouse IgG secondary Ab, Casein blocking buffer, Alexa Fluor 568 (AF568)-conjugated phalloidin, CellTracker Orange CMRA, poly-L-lysine, glutaraldehyde, cacodylate buffer, and lipofectamine 3000 were purchased from Thermo Fisher Scientific. AF647-conjugated Histone H3 recombinant rabbit mAb and AF647-conjugated goat anti-rabbit IgG secondary Ab were purchased from Invitrogen. The KIM127 mAb reporting the ectodomain extension was a gift from Dr. Klaus Ley at the La Jolla Institute for Immunology and was purified at the Lymphocyte Culture Center at the University of Virginia from hybridoma supernatant (ATCC). KIM127 was directly labeled by DyLight 550 or Dylight 650 using DyLight antibody labeling kits from Thermo Fisher Scientific according to the manufacturer’s instructions. Rabbit anti-MFN2 mAb was purchased from Cell Signaling. Anti-β-tubulin Ab (E7) was obtained from the Developmental Studies Hybridoma Bank. Roswell Park Memorial Institute medium 1640 (RPMI-1640) with or without phenol red, PBS without Ca2+ and Mg2+, goat serum, and puromycin were purchased from Gibco. Human serum albumin (HSA) and fetal bovine serum (FBS) were purchased from Gemini Bio Products. UltraComp eBeads for compensation controls were purchased from eBioscience. fMLP, PMA, polybrene, paraformaldehyde (PFA), and DMSO were purchased from Sigma–Aldrich. The e-Myco plus Mycoplasma PCR Detection Kit was purchased from Bulldog Bio. Poly/Bed 812 embedding kit was purchased from Polysciences. “Ready to use” lead citrate was purchased from Electron Microscopy Sciences.
2.2 ∣. Cell culture
The HL60 line was a generous gift from Dr. Orion D. Weiner (University of California San Francisco). HL60 cells were maintained in culture medium (RPMI-1640, 10% FBS, 100 mL−1 penicillin, 100 μg/mL streptomycin, and 250 ng/mL amphotericin B) at 37°C and 5% CO2. In most experiments, HL60 cells were differentiated with 1.3% DMSO for 7 days before assays. In the time-lapse assay of integrin activation maturation, HL60 cells were differentiated for 0 to 7 days before the assay. Cells were checked monthly for mycoplasma using the e-Myco plus Mycoplasma PCR Detection Kit.
2.3 ∣. Neutrophil isolation
Heparinized whole blood was obtained from healthy human donors after informed consent, as approved by the Institutional Review Board of UConn Health in accordance with the Declaration of Helsinki. Informed consent was obtained from all donors. Neutrophils were isolated by using Polymorphprep (a mixture of sodium metrizoate and Dextran 500) density gradient. Briefly, human blood was applied onto Polymorphprep, centrifuged at 500 × g for 35 min at 20-25°C, resulting in neutrophils concentrated in a layer between PBMCs and erythrocytes. After washing with PBS without Ca2+ and Mg2+ twice, the neutrophils were re-suspended in RPMI-1640 without phenol red plus 2% HSA and were used within 4 h.
2.4 ∣. Mice
All mice used in this study were obtained from the Jackson Laboratory (Bar Harbor, Maine, USA). Mice were fed a standard rodent chow diet and were housed in microisolator cages in a pathogen-free facility. Mice were euthanized by CO2 inhalation. All experiments followed guidelines of the UConn Health Animal Care and Use Committee, and approval for the use of rodents was obtained from UConn Health according to criteria outlined in the Guide for the Care and Use of Laboratory Animals from the National Institutes of Health. Conditional Mfn2 knockout mice (B6.129(Cg)-Mfn2tm3Dcc/J; 026525) were crossed to MRP8-Cre (B6.Cg-Tg(S100A8-cre, -EGFP)1Ilw/J; 021614) transgenic mice to obtain homozygous floxed Mfn2 alleles with or without MRP8-Cre (MFN2flox/floxMRP8-cre+ and MFN2flox/flox MRP8-cre−). Males at age 6–8 weeks were used for experiments.
2.5 ∣. RNA interference
To generate knockdown lines in HL60 cells, pLKO.1 lentiviral constructs with MFN2-targeting shRNA were obtained from Sigma-Aldrich (MFN2-sh1:TRCN0000082684 5′-gcaggtttactgcgaggaaat-3′, MFN2-sh2: TRCN0000082687 5′-gtcaaaggttacctatccaaa-3′), and SHC 003 was used as a non-targeting control. Lentiviral constructs together with pCMV-dR8.2 dvpr (Addgene #8455) and pCMV-VSV-G (Addgene #8454) were co-transfected into HEK293T cells with Lipofectamine 3000 to produce lentivirus. Virus supernatant was collected at both 48 and 72 h after transfection and further concentrated with Lenti-X concentrator (Clotech 631232). HL60 cells were infected in the complete medium supplemented with 4 μg/mL polybrene (Sigma TR-1003-G) and selected with 1 μg/mL puromycin (Gibco A1113803) to generate a stable line.
2.6 ∣. Western blots
Protein samples were separated using SDS-PAGE and transferred onto nitrocellulose membranes (LI-COR NC9680617). Membranes were blocked for ~30 min in PBST (PBS and 0.1% Tween 20) with 5% BSA. After blocking, membranes were incubated overnight with primary antibodies diluted 1:1,000 in PBST at 4°C, washed, and incubated with secondary antibodies diluted 1:10,000 in PBST at room temperature for 1 h. Blots were then washed, developed, and imaged. Odyssey (LI-Cor) was used to image membranes (Fig. 1A-B).
FIGURE 1. Mitofusin-2 knockdown reduces the adhesion of neutrophil-like HL60 cells.
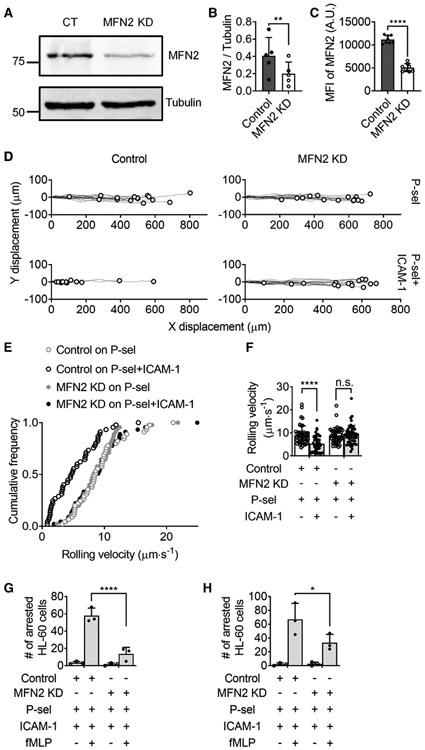
(A) Western blot showing protein expression of mitofusin-2 (MFN2) in HL60 cells transfected with control (CT) or MFN2 knockdown (KD) with shRNA. Tubulin is an internal control. (B) Mean ± SD of MFN2 versus tubulin expression from n = 5 independent western blot experiments. (C) Mean ± SD of MFN2 expression from n = 8 independent intracellular staining flow cytometry experiments. (D) Tracks of Control or MFN2 KD HL60 cells rolling on a substrate of P-selectin or P-selectin/ICAM-1 under a wall shear stress of 6 dyn/cm2. The recording time of rolling was 60 s. The rolling paths (line) and ending positions (circle) of n = 15 cells from 3 individual experiments per group are shown. (E and F) A cumulative histogram (E) and a bar graph (F, mean ± SD, n = 45 cells from 3 individual experiments per group) showing the rolling velocity of CT or MFN2 KD HL60 cells rolling on the substrate of P-selectin with or without ICAM-1 under a wall shear stress of 6 dyn/cm2. (G and H) The number of arrested CT or MFN2 KD HL60 cells on the substrate of P-selectin/ICAM-1 with or without the stimulation of fMLP (100 nM) under a wall shear stress of 6 dyn/cm2 (G) and 2 dyn/cm2 (H). Mean ± SD, n = 3 individual records. n.s., non-significant (P > 0.05). *P < 0.05, ****P < 0.0001 by paired Student’s t-tests (B and C) or 2-way ANOVA followed by Tukey’s multiple comparisons tests (F and H). HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
2.7 ∣. Microfluidic cell adhesion assay
10 mm plastic Petri dishes or 6-well plates were coated with recombinant human P-selectin-Fc (2 μg/mL) with or without recombinant human ICAM-1-Fc (10 μg/mL) for 2 h and then blocked with casein (1%) at room temperature for 1 h. After coating, a polydimethylsiloxane chip with microfluidic imprints was sealed to the coated area by vacuum to create flow chamber channels ~50 μm high and ~500 μm across. By modulating the pressure between the inlet well and the outlet reservoir, a 6 or 2 dyn/cm2 wall shear stress was applied during experiments as indicated in figure legends. HL60 cells (5×106 cells/mL in RPMI-1640 without phenol red plus 2% HSA) were perfused in the microfluidic chamber. The bright field rolling and arrest of cells was recorded by an Axiovert 100 inverted research microscope (Zeiss) with a 10× NA 0.3 air objective (Fig. 1D-H, Supplemental Movies 1 and 2). Cell arrest was induced by adding 100 nM or 10 nM fMLP for 6 or 2 dyn/cm2, respectively.
To assess the rolling and arrest of CD87high HL60 cells (Fig. 8O-Q), cells were incubated with CD87-PE antibody (4 μg/mL) at room temperature for 10 min, washed twice with PBS, and resuspended in RPMI-1640 without phenol red plus 2% HSA prior to the perfusion into the chamber. The fluorescence imaging was performed by using an iX83 Olympus inverted microscope equipped with the SAFe Light module (Abbelight, includes four color lasers, λ = 405 nm, 488 nm, 532 nm, and 640 nm), sCMOS fusion cameras (Hamamatsu), and a 10× NA 0.3 air objective. The threshold for CD87high population was based on gating the brightest ~20% of control cells. The same threshold was applied in analyzing MFN2 KD cells.
FIGURE 8. Mitofusin-2 knockdown inhibits β2 integrin activation and adhesion of HL60 cells at the same maturation level.
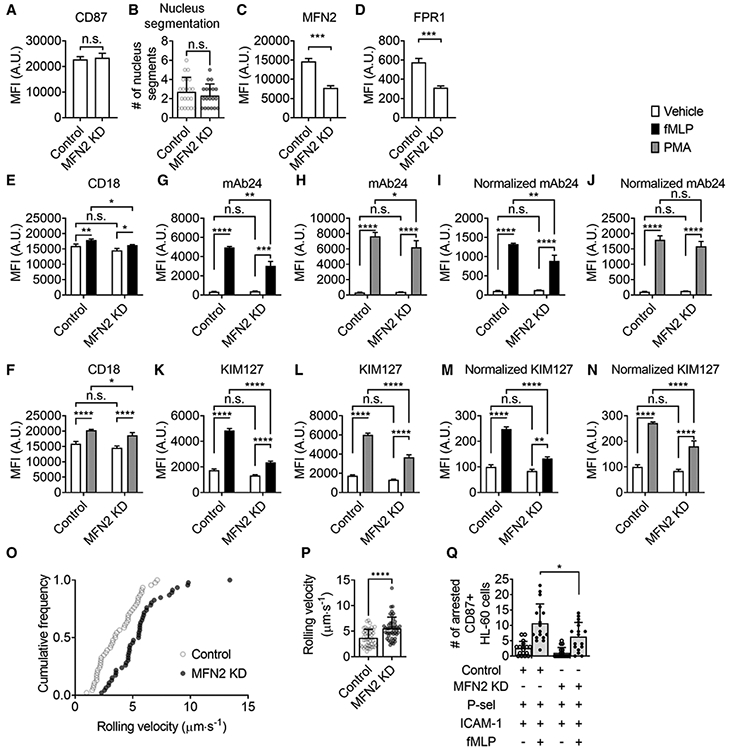
(A) Mean ± SD of surface CD87 expression on CD87high Control and MFN2-knockdown (MFN2 KD) HL60 cells from n = 6 individual experiments. (B) Mean ± SD of nucleus segment number of CD87high Control (n = 21) and MFN2 KD (n = 19) HL60 cells from 9 and 11 independent records, respectively. (C and D) Mean ± SD of MFN2 and FPR1 expression on CD87high Control and MFN2 KD HL60 cells from n = 3 individual experiments. (E and F) Mean ± SD of overall surface CD18 (β2 integrins) expression on CD87high control or MFN2 KD HL60 cells stimulated with FPR1-dependent fMLP (100 nM, at RT for 20 min, closed bars, E), receptor-independent PMA (100 nM, at RT for 20 min, gray bars, F), or vehicle control (open bars) from n = 3 individual experiments. (G-N) Mean±SD of high-affinity (H+, mAb24 staining, G-J) and extended (E+, KIM127 staining, K-N) β2 integrin expression on CD87high control or MFN2 KD HL60 cells stimulated with FPR1-dependent fMLP (100 nM, at RT for 20 min, closed bars, G, I, K, M), receptor-independent PMA (100 nM, at RT for 20 min, gray bars, H, J, L, N), or vehicle control (open bars) from n = 3 individual experiments. In (I and J) and (M and N), the MFI of mAb24 and KIM127 is normalized to overall expression of CD18 (E and F). (O and P) A cumulative histogram (O) and a bar graph (P, mean ± SD, n = 46 cells from 3 individual experiments per group) showing the rolling velocity of CD87high Control or MFN2 KD HL60 cells rolling on the substrate of P-selectin+ICAM-1 under a wall shear stress of 6 dyn/cm2. (Q) The number of arrested CD87high Control or MFN2 KD HL60 cells on the substrate of P-selectin/ICAM-1 with or without the stimulation of fMLP (100 nM) under a wall shear stress of 6 dyn/cm2. Mean ± SD, n = 17 individual records. n.s., non-significant (P > 0.05). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t-test (A-D, P) or 2-way ANOVA followed by Tukey’s multiple comparisons test (E-N, Q). HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
Cell rolling was tracked by the manual tracking plugin in FIJI-ImageJ258 to obtain rolling tracks and velocities. The numbers of arrested cells were counted in three separate recordings.
2.8 ∣. Flow cytometry
To assess MFN2 expression in MFN2 KD and control HL60 cells (Fig. 1C and 8C), 5×105 cells/mL cells were stained with PE-conjugated CD87 antibody (2 μg/mL) at room temperature for 20 min, then fixed with 1% PFA at room temperature for 10 min, washed twice with intracellular staining perm wash buffer plus 5% goat serum, and incubated with MFN2 antibody (1 μg/mL) in intracellular staining perm wash buffer plus 5% goat serum at room temperature for 30 min. After washing with PBS, cells were stained with AF647-conjugated goat anti-rabbit IgG secondary Ab (2 μg/mL) at room temperature for 30 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII (BD Biosciences) and analyzed with FlowJo software (v10.6.1).
To assess MFN2 expression in blood neutrophils from 3 MFN2flox/floxMRP8-cre− and 5 MFN2flox/floxMRP8-cre+ mice (Fig. S2A), 25 μL mouse blood was collected by retro-orbital bleeding with 2 μL heparin (1000 USP units/mL) in the collection tube and incubated with BV650-conjugated rat anti-mouse Ly6G antibody (1 μg/mL) at room temperature for 10 min, then fixed with 4% PFA at room temperature for 10 min, washed twice with intracellular staining perm wash buffer plus 5% goat serum, and incubated with MFN2 antibody (1 μg/mL) in intracellular staining perm wash buffer plus 5% goat serum at room temperature for 30 min. After washing with PBS, cells were stained with AF647-conjugated goat anti-rabbit IgG secondary antibody (2 μg/mL) at room temperature for 30 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and analyzed with FlowJo software.
To test expression of different molecules on MFN2 KD and control HL60 cells, 5 × 105 cells/mL cells were incubated with fMLP (100 nM) or PMA (100 nM) or vehicle control in the presence of the following Ab panels at room temperature for 20 min: (1) For Figures 2A,D-I, 8D, and S3B: APC-conjugated FPR1 Ab (1 μg/mL), PerCP-conjugated CD11a Ab (1 μg/mL), Pacific Blue-conjugated CD11b Ab (1 μg/mL), AF700-conjugated CD11c Ab (1 μg/mL), and PE-conjugated CD87 Ab (2 μg/mL); (2) For Figure 2B-C, 6K, 8A,E-F, and S3C-E: APC-conjugated CD18 Ab (1 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (3) For Figure 2J-M, 8G-J, and S3F-I: APC-conjugated mAb24 (0.5 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (4) For Figure 2N-Q, 8K-N, and S3J-M: Dylight 650-conjugated KIM127 (0.5 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (5) For Figure 2R-U: AF647-conjugated CBRM1/5 (0.5 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (6) For Figure 6I-J: AF647-conjugated CD35 Ab (1 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (7) For Supplemental Figure S1A: APC-conjugated PSGL-1 Ab (1 μg/ml). Cells were fixed with 1% PFA at room temperature for 10 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and analyzed with FlowJo software.
FIGURE 2. Mitofusin-2 knockdown reduces fMLP receptor expression as well as receptor-dependent and independent β2 integrin activation in neutrophil-like HL60 cells.
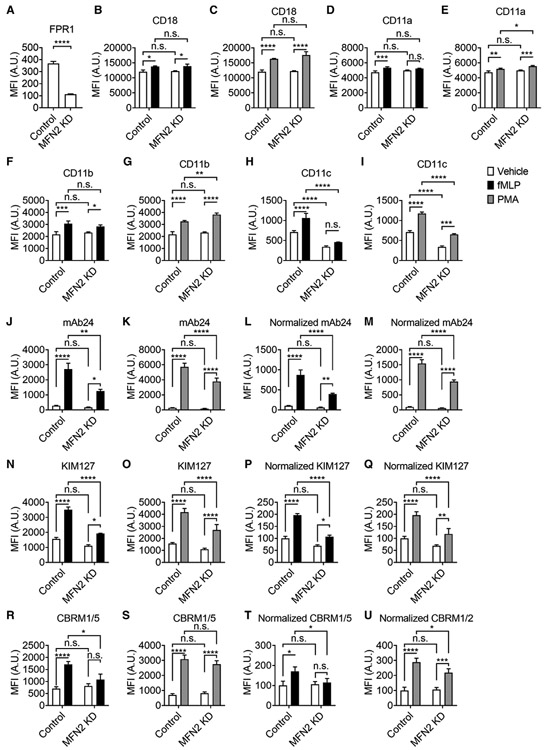
(A) Mean ± SD of surface FPR1 expression on Control or MFN2-knockdown (MFN2 KD) HL60 cells from n = 3 individual experiments. (B-I) Mean ± SD of overall surface CD18 (β2 integrins, B,C), CD11a (αL integrins, D,E), CD11b (αM integrins, F,G), CD11c (αX integrins, H,I) expression on Control or MFN2 KD HL60 cells stimulated with FPR1-dependent fMLP (100 nM, at RT for 20 min, closed bars, B, D, F, H), receptor-independent PMA (100 nM, at RT for 20 min, gray bars, C, E, G, I), or vehicle control (open bars) from n = 3 individual experiments. (J-U) Mean ± SD of high-affinity (H+, mAb24 staining, J-M) and extended (E+, KIM127 staining, N-Q) β2 integrin as well as H+ αM integrin (CBRM1/5 staining, R-U) expression on Control or MFN2 KD HL60 cells stimulated with FPR1-dependent fMLP (100 nM, at RT for 20 min, closed bars, J, L, N, P, R, T), receptor-independent PMA (100 nM, at RT for 20 min, gray bars, K, M, O, Q, S, U) or vehicle control (open bars) from n = 3 individual experiments. In L,M; P,Q; and T,U, the MFI of mAb24, KIM127, and CBRM1/5 is normalized to overall expression of CD18 (B,C), CD18 (B,C), and CD11b (F,G), respectively. n.s., non-significant (P > 0.05). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001 by Student’s t-test (A) or 2-way ANOVA followed by Tukey’s multiple comparisons test (B-U). HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
FIGURE 6. Mitofusin-2 knockdown impairs expression of β2-integrin-activation maturation markers during the DMSO-induced differentiation of HL60 cells.
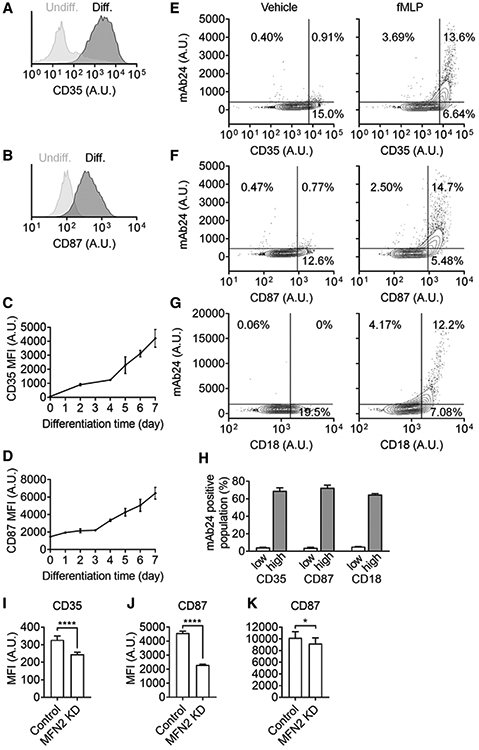
(A,B) Representative histograms showing expression of CD35 (A) and CD87 (B) on HL60 cells pre-differentiated for 5 days (dark gray) or not (light gray). (C-D) Expression of CD35 (C), and CD87 (D) on HL-60 cells after different days of DSMO-induced differentiation. Mean ± 95%CI for n = 3 individual experiments. (E-H) Contour plots showing expression of high-affinity (H+, mAb24 staining) β2 integrins on pre-differentiated HL60 cells with different expression of CD35 (E), CD87 (F), and overall CD18 (G) stimulated with fMLP (100 nM, right panels) or vehicle control (left panels). (H) The percentage of mAb24+ cells in the low-expression or high-expression population of different markers upon fMLP stimulation. (I-K) Expression of CD35 (I) and CD87 (J-K) on Control or MFN2-knockdown (MFN2 KD) HL60 cells after pre-differentiated for 5 (I-J) or 7 (K) days. Mean ± SD for n = 3 (I) or 9 (J-K) individual experiments, *P < 0.05, ****P < 0.0001 by Student’s t-test
For the time-lapse experiment showing the heterogeneous β2 integrin activation and the maturation of β2 integrin activation during neutrophil-directed differentiation of HL60 cells, purified human neutrophils (the positive control) and HL60 cells with different differentiation time (0-7 days) were incubated with fMLP (100 nM) or vehicle control at room temperature for 10 minutes in the presence of: (1) For Figure 5A-D: APC-conjugated mAb24 (1 μg/ml) and Dylight 550-conjugated KIM127 (1 μg/ml); (2) For Figure 5E: AF647-conjugated CD18 Ab (2 μg/ml) and PE-conjugated CD87 Ab (2 μg/ml); (3) For Figure 5F: APC-conjugated FPR1 Ab (1 μg/ml); (4) For Figure 6C-D: AF647-conjugated CD35 Ab (2 μg/ml), and PE-conjugated CD87 Ab (2 μg/ml). Cells were fixed with 1% PFA at room temperature for 10 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and was analyzed with FlowJo software.
FIGURE 5. The maturation of β2 integrin activation during the DMSO-induced differentiation of HL60 cells.
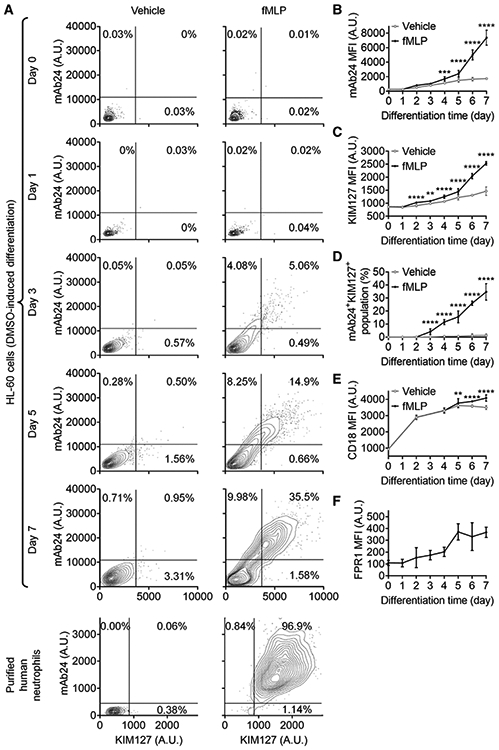
(A) Contour plots showing expression of high-affinity (H+, mAb24 staining) and extended (E+, KIM127 staining) β2 integrins on HL60 cells (upper panels) or primary human neutrophils (lower panel) stimulated with fMLP (1 mM, right panels) or vehicle control (left panels). HL60 cells were pre-differentiated with 1.3% DMSO for indicated days. (B,C) Expression of mAb24 (B) and KIM127 (C) on HL-60 cells after different days of DSMO-induced differentiation with or without fMLP stimulation. (D) The percentage of mAb24+KIM127+ HL-60 cells after different days of DSMO-induced differentiation with or without fMLP stimulation. (E) Expression of overall CD18 (conformation unspecific) on HL-60 cells after different days of DSMO-induced differentiation with or without fMLP stimulation. (F) FPR1 expression on HL-60 cells after different days of DSMO-induced differentiation. Mean ± 95%CI for n = 3 individual experiments in (B-G), **P < 0.01, ***P < 0.001, ****P < 0.0001 by comparing fMLP stimulated cells and vehicle control using 2-way ANOVA followed by Sidak’s multiple comparisons test
To assess the maturation markers of β2 integrin activation, 5 × 105 cells/mL undifferentiated HL60 cells incubated with the vehicle control or differentiated HL60 cells (day 5) incubated with fMLP (100 nM) or vehicle control were stained with AF488-conjugated mAb24 (1 μg/mL), PE-conjugated CD87 Ab (2 μg/mL), and AF647-conjugated CD35 Ab (1 μg/mL; Fig. 6A-B,E-F,H); or AF488-conjugated mAb24 (1 μg/mL) and AF647-conjugated CD18 Ab (2 μg/mL; Fig. 6G, H) at room temperature for 10 min. Cells were fixed with 1% PFA at room temperature for 10 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and analyzed with FlowJo software.
For intracellular CD18 staining (Fig. S1B), cells were first incubated with APC-conjugated CD18 Ab (1 μg/mL, surface CD18) and PE-conjugated CD87 Ab (2 μg/mL) at room temperature for 20 min, fixed with 1% PFA at room temperature for 10 min, washed twice with intracellular staining perm wash buffer plus 5% goat serum and stained with AF700-conjugated CD18 Ab (1 μg/mL, intracellular staining) in intracellular staining perm wash buffer plus 5% goat serum at room temperature for 30 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and analyzed with FlowJo software.
For the soluble ICAM-1 binding assay of blood neutrophils from MFN2flox/floxMRP8-cre− and 5 MFN2flox/floxMRP8-cre+ mice (Fig. S2A), 350 μL of mouse blood was collected by heart puncture directly after euthanasia with 10 μL heparin (1000 USP units/mL) in the collection tube. Each sample had 50 μL blood and was incubated with rat anti-mouse CD11a Ab (M17/4, 10 μg/mL) plus rat IgG2b isotype control (10 μg/mL), rat anti-mouse CD11b Ab (M1/70, 10 μg/mL) plus rat IgG2a isotype control (10 μg/mL), and rat IgG2b isotype control (10 μg/mL) plus rat IgG2a isotype control (10 μg/mL), respectively, at room temperature for 10 min. Then samples were incubated with recombinant mouse ICAM-1-Fc (10 μg/mL) and APC-conjugated mouse anti-human IgG Fc Ab (10 μg/mL) with or without the stimulation of PMA (200 nM) at room temperature for 30 min. Cells were fixed with 1% PFA at room temperature for 10 min. After 2 washes with PBS, cell fluorescence was assessed with an LSRII and analyzed with FlowJo software.
Zombie Yellow Fixable Viability Kit (1:1000 at room temperature for 15 min) was used to gate out dead cells. Compensations were performed before all experiments. Isotype controls were used to subtract the background fluorescence.
2.9 ∣. Actin polymerization imaging
Differentiated MFN2 KD and control HL60 cells (5 × 105 cells/mL) were incubated with fMLP (100 nM) or PMA (100 nM) or vehicle control at room temperature for 20 min. Cells were fixed with 1% PFA at room temperature for 10 min, washed twice with intracellular staining perm wash buffer plus 5% goat serum, and stained with AF568-conjugated phalloidin (5 U/mL) in intracellular staining perm wash buffer plus 5% goat serum at room temperature for 30 min. After 2 washes with PBS, cells were added into μ-Slide 8 Well Glass Bottom chamber (ibidi) pre-coated with 0.01% Poly-l-lysine (at 4°C overnight) and centrifuged at 500 × g at room temperature for 5 min to let the cells settle and adhere. Epifluorescence images (Fig. 3A)were acquired by using an iX83 Olympus inverted microscope equipped with the SAFe Light module (Abbelight, includes four color lasers, λ = 405 nm, 488 nm, 532 nm, and 640 nm), sCMOS fusion cameras (Hamamatsu), and a 100× NA 1.5 oil objective. The phalloidin median fluorescence intensity (MFI), which reflects the actin polymerization, was quantified by the “analyzing particles” function in FIJI-ImageJ258 (Fig. 3B).
FIGURE 3. Mitofusin-2 knockdown eliminates actin polymerization in neutrophil-like HL60 cells after receptor-dependent and independent stimulation.
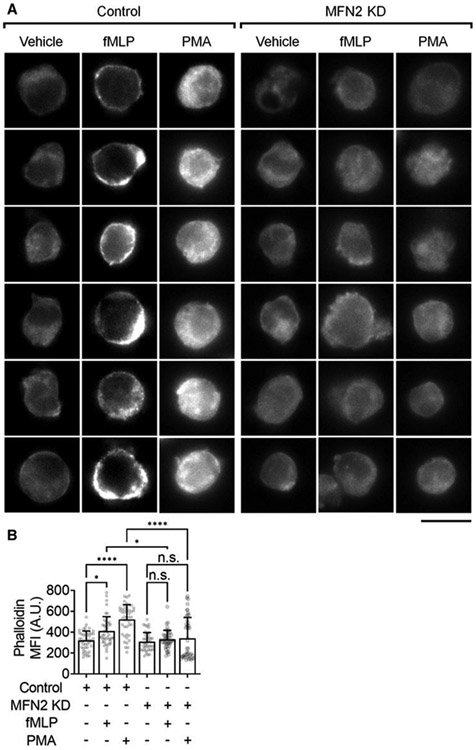
(A) Representative images of F-actin (phalloidin-AF568) in Control or MFN2-knockdown (MFN2 KD) HL60 cells stimulated with FPR1-dependent fMLP (100 nM, at RT for 20 min), receptor-independent PMA (100 nM, at RT for 20 min), or vehicle control. The scale bar is 10 μm. (B) Mean ± SD of phalloidin MFI quantified from n = 45 cells in 6 independent records per group. n.s., non-significant (P > 0.05) *P < 0.05, ****P < 0.0001 by 1-way ANOVA followed by Tukey’s multiple comparisons test. HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
2.10 ∣. Cell spreading assay
The μ-Slide 8 Well Glass Bottom chamber (ibidi) was coated with recombinant human ICAM-1-Fc (5 μg/mL) at room temperature for 3 h. Differentiated MFN2 KD and control HL60 cells (2 × 106 cells/mL) cells were incubated with CellTracker Orange CMRA (4 μM) at room temperature for 1 h. After 2 washes with PBS, cells were incubated with unconjugated mouse anti-human CD18 blocking Ab (TS1/18, 4 μg/mL) or mouse IgG1κ isotype control (4 μg/mL) at room temperature for 10 min. After 2 washes with PBS, cells were resuspended in PBS plus 1 μM Mn2+, added into the chamber, and centrifuged at 500 × g at room temperature for 5 min to induce spreading. Cells were fixed with 1% PFA at room temperature for 5 min and washed twice with PBS to remove unadhered cells. Total internal reflection fluorescence (TIRF) images (Fig. 4A) were acquired with an iX83 Olympus inverted microscope equipped with a SAFe Light module (Abbelight, includes four color lasers, λ = 405 nm, 488 nm, 532 nm, and 640 nm), sCMOS fusion cameras (Hamamatsu), and a 100× NA 1.5 oil objective. A TIRF incidence angle of θ = 70° was used. The area of cell footprint was quantified by the “analyzing particles” function in FIJI-ImageJ258 (Fig. 4B).
FIGURE 4. Mitofusin-2 knockdown inhibited Mn2+-induced integrin-outside-in-signaling-dependent spreading of neutrophil-like HL60 cells.
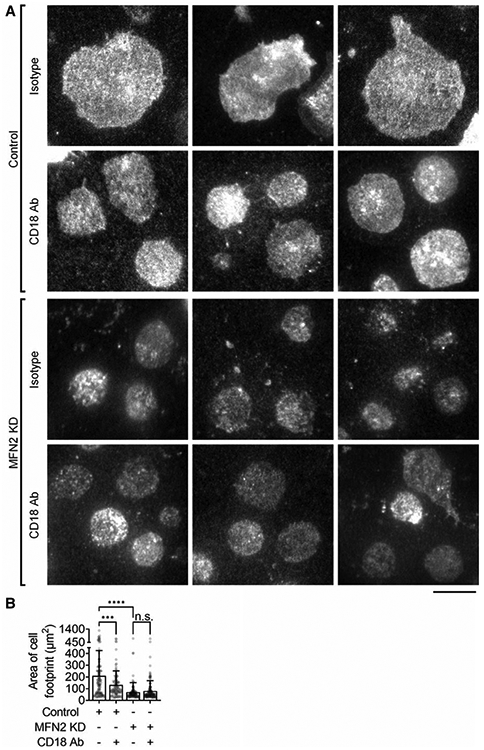
(A) Representative total internal reflection fluorescence images of spreading in Control or MFN2-knockdown (MFN2 KD) HL60 cell footprints pretreated with anti-CD18 blocking antibody or isotype control. Cells were stained with CellTracker Orange CMRA. Spreading was induced by 1 μM Mn2+ on coated ICAM-1. The scale bar is 10 μm. (B) Mean ± SD of the cell footprint area of n = 122 (control, isotype), n = 114 (control, CD18 Ab), n = 118 (MFN2 KD, isotype), and n = 134 (MFN2 KD, CD18 Ab) cells, respectively, from 6 independent records per group. n.s., non-significant (P > 0.05) ***P < 0.001, ****P < 0.0001 by 1-way ANOVA followed by Tukey’s multiple comparisons test. HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
2.11 ∣. Nucleus segmentation
Undifferentiated and differentiated MFN2 KD and control HL60 cells (5 × 105 cells/mL) cells were incubated with PE-conjugated CD87 Ab (1 μg/ml) at room temperature for 20 min, fixed with 1% PFA at room temperature for 10 min, washed twice with intracellular staining perm wash buffer plus 5% goat serum, and stained with AF647-conjugated Histone H3 recombinant rabbit monoclonal Ab (1 μg/ml) in intracellular staining perm wash buffer plus 5% goat serum at room temperature for 30 min. After two washes with PBS, cells were put into μ-Slide 8 Well Glass Bottom chamber (ibidi) pre-coated with 0.01% Poly-L-lysine (at 4 °C overnight) and centrifuged at 500 × g for 5 min to let the cells settle and adhere. Cell bright field and nucleus epifluorescence images (Fig. 7A) were acquired by using an iX83 Olympus inverted microscope equipped with a SAFe Light module (Abbelight, includes 4 color lasers, λ = 405 nm, 488 nm, 532 nm, and 640 nm), sCMOS fusion cameras (Hamamatsu), and a 100× NA 1.5 oil objective. The nucleus segment number (Fig. 7C) was counted manually by an investigator blinded to groups. To assess the nucleus segmentation of CD87high HL60 cells (Fig. 8B), CD87 MFI of cells in the above images was used to identify CD87high cells. The threshold for CD87high population was based on gating the brightest 20% of control cells. The same threshold was applied in analyzing MFN2 KD cells.
FIGURE 7. Mitofusin-2 knockdown limits nucleus segmentation during the DMSO-induced differentiation of HL60 cells.
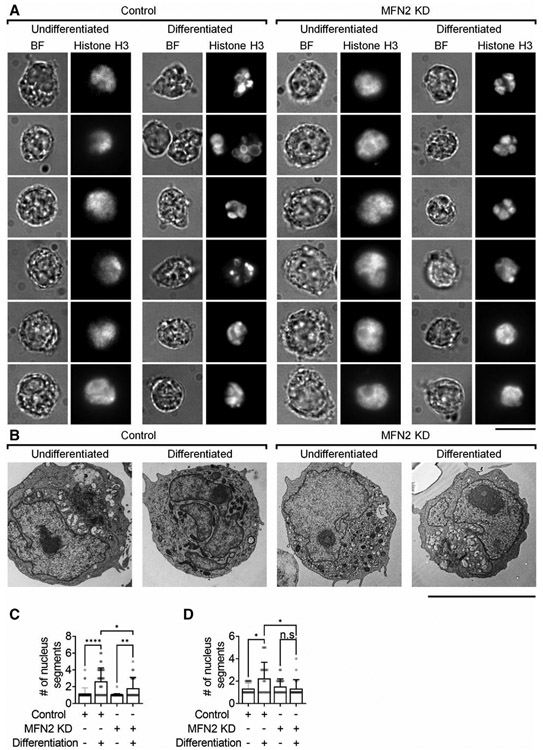
(A) Representative bright field (BF) and nucleus (anti-Histone H3-AF647) images of Control or MFN2-knockdown (MFN2 KD) HL60 cells before and after DMSO-induced differentiation. (B) Representative electron microscopy images of Control or MFN2 KD HL60 cells before and after the DMSO-induced differentiation. Scale bars are 10 μm. (C and D) Mean ± SD of nucleus segment number of n = 36 (control undifferentiated), n = 34 (control differentiated), n = 47 (MFN2 KD undifferentiated), and n = 24 (MFN2 KD differentiated) cells from 7, 9, 6, and 11 independent records, respectively, per group. n.s., non-significant (P > 0.05). *P < 0.05, **P < 0.01, ****P < 0.0001 by 1-way ANOVA followed by Tukey’s multiple comparisons test. HL60 cells were pre-differentiated with 1.3% DMSO for 7 days
For electron microscopy images (Fig. 7B), the 6-well plates were coated with recombinant human P-selectin-Fc (2 μg/mL) and recombinant human ICAM-1-Fc (10 μg/mL) for 2 h and then blocked with casein (1%) at room temperature for 1 h. Undifferentiated and differentiated MFN2 KD and control HL60 cells (5 × 106 cells/mL) cells were rolling on the substrate using the microfluidic device mentioned above. After observing stable rolling, 2.5% glutaraldehyde in 0.1 M cacodylate buffer was perfused to fix cells and was kept in the wells at room temperature for 1 h after detaching the microfluidic device. Further sample preparation and imaging were performed by the Central Electron Microscopy Facility of UConn Health: cells were fixed by 1% OsO4 and 0.8% ferricyanide in 0.1 M cacodylate buffer at room temperature for 1 h, rinsed 3 times with 0.1 M cacodylate buffer, and deionized water, respectively. En bloc staining was performed using freshly made 1% uranyl acetate in deionized water at room temperature for 1 h. After quick rinses with deionized water for 4 times, samples were dehydrated by using 50%, 75%, 95%, and 100% ethanol sequentially (10 min each, 3 times for 100% ethanol). The Poly/Bed 812 (resin) embedding kit was used to embed the samples by treatment with 1:1 100% ETOH/resin at room temperature for 1 h, 1:3 100% ETOH/resin at room temperature for 2 h, and resin at room temperature overnight, and then embedded in a 60°C oven for 48 h. Sections of 70–80 nm were cut by Leica EM UC7 ultramicrotome and stained with 6% uranyl acetate in methanol and lead citrate. Samples were examined in a Hitachi H-7650 transmission electron microscope operating at 80 kV. The nucleus segment number (Fig. 7D) was counted manually by an investigator blinded to groups.
2.12 ∣. Statistics
Statistical analysis was performed using PRISM software (version 8.30, GraphPad Software). Data analysis was performed using Student’s t-test, one-way ANOVA followed by Tukey’s multiple comparisons test, or 2-way ANOVA followed by Tukey’s multiple comparisons test or Sidak’s multiple comparisons test, which are indicated in figure legends. P-values < 0.05 were considered significant.
3 ∣. RESULTS
3.1 ∣. MFN2 is important for the slow-rolling and arrest of neutrophil-like HL60 cells
Our previous study showed that MFN2-deficient HL60 cells have a defect of adhesion to TNF-α-stimulated human umbilical vein endothelial cells (HUVECs) when measured under flow conditions.43 However, detailed molecular mechanisms need further investigation. Using a microfluidic assay20,25,59,60 on adhesion-protein-reconstituted substrates, we aimed to study the effect of MFN2 deficiency on PSGL-1-mediated rolling and β2-integrin-mediated slow-rolling and arrest individually.
First, RNA interference (shRNA, MFN2-sh1) was used to reduce protein expression of MFN2 in neutrophil-like differentiated HL60 (dHL60) cells, which is confirmed by using both western blot (Fig. 1A,B) and intracellular staining flow cytometry (Fig. 1C). We also tried another shRNA (MFN2-sh2) and found a slight knockdown (KD) (<10%) of MFN2 expression compared to the control (Fig. S3A). Thus, we used MFN2-sh1 in most of our experiments, and MFN2-sh2 in a few experiments later. In the microfluidic assay, control or MFN-knockdown (MFN2KD) dHL60 cells were perfused into the flow chamber with a substrate of P-selectin and showed similar rolling distance (Fig. 1D) and velocities (Fig. 1E, F), suggesting that the function of PSGL-1 may not be affected. As expected, control dHL60 cells showed a decreased velocity when rolling on the substrate of P-selectin/ICAM-1 compared to the substrate of P-selectin only (Fig. 1D-F), which is called slow-rolling and depends on β2 integrin extension that is induced by the interaction of PSGL-1 and selectins.16-20 MFN2 deficiency in dHL60 cells eliminated the slow-rolling (Fig. 1D-F), suggesting that MFN2 is important for β2 integrin extension. The perfusion of fMLP, a bacteria-derived chemotactic factor that can trigger neutrophil activation, 61 adhesion,24,62 and chemotaxis,63-67 induced the arrest of dHL60 on the P-selectin/ICAM-1 substrate. By stark contrast, we found that MFN2 deficiency reduced the number of arrested dHL60 cells by around 80% under a wall shear stress of 6 dyn/cm2 (Fig. 1G) and around 60% under a wall shear stress of 2 dyn/cm2 (Fig. 1H). These results suggest that MFN2 is important for the activation or surface expression of β2 integrins and affects the slow-rolling and arrest of dHL60 cells.
3.2 ∣. MFN2 deficiency inhibits fMLP receptor expression on neutrophil-like HL60 cells
Integrin activation and arrest of neutrophils require the integrin inside-out signaling initiated by the interaction of chemokines or chemotactic factors and their corresponding receptors.23,28,35,68-71 There are two fMLP receptors expressed on neutrophils – formyl peptide receptor 1 (FPR1)72,73 and formyl peptide receptor 2 (FPR2).74,75 To test whether the adhesion deficiency of dHL60 cells is due to reduced fMLP receptor expression, we used flow cytometry to test expression level of FPR1 and FPR2 on control or MFN2 KD dHL60 cells. We did not find FPR2 expression on our dHL60 cells (data not shown). We found that FPR1 expression was dramatically reduced by around 70% in MFN2 KD dHL60 cells compared to control (Fig. 2A). These results revealed that reduced MFN2 protein expression significantly reduced fMLP receptor expression, possibly contributing to the adhesion deficiency observed in dHL60 cells. Thus, we also need to introduce receptor-independent stimulators when assessing β2 integrin activation and expression. PMA can bypass the receptors of chemokines or chemotactic factors to directly activate protein kinase C isozymes (PKC), which are key components in the integrin inside-out signaling pathway,76 and active β2 integrins on leukocytes.
3.3 ∣. MFN2 regulates adhesion molecule expression on neutrophil-like HL60 cells
We next tested the expression of adhesion molecules PSGL-1 and β2 integrins on control and MFN KD dHL60 cells. PSGL-1 expression was significantly increased by around 40% in MFN2-deficient dHL60 cells (Fig. S1). However, this had no effect on the P-selectin-dependent rolling velocity of cells (Fig. 1D-F). For β2 integrins, we tested the expression of both the β2 subunit (CD18, Fig. 2B,C) and α subunits (αL, CD11a; αM, CD11b; αX, CD11c, Fig. 2D-I). Neutrophil degranulation after fMLP stimulation will deliver granular β2 integrins (CD18, CD11b, and CD11c) to the cell surface and increase surface expression,18 which may contribute to neutrophil adhesion. We observed this degranulation in dHL60 cells after both FPR-1-dependent fMLP stimulation (Fig. 2B) and receptor-independent PMA stimulation (Fig. 2C). We found that overall surface CD18 expression before and after FPR-1 dependent fMLP stimulation (Fig. 2B) or receptor-independent PMA stimulation (Fig. 2C) remained the same in MFN2-deficient dHL60 cells compared to controls. We also performed intracellular CD18 staining to assess the degranulation (Fig. S1B). The intracellular CD18 expression before and after stimulation remained the same in MFN2-deficient dHL60 cells compared to controls. This suggested that MFN2 KD does not affect the expression and degranulation of overall β2 integrins.
The condition became more complex when assessing the α subunits of β2 integrins. Surface CD11a expression before stimulation remained the same in MFN2 KD dHL60 cells (Fig. 2D-E) compared to controls. A slight increase of surface CD11a expression was observed in control but not MFN2 KD dHL60 cells after fMLP stimulation (Fig. 2D). However, in PMA stimulation experiments, a significantly higher increase of surface CD11a expression was observed in MFN2 KD dHL60 cells compared to control, suggesting that the loss of CD11a increase in fMLP-stimulated MFN2 KD dHL60 cells is possibly due to decreased FPR1 expression.
Surface CD11b expression before stimulation remained the same in MFN2 KD dHL60 cells (Fig. 2F,G) compared to controls. After fMLP stimulation, both MFN2 KD and control dHL60 cells showed an increase of surface CD11b expression (Fig. 2F). This increase in MFN2 KD dHL60 cells was slightly less but not significant. In PMA stimulation experiments, both MFN2 KD and control dHL60 cells showed an increase of surface CD11b expression (Fig. 2G). This increase in MFN2 KD dHL60 cells was significantly higher than in controls. These results suggest that MFN2 KD dHL60 has a higher disposition of CD11b degranulation, and this disposition is compensated by decreased FPR1 expression in fMLP stimulation.
Surface CD11c expression was dramatically decreased in MFN2 KD dHL60 cells (Fig. 2H,I) compared to controls. The increase of surface CD11c expression was observed in control dHL60 cells upon both fMLP and PMA stimulation. In MFN2 KD dHL60 cells, there is no significant increase of surface CD11c expression after fMLP stimulation (Fig. 2H). An increase of surface CD11c expression was observed in MFN2 KD dHL60 cells after PMA stimulation. But this increase is significantly less than in controls (Fig. 2I). These results suggest that MFN2 KD decreases both expression and degranulation of CD11c. Both a direct effect and an FPR1-dependent indirect effect contributed to this CD11c deficiency.
Overall, we can conclude that MFN2 influences the expression of adhesion molecules on dHL60 cells to different degrees, with the most apparent changes occurring in the CD11c/αX subunit.
3.4 ∣. MFN2 deficiency impairs β2 integrin activation on neutrophil-like HL60 cells
By using conformation-specific antibodies, we tested β2 integrin activation on dHL60 cells. When human β2 integrins acquire H+, a neoepitope in the β2 I-like domain49,50 is exposed, which is recognized by mAb24.51 E+ of β2 integrins is detected by the monoclonal antibody (mAb) KIM127,47 which recognizes a neoepitope77 that is hidden in the bent knee of human β2. Thus, mAb24 staining indicates H+ β2 integrins, and KIM127 staining indicates E+ β2 integrins.
We found that the mAb24 MFI was increased by about 10 times upon stimulation with fMLP in control dHL60 cells (Fig. 2J). In MFN2 KD dHL60 cells, the mAb24 MFI was also increased after fMLP stimulation, but the increase was reduced by more than 50% compared to that of controls (Fig. 2J). Since expression of fMLP receptor FPR1 was decreased in MFN2 KD dHL60 cells (Fig. 2A), we also performed receptor-independent integrin activation assay using PMA stimulation (Fig. 2K). The increase of mAb24 MFI was inhibited by about 30% in MFN2 KD dHL60 cells compared to controls upon PMA stimulation. Next, we normalized the mAb24 MFI to the MFI of overall β2 integrin (CD18, Fig. 2B,C). Similar results of normalized mAb24 were observed compared to mAb24 MFI results and the increase of mAb24 MFI was inhibited by around 50% (Fig. 2L) or around 40% (Fig. 2 M) after fMLP or PMA stimulation, respectively, owing to the lack of MFN2. This is because overall CD18 expression was not changed significantly between MFN2 KD and control dHL60 cells.
The KIM127 MFI was increased more than two times upon fMLP stimulation in control dHL60 cells (Fig. 2N). In MFN2 KD dHL60 cells, the KIM127 MFI was also increased after fMLP stimulation, but the increase was reduced by more than 60% compared to controls (Fig. 2N). In receptor-independent PMA stimulation, KIM127 MFI was increased to around 2.5 times upon PMA stimulation in control dHL60 cells (Fig. 2O). And this increase was inhibited by around 30% in MFN2 KD dHL60 cells. After normalizing to the overall β2 integrin staining, the increase of KIM127 MFI was inhibited by around 70% (Fig. 2P) or around 50% (Fig. 2Q) after fMLP or PMA stimulation, respectively, owing to the lack of MFN2.
We also tested the staining of another monoclonal antibody CBRM1/5, which reports the H+ of Mac-1 (αMβ2 integrins). In control dHL60 cells, CBRM1/5 MFI was increased to about 2.5-times or 4.5-times upon FPR1-dependent fMLP or receptor-independent PMA stimulation, respectively (Fig. 2R,S). This increase was eliminated in fMLP-stimulated MFN2 KD dHL60 cells (Fig. 2R) but was not significantly changed in PMA-stimulated MFN2 KD dHL60 cells (Fig. 2S). After normalization to overall CD11b expression, the fMLP-induced CBRM1/5 MFI increase was also abolished in MFN2 KD dHL60 cells compared to control (Fig. 2T). PMA-induced CBRM1/5 MFI increase was inhibited by around 30% (Fig. 2U). These results suggested that MFN2 KD affects Mac-1 H+ mainly through decreasing FPR1 expression. Other components in the Mac-1 activation signaling pathway may also be affected but were compensated by the increase of Mac-1 degranulation upon PMA stimulation (Fig. 2G).
To assess whether MFN2 is involved in integrin activation in primary neutrophils, we analyzed MFN2flox/floxMRP8-cre+ mice, in which MFN2 expression in neutrophils is ~50% of that in neutrophils from control MFN2flox/flox MRP8-cre− mice (Fig. S2A). We did a soluble ICAM-1 (sICAM-1) binding assay to evaluate β2 integrin activation and found that both MFN2flox/floxMRP8-cre+ and MFN2flox/floxMRP8-cre− neutrophils (control) have increased sICAM-1 binding after receptor-independent PMA stimulation (Fig. S2B). The increase of MFN2flox/floxMRP8-cre+ neutrophils is significantly less (~50%) than that of controls (Fig. S2B). Both CD11a and CD11b blockade eliminated this difference, suggesting both CD11a and CD11b are involved in MFN2-dependent β2 integrin activation (Fig. S2B).
3.5 ∣. MFN2 deficiency inhibits actin polymerization on neutrophil-like HL60 cells
Previous studies showed that actin polymerization inhibitor latrunculin B prevents β2 integrin activation, specifically H+ (reported by antibody MEM148 and ICAM-1 dependent arrest), on neutrophils.78 We also showed previously that MFN2 knockout (KO) in mouse embryonic fibroblasts showed reduced stress fibers in the cell body.43 Thus, we tested whether MFN2 KD in dHL60 cells prevents actin polymerization by using phalloidin to show F-actin amount and distribution (Fig. 3). We observed that both FPR1-dependent fMLP or receptor-independent PMA was able to increase actin polymerization in control dHL60 cells (Fig. 3A, left 3 panels, and B). Upon fMLP stimulation, F-actin was more located in the cell cortex close to the membrane (Fig. 3A, the second panel), which might be due to the local signaling generated by membrane FPR1. In contrast, PMA stimulation made F-actin spread throughout the cytoplasm (Fig. 3A, the third panel), which makes sense because PMA activates PKC throughout the cytoplasm. In MFN2 KD dHL60 cells, both fMLP and PMA-induced actin polymerizations were abolished (Fig. 3A, right 3 panels, Fig. 3B).
3.6 ∣. MFN2 deficiency abolishes β2 integrin outside-in-signaling-dependent spreading of neutrophil-like HL60 cells
Besides the G protein-coupled receptor (GPCR, such as FPR1)-initiated inside-out signaling of β2 integrins, β2 integrin outside-in-signaling is also crucial for neutrophil recruitment.79 We tested whether MFN2 was involved in the outside-in-signaling by using a Mn2+-induced spreading assay.80 In this assay, Mn2+ will force all β2 integrins into the E+H+ conformation and bind to ICAM-1 coated in the chamber to initiate the outside-in-signaling and spreading. As shown in Figure 4, control dHL60 cells were able to spread well on the immobilized ICAM-1, and the blockade of β2 integrins (CD18 blocking antibody TS1/18) led to reduced cell spreading (Fig. 4A, top two panels, Fig. 4B). MFN2 KD abolished dHL60 cell spreading with or without β2 integrin blockade (Fig. 4A, bottom two panels, Fig. 4B).
3.7 ∣. MFN2 deficiency limits the maturation of integrin activation during DMSO-induced differentiation
Integrins cannot be activated upon fMLP stimulation in undifferentiated HL60 cells (Fig. 5A, top panel). During the DMSO-induced neutrophil-like differentiation, the capacity of integrin activation is acquired (matured) over time (Fig. 5A-D). The maturation of fMLP-stimulated β2 integrin H+ (mAb24 staining, Fig. 5B) or E+ (KIM127 staining, Fig. 5C) was observed starting from four or two days, respectively, after incubating with 1.3% DMSO. These fMLP-stimulated β2 integrin H+ and E+ states became more significant when HL60 cells were differentiated for a longer time (Fig. 5B and C). The fMLP-induced mAb24+KIM127+ population was significantly increased compared to vehicle controls starting from three days of DMSO-differentiation (Fig. 5D). This population increased when HL60 cells were differentiated for a longer time (Fig. 5D). Thus, besides the reduced expression of fMLP receptors, the integrin activation defect observed in MFN2 KD dHL60 cells might be due to the influence of MFN2 on the maturation of integrin activation.
We further assessed the expression of overall β2 integrins (CD18, Fig. 3E) and fMLP receptors (Fig. 5E and F), which contribute to H+ and E+ β2 integrin expression, during the DMSO-induced neutrophil-like differentiation. Expression of overall β2 integrins increased to 3-fold after two-day differentiation compared to undifferentiated HL60 cells (Fig. 5E). The degranulation of β2 integrins, which may contribute to the increase of integrin H+ and E+, was observed starting from five days of DMSO-differentiation (Fig. 5E). FPR1 expression had a significant increase observed at day 5 and reached a plateau (Fig. 5F), which is the same time that dHL60 shows degranulation ability. Overall, expression and degranulation of β2 integrins, as well as FPR1 expression, showed different increasing patterns compared to β2 integrin H+ and E+, suggesting that there are other molecules involved in the process of integrin activation maturation.
It is well known that HL60 cells have heterogeneous populations after DMSO-induced differentiation. The integrin activation of dHL60 cells shows the same case – even after 7 days of differentiation, only around 36% of cells show KIM127 and mAb24 double-positive staining, and around 53% of cells are double-negative after 100 nM fMLP stimulation (Fig. 5A, D). By using primary human neutrophils as a positive control (Fig. 5A, bottom panel), around 97% of neutrophils show KIM127 and mAb24 double-positive staining after fMLP stimulation, Thus, finding markers to identify the maturation stages of integrin activation becomes a priority.
A previous study has shown the changes in antigen expression on HL60 cells upon differentiation.81 After testing several candidates, we determined that expression of CD35 (complement receptor 1, Fig. 6A) and CD87 (urokinase-type plasminogen activator receptor, uPAR, Fig. 6B) were increased after differentiation. Expression of both CD35 (Fig. 6C) and CD87 (Fig. 6D) increased over time during DMSO-induced neutrophil-like differentiation. Expression of CD35 and CD87 correlated with mAb24 staining (H+ β2 integrins) after fMLP stimulation in dHL60 cells (Fig. 6E and F, respectively). In all the dHL60 cells, around 17% of them showed mAb24-positive staining. About 67% of the CD35-high population (approximately 20% of total dHL60 cells) showed mAb24 positive staining, whereas around 5% of the CD35-low population showed mAb24 positive staining (Fig. 6E, H). About 73% of the CD87-high population (approximately 20% of total dHL60 cells) showed mAb24 positive staining, whereas around 3% of the CD87-low population showed mAb24 positive staining (Fig. 6F, H). As expected, we also found that β2 integrin expression (CD18, Fig. 6G) is also correlated with β2 integrin activation (mAb24 staining). About 63% of the CD18-high population (approximately 20% of total dHL60 cells) showed mAb24-positive staining, whereas around 5% of the CD18-low population showed mAb24-positive staining (Fig. 6G, H). These data suggested that CD35, CD87, or CD18 may be used as a maturation marker of integrin activation, with CD87 having the highest potential.
In the MFN2-deficient HL60 cells, we detected ~25% and ~55% decreases in expression of CD35 (Fig. 6I) and CD87 (Fig. 6J), respectively, after culturing with 1.3% DMSO for 5 days, compared to controls. The decrease of CD87 expression was also significant but became less (~10%) when differentiating cells for 7 days. These results suggest that MFN2 may be important for the maturation of integrin activation during HL60 differentiation.
Nucleus segmentation is another marker for the differentiation of HL60 cells.82 Thus, we assessed the nucleus segmentation of control and MFN2 KD HL60 cells with or without differentiation by both epifluorescence and electron microscopy nucleus imaging (Fig. 7). Nucleus segmentation was observed in both control and MFN2 KD HL60 cells after differentiation (Fig. 7A and B). However, the number of nucleus segments significantly decreased in MFN2 KD dHL60 cells compared to controls (Fig. 7C and D).
3.8 ∣. MFN2 deficiency impairs β2 integrin activation on matured neutrophil-like HL60 cells
Since MFN2 KD affects β2 integrin activation maturation during neutrophil-directed HL60 differentiation, it is important to assess the direct involvement of MFN2 in β2 integrin activation by analyzing dHL60 cells of the same maturation stage. Using CD87 as a maturation marker of β2 integrin activation, we were able to do this. After gating the CD87high population of dHL60 cells (21.6±3.6% in control cells, same gating yielding 14.7 ± 7.4% in MFN2 KD cells), we first confirmed that CD87high MFN2 KD and control dHL60 cells had similar CD87 expression (Fig. 8A) and nucleus segmentation (Fig. 8B), indicating that they were in the same differentiation stage or integrin activation maturation stage. Intracellular staining confirmed that MFN2 expression in CD87high MFN2 KD dHL60 cells was around 50% of control (Fig. 8C). FPR1 expression was decreased in CD87high MFN2 KD dHL60 cells compared to control (Fig. 8D). Thus, we needed to test both fMLP and PMA stimulation.
MFN2 deficiency does not affect overall β2 integrin (CD18) expression (Fig. 8E, F). The degranulation of CD18 was slightly inhibited upon both fMLP (Fig. 8E) and PMA (Fig. 8F) stimulation in CD87high MFN2 KD dHL60 cells compared to controls. When assessing β2 integrin H+ by mAb24, we observed that fMLP (Fig. 8G, I) and PMA (Fig. 8H, J) stimulation elevated mAb24 MFI in both MFN2 KD and control CD87high dHL60 cells. MFN2 KD led to an ~40% (Fig. 8G) and ~20% (Fig. 8H) decrease in mAb24 MFI elevation in this CD87high population upon fMLP and PMA stimulation, respectively. After normalization to overall CD18 expression, the decreases of mAb24 MFI elevation in CD87high MFN2 KD dHL60 cells were ~30% (Fig. 8I) and ~10% (but not significantly different, Fig. 8J) compared to CD87high control dHL60 cells upon fMLP and PMA stimulation, respectively. In the case of β2 integrin E+ assessed by KIM127, the effect of MFN2 was clearer. Although there were elevations of KIM127 MFI in CD87high MFN2 KD dHL60 cells, the elevations were significantly less compared to CD87high control dHL60 upon both fMLP (~70%, Fig. 8K) and PMA (~50%, Fig. 8L) stimulation. After normalization to overall CD18 expression, the decreases of KIM127 MFI elevation in CD87high MFN2 KD dHL60 cells were ~80% (Fig. 8I) and ~50% (Fig. 8J) compared to CD87high control dHL60 cells upon fMLP and PMA stimulation, respectively.
Another way to assess the role of MFN2 in dHL60 cells at the same maturation stage is using the MFN2-sh2-mediated KD cells (Fig. S3), which has <10% MFN2 KD, as we mentioned above (Fig. S3A). This is because these cells had no significant changes in expression of FPR1 (Fig. S3B), CD87 (Fig. S3C), or CD18 (Fig. S3D-E), as well as CD18 degranulation upon both fMLP (Fig. S3D) and PMA (Fig. S3E) stimulation, suggesting this slight KD of MFN2 did not affect β2 integrin activation maturation during neutrophil-directed HL60 differentiation. A slight but significant decrease of fMLP-induced (Fig. S3F, H, J, L) or PMA-induced (Fig. S3G, I, K, M) integrin activation (both mAb24 Fig. S3F-I, and KIM127 Fig. S3J-M) was observed in these MFN2-sh2 KD cells compared to controls. This further confirmed the role of MFN2, especially its direct involvement, in integrin activation.
To test whether the integrin activation defect in CD87high MFN2 KD dHL60 cells has functional consequences, we performed the microfluidic rolling and arrest assay combined with CD87 fluorescence staining (Fig. 8O-Q). We found that when rolling on the substrate of P-selectin plus ICAM-1, the rolling velocity of CD87high MFN2 KD dHL60 cells is significantly higher than that of CD87high control cells (Fig. 8O-P), suggesting the slow-rolling was impaired in CD87high MFN2 KDdHL60 cells, which is consistent with the β2 integrin E+ deficiency mentioned above. After the perfusion of fMLP to induce cell arrest, CD87high MFN2 KD dHL60 cells showed a significantly lower number of arrested cells than controls (Fig. 8Q).
4 ∣. DISCUSSION
MFN2 has been reported to regulate leukocyte adhesion under flow, although the mechanism for such regulation has yet to be defined. In the present study, we investigated the role of MFN2 in regulating fMLP receptors, adhesion molecules, as well as integrin activation (Fig. 9). Using a microfluidic assay on substrate reconstituted from purified adhesion proteins, we confirmed the adhesion deficiency of neutrophil-like HL60 cells in which MFN2 had been knocked down by RNA interference (Fig. 1A-C). Specifically, MFN2-deficient cells displayed a defect of β2 integrin-mediated slow-rolling and arrest, but not PSGL-1-mediated rolling (Fig. 1D-H). Flow cytometry staining showed that MFN2-deficient cells had lower expression of fMLP receptor FPR1 (Fig. 2A), which initiates integrin inside-out activation, and similar overall β2 integrin expression (Fig. 2B-C) with some variations in the expression of different β2 integrin α subunits (Fig. 2D-I). The activation of β2 integrins assessed by conformation-specific antibodies mAb2449-51 and KIM12747,48 was further observed to be inhibited on MFN2-deficient cells by using both FPR1-dependent fMLP stimulation and receptor-independent PMA stimulation (Fig. 2J-Q). These results suggest that MFN2 is essential for β2 integrin activation, not only through the indirect effect on FPR1 expression, but also a direct effect. We also observed actin polymerization, which is involved in leukocyte β2 integrin activation,78 was impaired in MFN2-deficient cells (Fig. 3). This might provide a mechanism for why integrin activation is inhibited in MFN2-deficient cells. Besides the β2 integrin inside-out signaling, we also showed that the β2 integrin outside-in signaling-dependent cell spreading is abolished in MFN2-deficient cells (Fig. 4). This might also link to the actin polymerization deficiency and strengthen the idea that MFN2 is critical for neutrophil trafficking and recruitment.
FIGURE 9. Schematics showing the function of MFN2 in HL60 adhesion.

(A) During the neutrophil-directed differentiation of HL60 cells, β2 integrin activation increases in response to fMLP stimulation, which is critical for cell adhesion. We call this process the maturation of β2 integrin activation. Expression of β2 integrins, fMLP receptor FPR1, CD87, and CD35 also increase during the neutrophil-directed differentiation of HL60 cells. The maturation of β2 integrin activation is associated with the expression of β2 integrins, CD87, and CD35. Thus, the expression of β2 integrins, CD87, and CD35 can serve as markers for the maturation of β2 integrin activation. (B) Knockdown of MFN2 inhibited the maturation of β2 integrin activation during the neutrophil-directed differentiation of HL60 cells, causing less expression of FPR1, CD87, and CD35, less activation of β2 integrins, and less cell adhesion underflow. (C) In β2 integrin activation matured cells (CD87high in this study), the loss of MFN2 expression decreases β2 integrin
Another key finding in our study is the maturation markers of β2 integrin activation in HL60 cells. HL60 is the most commonly used cell line model of human neutrophils.36,43,56,60,83-87 Because neutrophils are short-lived,1,5 hard to transfect, and sensitive to transfection, the cell line model is critical for studying the molecular mechanisms in neutrophil behaviors and functions, such as integrin activation and cell adhesion. We examined the maturation of β2 integrin activation over the time of neutrophil-directed differentiation of HL60 cells and observed a heterogeneous β2 integrin activation in response to fMLP (Fig. 5). Finding a maturation marker is critical for improving the signal-to-noise ratio in HL60 assays and understanding integrin activation by comparing populations in the same maturation stage. We have identified three such markers-CD35, CD87, and overall CD18 (Fig. 6). This is important for future studies using HL60 cells as a model of neutrophils, such as studies of integrin activation, cell adhesion, cell migration, and phagocytosis.
CD87 is also known as uPAR and has been reported to be associated with and regulate the functions of β2 integrins on the surface of leukocytes, including neutrophils.88 CD87 has been indicated as a neutrophil maturation marker that is expressed at the band and segmented neutrophil stage of development,82 which is consistent with our results in HL60 cells (Fig. 6-8). The association of CD87 and CD11b has been demonstrated by colocalization using confocal microscopy89 and fluorescence resonance energy transfer (FRET).89 Ab blockade of CD87 on human monocytes inhibits their adhesion to fibrinogen and keyhole limpet hemocyanin (KLH) upon PMA-stimulation by ~50%.89 The same blockade inhibits neutrophil adhesion to both ICAM-1 and fibrinogen.90 RNA interference of CD87 reduces cell adhesion to KLH by ~45%,89 and functional Ab blockade of CD87 inhibits neutrophil chemotaxis.91 Co-immunoprecipitation (Co-IP) of CD87 and β2 integrins have been shown as well.92 Domain 1 deletion of CD87 diminished its co-IP with β2 integrins.93 Mapping experiments showed that CD87 binds to the β-propeller domain of CD11b (amino acids 424–440).94 This deficiency can be restored by adding soluble recombinant CD87.89 CD87 KO mice showed a defect in β2-integrin-dependent leukocyte recruitment.95-99 The role of CD87 in integrin functions is ligand (urokinase-type plasminogen activator, uPA)-independent.91,96 The reporter antibody test showed that the CD87 association promotes hybrid domain swing-out (clone MEM148) but not the extension of the ectodomain (clone KIM127) of Mac-1.93 FRET assay showed that the CD87 association reorientates and separates the transmembrane domain of Mac-1.93 CD35 is also known as complement receptor 1, and has not been reported to be associated with integrin activation.
Here, we have shown that MFN2 KD leads to a deficiency in the maturation of β2 integrin activation, which is identified by decreased expression of CD35 and CD87 (Fig. 6I-K) as well as fewer nucleus segments (Fig. 7). This could provide some explanation for the deficiency of β2 integrin activation and cell adhesion. One of the maturation markers, CD87, directly promotes integrin activation, as described above. Whether there are other molecules involved remains to be investigated. The defect of integrin activation maturation in MFN2-deficient cells raises concerns about the involvement of MFN2 in neutrophil development. It has been shown that MFN2 is important for maintaining hematopoietic stem cells with extensive lymphoid potential by using mice with conditional deletion of the MFN2 gene (Mfn2) in the hematopoietic system (Mfn2fl/fl-Vav-Cre).57 This study briefly investigated the Mfn2 deletion in myeloid cell development and concluded only a minor effect. However, the myeloid populations, such as different stages of myeloid progenitors, neutrophils, monocytes, M<s, and dendritic cells, were not identified in this study. Thus, the effects of Mfn2 deletion in neutrophil development await further analysis.
Besides the indirect effect caused by maturation, we showed that MFN2 also has direct effects on integrin activation. First, we assessed MFN2-deficient and control cells at the same maturation stage (CD87high population, Fig. 8). We confirmed the same CD87 expression and nucleus segmentation of the 2 groups as well as the decreased MFN2 and FPR1 expression in the MFN2-deficient cells (Fig. 8A-D). In these CD87high cells, MFN2 deficiency showed the deficiency of integrin activation in response to both FPR1-dependent fMLP and receptor-independent PMA stimulation (Fig. 8E-M). Functional rolling and arrest assay showed that in CD87high cells, MFN2 deficiency led to defects of slow-rolling and arrest (Fig. 8O-Q). These results suggest that MFN2 has direct effects on integrin activation. Since we have shown that MFN2 deficiency increases the activation of ras-related C3 botulinum toxin substrate (Rac),43 which is important in the GPCR-initiated inside-out pathway of integrin activation,100-103 MFN2 deficiency must inhibit other components downstream of Rac, such as phospholipases,26,103,104 CalDag-GEFI,105 Rap1,104-107 Rap1-GTP-interacting adaptor molecule (RIAM),108 talins,19,109 or kindlins.19,110-112 Whether these molecules were involved in the MFN2-dependent β2 integrin activation needs further investigation.
MFN2 itself is a dynamin-like GTPase that plays a central role in regulating mitochondrial fusion and cell metabolism and uses its GTPase domain interface to form sustained dimers that account for membrane-tethering.113 The GTPase domain is also important for GTP-dependent membrane fusion.114 This suggests that MFN2 may be involved in granule-dependent integrin trafficking. This is possible because although MFN2 KD did not affect dHL60 overall CD18 degranulation (Fig. 2B-C, S1B), CD11a and CD11b degranulation were slightly enhanced (Fig. 2E, G), and CD11c degranulation was inhibited (Fig. 2I). The role of MFN2 in regulating trafficking and membrane fusion of different granules may need further investigation. Also, as mentioned above, several GTPases are involved in inside-out signaling of β2 integrin activation, such as Rho GTPases that can bind115 and activate phospholipases100-103 or Rap1 that recruits RIAM and talin to integrins.104-107 Thus, whether the GTP domain of MFN2 has active phospholipases, binds to RIAM or talin, or directly activates integrins would be interesting to pursue in further studies. This is possible because PXXP motifs that can bind phospholipase C were identified in MFN2 (amino acids 523–526, PLLP, and amino acids 595–609, PIPLTPANPSMPPLP).116 Another study focusing on Yap showed that Yap inhibition reduces both talin-1 and MFN2 expression, suggesting a correlation between talin and MFN2.117
In conclusion, MFN2 is important for β2 integrin activation and the β2 integrin-mediated adhesion of neutrophil-like dHL60 cells. MFN2 is required for FPR1 expression but also contributes to β2 integrin inside-out activation in a receptor-independent manner. MFN2 is required for actin polymerization, which is vital for integrin activation. MFN2 is also required for β2 integrin outside-in signaling. MFN2 is crucial for integrin activation maturation during neutrophil-directed differentiation of HL60 cells, which indirectly contributes to this β2 integrin activation phenotype. By comparing cells at the same maturation stage, we know that MFN2 also directly contributes to β2 integrin activation.
Supplementary Material
ACKNOWLEDGMENTS
The clonal HL-60 cell line was generated by Alba Diz-Munoz (EMBL Heidelberg) and was a generous gift from the laboratory of Orion Weiner (University of California San Francisco). We thank Dr. Evan Jellison and Ms. Li Zhu in the flow cytometry core at UConn Health for their assistance with flow cytometry, and Dr. Lynn Puddington in the Department of Immunology at UConn Health for her support of the instruments. We thank Maya Yankova and Dr. Stephen M. King from UConn Health Central Electron Microscopy Facility for their assistance in the sample preparation and transmission electron microscopy imaging. KIM127 antibody is a gift from Dr. Klaus Ley from the La Jolla Institute for Immunology. This research was supported by funding from the National Institutes of Health, USA (NIH, R01HL145454 and R35GM119787), a Purdue Research Foundation Grant from Purdue University, and a startup fund from UConn Health.
Abbreviations:
- AF647
Alexa Fluor 647
- APC
allophycocyanin
- Co-IP
Co-immunoprecipitation
- DMSO
dimethyl sulfoxide
- E+
extension
- FITC
fluorescein isothiocyanate
- fMLP
N-formylmethionyl-leucyl-phenylalanine
- FBS
fetal bovineserum
- FRET
fluorescence resonance energy transfer
- FPR1
formyl peptidereceptor 1
- FPR2
formyl peptidereceptor2
- GPCR
G protein-coupled receptor
- H+
high-affinity
- HSA
human serum albumin
- HUVECs
human umbilical vein endothelial cells
- ICAM-1
intercellular adhesion molecule 1
- KD
knockdown
- KLH
keyhole limpet hemocyanin
- KO
knockout
- mAb
monoclonal antibody
- MFN-2
mitofusin-2
- PBMC
peripheral blood mononuclear cells
- PE
phycoerythrin
- PBS
phosphate-buffered saline
- PFA
paraformaldehyde
- PKC
protein kinaseC isozymes
- PMA
phorbol-12-myristate-13-acetate
- PSGL-1
P-selectin glycoprotein ligand-1
- Rac
ras-related C3 botulinumtoxin substrate
- RIAM
Rap1-GTP-interactingadaptor molecule
- RPMI-1640
Park Memorial Institute medium 1640
- TIRF
total internal reflection fluorescence
- uPA
urokinase-type plasminogen activator
- uPAR
urokinase-type plasminogen activator receptor
Footnotes
DISCLOSURE
The authors declare no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon request.
REFERENCES
- 1.Ley K, Hoffman HM, Kubes P, et al. Neutrophils: new insights and open questions. Sci Immunol. 2018;3. [DOI] [PubMed] [Google Scholar]
- 2.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for β2 integrin-mediated leukocyte adhesion to endothelial cells. Nature medicine. 2009;15:300–305. 10.1038/nm.1921. [DOI] [PubMed] [Google Scholar]
- 3.Fagerholm SC, Guenther C, Llort Asens M, Savinko T, Uotila LM. Beta2-Integrins and interacting proteins in leukocyte trafficking, immune suppression, and immunodeficiency disease. Front Immunol. 2019;10:254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ley K, Laudanna C, Cybulsky MI, Nourshargh S. Getting to the site of inflammation: the leukocyte adhesion cascade updated. Nat Rev Immunol. 2007;7:678–689. [DOI] [PubMed] [Google Scholar]
- 5.Kolaczkowska E, Kubes P. Neutrophil recruitment and function in health and inflammation. Nat Rev Immunol. 2013;13:159–175. [DOI] [PubMed] [Google Scholar]
- 6.Alon R, Dustin ML. Force as a facilitator of integrin conformational changes during leukocyte arrest on blood vessels and antigen-presenting cells. Immunity. 2007;26:17–27. [DOI] [PubMed] [Google Scholar]
- 7.Alon R, Feigelson SW. Chemokine-triggered leukocyte arrest: force-regulated bi-directional integrin activation in quantal adhesive contacts. Curr Opin Cell Biol. 2012;24:670–676. [DOI] [PubMed] [Google Scholar]
- 8.Mcever RP, Cummings RD. Role of PSGL-1 binding to selectins in leukocyte recruitment. J Clin Invest. 1997;100:S97–103. [PubMed] [Google Scholar]
- 9.Sundd P, Gutierrez E, Pospieszalska MK, Zhang H, Groisman A, Ley K. Quantitative dynamic footprinting microscopy reveals mechanisms of neutrophil rolling. Nat Methods. 2010;7:821–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Xia L, Sperandio M, Yago T, et al. P-selectin glycoprotein ligand-1-deficient mice have impaired leukocyte tethering to E-selectin under flow. J Clin Invest. 2002;109:939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ramachandran V, Williams M, Yago T, Schmidtke DW, Mcever RP. Dynamic alterations of membrane tethers stabilize leukocyte rolling on P-selectin. Proc Natl Acad Sci U S A. 2004;101:13519–13524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sundd P, Gutierrez E, Koltsova EK, et al. ‘Slings’ enable neutrophil rolling at high shear. Nature. 2012;488:399–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stadtmann A, Germena G, Block H, et al. The PSGL-1-L-selectin signaling complex regulates neutrophil adhesion under flow. J Exp Med. 2013;210:2171–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marki A, Gutierrez E, Mikulski Z, Groisman A, Ley K. Microfluidics-based side view flow chamber reveals tether-to-sling transition in rolling neutrophils. Sci Rep. 2016;6:28870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Marki A, Buscher K, Mikulski Z, Pries A, Ley K. Rolling neutrophils form tethers and slings under physiologic conditions in vivo. J Leukoc Biol. 2018;103:67–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarbock A, Lowell CA, Ley K. Spleen tyrosine kinase Syk is necessary for E-selectin-induced alpha(L)beta(2) integrin-mediated rolling on intercellular adhesion molecule-1. Immunity. 2007;26:773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcR gamma to induce slow leukocyte rolling. J Exp Med. 2008;205:2339–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuwano Y, Spelten O, Zhang H, Ley K, Zarbock A. Rolling on E- or P-selectin induces the extended but not high-affinity conformation of LFA-1 in neutrophils. Blood. 2010;116:617–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefort CT, Rossaint J, Moser M, et al. Distinct roles for talin-1 and kindlin-3 in LFA-1 extension and affinity regulation. Blood. 2012;119:4275–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fan Z, Mcardle S, Marki A, et al. Neutrophil recruitment limited by high-affinity bent beta2 integrin binding ligand in cis. Nat Commun. 2016;7:12658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishida N, Xie C, Shimaoka M, Cheng Y, Walz T, Springer TA. Activation of leukocyte beta2 integrins by conversion from bent to extended conformations. Immunity. 2006;25:583–594. [DOI] [PubMed] [Google Scholar]
- 22.Graham GJ, Handel TM, Proudfoot AEI. Leukocyte adhesion: reconceptualizing chemokine presentation by glycosaminoglycans. Trends Immunol. 2019;40:472–481. [DOI] [PubMed] [Google Scholar]
- 23.Sanz M-J, Kubes P. Neutrophil-active chemokines in in vivo imaging of neutrophil trafficking. Eur J Immunol. 2012;42:278–283. [DOI] [PubMed] [Google Scholar]
- 24.Schaff UY, Dixit N, Procyk E, Yamayoshi I, Tse T, Simon SI. Orai1 regulates intracellular calcium, arrest, and shape polarization during neutrophil recruitment in shear flow. Blood. 2010;115:657–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fan Z, Kiosses WB, Sun H, et al. High-affinity bent beta2-integrin molecules in arresting neutrophils face each other through binding to ICAMs in cis. Cell Rep. 2019;26:119–130 e115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Block H, Stadtmann A, Riad D, et al. Gnb isoforms control a signaling pathway comprising Rac1, Plcbeta2, and Plcbeta3 leading to LFA-1 activation and neutrophil arrest in vivo. Blood. 2016;127:314–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Von Andrian UH, Chambers JD, Mcevoy LM, Bargatze RF, Arfors KE, Butcher EC. Two-step model of leukocyte-endothelial cell interaction in inflammation: distinct roles for LECAM-1 and the leukocyte beta 2 integrins in vivo. Proc Natl Acad Sci U S A. 1991;88:7538–7542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Divietro JA, Smith MJ, Smith BRE, Petruzzelli L, Larson RS, Lawrence MB. Immobilized IL-8 triggers progressive activation of neutrophils rolling in vitro on P-selectin and intercellular adhesion molecule-1. J Immunol. 2001;167:4017–4025. [DOI] [PubMed] [Google Scholar]
- 29.Smith CW, Marlin SD, Rothlein R, Toman C, Anderson DC. Cooperative interactions of LFA-1 and Mac-1 with intercellular adhesion molecule-1 in facilitating adherence and transendothelial migration of human neutrophils in vitro. J Clin Invest. 1989;83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts R, Hallett M. Neutrophil cell shape change: mechanism and signalling during cell spreading and phagocytosis. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li N, Yang H, Wang M, Lü S, Zhang Y, Long M. Ligand-specific binding forces of LFA-1 and Mac-1 in neutrophil adhesion and crawling. Mol Biol Cell. 2018;29:408–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Harding MG, Zhang K, Conly J, Kubes P. Neutrophil crawling in capillaries; a novel immune response to Staphylococcus aureus. PLoS Pathog. 2014;10:e1004379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phillipson M, Heit B, Parsons SA, et al. Vav1 is essential for mechanotactic crawling and migration of neutrophils out of the inflamed microvasculature. J Immunol. 2009;182:6870–6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phillipson M, Heit B, Colarusso P, Liu L, Ballantyne CM, Kubes P. Intraluminal crawling of neutrophils to emigration sites: a molecularly distinct process from adhesion in the recruitment cascade. J Exp Med. 2006;203:2569–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Honda M, Takeichi T, Hashimoto S, et al. Intravital imaging of neutrophil recruitment reveals the efficacy of FPR1 blockade in hepatic ischemia-reperfusion injury. J Immunol. 2017;198:1718–1728. [DOI] [PubMed] [Google Scholar]
- 36.Buffone A, Anderson NR, Hammer DA. Human neutrophils will crawl upstream on ICAM-1 if Mac-1 is blocked. Biophys J. 2019;117:1393–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Girbl T, Lenn T, Perez L, et al. Distinct compartmentalization of the chemokines CXCL1 and CXCL2 and the atypical receptor ACKR1 determine discrete stages of neutrophil diapedesis. Immunity. 2018;49:1062–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell D, Tauzin S, Hind LE, Deng Q, Beebe DJ, Huttenlocher A. Chemokine signaling and the regulation of bidirectional leukocyte migration in interstitial tissues. Cell Rep. 2017;19: 1572–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kienle K, Lämmermann T. Neutrophil swarming: an essential process of the neutrophil tissue response. Immunol Rev. 2016;273:76–93. [DOI] [PubMed] [Google Scholar]
- 40.Lämmermann T, Afonso PV, Angermann BR, et al. Neutrophil swarms require LTB4 and integrins at sites of cell death in vivo. Nature. 2013;498:371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chtanova T, Schaeffer M, Han S-Ji, et al. Dynamics of neutrophil migration in lymph nodes during infection. Immunity. 2008;29:487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hsu AY, Wang D, Liu S, et al. Phenotypical microRNA screen reveals a noncanonical role of CDK2 in regulating neutrophil migration. Proc Natl Acad Sci U S A. 2019;116:18561–18570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhou W, Hsu AY, Wang Y, et al. Mitofusin 2 regulates neutrophil adhesive migration and the actin cytoskeleton. J Cell Sci. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Deng Q, Sarris M, Bennin DA, Green JM, Herbomel P, Huttenlocher A. Localized bacterial infection induces systemic activation of neutrophils through Cxcr2 signaling in zebrafish. J Leukoc Biol. 2013;93:761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhou W, Pal AS, Hsu AYi-H, et al. MicroRNA-223 suppresses the canonical NF-kappaB pathway in basal keratinocytes to dampen neutrophilic inflammation. Cell Rep. 2018;22:1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Luo B-H, Carman CV, Springer TA. Structural basis of integrin regulation and signaling. Annu Rev Immunol. 2007;25:619–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robinson MK, Andrew D, Rosen H, et al. Antibody against the Leu-CAM beta-chain (CD18) promotes both LFA-1- and CR3-dependent adhesion events. J Immunol. 1992;148:1080–1085. [PubMed] [Google Scholar]
- 48.Lu C, Ferzly M, Takagi J, Springer TA. Epitope mapping of antibodies to the C-terminal region of the integrin beta 2 subunit reveals regions that become exposed upon receptor activation. J Immunol. 2001;166:5629–5637. [DOI] [PubMed] [Google Scholar]
- 49.Kamata T, Tieu KK, Tarui T, Puzon-Mclaughlin W, Hogg N, Takada Y. The role of the CPNKEKEC sequence in the beta(2) subunit I domain in regulation of integrin alpha(L)beta(2) (LFA-1). J Immunol. 2002;168:2296–2301. [DOI] [PubMed] [Google Scholar]
- 50.Lu C, Shimaoka M, Zang Q, Takagi J, Springer TA. Locking in alternate conformations of the integrin alphaLbeta2 I domain with disulfide bonds reveals functional relationships among integrin domains. Proc Natl Acad Sci U S A. 2001;98:2393–2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dransfield I, Hogg N. Regulated expression of Mg2+ binding epitope on leukocyte integrin alpha subunits. EMBO J. 1989;8:3759–3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Westermann B Mitochondrial fusion and fission in cell life and death. Nat Rev Mol Cell Biol. 2010;11:872–884. [DOI] [PubMed] [Google Scholar]
- 53.Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koshiba T Structural basis of mitochondrial tethering by mitofusin complexes. Science. 2004;305:858–862. [DOI] [PubMed] [Google Scholar]
- 55.De Brito OM, Scorrano L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature. 2008;456:605–610. [DOI] [PubMed] [Google Scholar]
- 56.Mazaki Y, Takada S, Nio-Kobayashi J, Maekawa S, Higashi T, Onodera Y, et al. Mitofusin 2 is involved in chemotaxis of neutrophil-like differentiated HL-60cells. Biochem Biophys Res Commun. 2019;513:708–713. [DOI] [PubMed] [Google Scholar]
- 57.Luchsinger LL, De Almeida MJ, Corrigan DJ, Mumau M, Snoeck H-W. Mitofusin 2 maintains haematopoietic stem cells with extensive lymphoid potential. Nature. 2016;529:528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schindelin J, Arganda-Carreras I, Frise E, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods. 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abadier M, Pramod AB, Mcardle S, et al. Effector and regulatory T cells roll at high shear stress by inducible tether and sling formation. Cell Rep. 2017;21:3885–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sun H, Fan Z, Gingras AR, Lopez-Ramirez MA, Ginsberg MH, Ley K. Frontline science: a flexible kink in the transmembrane domain impairs beta2 integrin extension and cell arrest from rolling. J Leukoc Biol. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Torres M, Hall FL, O’Neill K. Stimulation of human neutrophils with formyl-methionyl-leucyl-phenylalanine induces tyrosine phosphorylation and activation of two distinct mitogen-activated protein-kinases. J Immunol. 1993;150:1563–1577. [PubMed] [Google Scholar]
- 62.Černyšiov V, Mauricas M, Girkontaite I. Melatonin inhibits granulocyte adhesion to ICAM via MT3/QR2 and MT2 receptors. Int Immunol. 2015;27:599–608. [DOI] [PubMed] [Google Scholar]
- 63.Liu X, Ma Bo, Malik AB, et al. Bidirectional regulation of neutrophil migration by mitogen-activated protein kinases. Nat Immunol. 2012;13:457–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen C, Yang C, Barbieri JT. Staphylococcal Superantigen-like protein 11 mediates neutrophil adhesion and motility arrest, a unique bacterial toxin action. Sci Rep. 2019;9:4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Liu L, Aerbajinai W, Ahmed SM, Rodgers GP, Angers S, Parent CA. Radil controls neutrophil adhesion and motility through beta2-integrin activation. Mol Biol Cell. 2012;23:4751–4765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang M-J, Artemenko Y, Cai W-J, Iglesias PA, Devreotes PN. The directional response of chemotactic cells depends on a balance between cytoskeletal architecture and the external gradient. Cell Rep. 2014;9:1110–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yadav SK, Stojkov D, Feigelson SW, et al. Chemokine-triggered microtubule polymerization promotes neutrophil chemotaxis and invasion but not transendothelial migration. J Leukoc Biol. 2019;105:755–766. [DOI] [PubMed] [Google Scholar]
- 68.Leoni G, Gripentrog J, Lord C, et al. Human neutrophil formyl peptide receptor phosphorylation and the mucosal inflammatory response. J Leukoc Biol. 2015;97:87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lomakina EB, Waugh RE. Signaling and dynamics of activation of LFA-1 and Mac-1 by immobilized IL-8. Cell Mol Bioeng. 2010;3:106–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Yago T, Liu Z, Ahamed J, Mcever RP. Cooperative PSGL-1 and CXCR2 signaling in neutrophils promotes deep vein thrombosis in mice. Blood. 2018;132:1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schaff UY, Yamayoshi I, Tse T, Griffin D, Kibathi L, Simon SI. Calcium flux in neutrophils synchronizes beta2 integrin adhesive and signaling events that guide inflammatory recruitment. Ann Biomed Eng. 2008;36:632–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dorward DA, Lucas CD, Chapman GB, Haslett C, Dhaliwal K, Rossi AG. The role of formylated peptides and formyl peptide receptor 1 in governing neutrophil function during acute inflammation. Am J Pathol. 2015;185:1172–1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang S-C, Chung P-J, Ho C-M, et al. Propofol inhibits superoxide production, elastase release, and chemotaxis in formyl peptide-activated human neutrophils by blocking formyl peptide receptor 1. J Immunol. 2013;190:6511–6519. [DOI] [PubMed] [Google Scholar]
- 74.Vital SA, Becker F, Holloway PM, et al. Formyl-peptide receptor 2/3/Lipoxin A4 receptor regulates neutrophil-platelet aggregation and attenuates cerebral inflammation: impact for therapy in cardiovascular disease. Circulation. 2016;133:2169–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lind S, Gabl M, Holdfeldt A, et al. Identification of residues critical for FPR2 activation by the cryptic peptide mitocryptide-2 originating from the mitochondrial DNA-encoded cytochrome b. J Immunol. 2019;202:2710–2719. [DOI] [PubMed] [Google Scholar]
- 76.Laudanna C, Campbell JJ, Butcher EC. Elevation of intracellular cAMP inhibits RhoA activation and integrin-dependent leukocyte adhesion induced by chemoattractants. J Biol Chem. 1997;272:24141–24144. [DOI] [PubMed] [Google Scholar]
- 77.Lu C, Ferzly M, Takagi J, Springer TA. Epitope mapping of antibodies to the C-terminal region of the integrin beta2 subunit reveals regions that become exposed upon receptor activation. J Immunol. 2001;166:5629–5637. [DOI] [PubMed] [Google Scholar]
- 78.Shao B, Yago T, Coghill PA, et al. Signal-dependent slow leukocyte rolling does not require cytoskeletal anchorage of P-selectin glycoprotein ligand-1 (PSGL-1) or integrin alphaLbeta2. J Biol Chem. 2012;287:19585–19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abram CL, Lowell CA. The ins and outs of leukocyte integrin signaling. Annu Rev Immunol. 2009;27:339–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sun H, Fan Z, Gingras AR, Lopez-Ramirez MA, Ginsberg MH, Ley K. Frontline science: a flexible kink in the transmembrane domain impairs beta2 integrin extension and cell arrest from rolling. J Leukoc Biol. 2020;107:175–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Trayner ID, Bustorff T, Etches AE, Mufti GJ, Foss Y, Farzaneh F. Changes in antigen expression on differentiating HL60 cells treated with dimethylsulphoxide, all-trans retinoic acid, alpha1,25-dihydroxyvitamin D3 or 12-O-tetradecanoyl phorbol-13-acetate. Leuk Res. 1998;22:537–547. [DOI] [PubMed] [Google Scholar]
- 82.Elghetany MT, Patel J, Martinez J, Schwab H. CD87 as a marker for terminal granulocytic maturation: assessment of its expression during granulopoiesis. Cytometry B Clin Cytom. 2003;51:9–13. [DOI] [PubMed] [Google Scholar]
- 83.Hauert AB, Martinelli S, Marone C, Niggli V. Differentiated HL-60 cells are a valid model system for the analysis of human neutrophil migration and chemotaxis. Int J Biochem Cell Biol. 2002;34:838–854. [DOI] [PubMed] [Google Scholar]
- 84.Fleck RA, Romero-Steiner S, Nahm MH. Use of HL-60 cell line to measure opsonic capacity of pneumococcal antibodies. Clin Diagn Lab Immunol. 2005;12:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Skoge M, Wong E, Hamza B, et al. A worldwide competition to compare the speed and chemotactic accuracy of neutrophil-like cells. PLoS One. 2016;11:e0154491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rincón E, Rocha-Gregg BL, Collins SR. A map of gene expression in neutrophil-like cell lines. BMC Genomics. 2018;19:573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Birnie GD. The HL60 cell line: a model system for studying human myeloid cell differentiation. Br J Cancer Suppl. 1988;9:41–45. [PMC free article] [PubMed] [Google Scholar]
- 88.Smith HW, Marshall CJ. Regulation of cell signalling by uPAR. Nat Rev Mol Cell Biol. 2010;11:23–36. [DOI] [PubMed] [Google Scholar]
- 89.Sitrin RG, Todd RF, Albrecht E, Gyetko MR. The urokinase receptor (CD87) facilitates CD11b/CD18-mediated adhesion of human monocytes. J Clin Invest. 1996;97:1942–1951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu Xia, Borland G, Huang J, et al. Function of the lectin domain of Mac-1/complement receptor type 3 (CD11b/CD18) in regulating neutrophil adhesion. J Immunol. 2002;169:6417–6426. [DOI] [PubMed] [Google Scholar]
- 91.Gyetko MR, Sitrin RG, Fuller JA, Todd RF, Petty H, Standiford TJ. Function of the urokinase receptor (CD87) in neutrophil chemotaxis. J Leukoc Biol. 1995;58:533–538. [DOI] [PubMed] [Google Scholar]
- 92.Bohuslav J, Horejsí V, Hansmann C, et al. Urokinase plasminogen activator receptor, beta 2-integrins, and Src-kinases within a single receptor complex of human monocytes. J Exp Med. 1995;181:1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tang M-Li, Vararattanavech A, Tan S-M. Urokinase-type plasminogen activator receptor induces conformational changes in the integrin alphaMbeta2 headpiece and reorientation of its transmembrane domains. J Biol Chem. 2008;283:25392–25403. [DOI] [PubMed] [Google Scholar]
- 94.Simon DI, Wei Y, Zhang Li, et al. Identification of a urokinase receptor-integrin interaction site. Promiscuous regulator of integrin function. J Biol Chem. 2000;275:10228–10234. [DOI] [PubMed] [Google Scholar]
- 95.May AE, Kanse SM, Lund LR, Gisler RH, Imhof BA, Preissner KT. Urokinase receptor (CD87) regulates leukocyte recruitment via beta 2 integrins in vivo. J Exp Med. 1998;188:1029–1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gyetko MR, Sud S, Sonstein J, Polak T, Sud A, Curtis JL. Antigendriven lymphocyte recruitment to the lung is diminished in the absence of urokinase-type plasminogen activator (uPA) receptor, but is independent of uPA. J Immunol. 2001;167:5539–5542. [DOI] [PubMed] [Google Scholar]
- 97.Gyetko MR, Sud S, Kendall T, Fuller JA, Newstead MW, Standiford TJ. Urokinase receptor-deficient mice have impaired neutrophil recruitment in response to pulmonary Pseudomonas aeruginosa infection. J Immunol. 2000;165:1513–1519. [DOI] [PubMed] [Google Scholar]
- 98.Rijneveld AW, Levi M, Florquin S, Speelman P, Carmeliet P, Van Der Poll T. Urokinase receptor is necessary for adequate host defense against pneumococcal pneumonia. J Immunol. 2002;168:3507–3511. [DOI] [PubMed] [Google Scholar]
- 99.Van Zoelen MAD, Florquin S, De Beer R, et al. Urokinase plasminogen activator receptor-deficient mice demonstrate reduced hyperoxiainduced lung injury. Am J Pathol. 2009;174:2182–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Fan Z, Ley K. Leukocyte arrest: biomechanics and molecular mechanisms of beta2 integrin activation. Biorheology. 2015;52:353–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herter JM, Rossaint J, Block H, Welch H, Zarbock A. Integrin activation by P-Rex1 is required for selectin-mediated slow leukocyte rolling and intravascular crawling. Blood. 2013;121:2301–2310. [DOI] [PubMed] [Google Scholar]
- 102.Damoulakis G, Gambardella L, Rossman KL, et al. P-Rex1 directly activates RhoG to regulate GPCR-driven Rac signalling and actin polarity in neutrophils. J Cell Sci. 2014;127:2589–2600. [DOI] [PubMed] [Google Scholar]
- 103.Bolomini-Vittori M, Montresor A, Giagulli C, et al. Regulation of conformer-specific activation of the integrin LFA-1 by a chemokinetriggered Rho signaling module. Nat Immunol. 2009;10:185–194. [DOI] [PubMed] [Google Scholar]
- 104.Katagiri K, Shimonaka M, Kinashi T. Rap1-mediated lymphocyte function-associated antigen-1 activation by the T cell antigen receptor is dependent on phospholipase C-gamma1. J Biol Chem. 2004;279:11875–11881. [DOI] [PubMed] [Google Scholar]
- 105.Ghandour H, Cullere X, Alvarez A, Luscinskas FW, Mayadas TN. Essential role for Rap1 GTPase and its guanine exchange factor CalDAG-GEFI in LFA-1 but not VLA-4 integrin mediated human T-cell adhesion. Blood. 2007;110:3682–3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Chung K-J, Mitroulis I, Wiessner JR, et al. A novel pathway of rapid TLR-triggered activation of integrin-dependent leukocyte adhesion that requires Rap1 GTPase. Mol Biol Cell. 2014;25:2948–2955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lorenowicz MJ, Van Gils J, De Boer M, Hordijk PL, Fernandez-Borja M. Epac1-Rap1 signaling regulates monocyte adhesion and chemotaxis. J Leukoc Biol. 2006;80:1542–1552. [DOI] [PubMed] [Google Scholar]
- 108.Klapproth S, Sperandio M, Pinheiro EM, et al. Loss of the Rap1 effector RIAM results in leukocyte adhesion deficiency due to impaired beta2 integrin function in mice. Blood. 2015;126:2704–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Klapholz B, Brown NH. Talin - the master of integrin adhesions. J Cell Sci. 2017;130:2435–2446. [DOI] [PubMed] [Google Scholar]
- 110.Moser M, Bauer M, Schmid S, et al. Kindlin-3 is required for beta2 integrin-mediated leukocyte adhesion to endothelial cells. Nat Med. 2009;15:300–305. [DOI] [PubMed] [Google Scholar]
- 111.Wen L, Marki A, Roy P, et al. Kindlin-3 recruitment to the plasma membrane precedes high affinity beta2 integrin and neutrophil arrest from rolling. Blood. 2020. 10.1182/blood.2019003446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Manevich-Mendelson E, Feigelson SW, Pasvolsky R, et al. Loss of Kindlin-3 in LAD-III eliminates LFA-1 but not VLA-4 adhesiveness developed under shear flow conditions. Blood. 2009;114:2344–2353. [DOI] [PubMed] [Google Scholar]
- 113.Li Yu-J, Cao Yu-Lu, Feng J-X, et al. Structural insights of human mitofusin-2 into mitochondrial fusion and CMT2A onset. Nat Commun. 2019;10:4914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mcnew JA, Sondermann H, Lee T, Stern M, Brandizzi F. GTP-dependent membrane fusion. Annu Rev Cell Dev Biol. 2013;29:529–550. [DOI] [PubMed] [Google Scholar]
- 115.Jezyk MR, Snyder JT, Gershberg S, Worthylake DK, Harden TK, Sondek J. Crystal structure of Rac1 bound to its effector phospholipase C-beta2. Nat Struct Mol Biol. 2006;13:1135–1140. [DOI] [PubMed] [Google Scholar]
- 116.Karbowski M, Youle RJ. Dynamics of mitochondrial morphology in healthy cells and during apoptosis. Cell Death Differ. 2003;10:870–880. [DOI] [PubMed] [Google Scholar]
- 117.Yan H, Qiu C, Sun W, et al. Yap regulates gastric cancer survival and migration via SIRT1/Mfn2/mitophagy. Oncol Rep. 2018;39: 1671–1681. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon request.