

Abstract
Background
Emicizumab is a subcutaneously administered humanized, bispecific, monoclonal antibody approved for prophylaxis in people with hemophilia A.
Methods
HAVEN 5 (NCT03315455) is a randomized, open‐label, phase 3 study of individuals aged ≥12 years with severe hemophilia A without factor VIII (FVIII) inhibitors, or hemophilia A of any severity with FVIII inhibitors, across the Asia‐Pacific region. Participants were randomly assigned (2:2:1) to receive emicizumab 1.5 mg/kg once weekly (arm A), emicizumab 6 mg/kg every 4 weeks (arm B), or no prophylaxis (arm C). The primary end point was annualized bleeding rate (ABR) for treated bleeds; ABRs were compared between people receiving emicizumab prophylaxis versus those with no prophylaxis. Secondary end points included ABR for treated target joint bleeds. Safety was also evaluated.
Results
From April 26, 2018, to January 4, 2019, 70 of 76 screened participants were enrolled and randomized (arm A, n = 29; arm B, n = 27; arm C, n = 14). ABRs (95% confidence interval) for treated bleeds and treated target joint bleeds, respectively, were: arm A, 1.0 (0.53‐1.85) and 0.4 (0.18‐1.09); arm B, 1.0 (0.50‐1.84) and 0.3 (0.12‐0.85); arm C, 27.0 (13.29‐54.91) and 8.6 (3.15‐23.42). The most common adverse event, upper respiratory tract infection, was reported for 14 of 56 (25.0%; emicizumab) and 2 of 14 (14.3%; no prophylaxis) participants. No thrombotic events, thrombotic microangiopathies, or deaths were reported.
Conclusion
Emicizumab 1.5 mg/kg once weekly and 6 mg/kg every 4 weeks demonstrated bleed control in this study population, was well tolerated, and could improve use of prophylaxis in people with hemophilia A.
Keywords: clinical trials, emicizumab, factor VIII, hemophilia A, prophylaxis, randomized
Essentials.
Hemophilia A treatment is limited in developing countries of the Asia‐Pacific region.
HAVEN 5 evaluates emicizumab prophylaxis in people with hemophilia A in the Asia‐Pacific region.
Emicizumab demonstrated bleed control and was well tolerated in this study population.
Emicizumab could improve use of prophylaxis in people with hemophilia A from the Asia‐Pacific region.
1. INTRODUCTION
Hemophilia A is a congenital disorder associated with bleeding caused by a deficiency in coagulation factor FVIII (FVIII). 1 Prophylaxis is the current standard of care for people with hemophilia A with a severe phenotype 2 ; however, treatment burden with FVIII prophylaxis can be high due to the requirement for frequent intravenous administration (>1 per week), 3 and still, unsatisfactory bleed control. 4 Furthermore, up to 30% of people with hemophilia A treated with factor products develop FVIII inhibitors, neutralizing the function of infused FVIII and further complicating treatment management. 5 Disease burden and treatment challenges, therefore, can be greater in those who develop FVIII inhibitors. 6 Bypassing agents (BPAs) are options for people with hemophilia A with FVIII inhibitors 5 ; however, suboptimal efficacy and treatment burden can remain an issue, ultimately impacting patient management. 7
In developing countries of the Asia‐Pacific region, the disease burden associated with hemophilia A is greater compared with Western countries, with the limited use of prophylaxis being one of the main contributing factors. 8 , 9 The Asia‐Pacific Hemophilia Working Group has developed a Principles of Hemophilia Care document, taking into account variability in regional health care systems and the socioeconomic and cultural diversities, in an effort to drive forward hemophilia care in the region. 10 There remains, however, a high unmet need in reducing disease burden for people with hemophilia A in the Asia‐Pacific region.
Emicizumab is a humanized, bispecific, monoclonal antibody that bridges activated factor IX (FIXa) and factor X (FX), replacing the function of missing activated FVIII and restoring hemostasis in people with hemophilia A. 11 Its long half‐life 12 allows for dosing regimens of once weekly, every 2 weeks, or every 4 weeks. Coupled with a subcutaneous route of administration and high bioavailability, 13 these dosing regimens could improve adherence to prophylactic therapy and offer people with hemophilia A a less burdensome treatment relative to traditional factor products.
The efficacy and favorable safety profile of emicizumab in people with hemophilia A aged ≥12 years, regardless of FVIII inhibitor status, were demonstrated in 3 pivotal phase 3 studies (HAVEN 1 [once weekly], 14 HAVEN 3 [once weekly/every 2 weeks], 15 and HAVEN 4 [every 4 weeks] 16 ), with similar findings demonstrated for individuals aged <12 years (HAVEN 2 17 and HOHOEMI 18 ). A pooled analysis of the HAVEN 1 to 4 studies demonstrated long‐term efficacy and tolerability across a median (interquartile range [IQR]) efficacy period of 120 (89‐164) weeks. 19
HAVEN 5 (NCT03315455) was designed to evaluate the efficacy, safety, immunogenicity, and pharmacokinetic (PK) profile of 1.5 mg/kg once weekly and 6 mg/kg every 4 weeks emicizumab in people with hemophilia A in the Asia‐Pacific region. Here, we report primary and select secondary outcome data after ≥24 weeks of treatment during the ongoing HAVEN 5 trial.
2. METHODS
2.1. Study design
HAVEN 5 is a randomized, multicenter, open‐label, phase 3 clinical study (Figure 1) conducted at 13 sites across China, Malaysia, and Thailand; a full list of sites is provided in Table S1.
FIGURE 1.
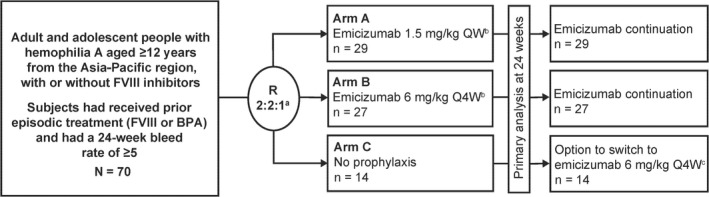
HAVEN 5 study design. BPA, bypassing agent; F, factor; QW, once weekly; Q4W, once every 4 weeks; R, randomization. aRandomization was stratified based on the number of bleeds in the 24 weeks before study start (<9 or ≥9). bEmicizumab was administered at a loading dose of 3 mg/kg once weekly for 4 weeks before the maintenance dose indicated. cEmicizumab was administered at a loading dose of 3 mg/kg once weekly for 4 weeks before 6 mg/kg every 4 weeks maintenance dosing
Study participants were randomized to 3 treatment arms: emicizumab 3 mg/kg once weekly for the first 4 weeks (loading dose) followed by a maintenance dose of either 1.5 mg/kg once weekly (arm A) or 6 mg/kg every 4 weeks (arm B), or no prophylaxis (arm C) (Figure 1). After completing 24 weeks of study, participants randomized to arm C could switch to emicizumab (3 mg/kg once weekly loading dose for 4 weeks followed by a maintenance regimen of 6 mg/kg every 4 weeks). After at least 24 weeks of emicizumab prophylaxis, participants could continue taking maintenance therapy (1.5 mg/kg once weekly or 6 mg/kg every 4 weeks) or, if they had suboptimal control of bleeding, defined as ≥2 spontaneous and clinically significant bleeding events during the prior 24 weeks of emicizumab administration, both occurring after the end of the loading‐dose period, change to an increased dose of 3 mg/kg once weekly.
The study was conducted in compliance with the International Conference on Harmonisation Guidelines for Good Clinical Practice, the principles of the Declaration of Helsinki, and all informed consent guidelines; the protocol was approved by the institutional review boards/independent ethics committees at each participating site and carried out in accordance with applicable regulations.
2.2. Participants
People with hemophilia A with or without FVIII inhibitors were enrolled in HAVEN 5. Since emicizumab efficacy is similar irrespective of FVIII inhibitor status and age, 19 and to ensure a representative range of people with hemophilia A in this study, enrollment of up to 55 noninhibitor participants was permitted.
Participants were aged ≥12 years with a diagnosis of severe hemophilia A (intrinsic FVIII level <1%) or hemophilia A with FVIII inhibitors, and were required to have documented ≥5 bleeds and use of episodic therapy (FVIII or BPAs) in the 24 weeks before study entry to be eligible for inclusion. Participants without FVIII inhibitors (<0.6 BU/mL) who successfully completed immune tolerance induction (ITI) therapy were required to have done so ≥5 years before screening and have no evidence of permanent/temporary inhibitor recurrence since ITI (>0.6 BU/mL). Those with inherited or acquired bleeding disorders other than hemophilia A, at high risk for thrombotic microangiopathy (TMA), and/or with signs of thrombotic events (TEs) or previous/current treatment for TE were excluded. Full inclusion/exclusion criteria are listed in Appendix S1.
All adult participants provided written informed consent before study start; parents/legally authorized representatives of adolescents provided written informed consent, and adolescents provided informed assent.
2.3. Randomization
Participants were randomly assigned centrally (2:2:1) to arm A, B, or C using an interactive voice/web response system. To ensure a balance of participants by bleed frequency across study arms, block‐based randomization was used to stratify participants according to the number of bleeds (<9 or ≥9) in the 24 weeks before study entry.
2.4. Procedures
Emicizumab was administered subcutaneously by/under the observation of a health care professional, after which self‐administration was performed at home. Bleed data (including start date and type) and hemophilia‐related medication use data were entered on an electronic handheld device (cell phone) by the participant via the Blood and Medication Questionnaire (BMQ; developed and validated by the sponsor). Participants were required to complete the BMQ at the start of the week 1 study visit and once a bleed was managed; data were recorded weekly (including confirmation of no bleeds) and could be also entered for the previous 7 days. This retrospective collection of data was considered acceptable in terms of recall bias and was permitted to optimize completeness of data collection.
Participant‐reported health‐related quality of life (HRQoL) and health status data were captured by an electronic handheld device (tablet) at weeks 1, 13, and 25. HRQoL was measured using the Haemophilia Quality of Life Questionnaire for Adults (Haem‐A‐QoL; for participants aged ≥18 years) and Haemophilia Quality of Life Questionnaire for Adolescents (Haemo‐QoL‐SF; for participants aged 12‐17 years). Health status was measured using the EuroQoL‐5 Dimensions‐5 Levels Questionnaire (EQ‐5D‐5L), which consists of two parts: (i) an Index Utility Scale (IUS) and (ii) a Visual Analog Scale (VAS). Details on Haem‐A‐QoL and EQ‐5D‐5L domains and scales are provided in Appendix S1.
Safety assessments consisted of monitoring adverse events (AEs), serious AEs (SAEs), injection‐site reactions (ISRs), and AEs of special interest (AESIs; specifically, TEs, TMA, and hypersensitivity/anaphylactic/anaphylactoid reaction), development of antidrug (emicizumab) antibodies (ADAs), and protocol‐specified vital signs, electrocardiograms, and laboratory assessments. AEs were recorded on an electronic case report form using the standardized Medical Dictionary for Regulatory Activities (version 22.0); terms were coded and tabulated by System Organ Class. Additional details are provided in the supplement.
Plasma samples were collected at prespecified time points for analysis of emicizumab exposure (once weekly regimen: every week during weeks 1‐4, every 2 weeks during weeks 5‐8, and every 4 weeks during weeks 9‐24; every‐4‐week regimen: every week during weeks 1‐4 and every 4 weeks during weeks 5‐24). Previously reported validated ELISAs were used to detect ADA development 20 and analyze emicizumab plasma concentrations 21 and were conducted by QPS Netherlands B.V (Groningen, the Netherlands).
2.5. Outcomes
The primary efficacy end point was annualized bleeding rate (ABR) for treated bleeds in people with hemophilia A receiving once‐weekly or every‐4‐weeks emicizumab prophylaxis or no prophylaxis (see Appendix S1 for definition of treated bleeds). Secondary efficacy end points were ABRs for all bleeds and treated spontaneous/joint/target joint bleeds in participants receiving once‐weekly or every‐4‐weeks emicizumab prophylaxis versus no prophylaxis. Bleeds were counted as one bleed if they were of the same type and occurred at the same anatomic location within 72 hours after stopping treatment for the first bleed (the “72‐hour rule”) 22 ; bleeds due to procedure/surgery were excluded. As per ISTH definition, target joints were defined as major joints in which ≥3 bleeding events occurred over a 24‐week period. 23 Change from baseline in HRQoL and health status after 24 weeks of emicizumab prophylaxis versus no prophylaxis was also evaluated.
Incidence of AEs, SAEs, AEs leading to treatment discontinuation/modification/interruption, local ISRs, AESIs, and ADAs were used to assess safety. Exposure (trough plasma concentration) following emicizumab once weekly and every 4 weeks was characterized via PK sampling.
2.6. Statistical analysis
Assuming a randomization ratio of 2:2:1, a sample size of N = 70 participants (28 participants each in arms A and B and 14 in arm C) would achieve a power of >90% at the two‐sided 0.05 level of significance. This calculation assumes mean ABRs of 4 and 18 for emicizumab prophylaxis (arms A and B) and no prophylaxis (arm C), respectively, representing an expected 78% reduction in the ABR with prophylaxis.
Participant demographics and clinical characteristics are reported using descriptive statistics including mean, standard deviation (SD), median, and IQR. Primary and secondary efficacy analyses were performed in the intent‐to‐treat (ITT) population, which comprised people with hemophilia A with/without FVIII inhibitors. Similar PK, and relationship between PK and bleed frequency, have been previously observed in inhibitor and noninhibitor populations. 14 , 15 , 24
Formal hypothesis testing was conducted for the randomized comparisons of arm A/B versus arm C; for primary and bleed‐related secondary end points, a model‐based comparison of the number of bleeds over the study period in arms A/B compared with arm C was performed using a negative‐binomial regression model, which takes into account the varying follow‐up time for each individual. Statistical testing at the prespecified α level was based on the Wald test. Bleed rates for emicizumab and no prophylaxis groups, and rate ratio (quantifies the risk of bleeding associated with emicizumab versus no prophylaxis) including 95% confidence intervals (CIs) are described. ABR = (number of bleeds/total number of days during the efficacy period) × 365.25 was used to calculate median (IQR) and mean (95% CI) ABRs. Type 1 errors for secondary end points were controlled through a hierarchical testing framework.
Safety data were collected throughout the study and are described using numbers and percentages, and laboratory data were summarized descriptively over time, although not all ADA and PK data were available at the clinical cutoff date. Immunogenicity data were summarized using standard language/terminology as per Shankar et al. 25 Trough emicizumab plasma concentrations were analyzed in participants who received ≥1 emicizumab dose and had ≥1 postbaseline concentration measurement. Details of the methodology for subgroup and HRQoL statistical analyses are provided in the supplement. Data were analyzed using SAS software, version 9.4 of the SAS System for Unix (SAS Institute, Cary, NC, USA).
All authors had access to the data and confirm adherence to the statistical analysis plan.
This study is registered with ClinicalTrials.gov (NCT03315455).
3. RESULTS
From April 26, 2018, to January 4, 2019, a total of 76 people with hemophilia A were screened for eligibility, and 70 were enrolled in this ongoing study (Figure 2). Of the six participants who failed screening, four did not meet inclusion criteria (diagnosis of severe congenital hemophilia A or hemophilia A with FVIII inhibitors, n = 2; documentation of episodic therapy and number of bleeds in the past 24 weeks, n = 1; adequate hepatic function, n = 1), and two were ineligible for other reasons. No individuals who met the eligibility criteria declined to participate in the study. Participants were randomized 2:2:1 to arms A (n = 29), B (n = 27), and C (n = 14), respectively. After completing 24 weeks of study, 13 participants in arm C switched to receive emicizumab 6 mg/kg every 4 weeks; one participant received emicizumab 1.5 mg/kg once weekly due to an AE of headache that occurred when receiving the 3‐mg/kg loading dose. This grade 1 (nonserious) AE was considered to be treatment related by the investigator. Treatment dose for one participant (arm B) was up‐titrated to 3 mg/kg once weekly at week 25 following suboptimal bleeding control (ie, ≥2 spontaneous and clinically significant bleeds within 24 weeks).
FIGURE 2.
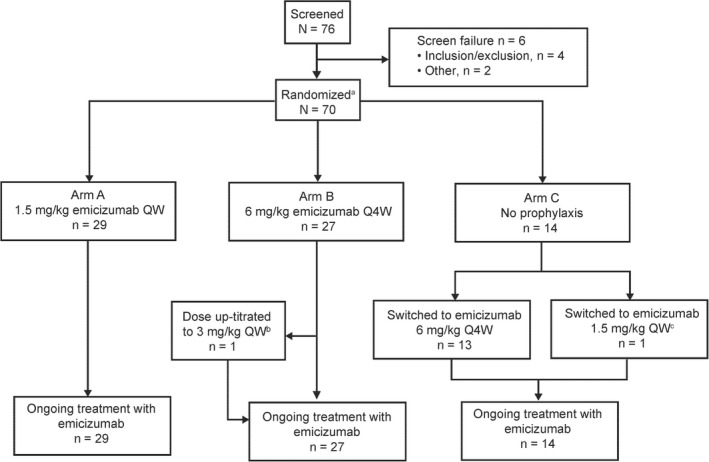
Participant disposition. QW, once weekly; Q4W, once every 4 weeks. aParticipants randomized (2:2:1) to arms A, B, and C. bThis participant was up‐titrated to 3 mg/kg once weekly at week 25 following suboptimal bleeding control (ie, ≥2 spontaneous and clinically significant bleeds within 24 weeks). cOne participant switched from emicizumab 6 mg/kg every 4 weeks to 1.5 mg/kg once weekly at the discretion of the investigator and medical director
As of the clinical cutoff date for the reported analysis (June 21, 2019), all randomized participants (n = 70) had completed at least 24 weeks on study and all continued emicizumab. The ITT and safety populations included all randomized participants. PK and ADA samples for six participants were not analyzed due to administrative reasons; as such, the PK‐/ADA‐evaluable populations included only 64 participants. The majority of participants in the ITT population (≥85.7% in all treatment arms) completed all scheduled assessments of the BMQ, Haem‐A‐QoL, and EQ‐5D‐5L. However, only 63.6% (7 of 11 participants aged 12‐17 years) completed all Haemo‐QoL‐SF assessments; therefore, these data are not presented.
Baseline demographics and disease characteristics are shown in Table 1. All participants were male, with a median (range) age of 29.0 (12‐66) years, and most were Chinese (85.7%). The majority of participants had severe hemophilia A (95.7%) and target joints (74.3%) at baseline. The median (range) number of bleeds in the 24 weeks before study entry was 14.5 (5‐39). At baseline, a total of 16 (22.9%) participants presented with FVIII inhibitors and 54 (77.1%) without inhibitors; further, 3 three participants with FVIII inhibitors had previously undergone ITI therapy (arm B, n = 2; arm C, n = 1).
TABLE 1.
Participant demographics and disease characteristics at baseline (all randomized participants)
| Arm A (emicizumab 1.5 mg/kg once weekly) (n = 29) | Arm B (emicizumab 6 mg/kg every 4 weeks) (n = 27) | Arm C (no prophylaxis) (n = 14) | Total (N = 70) | |
|---|---|---|---|---|
| Age, y | ||||
| Median (range) | 31.0 (12‐57) | 28.0 (13‐66) | 26.5 (13‐46) | 29.0 (12‐66) |
| <18, n (%) | 3 (10.3) | 6 (22.2) | 2 (14.3) | 11 (15.7) |
| ≥18‐64, n (%) | 26 (89.7) | 20 (74.1) | 12 (85.7) | 58 (82.9) |
| ≥65, n (%) | 0 (0.0) | 1 (3.7) | 0 (0.0) | 1 (1.4) |
| Race, n (%) | ||||
| Chinese | 25 (86.2) | 21 (77.8) | 14 (100) | 60 (85.7) |
| Non‐Chinese | 4 (13.8) | 6 (22.2) | 0 (0.0) | 10 (14.3) |
| Hemophilia severity at baseline, n (%) | ||||
| Mild | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Moderate a | 2 (6.9) | 0 (0.0) | 1 (7.1) | 3 (4.3) |
| Severe | 27 (93.1) | 27 (100) | 13 (92.9) | 67 (95.7) |
| FVIII inhibitor status, n (%) | ||||
| Yes | 6 (20.7) | 7 (25.9) | 3 (21.4) | 16 (22.9) |
| No | 23 (79.3) | 20 (74.1) | 11 (78.6) | 54 (77.1) |
| Participants with inhibitors previously treated with ITI, n (%) | 0 (0.0) | 2 (28.6) | 1 (33.3) | 3 (18.8) |
| Bleeds in 24 weeks before study entry | ||||
| Median (range) | 14.0 (5‐28) | 14.0 (5‐39) | 19.5 (6‐34) | 14.5 (5‐39) |
| <9, n (%) | 7 (24.1) | 6 (22.2) | 3 (21.4) | 16 (22.9) |
| ≥9, n (%) | 22 (75.9) | 21 (77.8) | 11 (78.6) | 54 (77.1) |
| Target joints, n (%) | ||||
| Yes | 20 (69.0) | 20 (74.1) | 12 (85.7) | 52 (74.3) |
| >1 | 13/20 (65.0) | 14/20 (70.0) | 8/12 (66.7) | 35/52 (67.3) |
FVIII, factor VIII; ITI, immune tolerance induction.
All participants with moderate phenotypes had FVIII inhibitors.
The median (IQR) efficacy period was 43.7 (36.14‐48.43) weeks for arm A (1.5 mg/kg once weekly), 46.1 (36.71‐49.29) weeks for arm B (6 mg/kg every 4 weeks), and 24.0 (24.00‐24.29) weeks for arm C (no prophylaxis). The primary efficacy end point was met for both emicizumab regimens; following emicizumab prophylaxis, the model‐based ABR (95% CI) for treated bleeds was 1.0 (0.53‐1.85) for arm A and 1.0 (0.50‐1.84) for arm B, compared with 27.0 (13.29‐54.91) for arm C. Therefore, a statistically significant and clinically meaningful reduction of 96% in ABR for treated bleeds was observed for both emicizumab once weekly and every 4 weeks compared with no prophylaxis (both P < .0001; Figure 3A). The participant in arm B whose dose was up‐titrated from 6 mg/kg every 4 weeks to 3 mg/kg once weekly had an ABR for treated bleeds of 4.4 before up‐titration and 0.0 after up‐titration. In arms A and B, 65.5% (n = 19/29) and 55.6% (n = 15/27) of participants had zero treated bleeds, respectively, versus 17.1% (n = 1/14) in arm C (Figure 3B). The proportion of participants with zero to three treated bleeds was 86.2% and 92.6% for arms A and B, respectively, compared with 7.1% in arm C. Subgroup analyses are detailed in Appendix S1 and in Figure S1.
FIGURE 3.
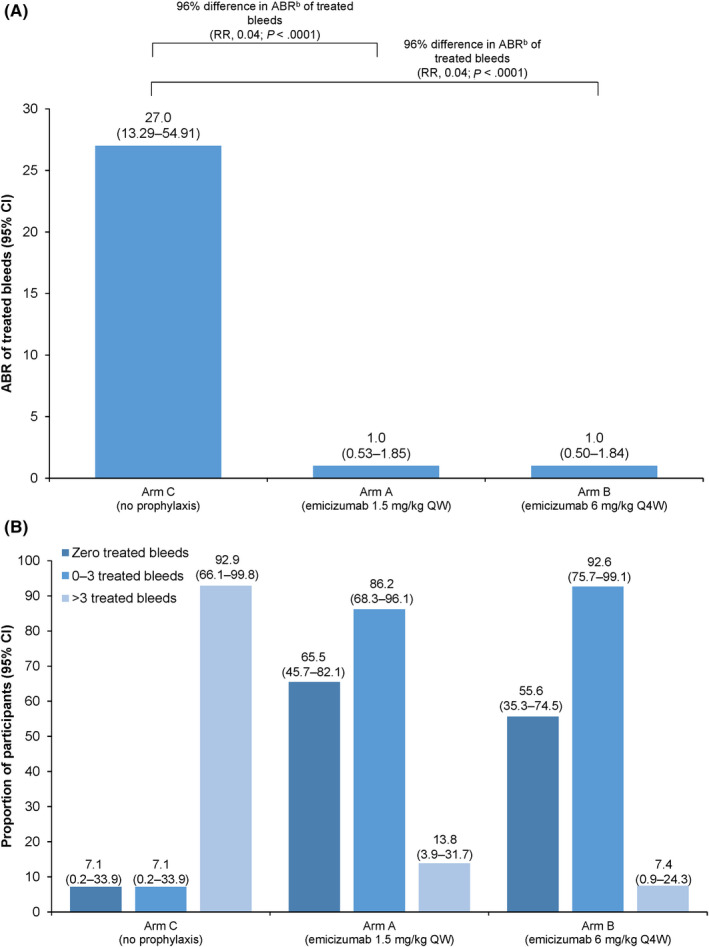
Randomized comparison of treateda bleeds. (A) Model‐based ABRs and (B) proportion of participants with 0, 0–3, and >3 bleeds (primary efficacy end point; ITT population). Model‐based ABRs were calculated using a negative binomial regression model. ABRs were calculated by (Number of bleeds/total number of days during the efficacy period) × 365.25. P values were obtained via a global model with a three‐level categorical effect for treatment. ABR, annualized bleeding rate; CI, confidence interval; ITT, intent‐to‐treat; QW, once weekly; Q4W, once every 4 weeks; RR, rate ratio. aTreated bleeds are defined as a bleed followed by treatment for a bleed; bleeds due to surgery/procedure were excluded. bModel‐based ABR
The other bleed‐related end points were also met for both emicizumab regimens compared with no prophylaxis (Table 2); statistically significant reductions of ≥95% in ABRs were observed (P < .0001). In both arms A and B, calculated median ABRs were zero for treated spontaneous bleeds, treated joint bleeds, and treated target joint bleeds (Table 2). A total of 24 (82.8%) participants in arm A and 19 (70.4%) in arm B had zero treated target joint bleeds, compared with 4 (28.6%) in arm C. Emicizumab was also efficacious among those with/without FVIII inhibitors compared with no prophylaxis (Table S2).
TABLE 2.
Bleed‐related secondary efficacy end points (ITT population)
| Arm A (emicizumab 1.5 mg/kg once weekly) (n = 29) | Arm B (emicizumab 6 mg/kg every 4 weeks) (n = 27) | Arm C (no prophylaxis) (n = 14) | |
|---|---|---|---|
| Median (IQR) efficacy period, weeks | 43.7 (36.1‐48.4) | 46.1 (36.7‐49.3) | 24.0 (24.0‐24.3) |
| All bleeds | |||
| Model‐based ABR a (95% CI) | 1.9 (1.23‐2.97) | 2.1 (1.33‐3.26) | 41.1 (26.37‐64.19) |
| % reduction vs arm C (P value) b | 95 (<.0001) | 95 (<.0001) | … |
| Median ABR c (IQR) | 1.5 (0.0‐4.2) | 1.9 (0.0‐5.6) | 56.7 (26.1‐70.8) |
| Mean ABR (95% CI) | 2.7 (0.51‐8.37) | 3.1 (0.67‐8.94) | 53.0 (39.71‐69.33) |
| % participants with zero bleeds (95% CI) | 37.9 (20.7‐57.7) | 33.3 (16.5‐54.0) | 0 (0.0‐23.2) |
| Treated spontaneous bleeds | |||
| Model‐based ABR a (95% CI) | 0.4 (0.18‐0.96) | 0.5 (0.20‐1.12) | 23.6 (9.28‐60.03) |
| % reduction vs arm C (P value) b | 98 (<.0001) | 98 (<.0001) | … |
| Median ABR c (IQR) | 0.0 (0.0‐0.0) | 0.0 (0.0‐1.0) | 21.8 (6.5‐52.2) |
| Mean ABR (95% CI) | 0.5 (0.0‐4.66) | 0.6 (0.0‐4.88) | 30.9 (20.95‐43.85) |
| % participants with zero bleeds (95% CI) | 82.8 (64.2‐94.2) | 74.1 (53.7‐88.9) | 14.3 (1.8‐42.8) |
| Treated joint bleeds | |||
| Model‐based ABR a (95% CI) | 0.7 (0.36‐1.46) | 0.6 (0.28‐1.22) | 17.7 (8.33‐37.57) |
| % reduction vs arm C (P value) b | 96 (<.0001) | 97 (<.0001) | … |
| Median ABR c (IQR) | 0.0 (0.0‐0.0) | 0.0 (0.0‐1.4) | 10.9 (8.7‐50.0) |
| Mean ABR (95% CI) | 1.0 (0.02‐5.57) | 0.8 (0.01‐5.20) | 25.5 (16.62‐37.56) |
| % participants with zero bleeds (95% CI) | 75.9 (56.5‐89.7) | 59.3 (38.8‐77.6) | 7.1 (0.2‐33.9) |
| Treated target joint bleeds | |||
| Model‐based ABR a (95% CI) | 0.4 (0.18‐1.09) | 0.3 (0.12‐0.85) | 8.6 (3.15‐23.42) |
| % reduction vs arm C (P value) b | 95 (<.0001) | 96 (<.0001) | … |
| Median ABR c (IQR) | 0.0 (0.0‐0.0) | 0.0 (0.0‐1.1) | 6.5 (0.0‐19.7) |
| Mean ABR (95% CI) | 0.7 (0.0‐5.06) | 0.5 (0.0‐4.76) | 15.6 (8.83‐25.47) |
| % participants with zero bleeds (95% CI) | 82.8 (64.2‐94.2) | 70.4 (49.8‐86.2) | 28.6 (8.4‐58.1) |
A treated bleed is defined as a bleed followed by treatment for a bleed; bleeds due to surgery/procedure were excluded. A target joint is defined as a joint in which ≥3 bleeds occurred during the 24 weeks before study entry; bleeds due to surgery/procedure were excluded.
ABR, annualized bleeding rate; CI, confidence interval; IQR, interquartile range; ITT, intent‐to‐treat.
Calculated using a negative binomial regression model.
P values were obtained via a global model with a three‐level categorical effect for treatment.
Calculated by (Number of bleeds/total number of days during the efficacy period) × 365.25.
Mean (95% CI) Haem‐A‐QoL physical health score and total score decreased from baseline with emicizumab prophylaxis, indicating improvement in HRQoL (physical health: arm A, −20.20 [−12.02 to −28.38]; arm B, −22.14 [−14.82 to −29.47]; arm C, −5.63 [−6.08 to −17.33]; total score: arm A, −10.14 [−3.46 to −16.81]; arm B, −17.61 [−10.96 to −24.25]; arm C, −2.50 [−3.74 to −8.75]). Mean (SD) VAS and IUS scores increased slightly from baseline for arm A (by 4.82 [19.14] and 0.08 [0.22], respectively) and arm B (by 7.40 [16.67] and 0.08 [0.21], respectively); smaller increases were observed for arm C (VAS, 2.00 [13.27]; IUS, 0.02 [0.09]). Mean physical health, total score, IUS, and VAS scores are shown in Figure S2; see Supporting Information for additional HRQoL and health status data.
In total, 185 AEs (arm A, n = 109; arm B, n = 76) were reported for a total of 44 participants following prophylaxis and 3 AEs for two participants in arm C following no prophylaxis (Table 3). The majority (78.6%) of emicizumab‐treated participants reported at least 1 AE compared with 14.3% of participants not taking prophylaxis (Table 3); the majority of AEs were grade 1 or 2. Upper respiratory tract infection was the most commonly reported AE (arm A, n = 9 participants [31.0%]; arm B, n = 5 [18.5%]; arm C, n = 2 [14.3%]). Local ISRs were reported for 13.8%, 18.5%, and 0% of participants in arms A, B, and C, respectively.
TABLE 3.
Safety summary
| Arm A (emicizumab 1.5 mg/kg once weekly) (n = 29) | Arm B (emicizumab 6 mg/kg every 4 weeks) (n = 27) | Arm C (no prophylaxis) (n = 14) | Arm C a (emicizumab 6 mg/kg every 4 weeks) (n = 14) | |
|---|---|---|---|---|
| Median (range) duration of exposure, weeks b | 43.1 (28.1‐60.1) | 44.1 (20.1‐56.6) | NA c | 18.3 (4.1‐32.1) |
| Total number of AEs | 109 | 76 | 3 | 26 |
| Participants with ≥1 AE, n (%) | 25 (86.2) | 19 (70.4) | 2 (14.3) | 10 (71.4) |
| AE with fatal outcome | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| SAE | 2 (6.9) | 1 (3.7) | 0 (0.0) | 0 (0.0) |
| AE leading to withdrawal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| AE leading to dose modification/interruption | 2 (6.9) | 0 (0.0) | 0 (0.0) | 2 (14.3) |
| Grade ≥3 AE | 3 (10.3) | 1 (3.7) | 0 (0.0) | 0 (0.0) |
| Treatment‐related AE d | 12 (41.4) | 10 (37.0) | 0 (0.0) | 5 (35.7) |
| Local ISR | 4 (13.8) | 5 (18.5) | 0 (0.0) | 0 (0.0) |
| Participants with ≥1 AESI, n (%) | ||||
| Systemic hypersensitivity/anaphylactic/anaphylactoid reaction e | 0 (0.0) | 1 (3.7) | 0 (0.0) | 0 (0.0) |
| TE | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| TMA | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
AE, adverse event; AESI, adverse event of special interest; ALT, alanine aminotransferase; AST, aspartate aminotransferase; ISR, injection‐site reaction; NA, not applicable; SAE, serious adverse event; TE, thromboembolic event; TMA, thrombotic microangiopathy.
Includes emicizumab prophylaxis period only.
Treatment duration is the date of the last dose of study medication minus the date of the first dose plus 1 day.
Participants in arm C (no prophylaxis) were monitored for ≈24 weeks before switching to emicizumab prophylaxis.
ISRs were the most common emicizumab‐related AEs (12.9% of all emicizumab‐treated participants), followed by elevated AST (8.6%), elevated ALT (8.6%), dizziness (4.3%), and headache (4.3%).
Assessed using Sampson criteria and includes all participants who experienced indicative symptoms. One participant in arm B was identified through algorithmic analysis as potentially having a systemic hypersensitivity/anaphylactic/anaphylactoid reaction; however, medical review of the case showed that Sampson criteria were not met.
The safety profile for all emicizumab‐treated participants, across arms A through C (after participants randomly assigned to arm C switched to emicizumab after 24 weeks of no prophylaxis), was generally consistent (Table 3). Treatment‐related AEs were reported for 38.6% of all emicizumab‐treated participants. AEs reported by ≥5% of all emicizumab‐treated participants are summarized in Table S3.
A total of four SAEs were reported for three participants (arm A, n = 2 [6.9%]; arm B, n = 1 [3.7%]), none of which were reported to be related to study treatment: intra‐abdominal hemorrhage and pancreatitis in one participant, and arthralgia and infective exacerbation of bronchiectasis. Two potential AESIs were reported, which were subsequently ruled out; additional details are presented in the Supporting Information. No deaths, TEs, TMAs, or clinically significant changes from baseline in vital signs were reported during the study (Table 3); hematology parameters were stable in most participants and did not increase beyond World Health Organization grade 1 (Table S4).
Treatment‐induced ADAs were detected for a total of 8 of 64 (12.5%) evaluable participants (arm A, n = 4/25 [16.0%]; arm B, n = 4/25 [16.0%]; arm C [after switch to emicizumab], 0/14 [0.0%]). Of these, one participant had ADAs with neutralizing potential (ie, associated with decreased emicizumab plasma concentrations). This participant first tested positive for ADAs at week 4, and a decline in emicizumab concentration was observed from week 14; however, there was an apparent recovery in exposure from week 34. For the other seven participants, no impact on PK or clinically relevant effect on bleeding was observed.
Once‐weekly and every‐4‐week emicizumab dosing regimens achieved sustained effective trough plasma concentrations; mean trough plasma concentrations increased with loading doses to 39.8 µg/mL (once weekly) and 43.5 µg/mL (every 4 weeks) until week 5, then were maintained at ≈38 µg/mL and 33 µg/mL with once‐weekly and every‐4‐week dosing, respectively, through week 49 (Figure 4).
FIGURE 4.
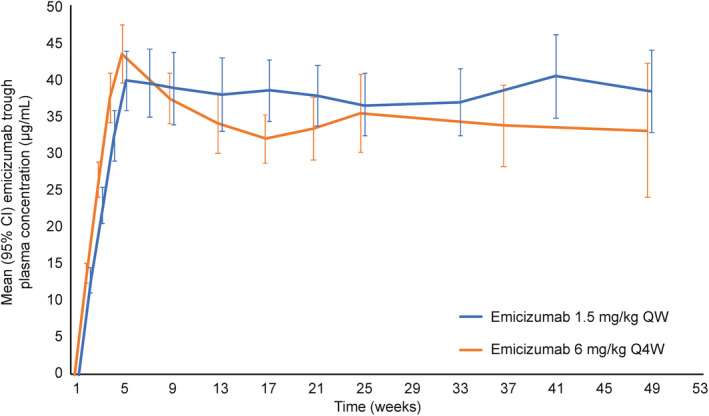
Emicizumab trough plasma concentration over time. Data points for emicizumab 1.5 mg/kg once weekly and 6 mg/kg every 4 weeks are offset on the x axis to aid visualization. CI, confidence interval; QW, once weekly; Q4W, once every 4 weeks
4. DISCUSSION
Data from this primary analysis of the HAVEN 5 study show that emicizumab (once weekly and every 4 weeks) significantly reduced ABRs in people with hemophilia A (aged ≥12 years) from the Asia‐Pacific region and was well tolerated, with a favorable safety profile. Emicizumab once weekly and every 4 weeks both reduced the ABR for treated bleeds by 96% and the ABR for all bleeds by 95% versus no prophylaxis (all P < .0001) at week 24. Overall, 65.5% (arm A) and 55.6% (arm B) of participants had zero treated bleeds, compared with 7.1% of those not receiving emicizumab. Moreover, efficacy data were generally consistent among those with or without FVIII inhibitors, and a ≥88% reduction in bleed rate was observed for all evaluable subgroups (number of bleeds in the 24 weeks before study start [<9 vs ≥9]; age at baseline; target joints), highlighting that the clinical benefit of emicizumab extends broadly across people with hemophilia A.
These positive outcomes are consistent with efficacy data reported for the other HAVEN studies and the HOHOEMI study (Table S5), with ABRs (treated bleeds) of 0.3 to 2.9 (once weekly), 0.2 to 1.3 (every 2 weeks), and 0.7 to 2.4 (every 4 weeks); similarly, 56% to 77% (once weekly), 33% to 90% (every 2 weeks; 33% is based on n = 2/6 individuals), and 56% to 71% (every 4 weeks) of participants reported zero treated bleeds. 14 , 15 , 16 , 17 , 18
The clinical benefit of emicizumab was further demonstrated by significantly reducing bleed rates by ≥95% versus no prophylaxis for treated target joint bleeds (ABR [95% CI]: arm A, 0.4 [0.18‐1.09]; arm B, 0.3 [0.12‐0.85]; P < .0001). More than 70% of participants receiving prophylaxis experienced zero treated target joint bleeds compared with less than one‐third not receiving prophylaxis. These data were consistent with the participants aged ≥12 years in the HAVEN studies. 14 , 15 , 16 This positive trend of reducing target joint bleeds following emicizumab prophylaxis was recently observed for a pooled, long‐term efficacy and safety analysis of the HAVEN program; ≈94% of 170 participants reported zero target joint bleeds during the final 24 weeks of the 144‐week analysis period. 19 These data were generally consistent among those with or without inhibitors.
Previous studies of emicizumab prophylaxis versus no prophylaxis have shown that markedly lower bleed rates translate into improvements in participants’ HRQoL and health status, regardless of FVIII inhibitor status. 14 , 15 , 16 This finding of lower ABRs coupled with a trend toward a clinically meaningful improvement in HRQoL (achieving a change higher than minimal responder thresholds described in the literature 26 , 27 ) was observed for both emicizumab regimens in HAVEN 5.
Both emicizumab regimens were generally well tolerated in this study population, and, overall, no new safety signals were observed during HAVEN 5. Safety data reported were generally consistent with those for the populations included in the HAVEN trials 14 , 15 , 16 , 17 and studies of emicizumab in Japanese people with hemophilia A. 18 , 28 , 29 ISRs were one of the most frequently reported AEs across the primary analyses of HAVEN 1 through 5 (15%, 31%, 25%, and 22% for HAVEN 1, 2, 3, and 4, respectively; and 13% for HAVEN 5). Other than the three TMAs and two TE events associated with concomitant activated prothrombin complex concentrate (aPCC) administration reported during the HAVEN 1 primary analysis 14 and two further TEs reported during a long‐term analysis of HAVEN 1 and 3 (n = 1 each), which were not associated with aPCC use, 19 there were no TMAs or TEs reported during the HAVEN 1 through 5 trials or studies of emicizumab in Japanese people with hemophilia A 18 , 29 Aside from the unrelated fatality due to rectal hemorrhage previously reported from HAVEN 1, there have been no deaths on any other trial in the emicizumab clinical development program.
Of the eight participants who developed ADAs during treatment in HAVEN 5, only one exhibited neutralizing ADAs associated with decreased emicizumab plasma concentrations, which were transient in nature. 30 No impact on efficacy or PK was observed for the remaining seven participants, and the presence of ADAs did not affect the safety profile of emicizumab.
In HAVEN 5, mean emicizumab trough plasma concentrations were, on average, 22% lower for a given dosing regimen or frequency than plasma concentrations observed in other HAVEN studies 14 , 15 , 16 ; despite this, emicizumab trough plasma concentrations were largely within range of those observed in other HAVEN studies and, importantly, remained efficacious, indicating that the decreased exposure is not clinically significant. This observation is in line with data from an emicizumab exposure‐response relationship analysis. 24 Despite comprehensive investigations, no operational reasons for the observed differences were identified. Furthermore, PK analyses of emicizumab in Asian and White people with hemophilia A indicate that this difference is unlikely to be associated with ethnicity, 21 , 31 signifying that these differences may be a study‐specific phenomenon. Of note, the PK profile and exposure resulting from a single 1‐mg/kg subcutaneous dose of emicizumab in healthy Chinese participants has been shown to be comparable with that of healthy Japanese and White individuals receiving the same dose. 12 , 13 , 31
Owing to participant variability, data from primary and subgroup analyses where the number is small should be interpreted with caution. Further, despite randomization, the median number of bleeds in the 24 weeks before study enrollment was numerically higher in arm C (19.5) versus arms A and B (both 14.0), which may exaggerate perceived efficacy. Follow‐up duration for evaluating prophylaxis was shorter for those who switched to emicizumab in arm C (24 weeks) than for those in arms A and B (44‐46 weeks), and an absence of participants <12 years of age limits the scope of the findings.
5. CONCLUSIONS
HAVEN 5 met its primary efficacy end point while demonstrating an overall favorable safety profile. Emicizumab prophylaxis achieved highly effective bleed control with a significant reduction in treated ABR versus no prophylaxis in adult and adolescent people with hemophilia A from the Asia‐Pacific region, regardless of FVIII inhibitor status. Prophylaxis was well tolerated, with no fatalities, TEs, TMAs, or new safety signals. Overall, the lower emicizumab exposure observed in this study did not influence the efficacy of emicizumab in people with hemophilia A from the Asia‐Pacific region.
These robust efficacy and safety data, coupled with observed clinically significant improvements in HRQoL, indicate that emicizumab may improve patient care by decreasing treatment burden, which in turn may enable improved adherence to effective prophylaxis, potentially decreasing the development of secondary complications for people with hemophilia A. These data indicate that clinical practice guideline updates and a paradigm shift in the provision of care for people with hemophilia A are warranted in the Asia‐Pacific region.
RELATIONSHIP DISCLOSURE
RY has received speaker and consultancy fees from Bayer, Novo Nordisk, Pfizer, F. Hoffmann‐La Roche Ltd, and Takeda. AC has received honoraria from Novo Nordisk, Grifols, and Takeda. SW, XW, JS, JZ, and XZ have no disclosures to make. CS is an employee of F. Hoffmann‐La Roche Ltd, holds stock in F. Hoffmann‐La Roche Ltd, and is an inventor on a patent related to an anti‐FIXa/FX bispecific antibody. WH, JX, and LL are employees of F. Hoffmann‐La Roche Ltd. TC is a former employee of Genentech, Inc.
AUTHOR CONTRIBUTIONS
RY: conception and design of the work; data acquisition, analysis, and/or interpretation; revised the manuscript critically and provided final approval of the version to be published. SW: data acquisition, analysis, and/or interpretation; revised the manuscript critically; and provided final approval of the version to be published. XW: data acquisition, analysis, and/or interpretation; enrollment and execution of the study; revised the manuscript critically; and provided final approval of the version to be published. JS: data acquisition, analysis, and/or interpretation; enrollment and execution of the study; revised the manuscript critically; and provided final approval of the version to be published. AC: data acquisition, analysis, and/or interpretation; enrollment and execution of the study; revised the manuscript critically; and provided final approval of the version to be published. JZ: data acquisition, analysis, and/or interpretation; enrollment and execution of the study; revised the manuscript critically; and provided final approval of the version to be published. CS: conception and design of the work; data acquisition, analysis, and/or interpretation; revised the manuscript critically; and provided final approval of the version to be published. WH: data acquisition, analysis, and/or interpretation, revised the manuscript critically; and provided final approval of the version to be published. JX: conception and design of the work; data acquisition, analysis, and/or interpretation; revised the manuscript critically; and provided final approval of the version to be published. LL: conception and design of the work; data acquisition, analysis, and/or interpretation; revised the manuscript critically; and provided final approval of the version to be published. TC: data acquisition, analysis, and/or interpretation; revised the manuscript critically; and provided final approval of the version to be published. XZ: data acquisition, analysis, and/or interpretation; enrollment and execution of the study; revised the manuscript critically; and provided final approval of the version to be published. All authors agree to be accountable for all aspects of the work.
Supporting information
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the study participants and their families, as well as the study investigators, research coordinators, and nurses. Initial study design and statistics considerations were provided by Elina Asikanius. Medical writing assistance for the development of this manuscript, under the direction of the authors, was provided by Adele Blair, PhD, of Ashfield MedComms, an Ashfield Health company, and funded by F. Hoffmann‐La Roche Ltd. The study was funded by F. Hoffmann‐La Roche Ltd. The study sponsors were involved in the study design; collection, analysis, and interpretation of the data; writing of the report; and the decision to submit the paper for publication. The corresponding author had full access to all study data and had final responsibility for the decision to submit the publication.
Yang R, Wang S, Wang X, et al. Prophylactic emicizumab for hemophilia A in the Asia‐Pacific region: A randomized study (HAVEN 5). Res Pract Thromb Haemost. 2022;6:e12670. doi: 10.1002/rth2.12670
Renchi Yang and Shujie Wang are co–first authors.
Handling Editor: Pantep Angchaisuksiri
Funding information
The study was funded by F. Hoffmann‐La Roche Ltd.
DATA AVAILABILITY STATEMENT
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.
REFERENCES
- 1. Mannucci PM, Tuddenham EG. The hemophilias–from royal genes to gene therapy. N Engl J Med. 2001;344:1773‐1779. [DOI] [PubMed] [Google Scholar]
- 2. Srivastava A, Santagostino E, Dougall A, et al. WFH guidelines for the management of hemophilia. Haemophilia. 2020;26(suppl 6):1‐158. [DOI] [PubMed] [Google Scholar]
- 3. Lambert T, Benson G, Dolan G, et al. Practical aspects of extended half‐life products for the treatment of haemophilia. Ther Adv Hematol. 2018;9:295‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ljung R, Gretenkort Andersson N. The current status of prophylactic replacement therapy in children and adults with haemophilia. Br J Haematol. 2015;169:777‐786. [DOI] [PubMed] [Google Scholar]
- 5. Witmer C, Young G. Factor VIII inhibitors in hemophilia A: rationale and latest evidence. Ther Adv Hematol. 2013;4:59‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Berntorp E, Shapiro A, Astermark J, et al. Inhibitor treatment in haemophilias A and B: summary statement for the 2006 international consensus conference. Haemophilia. 2006;12(suppl 6):1‐7. [DOI] [PubMed] [Google Scholar]
- 7. Nogami K, Shima M. New therapies using nonfactor products for patients with hemophilia and inhibitors. Blood. 2019;133:399‐406. [DOI] [PubMed] [Google Scholar]
- 8. Dou X, Poon M‐C, Yang R. Haemophilia care in China: achievements in the past decade. Haemophilia. 2020;26:759‐767. [DOI] [PubMed] [Google Scholar]
- 9. Song X, Zhong J, Xue F, et al. An overview of patients with haemophilia A in China: epidemiology, disease severity and treatment strategies. Haemophilia. 2021;27:e51‐e59. [DOI] [PubMed] [Google Scholar]
- 10. Dunkley S, Lam JCM, John MJ, et al. Principles of haemophilia care: the Asia‐Pacific perspective. Haemophilia. 2018;24:366‐375. [DOI] [PubMed] [Google Scholar]
- 11. Kitazawa T, Igawa T, Sampei Z, et al. A bispecific antibody to factors IXa and X restores factor VIII hemostatic activity in a hemophilia A model. Nat Med. 2012;18:1570‐1574. [DOI] [PubMed] [Google Scholar]
- 12. Uchida N, Sambe T, Yoneyama K, et al. A first‐in‐human phase 1 study of ACE910, a novel factor VIII‐mimetic bispecific antibody, in healthy subjects. Blood. 2016;127:1633‐1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kotani N, Yoneyama K, Kawakami N, et al. Relative and absolute bioavailability study of emicizumab to bridge drug products and subcutaneous injection sites in healthy volunteers. Clin Pharmacol Drug Dev. 2019;8:702‐712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oldenburg J, Mahlangu JN, Kim B, et al. Emicizumab prophylaxis in hemophilia A with inhibitors. N Engl J Med. 2017;377:809‐818. [DOI] [PubMed] [Google Scholar]
- 15. Mahlangu J, Oldenburg J, Paz‐Priel I, et al. Emicizumab prophylaxis in patients with hemophilia A without inhibitors. N Engl J Med. 2018;379:811‐822. [DOI] [PubMed] [Google Scholar]
- 16. Pipe S, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open‐label, non‐randomised phase 3 study. Lancet Haematol. 2019;6:E295‐E305. [DOI] [PubMed] [Google Scholar]
- 17. Young G, Liesner R, Chang T, et al. A multicenter, open‐label, phase 3 study of emicizumab prophylaxis in children with hemophilia A with inhibitors. Blood. 2019;134:2127‐2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shima M, Nogami K, Nagami S, et al. A multicentre, open‐label study of emicizumab given every 2 or 4 weeks in children with severe haemophilia A without inhibitors. Haemophilia. 2019;25:979‐987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Callaghan M, Negrier C, Paz‐Priel I, et al. Long‐term outcomes with emicizumab prophylaxis for hemophilia A with or without FVIII inhibitors from the HAVEN 1–4 studies. Blood. 2021;137:2231‐2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Paz‐Priel I, Chang T, Asikanius E, et al. Immunogenicity of emicizumab in people with hemophilia A (PwHA): results from the HAVEN 1‐4 studies. Blood. 2018;132:633. [Google Scholar]
- 21. Retout S, Schmitt C, Petry C, Mercier F, Frey N. Population pharmacokinetic analysis and exploratory exposure‐bleeding rate relationship of emicizumab in adult and pediatric persons with hemophilia A. Clin Pharmacokinet. 2020;59:1611‐1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Donadel‐Claeyssens S, European Paediatric Network for Haemophilia Management . Current co‐ordinated activities of the PEDNET (European Paediatric Network for Haemophilia Management). Haemophilia. 2006;12:124‐127. [DOI] [PubMed] [Google Scholar]
- 23. Blanchette VS, Key NS, Ljung LR, et al. Definitions in hemophilia: communication from the SSC of the ISTH. J Thromb Haemost. 2014;12:1935‐1939. [DOI] [PubMed] [Google Scholar]
- 24. Jonsson F, Schmitt C, Petry C, et al. Exposure‐bleeding count modeling of emicizumab for the prophylaxis of bleeding in persons with hemophilia A with/without inhibitors against factor VIII. Clin Pharmacokinet. 2021;60:931‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Shankar G, Cocea SAL, Devanarayan V, et al. Assessment and reporting of the clinical immunogenicity of therapeutic proteins and peptides‐harmonized terminology and tactical recommendations. AAPS J. 2014;16:658‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Antunes SV, Tangada S, Stasyshyn O, et al. Randomized comparison of prophylaxis and on‐demand regimens with FEIBA NF in the treatment of haemophilia A and B with inhibitors. Haemophilia. 2014;20:65‐72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wyrwich KW, Krishnan S, Poon JL, et al. Interpreting important health‐related quality of life change using the Haem‐A‐QoL. Haemophilia. 2015;21:578‐584. [DOI] [PubMed] [Google Scholar]
- 28. Shima M, Hanabusa H, Taki M, et al. Factor VIII–mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374:2044‐2053. [DOI] [PubMed] [Google Scholar]
- 29. Shima M, Hanabusa H, Taki M, et al. Long‐term safety and efficacy of emicizumab in a phase 1/2 study in patients with hemophilia A with or without inhibitors. Blood Adv. 2017;1:1891‐1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Schmitt C, Emrich T, Chebon S, et al. Low immunogenicity of emicizumab in persons with haemophilia A. Haemophilia. 2021;27:984‐992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li H, Zhang W, Petry C, et al. Evaluation of the pharmacokinetics, pharmacodynamics, and safety of a single dose of emicizumab in healthy Chinese subjects. Clin Pharmacol Drug Dev. 2020;10:30‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Data Availability Statement
Qualified researchers may request access to individual patient‐level data through the clinical study data request platform (https://vivli.org/). Further details on Roche’s criteria for eligible studies are available at https://vivli.org/members/ourmembers/. For further details on Roche’s Global Policy on the Sharing of Clinical Information and how to request access to related clinical study documents, see https://www.roche.com/research_and_development/who_we_are_how_we_work/clinical_trials/our_commitment_to_data_sharing.htm.