

Abstract
Efficiently finding maximal exact matches (MEMs) between a sequence read and a database of genomes is a key first step in read alignment. But until recently, it was unknown how to build a data structure in  space that supports efficient MEM finding, where r is the number of runs in the Burrows–Wheeler Transform. In 2021, Rossi et al. showed how to build a small auxiliary data structure called thresholds in addition to the r-index in space. This addition enables efficient MEM finding using the r-index. In this article, we present the tool that implements this solution, which we call MONI. Namely, we give a high-level view of the main components of the data structure and show how the source code can be downloaded, compiled, and used to find MEMs between a set of sequence reads and a set of genomes.
space that supports efficient MEM finding, where r is the number of runs in the Burrows–Wheeler Transform. In 2021, Rossi et al. showed how to build a small auxiliary data structure called thresholds in addition to the r-index in space. This addition enables efficient MEM finding using the r-index. In this article, we present the tool that implements this solution, which we call MONI. Namely, we give a high-level view of the main components of the data structure and show how the source code can be downloaded, compiled, and used to find MEMs between a set of sequence reads and a set of genomes.
Keywords: MEM finding, r-index, run-length-encoded Burrows–Wheeler transform, thresholds
1. INTRODUCTION
Indexing genomes in sublinear space in a manner that supports read alignment has been an open problem since short read aligners—such as BWA (Li and Durbin, 2009) and Bowtie (Langmead et al., 2009; Langmead, and Salzberg, 2012)—started to make use of the FM-index (Ferragina and Manzini, 2005). A significant breakthrough in this problem was made by Gagie et al. (2020a) when they showed most of the functionalities of the FM-index can be retained by only storing the run-length encoded Burrows–Wheeler Transform (RL BWT) with a suffix array (SA) sample at the beginning and end of each run of the BWT.
These data structured are called the r-index, which follows from the fact that occupies space, where r is the number of runs in the BWT. Table 1 shows how r can vary depending on the data and the size of the data. Yet, Gagie et al. did not demonstrate how to construct the r-index or use it to perform read alignment. The construction of the traditional r-index was later accomplished by Boucher et al. (2019) and Kunhle et al. (2020) using a technique called Prefix-Free Parsing (PFP). However, it was still unclear how to use the r-index for read alignment.
Table 1.
An Overview of How r Varies for Different Data sets
| Name | Description | σ | n/106 | n/r |
|---|---|---|---|---|
| cere | Baking yeast genomes | 5 | 461.28 | 39.85 |
| einstein.de.txt | Wikipedia articles in German | 117 | 92.21 | 915.04 |
| einstein.en.txt | Wikipedia articles in English | 139 | 465.24 | 1611.17 |
| Escherichia_Coli | Bacteria genomes | 15 | 112.68 | 7.49 |
| influenza | Virus genomes | 15 | 154.80 | 51.21 |
| kernel | Linux Kernel sources | 160 | 249.51 | 92.41 |
| para | Yeast genomes | 5 | 429.26 | 27.45 |
| world_leaders | CIA world leaders files | 89 | 46.90 | 81.89 |
| chr19 | Human chromosome 19 (1000 haplotypes) | 5 | 60110.54 | 1287.38 |
| Salmonella | Salmonella genomes database (10,000 genomes) | 4 | 51820.38 | 36.61 |
| SARS-CoV2 | SARS-CoV2 genomes database (400,000 genomes) | 5 | 11930.96 | 1302.87 |
In this study, we give the name and description of the data sets (column 1 and 2), the alphabet size (column 3), the length of the file (column 4), and the ratio of the length to the number of runs in the BWT (column 5).
To apply the r-index to read alignment, Bannai et al. (2020) proposed the idea of finding maximal exact matches (MEMs) between a query string (read) and the index of genomes by first calculating the query's matching statistics. MEMs can then be extended to find full alignments between a query string and sequences in the index by dynamic programming or other chaining techniques (Li, 2013). Matching statistics for a query string are computed by using a set of thresholds that are defined for each subsequent runs of the same character in the BWT.
In particular, given two runs of character c in the BWT that are at positions i to and , the position within the interval that has a smallest longest common prefix (LCP) value is stored as the threshold for those runs, where the LCP value is the LCP between consecutive suffixes in the SA. Bannai et al. did not describe how to construct the thresholds efficiently—this was accomplished later by Rossi et al. (2021) by modifying the PFP algorithm to construct thresholds along with the other components of the r-index. The implementation of their method is referred to as MONI. In this study, we describe the practicalities of this tool, that is, how to build and use the r-index of Rossi et al. to find MEMs between a set of sequence reads and an index of genomes.
2. INSTALLATION
We begin by giving instructions on how to download and install MONI. We assume that users are on a system with a standard Linux or Unix installation that has a c++17 or higher compiler, and that apt is their default package manager. If another Unix-like system is being used, then the appropriate package manager for apt should be substituted.
Users should first download some prerequisite packages, and the source code from the github repository:
| $ apt-get update |
| $ apt-get install -y build-essential cmake git python3 zlib1g-dev |
| $ pip3 install psutil |
| $ git clone https://github.com/maxrossi91/moni |
Users can compile the code as follows; any missing dependencies will be automatically downloaded and compiled locally.
| $ cd moni |
| $ mkdir build |
| $ cd build |
| $ cmake .. |
| $ make |
| $ make install |
| $ cd .. |
These commands will also install the binaries of MONI in the default binary location of the system (e.g., /usr/local/bin for Ubuntu users), together with the moni pipeline. MONI binaries include BigBWT (Boucher et al., 2019), BigRePair (Gagie et al., 2019), and ShapedSLP (Gagie et al., 2020b). In addition, MONI uses sdsl-lite (Gog et al., 2014), ksw2 (Li, 2018; Suzuki and Kasahara, 2018), gsacak (Louza et al., 2017), and r-index (Mun et al., 2020). If the users want the binaries in a different location, then they should replace the cmake .. command mentioned with
| $ cmake –DCMAKE_INSTALL_PREFIX = < dir> .. |
where <DIR> is the desired destination directory.
3. CONSTRUCTION OF INDEX
Users can build a MONI index using the moni build command. In particular, the index can be built for a collection of genomic sequences in a FASTA file format. The provided input file can also be gzipped. In the data/SARS-CoV2 directory we provide an example gzipped data set of 1000 SARS-CoV2 genomes from EBIs COVID-19 data portal (The COVID-19 Data Portal, 2021) occupying 3.9 MB. To build the index, users can use the following command:
| $ moni build -r data/SARS-CoV2/SARS-CoV2.1k.fa.gz -o sars-cov2 -f |
Since MONI can handle also plain texts, the −f flag is used to specify that the input text is in FASTA format. The command's output consists of four files prefixed by sars−cov2 with different extensions, namely
– sars−cov2.idx (33 KB): the file containing the starting position and name of each fasta sequence in the reference file.
– sars−cov2 thrbv.ms (845 KB): the file containing the r-index of the concatenation of the sequences in the reference file.
– sars−cov2 plain.slp (355 KB): the file containing the straight-line program (grammar) generating the concatenation of the sequences in the reference file.
– sars−cov2.moni.log: the file containing log information of the construction process.
The user can configure the file prefix with the −o parameter. In this example, the total size of the final index is 1.2 MB. By default MONI builds a plain encoded straight-line program, however, it is also able to build a “shaped” straight-line program using the −g shaped option. A shaped straight-line program is about half the size of the plain version, but is over two times slower to query. In our example, sars-cov2.slp uses 164 KB of space.
On an Intel(R) Xeon(R) CPU E5-2640 v4 @ 2.40 GHz server, building the index for data/SARS-CoV2/SARS-CoV2.1k.fa.gz takes ∼6 seconds and 34 MB of RAM. On the same server, we built the index for 200 human genomes from The 1000 Genomes Project Consortium (2015), which takes 580 GB uncompressed, in 41 hours with 418 GB of memory using 32 threads.
4. COMPUTING MATCHING STATISTICS
Users can compute the matching statistics of a set of patterns stored in a FASTA or FASTQ file using moni ms. In our running example, we provide data/SARS-CoV2/reads.fastq.gz, which is a set of 10,000 100-bp reads simulated with Mason2 (Holtgrewe, 2010) from the first genome in the reference file using 0.001 error rate for both SNPs and small insertions/deletions (indels). The matching statistics can be computed with the following command:
| $ moni ms -i sars-cov2 -p data/SARS-CoV2/reads.fastq.gz -o reads |
On the same Intel(R) Xeon(R) CPU E5-2640 v4 @ 2.40 GHz server, computing the matching statistics for the input reads takes 2.2 seconds with an average of 0.22 ms per read.
5. FINDING MAXIMAL EXACT MATCHES
Users can compute the MEMs of a set of patterns stored in a FASTA or FASTQ file using moni mems. Using our running example, the MEMs can be computed with the following command:
| $ moni mems -i sars-cov2 -p data/SARS-CoV2/reads.fastq.gz -o reads |
On the same Intel(R) Xeon(R) CPU E5-2640 v4 @ 2.40 GHz server, computing the MEMs for the input reads takes 2.3 seconds with an average of 0.23 ms per read.
6. EXTENDING MAXIMAL EXACT MATCHES
Users can compute the extension of MEMs of a set of patterns stored in a FASTA or FASTQ file using moni extend. For each read of length , the algorithm works as follows: first, all MEMs are found, then an occurrence of the longest MEM is considered, a −L-length segment of the reference is extracted before and after the occurrence of the MEM, and the alignment is extended in this extracted region using ksw2 extension functions. MONI reports the extension if the score is greater than , as in Bowtie2. The −L parameter can be modified; it is 100 bp by default. We apply the same algorithm also to the reverse complement of the read.
If both of the read and its reverse complement have an MEM that has an extension with a valid score, both are reported. The default parameters for ksw2 are 2 as match score, 4 as mismatch penalty, 4 and 13 as gap open penalties, and 2 and 1 as gap extension penalties. We refer the users to Suzuki and Kasahara (2018) and Li (2018) for an in-depth explanation of the ksw2 parameters and the two-piece affine gap cost function. These parameters can be changed by the user using the −A flag for the match score, the −B flag for the mismatch penalty, the −O flag for the gap open penalty specifying the two values separated by a coma, and the −E flag for the gap extension penalty specifying the two values separated by a comma.
Using our running example, the MEM extensions can be computed with the following command:
| $ moni extend -i sars-cov2 -p data/SARS-CoV2/reads.fastq.gz -o reads |
With a verbose command:
| $ moni extend -i sars-cov2 -p data/SARS-CoV2/reads.fastq.gz -o reads -L 100 -A 2 -B 4 -O 4,13 -E 2,1 -g plain |
On the same Intel(R) Xeon(R) CPU E5-2640 v4 @ 2.40 GHz server, computing the extensions of the MEMs for the input reads takes 3.6 seconds with an average of 0.36 ms per read.
7. INTERPRETING THE OUTPUT
In this section, we describe the output of the three tasks performed in the previous sections.
7.1. Matching statistics
The output of moni ms is a pair of files with extensions .lengths and .pointers storing the lengths and the pointers of the matching statistics, respectively. Both files have the same format; for each read there are two lines: the first contains the name of the read preceded by a’>’ character, and the second contains a list of space-separated integers, one for each nucleotide in the read. The i-th integer of the list from the .lengths file represents the length of the longest prefix of the i-th suffix of the read that occurs in the reference, and the i-th integer of the list from the .pointers file represents the position of that longest prefix with respect to the reference being the concatenation of all the sequences in the input file. In our running example, the moni ms output is as follows (with ellipses used for conciseness):
| $ head reads.lengths $ head reads.pointers |
| > >simulated.1 > >simulated.1 |
| > 28 27 26 … 1> > 8004707 8004708 8004709 … 4675811 |
| > >simulated.2 > >simulated.2 |
| > 19 18 17 … 1 > 13019758 13019759 13019760 … 28479947 |
| > >simulated.3 > >simulated.3 |
| > 8 8 8 … 1 > 2891013 23134920 3596962 … 20281425 |
| > >simulated.4 > >simulated.4 |
| > 9 9 8 … 1 > 25840878 28883051 28883052 … 20281425 |
| > >simulated.5 > >simulated.5 |
| > 7 8 7 … 1 > 17796048 23730017 23730018 … 28479947 |
7.2. Maximal exact matches
The output of moni mems is a file with extension .mems storing the MEMs. For each read, the MEM's position in the read and its length is stored as a pair. The file reports two lines per read. The first contains the name of the read preceded by a’>’ character, and the second contains a list of space-separated pairs of integers, where the i-th pair consists of the starting position in the read of the i-th MEM, and its length. In our running example:
| $ head reads.mems |
| > >simulated.1 |
| > (0,28) (21,8) (24,6) (25,6) (26,8) (27,9) (28,9) (29,71) |
| > >simulated.2 |
| > (0,19) (12,8) (15,8) (17,7) (18,8) (19,81) |
| > >simulated.3 |
| > (0,8) (1,8) (2,8) … (89,8) (90,8) (91,9) |
| > >simulated.4 |
| > (0,9) (1,9) (3,9) … (89,8) (91,7) (92,8) |
| > >simulated.5 |
| > (0,7) (1,8) (3,9) … (85,10) (88,11) (93,7) |
7.3. Extension of maximal exact matches
The output of moni extend is in a SAM file (Li et al., 2009). The SAM format is a tab-separated file that begins with comment lines starting with the symbol “@.” The first line specifies the version of the SAM file, followed by one line for each sequence in the reference genome (1000 SARS-CoV2), and a line specifying the program used for processing the reads.
After these, the file consists of a line for each read in the query file. Each line consists of the following tab-separated fields: read name, flag, reference name, position, MAPQ score, CIGAR string, reference name for the next read in the read-pair, position of the next read in the read-pair, observed template length, read sequence, read quality scores, and miscellaneous tags, for example, alignment score (AS), number of mismatches (NM), and a string encoding mismatched and deleted reference bases (MD). In our running example:
| $ cat reads.sam |
| > @HD VN:1.6 SO:unknown |
| > @SQ SN:ENA|MW565758|MW565758.1 LN:29850 |
| > @SQ SN:ENA|MW565759|MW565759.1 LN:29847 |
| > @SQ SN:ENA|MW565760|MW565760.1 LN:29816 |
| … |
| > @SQ SN:ENA|MW566845|MW566845.1 LN:29842 |
| > @SQ SN:ENA|MW549699|MW549699.1 LN:29482 |
| > @SQ SN:ENA|MT704034|MT704034.1 LN:29791 |
| > @PG ID:moni PN:moni VN:0.2.0 |
| > simulated.1 0 ENA|LR899017|LR899017.1 17789 43 100M * 0 0 |
| AGA…TGA IHH…AHD AS:i:194 NM:i:1 MD:Z:28A71 |
| > simulated.2 0 ENA|MW566485|MW566485.1 25291 43 19M1D81M * 0 0 |
| TGA…AGG IIH…IEA AS:i:194 NM:i:1 MD:Z:19^G81 |
| > simulated.3 16 ENA|MW064948|MW064948.1 19019 43 100M * 0 0 |
| ATG…CAC FHI…IHI AS:i:200 NM:i:0 MD:Z:100 |
| > simulated.4 16 ENA|FR990399|FR990399.1 11322 43 100M * 0 0 |
| ACT…TTT IDI…IHH AS:i:200 NM:i:0 MD:Z:100 |
| … |
8. CONCLUSIONS AND FUTURE STUDY
In this article, we illustrated how to use MONI to find MEMs between sequence reads and a set of genomes. However, we note that there are several applications of our data structure that builds thresholds alongside the standard r-index.
Homopolymer compression has been proven to be effective in improving alignment accuracy of third generation sequencing long reads (Au et al., 2012; Li, 2018). In the homopolymer compressed representation of a sequence, homopolymers are contracted in a single base, for example, the homopolymer compression of GGGAAATTAA is GATA. MONI can be used also to find MEMs in homopolymer compressed space, which is suitable for removing homopolymer indel errors in PacBio long reads, for example. This can be done by homopolymer compressing the reference, building MONI index, and querying the reference with homopolymer compressed reads.
Recently, Khorsand et al. (2021) defined the problem of finding Substring-Free Sample-specific (SFS) strings and showed it can be effective for comparing individuals in a population to detect structural variants [see also Bannai et al. (2020), Section 5.3]. Given a set 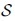 of strings and a query string T, an SFS in T, or T-specific string, is a substring s of T that does not occur as a substring in but any proper substring of s is a substring in the set . The main intuition is that an SFS does not occur in the set and hence, it captures variations in T with respect to the sample .
of strings and a query string T, an SFS in T, or T-specific string, is a substring s of T that does not occur as a substring in but any proper substring of s is a substring in the set . The main intuition is that an SFS does not occur in the set and hence, it captures variations in T with respect to the sample .
For example, an SFS in T may include a single nucleotide polymorphisms or a portion of a structural variation in T with respect to sample . We note that finding an SFS can be solved efficiently using MONI by building an index for , calculating the matching statistics for T, and finding MEMs between T and . Any SFS can be found by analyzing the overlap of consecutive MEMs. A clear advantage of the r-index is it can extend this approach by comparing of a query string T against a large population of genomes. Although this problem can be solved relatively efficiently using MONI, the more general version of the SFS problem where there is allowed to be k corrections in a substring x of T and still get a T-specific string cannot obviously be solved using MONI. We leave this problem for future study.
9. AVAILABILITY
MONI is publicly available under MIT license at (https://github.com/maxrossi91/moni).
AUTHORS’ CONTRIBUTIONS
M.R., T.G., and C.B. conceptualized the idea and developed the algorithmic contributions of this study. M.R. implemented the MONI tool. M.R. and M.O. conducted the experiments. M.R., M.O., C.B., and B.L. assisted and oversaw the experiments and implementation. P.B. suggested the connection between MONI and solving efficiently the SFS problem. All authors contributed to the writing of this article.
AUTHOR DISCLOSURE STATEMENT
The authors declare they have no competing financial interests.
FUNDING INFORMATION
M.R., M.O., T.G., B.L., and C.B. are funded by National Science Foundation NSF IIBR (Grant No. 2029552) and National Institutes of Health (NIH) NHGRI (Grant No. HG011392). M.R., M.O., and C.B. are funded by NSF IIS (Grant No. 1618814) and NIH NIAID (Grant No. R01AI141810). T.G. is funded by NSERC Discovery Grant (Grant No. RGPIN-07185-2020). P.B. is funded by the European Unions Horizon 2020 research and innovation program under the Marie Skodowska-Curie grants agreements numbers [872539] and [956229].
REFERENCES
- Au, K.F., Underwood, J.G., Lee, L., et al. 2012. Improving PacBio long read accuracy by short read alignment. PLoS ONE 7, e46679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannai, H., Gagie, T., and Tomohiro, I.. 2020. Refining the r-index. Theor. Comput. Sci. 812, 96–108. [Google Scholar]
- Boucher, C., Gagie, T., Kunhle, A., et al. 2019. Prefix-free parsing for building big BWTs. Algorithms Mol. Biol. 14, 13:1–13:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferragina, P., and Manzini, G.. 2005. Indexing compressed text. J. ACM 52, 552–581. [Google Scholar]
- Gagie, T., Navarro, G., and Prezza N.. 2020a. Fully functional suffix trees and optimal text searching in BWT-Runs Bounded Space. J. ACM 67, 2:1–2:54. [Google Scholar]
- Gagie, T., Tomohiro, I., Manzini, G., et al. 2019. Rpair: Rescaling RePair with Rsync. In Proceedings of the 26th International Symposium on String Processing and Information Retrieval, SPIRE 2019, Segovia, Spain, pp. 35–44. [Google Scholar]
- Gagie, T., Tomohiro, I., Manzini, G., et al. 2020b. Practical random access to SLP-Compressed Texts. In Proceedings of the 27th International Symposium on String Processing and Information Retrieval, SPIRE 2020, Orlando, FL, pp. 221–231. [Google Scholar]
- Gog, S., Beller, T., Moffat, A., et al. 2014. From theory to practice: Plug and play with succinct data structures. In Proceedings of the 13th International Symposium on Experimental Algorithms. SEA 2014, Copenhagen, Denmark, pp. 326–337. [Google Scholar]
- Holtgrewe M. 2010. Mason: A read simulator for second generation sequencing data. https://www.seqan.de/apps/mason.html.
- Khorsand, P., Denti, L., Bonizzoni, P., et al. 2021. Comparative genome analysis using sample-specific string detection in accurate long reads. Bioinformatics Adv. 1, vbab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunhle, A., Mun, T., Boucher, C., et al. 2020. Efficient construction of a complete index for pan-genomics read alignment. J. Comput. Biol. 27, 500–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B., and Salzberg, S.L.. 2012. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B., Trapnell, C., Pop, M., et al. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 1303.3997. [Google Scholar]
- Li, H. 2018. Minimap2: Pairwise alignment for nucleotide sequences. Bioinformatics 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., and Durbin, R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler Transform. Bioinformatics 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H., Handsaker, B., Wysoker, A., et al. 2009. The sequence alignment/map format and SAM-tools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louza, F.A., Gog, S., and Telles, G.P.. 2017. Inducing enhanced suffix arrays for string collections. Theor. Comput. Sci. 678, 22–39. [Google Scholar]
- Mun, T., Kunhle, A., Boucher, C., et al. 2020. Matching reads to many genomes with the r-index. J. Comput. Biol. 27, 514–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi, M., Oliva, M., Langmead, B., et al. 2022. MONI: A Pangenomics Index for finding Maximal Exact Matches. J. Comput. Biol. 29, 169–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, H., and Kasahara, M.. 2018. Introducing difference recurrence relations for faster semi-global alignment of long sequences. BMC Bioinformatics 19, 33–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium. 2015. A global reference for human genetic variation. Nature 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The COVID-19 Data Portal. Available at: https://www.covid19dataportal.org. Accessed May 17, 2021.