

Abstract
Keloid is a fibrous hyperplastic disease of the skin characterized by excessive collagen deposition. Keloid patients suffer from severe facial damage and psychological burden, but the underlying pathologic mechanism remains unclear. Keloid fibroblasts are often considered the key cell of keloid formation, but the regulation of the immune microenvironment of keloid fibroblasts is poorly understood. The pathogenic roles of macrophages, Tregs, CD8+ T cells, dendritic cells, and natural killer cells in keloids are reviewed and further directions proposed, which may provide a novel window of opportunity for immunotherapy of keloids. Considering the dearth of studies on the function of immune cells related to keloids, the mechanisms of these immune cells in other diseases are further examined herein to provide a reference for future research on the immune microenvironment of keloids.
Keywords: Keloids, macrophage, Treg, CD8+ T cells, dendritic cells, natural killer cells
Introduction
The keloid is a fibrous hyperplastic disease of the skin characterized by excessive deposition of collagen [1,2]. It is believed that the development and progression of keloids frequently begin following abnormal wound healing, leading to a fibroproliferative inflammatory process in which fibrin plays a major role [3]. Keloid tissue is the final product of this process. Keloids have histologic features similar to those of hypertrophic scars, but they have unique growth characteristics. They are characterized by persistent scar hyperplasia beyond the wound edge and generally do not resolve by themselves; they are often caused by skin diseases and local injuries [4]. Keloids tend to occur in areas of the body susceptible to tension and are commonly found in the chest, shoulders, and other easily exposed areas of the skin [5]. The prevalence of keloids is highest in people of African ethnicity, followed by Asians and Hispanics, with the lowest prevalence in individuals of white ethnicity [1]. Keloids can occur at any age but are most common in young people [3]. The pathogenesis of keloids is very complex [6]; keloids are associated with inflammatory responses to tissue injury and with clinical manifestations including pain, itching, and burning [3]. The site of the lesion exhibits unique histologic characteristics, with a large number of irregularly oriented, thickened transparent collagen fibers, commonly known as keloid tissue, and a large number of locally infiltrative inflammatory cells and cytokines [1]. Immune cells infiltrate the microenvironment, regulating it by secreting cytokines [1,6]. Keloid fibroblasts are often considered a key type of cell in keloid formation, but regulation of the immune microenvironment of keloid fibroblasts has not been fully explored. The role of immune cells in the pathogenesis of keloids remains unclear. In this review, we discuss the role of immune cells in keloid formation, which may provide a window of opportunity for keloid immunotherapy. A literature review was performed from 1981 to December 2020 using the PubMed database of the National Center of Biotechnology Information, Google Scholar, MEDLINE, Cochrane database, Embase, and Biosis. The terms “keloid”, “CD8+ T cells”, “macrophage”, “dendritic cells”, “Treg”, “natural killer cells”, and “immune microenvironment” were used, together with all the synonyms of these terms. Keloid fibroblasts are often considered an important cause of keloid formation, but the regulation of the immune microenvironment of keloid fibroblasts is poorly understood. The microenvironment is mainly composed of keloid fibroblasts and their surrounding immune cells, which have an important influence on the development of keloids. In this study, the role of various immune cells in keloid formation is discussed from the perspective of the keloid immune microenvironment (Figure 1).
Figure 1.
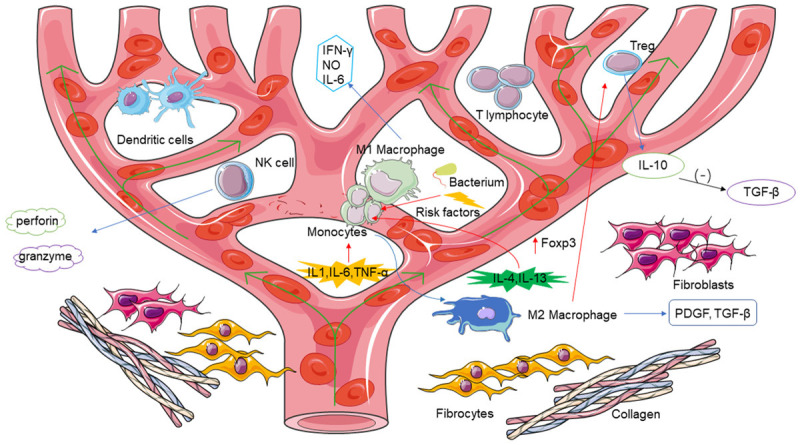
The immune microenvironment of keloids [1-8,12,33,36,49]. (-): Inhibition; green arrow: blood flow direction; red arrow: promotion; blue arrow: production.
The role of macrophages in keloid pathogenesis
During the early stages, macrophages play a proinflammatory role, which includes antigen presentation, phagocytosis, and the production of inflammatory cytokines and growth factors, thus promoting an inflammatory reaction. According to their different activation states and functions, macrophages can be divided into classically activated macrophages (M1) and alternatively activated macrophages (M2) [7]. During the proliferation stage, macrophages directly or indirectly stimulate the proliferation of connective tissue, endothelial tissue, and epithelial tissue, inducing the formation, re-epithelialization, and neovascularization of the extracellular matrix. Subsequently, macrophages can change the extracellular matrix during the angiogenesis and remodeling stage by releasing degrading enzymes and extracellular matrix molecules, suggesting that M2-type macrophages play an important role in this stage of wound healing [8]. The function of macrophages in wound healing can lead to the derailment of wound healing, culminating in ulcers, chronic wounds, hypertrophic scars, or keloid formation. The role played by macrophages, particularly their functional phenotypes, in keloid formation should be further investigated, to seek additional treatment options for keloids [1].
Typical monocytes are derived from bone marrow, denoted CD14++ CD16-, and circulate in the blood. Monocytes are recruited to target sites in response to pathogen-associated molecular patterns (PAMPs), IL-6, TNF-α, IL-1, and CCL2 [9]. When tissue damage occurs, the presence of inflammatory cytokines (e.g., TNF-α and IFN-γ) promotes the recruitment of circulating monocytes, their adherence to endothelial cells, and their translocation into the damaged tissue. CD14+ monocytes are transformed into macrophages upon entering a wound. Macrophages not only engulf pathogens and cell debris but also produce cytokines, which stimulate collagen generation and angiogenesis and start the healing process [1]. Macrophages can change their phenotypes according to their cytokine/chemokine microenvironment. IL-4, IL-13, or apoptotic neutrophils can transform macrophages into M2 macrophages [9]. Activation of M2 macrophages results in the production of cytokines, such as TGF-β and platelet-derived growth factor (PDGF), which stimulate the proliferation of fibroblasts. M2 macrophages have been divided into four subgroups: M2a, M2b, M2c, and M2d [10]. M2a macrophages produce arginase-1 (Arg1), PDGF, and IGF-1 under the stimulation of IL-4 and/or IL-13 [11]. M2a macrophages can stimulate the proliferation of fibroblasts. M2b can be stimulated by toll-like receptors to produce a regulatory effect. M2c macrophages can remodel the extracellular matrix under the stimulation of glucocorticoids, IL-10, and TGF cytokines [12] M2d macrophages produce IL-10 and VEGF [13]. They also inhibit the proinflammatory effects of M1 macrophages by downregulating TGF-β and IL-12.
The occurrence of keloids in different parts of the body is related to the number and subtype of macrophages. Butzelaar et al. measured macrophages in skin samples from susceptible sites (e.g., earlobes, mandible, neck, and shoulders) and non-susceptible sites (e.g., upper eyelids, cheeks, and abdomen) [14]. The number of M1 macrophages (CD40+) was significantly reduced in sites susceptible to keloid formation, while the number of M2 macrophages (CD163+) was equal in susceptible [15] and non-susceptible sites. These results were the same with the results observed by Vidyarthi et al. in gliomas [15]. Although the mechanism of susceptibility to keloids has not been fully elucidated, a change in M1/M2 macrophages ratio in the microenvironment seems to play an important role in keloid susceptibility as in other tumors [16].
One of the characteristics of malignant tumors is the polarization of tumor-associated macrophages from M1 macrophages (immune-promoting cells) to M2 macrophages (immunosuppressive cells) [17]. In tumor immunotherapy, M0- or M2-type tumor-related macrophages can be reprogrammed to become M1-type by targeting microRNAs related to macrophage activation and function [18], or the expression of M1-type cytokines that regulate polarization can be promoted by a feedback loop. De Felice et al. found that treatment with Ingenol mebutate was effective in reducing the growth of keloid fibroblasts [19]. The expression of miR-34 regulates the expression of proapoptotic genes following Ingenol mebutate treatment [19]. Currently, clinical trials to investigate changes in the polarized state of macrophages to treat keloids are lacking.
The M1/M2 ratio describes two major and opposite activities of macrophages [20]. M1 macrophages are the major macrophage population during the early stages of scar formation (early inflammation and proliferation), while M2 macrophages are the major macrophage population during the late stages of scar formation (late proliferation and remodeling) [20]. M1 macrophages specialize in secreting proinflammatory factors and chemokines, presenting antigens, participating in positive immune reactions, and playing a role in immune monitoring. M2-type macrophages are important immune cells that downregulate the immune response [21]. Damage-associated molecular patterns (DAMPs), bacterial products (e.g., lipopolysaccharide, LPS), and inflammatory cytokines (e.g., TNF-α, IFN-γ) stimulate macrophage differentiation into M1 macrophages, which exhibit antimicrobial properties by releasing inflammatory mediators such as IFN-γ, NO, and IL-6. Although important for host defense, these products can still cause severe tissue damage. Conversely, IL-4 and IL-13 stimulate the transformation of macrophages into M2-type macrophages that inhibit inflammatory and adaptive immune responses [10].
M1 and M2 macrophages promote Th1 and Th2 responses, respectively. Downstream products of Th1 and Th2 cells (such as IFN-γ and IL-4) also downregulate the activity of M2 and M1 macrophages, respectively [22]. Therefore, the M1/M2 ratio is crucial for immune regulation and is an important link in maintaining a perfect balance between innate and adaptive immunity [8]. In the future, the balance parameters of M1/M2 macrophages must be established. These could then be used as predictors of the risk assessment of pathologic scar formation.
The role of Tregs in keloid pathogenesis
Regulatory T cells (Tregs) play a central role in maintaining immune homeostasis and self-tolerance. Tregs are characterized by high levels of the forkhead family transcription factor Foxp3, which is also considered to be a reliable Treg marker. The subsets of CD4+ T cells mainly comprise type 1 helper Tregs, type 2 helper Tregs, type 17 helper Tregs [23], follicular helper Tregs, and regulatory Tregs [24]. Tregs are an important subgroup of cells with immunosuppressive functions. They can be differentiated in two ways: thymic differentiation, resulting in what are known as natural regulatory T cells (nTregs); peripheral differentiation, resulting in induced regulatory T cells (iTregs) [24]. There are many subtypes of Tregs, of which CD4+CD25+ Tregs are the most reported [25]. CD4+CD25+ Tregs must be activated by the T cell receptor to exert their inhibitory functions. Once activated, Tregs inhibit effector cells in a nonantigenic and nonspecific manner.
Studies have shown that Tregs directly inhibit the production of TGF-β by the release of IL-10 or indirectly inhibit the production of TGF-β by reducing the number of macrophages. Using coculture experiments of Tregs and keloid fibroblasts, Murao et al. [26] found that Tregs are present in keloid tissue and that the proportion of Tregs in the keloid dermis is relatively low. Using coculture experiments of Tregs and keloid fibroblasts, the presence of large quantities of Tregs reduces the expression of type I collagen and TGF-β mRNA in keloids. Jin et al. found that macrophages can promote Treg differentiation by upregulating Foxp3 expression [7]. Other studies have suggested that macrophages and Tregs act together during keloid formation. Macrophages in scar tissue are highly activated and polarized toward the M2 subtype [7].
The pathogenic process of keloids may be influenced by Tregs. Based on existing studies of Tregs, it is believed that the biochemical activity, physiologic function, and regulatory molecular mechanism of the transcription factor Foxp3 may play a crucial role in the occurrence and development of keloids. Although the pathogenic mechanism of Tregs is starting to be noticed, further research and exploration are required.
The role of CD8+ T cells in keloid pathogenesis
In the tumor immune microenvironment, decrease or disorder of anticancer immune function is a sign of tumor development. CD8+ T cells play an important role in the tumor microenvironment. Recent studies have found that when CD8+ T cells infiltrate tumor tissue, they gradually enter an undesirable state, known as T-cell exhaustion. Exhausted CD8+ T cells in tumors are characterized by reduced expression of the effector cytokines IL-2, TNF-α, and IFN-γ. Moreover, their cytolytic ability, cell viability, and proliferative capacity are also reduced. CD8+ T cell exhaustion may be attributed to the complex local microenvironment, such as the role of immunosuppressive cells and the state of physical and chemical imbalance [27].
Lymphoid aggregates of T and B cells, such as CD4+ and CD8+ T cells, are present in keloid tissue. Chen et al. used flow cytometry to analyze the subsets and functions of cells in keloid tissue and found that the number of effector memory CD8+ T cells and CD103+CD8+ resident memory T cells was increased in keloid tissue [28]. Studies investigating the role of CD8+ T cells in the pathogenesis of keloids are lacking. Therefore, the spatial distribution of CD8+ T cells in keloid tissue and the interactions of CD8+ T cells with other immune cells in the immune microenvironment should be investigated.
The role of dendritic cells in keloid pathogenesis
Dendritic cells (DCs) are the most effective antigen-presenting cells [29]. DCs can absorb and express costimulatory molecules of the presented antigen and then migrate to lymphoid organs to activate T cells, thereby initiating an immune response. DCs can not only induce immune responses but also induce immune tolerance during disease development [30]. As specialized antigen-presenting cells, they not only present antigens but also provide costimulatory molecules and cytokines for the activation and differentiation of T cells, thereby forming an immune response. DCs also interact with natural killer (NK) cells and B cells. DCs include Langerhans cells, monocyte-derived DCs (CD14+ DCs), myeloid dendritic cells, and plasma DCs [31]. Myeloid DCs have a stimulating effect, while lymphoid or plasmacytoid DCs are involved in regulation or tolerance. However, some myeloid DCs have been reported to have immunosuppressive activity and present antigens through plasmacytoid DCs [32]. DCs are thought to originate from myeloid progenitor cells shared with macrophages, known as macrophage-dendritic cell progenitors, which in turn produce common dendritic cell progenitors. Human plasmacytoid DCs express the surface markers CD123, CD303, or BDCA-2 and CD304 or BCDA-4. Human myeloid DCs are defined largely by CD1c/BDCA-1+ or CD141/BDCA-3+ expression [33].
DCs are most frequently expressed in blood and lymphoid tissue but are also widely present in other parts of the body [31]. The characteristics of human tumor-infiltrating DCs in skin melanoma have been studied. However, because patients are heterogeneous, it is difficult to clearly describe the characteristics of human tumor-infiltrating DCs. In melanoma, large numbers of DCs, which show a more mature phenotype, cluster in the area surrounding the tumor, while more immature, tumor-infiltrating DCs are found in the center of the tumor. However, the relationship between these results and clinical outcomes is not clear, as tumor-infiltrating DCs require interactions with other cells in the tumor immune microenvironment rather than just among themselves [34]. In general, mature DCs are considered to play a role in immune stimulation, while immature DCs are considered to play a role in immunosuppression and tolerance effects.
DCs play an important role in the tumor microenvironment, which is known to affect the progression of many malignant human tumors. Mature, active DCs infiltrate tumors, increasing immune activation and recruitment of disease-resistant immune effector cells and pathways. However, tumor cells have the ability to inhibit the function of DCs or alter the tumor microenvironment, allowing the recruitment of immunosuppressive DCs [35].
DCs are also involved in the initiation of keloid formation, with increased DC infiltration observed in keloid tissue [2]. A study by Onodera et al. showed that the number of FXIIIa-positive dermal DCs in the dermis covering an area of keloids was significantly higher than the corresponding hypertrophic or mature scar area. This finding suggested that FXIIIa-positive dermal DCs involved in dermal-epidermal interactions play an active role in the formation of keloids [36]. The role of DCs in the tumor immune microenvironment could be used as a reference to conduct more in-depth analyses of the pathogenic role of DCs in keloids.
The role of NK cells in keloid pathogenesis
The immune system can be divided into innate immunity and adaptive immunity. NK cells are an important part of the innate immune system and the first line of defense against viral infections and malignant tumors. NK cells are cytotoxic and can secrete various immunomodulatory cytokines and chemokines [23], mediating cytotoxicity by secreting IFN-γ and expressing TGF-β. NK cells account for between 5% and 15% of cells in the peripheral blood lymphatic circulation pool of healthy adults [23].
From a morphologic perspective, NK cells are a type of large, granular lymphocyte [37]. From the phenotypic perspective, they are mainly defined by the expression of CD56 and no expression of CD3. Among them, CD3-CD56dimCD16+ NK cells express a low-affinity receptor for the constant region of immunoglobulin G, Fc c RIIIa (CD16). Approximately 90% of peripheral blood NK cells are mature cells and mainly mediate toxic reactions. In contrast, NK cells with the CD3-CD56brightCD16- phenotype are immature cells; they are mainly present in tissues and only account for approximately 10% of total peripheral blood NK cells, with high levels of cytokine secretion and low cytotoxicity [38,39]. The different subtypes of NK cell surface receptors are quite different; therefore, their functions are not the same. Among them, CDCD56bright NK cells highly express CD94/NKG2A inhibitory receptors and NKp46 activation receptors, while CD56dim NK cells show the opposite effect [40]. CD56dim NK cells highly express CD16, perforin, granzyme, killer cell inhibitory receptor (KIR), and CD94/NKG2C activation receptors. This expression provides a material basis for the cytotoxic effects of these cells, but it is not detected or is minimally expressed in CDCD56bright NK cells [41]. It is interesting to note that CDCD56bright NK cells specifically express CC motif chemokine receptor 7 (CCR7), C-X-C motif chemokine receptor 3 (CXCR3), and CD62, so they have the ability to move to secondary lymphoid organs. CD56dim NK cells lack CCR7 expression but have high levels of CXCR1, CXCR2, CXCR3, CX3CR1, and sphingosine 1-phosphate-5 (S1P5) expression, which can lead to their entry into peripheral tissues to play an inflammatory role [42].
When the immune system detects viruses or tumor cells, NK cells bind to target cells and release cytotoxic particles containing perforin and granzyme to lyse target cells [43]. NK cells can also induce apoptosis in target cells. NK cells, through their surface Fas ligand (FasL) and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), bind to Fas and TRAIL receptors on the surface of target cells, respectively [43]. Finally, NK cells bind to target cells through CD16 and kill these cells by signal transduction, which exerts antibody-dependent cell-mediated cytotoxicity (ADCC) [44].
NK cells can also rely on certain functional receptors on the surface of NK cells to exert antitumor cytotoxicity, such as NKG2 and the KIR family [45]. In addition, NK cells produce a series of cytokines and chemokines to help regulate the immune response. By releasing CCL5, XCL1, and XCL2, NK cells can promote the accumulation of DCs in solid tumors and the antitumor activity of CD8+ T cells [46]. Following the inflammatory response, NK cells enter the tumor-draining lymph nodes to indirectly affect a T-cell response or regulate T-cell immunity by regulating DCs.
Although NK cells can identify malignantly transformed tumor cells early on and have a powerful ability to kill tumor cells, studies have shown that the ability of NK cells to target and lyse cancer cells in tumor tissues is significantly lower than that of NK cells in normal tissues. These findings suggest that the function of NK cells is impaired in the tumor microenvironment, which helps cancer cells to escape the immune response mediated by NK cells and promotes tumorigenesis and development [47,48].
NK cells may be involved in the formation of keloids. Zhou et al. found that interference with Smad7 can promote the proliferation and migration of NK cells and keloid cutaneous cells. Overexpression of Smad7 can inhibit the proliferation and migration of NK cells [49]. The role of NK cells in keloid formation remains to be further studied in the future.
Immune microenvironment of keloid fibroblasts
Keloid fibroblasts are flat and long spindle shaped, with obvious proliferation of spinous cells and increased cell layers in vivo [50]. These fibroblasts are disordered and their polarity disappears. Fibroblasts proliferate and secrete a large amount of extracellular matrix, resulting in excessive collagen synthesis and deposition [51]. Fibroblasts are the main cell population in the lesional area. Interaction between fibroblasts and other cells is thought to play an important role in the pathogenesis of keloid. Macrophages were observed to contact fibroblasts [52]. Macrophages secrete a variety of secretory products, including cytokines such as IL-1, IL-6, IL-8, tumor necrosis factor, fibroblast growth factor, and TGF-β. Monocytes cocultured with fibroblasts were skewed to an M2 macrophage-like phenotype [53]. Many factors and pathways have been found involved in fibrosis.
TGF-β can active the SMAD signaling pathway to induce fibrosis [54]. Increased expression of fibrosis-related genes such as fibronectin, CTGF, MMPs and α-SMA leads to fibroblast proliferation and extracellular matrix (ECM) deposition [55,56]. The up-regulation of growth factors VEGF and PDGF, the up-regulation of growth factors of proteases MMP2, TIMP-1 and the down-regulation of MMP8, and the up-regulation of cytokines TGF-β and TNF-α play an important role in the formation of keloids (Figure 2). In normal wound healing, IFN-γ usually antagonizes the fibrotic effect of TGF-β by increasing collagenase synthesis [55,56].
Figure 2.
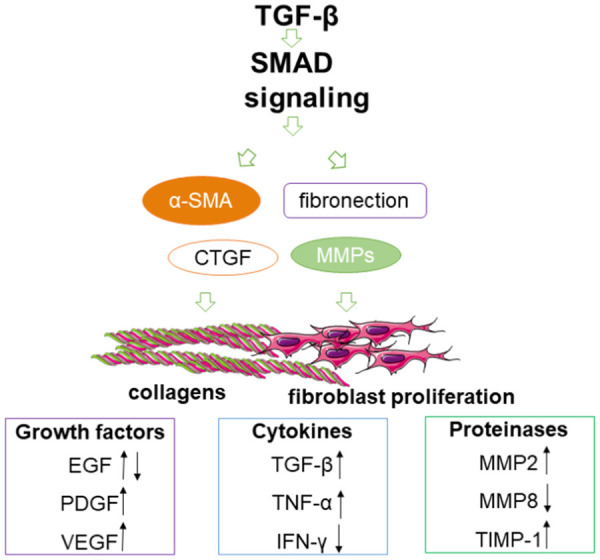
Immune microenvironment of keloid fibroblasts [52,54-56]. α-SMA, alpha smooth muscle actin; CTGF, connective tissue growth factor; MMPs, matrix metalloproteinases; TGF-β, transforming growth factor-beta; EGF, epidermal growth factor; PDGF, platelet derived growth factor; VEGF, vascular endothelial growth factor; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; TIMPs, tissue inhibitors of metalloproteinases; TIMP-1, tissue inhibitor of metalloproteinase 1.
Stem cells in keloid immune microenvironment
Stem cells are cells with unlimited self-renewal capacity and can produce highly differentiated progeny cells. Increased expression of TGF-β1 leads to vigorous proliferation of fibroblasts. These abnormal fibroblasts may be mesenchymal stem cells (MSCs) derived from epithelial-mesenchymal transition or endothelial-mesenchymal transition (EndoMT) [57,58]. In vitro, it can be differentiated into various phenotypic mature and functional pluripotent stem cells with limited differentiation ability. MSCs differentiate into abnormal fibroblasts, which are characterized by excessive fibrosis and collagen production. The inflammatory microenvironment can trigger the production of MSC intermediates that can differentiate into keloid fibroblasts.
Macrophages, lymphocytes and other inflammatory cells in the immune microenvironment secrete a variety of cytokines by up-regulating transcription factors (such as snail, slug, twist) [59]. After the newly formed mesenchymal cells lose intercellular adhesion, they have a high degree of mobility and can infiltrate surrounding tissues.
The expression of CD73, CD90, and CD105 increase in MSCs, but endothelial surface markers such as CD34 are lacking [60]. Zhang et al. [61] found a group of cells expressing higher levels of stage-specific embryonic antigen-4 (OCT4) and Octmer-4 in the dermis of scar tissue. These embryonic stem cell (ESC) markers indicate that there are keloid-derived stem cells. After injury, ESC on microvascular endothelial cells induces endothelial to mesenchymal transformation through cytokines such as TGF-β. These fibroblasts of keloids may also come from circulating bone marrow mesenchymal stem cells that migrate to target tissues through EndoMT. Elevated levels of cytokines IL-6 and IL-17 have been shown to increase the number of benign tumor-like stem cells [61].
Age-related immune microenvironment in keloid
In aging skin, the number and functions of antigen-presenting cells (such as Langerhans cells, mast cells) decrease with age [33,62]. The content of collagen in dermis will also decrease with age. In elderly animals, fibroblasts, vascular endothelial cells and keratinocytes have reduced ability to react to prolife rative responses [63,64]. Senescent fibroblasts have a weakened response to growth factors [65]. Re-epithelialization, collagen synthesis, and angiogenesis show a downward trend with aging [65]. Clarifying the changes in the age-related immune microenvironment in the pathogenesis of keloids may help to explain the low incidence and mild symptoms of keloids in the elderly and disclose the mechanism in young patients.
Conclusion
The immune microenvironment is mainly composed of keloid fibroblasts and surrounding immune cells. Immune cells infiltrate the microenvironment and regulate keloid fibroblasts by secreting cytokines. The genesis and development of keloids is the result of the coevolution of keloid fibroblasts and their surrounding immune cells. Immune cells in the microenvironment interact with each other to regulate the immune process and play a role in promoting or inhibiting the development of keloids. In this article, the immune cells commonly found in the keloid tumor microenvironment have been reviewed with a focus on the influence of these immune cells on the pathogenesis of keloids.
Future direction
Current studies have mostly focused on changes in the number of single types of immune cells and the effects of these changes. In the future, studies should be conducted that focus on the mechanism by which multiple immune cells interact, more in line with the immune regulation that occurs in humans. The exploration of immune cells that may play an important role in keloid formation will be beneficial for the development of keloid immunotherapy.
Acknowledgements
This study was supported by The National Natural Science Foundation of China (81871538) and Beijing Municipal Commission of Science and Technology (Z191100006619009).
Disclosure of conflict of interest
None.
References
- 1.Andrews JP, Marttala J, Macarak E, Rosenbloom J, Uitto J. Keloids: the paradigm of skin fibrosis - Pathomechanisms and treatment. Matrix Biol. 2016;51:37–46. doi: 10.1016/j.matbio.2016.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wu J, Del Duca E, Espino M, Gontzes A, Cueto I, Zhang N, Estrada YD, Pavel AB, Krueger JG, Guttman-Yassky E. RNA sequencing keloid transcriptome associates keloids With Th2, Th1, Th17/Th22, and JAK3-skewing. Front Immunol. 2020;11:597741. doi: 10.3389/fimmu.2020.597741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ogawa R. Keloid and hypertrophic scars are the result of chronic inflammation in the reticular dermis. Int J Mol Sci. 2017;18:606. doi: 10.3390/ijms18030606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jaloux C, Bertrand B, Degardin N, Casanova D, Kerfant N, Philandrianos C. Keloid scars (part II): treatment and prevention. Ann Chir Plast Esthet. 2017;62:87–96. doi: 10.1016/j.anplas.2016.04.006. [DOI] [PubMed] [Google Scholar]
- 5.Bayat A, Arscott G, Ollier WE, McGrouther DA, Ferguson MW. Keloid disease: clinical relevance of single versus multiple site scars. Br J Plast Surg. 2005;58:28–37. doi: 10.1016/j.bjps.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 6.Tsai CH, Ogawa R. Keloid research: current status and future directions. Scars Burn Heal. 2019;5:2059513119868659. doi: 10.1177/2059513119868659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jin Q, Gui L, Niu F, Yu B, Lauda N, Liu J, Mao X, Chen Y. Macrophages in keloid are potent at promoting the differentiation and function of regulatory T cells. Exp Cell Res. 2018;362:472–476. doi: 10.1016/j.yexcr.2017.12.011. [DOI] [PubMed] [Google Scholar]
- 8.Tardito S, Martinelli G, Soldano S, Paolino S, Pacini G, Patane M, Alessandri E, Smith V, Cutolo M. Macrophage M1/M2 polarization and rheumatoid arthritis: a systematic review. Autoimmun Rev. 2019;18:102397. doi: 10.1016/j.autrev.2019.102397. [DOI] [PubMed] [Google Scholar]
- 9.Tang Y, Le W. Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol Neurobiol. 2016;53:1181–1194. doi: 10.1007/s12035-014-9070-5. [DOI] [PubMed] [Google Scholar]
- 10.Martinez FO, Sica A, Mantovani A, Locati M. Macrophage activation and polarization. Front Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 11.Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262:153–166. doi: 10.1111/imr.12218. [DOI] [PubMed] [Google Scholar]
- 12.Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223:383–396. doi: 10.1016/j.imbio.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huang X, Li Y, Fu M, Xin HB. Polarizing macrophages in vitro. Methods Mol Biol. 2018;1784:119–126. doi: 10.1007/978-1-4939-7837-3_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Butzelaar L, Niessen FB, Talhout W, Schooneman D, Ulrich MM, Beelen R, Mink van der Molen AB. Different properties of skin of different body sites: the root of keloid formation. Wound Repair Regen. 2017;25:758–766. doi: 10.1111/wrr.12574. [DOI] [PubMed] [Google Scholar]
- 15.Vidyarthi A, Agnihotri T, Khan N, Singh S, Tewari MK, Radotra BD, Chatterjee D, Agrewala JN. Predominance of M2 macrophages in gliomas leads to the suppression of local and systemic immunity. Cancer Immunol Immunother. 2019;68:1995–2004. doi: 10.1007/s00262-019-02423-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Genin M, Clement F, Fattaccioli A, Raes M, Michiels C. M1 and M2 macrophages derived from THP-1 cells differentially modulate the response of cancer cells to etoposide. BMC Cancer. 2015;15:577. doi: 10.1186/s12885-015-1546-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44:450–462. doi: 10.1016/j.immuni.2016.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mantovani A, Sica A, Locati M. Macrophage polarization comes of age. Immunity. 2005;23:344–346. doi: 10.1016/j.immuni.2005.10.001. [DOI] [PubMed] [Google Scholar]
- 19.De Felice B, Manfellotto F, Garbi C, Santoriello M, Nacca M. miR-34 modulates apoptotic gene expression in Ingenol mebutate treated keloid fibroblasts. Mol Med Rep. 2018;17:7081–7088. doi: 10.3892/mmr.2018.8749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Wang SY, Kwak G, Yang Y, Kwon IC, Kim SH. Exosome-guided phenotypic switch of M1 to M2 macrophages for cutaneous wound healing. Adv Sci (Weinh) 2019;6:1900513. doi: 10.1002/advs.201900513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fukui S, Iwamoto N, Takatani A, Igawa T, Shimizu T, Umeda M, Nishino A, Horai Y, Hirai Y, Koga T, Kawashiri SY, Tamai M, Ichinose K, Nakamura H, Origuchi T, Masuyama R, Kosai K, Yanagihara K, Kawakami A. M1 and M2 monocytes in rheumatoid arthritis: a contribution of imbalance of M1/M2 monocytes to osteoclastogenesis. Front Immunol. 2017;8:1958. doi: 10.3389/fimmu.2017.01958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li X, Wang Y, Yuan B, Yang H, Qiao L. Status of M1 and M2 type macrophages in keloid. Int J Clin Exp Pathol. 2017;10:11098–11105. [PMC free article] [PubMed] [Google Scholar]
- 23.Björkström NK, Ljunggren HG, Michaëlsson J. Emerging insights into natural killer cells in human peripheral tissues. Nat Rev Immunol. 2016;16:310–320. doi: 10.1038/nri.2016.34. [DOI] [PubMed] [Google Scholar]
- 24.Tanaka A, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Cell Res. 2017;27:109–118. doi: 10.1038/cr.2016.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walker LS. CD4+ CD25+ Treg: divide and rule. Immunology. 2004;111:129–137. doi: 10.1111/j.0019-2805.2003.01788.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murao N, Seino K, Hayashi T, Ikeda M, Funayama E, Furukawa H, Yamamoto Y, Oyama A. Treg-enriched CD4+ T cells attenuate collagen synthesis in keloid fibroblasts. Exp Dermatol. 2014;23:266–271. doi: 10.1111/exd.12368. [DOI] [PubMed] [Google Scholar]
- 27.Zander R, Schauder D, Xin G, Nguyen C, Wu X, Zajac A, Cui W. CD4(+) T cell help is required for the formation of a cytolytic CD8(+) T cell subset that protects against chronic infection and cancer. Immunity. 2019;51:1028–1042. e4. doi: 10.1016/j.immuni.2019.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen Z, Zhou L, Won T, Gao Z, Wu X, Lu L. Characterization of CD45RO(+) memory T lymphocytes in keloid disease. Br J Dermatol. 2018;178:940–950. doi: 10.1111/bjd.16173. [DOI] [PubMed] [Google Scholar]
- 29.Reis e Sousa C. Dendritic cells in a mature age. Nat Rev Immunol. 2006;6:476–483. doi: 10.1038/nri1845. [DOI] [PubMed] [Google Scholar]
- 30.Collin M, Bigley V. Human dendritic cell subsets: an update. Immunology. 2018;154:3–20. doi: 10.1111/imm.12888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5:296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- 32.Ma Y, Shurin GV, Peiyuan Z, Shurin MR. Dendritic cells in the cancer microenvironment. J Cancer. 2013;4:36–44. doi: 10.7150/jca.5046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y, Jin Q, Fu X, Qiao J, Niu F. Connection between T regulatory cell enrichment and collagen deposition in keloid. Exp Cell Res. 2019;383:111549. doi: 10.1016/j.yexcr.2019.111549. [DOI] [PubMed] [Google Scholar]
- 34.Klarquist JS, Janssen EM. Melanoma-infiltrating dendritic cells: limitations and opportunities of mouse models. Oncoimmunology. 2012;1:1584–1593. doi: 10.4161/onci.22660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tran Janco JM, Lamichhane P, Karyampudi L, Knutson KL. Tumor-infiltrating dendritic cells in cancer pathogenesis. J Immunol. 2015;194:2985–2991. doi: 10.4049/jimmunol.1403134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Onodera M, Ueno M, Ito O, Suzuki S, Igawa HH, Sakamoto H. Factor XIIIa-positive dermal dendritic cells in keloids and hypertrophic and mature scars. Pathol Int. 2007;57:337–342. doi: 10.1111/j.1440-1827.2007.02105.x. [DOI] [PubMed] [Google Scholar]
- 37.Mandal A, Viswanathan C. Natural killer cells: in health and disease. Hematol Oncol Stem Cell Ther. 2015;8:47–55. doi: 10.1016/j.hemonc.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 38.Rizzo R, Zatelli MC, Rotola A, Cassai E, Degli Uberti E, Di Luca D, Caselli E. Increase in peripheral CD3-CD56brightCD16- natural killer cells in Hashimoto’s thyroiditis associated with HHV-6 infection. Adv Exp Med Biol. 2016;897:113–120. doi: 10.1007/5584_2015_5010. [DOI] [PubMed] [Google Scholar]
- 39.Becker PS, Suck G, Nowakowska P, Ullrich E, Seifried E, Bader P, Tonn T, Seidl C. Selection and expansion of natural killer cells for NK cell-based immunotherapy. Cancer Immunol Immunother. 2016;65:477–484. doi: 10.1007/s00262-016-1792-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Della Chiesa M, Pesce S, Muccio L, Carlomagno S, Sivori S, Moretta A, Marcenaro E. Features of memory-like and PD-1(+) human NK cell subsets. Front Immunol. 2016;7:351. doi: 10.3389/fimmu.2016.00351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carrega P, Bonaccorsi I, Di Carlo E, Morandi B, Paul P, Rizzello V, Cipollone G, Navarra G, Mingari MC, Moretta L, Ferlazzo G. CD56(bright)perforin(low) noncytotoxic human NK cells are abundant in both healthy and neoplastic solid tissues and recirculate to secondary lymphoid organs via afferent lymph. J Immunol. 2014;192:3805–3815. doi: 10.4049/jimmunol.1301889. [DOI] [PubMed] [Google Scholar]
- 42.Michel T, Poli A, Cuapio A, Briquemont B, Iserentant G, Ollert M, Zimmer J. Human CD56bright NK cells: an update. J Immunol. 2016;196:2923–2931. doi: 10.4049/jimmunol.1502570. [DOI] [PubMed] [Google Scholar]
- 43.Prager I, Watzl C. Mechanisms of natural killer cell-mediated cellular cytotoxicity. J Leukoc Biol. 2019;105:1319–1329. doi: 10.1002/JLB.MR0718-269R. [DOI] [PubMed] [Google Scholar]
- 44.Poli A, Michel T, Thérésine M, Andrès E, Hentges F, Zimmer J. CD56bright natural killer (NK) cells: an important NK cell subset. Immunology. 2009;126:458–465. doi: 10.1111/j.1365-2567.2008.03027.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pegram HJ, Andrews DM, Smyth MJ, Darcy PK, Kershaw MH. Activating and inhibitory receptors of natural killer cells. Immunol Cell Biol. 2011;89:216–224. doi: 10.1038/icb.2010.78. [DOI] [PubMed] [Google Scholar]
- 46.Böttcher JP, Bonavita E, Chakravarty P, Blees H, Cabeza-Cabrerizo M, Sammicheli S, Rogers NC, Sahai E, Zelenay S, Reis E Sousa C. NK cells stimulate recruitment of cDC1 into the tumor microenvironment promoting cancer immune Control. Cell. 2018;172:1022–1037. e14. doi: 10.1016/j.cell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.MacFarlane AW 4th, Jillab M, Smith MR, Alpaugh RK, Cole ME, Litwin S, Millenson MM, Al-Saleem T, Cohen AD, Campbell KS. NK cell dysfunction in chronic lymphocytic leukemia is associated with loss of the mature cells expressing inhibitory killer cell Ig-like receptors. Oncoimmunology. 2017;6:e1330235. doi: 10.1080/2162402X.2017.1330235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gill S, Vasey AE, De Souza A, Baker J, Smith AT, Kohrt HE, Florek M, Gibbs KD Jr, Tate K, Ritchie DS, Negrin RS. Rapid development of exhaustion and down-regulation of eomesodermin limit the antitumor activity of adoptively transferred murine natural killer cells. Blood. 2012;119:5758–5768. doi: 10.1182/blood-2012-03-415364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhou XF, Zhao YN, Xiao MQ, Xiao H, Liu Y. Effects of regulating Smad7 gene on epithelial-mesenchymal transition in keloid keratinocyte. Sichuan Da Xue Xue Bao Yi Xue Ban. 2020;51:790–796. doi: 10.12182/20201160104. [DOI] [PubMed] [Google Scholar]
- 50.Wang Q, Wang P, Qin Z, Yang X, Pan B, Nie F, Bi H. Altered glucose metabolism and cell function in keloid fibroblasts under hypoxia. Redox Biol. 2021;38:101815. doi: 10.1016/j.redox.2020.101815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Euler T, Valesky EM, Meissner M, Hrgovic I, Kaufmann R, Kippenberger S, Zöller NN. Normal and keloid fibroblasts are differentially influenced by IFN-γ and triamcinolone as well as by their combination. Wound Repair Regen. 2019;27:450–461. doi: 10.1111/wrr.12722. [DOI] [PubMed] [Google Scholar]
- 52.Shaker SA, Ayuob NN, Hajrah NH. Cell talk: a phenomenon observed in the keloid scar by immunohistochemical study. Appl Immunohistochem Mol Morphol. 2011;19:153–159. doi: 10.1097/PAI.0b013e3181efa2ef. [DOI] [PubMed] [Google Scholar]
- 53.Limandjaja GC, Waaijman T, Roffel S, Niessen FB, Gibbs S. Monocytes co-cultured with reconstructed keloid and normal skin models skew towards M2 macrophage phenotype. Arch Dermatol Res. 2019;311:615–627. doi: 10.1007/s00403-019-01942-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nguyen JK, Austin E, Huang A, Mamalis A, Jagdeo J. The IL-4/IL-13 axis in skin fibrosis and scarring: mechanistic concepts and therapeutic targets. Arch Dermatol Res. 2020;312:81–92. doi: 10.1007/s00403-019-01972-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Boyce DE, Ciampolini J, Ruge F, Murison MS, Harding KG. Inflammatory-cell subpopulations in keloid scars. Br J Plast Surg. 2001;54:511–516. doi: 10.1054/bjps.2001.3638. [DOI] [PubMed] [Google Scholar]
- 56.Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010;18:139–153. doi: 10.1111/j.1524-475X.2009.00553.x. [DOI] [PubMed] [Google Scholar]
- 57.Lee WJ, Park JH, Shin JU, Noh H, Lew DH, Yang WI, Yun CO, Lee KH, Lee JH. Endothelial-to-mesenchymal transition induced by Wnt 3a in keloid pathogenesis. Wound Repair Regen. 2015;23:435–442. doi: 10.1111/wrr.12300. [DOI] [PubMed] [Google Scholar]
- 58.Lim KH, Itinteang T, Davis PF, Tan ST. Stem cells in keloid lesions: a review. Plast Reconstr Surg Glob Open. 2019;7:e2228. doi: 10.1097/GOX.0000000000002228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Potenta S, Zeisberg E, Kalluri R. The role of endothelial-to-mesenchymal transition in cancer progression. Br J Cancer. 2008;99:1375–1379. doi: 10.1038/sj.bjc.6604662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nombela-Arrieta C, Ritz J, Silberstein LE. The elusive nature and function of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2011;12:126–131. doi: 10.1038/nrm3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang Q, Yamaza T, Kelly AP, Shi S, Wang S, Brown J, Wang L, French SW, Shi S, Le AD. Tumor-like stem cells derived from human keloid are governed by the inflammatory niche driven by IL-17/IL-6 axis. PLoS One. 2009;4:e7798. doi: 10.1371/journal.pone.0007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gosain A, DiPietro LA. Aging and wound healing. World J Surg. 2004;28:321–326. doi: 10.1007/s00268-003-7397-6. [DOI] [PubMed] [Google Scholar]
- 63.Reed MJ, Ferara NS, Vernon RB. Impaired migration, integrin function, and actin cytoskeletal organization in dermal fibroblasts from a subset of aged human donors. Mech Ageing Dev. 2001;122:1203–1220. doi: 10.1016/s0047-6374(01)00260-3. [DOI] [PubMed] [Google Scholar]
- 64.Puolakkainen PA, Twardzik DR, Ranchalis JE, Pankey SC, Reed MJ, Gombotz WR. The enhancement in wound healing by transforming growth factor-beta 1 (TGF-beta 1) depends on the topical delivery system. J Surg Res. 1995;58:321–329. doi: 10.1006/jsre.1995.1050. [DOI] [PubMed] [Google Scholar]
- 65.West MD. The cellular and molecular biology of skin aging. Arch Dermatol. 1994;130:87–95. [PubMed] [Google Scholar]