

Abstract
Background: Stiff-person syndrome (SPS) manifests by progressive rigidity along with muscle spasms that affect the axial and limb muscles. First discovered in 1956, significant progress has been made in its clinical characterization, comprehension of pathogenesis, as well as effective treatment therapy. Case presentation: A 67-year old female patient presented with a 2-year history of progressive stiffness along with painful spasms in both legs, with her condition worsening over the previous year making it considerably difficult for her to stand and walk. Here, we report a Stiff-person syndrome patient (SPS) with lung adenocarcinoma who was positive for anti-glutamate decarboxylase (anti-GAD) antibodies. Treatment with hormones and gamma-globulin improved her symptoms. In addition, we present a literature review of SPS patients with tumors. Conclusions: The diagnosis of autoimmune SPS was on the basis of clinical, electrophysiological, as well as immunological findings. Early SPS detection is critical to preventing long-term disability.
Keywords: Muscle stiffness, stiff person syndrome, rigidity, anti-glycine receptor antibody, paraneoplastic neurological syndrome, lung adenocarcinoma
Introduction
Stiff-person syndrome (SPS) is a rarely disabling central nervous system disorder that manifests with muscle stiffness, rigidity, and episodic painful spasms of the proximal and axial limb muscles. Circulating anti-GAD65 (glutamic acid decarboxylase) antibodies are characteristic of the disorder and exhibit distinct epitope specificity, potentially inhibiting glutamate decarboxylase and GABA synthesis. GABA is the primary suppressive neurotransmitter in the brain and reduced GABA levels may cause muscle hyperfunction [1]. Some cases of paraneoplastic SPS occur in association with antiamphiphysin antibodies. In paratumor SPS, the cross-reactive binding of serum antibodies to malignant cells expressing neuronal antigens (e.g. GAD and amphoteric fibrin) may trigger an autoimmune response. Although SPS research has made great progress in recent years, due to its variable clinical manifestations the workload of differential diagnosis is large, increasing misdiagnosis. Additionally, SPS is sometimes accompanied by tumors, including thymoma [2,3], Hodgkin’s lymphoma [4-6], small cell lung cancer [6,7], and breast cancer [8]. Herein, we report a case of GAD antibody-positive SPS associated with lung adenocarcinoma.
Case report
This SPS case report involves a 67-year-old female patient who was admitted to our hospital’s neurology clinic with the complaint of painful muscle contractions in 2018. She had experienced sudden, occasional pain in both heels for about 2 years, before the pain spread upwards. Symptoms first appeared in 2016, with a one-year history of irregular right lower limb stiffness that the patient described as ‘painful spasms’, resulting in poor sleep at night. She experienced reflex contraction of the muscles of the right lower limb, with the right foot held in plantar flexion. She then began walking unsteadily and was unable to turn over independently, leading to admission at another hospital. Except for muscle stiffness, there was no obvious numbness or weakness in lower limbs. The rest of her neurological assessment was normal. The patient had no medical or medication history for the condition. She felt stiff from the groin to feet. During that time, spasm and spasticity of her lower limbs gradually deteriorated, making it considerably difficult for her to stand and walk. The patient denied having any trauma, infection, poisoning, drugs, mental illness, and family history of the condition. Due to misdiagnosis as osteoporosis and Parkinson’s disease, the patient was initially treated with glucosamine, sulfate, elcatonin, celebrex, levodopa, and gabapentin but these were ineffective. Within the year prior to the initial hospital visit, she had fallen down several times due to muscle spasms. The main focus during the patient’s visit was SPS, which she reported was exacerbated by tension beyond her control and vice versa. Upon examination, her pulse rate, oxygen saturation, blood pressure, temperature and respiratory rate, and were 78 beats/min, 100%, 145/83 mmHg, 36.8°C, and 18 breaths/min, respectively. Her breath sounds were clear on chest auscultation. Her painful spasms included lumbar stiffness due to agonist and antagonistic muscle contractions and high lumbar hyperactivity. During neurological examination, her lower limb strength was measured by manual muscle test (MMT) of 4/5, with severe spasms and hyper-reflexes. Babinski and Chaddock’s reflexes were hard to assess due to spasms. The patient had no sensory disturbances. Initial laboratory examinations revealed: white blood cell count = 4,700/mm3; hemoglobin = 11.9 mg/dL, potassium = 4.0 mEq/L, platelet count = 23.4×103, C-reactive protein = 0.8 mg/dL, ALT (alanine aminotransferase) = 14 IU/L, lactate dehydrogenase = 180.3 IU/L, creatine = 50.4 umol/L, uric acid = 260 umol/L, sodium = 142 mEq/Laspartate aminotransferase = 17 international units (IU)/L, chloride = 104.9 mEq/L, glucose = 4.5 mmol/L, HbA1c = 5.5%, IgA = 2.94 g/L, IgG = 13.69 g/L, IgM = 0.84 g/L, prothrombin time = 11.7 sec, INR = 1.01 (normal INR = 1-2, activated partial thromboplastin time = 27.3 sec, and vitamin B12 = 815 pg/mL. The exception was positive anti-GAD antibodies in serum titers at 1:10. Spinal MRI findings were normal. Electromyography (EMG) evaluation showed continuous motor unit activity in both agonist and antagonist muscles. Electroencephalography (EEG) revealed no epileptic discharge. Brain MRI (magnetic resonance imaging) was normal. Cancer screening tests were all negative. On the basis of the medical history and auxiliary examination results, the patient was diagnosed with SPS based on clinical SPS diagnosis criteria by Dalakas et al.
Based on clinical history and the significant muscle spasms with stimulation, 0.5 mg clonazepam, twice daily, was added to her medication regimen. In the next days, the patient reported that clonazepam reduced stiffness onset. The patient also received intravenous immune globulin (10 g/day) for 3 days. Upon clonazepam dose increase to 3 times daily, she reported reduced frequency of anxiety-triggered spasms and less stiffness. During her first hospitalization a CT scan of the thorax revealed a 1.0-cm mass in the left inferior lobe anterior segment. Chest CT scan revealed ground-glass opacities, but the patient refused further PET-CT scan due to cost and improved symptoms.
After discharge, the patient remained on 1 mg oral clonazepine before bed. During follow-up, compatible with the last CT results, a CT scan done in July 2020 revealed a ground glass nodule in the lungs suspicious for early adenocarcinoma (Figure 1). The patient underwent left inferior lobectomy in August 2020 with pathological result revealing a well-differentiated adenocarcinoma with bronchoalveolar features (Figure 2). A molecular evaluation of genetic changes in EGFR (exon 21, 20, 19, and 18), ROS-1 and ALK genes. PD-L1 immunohistochemistry (ICH) analysis of the primary tumor as a new biopsy was not feasible technically. Rare complex EGFR mutations (exon 20 V769M and exon 18 G719A) were detected by PCR-based Sanger sequencing. No alterations were reported as EGFR, ROS-1 D4D6 ICH and DAKO ALK were all negative. Similarly, DAKO PDL-1 ICH 22 C3 was negative (Tumor proportion score 0%). The patient’s symptoms markedly improved in December 2020.
Figure 1.
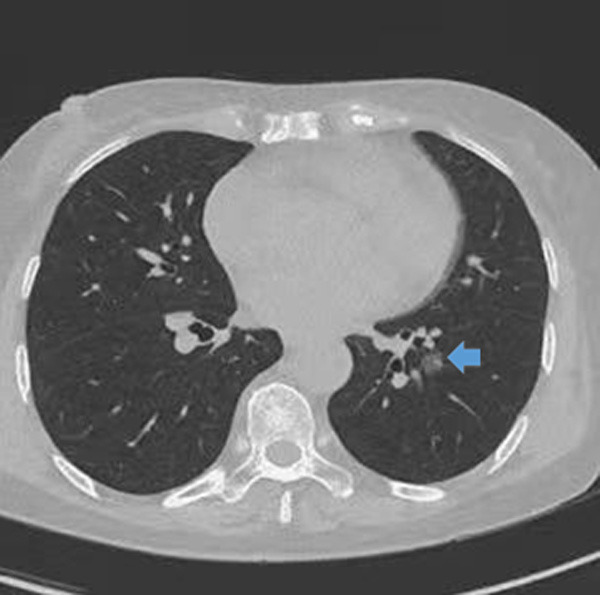
Computed tomography scan of the thorax showing a 1 cm nodular focus of ground-glass attenuation left inferior lobe anterior segment.
Figure 2.
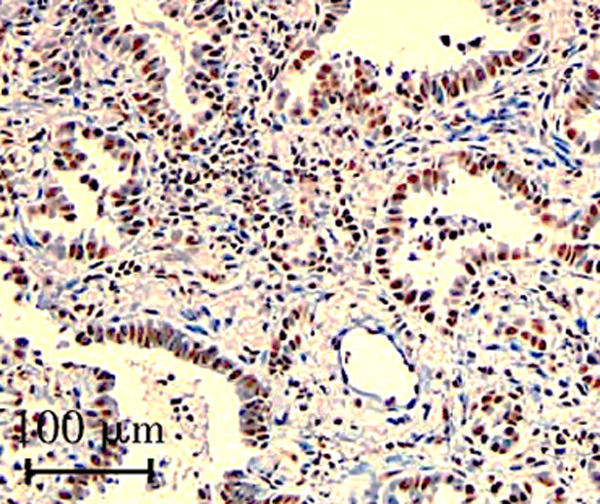
Pathologic findings of left inferior lobe specimen showing well-differentiated adenocarcinoma (Hematoxylin and Eosin, ×200). Black scale bar, 100 µm.
Discussion
SPS is an uncommon neurological auto-immune disorder that manifests with stiffness, rigidity along with spontaneous reflex- or action-triggered painful spasms in axial and proximal limb muscles. Initially reported in 1956 by Moersch and Woltman who described fourteen patients with fluctuating, but progressive rigidity along with painful-muscle spasms, causing gait difficulties, falls, and a ‘wooden-man’ appearance, hence the term ‘stiff man syndrome’. SPS prevalence is unknown but its incidence is estimated at about 1 in 1,000,000 individuals in both genders although it predominantly affects females [9]. SPS symptoms become apparent at 30-60 years of age [10]. SPS is clinically diagnosed based on high suspicion. Although this case did not meet the 2 minor criteria, SPS was diagnosed based on the main clinical manifestations and clear continuous motor activity of the agonist along with antagonist muscles on EMG [11]. SPS diagnosis is on the basis of the following clinical criteria: 1) muscular rigidity in the limbs, as well as the axial (trunk) muscles, dominant in the thoracolumbar and abdominal paraspinals, 2) persistent co-contraction of agonist, as well as antagonist muscles, verified electrophysiologically and clinically, 3) positive anti-GAD65 (glutamic acid decarboxylase-65, or amphiphysin) antibodies as per radioimmunoassay, immunocytochemistry, or western blot, 4) absence of any other neurologic disease to account for the rigidity and stiffness, and 5) episodes of spasms caused by unexpected noise, emotional upset, or tactile stimuli [12]. Clinical assessment reveals rigidity of the limbs with no extrapyramidal or pyramidal tract manifestations. SPS is often linked to other autoimmune diseases, as well as autoantibodies. In 5% of the patients, SPS has a paraneoplastic symptom. Such patients might have more pronounced stiffness in the arms and neck [1]. Diagnosis errors are common, including misdiagnosis as Parkinson’s disease, multiple sclerosis, or primary lateral sclerosis.
However, few cases of SPS patient with early lung cancer characterized by lung GGO change are known. Clinical SPS diagnosis strongly relies on understanding the major clinical manifestations of rigidity, as well as normal motor unit potential (MUPs), typical EMG (EMG), continued involuntary discharge performance, and autoantibody-positive serology. Important diagnostic basis for this disease include EMG characteristics and clinical manifestations. At rest, EMG can detect sustained normal motor unit activity, with significant increase in seizures. The disease usually begins insidiously, with intermittent muscle tension or pain, and eventually develops into increased muscle tension or rigidity associated with continuous co-contraction of the antagonistic muscle groups. This leads to limited range of motion, slow spontaneous movement, muscle hypertrophy, and characteristic lumbar anterior protrusion postures. SPS often develops from the trunk to the near end, and then to the distal limb muscles, affecting gait and balance. Patients may also experience muscle spasms due to emotional distress, shock (triggered by tactile, visual, and auditory, stimuli), or sudden contraction of neighboring muscles [13]. Five percent of SPS cases are associated with malignancies, especially neoplasms of the breast, thymus, colon, and lung, as well as Hodgkin’s lymphoma [14]. In patients presenting SMS without particular causes, general examinations, especially chest examinations, are essential for diagnosis. In this case, a chest CT scan revealed ground glass opacities in both lungs, but the patient refused further PET-CT scan and lumbar puncture. The initial anti-GAD antibodies autoantibody panel was positive. Nevertheless, we finally confirmed that the patient had evidence of paraneoplastic syndrome based on biological tests and imaging.
SPS treatment is based on symptomatic treatment of triads, immunotherapy, and cancer treatment as appropriate. Treatment involves drugs like diazepam (5-75 mg orally, 4 times a day) to enhance γ-GABAergic transmission. Baclofen, vigabatrin, sodium valproate, and gabapentin may also help treat refractory or severe cases. Intravenous immunoglobulin therapy is sometimes effective for patients [14].
By the end of treatment, the patient had significantly better mobility with resolved muscle spasms and was discharged to inpatient rehabilitation. Had the patient not responded to first-line treatment, IVIg [15] and therapeutic plasma exchange (TPE) [16] immunomodulators like rituximab were likely to be the next best steps in management. The latest evidence from case reports and case series suggests that about half of SPS patients benefit from TPE treatment. Interestingly, manifestations have been documented to improve or even disappear with cancer treatment [17]. For patients who paraneoplastic syndromes cannot be completely ruled out, regular follow ups should be used to assess their clinical state and surveillance of cancer occurrence.
Conclusion
Stiff person syndrome (SPS) ranges from mild to severe and can be disabling if untreated. Despite advances in comprehending and treating SPS, it remains underdiagnosed, delaying treatment. Thus, increased awareness of SPS among practicing physicians is necessary.
Disclosure of conflict of interest
Written consent was obtained from the patient. The authors certify that they have obtained all appropriate patient consent forms. The patient gave consent for the images and other clinical information to be reported in the journal. The patient understands that their name and initials will not be published and due efforts will be made to conceal their identity, but anonymity cannot be guaranteed.
References
- 1.Dalakas MC. Stiff person syndrome: advances in pathogenesis and therapeutic interventions. Curr Treat Options Neurol. 2009;11:102–110. doi: 10.1007/s11940-009-0013-9. [DOI] [PubMed] [Google Scholar]
- 2.Morise S, Nakamura M, Morita JI, Miyake K, Kunieda T, Kaneko S, Kusaka H. Thymoma-associated progressive encephalomyelitis with rigidity and myoclonus (PERM) with myasthenia gravis. Intern Med. 2017;56:1733–1737. doi: 10.2169/internalmedicine.56.7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ozaki K, Ohkubo T, Yamada T, Yoshioka K, Ichijo M, Majima T, Kudo S, Akashi T, Honda K, Ito E, Watanabe M, Sekine M, Hamagaki M, Eishi Y, Sanjo N, Ishibashi S, Mizusawa H, Yokota T. Progressive encephalomyelitis with rigidity and myoclonus resolving after thymectomy with subsequent anasarca: an autopsy case. Intern Med. 2018;57:3451–3458. doi: 10.2169/internalmedicine.1238-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Borellini L, Lanfranconi S, Bonato S, Trezzi I, Franco G, Torretta L, Bresolin N, Di Fonzo AB. Progressive encephalomyelitis with rigidity and myoclonus associated with Anti-GlyR antibodies and hodgkin’s lymphoma: a case report. Front Neurol. 2017;8:401. doi: 10.3389/fneur.2017.00401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tchapyjnikov D, Borst AJ. Immune-related neurological symptoms in an adolescent patient receiving the checkpoint inhibitor nivolumab. J Immunother. 2017;40:286–288. doi: 10.1097/CJI.0000000000000177. [DOI] [PubMed] [Google Scholar]
- 6.Spitz M, Ferraz HB, Barsottini OG, Gabbai AA. Progressive encephalomyelitis with rigidity: a paraneoplastic presentation of oat cell carcinoma of the lung. Case report. Arq Neuropsiquiatr. 2004;62:547–549. doi: 10.1590/s0004-282x2004000300033. [DOI] [PubMed] [Google Scholar]
- 7.Kyskan R, Chapman K, Mattman A, Sin D. Antiglycine receptor antibody and encephalomyelitis with rigidity and myoclonus (PERM) related to small cell lung cancer. BMJ Case Rep. 2013;2013:bcr2013010027. doi: 10.1136/bcr-2013-010027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dubey D, Jitprapaikulsan J, Bi H, Do Campo RV, McKeon A, Pittock SJ, Engelstad JK, Mills JR, Klein CJ. Amphiphysin-IgG autoimmune neuropathy: a recognizable clinicopathologic syndrome. Neurology. 2019;93:e1873–e1880. doi: 10.1212/WNL.0000000000008472. [DOI] [PubMed] [Google Scholar]
- 9.Smith SR, Fu JB. Paraneoplastic stiff person syndrome: Inpatient rehabilitation outcomes of a rare disease from two cancer rehabilitation programmes. J Rehabil Med. 2016;48:639–642. doi: 10.2340/16501977-2089. [DOI] [PubMed] [Google Scholar]
- 10.Hadavi S, Noyce AJ, Leslie RD, Giovannoni G. Stiff person syndrome. Pract Neurol. 2011;11:272–282. doi: 10.1136/practneurol-2011-000071. [DOI] [PubMed] [Google Scholar]
- 11.Rakocevic G, Floeter MK. Autoimmune stiff person syndrome and related myelopathies: understanding of electrophysiological and immunological processes. Muscle Nerve. 2012;45:623–634. doi: 10.1002/mus.23234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McCabe DJ, Turner NC, Chao D, Leff A, Gregson NA, Womersley HJ, Mak I, Perkin GD, Schapira AH. Paraneoplastic “stiff person syndrome” with metastatic adenocarcinoma and anti-Ri antibodies. Neurology. 2004;62:1402–1404. doi: 10.1212/01.wnl.0000123694.64121.d5. [DOI] [PubMed] [Google Scholar]
- 13.Cardoso F, Eduardo C, Silva AP, Mota CC. Chorea in fifty consecutive patients with rheumatic fever. Mov Disord. 1997;12:701–703. doi: 10.1002/mds.870120512. [DOI] [PubMed] [Google Scholar]
- 14.Damato V, Balint B, Kienzler AK, Irani SR. The clinical features, underlying immunology, and treatment of autoantibody-mediated movement disorders. Mov Disord. 2018;33:1376–1389. doi: 10.1002/mds.27446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dalakas MC. Invited article: inhibition of B cell functions: implications for neurology. Neurology. 2008;70:2252–2260. doi: 10.1212/01.wnl.0000313840.27060.bf. [DOI] [PubMed] [Google Scholar]
- 16.Sarva H, Deik A, Ullah A, Severt WL. Clinical spectrum of stiff person syndrome: a review of recent reports. Tremor Other Hyperkinet Mov (N Y) 2016;6:340. doi: 10.7916/D85M65GD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, Pittock SJ. Stiff-man syndrome and variants: clinical course, treatments, and outcomes. Arch Neurol. 2012;69:230–238. doi: 10.1001/archneurol.2011.991. [DOI] [PubMed] [Google Scholar]