

Abstract
One of the main challenges in drug discovery is predicting protein-ligand binding affinity. Recently, machine learning approaches have made substantial progress on this task. However, current methods of model evaluation are overly optimistic in measuring generalization to new targets, and there does not exist a standard dataset of sufficient size to compare performance between models. We present a new dataset for structure-based machine learning, the CrossDocked2020 set, with 22.5 million poses of ligands docked into multiple similar binding pockets across the Protein Data Bank, and perform a comprehensive evaluation of grid-based convolutional neural network (CNN) models on this dataset. We also demonstrate how the partitioning of the training data and test data can impact the results of models trained with the PDBbind dataset, how performance improves by adding more lower-quality training data, and how training with docked poses imparts pose sensitivity to the predicted affinity of a complex. Our best performing model, an ensemble of five densely connected CNNs, achieves a root mean squared error of 1.42 and Pearson R of 0.612 on the affinity prediction task, an AUC of 0.956 at binding pose classification, and a 68.4% accuracy at pose selection on the CrossDocked2020 set. By providing data splits for clustered cross-validation and the raw data for the CrossDocked2020 set, we establish the first standardized dataset for training machine learning models to recognize ligands in non-cognate target structures while also greatly expanding the number of poses available for training. In order to facilitate community adoption of this dataset for benchmarking protein-ligand binding affinity prediction, we provide our models, weights, and the CrossDocked2020 set at https://github.com/gnina/models.
Graphical Abstract
Introduction
Protein-ligand scoring is a key component of the drug-discovery pipeline, as it provides a way to narrow the scope of all of chemical space down to a much more feasible set of compounds to evaluate. A common approach is to utilize structure-based methods to score potential molecules with respect to the binding site of a given target protein structure to produce a ranked list of hits.1–5 The scoring function is responsible for evaluating the correctness of the pose of the molecule in the binding site and predicting its binding affinity. Traditionally, scoring functions fall into one of three categories: force-field based,6–9 empirical,10,11 or knowledge-based.12,13 Force-field based methods utilize parameters estimated from experimental and simulated data that aim to model the intermolecular potential energies through bonded and nonbonded terms.14 Empirical scoring functions are constructed from manually selected interaction terms, such as hydrophobicity and hydrogen bonding, that are parameterized to available data. Knowledge-based methods are constructed from entirely non-physical statistical potentials derived from known protein-ligand complexes. Each of these approaches commonly uses a linear fit of the input features to its target prediction. Lately, machine learning (ML) models have emerged as their own class of scoring which fit a non-linear function of their input to the target prediction.15–20
Often, ML approaches to scoring rely on predefined features to characterize protein-ligand binding, similar to knowledge-based empirical scoring functions. This overt featurization requirement possibly limits the performance of these scoring functions. This limitation can be avoided by using a direct representation of the protein-ligand structure as the input to a machine learning model. One such representation is a 3D grid (i.e., a 3D ‘picture’ of the complex) where the only features are the choice of atom types and how atom occupancy is represented in the grid. Several recent efforts have demonstrated success combining grid representations with convolutional neural networks (CNNs)21–25 which allow the model to learn its own representation of the protein-ligand interaction in order to determine what makes a strong binder.
Our previous work with CNNs21 showed good performance in identifying correct ligand poses, but did not directly predict binding affinity. Successive methods22–24 directly predict the binding affinity. DeepDTA22 showed reasonable performance with representations of the input complex consisting of the protein’s sequence and the ligand’s SMILE string. KDeep23 uses a 3D grid of chemical descriptor channels, rather than simple atom identities. Pafnuncy24 extends an atom type representation with additional atomic properties such as partial charge, SMARTS patterns, and hybridization. Imrie et al.25 improved on our original work by utilizing densely connected CNNs,26 transfer learning, target-specific models, and model ensembles to great effect on the DUD-E set.27 In addition to these approaches, there have been considerable advancements in utilizing graph-based representations and other ML models to predict protein-ligand binding affinity.28,29
Partially inspired by these successes, here we extend our original network21 to jointly train for pose selection, i.e. classifying poses as having a low root mean squared deviation (RMSD) to the true crystal pose or not, and affinity prediction, a regression problem. We expect that these two outputs should be related as both are ultimately a function of molecular interactions. Machine learning models for pose selection and affinity prediction have largely relied on the PDBbind dataset30 which curates the Protein Data Bank for high quality protein-ligand structures with published binding affinities. However, the ultimate goal of protein-ligand pose scoring is not to recapitulate the known pose of a ligand with respect to its cognate structure (redocking) but to predict the poses of novel ligands in a given structure. Here we present a new CrossDocked2020 training set that both augments and expands available data and better mimics the drug discovery process by including ligand poses cross-docked against non-cognate receptor structures as well as poses purposely generated to be counterexamples.
Additionally, it is important to note that the available data for protein-ligand binding is inherently biased and does not span all of available chemical space. Cleves and Jain31 looked into the inductive bias present for ligand-based modeling methods and found notable differences between 2D and 3D methods. They also note that the available data is the result of specific human design choices, e.g. drug campaigns for a specific receptor.31 Xia et al.32 provides a review of common biases encountered in virtual screening datasets: ‘analogue bias’ (highly similar active compounds), ‘artificial enrichment’ (poor property matching between actives and decoys leading to easier classification), and ‘false negatives’ (assumed decoys that were later experimentally verified to be active). With the recent successes of machine-learning based methods, there has been renewed interest in controlling for the biases in available datasets.33–37 Sieg et al.33 report that ML-based methods tend to fit to the initial biases of their training data and report on the importance of domain biases. Chen et al.36 show that there are numerous biases still present in the DUD-E dataset,38 and that ligand-only models achieve comparable performance to 3D CNNs on DUD-E, despite the lack of a receptor to inform the model’s predictions.
To partially address and measure bias, we evaluate a ligand-only version of our models, which are trained without a receptor structure. These versions of our models allow for a general model trained exclusively on ligand features and indicate to what extent our models are making predictions from purely cheminformatic information, including analogue bias and artificial enrichment, versus protein-ligand interactions. We further include limited comparisons with a variety of other common regression methods (linear regression with Lasso regularization, K-nearest neighbors, a decision tree, a random forest, gradient boosted decision trees, and support vector regression) that use simple chemical descriptors as input and are assessed on the same training and test sets. We also adopt clustered cross-validation34,39 on the PDBbind datasets, as well as our CrossDocked2020 set, to more rigorously evaluate generalization error. This is necessary since random data set splits do not measure a model’s ability to generalize to a new target, and, in the best case, are only appropriate for evaluating targets with a significant amount of known ligands.40
In order to assess a grid-based 3D CNN’s ability to predict protein-ligand binding affinity and perform pose selection, we designed a series of experiments. First, in order to help compare our method with other approaches, we tested on the PDBbind Core set using a variety of training sets. Next, we evaluated generalization to new targets using a clustered-cross validation analysis of the PDBbind. Additionally, we evaluated the impact of increasing training data by including lower quality structures as this strategy has been shown to be effective for random forest models.41 We also examined the impact on our models of including cross-docked poses, as well as counterexamples, in the CrossDocked2020 training set. We then assess pose sensitivity of our models by examining models trained with a ligand-only version of our training data and different pose selection approaches for affinity prediction. Lastly, we examined how an ensemble of models gives a boost to performance, as well as how a different Dense architecture performs at pose selection and protein-ligand binding affinity prediction. All data splits, trained models, and evaluation scripts are available at https://github.com/gnina.
Methods
Here we describe our grid-based 3D CNN model architectures, the construction of our datasets, our training procedure, and our evaluation metrics.
Model Architectures
We evaluate five distinct CNN model architecture variations shown in Figure 1. All models take a 3D grid of Gaussian-like atom type densities as input that is generated using our libmolgrid CUDA-accelerated library for molecular grid generation.42 There are 14 ligand atom types and 14 receptor atom types, including distinct types for oxygen/nitrogen hydrogen donor/acceptors and aliphatic/aromatic carbons. A cubic grid with dimension 23.5Å and 0.5Å resolution is used. Default 2017 (Def2017) is our originally proposed CNN architecture.21 Default 2018 (Def2018), HiRes Pose, and HiRes Affinity were discovered via an extensive hyper-parameter search where the goal was to maximize performance on clustered cross-validated splits of the PDBbind Refined set. The HiRes models were optimized strictly for a specific task and without particular regard to run-time performance, resulting in substantially more parameters (see Table 1). The Def2018 model was chosen based on its combined performance on affinity prediction, pose selection, and evaluation time. Additional details of the semi-automatic hyperparameter search used to discover these models are provided in the Supporting Information. Dense is a densely connected CNN26 derived from a model previously used for the virtual screening task.25
Figure 1:
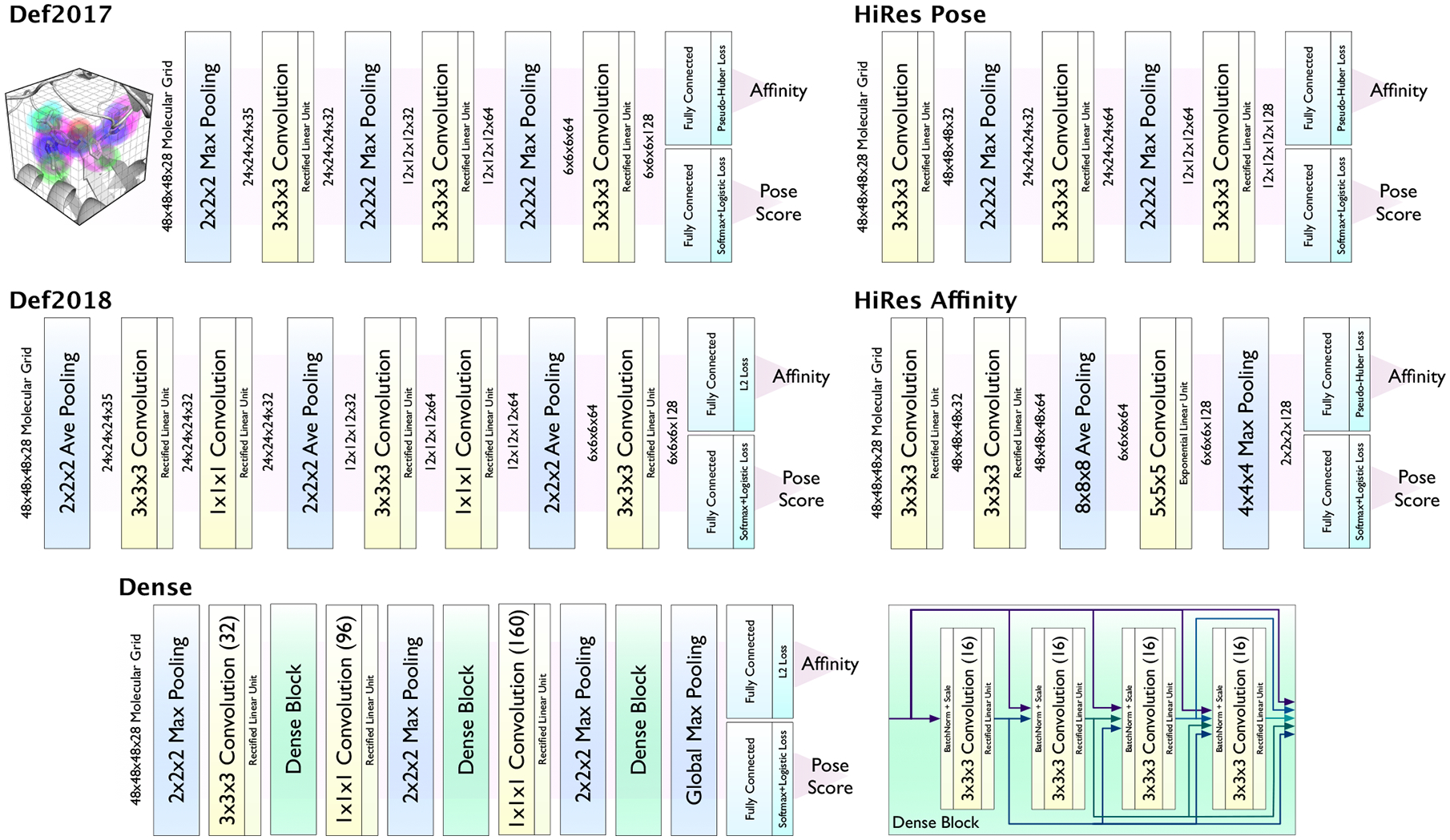
CNN model architectures. Code is available at http://github.com/gnina.
Table 1:
Number of parameters and time for a forward pass and backwards pass on a NVIDIA TITAN Xp for each model. The reported time is the average time per a single input complex averaged across 10 runs where each run consisted of 1000 iterations of batch size 50.
| 383,616 | 1.110 ± 0.0259 | 1.151 ± 0.0286 |
| 388,736 | 1.147 ± 0.0334 | 1.369 ± 0.0363 |
| 1,106,560 | 10.375 ± 0.181 | 20.640 ± 0.360 |
| 964,224 | 5.452 ± 0.0597 | 8.381 ± 0.918 |
All models consist of a series of 3D convolutional and/or pooling layers followed by two separate fully connected layers whose outputs are the pose score and affinity prediction. Pose selection (classification) is trained with a logistic loss to distinguish between low RMSD (< 2Å) and high RMSD (> 2Å) poses. Affinity prediction is trained using an L2-like pseudo-Huber loss that is hinged when evaluating high RMSD poses. That is, the model is penalized for predicting both a too low or too high affinity of a low RMSD pose, but only penalized for predicting a too high affinity for a high RMSD pose.
PDBbind Datasets
Traditionally, machine learning models have been evaluated using the predefined ‘Core’ set as a test set and the remainder of the PDBbind as the training data. As the PDBbind consists of both a curated (Refined) and expansive (General) set, we created several partitions of PDBbind v2016 for training and evaluation purposes: Refined\Core, General\Core, clustered cross-validated (CCV) Refined, and CCV General. Complexes were discarded if the ligand molecular weight was greater than 1000Da or if the ligand name was ambiguous. Each receptor and ligand was downloaded directly from the PDB as an SDF through the downloadLigandFiles service to avoid ambiguities in bond orders and protonation states present in the full PDB file. The receptor had its water and all atoms identified by the HETATM tag stripped via the ProDy Python package.43
Docked poses were generated by docking ligands into their cognate receptor with smina.44 Up to 20 poses were generated per receptor-ligand pair and poses were docked in a box defined using the autobox option with the crystal ligand. Otherwise, default settings were used. In order to increase the likelihood of each complex having a low RMSD pose in the training set while still ensuring that all training poses have the same geometric properties of a docked pose, an energy minimized crystal ligand was included in the training set. The crystal ligand was refined using the UFF force-field45 in RDKit,46 which is the same force-field used when generating conformers for docking, and then minimized with respect to the receptor structure using the Vina scoring function as implemented in smina, just as with the docked poses. Thus, there are two sets of poses when utilizing the PDBbind data: crystal poses and generated poses. The direct crystal pose is utilized in models trained with the “Crystal” dataset, and the generated poses (e.g. docked, and energy-minimized crystal pose) are utilized in models trained with the “Docked” data.
This filtering process resulted in the Refined set, with 3,805 complexes and 66,953 generated poses, the General set, with 11,324 complexes and 201,839 generated poses, and the Core set, with 280 complexes and 4,618 generated poses. We trained for affinity prediction using the pK reported in the PDBbind. Dataset information is shown in Table 2.
Table 2:
Composition of the datasets used in this paper. ReDocked2020 and CrossDocked2020 both have model-generated counterexample. CrossDocked Iteration 0 is the CrossDocked2020 set without any counterexamples added. ReDocked2020 and CrossDocked Only form a non-overlapping partition of CrossDocked2020 into redocked and cross-docked poses. Affinity Data refers to the percentage of poses with associated binding affinities from the PDBbind.
| Dataset | Pockets | Complexes | Poses | Ligands | Affinity Data % |
|---|---|---|---|---|---|
| PDBbind Core | – | 280 | 4,618 | 280 | 100 |
| PDBbind Refined | – | 3,805 | 66,953 | 2,972 | 100 |
| PDBbind General | – | 11,324 | 201,839 | 8,757 | 100 |
| ReDocked2020 | 2,916 | 18,369 | 786,960 | 13,780 | 32.7 |
| CrossDocked Iteration 0 | 2,922 | 18,450 | 10,691,929 | 13,839 | 39.9 |
| CrossDocked Iteration 1 | 2,922 | 18,450 | 19,182,423 | 13,839 | 41.3 |
| CrossDocked Only | 2,767 | 18,293 | 21,797,142 | 13,786 | 42.2 |
| CrossDocked2020 | 2,922 | 18,450 | 22,584,102 | 13,839 | 41.9 |
In order to test for model generalizability, we need to avoid similar protein structures as well as similar chemotypes existing in both the training and the test sets. To achieve this, we create splits for clustered cross-validation. Thus Clusters were created by grouping together receptors with over 50% sequence similarity or with over 40% sequence similarity and 90% ligand similarity, as computed with RDKit’s46 FingerprintMols. That is, complexes with highly similar ligands will only be placed in distinct clusters if the receptors have less than 40% sequence similarity. Clusters were then randomly assigned to folds for 3-fold cross-validation.
CrossDocked2020 Dataset
The CrossDocked2020 set was generated by downloading the PDB structures specified by Pocketome v17.12.47 Pocketome groups structures from the PDB based on the similarity of their ligand binding sites, with all identified receptors and ligands forming a “pocket.” Ligands with over 1000Da molecular weight were removed, and structures were stripped of water and aligned to the Pocketome binding sites using ProDy.43 Ions, as identified by ProDy, were retained and assigned as receptor atoms, unlike with the PDBbind data. The ligands associated with a given pocket were then docked into each receptor assigned to that pocket by Pocketome using smina,44 as previously described, resulting in a combinatorial expansion of docked poses. Binding data (pK) for the CrossDocked2020 set was taken from PDBbind v2017, where we assumed that a given ligand’s binding affinity would be the same for all receptors of a given pocket. We also assume the aligned co-crystal can be used to evaluate the pose quality of cross-docked ligand structures. Although these assumptions are commonly made during structure-based virtual screening, they are not necessarily valid and, as a result the labels of cross-docked poses are inherently noisier.
Unlike the PDBbind based data, the CrossDocked2020 dataset has already defined cluster centers of similar protein-ligand interactions from the Pocketome database. Leveraging this, we can compare across the already defined protein-ligand complex groupings sharing the same pocket. Clustered cross-validated sets were generated by grouping pockets into clusters using the ProBiS48 algorithm with the z-score parameter set to 3.5. Clusters were randomly assigned to folds for cross-validation. In total, the CrossDocked2020 set contains 13,780 unique ligands, 41.9% of which have binding affinity data, 2,922 pockets, and 18,450 complexes. Note that we grouped this data into complexes labeled by ‘pocket-ligand’ pairs, meaning the same ligand can be found among multiple complexes. The CrossDocked2020 set contains a total of 22,584,102 poses, 11,892,173 of which are counterexamples added through our iterative training set approach (outlined in the next section). A ReDocked subset was created by only including poses where the ligand was docked to its cognate receptor. The ReDocked set contains the same pockets and ligands as the CrossDocked2020 set, but only has 18,369 complexes and 786,960 poses of which 391,137 are counterexamples (see Table 2).
Iterative Training Set Preparation
We have shown that an iterative approach to the generation of training data improves the robustness of the trained model.49 In this approach, we train a model utilizing all of the available training data and use it to optimize the docked poses from the training data with respect to the newly trained model. This results in poses that the model generally considers to be good. Since we have the true crystal structure, we can identify those poses the model is most challenged by. We update the dataset with poses that scored high (above 0.9) while being more than 2Å RMSD away from the crystal pose, or scored low (below 0.5) while being less than 2Å RMSD away from the crystal pose. This provides a set of counterexamples that are specifically designed to confuse the model.50 Only unique counterexample poses are added to the training set (a new pose must be more than 0.25Å RMSD different relative to any pose already in the training set). This process was performed twice on the CrossDocked2020 set. Each iteration added fewer poses (Table 2) and the impact of the second iteration did not justify the computational expense of an additional iteration (see Supporting Information for more details). Tools for generating these counterexamples are provided at http://github/gnina/scripts.
Training Procedure
Models were trained using our custom fork of the Caffe deep learning framework51 with libmolgrid integration42 using the train.py script available at https://github.com/gnina/scripts. Training examples were randomly shuffled and each training iteration used a batch size of 50. Batches were balanced with respect to class labels (low RMSD vs high RMSD poses) and examples were stratified with respect to their receptor so that targets are sampled uniformly during training regardless of the number of docked poses per a target. Since grids are inherently coordinate frame dependent, input structures were randomly rotated and translated (up to 6Å as long as the ligand did not translate outside the box). This data augmentation by pose modification is essential to achieve good performance with grid-based CNNs.21 The stochastic gradient descent (SGD) optimizer with an initial learning rate of 0.01 and momentum 0.9 was used with a weight decay of 0.001.
Models were trained utilizing an early stopping criteria that seeks to dynamically reduce the learning rate and terminate training when the model appears to be converging. Early stopping hyperparameters for each training set are provided in Table S1. Early stopping evaluates the loss of the trained network every 1000 iterations on a sample of the training set. The size of this sample is determined by the percent_reduced parameter to train.py. The training set is used for the stopping criterion instead of a test set since we use the same procedure when training on the entire dataset with no held-out information. If there is no reduction in the training loss during the last step_when evaluations, then the learning rate is lowered by a factor of 10. We select this parameter so that the network will see the entire training set or 200,000 examples, whichever is smaller, before updating the learning rate. The learning rate is lowered step_end_cnt times.
For each dataset, we trained 5 models using 5 different random seeds to assess the variability of model performance. Additionally, for the clustered cross-validated PDBbind data, each seed utilized a different 3-fold split of the data. This was not the case for the CrossDocked2020 and ReDocked2020 datasets where the 5 different seeds were tested on the same 3-fold split of the data, due to the computational cost and time required to create a split of this much larger dataset. Cross-validated model performance is reported as the average of the three validation sets. We emphasize that validation sets used for reporting performance never overlap with the training set of the model being evaluated.
Evaluation Metrics
To evaluate pose selection performance we consider both the area under the curve (AUC) of the receiver operating characteristic (ROC) curve and the ‘Top1’ percentage. The AUC indicates how well the model separates low RMSD poses from high RMSD poses overall and provides a measure of inter-target ranking power, while Top1 is the percentage of low-RMSD (< 2Å) poses among the top-ranked poses, and is a measure of intra-target ranking power (i.e., how often docking is successful). The meaning of Top1 depends significantly on the underlying ratio. If not all complexes have a low RMSD pose, which can happen when evaluating docked poses, the best possible Top1 will be less than 100%. Additionally, the expected Top1 of a random classifier will vary depending on the percentage of low RMSD poses sampled. To provide context for our Top1 results, we provide the best possible Top1 and the expected performance of random selection. When evaluating cross-docked poses, we consider all docked poses of a ligand across multiple receptor structures as a single set, emulating ensemble docking.52
Our models aim to both score the quality of binding pose and predict its affinity. In order to evaluate affinity prediction, we must first select which docked pose of a ligand to evaluate. Unless stated otherwise, we select a pose for a given complex (receptor:ligand for PDBbind, or pocket:ligand for Pocketome) by taking the pose with the highest pose score (the same pose used to generate the ‘Top1’ statistic) or best Vina score when evaluating the Vina scoring function. The predicted affinity of this pose is then used to calculate the Pearson’s R and root mean squared error (RMSE) with the experimental affinity in pK units. We also analyzed the effect of selecting the pose with the highest predicted affinity, the best pose (lowest RMSD to the crystal), the worst scoring pose, or a random pose on affinity prediction.
As a baseline, we evaluate poses using the Autodock Vina53 scoring function. In order to compare to binding affinities from PDBbind, we convert the Vina score, which is in units of kcal/mol, to pK units using the formula
. Where T = 295K and R = 1.98720 · 10−3kcal mol−1K−1 is the ideal gas constant.
Results
We show that our models achieve comparable performance to other ML models for protein-ligand scoring. We then show the benefits of training and evaluating on larger and more sophisticated datasets, while evaluating how well models generalize across different datasets. We also evaluate ligand-only models, model ensembles, and the architecturally distinct Dense model.
Our models achieve comparable performance to other ML models
In Table 3 we compare our models with previously published work. Models are trained on the PDBBind (with the Core set removed) and tested on the Core set. Pafnucy24 and KDeep23 are both CNNs based on grids similar to our models, RF-Score16 is a random forest, and 1D2D CNN29 is a CNN with a distinct input representation based off of the topology of the input. Vina is included as a representative of a traditional scoring function. Our best performing models show similar results to the previous grid-based CNN methods as well as RF score, although precise comparisons are not possible due to differences in the training and test sets.
Table 3:
Affinity prediction performance on PDBbind Core (N=280) for a variety of models.
| Model | RMSE | R |
|---|---|---|
| Def2018 Refined Crystal | 1.50 | 0.73 |
| Def2018 Refined | 1.50 | 0.72 |
| Def2018 General | 1.38 | 0.79 |
| Def2018 General Ensemble | 1.37 | 0.80 |
| Dense General | 1.49 | 0.73 |
| Dense General Ensemble | 1.35 | 0.79 |
| Pafnucy24* | 1.42 | 0.78 |
| KDeep23† | 1.27 | 0.82 |
| RF Score16‡ | 1.39 | 0.80 |
| 1D2D CNN29† | 1.64 | 0.848 |
| Vina | 2.22 | 0.41 |
Train: PDBbind General and Refined v2016 crystal structures (N=11,906). Removed Nucleic Acid+Protein, Protein+Protein, and Nucelic Acid+Ligand from all sets. Test: remaining Core set (N=290).
Train: PDBbind Refined v2016 crystal structures (N=3767). Test: PDBbind Core set crystal structures (N=290)
Train: PDBbind Refined v2007 crystal structures (N=1300). Test: PDBbind Core set crystal structures (N=195)
Training on docked poses has little effect on affinity prediction
We consider the effect of using docked poses versus crystal poses for affinity prediction using the PDBbind Refined-Core set and testing on Core. We trained on two versions of this data: one with only the crystal poses, Crystal, and another with only docked poses, Docked. When training with Crystal the pose score layer of the model is omitted and the only loss computed is the L2-like loss of affinity prediction. The Docked set includes both low (< 2Å) and high (> 2Å) RMSD poses where the high RMSD poses are trained using a hinge loss as described in the Methods.
Affinity prediction performance on these two sets, as measured by Pearson’s R and RMSE, is shown in Figure S8. All four models achieve comparable performance on both the Crystal and Docked datasets, with average R values in the range of 0.72 to 0.75. This shows that the inclusion of docked poses does not reduce affinity prediction performance, despite the presence of high RMSD poses. We also demonstrate that models trained on the Crystal dataset and evaluated on the Docked dataset, and vice versa, display only minimal differences in performance. This indicates these models are insensitive to small perturbations of ligand positions. In other words, a low RMSD pose is scored similarly to a crystal pose. This stands in contrast to the performance of the empirical AutoDock Vina scoring function, which performed particularly poorly on the Crystal data. This is due to several crystal structures with short intermolecular distances that result in a large repulsion term. Vina does significantly better predicting the affinity of the low RMSD docked poses, which are all at local minima with respect to the Vina scoring function and so do not have these artifacts. This is especially apparent when isolating the minimized crystal poses out of all docked poses, upon which Vina performs the best.
Extensive hyperparameter tuning yielded limited benefit
As shown in Figure S8, the hyperparameter optimized models behave similarly to the Def2017 model. In successive analyses we find that the hyperparameter optimized models generally exhibit a modest performance improvement relative to the original Default 2017 model. The HiRes models are the best at the task and dataset (Refined) they were optimized for, but this pattern is not conserved across different training and test sets. This suggests these models may have been selected based on their ability to overfit a specific training regime. The consideration of model complexity and run-time performance in the selection of Def2018 may have had the effect of regularizing the hyperparameter search as this model is consistently close to the best model in all the evaluations. As all four models demonstrate similar trends and the Def2018 model generally performs best, for the remaining evaluations we present results only for the Def2018 model, with evaluations for all four models available in Figures S9, S10, S11, S13, and S14. The limited improvement achieved via hyperparameter search with these models motivated the evaluation of a substantially different network architecture, the Dense model, which we evaluate in Figure 11.
Figure 11:
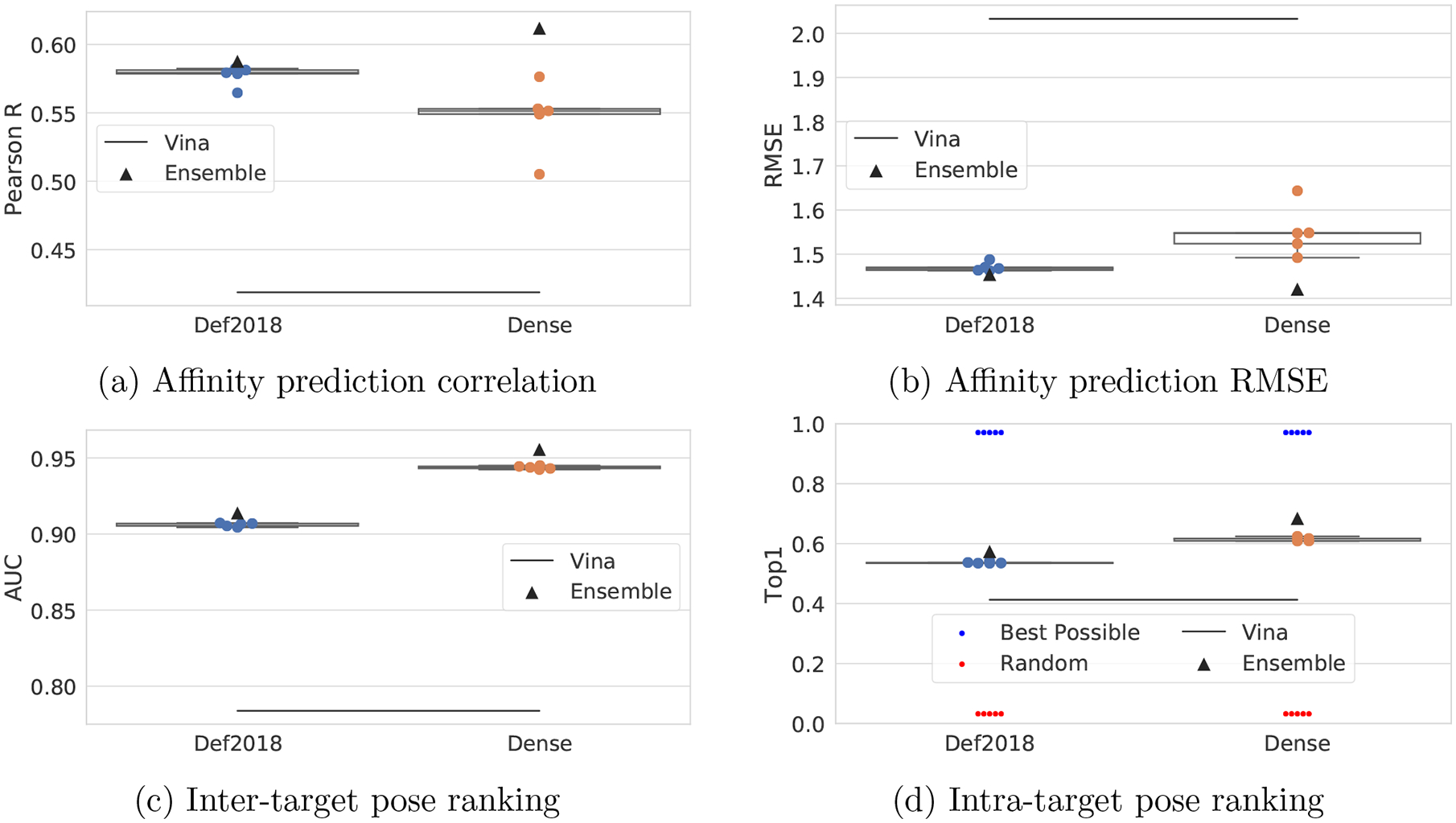
Dense model compared to Def2018 on the CrossDocked2020 set. The performance of the ensemble of both sets of five models is also shown.
Training on docked poses increases pose sensitivity
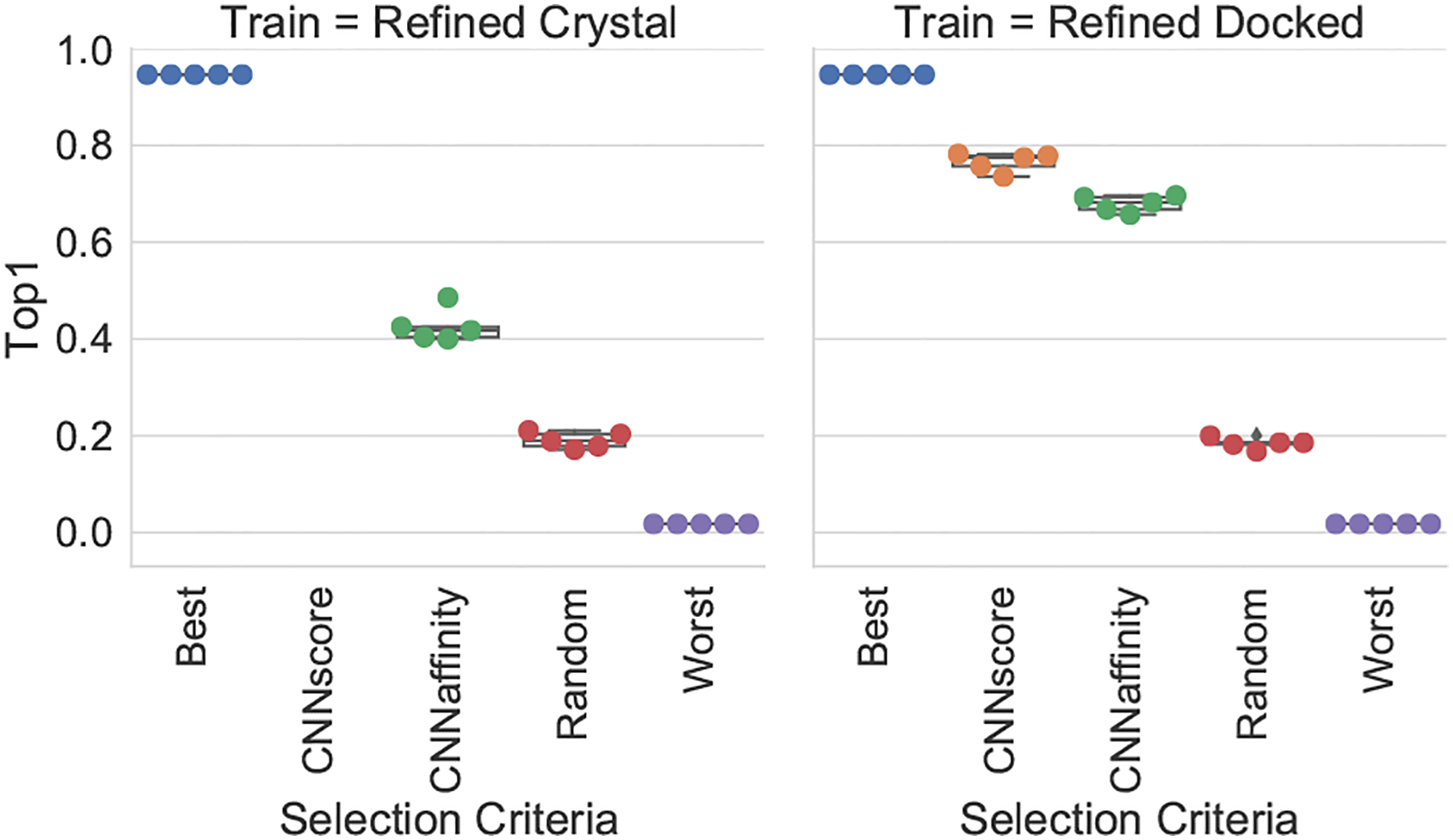
As training on docked poses has little effect on affinity prediction (Figure S8), we sought to evaluate how sensitive affinity prediction is to the choice of pose selected for evaluation. We evaluated 5 different pose selection methods: Best (selecting the pose with the lowest RMSD to the crystal pose), CNNscore (selecting the pose with the highest predicted CNNscore, the default), CNNaffinity (selecting the pose with the highest predicted affinity), Random (selecting a random pose), and Worst (selecting the pose with the highest RMSD to the crystal pose). Figure 2 shows the results of this analysis for the Def2018 model trained with either the Refined\Core Crystal set or the Refined\Core Docked set and tested on the Core set made up of docked poses. Note that Crystal trained models do not generate a pose score (CNNscore) since they are not trained for this task.
Figure 2:
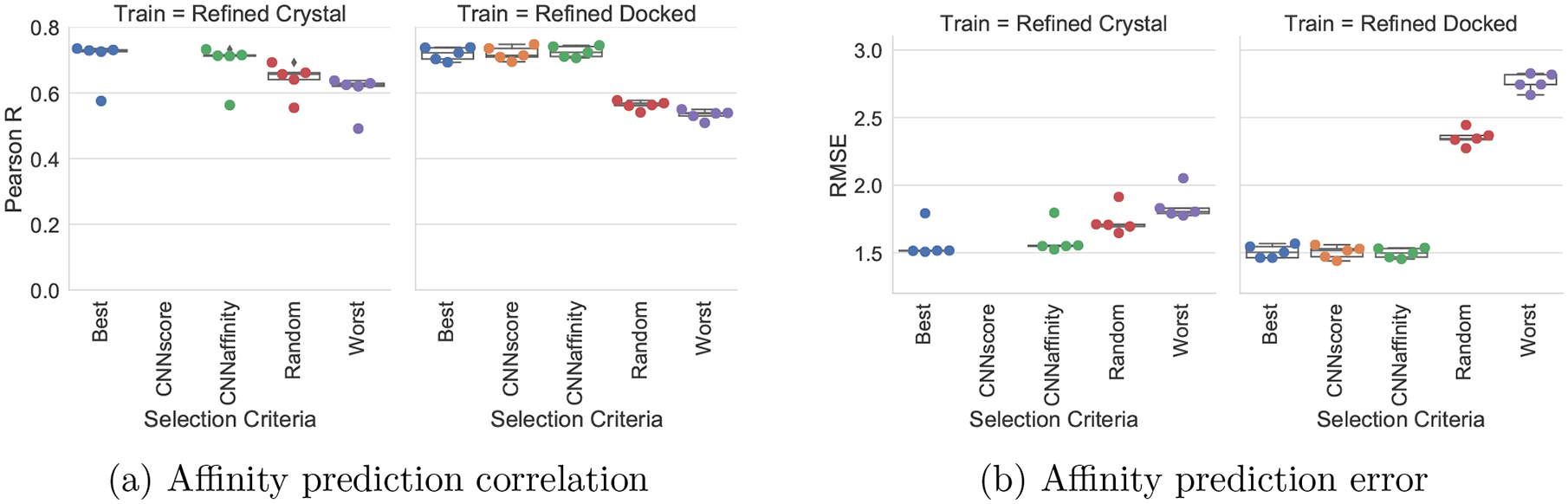
Affinity prediction performance for Def2018 model with different pose selection methods when trained on Crystal or Docked poses of PDB Refined and tested on Core. Best is the lowest RMSD pose to the crystal pose, CNNscore is the highest predicted scoring pose (not applicable for Crystal trained models), CNNaffinity is the highest predicted affinity, Worst is the highest RMSD pose to the crystal pose, and Random is taking a pose at random.
As the quality of the selected pose decreases, both the correlation and RMSE performance of affinity prediction worsens. This effect is more pronounced for the Docked trained models, and the impact on RMSE is particularly notable. With the Docked trained models, high RMSD poses are assigned significantly lower affinity predictions on average, so that while some correlation is retained (Figure S12), the RMSE increases significantly. Interestingly, while using the highest RMSD pose reduces affinity prediction performance, the Crystal trained Def2018 model still achieves an R of 0.60 compared to 0.70 with the best possible pose. This suggests the model is making minimal use of protein-ligand interactions in making its affinity prediction. In contrast, models trained on Docked poses are more pose sensitive. Not only does affinity prediction quality better correlate with pose quality (Figure 2), the affinity prediction by itself is significantly better at selecting low RMSD poses. This is shown in Figure 3 where the CNNaffinity selection is only 43% successful at selecting a low RMSD pose when trained on Crystal poses compared to the 68% success rate of this selection method using the Docked trained model.
Figure 3:
Intra-target pose ranking performance of various pose selection methods with the Def2018 model when trained on Crystal or Docked poses of PDB Refined and tested on Core.
Expanding training data with PDBbind General improves performance
The PDBbind Refined set is filtered from the PDBbind General set to exclude complexes where there are concerns about the quality of the structure or the binding data.54 In order to investigate the effect of adding more, but lower quality, training data on predicting the Core set, we compare models trained using PDBbind Refined\Core to those trained using PDBbind General\Core in Figure 4. For all models and metrics, training on PDBbind General improves Core set predictions, suggesting the quality controls used to create the Refined set may be overly strict for the purposes of training machine learning models.
Figure 4:
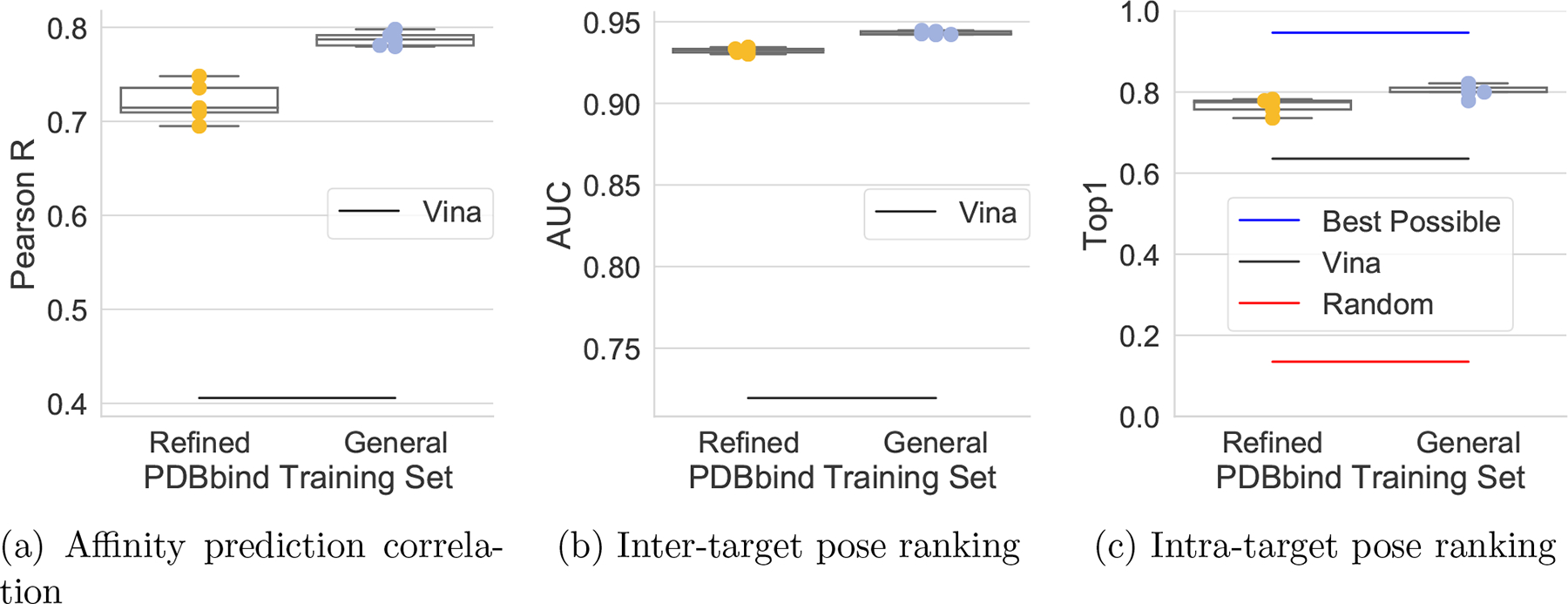
Performance on Core when the training set is expanded from PDB Refined to General.
Clustered cross-validation reveals Core set evaluations are overly optimistic
We compare the performance of models trained on the PDBbind General set and tested on the Core set with models trained and evaluated using 3 fold clustered cross-validation of the entire PDBbind General set in Figure 5. For each of our three metrics, the clustered cross-validated models perform substantially worse. Pearson R drops from 0.78 to 0.56, AUC from 0.94 to 0.89, and Top1 from 0.77 to 0.62. This drop is not due to the reduced size of the training set in each of the cross-validation folds, as these sets are still substantially larger than the PDBbind Refined set (Table 2), and yet the clustered cross-validation metrics are also substantially worse than Refined set performance (Figure 4). As clustered cross-validation measures the performance of models on new target classes, the most likely explanation for this performance difference is that testing on the Core set is a poor measure of a model’s ability to generalize. The Core set is embedded in the same chemical and target space as the Refined/General set by design, and, by virtue of its intentionally reduced size, only samples a portion of the full protein-ligand landscape. Furthermore, the Core set is constructed so there are low/medium/high affinity examples for each target class. This results in a distinctly different distribution of affinity values that produces artificially high correlations of affinity prediction performance (as shown in Figure 5). These factors suggest a significant portion of the measured performance on the Core set is due to overfitting to the training data, or at least not generalizing beyond a narrow domain of applicability. Machine learning models that seek to generalize to new targets and chemotypes should use a more rigorously constructed test set than the Core set.
Figure 5:
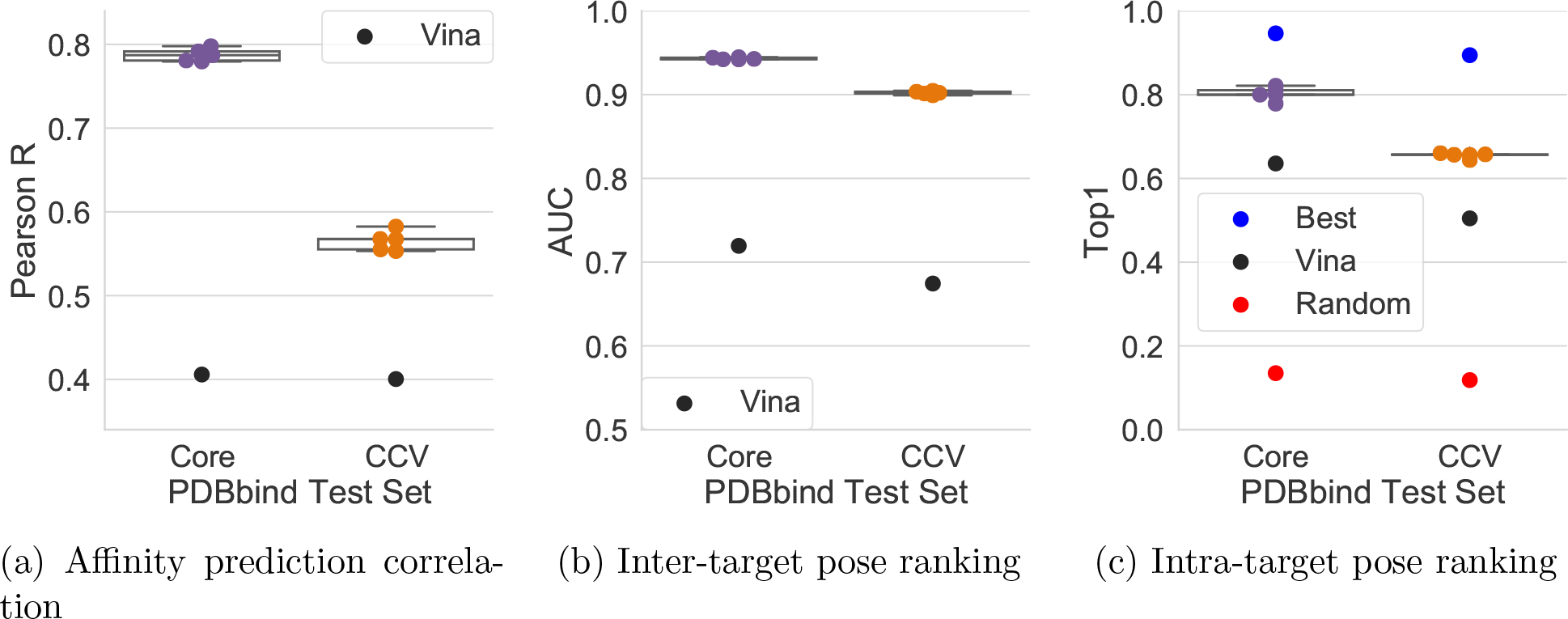
Performance when utilizing different train/test splits. Models were either trained on PDBbind General and tested on PDBbind Core (Core) or trained with clustered cross-validation splits of the PDBbind General. Note the same data is in both sets, but is divided differently among train and test.
Expanding training data beyond PDBbind improves performance
Motivated by the improved performance of the larger, but lower quality, PDBbind General set, and with the goal of training models that are appropriate to use in prospective docking studies, we created the even larger CrossDocked2020 set (Table 2). The CrossDocked2020 set is greatly expanded by including cross-docked poses, by including complexes that lack affinity data, and by including counterexamples. For a more apt comparison to the PDBbind datasets, we also consider models trained only on the ReDocked2020 subset when evaluating clustered cross-validation performance. As shown in Figure 6, as more redocked poses are added, performance increases for all metrics. Notably, affinity prediction performance increases despite the inclusion of complexes without affinity data, which are omitted from the loss calculation. However, we caution that as the underlying datasets are different, it isn’t possible to definitively conclude that the improvement is due to the volume of data. In fact, Vina also sees a significant improvement on the ReDocked2020 dataset. Nonetheless, it is reassuring that expanding the dataset with additional redocked poses does not reduce performance, despite the minimal filtering applied.
Figure 6:
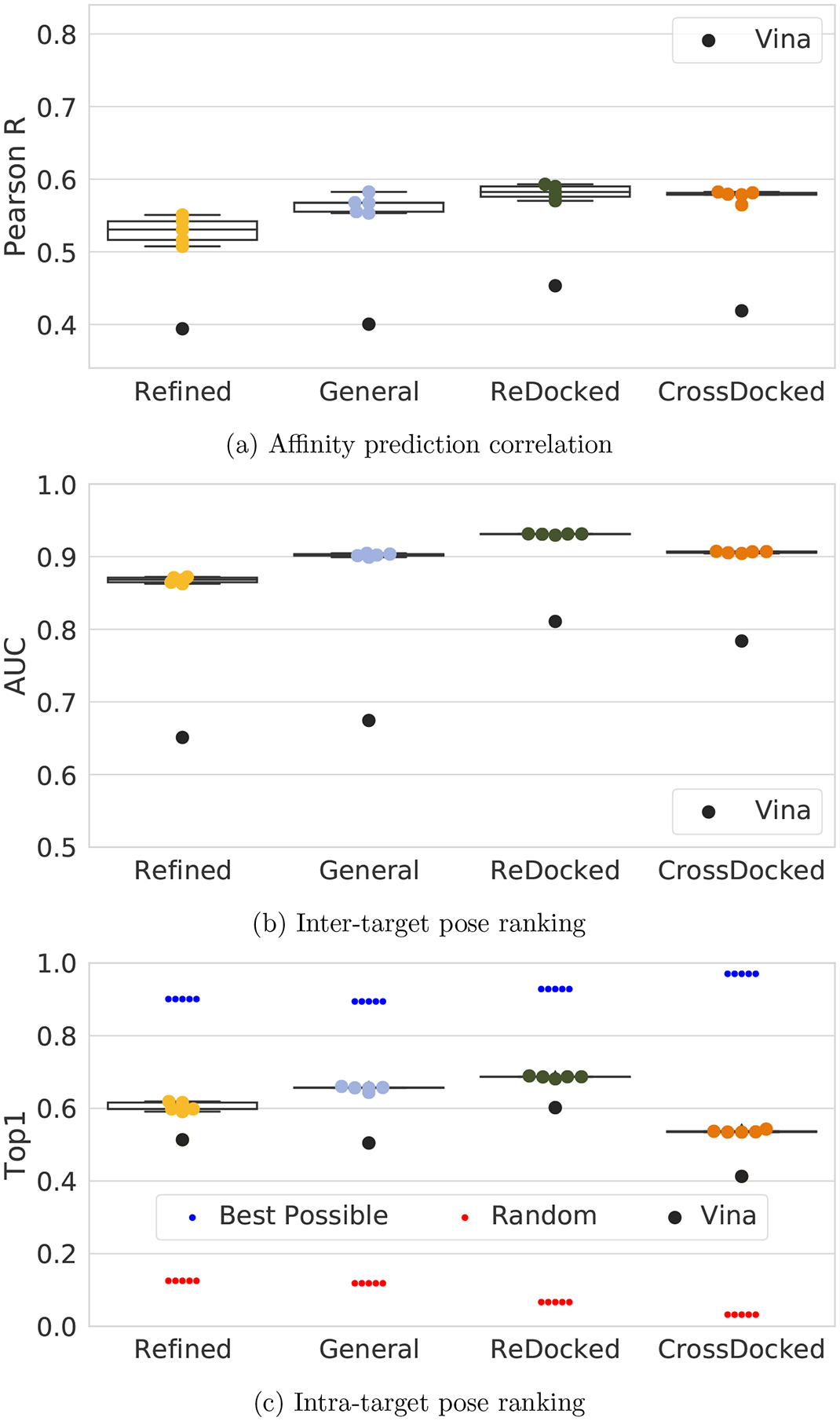
Clustered cross-validation performance of the Def2018 model trained with our various datasets. Training and testing set size increases along the horizontal axis. Note, as each test set is distinct the performance of each method cannot be directly compared. Instead compare with performance relative to Vina
As expected, pose selection performance on the full CrossDocked2020 set is substantially reduced. Cross-docked poses are inherently noisier and there are many more poses to select from. This results in a more challenging task. However, it is also a more realistic assessment of a model’s performance in a prospective docking experiment, and the drop in docking accuracy for the CNN model is less than that exhibited by Vina. Interestingly, affinity prediction performance is less affected by the inclusion of cross-docked poses in the training set. Since there is not a performance decrease, we can conclude that the model’s predictive power for affinity is not hampered by the inclusion of extra negative examples and noisier pose labels. We investigated this further by evaluating models trained on Redocked2020 and tested on CrossDocked2020 and vice-versa in Figure 8. The difference in Pearson R between models is not statistically significant (p > 0.05, Student’s t-test), and the CrossDocked2020 trained model has a slightly better AUC and slightly worse Top1 than the ReDocked2020 model. However, models trained with CrossDocked2020 have a performance boost when evaluated on the ReDocked2020 test set, whereas models trained with ReDocked2020 have a performance drop when evaluated with CrossDocked2020. This suggests that models trained with the cross-docked poses are more robust.
Figure 8:
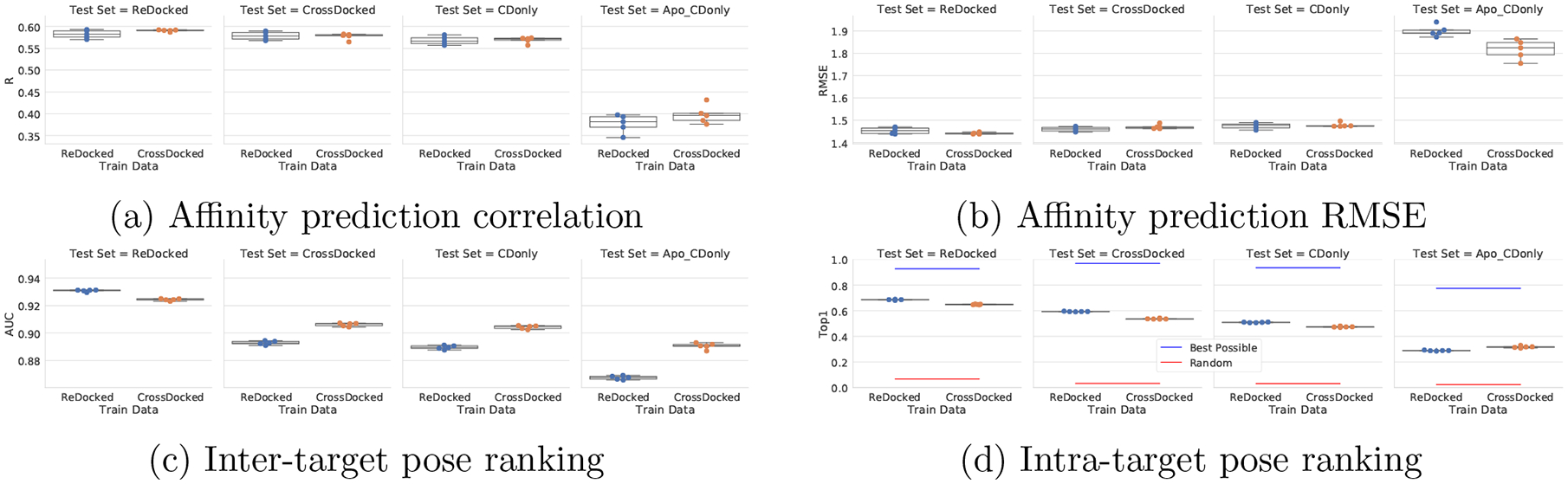
Performance of training and testing with and without cross-docked poses. Def2018 models were trained on either the ReDocked2020 set or the CrossDocked2020 set. They were then evaluated on either the ReDocked2020 set, the CrossDocked2020 set, only the cross-docked poses in the CrossDocked2020 set (CDonly), or only the apo receptors of the CDonly set.
Training on redocked poses is less effective when targeting apo receptor conformations than training on cross-docked poses
In order to asses the impact of training with cross-docked poses, we tested our models trained with the clustered cross validated PDBbind General or on CrossDocked2020 on the other’s test sets, as well as CrossDocked2020 without the counterexamples (it0). In addition, we compared models trained with only the redocked poses of CrossDocked2020 and models trained on the whole set on various splits of CrossDocked2020. Note that since the data splits of the PDBbind General set are different per seed, each corresponding swapped test set has a different amount removed in order to avoid test-on-train. This is the reason for the varying Best and Random performances of the swapped test sets in Figure 7, and means that each column cannot be directly compared. Thus we can only comment on the trends of the results. As shown in Figure 7, models trained with PDBbind data alone are fooled by the counterexamples provided in CrossDocked2020, whereas models trained on CrossDocked2020 generalize well to the PDBbind data. If the counterexamples are removed, then the PDBbind trained model generalizes well for affinity prediction, but the performance drop on pose selection is only slightly alleviated showing that pose selection is sensitive to the inclusion of cross-docked poses, as expected, unlike affinity prediction. Additionally, when comparing the CrossDocked2020 test set without the iteratively generated poses (the grey column) models trained with the CrossDocked2020 data exhibit a small performance gain on AUC, Pearson’s R, and Top1, while also having a substantial improvement on RMSE. This suggests that models trained with only the redocked PDBbind data are not as equipped to handle the cross-docking tasks.
Figure 7:
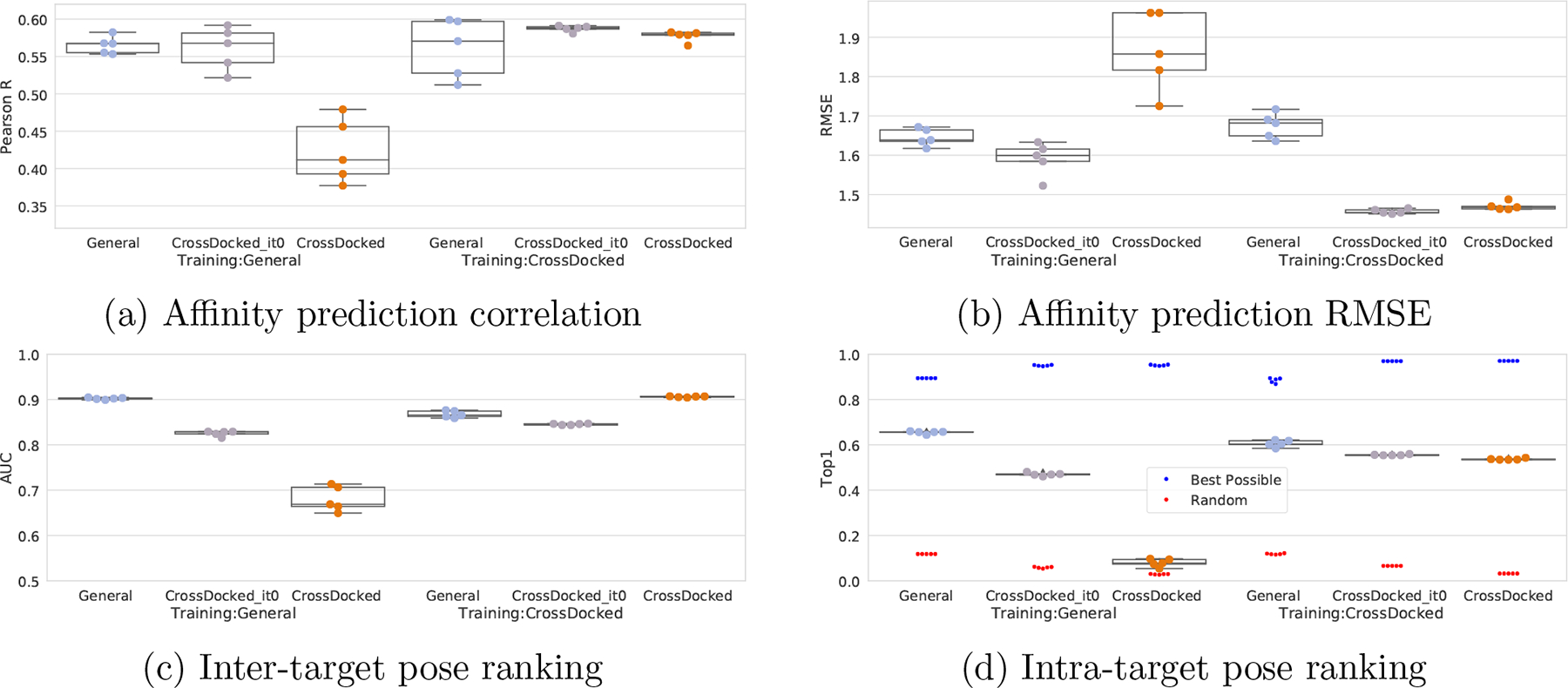
Performance of training and testing with and without cross-docked poses. Def2018 models were trained on either the clustered cross-validated PDBbind General set or the CrossDocked2020 set. They were then evaluated on either the PDBbind General set, the CrossDocked2020 set without counterexamples, or only the full CrossDocked2020 set. Note that each test set here is unique, due to varying splits of PDBbind General having different overlap with CrossDocked2020
In order to investigate if the performance differences in Figure 7 were caused by differences in model performance due to the inclusion of cross-docked poses or just from differences between the PDBbind based data and CrossDocked2020 data, we tested models trained on the redocked subset of CrossDocked2020 and compared them to models trained on all of CrossDocked2020. Figure 8 reports the performance of these models on test sets escalating in receptor deviation from the cognate receptor. We expect that the apo structures present in the CrossDocked2020 dataset represent both the most challenging examples, as we are trying to fit a ligand into a structure with no ligand present, as well as a common use case scenario for a drug discovery pipeline. We show that training on CrossDocked poses allows for a small performance boost on affinity prediction for the apo structures (R from 0.378 to 0.398, and RMSE from 1.90 to 1.82), AUC (0.867 to 0.891), and Top1 (0.289 to 0.317). Additionally R and RMSE give similar performance on the other sets, whereas the CrossDocked2020 trained models perform better than the ReDocked models on CrossDocked data, while only having a minor loss of performance on ReDocked data. Lastly, CrossDocked2020 trained models have a small performance drop in Top1 for all but the most challenging dataset.
Counterexamples yield limited performance benefit but are necessary to support pose optimization
The CrossDocked2020 set included two iterations of counterexample generation. To evaluate the influence of these counterexample poses, we consider models trained using our initial “Iteration 0” CrossDocked2020 set, without any counterexamples, and our full CrossDocked2020 set, “Iteration 2” in Figure 9.
Figure 9:
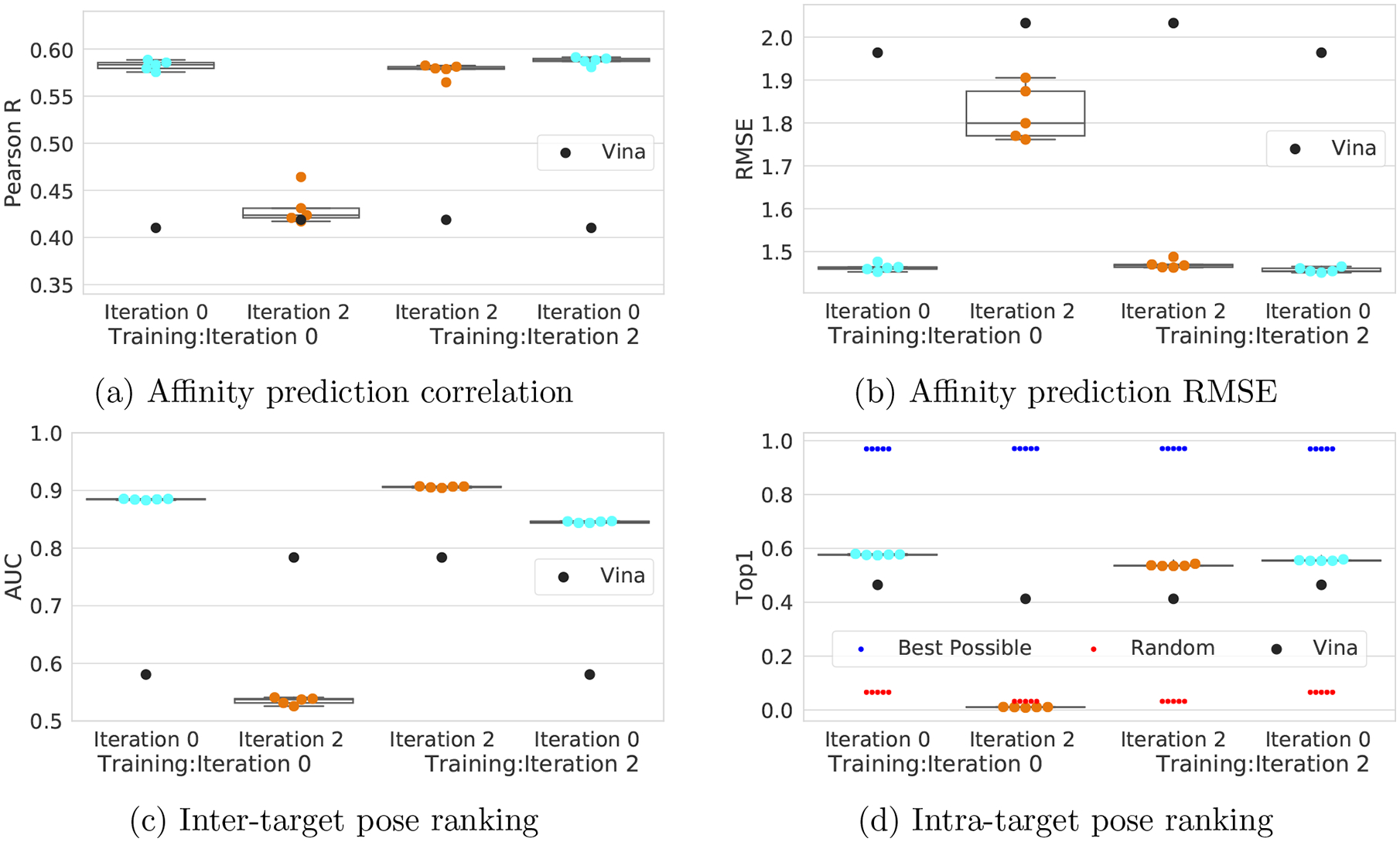
Effect of counterexamples on Def2018 clustered cross-validated performance. The models were trained on the CrossDocked2020 set either without counterexamples (Iteration 0) or with counterexamples (Iteration 2). They were then evaluated on the test set without or with the counterexamples. Note same colors indicates the same test set.
When evaluating a test set without counterexamples, models trained with counterexamples perform worse than their counterparts trained without them at the pose selection task (0.885 to 0.845 AUC and 0.577 to 0.556 Top1), indicating that the counterexamples hurt absolute pose predictive power. In contrast to this worse pose selection performance, models trained with counterexamples exhibit slight performance improvements in affinity prediction (0.577 to 0.587 Pearson’s R and 1.463 to 1.457 RMSE) when evaluated on the initial set (light blue Figure 9), indicating that the inclusion of the confusing counterexamples is potentially beneficial to affinity prediction. Taken together, these two results suggest that adding counterexamples into the training regime does not strictly improve model performance on the original data.
However, the motivation for including counterexamples was not primarily to improve model performance; rather, it is to improve the model’s robustness to out-of-distribution poses. Training without counterexamples and testing on a set containing them (orange columns in Figure 9) results in a drastic performance dropoff across all metrics for both pose selection and affinity prediction. We expected the pose selection performance drop as more than half of the poses (Table 2) are specifically selected to confuse the initial model. Additionally, we expected a small performance drop in affinity prediction, as the model first has to select a pose for its affinity prediction. The performance drop we observed was higher than expected, which indicates that there is a link between the pose of a complex and its predicted affinity, as desired, and that the confusing pose counterexamples also serve to confound the affinity prediction task. Taken together, these results imply that the inclusion of our counterexamples have little effect on the absolute performance of the models as long as they are evaluated on poses generated using the same protocol as the training set. The main benefit of including counterexamples when training a model is their effect on pose optimization. This is shown by the improved distribution in RMSD from the crystal structure exhibited after optimizing poses with a model trained with counterexamples (see Figure S4). Without counterexample training, most poses get optimized to a significantly worse pose, whereas with counterexample training poses improve on average. Counterexamples constructed from one model (Default2018) also improve optimization performance when used to train a different model architecture (Dense), as shown in Figure S5. However, this improvement in optimization is not as significant as when training with counterexamples generated using an identical network architecture, suggesting that for best results when training models for pose optimization the CrossDock2020 set should be extended with custom counterexamples. Tools for generating such model-specific counterexamples are provided at http://github/gnina/scripts.
Ligand-only information is a significant factor in affinity prediction
The expectation in training a structure-based model is that its output is primarily a function of protein-ligand interactions, as is the case with classical scoring functions. However, recent work33,36,55 demonstrated that ligand-only, cheminformatic information can explain much of the performance of structure-based machine learning methods. We investigate this effect in Figure 10 (CrossDock) and Figure S15 (General) by comparing results between versions of our model trained using only ligand information with models trained with the full complex. Note that models trained with no receptor information will have the weights that would deal with those channels set to zero during training due to weight regularization. Additionally,when evaluating a model trained with receptor and ligand information, but testing on only ligand information, we zeroed out the receptor, which eliminates the weights that would interact with the receptor channels as we are multiplying them by zero. We seek to quantify the amount of performance that can be gained by a general model using only the ligand information available in our datasets. Unsurprisingly, models trained without the receptor information have unchanged performance when tested on a set with the receptors. Models trained without the receptor performed better than models trained with the receptor on the ligand-only test set, indicating that receptor-trained models are using receptor information to make their predictions. Consistent with previously reported results,35,36,55 models trained without any receptor information still achieve a significant correlation in affinity prediction, although there is a significant decrease in the average Pearson’s correlation coefficient, with the PDBbind General set decreasing from 0.56 to 0.52 and the CrossDocked2020 set from 0.58 to 0.49. This suggests that protein-ligand interactions play more of a role in affinity prediction for models trained with the CrossDocked2020 data. Further evidence suggesting that cheminformatic information has a greater role in driving good performance for the PDBbind datasets than the CrossDocked2020 set is shown in Table S3; a variety of models trained on simple descriptors can obtain a correlation of 0.51 on average for the PDBbind General set, but only a correlation of 0.27 on CrossDocked2020.
Figure 10:
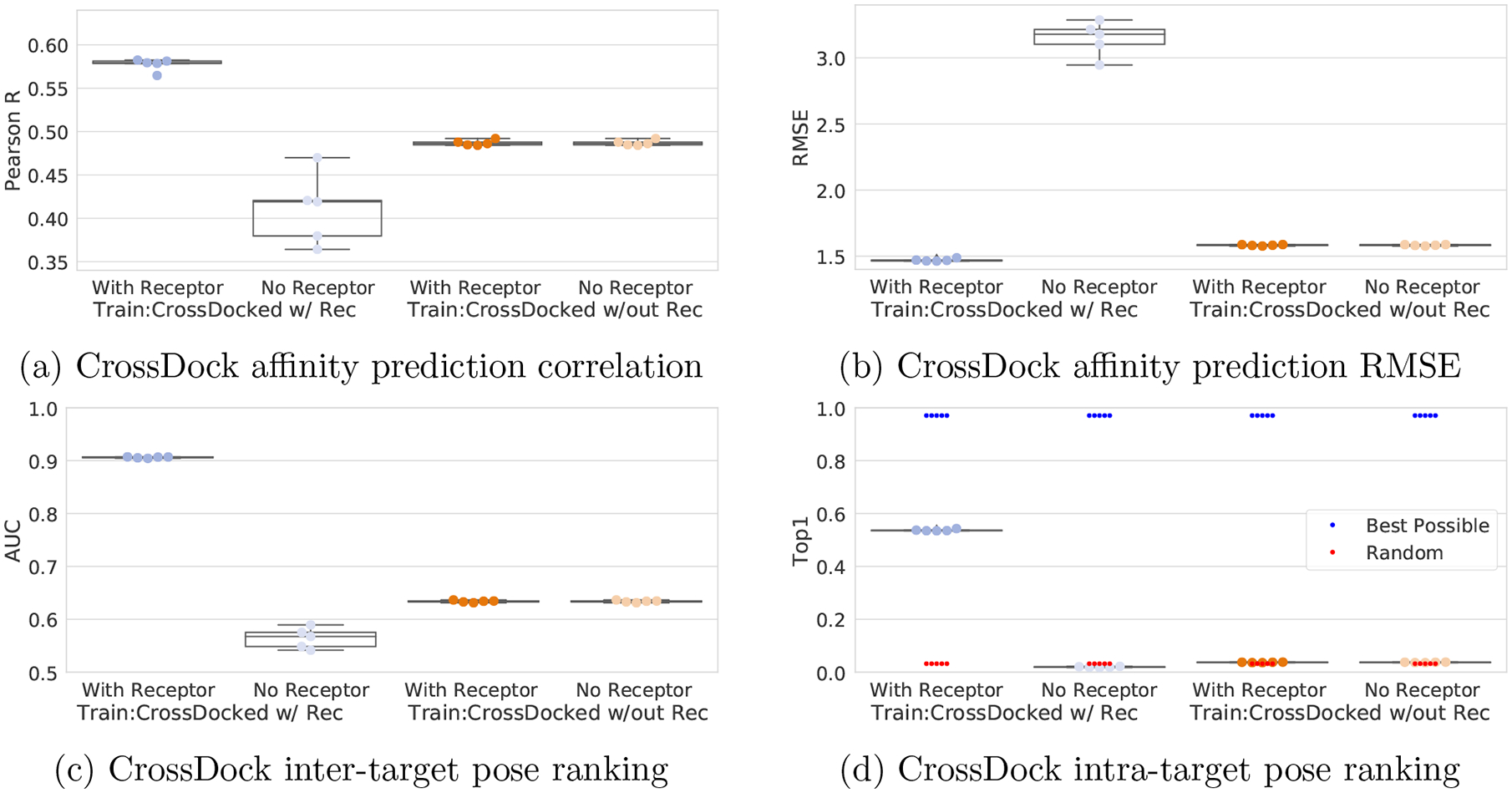
Ligand-only model performance. Def2018 models were trained with or without receptors (w/ Rec or w/out Rec) and evaluated on test sets with or without receptors (With Receptor or No Receptor).
As expected, since pose selection is inherently a function of the pose of the ligand relative to the receptor, models trained without a receptor have a Top1 metric equal to random performance. Models trained with a receptor exhibited close to random AUCs and substantially reduced correlations when evaluated on the ligand-only test set. However, surprisingly, the AUC of ligand-only models is significantly higher than the expected 0.5 of a random classifier, with the PDBbind General trained model exhibiting an AUC of 0.59. As there is no receptor, this non-random performance must be due to differences in ligand conformation or a general cheminformatic descriptor. Scoring poses using their internal energy, as calculated by the UFF force field, classified poses from the General set with an AUC of 0.55 (Figure S16). This suggests that ligand strain may provide some signal into the quality of a docked pose, but does not fully explain our ligand-only models’ performance. We also evaluated using a 2D-only Morgan fingerprint of the molecule with a linear regression model, which resulted in an AUC of 0.60 on the General set, matching the performance of our ligand-only model. As fingerprints are independent of ligand conformation, this result is achieved despite different poses of the same ligand producing identical scores. The reason this does not result in an AUC of 0.5 is that not all ligands have the same percentage of low RMSD poses. For example, rigid molecules that bind to fully enclosed protein pockets have fewer high RMSD poses, as the steric constraints of the system prevent them from being sampled during docking. It appears the model can identify these ‘highly dockable’ chemotypes resulting in the non-random AUC. Since Top1 strictly evaluates ordering of the poses of the same ligand, unlike AUC which evaluates pose ordering across the entire dataset, it is not affected by this source of artificial enrichment. See the Supporting Information for additional analysis of this effect.
Individual Dense models improve pose selection but not affinity prediction
Finally, we evaluate the computationally demanding Dense model on the CrossDocked2020 dataset in Figure 11. The Dense model has nearly twice as many parameters as Def2018 and takes ten times as long to evaluate (Table 1). This extra computation enables a significant improvement in pose selection performance, with an average Top1 percentage on the CrossDocked2020 set of 61.5% compared to 53.7% for the Def2018 model. Interestingly, individual models perform worse at affinity prediction, with an average R of 0.55 compared to 0.58 with Def2018. However, as we see next, the use of an ensemble of models, rather than individual models, rescues this decrease in affinity prediction performance.
An ensemble of models improves performance.
Typically training an ensemble of deep learning models results in a small performance gain. This is indeed the case for our models, as shown in Table 4 and Figure 11. The ensemble model predicts the average of the five individual models, each of which was trained using a different random seed on the same training set. We evaluated ensembles of up to ten Def2018 models on the CrossDocked2020 dataset, but found diminishing returns after five models were used, as shown in Figure S17. An alternative means of generating an ensemble that is potentially more efficient than using multiple models is to exploit the coordinate frame dependent nature of the grid representation and evaluate an ensemble of random rotations of the input using the same model. However, as shown in Figure S18, this strategy does not meaningfully improve model performance. In all cases, an ensemble of models is equal or superior to the average performance of the individual models. Smaller training sets tend to benefit more from the use of an ensemble than the larger datasets, with the notable exception of the Dense model, which had the best gains via an ensemble approach. This is not necessarily surprising, as the much larger number of parameters suggests that the Dense models are more overfit to their training data and ensembling is an effective method for compensating for overfitting.56 Our best performance overall is achieved with an ensemble of Dense models, which has a Top1 of 0.684 and R of 0.612 on the CrossDocked2020 set, compared to 0.413 and 0.419 respectively for Vina.
Table 4:
Effect of using an ensemble of models compared to average of individual model performance. BP: Best possible fraction of low RMSD poses; Rand: expected fraction of randomly sampled low RMSD poses.
| Train | Test | Model | Evaluation | RMSE | R | AUC | Top1 | BP | Rand |
|---|---|---|---|---|---|---|---|---|---|
| CrossDock | CCV | Dense | |||||||
| Ensemble | 1.42 | 0.612 | 0.956 | 0.684 | 0.970 | 0.0321 | |||
| CrossDock | CCV | Def2018 | |||||||
| Ensemble | 1.45 | 0.587 | 0.914 | 0.574 | 0.970 | 0.0321 | |||
| General | Core | Dense | |||||||
| Ensemble | 1.348 | 0.788 | 0.960 | 0.836 | 0.946 | 0.135 | |||
| General | Core | Def2018 | |||||||
| Ensemble | 1.368 | 0.796 | 0.946 | 0.814 | 0.946 | 0.135 | |||
| Refined | Core | Def2018 | |||||||
| Ensemble | 1.438 | 0.749 | 0.941 | 0.800 | 0.946 | 0.135 |
Discussion
We have presented several grid-based 3D CNN model architectures for affinity prediction and pose selection and trained and evaluated them using a variety of datasets. The 3D grid representation has the advantage that it does not require overt featurization (although the inclusion of additional chemical information may improve performance23) and, since a CNN is differentiable, poses can be optimized with respect to the model using standard optimization techniques. Our models exhibit competitive performance relative to similar methods (Table 3), although an exact comparison is complicated by differences in test set selection, even when the same PDBbind Core set is used. Our best performing single model at affinity prediction is the Def2018 models trained with the PDBbind General Set with a Pearson’s R of 0.79. This can be compared to KDeep, the best performing grid-based CNN model with a Pearson’s R of 0.82, as Jiménez Luna et al.23 reported that training on the General set did not improve KDeep’s performance.23 We note that KDeep uses 1,340,769 parameters, which is about triple that of the Def2018 model. We substantially outperform a popular empirical scoring function, AutoDock Vina. When evaluated on cross-docked poses, our best model successfully selects a low RMSD pose as its best pose 65% more often than Vina (0.684 Top1 vs. 0.413).
Consistent with conventional wisdom that machine learning models struggle to extrapolate beyond the domain they are trained for, we have shown the difficulty these models have generalizing. Models trained to predict binding affinity using only crystal structures will struggle to correctly select low RMSD docked poses (Figure 3), despite performing well on crystal poses (Figure S8). Using Docked poses and jointly training with pose selection does not improve affinity prediction performance (Figure S8), but it does make the affinity prediction model more pose sensitive (Figure 2). Models trained with docked poses can perform well at the pose selection task as long as poses are generated using the same method as during training. However, if the model is integrated into the sampling strategy of a typical docking routine and is driving the generation of new poses, it will likely fail unless it is iteratively trained with counterexamples generated by this sampling strategy (Figure S4, Figure 9). This strongly cautions that the performance of models trained using crystal poses and affinity prediction in truly prospective structure-based drug discovery efforts may fall far short of what is expected from validation set performance.
A significant trend across all our evaluations is that expanding the training set, both with more data and with more diverse representations of the underlying data, expands the domain of applicability of the model and makes it more robust to variations in the construction of the input (e.g., docked vs. crystal or redocked vs. cross-docked poses). We believe our CrossDocked2020 dataset provides a close approximation of the desired domain of a general-purpose model for structure-based drug design. Importantly, although the set is large, with more than 22 million protein-ligand poses, every ligand is associated with an experimental structure so labels can be accurately assigned (modulo inconsistencies introduced by cross-docking). However, we suspect even CrossDocked2020 trained models would struggle if presented with ligand poses whose geometries were optimized using a different force field or were docked with a different algorithm. Care should be taken to ensure the prospective application of a model matches as closely as possible to the training domain.
We have similar concerns about the generalizability of affinity prediction. The substantial performance drop when evaluating affinity prediction using clustered cross-validation compared to the Core set (Figure 5), the minimal importance of the receptor structure (Figure 10), and the relative success of affinity prediction even using high RMSD poses (Figure 2) strongly suggest these models will not generalize well to new chemotypes. Since it is not possible to augment affinity training sets to the same degree as when training for pose selection, where additional poses can be generated and labeled, expanding the domain of applicability of structure-based affinity prediction models remains an open challenge. One possibility is to use binding affinity data from ligands without a known structure. However, previously we have shown that training models on such data by simply using the top-ranked docked poses results in entirely pose-insensitive (i.e. cheminformatic) models.21 A more careful process for generating putative ligand poses, such as template-based docking,57 may yield better results. Alternatively, a different input representation, model architecture, or training regime might force a model to only predict using protein-ligand interaction information,58 although it is not clear this necessarily results in a more generalizable model as ligand-only information is embedded in protein-ligand interaction information.
Protein-ligand scoring functions traditionally struggle to balance pose selection and affinity prediction performance.59–63 A unique feature of our models is the affinity prediction and pose selection score share a significant amount of computation (all but the last fully connected layer - see Figure 1), but are still uniquely computed outputs. This results in a affinity predictions that both seem to be more robust to perturbed poses while still being sensitive to the ligand pose. Figures 2 and 7 suggest that our models’ affinity predictive power is unaffected by the inclusion of negative poses in the training data. Additionally, all of our experiments on swapped test sets (Figures 7, 9, S8, 8, and S15) show that performance losses on affinity prediction metrics were less severe than on pose prediction metrics. Specifically, in Figure S15 we show that when trained with receptor information, performance is worse than when trained on ligands alone when evaluated on ligand-only structures. This demonstrates the model predictions are dependent on the receptor input. The use of receptor information improves affinity prediction (0.577 vs 0.487) and CNN ligand-only models outperform ligand-only models trained on simple descriptors (STable S3). This shows that, although ligand-only information substantially contributes to affinity prediction performance, CrossDock2020 performance can be improved through the inclusion of structural and receptor information.
We show that our models’ affinity predictions are indeed pose sensitive in Figure S12, as using the worst available pose has a large negative impact on affinity prediction correlation. Additionally, Figures 3 and S11 show that selecting a pose by taking the highest predicted CNNaffinity can recover most of the pose predictive performance of selecting by the highest CNNscore for the complex. We suspect that an underlying reason for the affinity predictions to behave this way is due to our training procedure. During training, we penalize the affinity prediction in two ways: for getting the affinity incorrect on a good pose or over-predicting the affinity on a bad pose. Thus, the model is not penalized for under-predicting the affinity of a bad complex, whereas correct complexes need to get the affinity correct. This allows the predicted affinity to have a great deal of pose predictive power, as the best predicted affinity would tend to be good poses as they are penalized for both under and over predicting the binding affinity.
We find there are several subtleties involved in training and evaluating these models. We were surprised to see a better than random AUC on pose selection using ligand-only models. Unlike affinity prediction, where cheminformatic models are routinely successful, pose selection seems like it should be entirely dependent on the receptor structure, and there should be no relevant information available from the ligand alone. This better than random performance is in part due to the construction of the training set. Some ligands had a higher percentage of low RMSD poses than others, so a model can learn a ‘dockablity’ index for each ligand. This may be a useful prior to learn since ligands that are inherently easier to dock, e.g. rigid ligands, will be scored more confidently, but, if desired, it should be possible to eliminate this effect by resampling the training set so that every ligand has the same ratio of low to high RMSD poses. This effect is also due to the use of AUC as a metric, which measures how well a classifier separates positive from negative poses across the entire set of ligands. Typically, when docking, the goal is to correctly rank poses for the same ligand, and comparing the scores of different ligands is irrelevant for the purposes of pose prediction. This intra-target ligand ranking is precisely what the Top1 metric measures and is a better representation of docking performance. Unfortunately, the significance of both the AUC and Top1 metrics is highly dependent on the construction of the test set. For example, although a random classifier has an expected AUC of 0.5, its Top1 will depend on the average fraction of low RMSD poses available for each ligand. Conversely, expanding the set with trivial-to-predict high RMSD poses (e.g., ligand poses that don’t interact with the protein or overlap it completely) will artificially increase the AUC while leaving the Top1 metric unchanged. Thus, for pose selection, precise comparisons between methods can only be made if the identical test set is used. As an example of the difficulty in comparing pose selection performance, consider the PDBbind-based evaluation of the graph-based model of Lim et al.28. This model achieves an AUC of 0.968, which is higher than any of our PDBbind evaluations, but also exhibits a Top1 of less than 50%, which is substantially lower than our worse Top1 PDBbind statistic (77%, Figure 4). However, we do not know the underlying distribution of positive to negative examples in their test set, making a direct comparison between our Top1 and their Top1 impossible as we do not know what the best possible performance nor the performance of a random classifier on their test set is. As an example, if only half of their test set had sampled a positive example, then a Top1 of 50% is actually perfect performance.
In an effort to help standardize comparisons of structure-based machine learning models, we provide all of our training and test sets (Table 2) both as standard SDF/PDB files and in a custom ‘molcache’ format that can be efficiently used via libmolgrid42 (https://github.com/gnina/libmolgrid) to enable replication of and direct comparisons to our results. Our recommendation is that practitioners train and evaluate using the CrossDocked2020 set without the iteratively generated poses (unless the model will be used for pose optimization) as this best emulates real-world usage for scoring a ligand’s pose and predicting its affinity. However, if this is too computationally demanding, we provide the ReDocked and PDBbind datasets as well as an intelligently downsampled version of the CrossDocked2020 set. The DownsampledCD2020 set contains up to ten randomly selected positive examples and up to twenty randomly sampled negative examples per Pocket:Ligand pair that sampled at least one positive pose for each train/test fold. This results in a dataset that is 2.25% of the size of CrossDocked2020. Table S2 shows that the reduced set achieves similar performance on RMSE (1.45 vs 1.47), Pearson’s R (0.59 vs 0.58), and AUC (0.91 vs 0.89), but has a much higher Top1 (0.89 vs 0.54). The Top1 metric is not comparable due to the different distribution of positive and negative poses in the reduced set. Note that the purpose of our CrossDocked2020 set is orthogonal to cross-docking benchmark datasets64 as the goal is not to evaluate docking algorithms, but to provide a standard set of already generated poses for training, evaluating, and comparing machine learning models. In all cases, the clustered cross-validation train/test splits should be used, as we have shown (Figure 5) that using PDBbind Core as a test set results in an over-optimistic evaluation of performance on new targets. Additionally, we recommend our iterative training procedure for generating new poses as counterexamples for a model if that model will be used to optimize ligand poses. When training a new model architecture or for a different training task, we recommend this counterexample generation procedure be repeated. While our provided counterexamples show some benefit with different model architectures, and therefore serve as a good starting point, the overall benefit is less than using model-specific counterexamples (Figure S5). We also do not expect our datasets and splits to be appropriate for every task. For example, our clustered cross-validation splits primarily use target identity for creating clusters, since our primary concern is the ability of models to generalize to new targets, but this may result in an undesirable amount of ligand similarity between train and test sets for some applications. We also purposely retain unequal ratios of low to high RMSD poses among ligands, resulting in a ligand-specific ‘dockability’ prior. Nonetheless, our hope is that a standard dataset for structure-based machine learning will aid the development of more effective models while also illuminating additional improvements that are needed in such a dataset.
We have shown that 3D CNN models can substantially outperform a conventional empirical scoring function (Vina) at affinity prediction and pose selection, but do not necessarily generalize beyond the domain they are trained on. To partially address this issue and to provide a resource for structure-based machine learning models, we created the CrossDocked2020 set of more than 10 million poses. This dataset better approximates the domain of prospective structure-based drug design where a ligand is evaluated against a non-cognate structure. In Figure S15 we demonstrate that models trained with CrossDocked2020 are more pose-sensitive, as their performance drop is more substantial when the receptor is removed. We additionally showed that models trained with CrossDocked2020 are more robust (Figure 8, Figure 7, and Figure 9). We have deployed models trained on the entire CrossDocked2020 set within the latest version of our open source gnina (https://github.com/gnina) deep learning framework for molecular docking so they can be easily used for pose scoring and minimization.
Supplementary Material
Acknowledgement
The authors thank Matthew Ragoza, Jonathan King, and Andrew McNutt for their insightful contributions during the preparation of this manuscript.
This work is supported by R01GM108340 from the National Institute of General Medical Sciences, by the University of Pittsburgh Center for Research Computing through the resources provided, by the Extreme Science and Engineering Discovery Environment (XSEDE) Bridges GPU resource at the Pittsburgh Supercomputing Center (allocation TG-MCB190049), which is supported by National Science Foundation grant number ACI-1548562, support from Google Cloud, and by a GPU donation from the NVIDIA corporation.
Footnotes
Supporting Information Available
Supporting Information Available: Supplementary Figures (S1–S18), Tables (S1–S2), and Methods. This material is available free of charge via the Internet at http://pubs.acs.org/.
References
- (1).Gau D; Lewis T; McDermott L; Wipf P; Koes D; Roy P Structure-based virutal screening identifies a small-molecule inhibitor of the profilin 1-actin interaction. J. Biol. Chem 2018, 293, 2606–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Fradera X; Babaoglu K Overview of Methods and Strategies for Conducting Virtual Small Molecule Screening. Curr. Protoc. Chem. Biol 2017, 9, 196–212. [DOI] [PubMed] [Google Scholar]
- (3).Bajusz D; Ferenczy GG; Keseru GM Structure-Based Virtual Screening Approaches in Kinase-directed Drug Discovery. Curr. Trends Med. Chem 2017, 17, 2235. [DOI] [PubMed] [Google Scholar]
- (4).Cheng T; Li Q; Zhou Z; Wang Y; Bryant SH Structure-based virtual screening for drug discovery: a problem-centric review. AAPS J 2012, 14, 133–41, [doi: 10.1208/s12248-012-9322-0]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ripphausen P; Nisius B; Peltason L; Bajorath J Quo vadis, virtual screening? A comprehensive survey of prospective applications. J. Med. Chem 2010, 53, 8461–8467. [DOI] [PubMed] [Google Scholar]
- (6).Cornell WD; Cieplak P; Bayly CI; Gould IR; M. KM Jr.; Ferguson DM; Spellmeyer DC; Fox T; Caldwell JW; Kollman PA A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules. J. Am. Chem. Soc 1995, 117, 5179–5197. [Google Scholar]
- (7).M. AD Jr. et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- (8).Simonsen T; Archontis G; Karplus M Free Energy Simulations Come of Age: Protein-Ligand Recognition. Acc. Chem. Res 2002, 35, 430–437. [DOI] [PubMed] [Google Scholar]
- (9).Koes DR; Baumgartner MP; Camacho CJ Lessons Learned in Empirical Scoring with smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model 2013, 53, 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Friesner RA; Banks JL; Murphy RB; Halgren TA; Klicic JH; Mainz DT;Repasky MP; Knoll EH; Shelley M; Perry JK; Shaw DE; Francis P; Shenkin PS Glide: A New Approach for Rapid, Accurate Docking and Scoring. 1. Method and Assessment of Docking Accuracy. J. Med. Chem 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- (11).Wang R; Lai L; Wang S Further development and validation of empirical scoring functions for structure-based affinity prediction. J. Comput.-Aided Mol. Des 2002, 16, 11–26. [DOI] [PubMed] [Google Scholar]
- (12).Muegge I A knowledge-based scoring function for protein-ligand interactions: Probing the reference state. Perspect. Drug Discovery Des 2000, 20, 99–114. [Google Scholar]
- (13).Gohlke H; Hendlich M; Klebe G Knowledge-based scoring function to predict protein-ligand interactions. J. Mol. Biol 2000, 295, 337–356. [DOI] [PubMed] [Google Scholar]
- (14).Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov 2004, 3, 935–49, [doi: 10.1038/nrd1549]. [DOI] [PubMed] [Google Scholar]
- (15).Durrant JD; McCammon JA NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf. Model 2011, 51, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Ballester PJ; Mitchell JBO A machine learning approach to predicting protein-ligand binding affinity with applications to molecular docking. Bioinformatics 2010, 26, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Hassan MM; Mogollon DC; Fuentes O; Sirimulla S DLSCORE: A Deep Learning Model for Predicting Protein-Ligand Binding Affinities. ChemRxiv 2018, [Google Scholar]
- (18).Wojcikowski M; Kikielka M; Stepniwska-Dziubinska MM; Siedlecki P Development of a protein-ligand extended connectivity (PLEC) fingerprint and its application for binding affinity predictions. Bioinformatics 2018, bty757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Shen C; Ding J; Wang Z; Cao D; Ding X; Hou T From machine learning to deep learning: Advances in scoring functions for proteinligand docking. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2020, 10, e1429. [Google Scholar]
- (20).Li H; Sze K-H; Lu G; Ballester PJ Machine-learning scoring functions for structure-based drug lead optimization. Wiley Interdiscip. Rev.: Comput. Mol. Sci n/a, e1465. [Google Scholar]
- (21).Ragoza M; Hochuli J; Idrobo E; Sunseri J; Koes DR Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Ozturk H; Ozgur A; Ozkirimli E DeepDTA: deep drug-target binding affinity prediction. Bioinformatics 2018, 34, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Jiménez Luna J; Skalic M; Martinez-Rosell G; De Fabritiis G K DEEP: Protein-ligand absolute binding affinity prediction via 3D-convolutional neural networks. J. Chem. Inf. Model 2018, [DOI] [PubMed] [Google Scholar]
- (24).Stepniewska-Dziubinska MM; Zielenkiewicz P; Siedlecki P Development and evaluation of a deep learning model for protein-ligand binding affinity prediction. Bioinformatics 2018, 34, 3666–3674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Imrie F; Bradley AR; van der Schaar M; Deane CM Protein Family Specific Models Using Deep Neural Networks and Transfer Learning Improve Virtual Screening and Highlight the Need for More Data. J. Chem. Inf. Model 2018, 58, 2319–2330. [DOI] [PubMed] [Google Scholar]
- (26).Huang G; Liu Z; Van Der Maaten L; Weinberger KQ Densely connected convolutional networks. Proceedings of the IEEE conference on computer vision and pattern recognition. 2017; pp 4700–4708. [Google Scholar]
- (27).Mysinger MM; Carchia M; Irwin JJ; Shoichet BK Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Lim J; Ryu S; Park K; Choe YJ; Ham J; Kim WY Predicting drug-target interaction using 3D structure-embedded graph representations from graph neural networks. J. Chem. Inf. Model 2019, 59, 3981–3988. [DOI] [PubMed] [Google Scholar]
- (29).Cang Z; Mu L; Wei G-W Representability of algebraic topology for biomolecules in machine learning based scoring and virtual screening. PLoS Comput. Biol 2018, 14, 1–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Liu Z; Su M; Han L; Liu J; Yang Q; Li Y; Wang R Forging the Basis for Developing ProteinLigand Interaction Scoring Functions. Acc. Chem. Res 2017, 50, 302–309. [DOI] [PubMed] [Google Scholar]
- (31).Cleves A; Jain A Effects of inductive bias on computational evaluations of ligand-based modeling and on drug discovery. J. Comput.-Aided Mol. Des 2008, 22, 147–159. [DOI] [PubMed] [Google Scholar]
- (32).Xia J; Tilahun EL; Reid T-E; Zhang L; Wang XS Benchmarking Methods and Data Sets for Ligand Enrichment Assessment in Virutal Screening. Methods 2015, 71, 146–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Sieg J; Flachsenberg F; Rarey M In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model 2019, 59, 947–961. [DOI] [PubMed] [Google Scholar]
- (34).Lopez-del Rio A; Nonell-Canals A; Vidal D; Perera-Lluna A Evaluation of Cross-Validation Strategies in Sequence-Based Binding Prediction Using Deep-Learning. J. Chem. Inf. Model 2019, 59, 1645–1657. [DOI] [PubMed] [Google Scholar]
- (35).Wallach I; Heifets A Most Ligand-Based Classification Benchmarks Reward Memorization Rather than Generalization. J. Chem. Inf. Model 2018, 58, 916–932. [DOI] [PubMed] [Google Scholar]
- (36).Chen L; Cruz A; Ramsey S; Dickson CJ; Duca JS; Hornak V; Koes DR; Kurtzman T Hidden bias in the DUD-E dataset leads to misleading performance of deep learning in structure-based virtual screening. PLOS ONE 2019, 14, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tran-Nguyen V-K; Jacquemard C; Rognan D LIT-PCBA: An Unbiased Data Set for Machine Learning and Virtual Screening. J. Chem. Inf. Model 2020, [DOI] [PubMed] [Google Scholar]
- (38).Mysinger MM; Carchia M; Irwin JJ; Shoichet BK Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking. J. Med. Chem 2012, 55, 6582–6594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Kramer C; Gedeck P Leave-Cluster-Out Cross-Validation Is Appropriate for Scoring Functions Derived from Diverse Protein Data Sets. J. Chem. Inf. Model 2010, 50, 1961–1969. [DOI] [PubMed] [Google Scholar]
- (40).Ballester PJ; Mitchell JBO Comments on Leave-Cluster-Out Cross-Validation Is Appropriate for Scoring Functions Derived from Diverse Protein Data Sets: Significance for the Validation of Scoring Functions. J. Chem. Inf. Model 2011, 51, 1739–1741. [DOI] [PubMed] [Google Scholar]
- (41).Li H; Leung K-S; Wong M-H; Ballester PJ Low-Quality Structural and Interaction Data Improves Binding Affinity Prediction via Random Forest. Molecules 2015, 20, 10947–10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Sunseri J; Koes DR libmolgrid: Graphics Processing Unit Accelerated Molecular Gridding for Deep Learning Applications. J. Chem. Inf. Model 2020, 60, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bakan A; Dutta A; Mao W; Liu Y; Chennubhotla C; Lezon TR; Bahar I Evol and ProDy for bridging protein sequence evolution and structural dynamics. Bioinformatics 2014, 30, 2681–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Koes DR; Baumgartner MP; Camacho CJ J. Chem. Inf. Model 2013, [doi: 10.1021/ci300604z]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Coupry DE; Addicoat MA; Heine T Extension of the Universal Force Field for MetalOrganic Frameworks. J. Chem. Theory Comput 2016, 12, 5215–5225. [DOI] [PubMed] [Google Scholar]
- (46).RDKit: Open-Source Cheminformatics. http://www.rdkit.org, accessed November 6, 2017.
- (47).Kufareva I; Ilatovskiy AV; Abagyan R Pocketome: an encyclopedia of small-molecule binding sites in 4D. Nucleic Acids Res. 2012, 40, 535–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Konc J; Janežič D ProBiS algorithm for detection of structurally similar protein binding sites by local structural alignment. Bioinformatics 2010, 26, 1160–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Ragoza M; Turner L; Koes DR Ligand Pose Optimization with Atomic Grid-Based Convolutional Neural Networks. Machine Learning for Molecules and Materials NIPS 2017 Workshop. 2017; arXiv preprint arXiv:1710.07400. [Google Scholar]
- (50).Dreossi T; Ghosh S; Yue X; Keutzer K; Sangiovanni-Vincentelli A; Seshia SA Counterexample-guided data augmentation. arXiv preprint arXiv:1805.06962 2018, [Google Scholar]
- (51).Jia Y; Shelhamer E; Donahue J; Karayev S; Long J; Girshick R; Guadarrama S; Darrell T Caffe: Convolutional Architecture for Fast Feature Embedding. arXiv preprint arXiv:1408.5093 2014, [Google Scholar]
- (52).Huang S-Y; Zou X Ensemble docking of multiple protein structures: considering protein structural variations in molecular docking. Proteins: Struct., Funct., Bioinf 2007, 66, 399–421. [DOI] [PubMed] [Google Scholar]
- (53).Trott O; Olson AJ AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Li Y; Liu Z; Li J; Han L; Liu J; Zhao Z; Wang R Comparative assessment of scoring functions on an updated benchmark: 1. Compilation of the test set. J. Chem. Inf. Model 2014, 54, 1700–1716. [DOI] [PubMed] [Google Scholar]
- (55).Boyles F; Deane CM; Morris GM Learning from the ligand: using ligand-based features to improve binding affinity prediction. Bioinformatics 2020, 36, 758–764. [DOI] [PubMed] [Google Scholar]
- (56).Sollich P; Krogh A Learning with ensembles: How overfitting can be useful. Advances in neural information processing systems. 1996; pp 190–196. [Google Scholar]
- (57).Ye Z; Baumgartner MP; Wingert BM; Camacho CJ Optimal strategies for virtual screening of induced-fit and flexible target in the 2015 D3R Grand Challenge. J. Comput.-Aided Mol. Des 2016, 30, 695–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Morrone JA; Weber JK; Huynh T; Luo H; Cornell WD Combining docking pose rank and structure with deep learning improves protein-ligand binding mode prediction. arXiv preprint arXiv:1910.02845 2019, [DOI] [PubMed] [Google Scholar]
- (59).Li H; Leung K-S; Wong M-H; Ballester PJ Correcting the impact of docking pose generation error on binding affinity prediction. BMC Bioinf. 2016, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Ashtawy HM; Mahapatra NR Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model 2018, 58, 119–133. [DOI] [PubMed] [Google Scholar]
- (61).Wang C; Zhang Y Improving scoring-docking-screening powers of proteinligand scoring functions using random forest. J. Comput. Chem 2017, 38, 169–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Damm-Ganamet KL; Smith RD; Dunbar JB; Stuckey JA; Carlson HA CSAR Benchmark Exercise 20112012: Evaluation of Results from Docking and Relative Ranking of Blinded Congeneric Series. J. Chem. Inf. Model 2013, 53, 1853–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Yuriev E; Ramsland PA Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit 2013, 26. [DOI] [PubMed] [Google Scholar]
- (64).Wierbowski SD; Wingert BM; Zheng J; Camacho CJ Cross Docking Benchmark for automated Pose and Ranking prediction of ligand binding. Protein Sci. 2019, [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.