

Abstract
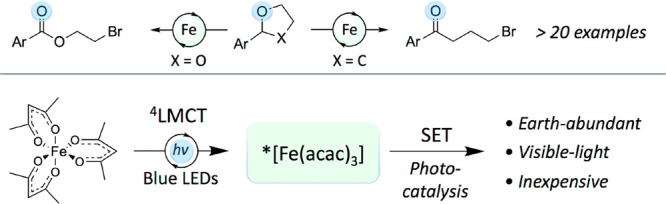
A new method employing iron(III) acetylacetonate along with visible light is described to effect oxidative ring opening of cyclic ethers and acetals with unparalleled efficiency. The method allows for a photocatalytic radical chemistry approach to functionalize relatively inert cyclic ethers into useful synthetic intermediates. The methodology sheds further light on the use of underexplored iron complexes in visible-light photochemical contexts and illustrates that simple Fe(III) complexes can initiate redox processes from 4LMCT excited states.
Cyclic ethers are used in most scientific disciplines utilizing organic compounds. Despite being ubiquitous structural moieties, synthetically useful transformations employing these structural features as synthons are scarce. Attempts at achieving oxidative ring opening of cyclic ethers has essentially been limited to epoxides and oxetanes, relying on strain relief mediating the transformations.1−3 Among the few examples known for nonstrained cyclic ethers, e.g. tetrahydrofurans, harsh conditions such as aqueous molecular bromine has been used to oxidatively ring-open THF to give 4-hydroxybutanal in only 20% yield (Scheme 1b).4 A more recent paper by Leadbeater and co-workers pursued the transformation of 2-phenyltetrahydrofurans into 4-hydroxy-1-phenylbutane-1-ones by employing oxoammonium salts as oxidants (Scheme 1b).3 The transformation is achieved by a hydride abstraction generating a stabilized carbocation. The authors concluded that the cationic intermediates were too stable resulting in multiple reaction pathways, e.g. polymerizations, giving poor yields with the best example reaching only 45% yield. Moreover, the method was unable to use any other nucleophile than water due to nucleophile oxidation by the oxoammonium cation. These examples illustrate the typical difficulties associated with oxidative approaches for fragmentative diversification of the ether functionality.
Scheme 1. Oxidative Ring Opening of THF-Core and Methods of Halogenated Ketone Syntheses.

The past decade has seen a surge in catalytic visible-light mediated methodologies demonstrating its success in achieving novel radical transformations under mild conditions.5−7 As part of our ongoing research activities, we aimed to explore if photoredox catalysis could provide opportunities for developing a reliable and robust approach engaging ether functionalities oxidatively, and thereby generating structural features of broader synthetic value. Our initial ambition was to target the generation of γ-bromo ketones, which are valuable building blocks for various chemical transformations and utilized in the syntheses of many biologically relevant compound classes (Scheme 1a).8−10 Recent work from the Knowles11 and Zhu12 groups provide elegant examples of PCET mediated fragmentative functionalization of cyclic alcohols to access γ- or δ-bromo ketones.13−15 Also noteworthy is the work by Almqvist, König and co-workers that recently disclosed the generation of β-chloroketones via a light-mediated oxidative fragmentation using aryl cyclopropanes as starting material (Scheme 1c).16
We hypothesized that hydrogen atom transfer, HAT, not only could be used for chemoselective generation of carbon-centered radicals from ethers, but that such an approach toward oxidative fragmentation might overcome the limited substrate scope and competing polymerization as previously reported for methods involving polar pathways. Herein, we report the use of simple Fe(acac)3 as an inexpensive, efficient, and earth abundant based catalyst in a visible-light driven oxidative ring opening of tetrahydrofurans, tetrahydropyrans, and acetals to give synthetically useful bromo-substituted ketones or esters (Scheme 1d). Recently, other noteworthy examples in photoredox catalysis reported the use of simple catalysts based on iron.17−23 To the best of our knowledge, the present method constitutes the first high-yielding oxidative ring opening of nonstrained cyclic ethers and, furthermore, the process is presumably initiated via an unprecedented SET from an excited state Fe(III) species by an initial 4LMCT absorption.
In our initial investigation we explored the use of Ru(bpy)3(PF6)2 as a photocatalyst to oxidatively ring-open 2-(4-chlorophenyl)tetrahydrofuran (1aa) to the target 4-bromo-1-(4-chlorophenyl)butan-1-one (1a). To our dismay, these conditions provided irreproducible results, ranging from 0% to quantitative yields based on 1H NMR (Table 1, entry 1). Even more surprisingly, we found the reaction to behave similarly in our control experiment without any catalyst present. We suspected that trace metal impurities possibly could be responsible for the irreproducibility issues, which prompted us to screen various transition metal additives (see Supporting Information (SI)). Indeed, when using new glassware or glassware cleaned with aqua regia, conversion to the product was reproducible (Table 1, entry 2). We were pleased to find that 1a was consistently formed in 90% yield using 1 mol % of Fe(acac)3 and 3 equiv of BrCCl3 in dichloroethane irradiated with blue LEDs (Table 1, entry 3).
Table 1. Deviation from Standard Conditions.

| Entry | Catalyst | BrCCl3 | Solvent | Yield (%)a |
|---|---|---|---|---|
| 1b | Ru(bpy)3(PF6)2 1–5 mol % | 2–10 equiv | Solvents | 0-quant |
| 2 | Ru(bpy)3(PF6)2 1 mol % | 3 equiv | DCE | 31 (full convc)g |
| 3 | Fe(acac)31 mol % | 3 equiv | DCE | 89 (90d) |
| 4 | Fe(acac)3 1 mol % | CBr4, 3 equiv | DCE | 64 |
| 5 | FeBr3 1 mol % | 3 equiv | DCE | 55 |
| 6e | Fe(acac)3 1 mol % | 3 equiv | DCE | Trace |
| 7f | Fe(acac)3 1 mol % | 3 equiv | DCE | No reaction |
| 8g | No catalyst | 3 equiv | DCE | Trace |
Isolated yields conducted at 0.1 mmol scale.
Note, irreproducible yields were consistently obtained also when keeping all parameters constant. Yields were determined by 1H NMR using dimethyl sulfone or ethylene carbonate as internal standard.
Reaction run for 18 h.
Average isolated yield of two runs at 0.2 mmol scale.
Heat control (80 °C).
Control experiment in the dark.
Reaction conducted in either a brand new vial or a vial cleaned with aqua regia.
It should be pointed out that these conditions provided the product much more efficiently than the corresponding conditions using Ru(bpy)3(PF6)2 as catalysts (6 h and 89% yield vs 6 h and 31% yield). It should be noted that a catalyst loading of 0.1 mol % also promotes the reaction with a slightly diminished efficacy (SI, Table 1, entry 12), whereas decreasing the loading even further down to ppm levels generates conditions that provide 50–60% conversion when irradiated for 18 h. These results may shed some light on recently published methods in which BrCCl3 is claimed to be engaged in photo-mediated processes under catalyst-free conditions.24,25 Our control experiments show that the reaction is not thermally promoted and both Fe(acac)3 and light are essential for the reaction to progress (Table 1, entries 7–8).
With optimized conditions at hand we started our scope exploration by varying the aromatic moiety of the tetrahydrofuran derivative. As can be seen in Scheme 2, the method is compatible with a vast range of electronic properties associated with the aromatic functionality (2a–2k). Going from methoxy (2d) to a nitrile (2c) substituent in the para-position practically does not affect the efficiency of the reaction giving a 79% and 81% yield, respectively. The reaction also progressed well with a sterically hindered isopropyl substituent in the ortho-position (2f, 92%). Expanding the scope to also include electron-rich N-heteroaromatics typically provided trichloromethyl functionalized THF derivatives, as exemplified by the formation of 2l, along with unreacted starting material. These observations can be rationalized by the basic nature of these moieties, which are localized in close proximity to the β-carbon of the THF functionality. This setup can possibly mediate an elimination of the so formed cationic intermediate (Scheme 6) providing the corresponding 2,3-dihydrofuran derivatives.
Scheme 2. Scope of Aromatic Moiety of THF-Derivative.

Reactions were conducted at 0.2 mmol scale and 0.1 mol/dm3, 3 eq. BrCCl3 and 1 mol % Fe(acac)3. Yields are reported as average isolated yield of two runs, except for (2j) with only one run.
Scheme 6. Proposed Mechanism.

Next we turned to investigate congeners to the THF functionality (Scheme 3a). Introducing a methyl group in the 3-position of the THF-core gave an exceptionally clean reaction in 95% yield (3a). Enlarging the ring size to a tetrahydropyran was also compatible with the reaction conditions, yielding 5-bromo-1-phenylpentan-1-one (3b) in 84% yield. Surprisingly, when the dioxane core (3cc) was subjected to the reaction conditions, no product or only traces could be detected with close to total recovery of the starting material.
Scheme 3. Scope of Ether and Acetal.

Reactions were conducted at 0.2 mmol scale and 0.1 mol/dm3, 3 equiv of BrCCl3 and 1 mol % Fe(acac)3. Yields are reported as average isolated yield of two runs.
Reaction conducted at 1 mmol scale.
Regioisomeric ratio.
Percentage refers to conversion as determined by 1H NMR; 10 equiv of BrCCl3 were used.
Acetals have previously been reported to oxidatively convert to esters under various conditions, and we consequently wondered if such a transformation would also be accessible with our iron-catalyzed photodriven conditions.26−29 To our delight, benzaldehyde dimethyl acetal neatly transformed to methyl benzoate (3d) in good yield (71%). Cyclic acetals, 1,3-dioxolanes, were all converted to the expected products with high yields ranging from 78% to 95% (3e–3h). 2-(3-Bromophenyl)-1,3-dioxolane was subjected to the reaction conditions on a 1 mmol scale, resulting in only a minor decrease in yield, providing 82% as compared to 86% (3e). When 4-methyl-2-phenyl-1,3-dioxolane was subjected to the reaction conditions, an inseparable mixture of two regioisomers was formed in a 1:24 ratio with a total yield of 95% (3f). We continued examining the compatibility with heteroaromatic functionalities of our system on cyclic acetals and contentedly found that benzo[b]thiophene- and benzofuran cores are successfully tolerated providing 3g and 3h in 78% and 85% yield, respectively. Furthermore, applying more forcing conditions by increasing the equivalents of BrCCl3 also converted a nonaromatic THF-derivative with a pendant alkyl chain to the γ-bromo ketone.
Next, exploring the engagement of acyclic ethers provides a method for oxidative dealkylative fragmentation to yield aldehydes or ketones (Scheme 3b). Benzylmethyl ether and (3-(benzyloxy)propyl)benzene both converted to benzaldehyde (3j) in good yields (85% and 81% respectively). The latter result, showing selectivity for the benzyl ether hydrogen over the benzylic hydrogen, sheds light on the possibility of brominative deprotection of benzyl ethers. Lastly, introducing a methyl group on the benzylic position gives access to the corresponding acetophenone (3k) in 80% yield.
To further demonstrate the utility of our system, we addressed the production of a key intermediate in the synthesis of H1 receptor antagonist fexofenadine.10 The key intermediate is a 4-bromo-1-phenylbutane-1-one with an isobutyronitrile group in the para-position. We envisioned a two-step synthesis of this compound starting with the production of the corresponding tetrahydrofuran precursor followed by our iron-catalyzed visible-light driven reaction (Scheme 4). The one-pot Heck and hydrogenation reaction described by Evans and co-workers gave the desired 2-methyl-2(4-(tetrahydrofuran-2-yl)phenyl)propanenitrile (4aa) in a satisfactory 68% yield.30 Next, subjected to our conditions the key intermediate (4a) was formed in 82% yield giving a total isolated yield of 56%, which should be compared to 53% yield over 3 steps as previously reported.10
Scheme 4. Synthesis of Key Intermediate to Fexofenadine.

To gain some insight into the mechanism of this reaction we carried out a set of reactions under different conditions to probe for the role of Fe(acac)3. It has been described that Fe(acac)3 under irradiation of visible light undergoes 4LMCT resulting in release of an acac radical (Scheme 5).31,32 One conceiving mechanism could be that an extruded acac radical might act as the hydrogen atom acceptor, initiating a propagation based generation of the product. To probe this possibility two experiments were devised (i–ii in Scheme 5).
Scheme 5. Mechanistic Experiments of acac Radical Involvement.

In experiment (i), equimolar amounts of Fe(acac)3 and 1aa were irradiated under optimized conditions. If acac radicals were to be extruded, degradation of 1aa with potential formation of dimers or the dihydrofuran derivative would be expected. However, after 6 h of irradiation, no reaction could be observed. Experiment (ii) served the same purpose, to probe the formation of acac radicals. By adding an electrophilic reaction partner an opportunity is provided to more efficiently engage the hypothetical THF radical in a Giese reaction. However, as for the previous experiment, no reaction occurred. Experiment (iii) served the purpose of probing for any potential ionic intermediates formed in the absence of BrCCl3. Again, only starting material was observed after 6 h of irradiation. Taken together, these findings are in agreement with previous studies that reported on the high photostability of Fe(acac)3.33 We therefore find it unlikely that acac radicals initiate the reaction.
Instead, we turned our attention to Fe(acac)3 as an activator of BrCCl3. Because Fe(acac)3 does not fluoresce within the visible region, the typical Stern–Volmer quenching studies were not a viable option to probe such activation. That said, if Fe(acac)3 could induce a mesolytic cleavage of BrCCl3, this process should be able to initiate a dimerization of the so formed trichloromethyl radicals. Indeed, when irradiating an equimolar solution of BrCCl3 and Fe(acac)3, hexachloroethane was detected as a major constituent by GC-MS (iv in Scheme 5). This result strongly suggests that *[Fe(acac)3] is quenched by BrCCl3 either by energy transfer or by SET. Furthermore, the reaction was almost completely impeded by addition of TEMPO.
Besides quenching of *[Fe(acac)3] by BrCCl3, there is a possibility of forming an EDA-complex between Fe(acac)3 and BrCCl3 for which UV–vis titration experiments were conducted, but these ruled out any potential absorption complex formation (see SI). Energy transfer based initiation via homolytic cleavage can also be ruled out due to the energy discrepancy between the relaxed 4LMCT state of *[Fe(acac)3] (220 kJ/mol)34 and that of the 1[BrCCl3] → 3[BrCCl3] transition (571 kJ/mol; see SI). On the other hand, the estimated redox potential of the relaxed 4LMCT state of *[Fe(acac)3] (E1/2red = −0.68 V vs SCE)34,35 clearly provides an exergonic pathway for SET based quenching with BrCCl3 (E1/2red = −0.18 V vs SCE).5
The absorption characteristics and the excited state dynamics of Fe(acac)3 have been thoroughly studied experimentally.33 This study together with early work by Wilkinson and Farmilo34 clearly indicates that Fe(acac)3 harbors at least two metal centered excited states lower in energy than the initial vibrationally relaxed 4LMCT state of *[Fe(acac)3] (λmax = 440 nm). The estimated excited state redox potential for one of these states do allow for an exergonic SET to BrCCl3 (see SI Figure S2). Upon excitation at 440 nm, Fe(acac)3 is known to regain the absorption characteristics of its fully decayed ground state after ca. 60 ps.33 As such, Fe(acac)3 decay back to its ground state roughly 1 order of magnitude faster than [Fe(bpy)3]2+, a complex known to efficiently initiate propagation based processes via the excited state.18
With these experimental results at hand, we propose a mechanism starting with Fe(acac)3 absorbing a photon resulting in a 4LMCT state of *[Fe(acac)3] (Scheme 6). Oxidative quenching of the excited catalyst by BrCCl3 causes mesolytic cleavage to give a trichloromethyl radical and a bromide ion. The trichloromethyl radical abstracts the α-hydrogen, forming chloroform. From here a propagation mechanism starts where the radical intermediate abstracts a bromine from BrCCl3 forming another trichloromethyl radical. The bromine intermediate fragmentizes to a stabilized carbocation intermediate that ring-opens irreversibly by action of the bromide ion. The [Fe(acac)3]+ formed is highly oxidizing (+1.60 V vs SCE)35 and, from a thermodynamic perspective, capable of oxidizing the nucleophilic radical (<+0.16 V vs SCE).36 This SET event closes the catalytic cycle for Fe(acac)3. There is also the possibility of the so formed electron-rich radical to reduce Fe(acac)3 to Fe(II) from which another mode of initiation could act in parallel.
In summary we have developed an efficient method for the oxidative ring opening of cyclic ethers as well as acetals with good to excellent yields. The method employs the novel reactivity of Fe(acac)3 in conjunction with visible light. There is still some uncertainties regarding the detailed mechanism, and a more in-depth understanding of light-induced iron chemistry is needed.
Acknowledgments
The Swedish Research Council is acknowledged for financial support. We acknowledge the support from the Swedish NMR Centre at the University of Gothenburg for running 13C-NMR samples. A special recognition to August Runemark (University of Gothenburg), Daniel Tietze (Chalmers University of Technology), and Jurgen Grafenstein (University of Gothenburg) for assistance with UV–vis, LC-(HR)MS, and computation, respectively.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.orglett.2c00231.
Synthetic procedures and characterization data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Arjun Reddy M.; Bhanumathi N.; Rama Rao K. A Mild and Efficient Biomimetic Synthesis of α-Hydroxymethylarylketones from Oxiranes in the Presence of β-Cyclodextrin and NBS in Water. Tetrahedron Lett. 2002, 43 (17), 3237–3238. 10.1016/S0040-4039(02)00433-1. [DOI] [Google Scholar]
- Surendra K.; Krishnaveni N. S.; Reddy M. A.; Nageswar Y. V.D.; Rao K. R. Highly Selective Oxidative Cleavage of β-Cyclodextrin - Epoxide/Aziridine Complexes with IBX in Water. J. Org. Chem. 2003, 68 (23), 9119–9121. 10.1021/jo034079c. [DOI] [PubMed] [Google Scholar]
- Loman J. J.; Carnaghan E. R.; Hamlin T. A.; Ovian J. M.; Kelly C. B.; Mercadante M. A.; Leadbeater N. E. A Combined Computational and Experimental Investigation of the Oxidative Ring-Opening of Cyclic Ethers by Oxoammonium Cations. Org. Biomol. Chem. 2016, 14 (16), 3883–3888. 10.1039/C6OB00347H. [DOI] [PubMed] [Google Scholar]
- Deno N. C.; Potter N. H. The Mechanism and Synthetic Utility of the Oxidative Cleavage of Ethers by Aqueous Bromine. J. Am. Chem. Soc. 1967, 89 (14), 3550–3554. 10.1021/ja00990a035. [DOI] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113 (7), 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staveness D.; Bosque I.; Stephenson C. R. J. Free Radical Chemistry Enabled by Visible Light-Induced Electron Transfer. Acc. Chem. Res. 2016, 49 (10), 2295–2306. 10.1021/acs.accounts.6b00270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsui J. K.; Lang S. B.; Heitz D. R.; Molander G. A. Photoredox-Mediated Routes to Radicals: The Value of Catalytic Radical Generation in Synthetic Methods Development. ACS Catal. 2017, 7 (4), 2563–2575. 10.1021/acscatal.7b00094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y.; Liu X. Y.; Wang Z. H.; Tang L. F. Synthesis of 3-Acyl, Methylene and Epoxy Substituted Isoindolinone Derivatives via the Ortho-Lithiation/Cyclization Procedures of Aromatic Imines with Carbon Monoxide. Tetrahedron 2017, 73 (52), 7245–7253. 10.1016/j.tet.2017.11.001. [DOI] [Google Scholar]
- Hu H.; Dyke J. C.; Bowman B. A.; Ko C. C.; You W. Investigation of Dopamine Analogues: Synthesis, Mechanistic Understanding, and Structure-Property Relationship. Langmuir 2016, 32 (38), 9873–9882. 10.1021/acs.langmuir.6b02141. [DOI] [PubMed] [Google Scholar]
- Huang J.; Wang W.; Wang L. X. Novel Preparation of H1 Receptor Antagonist Fexofenadine. Org. Process Res. Dev. 2010, 14 (6), 1464–1468. 10.1021/op100090j. [DOI] [Google Scholar]
- Yayla H. G.; Wang H.; Tarantino K. T.; Orbe H. S.; Knowles R. R. Catalytic Ring-Opening of Cyclic Alcohols Enabled by PCET Activation of Strong O-H Bonds. J. Am. Chem. Soc. 2016, 138 (34), 10794–10797. 10.1021/jacs.6b06517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D.; Mao J.; Zhu C. Visible Light-Promoted Ring-Opening Functionalization of Unstrained Cycloalkanols via Inert C-C Bond Scission. Chem. Sci. 2018, 9 (26), 5805–5809. 10.1039/C8SC01763H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao R.; Yao Y.; Zhu D.; Chang D.; Liu Y.; Shi L. Visible-Light-Enhanced Ring Opening of Cycloalkanols Enabled by Brønsted Base-Tethered Acyloxy Radical Induced Hydrogen Atom Transfer-Electron Transfer. Org. Lett. 2018, 20 (4), 1228–1231. 10.1021/acs.orglett.8b00161. [DOI] [PubMed] [Google Scholar]
- Fan X.; Zhao H.; Yu J.; Bao X.; Zhu C. Regiospecific Synthesis of Distally Chlorinated Ketones via C-C Bond Cleavage of Cycloalkanols. Org. Chem. Front. 2016, 3 (2), 227–232. 10.1039/C5QO00368G. [DOI] [Google Scholar]
- Huan L.; Zhu C. Manganese-Catalyzed Ring-Opening Chlorination of Cyclobutanols: Regiospecific Synthesis of γ-Chloroketones. Org. Chem. Front. 2016, 3 (11), 1467–1471. 10.1039/C6QO00443A. [DOI] [Google Scholar]
- Petzold D.; Singh P.; Almqvist F.; König B. Visible-Light-Mediated Synthesis of β-Chloro Ketones from Aryl Cyclopropanes. Angew. Chemie - Int. Ed. 2019, 58, 8577–8580. 10.1002/anie.201902473. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Li L.; Ji H.; Ma W.; Chen C.; Zhao J. Iron(III)-Mediated Photocatalytic Selective Substitution of Aryl Bromine by Chlorine with High Chloride Utilization Efficiency. Chem. Commun. 2014, 50 (18), 2344–2346. 10.1039/c3cc48246d. [DOI] [PubMed] [Google Scholar]
- Gualandi A.; Marchini M.; Mengozzi L.; Natali M.; Lucarini M.; Ceroni P.; Cozzi P. G. Organocatalytic Enantioselective Alkylation of Aldehydes with [Fe(Bpy)3]Br2 Catalyst and Visible Light. ACS Catal. 2015, 5 (10), 5927–5931. 10.1021/acscatal.5b01573. [DOI] [Google Scholar]
- Li S.; Zhu B.; Lee R.; Qiao B.; Jiang Z. Visible Light-Induced Selective Aerobic Oxidative Transposition of Vinyl Halides Using a Tetrahalogenoferrate(III) Complex Catalyst. Org. Chem. Front. 2018, 5 (3), 380–385. 10.1039/C7QO00798A. [DOI] [Google Scholar]
- Ye J. H.; Miao M.; Huang H.; Yan S. S.; Yin Z. B.; Zhou W. J.; Yu D. G. Visible-Light-Driven Iron-Promoted Thiocarboxylation of Styrenes and Acrylates with CO2. Angew. Chemie - Int. Ed. 2017, 56 (48), 15416–15420. 10.1002/anie.201707862. [DOI] [PubMed] [Google Scholar]
- Huang B.; Li Y.; Yang C.; Xia W. Three-Component Aminoselenation of Alkenes: Via Visible-Light Enabled Fe-Catalysis. Green Chem. 2020, 22 (9), 2804–2809. 10.1039/C9GC04163J. [DOI] [Google Scholar]
- Li Z.; Wang X.; Xia S.; Jin J. Ligand-Accelerated Iron Photocatalysis Enabling Decarboxylative Alkylation of Heteroarenes. Org. Lett. 2019, 21 (11), 4259–4265. 10.1021/acs.orglett.9b01439. [DOI] [PubMed] [Google Scholar]
- Xia S.; Hu K.; Lei C.; Jin J. Intramolecular Aromatic C-H Acyloxylation Enabled by Iron Photocatalysis. Org. Lett. 2020, 22 (4), 1385–1389. 10.1021/acs.orglett.0c00002. [DOI] [PubMed] [Google Scholar]
- Nauth A. M.; Orejarena Pacheco J. C.; Pusch S.; Opatz T. Oxidation of Trialkylamines by BrCCl3: Scope, Applications and Mechanistic Aspects. Eur. J. Org. Chem. 2017, 2017 (46), 6966–6974. 10.1002/ejoc.201701320. [DOI] [Google Scholar]
- Franz J. F.; Kraus W. B.; Zeitler K. No Photocatalyst Required-Versatile, Visible Light Mediated Transformations with Polyhalomethanes. Chem. Commun. 2015, 51 (39), 8280–8283. 10.1039/C4CC10270C. [DOI] [PubMed] [Google Scholar]
- Huyser E. S.; Garcia Z. Peroxide-Induced Conversions of Cyclic Acetals of Benzaldehyde to Benzoate Esters. J. Org. Chem. 1962, 27 (8), 2716–2719. 10.1021/jo01055a002. [DOI] [Google Scholar]
- Hai-Xia L.; Liang-Heng X.; Nai-Ju H. A New Synthetic Method for Haloalkyl Carboxylic Esters from the Radical Ring Cleavage of Cyclic Acetals with Haloform. Synth. Commun. 1997, 27 (2), 303–306. 10.1080/00397919708005032. [DOI] [Google Scholar]
- Prugh J. D.; McCarthy W. C. The Oxidation of Acetals with N-Bromosuccinimide. Tetrahedron Lett. 1966, 7 (13), 1351–1356. 10.1016/S0040-4039(01)99721-7. [DOI] [Google Scholar]
- Elad D.; Youssefyeh R. The Photochemical Conversion of Acetals to Carboxylic Esters. Tetrahedron Lett. 1963, 4 (30), 2189–2191. 10.1016/S0040-4039(01)90993-1. [DOI] [Google Scholar]
- Geoghegan K.; Kelleher S.; Evans P. An Investigation into the One-Pot Heck Olefination-Hydrogenation Reaction. J. Org. Chem. 2011, 76 (7), 2187–2194. 10.1021/jo200023r. [DOI] [PubMed] [Google Scholar]
- Lintvedt R. L.; Kernitsky L. K. Ligand Field Information from Charge-Transfer Spectra of Substituted Tris(1,3-Diketonato)Iron(III) Chelates. Spectrochemical Series for 1,3-Diketones. Inorg. Chem. 1970, 9 (3), 491–494. 10.1021/ic50085a012. [DOI] [Google Scholar]
- Lang K.; Luňák S. Photocatalytic Degradation of 4-Chlorophenoxyacetic Acid in the Presence of an Iron Complex and Hydrogen Peroxide. Photochem. Photobiol. Sci. 2002, 1 (8), 588–591. 10.1039/B203147G. [DOI] [PubMed] [Google Scholar]
- Maçôas E. M. S.; Kananavicius R.; Myllyperkiö P.; Pettersson M.; Kunttu H. Ultrafast Electronic and Vibrational Energy Relaxation of Fe(Acetylacetonate)3 in Solution. J. Phys. Chem. A 2007, 111 (11), 2054–2061. 10.1021/jp066271z. [DOI] [PubMed] [Google Scholar]
- Wilkinson F.; Farmilo A. Mechanism of Quenching of the Triplet States of Organic Compounds by Tris-(Beta-Diketonato) Complexes of Iron (III), Rhuthenium(III) and Aluminium(III). J. Chem. Soc., Faraday Trans. 2 1976, 72 (72), 604–618. 10.1039/F29767200604. [DOI] [Google Scholar]
- Richert S. A.Iron PhD Thesis. PhD Thesis, Texas A&M Univ., 1989. [Google Scholar]
- Wayner D. D. M.; McPhee D. J.; Griller D. Oxidation and Reduction Potentials of Transient Free Radicals. J. Am. Chem. Soc. 1988, 110 (1), 132–137. 10.1021/ja00209a021. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.