

Abstract
For decades, anticancer targeted therapies have been designed to inhibit kinases or other enzyme classes and have profoundly benefitted many patients. Novel approaches are required, however, to target transcription factors, scaffolding proteins and other proteins central to cancer biology that typically lack catalytic activity and have remained mostly recalcitrant to drug development. The selective degradation of target proteins is an attractive approach to expand the druggable proteome, and the selective oestrogen receptor degrader fulvestrant served as an early example of this concept. Following a long and tragic history in the clinic, the immunomodulatory imide drug (IMiD) thalidomide was discovered to act by a novel and unexpected mechanism of action: targeting proteins to an E3 ubiquitin ligase for subsequent proteasomal degradation. This discovery has paralleled and directly catalyzed myriad breakthroughs in drug development, leading to the rapid maturation of generalizable chemical platforms for the targeted degradation of previously undruggable proteins. Decades of clinical experience have established frontline roles for thalidomide analogues in the treatment of haematological malignancies. With a new generation of ‘degrader’ drugs currently in development, this experience provides crucial insights into class-wide features of degraders, including a unique pharmacology, mechanisms of resistance and emerging therapeutic opportunities. Herein, we review these past experiences and discuss their application in the clinical development of novel degrader therapies.
Introduction
Thalidomide was introduced to the clinic in the late 1950s as a sedative and antiemetic and was marketed as a treatment for morning sickness in pregnant women1. Tragically, in utero exposure to thalidomide caused an embryopathy characterized by foreshortened limb anomalies, known as phocomelia, in thousands of children worldwide2,3. In the early 1960s, Frances Kelsey at the FDA played a crucial part in preventing the marketing approval and thus the widespread use of thalidomide in the USA until the association of this drug with teratogenicity was publically exposed in 19624 and the agency’s annual drug safety excellence award is named in her honour. Although broadly withdrawn from clinical use, thalidomide remained on the market for decades as a treatment for erythema nodosum leprosum (an immune-mediated complication of leprosy)5,6. In the 1990s, thalidomide was reconsidered as an anticancer agent7. Clinical testing revealed the efficacy and tolerability of thalidomide in patients with multiple myeloma (MM)8–10, and this success spurred the development of chemically similar analogues, first lenalidomide and then pomalidomide, as cancer therapies.
In 2013, thalidomide analogues were discovered to exert their therapeutic effects by acting as a molecular glue to recruit disease-relevant neosubstrate proteins to an E3 ubiquitin ligase, resulting in neosubstrate ubiquitination and, ultimately, proteasomal degradation11–13. This molecular mechanism of action and the clinical experience with thalidomide analogues highlight unique features of ‘degrader’ therapeutics, including a distinct pharmacology, mechanisms of resistance and the integrated effect of degrading multiple substrates on efficacy and toxicity profiles.
Herein, we review the wide and expanding use of thalidomide analogues in the treatment of multiple cancers, including MM, non-Hodgkin lymphoma (NHL) and myelodysplastic syndrome (MDS). With novel degrader drugs poised to expand the druggable proteome, we also outline the lessons learned from thalidomide analogues — particularly lenalidomide — that serve as an essential guide to the clinical development of targeted protein degradation platforms.
Thalidomide analogues
Mechanism of action and chemistry
Thalidomide and its derivatives were used clinically for decades prior to elucidation of the molecular basis of their activity. In 2010, thalidomide was found to directly bind to the protein cereblon (CRBN)14, which serves as a substrate receptor component of the cullin-RING E3 ubiquitin ligase complex 4 (CRL4) comprising cullin-4 (CUL4), DNA damage-binding protein 1 (DDB1) and E3 ubiquitin-protein ligase RBX1 (FIG. 1a,b and BOX 1). Subsequently, our group and others concurrently discovered that thalidomide and its analogues do not inhibit the enzymatic activity of CRL4CRBN, but instead confer neomorphic activity on CRBN to mediate the selective ubiquitination and resultant proteasomal degradation of the transcription factors Ikaros family zinc finger protein 1 (IKZF1) and IKZF3 (Aiolos) in MM cells and T cells11–13. Similarly, in del(5q) MDS cells, lenalidomide mediates the CRBN-dependent ubiquitination and degradation of casein kinase I isoform α (CK1α)15. IKZF1, IKZF3 and CK1α are required for MM or MDS cell survival (BOX 2), thereby establishing selective degradation of disease-relevant proteins as the unexpected mechanism underlying the anticancer effects of thalidomide analogues16.
Fig. 1 |. Mechanisms of targeted protein degradation.
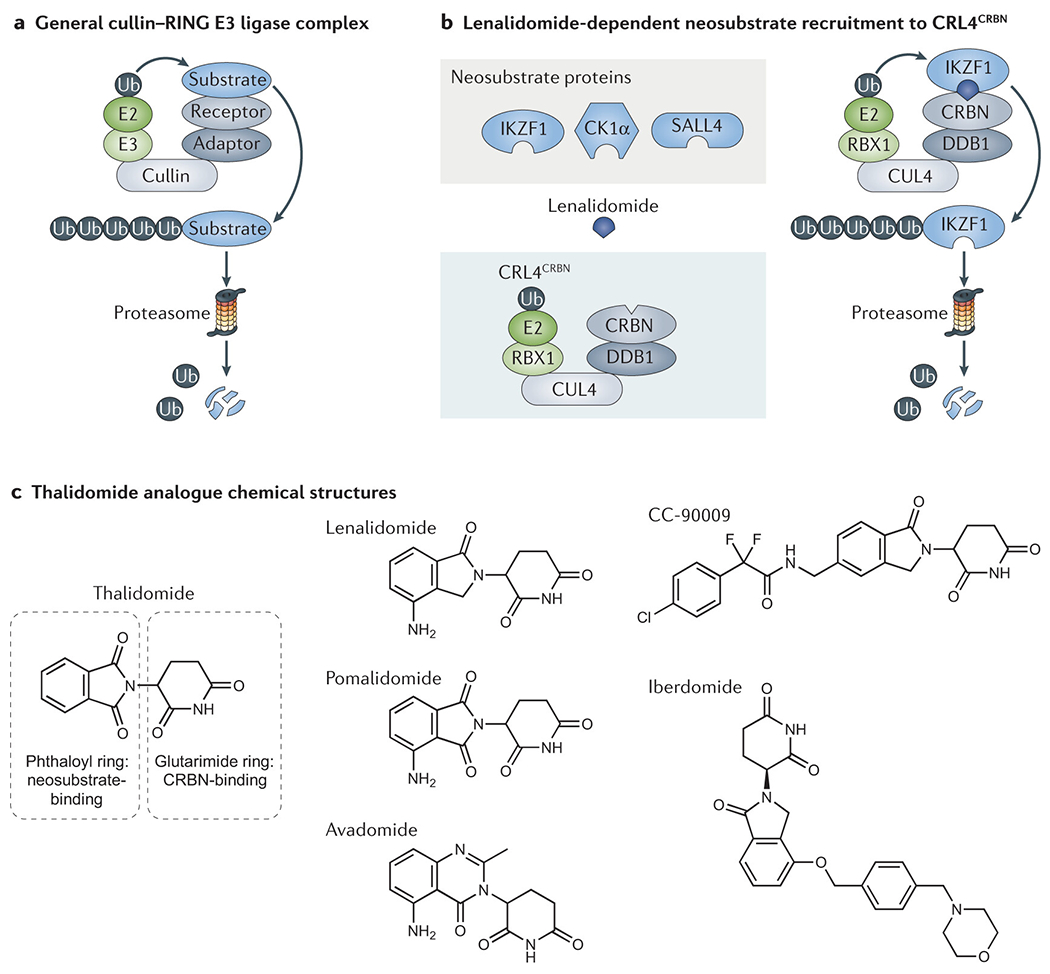
a | Cullin-RING ligases (CRLs) constitute the largest family of E3 ubiquitin ligases. CRL complexes mediate the selective transfer of ubiquitin (Ub) directly from an E2 enzyme to a receptor-bound substrate protein. This marking with Ub targets the substrate protein for proteasomal degradation. b | Lenalidomide acts as a ‘molecular glue’ to mediate drug-dependent recruitment of neosubstrate proteins to the cereblon (CRBN) receptor component of the cullin-RING ligase CRL4CRBN, which results in neosubstrate ubiquitination and degradation. c | Thalidomide analogues all share a glutarimide ring that binds to CRBN; however, these agent all vary subtly, in the case of lenalidomide, pomalidomide, and avadomide, or more substantially, in the case of iberdomide and CC-90009, in their neosubstrate-binding moiety. Owing to these chemical variations, different thalidomide analogues promote the degradation of overlapping but distinct sets of neosubstrate proteins.
Box 1 |. An overview of the ubiquitin-proteasome system.
The ubiquitin-proteasome system (UPS) is a major cellular regulator of protein turnover and quality control209.
Ubiquitin is a 76-amino-acid protein that is activated and covalently linked to proteins in the following steps: 1) ATP-dependent conjugation of ubiquitin to an E1 ubiquitin-activating enzyme; 2) transfer of the activated ubiquitin to an E2 ubiquitin-conjugating enzyme; and 3) E3 ubiquitin ligase-mediated transfer of ubiquitin from the E2 enzyme to the substrate protein.
The human genome contains two genes encoding E1 enzymes, 41 genes encoding E2 enzymes and >600 genes encoding E3 ubiquitin ligases.
The various E3 ubiquitin ligases mediate selective protein ubiquitination by recognizing particular degradation signals in their substrate proteins, termed ‘degron’ motifs, which are most commonly linear sequences or, alternatively, conformational shapes. Each ligase might recognize multiple substrate proteins210.
A single ubiquitin protein can be linked at its C-terminus to lysine residues or the N-terminal methionine of substrate proteins, either at one target site (monoubiquitination) or successively at multiple sites (multimonoubiquitination).
Successive ubiquitination can also form ubiquitin–ubiquitin linkages to generate polyubiquitin chains that can be differentiated according to which lysine residue (K6, K11, K27, K29, K33, K48 or K63) or N-terminal methionine of each ubiquitin monomer is covalently attached to the C-terminus of the subsequent monomer.
Ubiquitination can be reversed by deubiquitination enzymes (DUBs). The human genome encodes approximately 100 DUBs.
Patterns of ubiquitination constitute an extraordinarily complex ‘ubiquitin code’ and act as signals to elicit diverse functional outcomes, including protein transport, signal transduction or protein degradation211. For example, K11-linked and K48-linked polyubiquitin chains target proteins for proteasomal degradation.
The 26S proteasome is the major machinery for selective, ubiquitin-mediated protein degradation. The proteasome is a large enzymatic protein complex that first removes the attached ubiquitin groups and then unfolds the target protein into a linear polypeptide chain, which is successively fed into the barrel-shaped core of the proteasome for processive proteolysis.
Box 2 |. Target proteins implicated in thalidomide analogue efficacy and toxicities.
The proteasomal degradation of specific neosubstrate proteins underlies the clinical efficacy of thalidomide analogues in the treatment of cancer16,212. Various neosubstrate proteins, although unrelated in sequence, contain a structurally conserved degron motif composed of a β-hairpin with a pinnacle glycine residue, most often as part of a Cys2His2 (C2H2) zinc finger domain. This degron motif can bind to a composite thalidomide analogue–cereblon (CRBN) surface, ultimately resulting in neosubstrate ubiquitination and degradation.
IKZF1 and IKZF3 are pan-thalidomide analogue targets and zinc finger-containing transcription factors that have crucial roles in B cell, T cell and plasma cell development88,213,214. The anti-myeloma effects of lenalidomide are mediated by depletion of IKZF1 and IKZF311,12, which in turn downregulates the expression of IRF4, an oncogenic transcription factor that drives multiple myeloma and other mature B cell neoplasms81,215. In T cells, IKZF3 directly binds to and represses the IL2 promoter216,217, and thus thalidomide analogues mediate immunomodulatory effects in part by derepression of IL-2 expression11,13.
CSNK1A1 is located in the chromosomal region commonly deleted in del(5q) myelodysplastic syndrome (MDS) and encodes the serine/threonine kinase CK1α, which is a lenalidomide target. Haploinsufficiency of CSNK1A1 provides haematopoietic stem and progenitor cells (HSPCs) with a clonal advantage via derepression of β-catenin96, whereas homozygous loss of CSNK1A1 induces p53 and HSPC apoptosis96,218. Thus, CK1α is the primary target for lenalidomide-mediated activity in del(5q) MDS15. Indeed, lenalidomide is the first clinically approved drug exploiting a therapeutic opportunity presented by haploinsufficiency of a protein219, via selective induction of p53 in del(5q) cells haploinsufficient for CSNK1A115.
GSPT1 (eRF3a) is a target of thalidomide analogues CC-885 and the related compound CC-90009 and is a translation termination release factor required for the G1 to S phase cell cycle transition220,221. Hence, GSPT1 is an essential protein, depletion of which is especially toxic to acute myeloid leukaemia cells121.
SALL4 is a pan-thalidomide analogue target and a developmental zinc-finger transcription factor implicated in thalidomide embryopathy165,222. Indeed, germline mutations in SALL4 cause syndromes with multifaceted phenotypic overlap with thalidomide syndrome222,223.
The distinct activity of each thalidomide derivative reflects differences in the specific set of proteins that are targeted for degradation by each drug. All pharmacologically active derivatives of thalidomide contain a glutarimide ring that directly binds within a conserved pocket on the surface of CRBN17 (FIG. 1c). Each thalidomide analogue is distinguished by variations on a phthaloyl ring moiety that dictate binding to a limited number of neosubstrate proteins. Many of the degraded neosubstrates, such as zinc finger transcription factors, do not contain ligand-binding sites that could be targeted using traditional therapeutic agents. Developed from the lead compound thalidomide, lenalidomide and pomalidomide are more potent degraders of IKZF1 and IKZF3, and all of these agents have distinct patterns of neosubstrate degradation18. Thus, the activity and toxicity profiles of each thalidomide analogue are determined by their unique polypharmacology: the combinatorial degradation of multiple neosubstrates by the ubiquitin-proteasomal system (UPS; BOX 1 and BOX 2).
Thalidomide analogues in cancer therapy
Thalidomide, lenalidomide and pomalidomide are currently FDA approved in a variety of clinical settings, including MM, NHL and MDS as well as Kaposi sarcoma (KS)19–22. Next generation analogues that may overcome therapeutic resistance or that have activity in new disease indications are in clinical development.
Lenalidomide in MM.
In 2002, a phase I trial of lenalidomide monotherapy revealed promising signs of efficacy in patients with relapsed and/or refractory (R/R) MM, about 50% of whom had previously received thalidomide23. Lenalidomide doses of up to 50 mg daily were tested and no dose-limiting toxicities were observed, although all patients treated at this dose developed severe myelosuppression after 1–2 cycles and thus 25 mg daily was suggested as the maximum tolerated dose23Data from a subsequent phase II study established 30 mg daily as a safe and active dose of lenalidomide in patients with R/R MM, with a single-agent objective response rate (ORR) of 25%; a further 29% of patients responded following the addition of dexamethasone19Again, responses were seen even in patients who had disease progression on prior thalidomide therapy and the combination of lenalidomide and dexamethasone in patients with one or more prior lines of therapy was FDA approved in 200624,25.
In patients with newly diagnosed MM, ORRs are even higher, with up to 90% of patients responding to the combination of lenalidomide and dexamethasone26. A randomized study comparing lenalidomide and dexamethasone to melphalan, thalidomide and prednisone in newly diagnosed patients demonstrated superior 4-year OS (59% versus 51%; HR 0.78, p=0.02) and led to FDA approval in 201527. Subsequent research has focused on demonstrating the safety and efficacy of lenalidomide and dexamethasone in combination with various other anti-myeloma agents, leading to the establishment of a variety of triplet and quadruplet induction therapy regimens as the current standard of care for patients with newly diagnosed MM. In this population, lenalidomide and dexamethasone are typically combined with a proteasome inhibitor, such as bortezomib28, and/or the CD38-targeting monoclonal antibody daratumumab29. For older patients and/or those unfit for autologous transplantation, the preferred induction approach remains lenalidomide in combination with dexamethasone with or without daratumumab, regimens that are effective and well tolerated in this patient population27,29.
Maintenance treatment with lenalidomide.
Maintenance lenalidomide following induction therapy with or without autologous haematopoietic stem cell transplantation (auto-HSCT) is also a standard of care for patients with MM30,31. Such maintenance therapy was first attempted with thalidomide, resulting in an increase in complete remission (CR) rates (62% versus 43% without maintenance; P <0.001) and an event-free survival (EFS) benefit (56% versus 44% at 5 years; P = 0.01), but no overall survival (OS) benefit (5-year OS ~65% in both groups; P = 0.90)32. By contrast, lenalidomide has demonstrated a clear OS advantage (~88% versus 80% at 3 years) with a hazard ratio for death of approximately 0.6030,31,33,34. Patients with high-risk cytogenetic features might be the only exception, although they do derive a progression-free survival (PFS) benefit34,35. In this subset of patients, maintenance therapy with proteasome inhibitors alone or in combination with lenalidomide seems to be more effective than single-agent lenalidomide34,35. The most worrisome adverse effect observed in studies of lenalidomide maintenance therapy is an up to six-fold increase in the risk of second primary malignancies, in particular, MDS and acute myeloid leukaemia (AML): 5.3% versus 0.8% with placebo or observation34. Efforts to limit the risk of this toxicity have fueled debate about whether maintenance should continue until disease progression or be limited to 1–2 years. Meta-analyses suggest that indefinite maintenance therapy is superior in terms of survival outcomes36, although the optimal maintenance duration is a subject of an ongoing randomized trial (NCT01863550).
Current roles of thalidomide analogues in R/R MM.
Patients who have disease progression while not on treatment with lenalidomide can often be re-treated effectively with this agent37,38, sometimes multiple times, and typically in combination with proteasome inhibitors such as carfilzomib39 and ixazomib40 or with daratumumab41. The success of lenalidomide led to the development of pomalidomide, which has demonstrated efficacy in around a third of patients with lenalidomide-refractory MM42–44. Pomalidomide is most often used in combination with dexamethasone, either alone or as part of triplet regimens with a variety of other anti-myeloma agents, including bortezomib45, daratumumab46 or elotuzumab47.
Synergy with other anti-myeloma agents.
Response rates to single-agent lenalidomide have consistently been surpassed by those achieved with doublet or triplet combinations, which has been taken as evidence for synergistic activity of this agent with other anti-myeloma agents48. Data from a number of in vitro studies support the hypothesis that thalidomide analogues synergize with dexamethasone49,50, proteasome inhibitors51,52, histone deacetylase inhibitors53,54 and a variety of other agents; however, whether such combinations have additive or synergistic clinical activity remains unclear.
The two drugs most commonly combined with thalidomide analogues, bortezomib and carfilzomib, are proteasome inhibitors. The discovery that thalidomide analogues induce proteasomal degradation of their target proteins is therefore surprising, given that proteasome inhibitors antagonize lenalidomide-induced protein degradation11,12,55. Indeed, the pharmacokinetic characteristics of these drug combinations emphasize independent actions, in keeping with the hypothesis that independent drug effects against differentially sensitive cell populations explain the efficacy of most combination cancer therapies56. Specifically, whereas lenalidomide is dosed daily, bortezomib and carfilzomib are administered once or twice a week and are rapidly metabolized57, resulting in a background of prolonged selective, lenalidomide-induced protein degradation punctuated by short periods of proteasome inhibition. Notably, targeted protein degraders have the pharmacological advantage of extended target suppression until protein re-synthesis (FIG. 2)58 which might negate the effects of transient disruption of protein degradation and contribute to the efficacy of the counterintuitive combination of targeted protein degraders and proteasome inhibitors.
Fig. 2 |. Event-driven versus occupancy-driven pharmacology.
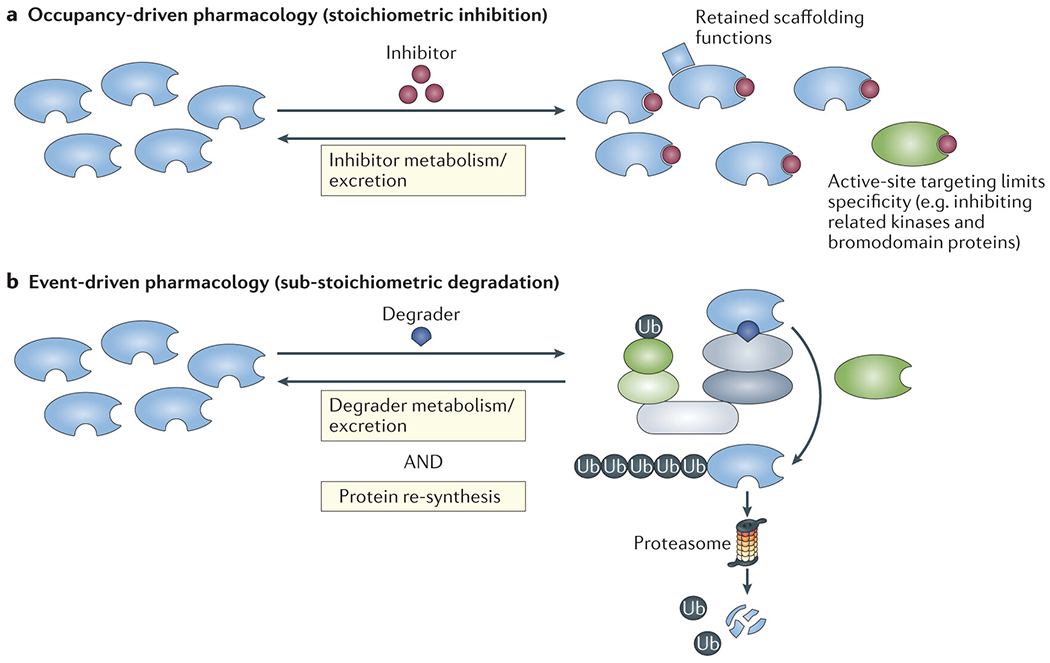
a | According to the occupancy-driven pharmacological paradigm, small-molecule inhibitors must maintain target-protein occupancy for a sustained antagonistic effect. Moreover, noncatalytic, scaffolding functions of the target protein can be retained and thus continue to contribute to oncogenesis. Furthermore, specific catalytic inhibition of a target protein at the active site, without cross-reactivity within the same protein family, is often challenging. In the figure, the blue shapes represent target proteins, with the active site at the right side and degrader-binding site at the bottom. The blue square represents a separate protein that is able to bind to the target protein regardless of inhibitor occupancy at the active site. The green shape illustrates a protein of the same family as the target that is subject off-target effects of the inhibitor, owing to active site homology, but lacks the degrader-binding site and would, therefore, be spared from off-target degrader effects. b | Degrader drugs deplete the target protein, and functional target inhibition persists until the protein is re-synthesized, constituting an example of event-driven pharmacology. Degraders have a selectivity advantage by mediating ligase–drug–target ternary complex formation using surface interfaces that are generally broader and less conserved than active sites.
Immunomodulatory effects of thalidomide analogues.
A constellation of immunomodulatory effects resulting from IKZF1 and IKZF3 degradation have been attributed to thalidomide analogues18 and are proposed to contribute to the activity of these agents. These effects include alterations of immune synapse formation59, mimicry of interferon signaling in large B cell lymphoma cells60, enhanced T cell co-stimulation61, cytokine release and function, as well as inhibition of TNF secretion by stimulated human monocytes62. Indeed, early thalidomide analogues were developed for optimized TNF inhibition, thus leading to their classification as IMiDs18. These immunomodulatory effects have been hypothesized to have a major role in the therapeutic activity of thalidomide analogues18; however, in no malignancies are these agents active without evidence of direct cytotoxicity to the cancer cells. Notably, no relevant clinical activity has been observed in trials encompassing a variety of solid tumour types, including immunotherapy-sensitive tumours such as renal cell carcinoma63 and melanoma64. Thus, despite clear effects on immune cell function and clinical activity against erythema nodosum leprosum and KS22, no strong data support a primarily immunomodulatory mechanism of antitumour action of thalidomide analogues. This scenario might be explained by both immunostimulatory and immunosuppressive effects of these agents, which might balance each other in the setting of solid tumour biology.
Synergistic immunomodulatory mechanisms have also been proposed for combinations of thalidomide analogues with monoclonal antibodies used in the treatment of MM. The anti-SLAMF7 antibody elotuzumab has no anti-myeloma activity as monotherapy65, but the addition of this agent to lenalidomide or pomalidomide and dexamethasone improved ORRs, PFS and OS in patients with R/R MM47,66,67. Elotuzumab has been proposed to function at least partially through upregulation of IL-2 production by T cells and stimulation of natural killer (NK) cell-mediated antibody-dependent cellular cytotoxicity (ADCC)68–70. Lenalidomide also enhances T cell and NK cell functions, probably via degradation of IKZF3, raising the possibility of mechanistic synergy between these two agents. Similar mechanisms of synergy between lenalidomide and daratumumab have been proposed71; however, the combination of daratumumab with either lenalidomide or bortezomib results in similar ORRs41,72, suggesting that potential mechanistic synergy does not translate into substantial clinical advantages.
Lenalidomide in B cell lymphomas and leukaemias.
In addition to MM, other mature B cell malignancies seem to depend upon IKZF1 and IKZF3 for maintenance of cancer cell phenotype and survival. In relapsed mantle cell lymphoma single agent lenalidomide produced a significant increase in median PFS (8.7 versus 5.2 months; HR 0.61; p=0.004) compared to provider’s choice chemotherapy in patients with relapsed or refractory disease, leading to its FDA approval in 201373,74. Lenalidomide has been combined with the anti-CD20 antibody rituximab in the so-called ‘R2’ regimen, a chemotherapy-free treatment, which was approved by FDA in 2019 for the treatment of R/R marginal zone lymphoma (MZL) or follicular lymphoma (FL)75,76 based on high ORRs (65% in MZL and up to 80% in FL versus 55% and 44%, respectively with placebo plus rituximab) and a substantial improvement in PFS (39.4 months versus 14.1 months; HR 0.46; 95% CI 0.34–0.62; P <0.0001). Frontline treatment with this chemotherapy-free regimen has also produced promising results in patients with FL, with CR and 3-year PFS rates similar to those achieved with rituximab plus chemotherapy (48% versus 53% and 77% versus 78%, respectively) but with a lower incidence of grade 3–4 neutropenia (32% versus 50%)75. In patients with treatment-naive MCL, the R2 regimen has produced an ORR of 92%, a CR rate of 64%, and 2-year PFS and OS of 85% and 97%, respectively77. In addition, patients with R/R chronic lymphocytic leukaemia (CLL) receiving R2 have a ORR of >65%78, and lenalidomide monotherapy triples PFS when used as a maintenance regimen following standard second-line chemotherapy (median PFS 33.9 months versus 9.2 months with placebo; HR 0.40, 95% CI 0.29–0.55; P <0.0001)79. Lenalidomide has also safely been combined with other anti-CD20 antibodies, such as obinutuzumab, in the treatment of B cell lymphomas (for example, R/R FL)80. Nevertheless, adoption of lenalidomide for the treatment of indolent lymphomas and CLL has been limited, largely owing to the availability of other highly active therapies for these diseases.
Lenalidomide has been less successful in the treatment of patients with aggressive lymphomas, such as diffuse large B cell lymphoma (DLBCL). Data from early phase clinical trials and in vitro models indicate that this agent might be active in patients with activated B cell-like DLBCL81,82; however, a phase III study showed no benefit of adding lenalidomide to chemoimmunotherapy with rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone (R-CHOP) in newly diagnosed patients with this disease subtype83. Lenalidomide maintenance might have a role in patients with R/R DLBCL who are not fit for potentially curative treatments84, such as auto-HSCT or chimeric antigen receptor (CAR) T cell therapies, and more potent, next-generation thalidomide analogues (such as avadomide) might have greater activity in this disease85. Lenalidomide is unlikely to have utility in patients with early B cell malignancies, such as pre-B cell acute lymphoblastic leukaemia (ALL), considering that deletions and loss of function mutations in IKZF1 promote the development of these cancers86,87.
Lenalidomide in T cell lymphomas.
In mice, both Ikzf1 and Ikzf3 are required for normal T cell development and maintenance of mature T cell populations88,89, which provides a rationale for targeting these proteins using lenalidomide in patients with T cell malignancies. In early phase studies, single-agent lenalidomide typically induced responses in ~30% (ORRs of 22–42%) of patients with R/R peripheral T cell lymphomas90–92, mycosis fungoides or Sézary syndrome93. Clinical studies combining lenalidomide with conventional CHOP chemotherapy (NCT01553786), antibody–drug conjugates (such as brentuximab vedotin; NCT03409432) or other, novel agents (such as histone deacetylase inhibitors [NCT02232516] and immune-checkpoint inhibitors [NCT04038411]) in these diseases are ongoing.
Lenalidomide in myeloid malignancies.
Lenalidomide was first approved by the FDA in 2005 for the treatment of transfusion-dependent anaemia resulting from MDS with del(5q), an indication in which it is associated with ORRs of 76–83%, with 45–50% of patients also having a cytogenetic CR21,94. Del(5q) is the most common cytogenetic abnormality in MDS; patients with this genetic aberration often present with a consistent clinical phenotype, termed the ‘5q− syndrome’, which is more common in women than in men and has a relatively indolent course characterized by hypoplastic anaemia95. Lenalidomide has activity in this disease by inducing degradation of CK1α, the protein product of the haploinsufficient gene CSNK1A1 that is located on the common deleted region of chromosome 5q, leading to activation of p53 and subsequent apoptosis15,96. Accordingly, mutations in TP53 lead to resistance to lenalidomide in patients with MDS97,98 (FIG. 3). Co-occurring TP53 mutations probably also explain why patients with del(5q) in combination with a complex karyotype or in the setting of AML do not respond to lenalidomide99. Of note, IKZF1 is expressed throughout haematopoiesis and regulates myeloid cell development and function100. Thus, altered regulation of gene expression as a result of IKZF1 degradation might also contribute to responsiveness of myeloid malignancies to lenalidomide97,101.
Fig. 3 |. Mechanisms of resistance to targeted protein degradation.
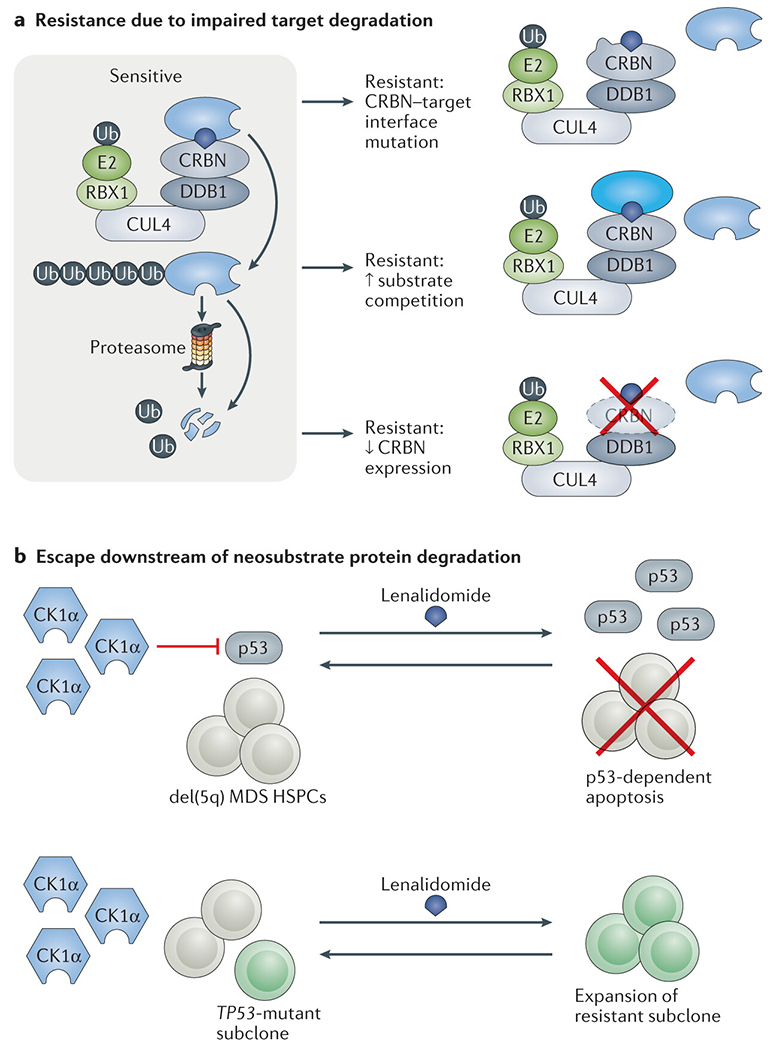
a | Tumour cells can escape from thalidomide analogue-induced degradation of key E3 ubiquitin ligase neosubstrate proteins via mutations affecting the cereblon (CRBN)–neosubstrate interface, reduced expression of CRBN or overexpression of competing substrates for the same ligase. b | Tumour cells can escape from the tumour-suppressive consequences of neosubstrate protein degradation through alterations affecting the function of downstream mediators of the antitumour effects. For example, TP53-mutant subclones of del(5q) myelodysplastic syndrome (MDS) haematopoietic stem and progenitor cells (HSPCs) are resistant to p53-mediated apoptosis induce by lenalidomide-mediated degradation of CK1α.
Lenalidomide monotherapy has produced CR rates of almost 20% in the settings of both newly diagnosed and R/R AML102,103. In patients with R/R AML, the combination of high-dose lenalidomide (50 mg daily) with mitoxantrone, etoposide and cytarabine (MEC) resulted in a CR or CR with incomplete haematological recovery (CR/CRi) rate of 41%104, which is considerably higher than expected with MEC alone (17–28%) based on data from historical cohorts105,106. A phase II study evaluating lenalidomide with MEC in this patient population is ongoing (NCT03118466). Lenalidomide provides limited benefit when combined with azacytidine in patients with newly diagnosed AML107; however, the results are more encouraging when this combination is used following disease relapse after allogeneic HSCT, with almost 50% of patients having a major clinical response, possibly owing to stimulation of graft versus leukaemia immune responses108. Lenalidomide also has activity in patients with myelofibrosis, with responses occurring in up to a third of patients when used in combination with prednisone, some of which have lasted as long as 9 years109.
Kaposi sarcoma.
In 2020, pomalidomide was approved by the FDA for the treatment of KS, an endothelial cell-derived solid tumour caused by human herpesvirus 8 (HHV8) infection110, based on data from a phase I/II trial that demonstrated an ORR of 73% in patients with newly diagnosed KS22. The mechanistic basis underlying the efficacy of pomalidomide in this disease is not well characterized. Interestingly, two other HHV8-associated neoplasms, primary effusion lymphoma111 and Castleman disease112, have also been shown to be responsive to treatment with thalidomide analogues.
Next-generation thalidomide analogues
New thalidomide analogues are being developed both to overcome resistance by inducing deeper degradation of known targets and also to degrade new targets. These agents include avadomide, iberdomide, CC-92480 and CC-90009, which are rapidly making their way toward the clinic (TABLE 1).
Table 1 |.
Selected ongoing clinical trials of next-generation thalidomide analogues
| Agent | Disease | Treatment approach | Study phase | ClinicalTrials.gov identifier |
|---|---|---|---|---|
| CC-90009 | R/R high-risk MDS or R/R AML | Single agent | Phase I | NCT02848001 |
| Newly diagnosed or R/R AML | In combination with venetoclax (BCL2 inhibitor) and azacitidine (HMA) or with gilteritinib (FLT3 inhibitor) | Phase I/II | NCT04336982 | |
| Avadomide (CC-122) | Advanced-stage melanoma (naive or refractory to PD-1 inhibition) | In combination with nivolumab (anti-PD-1 antibody) | Phase II | NCT03834623 |
| Advanced-stage HCC (after one prior line of therapy) | In combination with nivolumab | Phase I/II | NCT02859324 | |
| Newly diagnosed DLBCL with poor-risk factors | In combination with R-CHOP chemotherapy | Phase I | NCT03283202 | |
| Treatment-naive or R/R CLL or SLL | Single-agent, or in combination with ibrutinib (BTK inhibitor) or obinutuzumab (anti-CD20 antibody) | Phase I/II | NCT02406742 | |
| R/R DLBCL or R/R FL (naive or refractory to lenalidomide) | In combination with obinutuzumab | Phase Ib | NCT02417285 | |
| R/R DLBCL or R/R FL (naive or refractory to lenalidomide) | In combination with rituximab (anti-CD20 antibody) and/or either onatasertib (CC-223; mTOR inhibitor) or spebrutinib (CC-292; BTK inhibitor) | Phase Ib | NCT02031419 | |
| R/R B cell malignancies | In combination with JCAR017 (autologous anti-CD19 4-1BB CAR T cell product) | Phase I/II | NCT03310619 | |
| R/R advanced-stage solid tumours, NHL or MM | Single agent | Phase I | NCT01421524 | |
| Advanced-stage solid tumours or NHL | Single agent | Phase I | NCT02509039 | |
| Iberdomide (CC-220) | R/R MM | Single agent, or in combination with daratumumab (anti-CD38 antibody), bortezomib (PI), carfilzomib (PI) and/or dexamethasone (corticosteroid) | Phase Ib/IIa | NCT02773030 |
| R/R MM | In combination with low-dose cyclophosphamide (chemotherapy) and dexamethasone | Phase II | NCT04392037 | |
| Newly diagnosed MM | Single-agent maintenance after ASCT | Phase II | NCT04564703 | |
| R/R lymphomas | Single agent, or in combination with rituximab or obinutuzumab | Phase I/II | NCT04464798 | |
| R/R B cell malignancies | In combination with JCAR017 (autologous anti-CD19 4-1BB CAR T cell product) | Phase I/II | NCT03310619 | |
| CC-92480 | R/R MM | Single agent, in combination with dexamethasone | Phase I | NCT03374085 |
| Newly diagnosed or R/R MM | In combination with dexamethasone plus either daratumumab, bortezomib or carfilzomib | Phase I/II | NCT03989414 |
Trials involving patients with cancer and noted as being ‘not yet recruiting’, ‘active, not yet recruiting’, ‘recruiting’ or ‘completed’ with no results posted were retrieved from the ClinicalTrials.gov database. AML, acute myeloid leukaemia; ASCT, autologous haematopoietic stem cell transplantation; CLL, chronic lymphocytic leukaemia; DLBCL, diffuse large B cell lymphoma; FL, follicular lymphoma; HCC, hepatocellular carcinoma; HMA, hypomethylating agent; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; PI, proteasome inhibitor; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine and prednisone; R/R, relapsed and/or refractory; SLL, small lymphocytic leukaemia.
Avadomide.
Avadomide (previously known as CC-122) is chemically similar to pomalidomide (FIG. 1c), and accordingly, has similar substrate specificity and potency of degradation113. Avadomide has been tested in clinical trials involving patients with R/R DLBCL, demonstrating promising single-agent activity (ORR 29% and CR rate 11%)85. This agent has also been tested in patients with a variety of solid tumours; although no objective responses were seen in an initial report114, five of six patients with brain tumours had stable disease for at least 6 months. Additional clinical studies of avadomide are ongoing in other solid tumour settings, including advanced-stage melanoma and hepatocellular carcinoma, and in various combinations (TABLE 1).
Iberdomide.
Iberdomide (CC-220) is a thalidomide derivative with an increased binding affinity for CRBN and greater potency than pomalidomide in multiple myeloma cell lines, with activity observed against some pomalidomide-resistant cell lines115,116. Iberdomide has a distinct chemical structure compared with other thalidomide analogues (FIG. 1c) and might have a broader target specificity117. This agent has promising activity in combination with dexamethasone in patients with R/R MM, with an ORR of 31% and a disease-control rate of 88%118, and its use in combination with other agents is being explored in this disease (TABLE 1). Iberdomide has also shown efficacy in models of systemic lupus erythematosus in vitro where treatment can decrease production of auto-antibodies, and trials are ongoing in this disease setting119.
CC-92480.
CC-92480 is currently being tested in patients with MM (TABLE 1). This agent has activity across a variety of MM cell lines resistant to lenalidomide and pomalidomide, even those with low levels of CRBN expression, which has been proposed as a mechanism of drug resistance (FIG. 3)120.
CC-90009.
CC-90009 differs from the other thalidomide analogues in that it targets the substrates G1 to S phase transition protein 1 homologue (GSPT1) and GSPT2, which are also known as eukaryotic peptide chain release factor GTP-binding subunits ERF3A and ERF3B, respectively, reflecting their roles as essential regulators of translation termination113,121. CC-90009 has been tested in patients with R/R MDS or AML and demonstrated promising single-agent activity: a total of 49 were treated, three of whom had a CRi or better with a morphological leukaemia-free state achieved in one additional patient122. Toxicities included manageable grade 3–4 hypotension (in 13% of patients) and on-target, drug-induced hypocalcaemia (in 22%)122. CC-90009 is currently being investigated as a single agent as well as in combination with hypomethylating agents, venetoclax or the FLT3 inhibitor gilteritinib in patients with MDS and AML (TABLE 1).
Clinical toxicities
Effects on haematopoiesis.
Thalidomide analogues often lead to peripheral blood cytopenias, most commonly neutropenia (28% grade 3/4) and thrombocytopenia (8% grade 3/4)27. Thalidomide itself is an exception, given that it rarely causes thrombocytopenia10, making it a preferred option for some patients. Aside from neutropenia, data suggest that lenalidomide use is associated with limited immune dysfunction beyond that associated with the underlying malignancies. Both IKZF1 and CK1α have important roles in haematopoietic stem and progenitor cell (HSPC) function and differentiation, although the precise effects of thalidomide analogues on haematopoiesis have not been well studied. Lenalidomide therapy decreases the efficiency of growth factor-stimulated stem cell mobilization and collection for auto-HSCT, thus limiting its use prior to stem cell harvesting123,124. This issue can often be overcome using high-dose cyclophosphamide or the CXCR4 antagonist plerixafor125.
Second primary malignancies.
Long-term use of lenalidomide, especially following high-dose melphalan and autologous stem cell rescue, has been associated with an increased incidence of second solid tumours and haematopoietic malignancies, primarily MDS and AML34,126. Effects on HSPC function probably contribute to the increased risk of secondary myeloid malignancies, although these effects might also underlie the activity of lenalidomide in these same malignancies. Solid tumours associated with lenalidomide use are most commonly non-melanoma skin cancers, which can usually be managed conservatively127.
Thrombosis.
Thalidomide analogues are associated with an increased risk of primarily venous thromboembolism128–130 (OR 3.51; p<0.001), although arterial events can also rarely occur131. Concomitant use of anti-platelet or anti-thrombotic agents is, therefore, a routine component of clinical care132. Whether the thrombotic phenotype occurs through on-target protein degradation remains unclear. The increased risk seen with concomitant use of corticosteroids, such as dexamethasone, and chemotherapeutic agents, such as anthracyclines, raises the possibility of thrombosis secondary to endothelial cell injury133.
Teratogenicity.
The potential for teratogenic effects of analogues other than thalidomide has not been assessed. Nevertheless, this adverse effect is assumed to be common to all thalidomide analogues, and appropriate precautions are recommended for all drugs of this class.
Other adverse effects.
Other toxicities commonly associated with thalidomide analogues include rashes, fatigue and diarrhoea. The rash associated with lenalidomide use occurs in up to 30% of patients, is unpredictable and can occur at any time during treatment134. Moreover, this adverse effect can range in severity, from mild erythema to toxic epidermal necrolysis, but is often easily managed with corticosteroids and drug cessation135. Mechanistically, the rash might be related to immune activation. Most patients can be rechallenged with lenalidomide without recurrence of rash; in rare cases in which rashes recur, management with desensitization strategies involving gradual titration from low starting doses has been effective136. Fatigue is often a dose-limiting toxicity of thalidomide analogues, but its mechanism is unclear. Finally, long-term use of lenalidomide can cause diarrhoea related to fat malabsorption, which can be effectively treated with the bile acid-binder colestipol137.
The distinct pharmacology of degraders
Drugs that induce targeted protein degradation, such as thalidomide analogues, have a number of unique pharmacological features in comparison with conventional enzyme inhibitors58,138–140. These features can in turn provide unique clinically beneficial properties.
Event-driven vs occupancy-driven action
Classical inhibitors – covalent or non-covalent - must stoichiometrically occupy the binding site on the target enzyme, and therefore, sustained target inhibition requires maintenance of an effective drug concentration (FIG. 2a). By contrast to this occupancy-driven action, degrader pharmacology is event-driven; transient target–drug–ligase binding events mediate irreversible target degradation138,139 (FIG. 2b). Moreover, a single degrader molecule can cycle through multiple binding and degradation events, enabling sub-stoichiometric activity141. Effective target protein suppression can be prolonged and is reversed only following protein re-synthesis, thereby uncoupling pharmacokinetic and pharmarcodynamic relationships. Furthermore, any noncatalytic, scaffolding functions of degraded proteins are also abolished, potentially broadening anti-cancer effects beyond enzymatic inhibitors that typically spare scaffolding functions (FIG. 2a).
Resistance mechanisms
Downregulation of E3 ligase activity.
CRBN expression is required for the anti-myeloma activity of lenalidomide142. In genome-wide genetic screens of MM cells and lymphoma cells, disruption of CRL4CRBN components and the associated cullin-RING ligase machinery was found to result in resistance to lenalidomide143–145. Mutations in CRBN, including missense mutations in the thalidomide analogue binding domain, are associated with progressive thalidomide analogue exposure, and ultimately occur in up to a third of patients with pomalidomide-refractory disease146–148 (FIG. 3a). CRBN expression levels in patient samples has been positively associated with sensitivity to thalidomide analogues in some studies149–154, but not in others155,156. Future studies in this area will benefit from the use of standardized, direct measurements of CRBN expression156. Data from our group113 and others157 suggest that overexpression of CRBN can sensitize previously resistant MM cell lines to thalidomide analogues. Therefore, efforts to modulate CRBN expression might yield therapeutics that enhance the activity of thalidomide analogues.
Targeted protein degradation is influenced by multiple layers of competition for UPS components. In a study using a quantitative multiplex mass spectrometry-based assay to quantitate multiple substrate proteins, a stereotyped order of polysubstrate degradation was identified for various thalidomide analogues113, suggesting that substrates with higher affinity and/or greater abundance can outcompete others. Indeed, induced overexpression of one substrate reduced the degradation of other substrates and induced drug resistance in cellular models113. Therefore, cancer cells might develop resistance by upregulating the expression of unrelated substrates, thereby stabilizing substrates critical for oncogenesis, or alternatively upregulating critical targets such as IKZF1/3 directly158 (FIG. 3a); however, quantitative proteomic datasets from clinical specimens are required to confirm these hypothesis and are currently lacking. Beyond substrate competition for CRBN, CRBN itself competes with other substrate receptors for access to limiting concentrations of CRL4159. Thus, the expression profile of the entire set of substrates, substrate receptors and the CRL4 backbone probably determines the efficiency of drug-induced degradation (FIG. 3a), which might vary not only between different cell types and states but also between tumour subclones. Given this emerging portrait of the highly complex epigenetic determinants of degrader pharmacodynamics, efforts to develop biomarkers predictive of degrader activity remain in their infancy.
Escape downstream of neosubstrate protein degradation.
Del(5q) MDS HSPCs are typically sensitive to lenalidomide-mediated depletion of CK1α, which activates p53-dependent cell death15,96. Accordingly, loss of p53 leads to resistance of mouse HSPCs to Ck1α targeting with either lenalidomide, a direct inhibitor or genetic knockdown (FIG. 3b)15,160. Consistent with these experimental findings, resistance to lenalidomide and disease progression are associated with an increased frequency of TP53 mutations in patients with del(5q) MDS98,161,162. Similarly, lenalidomide-mediated depletion of IKZF1 in del(5q) MDS cells drives RUNX1 upregulation and megakaryocyte differentiation, and sequencing of matched clinical specimens has identified RUNX1 mutations that are associated with lenalidomide resistance97. Analogous mechanisms conferring resistance to degrader-mediated depletion of IKZF1 and IKZF3 in lymphoid malignancies are an active area of research.
Clinical pharmacodynamic monitoring
The detailed investigation of thalidomide analogue function in patients has been limited by the semi-quantitative nature of protein detection methodologies and limited deployment of such assays to clinical settings. In the past few years, our group and others have used targeted mass spectrometry to perform detailed quantitative analyses of thalidomide analogue neosubstrate proteins and the UPS113,159. These approaches have the advantages of enabling detection of target proteins from small sample inputs and rapid processing times that in turn enable high-resolution kinetic analyses. Such analyses have revealed the importance of competition among both substrates and E3 ubiquitin ligase complexes in determining neosubstrate abundance during thalidomide analogue therapy. Targeted proteomic assays have been translated to the clinical laboratory setting163,164, and such assays present a potentially appealing approach for the development of clinical biomarkers of activity and resistance to degrader drugs.
Emerging opportunities with degraders
Over the decades, the story of thalidomide has traversed phases of tragic teratogenicity, followed by renewed optimism as an anticancer agent and the subsequent discovery of an unexpected mechanism of action. In parallel to the discovery that thalidomide analogues act as ‘molecular glue’ degraders, heterobifunctional proteolysis targeting chimeras (PROTACs), which are based on a similar principle and consist of an E3 ubiquitin ligase-recruiting element linked to a target-binding moiety, have advanced from being proof-of-concept molecules to investigational anticancer drugs (BOX 3)140. The next phases of drug discovery focused on targeted protein degradation will benefit from mechanism-guided drug design and clinical development; next-generation degraders will be designed to more efficiently and selectively target novel disease-relevant neosubstrate proteins, and biomarkers of activity and resistance will be used to inform clinical decision-making. More broadly, lenalidomide and the other approved thalidomide analogues provide clinical validation for the therapeutic concept of targeted protein degradation, which has increased both interest and investment in a range of related technologies. In the following sections, we will highlight new paths to degrader therapeutics with potential clinical utility, as well as the possible use of degraders in synthetic biology approaches to controlling adoptive cell-based immunotherapies.
Box 3. Prospective development of targeted protein degraders.
The development of proteolysis targeting chimeras (PROTACs) over the past two decades has been expertly reviewed elsewhere58,138–140,224, particularly for a drug-discovery audience.
PROTACs leverage the concept of using bifunctional small molecules to bring proteins into close proximity225,226.
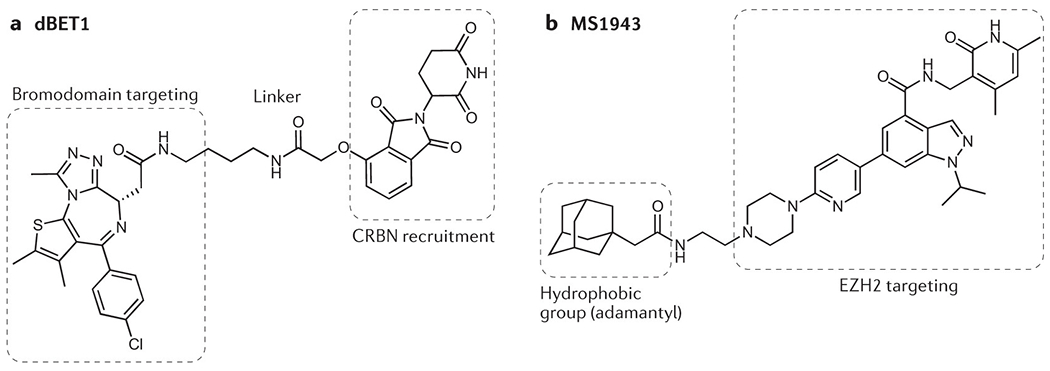
PROTACs are composed of three parts: an E3 ubiquitin ligase-recruiting ligand, a linker and a target-binding ‘warhead’ (see example of dBET1 in figure part a). PROTACs are therefore relatively large molecules that do not conform to typical rules of small-molecule drug design.
In contrast to thalidomide analogues, PROTACs can be prospectively designed for many liganded targets. PROTAC development remains empiric and challenging, although scientific breakthroughs and investment have spurred the development of PROTACs for many targets in oncology.
The first clinical trial of a PROTAC, ARV-110, opened in 2019 (NCT03888612); ARV-110 targets the androgen receptor and is being tested in patients with prostate cancer.
In many cases, the target-binding warhead is derived from existing inhibitors. In comparisons of chemically related inhibitors, PROTACs have been found to have higher target specificity, owing to the unique requirement for drug-induced stabilization of two proteins that have not evolved to interact with each other227–229.
Degraders can overcome point mutation-based resistance to target inhibition, for example, the BTK C481S resistance mutation to ibrutinib, using warheads that retain affinity for the mutated active site230,231
Only a few of the >600 E3 ubiquitin ligases have been leveraged for targeted protein degradation to date, including the those with cereblon (CRBN), VHL, MDM2, cIAP or ß-TRCP as substrate-recognition components. Thus, extraordinary potential exists to use additional ligases to exploit crucial features, including cell type-specific activity and target selectivity232.
Related bifunctional molecule concepts extend to the recruitment of target proteins to effectors other than ligases, such as lysosomes and phosphatases233,234.
Hydrophobic tagging is a related approach in which a hydrophobic small molecule binds to a target protein, thus mimicking a partially unfolded state and inducing degradation of the target by cellular quality control machinery198,235. The hydrophobic-tag EZH2 degrader MS1943 (see figure part b), for example, has demonstrated activity against triple-negative breast cancer cell lines that are refractory to catalytic EZH2 inhibitors236.
Identification of thalidomide analogue neosubstrates
The full complement of proteins targeted for degradation by thalidomide analogues remains to be elucidated. Most known neosubstrate proteins have been identified through proteomic analyses of different cell lineages11,15,165,166. Each target protein has been found to contain a structural degron composed of a β-hairpin with a pinnacle glycine motif, most often in the form of a Cys2-His2 (C2H2) zinc finger domain, that binds to a composite thalidomide analogue–CRBN surface17,117,121,167. The approximately 700 C2H2 zinc finger-containing transcription factors constitute the largest class of transcription factors in the human genome168, but have previously not been amenable to drug development. Functional genomics approaches coupled with computational docking and in vitro binding analyses have been used to interrogate all human C2H2 zinc finger domains in order to define the zinc finger ‘degrome’ of thalidomide analogues117. These studies resulted in the identification of 11 individual zinc finger domains and six full-length zinc finger-containing that are subject to drug-mediated degradation, as well as a larger number of zinc finger domains that are capable of binding to pomalidomide-engaged CRBN in vitro117. Notably, different thalidomide analogues mediate the degradation of overlapping but distinct sets of zinc finger domains117, implying that further chemical diversification will provide unique opportunities to target additional members of this important class of once-undruggable proteins. Crystallographic and saturation mutagenesis studies have identified the amino acid residues of particular zinc finger domains that are crucial for their drug-induced recruitment to CRL4CRBN and subsequent degradation117; such insights might facilitate further cycles of systematic neosubstrate protein discovery.
Endogenous substrates of CRBN
CRBN was first described as a gene that, when mutated, causes a monogenetic intellectual disability169. A number of endogenous CRBN-interacting proteins have been reported, including MEIS2, AMPK, MCT1, and GS17,170–172, some of which compete with thalidomide analogue-targeted neosubstrates for CRL4CRBN-dependent ubiquitination and degradation. The full range of endogenous CRL4CRBN substrates and the relevance of thalidomide analogue-induced modulation of these substrates for the efficacy and toxicities of such treatments are areas of active investigation.
Novel classes of degrader therapeutics
Two additional, entirely distinct classes of small-molecule molecular glue degraders have been discovered. The first class comprises aryl sulfonamides, including indisulam, tasisulam, E7820 and chloroquinoxaline, which to date have produced modest response rates as monotherapy in clinical trials involving patients with advanced-stage solid tumours173–176. Two independent groups identified RNA-binding motif protein 39 (RBM39, also known as CAPERα), a nuclear protein involved in pre-mRNA splicing, as the direct molecular target responsible for the antitumour effect of these sulfonamides, which recruit this splicing factor to the CRL4DCAF15 E3 ubiquitin ligase for polyubiquitination and subsequent proteasomal degradation177–179. Kinetic and structural analyses revealed relatively weak drug–protein interactions that are stabilized by ternary docking across broad protein–protein interfaces, which results in highly selective drug-dependent protein degradation of RBM39 and its paralogue RBM23179–181.
With regard to the second novel class of agents, three independent chemogenomic screening studies resulted in the identification of structurally diverse molecular glue degraders of cyclin K. Firstly, by mining correlations between drug sensitivity and the gene-expression profiles of E3 ubiquitin ligase components in hundreds of cell lines, it was discovered that CR8, which was originally developed as a cyclin-dependent kinase (CDK) inhibitor, acts as a first-in-class molecular glue degrader of cyclin K182. Secondly, comparative chemical screening of hyponeddylated and well-neddylated cell lines revealed three destabilizers of cyclin K183. Finally, a novel cyclin K-targeting cytotoxic compound, HQ461, was identified during high-throughput screening for chemical suppressors of NRF2 activity184. Remarkably, these structurally divergent small molecules all directly engage a substrate receptor-less CUL4 E3 ligase complex by binding to its substrate adaptor component, DDB1, whilst simultaneously interacting with and thereby recruiting CDK12 in complex with cyclin K. Thus, selective complementarity between the surfaces of DDB1 and CDK12 hold promise for the future development of cyclin K degrader therapeutics182. The discovery of multiple classes of molecular glue degraders raises the exciting possibility that additional existing drugs also work through this mechanism. Indeed, the full complement of molecular glue degraders is not yet known.
The molecular glue-based mechanism of action of thalidomide analogues and anticancer sulfonamides have inspired efforts to design molecular glue degraders prospectively. Attractive targets include oncogenic forms of β-catenin that harbor hotspot mutations in a phosphodegron sequence normally recognized by the E3 ligase SCFβ–TrCP, which result in the accumulation of β-catenin protein and thus constitutive Wnt pathway activation185. Simonetta and colleagues186 prospectively identified and performed subsequent rational structure-guided optimization of drug-like small molecules that profoundly enhance the interaction between SCFβ–TrCP and such mutant forms of β-catenin. Future efforts are expected to target otherwise undruggable oncoproteins with prospectively designed molecular glue degraders.
Beyond molecular glue degraders, drug-induced polymerization is an emerging mechanism of targeted protein degradation. The transcription factor BCL6 is a therapeutic target in certain B cell malignancies, and small-molecule screening for novel inhibitors of this protein resulted in the identification of BI-3802 as a compound that binds directly to the BTB dimerization domain of BCL6 and acts as a specific BCL6 degrader187. Cryoelectron microscopy revealed the polymerization of BCL6–BI-3802 complexes into helical filaments in vitro188. In cells, BI-3802 induced the formation of BCL6 protein foci that were then rapidly degraded by the UPS188. Drug-induced polymerization promoted BCL6 degradation by the E3 ubiquitin ligase SIAH1, which recognizes a linear degron sequence distal to the drug-binding domain188. Such small-molecule inducers of targeted protein degradation via polymerization present an exciting new approach to drugging previously undruggable proteins. Generalizable rules governing target protein polymerization remain to be defined.
Synthetic biology and cell therapies
Beyond direct antitumour effects, degrader therapeutics are under development as chemical biology tools to regulate engineered proteins in the context of genetically modified cell therapies. Such cellular immunotherapies are emerging as powerful anticancer agents, but these autonomous, ‘living drugs’ are capable of inducing severe toxicities189. For example, chimeric antigen receptor (CAR) T cells are highly efficacious in patients with B cell malignancies but can cause severe T cell hyperactivation-related toxicities190. Precision control is therefore required to enhance the safety, efficacy and accessibility of such therapies191, and indeed small-molecule-regulated kill switches for cell therapies192 and transcriptional control systems for gene vectors193 have entered clinical testing for this purpose. Multiple chemical–genetic systems have been devised to enable tunable regulation of protein stability using small molecules194, including FKBP12-based destabilization domains195, auxin-inducible degrons196 and small-molecule-assisted shutoff (SMASh)197. With each platform, a heterologous protein of interest is fused to a drug-regulated domain in order that the fusion protein can either be stabilized or degraded in the presence of the small-molecule controller. Likewise, fusion proteins incorporating the bacterial dehalogenase-derived HaloTag protein can be degraded upon addition of either hydrophobic tag ligands that mimic a partially denatured protein state198 or HaloPROTACs that recruit VHL (which serves as a substrate receptor component of the CRL2 E3 ubiquitin ligase)199. In addition, Nabet and colleagues200,201 have developed the dTag system that uses PROTACs composed of a CRBN or VHL ligand, a linker and an AP1867 derivative. AP1867 and FKBP12F36V are a bump-and-hole engineered small molecule–protein pair orthogonal to rapamycin and wild-type FKBP12202. Accordingly, AP1867-derivative PROTACs can mediate the selective degradation of heterologous FKBP12F36V-tagged proteins, enforcing rapid and potent depletion in vitro and in vivo200,201. CARs fused to an additional domain allowing for PROTAC-mediated degradation have been designed as a reversible chemical safety switch203. In aggregate, these approaches to rapidly and reversibly tune protein stability are invaluable research tools. However, with the exception of newer generations of destabilization domains204, these systems are currently difficult to directly translate into clinical cellular immunotherapies because they use non-human proteins, immunosuppressive controller drugs or non-approved small molecules.
A molecular switch consisting of a thalidomide analogue and a variable protein construct with a human zinc finger degron tag would probably be suitable for clinical use. Notably, Koduri and colleagues205 have demonstrated inducible degradation of multiple fusion proteins linked to a 25-amino acid zinc finger degron tag derived from IKZF3 and a >50% reduction in luminescence from tumour cells expressing a degron-tagged luciferase transgene upon treatment with thalidomide analogues. Carbonneau and colleagues206 fused a CAR with a 60-amino acid zinc finger degron derived from IKZF3, enabling reversible thalidomide analogue-mediated CAR depletion, partial inhibition of CAR T cell effector function in vitro, and suppression of anti-tumor function in vivo. To further develop this concept, ‘super-degron’ tags composed of hybrid zinc finger degrons have been engineered to enhance the efficiency of drug-mediated degradation117,207. These super-degron tags have been incorporated into thalidomide analogue-mediated off-switch CARs, which can be degraded and thus functionally inhibited at sub-therapeutic drug concentrations (FIG. 4)207. Furthermore, a thalidomide analogue-inducible dimerization domain has been generated from a fragment of CRBN and a zinc finger with all lysines substituted to arginines (K0), and with this system split CARs have been built that require drug treatment for dimerization and subsequent activation207. Such control systems might prove broadly useful to enhance the safety and the efficacy of future cellular immunotherapies, for example, by acting as safety switches or by enabling the measured use of therapeutic proteins that are toxic when constitutively expressed, such as the pro-inflammatory cytokine IL-12208. This early success in developing custom degrons suggests that further engineering of thalidomide analogue-based degron tags is possible using structure-guided or directed-evolution approaches. These efforts might greatly expand the toolkit of clinically suitable synthetic biology approaches for the design of next-generation cellular immunotherapies.
Fig. 4 |. Genetically engineered cell therapies regulated by targeted protein degraders.
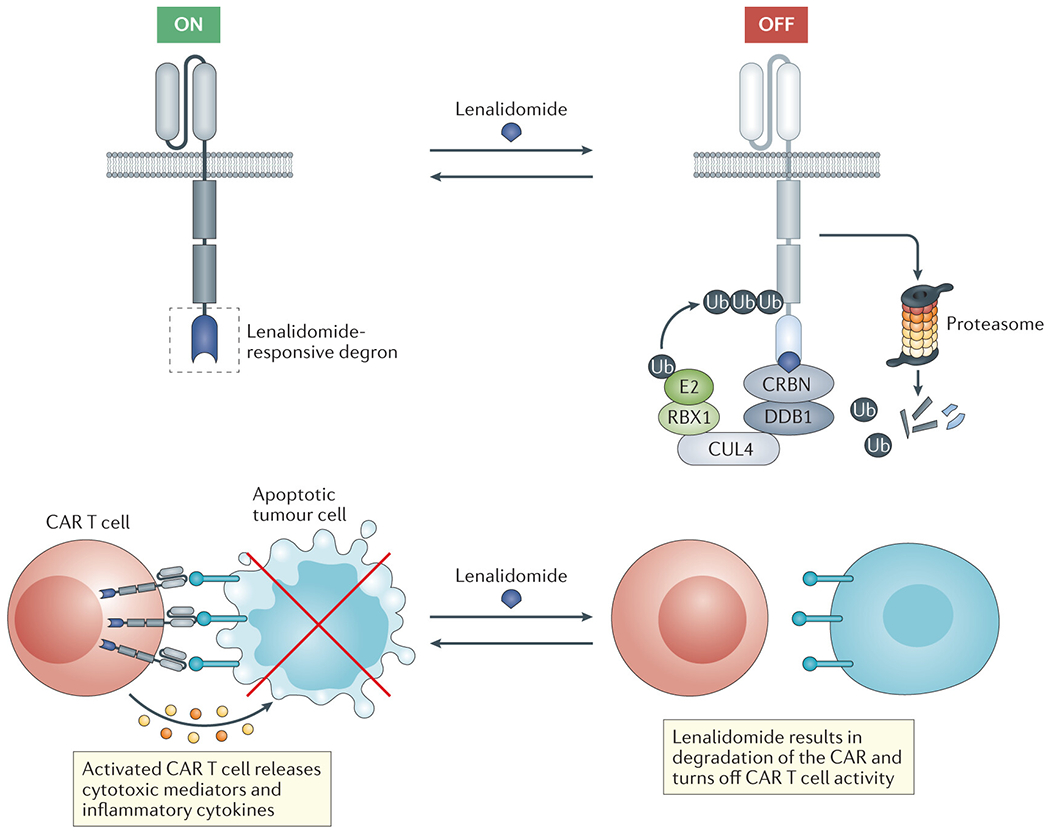
The figure outlines an example of the use of lenalidomide as a molecular ‘switch’ to dynamically control the activity of chimeric antigen receptor (CAR) cells. Specifically, a lenalidomide-responsive ‘degron’ moiety (for example, a zinc finger domain derived from IKZF3 can be incorporated in the CAR construct. These degradable constructs can then be used to generate CAR T cells that can be rapidly and reversibly turned ‘off’ through lenalidomide treatment in order to tune CAR signalling or mitigate toxicities associated with T cell hyperactivation. HaloTags or dTags, which also confer the ability to control expression of the engineered protein using small-molecule degraders, could be substituted for the lenalidomide-responsive degron.
Conclusions
Targeted protein degradation in oncology has reached an extraordinary juncture. Further chemical diversification of thalidomide analogues will expand the range of degradable target proteins. Two decades of development have culminated in the clinical testing of the first of many small-molecule PROTAC drugs, ARV-110 (NCT03888612). Additional classes of molecular glue and heterobifunctional degrader molecules will continue to be designed and discovered, building on the experience gained with such agents to date and exploiting the hundreds of E3 ubiquitin ligases. Chemical switches engineered from an expanding list of degrader–substrate pairs will render gene therapies and cell therapies more controllable. As new generations of degrader therapeutics reach the clinic, the lessons learned with lenalidomide and other thalidomide analogues can serve as a guide. Once-undruggable proteins that are amenable to therapeutic degradation owing to cancer dependency, synthetic lethality and/or aneuploidy must be identified, prioritized and targeted. Extensive testing of novel preclinical degraders across multiple cell types will be required to elucidate target substrates with the potential for efficacy and toxicity. Clinical proteomics will enable the monitoring of substrate protein dynamics. Mechanisms of relapse to drugs leveraging E3 ubiquitin ligases need to be anticipated and counteracted. Ultimately, insights into molecular glue chemistry, structural biology and proteomics will prove essential to realizing the extraordinary breadth of medical opportunities unlocked by the expanding range of degradable proteins.
Supplementary Material
Table 2 |.
FDA approved anticancer indications of thalidomide analogues
| Agent | Disease | Treatment approach | Approval year | Studies used for approval |
|---|---|---|---|---|
| Thalidomide | Newly Diagnosed Multiple Myeloma (NDMM) | In combination with dexamethasone | 2006 | PMID: 16365178 |
| NDMM (transplant eligible) | In combination with daratumumab, bortezomib and dexamethasone | 2019 | PMID: 31171419 | |
| Lenalidomide | Del(5q) MDS, low or intermediate risk | Single agent | 2005 | PMID: 17021321 |
| R/R MM | In combination with dexamethasone | 2006 | PMID: 18032763 PMID: 18032762 |
|
| R/R MM | In combination with carfilzomib and dexamethaonse | 2018 | PMID: 25482145 | |
| R/R MM | In combination with ixazomib and dexamethasone | 2015 | PMID: 27119237 | |
| R/R MM | In combination with daratumumab and dexamethasone | 2016 | PMID: 27705267 | |
| R/R MM | In combination with elotuzumab and dexamethasone | 2017 | PMID: 26035255 | |
| NDMM | In combination with dexamethasone | 2015 | PMID: 25184863 | |
| NDMM (transplant ineligible) | In combination with daratumumab and dexamethasone | 2019 | PMID: 31141632 | |
| MM maintenance after ASCT | Single agent | 2017 | PMID: 22571201 PMID: 22571202 |
|
| MCL relapsed after two or more prior lines, one of which was with bortezomib | Single agent | 2013 | PMID: 24002500 | |
| R/R FL | In combination with rituximab | 2019 | PMID: 30897038 NCT01996865 |
|
| R/R MZL | In combination with rituximab | 2019 | PMID: 30897038 NCT01996865 |
|
| Pomalidomide | MM after two prior lines | In combination with dexamethasone | 2013 | PMID: 24421329 PMID: 24007748 |
| MM after two prior lines | In combination with daratumumab and dexamethasone | 2017 | PMID: 28637662 | |
| MM after two prior lines | In combination with isatuximab and dexamethasone | 2020 | PMID: 31735560 | |
| Kaposi Sarcoma after failure of HAART in HIV+ patients or in non-HIV patients | Single agent | 2020 | PMID: 27863194 |
MM, multiple myeloma; MDS, myelodysplastic syndrome; ASCT, autologous stem cell transplant; MCL, mantle cell lymphoma; FL, follicular lymphoma; MZL, marginal zone lymphoma; HAART, highly active anti-retroviral therapy
Key points.
Thalidomide analogues including lenalidomide and pomalidomide are frontline anticancer agents against multiple hematologic malignancies. The dominant mechanism of action of thalidomide analogues is via targeted protein degradation of disease-relevant proteins.
Degrader therapeutics exhibit catalytic, event-driven pharmacology, and their biologic effects are a summation of the polypharmacology of multiple degraded neosubstrate proteins.
Cancers acquire resistance to thalidomide analogues by circumventing target protein degradation or by rewiring pathways downstream of target protein degradation.
Quantitative clinical proteomic assays can be used for pharmacodynamic monitoring of degrader therapeutics.
Molecular switches gated by degrader therapeutics can be used to control anticancer cellular immunotherapies.
Multiple classes of degrader therapeutics are expanding the draggable proteome to target non-catalytic cancer-relevant proteins including transcription factors, mRNA-splicing factors, and cyclins.
Acknowledgements
The work of M.J. is supported by a NIH T32 grant to the Massachusetts General Hospital Department of Pathology (NIH-5T32CA009216-39). The work of A.S.S. is supported by NIH grant K08CA252174. The work of B.L.E is supported by NIH grants R01HL082945 and P01CA108631, the Howard Hughes Medical Institute, the Edward P. Evans Foundation, the Leukemia and Lymphoma Society and the Adelson Medical Research Foundation.
Competing interests
B.L.E. has received research funding from Celgene, Deerfield, and Novartis and consulting fees from GRAIL. He serves on the scientific advisory boards and holds equity in Skyhawk Therapeutics, Exo Therapeutics, and Neomorph Therapeutics. The other authors declare no competing interests.
Glossary terms
- Molecular glue
A small molecule that directly bridges protein-protein interactions.
- Neosubstrates
Proteins that are conditionally targeted to a ubiquitin ligase in the presence of a small molecule.
- High-risk cytogenetic features
Chromosomal alterations that are associated with aggressive disease and unfavourable survival outcomes; in myeloma, these features include t(4;14), t(14;16) and del(17p) alterations.
- Antibody-dependent cellular cytotoxicity (ADCC)
A mechanism of cell killing that requires recognition of the antibody-bound cell by immune effector cells, such as natural killer cells.
- Activated B cell-like DLBCL
A subtype of DLBCL that is defined by a specific transcriptional signature and is associated with high risk of relapse following standard R-CHOP chemoimmunotherapy.
References
- 1.Stephens T & Brynner R Dark Remedy: The Impact of Thalidomide and its Revival as a Vital Medicine. (Perseus Publishing, 2009). [Google Scholar]
- 2.Lenz W, Pfeiffer RA, Kosenow W & Hayman DJ THALIDOMIDE AND CONGENITAL ABNORMALITIES. Lancet 279, 45–46 (1962). [Google Scholar]
- 3.Mcbride WG THALIDOMIDE AND CONGENITAL ABNORMALITIES. Lancet 278, 1358 (1961). [Google Scholar]
- 4.Mintz M “Heroine” of FDA Keeps Bad Drug Off of Market. The Washington Post (1962). [Google Scholar]
- 5.Kaur I, Dogra S, Narang T & De D Comparative efficacy of thalidomide and prednisolone in the treatment of moderate to severe erythema nodosum leprosum: A randomized study. Australas J Dermatol 50, 181–185 (2009). [DOI] [PubMed] [Google Scholar]
- 6.PEARSON JMH & VEDAGIRI M Treatment of Moderately Severe Erythema Nodosum Leprosum with Thalidomide - A Double-blind Controlled Trial. Leprosy Rev 40, 111–6 (1969). [DOI] [PubMed] [Google Scholar]
- 7.D’Amato RJ, Loughnan MS, Flynn E & Folkman J Thalidomide is an inhibitor of angiogenesis. Proc National Acad Sci 91, 4082–4085 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Singhal S et al. Antitumor Activity of Thalidomide in Refractory Multiple Myeloma. New Engl J Med 341, 1565–1571 (1999). [DOI] [PubMed] [Google Scholar]
- 9.Rajkumar SV et al. Combination Therapy With Thalidomide Plus Dexamethasone for Newly Diagnosed Myeloma. J Clin Oncol 20, 4319–4323 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Rajkumar SV et al. Phase III Clinical Trial of Thalidomide Plus Dexamethasone Compared With Dexamethasone Alone in Newly Diagnosed Multiple Myeloma: A Clinical Trial Coordinated by the Eastern Cooperative Oncology Group. J Clin Oncol 24, 431–436 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Krönke J et al. Lenalidomide causes selective degradation of IKZF1 and IKZF3 in multiple myeloma cells. Sci New York N Y 343, 301–5 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lu G et al. The myeloma drug lenalidomide promotes the cereblon-dependent destruction of Ikaros proteins. Sci New York N Y 343, 305–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gandhi AK et al. Immunomodulatory agents lenalidomide and pomalidomide co-stimulate T cells by inducing degradation of T cell repressors Ikaros and Aiolos via modulation of the E3 ubiquitin ligase complex CRL4 CRBN. Brit J Haematol 164, 811–821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ito T et al. Identification of a Primary Target of Thalidomide Teratogenicity. Science 327, 1345–1350 (2010). [DOI] [PubMed] [Google Scholar]
- 15.Krönke J et al. Lenalidomide induces ubiquitination and degradation of CK1α in del(5q) MDS. Nature 523, 183–188 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fink EC & Ebert BL The novel mechanism of lenalidomide activity. Blood 126, 2366–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fischer ES et al. Structure of the DDB1–CRBN E3 ubiquitin ligase in complex with thalidomide. Nature 512, 49–53 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bartlett JB, Dredge K & Dalgleish AG The evolution of thalidomide and its IMiD derivatives as anticancer agents. Nat Rev Cancer 4, 314–322 (2004). [DOI] [PubMed] [Google Scholar]
- 19.Richardson PG et al. A randomized phase 2 study of lenalidomide therapy for patients with relapsed or relapsed and refractory multiple myeloma. Blood 108, 3458–3464 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leonard JP et al. AUGMENT: A Phase III Study of Lenalidomide Plus Rituximab Versus Placebo Plus Rituximab in Relapsed or Refractory Indolent Lymphoma. J Clin Oncol 37, JCO.19.00010 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.List A et al. Lenalidomide in the Myelodysplastic Syndrome with Chromosome 5q Deletion. New Engl J Med 355, 1456–1465 (2006). [DOI] [PubMed] [Google Scholar]
- 22.Polizzotto MN et al. Pomalidomide for Symptomatic Kaposi’s Sarcoma in People With and Without HIV Infection: A Phase I/II Study. J Clin Oncol 34, 4125–4131 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Richardson PG et al. Immunomodulatory drug CC-5013 overcomes drug resistance and is well tolerated in patients with relapsed multiple myeloma. Blood 100, 3063–3067 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Weber DM et al. Lenalidomide plus Dexamethasone for Relapsed Multiple Myeloma in North America. New Engl J Medicine 357, 2133–2142 (2007). [DOI] [PubMed] [Google Scholar]
- 25.Dimopoulos M et al. Lenalidomide plus Dexamethasone for Relapsed or Refractory Multiple Myeloma. New Engl J Medicine 357, 2123–2132 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Rajkumar SV et al. Combination therapy with lenalidomide plus dexamethasone (Rev/Dex) for newly diagnosed myeloma. Blood 106, 4050–4053 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Benboubker L et al. Lenalidomide and Dexamethasone in Transplant-Ineligible Patients with Myeloma. New Engl J Med 371, 906–917 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Richardson PG et al. Lenalidomide, bortezomib, and dexamethasone combination therapy in patients with newly diagnosed multiple myeloma. Blood 116, 679–686 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Facon T et al. Daratumumab plus Lenalidomide and Dexamethasone for Untreated Myeloma. New Engl J Med 380, 2104–2115 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McCarthy PL et al. Lenalidomide after stem-cell transplantation for multiple myeloma. New Engl J Medicine 366, 1770–81 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Attal M et al. Lenalidomide maintenance after stem-cell transplantation for multiple myeloma. New Engl J Medicine 366, 1782–91 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Barlogie B et al. Thalidomide and Hematopoietic-Cell Transplantation for Multiple Myeloma. New Engl J Med 354, 1021–1030 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Palumbo A et al. Autologous Transplantation and Maintenance Therapy in Multiple Myeloma. New Engl J Med 371, 895–905 (2014). [DOI] [PubMed] [Google Scholar]
- 34.McCarthy PL et al. Lenalidomide Maintenance After Autologous Stem-Cell Transplantation in Newly Diagnosed Multiple Myeloma: A Meta-Analysis. J Clin Oncol Official J Am Soc Clin Oncol 35, 3279–3289 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goldschmidt H et al. Bortezomib before and after high-dose therapy in myeloma: long-term results from the phase III HOVON-65/GMMG-HD4 trial. Leukemia 32, 383–390 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Palumbo A et al. Continuous Therapy Versus Fixed Duration of Therapy in Patients With Newly Diagnosed Multiple Myeloma. J Clin Oncol 33, 3459–3466 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Madan S et al. Efficacy of retreatment with immunomodulatory drugs (IMiDs) in patients receiving IMiDs for initial therapy of newly diagnosed multiple myeloma. Blood 118, 1763–1765 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang M et al. Lenalidomide plus dexamethasone is more effective than dexamethasone alone in patients with relapsed or refractory multiple myeloma regardless of prior thalidomide exposure. Blood 112, 4445–4451 (2008). [DOI] [PubMed] [Google Scholar]
- 39.Stewart AK et al. Carfilzomib, Lenalidomide, and Dexamethasone for Relapsed Multiple Myeloma. New Engl J Medicine 372, 142–152 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Moreau P et al. Oral Ixazomib, Lenalidomide, and Dexamethasone for Multiple Myeloma. New Engl J Med 374, 1621–1634 (2016). [DOI] [PubMed] [Google Scholar]
- 41.Dimopoulos MA et al. Daratumumab, Lenalidomide, and Dexamethasone for Multiple Myeloma. New Engl J Med 375, 1319–1331 (2016). [DOI] [PubMed] [Google Scholar]
- 42.Siegel DS et al. Pomalidomide plus low-dose dexamethasone in relapsed refractory multiple myeloma after lenalidomide treatment failure. Brit J Haematol 188, 501–510 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Richardson PG et al. Pomalidomide alone or in combination with low-dose dexamethasone in relapsed and refractory multiple myeloma: a randomized phase 2 study. Blood 123, 1826–1832 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lacy MQ et al. Pomalidomide plus low-dose dexamethasone in myeloma refractory to both bortezomib and lenalidomide: comparison of 2 dosing strategies in dual-refractory disease. Blood 118, 2970–5 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson PG et al. Pomalidomide, bortezomib, and dexamethasone for patients with relapsed or refractory multiple myeloma previously treated with lenalidomide (OPTIMISMM): a randomised, open-label, phase 3 trial. Lancet Oncol 20, 781–794 (2019). [DOI] [PubMed] [Google Scholar]
- 46.Chari A et al. Daratumumab plus pomalidomide and dexamethasone in relapsed and/or refractory multiple myeloma. Blood 130, 974–981 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dimopoulos MA et al. Elotuzumab plus Pomalidomide and Dexamethasone for Multiple Myeloma. New Engl J Med 379, 1811–1822 (2018). [DOI] [PubMed] [Google Scholar]
- 48.Stewart AK, Richardson PG & San-Miguel JF How I treat multiple myeloma in younger patients. Blood 114, 5436–5443 (2009). [DOI] [PubMed] [Google Scholar]
- 49.Qian Z et al. Lenalidomide synergizes with dexamethasone to induce growth arrest and apoptosis of mantle cell lymphoma cells in vitro and in vivo. Leukemia Res 35, 380–386 (2011). [DOI] [PubMed] [Google Scholar]
- 50.Rychak E et al. Pomalidomide in combination with dexamethasone results in synergistic anti-tumour responses in pre-clinical models of lenalidomide-resistant multiple myeloma. Brit J Haematol 172, 889–901 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Das DS et al. Synergistic anti-myeloma activity of the proteasome inhibitor marizomib and the IMiD ® immunomodulatory drug pomalidomide. Brit J Haematol 171, 798–812 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chauhan D et al. Combination of novel proteasome inhibitor NPI-0052 and lenalidomide trigger in vitro and in vivo synergistic cytotoxicity in multiple myeloma. Blood 115, 834–845 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ocio EM et al. In vitro and in vivo rationale for the triple combination of panobinostat (LBH589) and dexamethasone with either bortezomib or lenalidomide in multiple myeloma. Haematologica 95, 794–803 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Siegel DS et al. Vorinostat in combination with lenalidomide and dexamethasone in patients with relapsed or refractory multiple myeloma. Blood Cancer J 4, e202 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shi C-X et al. Proteasome inhibitors block Ikaros degradation by Lenalidomide in Multiple Myeloma. Haematologica 100, e315–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Palmer AC & Sorger PK Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 171, 1678–1691.e13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Orlowski RZ et al. Phase I Trial of the Proteasome Inhibitor PS-341 in Patients With Refractory Hematologic Malignancies. J Clin Oncol 20, 4420–4427 (2002). [DOI] [PubMed] [Google Scholar]
- 58.Chamberlain PP & Hamann LG Development of targeted protein degradation therapeutics. Nat Chem Biol 15, 937–944 (2019). [DOI] [PubMed] [Google Scholar]
- 59.Ramsay AG et al. Chronic lymphocytic leukemia T cells show impaired immunological synapse formation that can be reversed with an immunomodulating drug. J Clin Invest 118, 2427–2437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hagner PR et al. CC-122, a pleiotropic pathway modifier, mimics an interferon response and has antitumor activity in DLBCL. Blood 126, 779–789 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luptakova K et al. Lenalidomide enhances anti-myeloma cellular immunity. Cancer Immunol Immunother 62, 39–49 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Muller GW et al. Structural Modifications of Thalidomide Produce Analogs with Enhanced Tumor Necrosis Factor Inhibitory Activity. J Med Chem 39, 3238–3240 (1996). [DOI] [PubMed] [Google Scholar]
- 63.Choueiri TK et al. Phase II study of lenalidomide in patients with metastatic renal cell carcinoma. Cancer 107, 2609–2616 (2006). [DOI] [PubMed] [Google Scholar]
- 64.Eisen T et al. Results of a multicenter, randomized, double-blind phase 2/3 study of lenalidomide in the treatment of pretreated relapsed or refractory metastatic malignant melanoma. Cancer 116, NA-NA (2009). [DOI] [PubMed] [Google Scholar]
- 65.Zonder JA et al. A phase 1, multicenter, open-label, dose escalation study of elotuzumab in patients with advanced multiple myeloma. Blood 120, 552–559 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Gavriatopoulou M, Terpos E & Dimopoulos MA The extended 4-year follow-up results of the ELOQUENT-2 trial. Oncotarget 10, 82–83 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lonial S et al. Elotuzumab Therapy for Relapsed or Refractory Multiple Myeloma. New Engl J Med 373, 621–631 (2015). [DOI] [PubMed] [Google Scholar]
- 68.Balasa B et al. Elotuzumab enhances natural killer cell activation and myeloma cell killing through interleukin-2 and TNF-α pathways. Cancer Immunol Immunother Cii 64, 61–73 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tai Y-T et al. Anti-CS1 humanized monoclonal antibody HuLuc63 inhibits myeloma cell adhesion and induces antibody-dependent cellular cytotoxicity in the bone marrow milieu. Blood 112, 1329–1337 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Collins SM et al. Elotuzumab directly enhances NK cell cytotoxicity against myeloma via CS1 ligation: evidence for augmented NK cell function complementing ADCC. Cancer Immunol Immunother Cii 62, 1841–9 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Veer M. S. van der et al. Towards effective immunotherapy of myeloma: enhanced elimination of myeloma cells by combination of lenalidomide with the human CD38 monoclonal antibody daratumumab. Haematologica 96, 284–90 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Palumbo A et al. Daratumumab, Bortezomib, and Dexamethasone for Multiple Myeloma. New Engl J Med 375, 754–766 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Trnêný M et al. Lenalidomide versus investigator’s choice in relapsed or refractory mantle cell lymphoma (MCL-002; SPRINT): a phase 2, randomised, multicentre trial. Lancet Oncol 17, 319–331 (2016). [DOI] [PubMed] [Google Scholar]
- 74.Goy A et al. Single-Agent Lenalidomide in Patients With Mantle-Cell Lymphoma Who Relapsed or Progressed After or Were Refractory to Bortezomib: Phase II MCL-001 (EMERGE) Study. J Clin Oncol 31, 3688–3695 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Morschhauser F et al. Rituximab plus Lenalidomide in Advanced Untreated Follicular Lymphoma. New Engl J Med 379, 934–947 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Leonard JP et al. Randomized Trial of Lenalidomide Alone Versus Lenalidomide Plus Rituximab in Patients With Recurrent Follicular Lymphoma: CALGB 50401 (Alliance). J Clin Oncol 33, 3635–3640 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ruan J et al. Lenalidomide plus Rituximab as Initial Treatment for Mantle-Cell Lymphoma. New Engl J Med 373, 1835–1844 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Badoux XC et al. Phase II Study of Lenalidomide and Rituximab As Salvage Therapy for Patients With Relapsed or Refractory Chronic Lymphocytic Leukemia. J Clin Oncol 31, 584–591 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chanan-Khan AA et al. Lenalidomide maintenance therapy in previously treated chronic lymphocytic leukaemia (CONTINUUM): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Haematol 4, e534–e543 (2017). [DOI] [PubMed] [Google Scholar]
- 80.Morschhauser F et al. Obinutuzumab combined with lenalidomide for relapsed or refractory follicular B-cell lymphoma (GALEN): a multicentre, single-arm, phase 2 study. Lancet Haematol 6, e429–e437 (2019). [DOI] [PubMed] [Google Scholar]
- 81.Yang Y et al. Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma. Cancer Cell 21, 723–737 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Nowakowski GS et al. Lenalidomide Combined With R-CHOP Overcomes Negative Prognostic Impact of Non–Germinal Center B-Cell Phenotype in Newly Diagnosed Diffuse Large B-Cell Lymphoma: A Phase II Study. J Clin Oncol 33, 251–257 (2015). [DOI] [PubMed] [Google Scholar]
- 83.Vitolo U et al. ROBUST: First report of phase III randomized study of lenalidomide/R-CHOP (R 2 -CHOP) vs placebo/R-CHOP in previously untreated ABC-type diffuse large B-cell lymphoma. Hematol Oncol 37, 36–37 (2019). [Google Scholar]
- 84.Ferreri AJM et al. Lenalidomide maintenance in patients with relapsed diffuse large B-cell lymphoma who are not eligible for autologous stem cell transplantation: an open label, single-arm, multicentre phase 2 trial. Lancet Haematol 4, e137–e146 (2017). [DOI] [PubMed] [Google Scholar]
- 85.Carpio C et al. Avadomide monotherapy in relapsed/refractory DLBCL: Safety, efficacy, and a predictive gene classifier. Blood (2020) doi: 10.1182/blood.2019002395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mullighan CG et al. BCR–ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature 453, 110–114 (2008). [DOI] [PubMed] [Google Scholar]
- 87.Mullighan CG et al. Deletion of IKZF1 and Prognosis in Acute Lymphoblastic Leukemia. New Engl J Med 360, 470–480 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Georgopoulos K et al. The ikaros gene is required for the development of all lymphoid lineages. Cell 79, 143–156 (1994). [DOI] [PubMed] [Google Scholar]
- 89.Wang J-H et al. Aiolos Regulates B Cell Activation and Maturation to Effector State. Immunity 9, 543–553 (1998). [DOI] [PubMed] [Google Scholar]
- 90.Morschhauser F et al. A phase 2, multicentre, single-arm, open-label study to evaluate the safety and efficacy of single-agent lenalidomide (Revlimid®) in subjects with relapsed or refractory peripheral T-cell non-Hodgkin lymphoma: The EXPECT trial. Eur J Cancer 49, 2869–2876 (2013). [DOI] [PubMed] [Google Scholar]
- 91.Ishida T et al. Multicenter Phase II Study of Lenalidomide in Relapsed or Recurrent Adult T-Cell Leukemia/Lymphoma: ATLL-002. J Clin Oncol 34, 4086–4093 (2016). [DOI] [PubMed] [Google Scholar]
- 92.Toumishey E et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma: Lenalidomide Therapy for T-Cell Lymphoma. Cancer 121, 716–723 (2014). [DOI] [PubMed] [Google Scholar]
- 93.Querfeld C et al. Results of an open-label multicenter phase 2 trial of lenalidomide monotherapy in refractory mycosis fungoides and Sézary syndrome. Blood 123, 1159–1166 (2014). [DOI] [PubMed] [Google Scholar]
- 94.List A et al. Efficacy of Lenalidomide in Myelodysplastic Syndromes. New Engl J Med 352, 549–557 (2005). [DOI] [PubMed] [Google Scholar]
- 95.List A, Ebert BL & Fenaux P A decade of progress in myelodysplastic syndrome with chromosome 5q deletion. Leukemia 32, 1493–1499 (2018). [DOI] [PubMed] [Google Scholar]
- 96.Schneider RK et al. Role of Casein Kinase 1A1 in the Biology and Targeted Therapy of del(5q) MDS. Cancer Cell 26, 509–520 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Martinez-Høyer S. et al. Loss of lenalidomide-induced megakaryocytic differentiation leads to therapy resistance in del(5q) myelodysplastic syndrome. Nat Cell Biol 22, 526–533 (2020). [DOI] [PubMed] [Google Scholar]
- 98.Jädersten M et al. TP53 Mutations in Low-Risk Myelodysplastic Syndromes With del(5q) Predict Disease Progression. J Clin Oncol 29, 1971–1979 (2011). [DOI] [PubMed] [Google Scholar]
- 99.Sekeres MA et al. A phase 2 study of lenalidomide monotherapy in patients with deletion 5q acute myeloid leukemia: Southwest Oncology Group Study S0605. Blood 118, 523–528 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Boutboul D et al. Dominant-negative IKZF1 mutations cause a T, B, and myeloid cell combined immunodeficiency. J Clin Invest 128, 3071–3087 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fang J et al. A calcium- and calpain-dependent pathway determines the response to lenalidomide in myelodysplastic syndromes. Nat Med 22, 727–734 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Fehniger TA et al. A phase 2 study of high-dose lenalidomide as initial therapy for older patients with acute myeloid leukemia. Blood 117, 1828–33 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Blum W et al. Dose Escalation of Lenalidomide in Relapsed or Refractory Acute Leukemias. J Clin Oncol 28, 4919–4925 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.DeAngelo DJ et al. A phase I study of lenalidomide plus chemotherapy with mitoxantrone, etoposide, and cytarabine for the reinduction of patients with acute myeloid leukemia. Am JHematol 93, 254–261 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Greenberg PL et al. Mitoxantrone, Etoposide, and Cytarabine With or Without Valspodar in Patients With Relapsed or Refractory Acute Myeloid Leukemia and High-Risk Myelodysplastic Syndrome: A Phase III Trial (E2995). J Clin Oncol 22, 1078–1086 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Feldman EJ et al. Phase III Randomized Multicenter Study of a Humanized Anti-CD33 Monoclonal Antibody, Lintuzumab, in Combination With Chemotherapy, Versus Chemotherapy Alone in Patients With Refractory or First-Relapsed Acute Myeloid Leukemia. J Clin Oncol 23, 4110–4116 (2005). [DOI] [PubMed] [Google Scholar]
- 107.Narayan R et al. Sequential azacitidine plus lenalidomide in previously treated elderly patients with acute myeloid leukemia and higher risk myelodysplastic syndrome. Leukemia Lymphoma 57, 609–615 (2015). [DOI] [PubMed] [Google Scholar]
- 108.Craddock C et al. Combination Lenalidomide and Azacitidine: A Novel Salvage Therapy in Patients Who Relapse After Allogeneic Stem-Cell Transplantation for Acute Myeloid Leukemia. J Clin Oncol 37, 580–588 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chihara D et al. Long-term results of a phase II trial of lenalidomide plus prednisone therapy for patients with myelofibrosis. Leukemia Res 48, 1–5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Bhutani M, Polizzotto MN, Uldrick TS & Yarchoan R Kaposi Sarcoma–Associated Herpesvirus-Associated Malignancies: Epidemiology, Pathogenesis, and Advances in Treatment. Semin Oncol 42, 223–246 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shimada K, Hayakawa F & Kiyoi H Biology and management of primary effusion lymphoma. Blood 132, 1879–1888 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Zhang L et al. Phase 2 study using oral thalidomide-cyclophosphamide-prednisone for idiopathic multicentric Castleman disease. Blood 133, 1720–1728 (2019). [DOI] [PubMed] [Google Scholar]
- 113.Sperling AS et al. Patterns of substrate affinity, competition, and degradation kinetics underlie biological activity of thalidomide analogs. Blood 134, 160–170 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rasco DW et al. A First-in-Human Study of Novel Cereblon Modulator Avadomide (CC-122) in Advanced Malignancies. Clin Cancer Res 25, 90–98 (2018). [DOI] [PubMed] [Google Scholar]
- 115.Matyskiela ME et al. A Cereblon Modulator (CC-220) with Improved Degradation of Ikaros and Aiolos. J Med Chem 61, 535–542 (2017). [DOI] [PubMed] [Google Scholar]
- 116.Bjorklund CC et al. Iberdomide (CC-220) is a potent cereblon E3 ligase modulator with antitumor and immunostimulatory activities in lenalidomide- and pomalidomide-resistant multiple myeloma cells with dysregulated CRBN. Leukemia 34, 1197–1201 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sievers QL et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 362, eaat0572 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lonial S et al. First clinical (phase 1b/2a) study of iberdomide (CC-220; IBER), a CELMoD, in combination with dexamethasone (DEX) in patients (pts) with relapsed/refractory multiple myeloma (RRMM). J Clin Oncol 37, 8006–8006 (2019). [Google Scholar]
- 119.Schafer PH et al. Cereblon modulator iberdomide induces degradation of the transcription factors Ikaros and Aiolos: immunomodulation in healthy volunteers and relevance to systemic lupus erythematosus. Ann Rheum Dis 77, 1516–1523 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Lopez-Girona A et al. CC-92480 Is a Novel Cereblon E3 Ligase Modulator with Enhanced Tumoricidal and Immunomodulatory Activity Against Sensitive and Resistant Multiple Myeloma Cells. Blood 134, 1812–1812 (2019). [Google Scholar]
- 121.Matyskiela ME et al. A novel cereblon modulator recruits GSPT1 to the CRL4CRBN ubiquitin ligase. Nature 535, 252–257 (2016). [DOI] [PubMed] [Google Scholar]
- 122.Uy GL et al. Clinical Activity of CC-90009, a Cereblon E3 Ligase Modulator and First-in-Class GSPT1 Degrader, As a Single Agent in Patients with Relapsed or Refractory Acute Myeloid Leukemia (R/R AML): First Results from a Phase I Dose-Finding Study. Blood 134, 232–232 (2019). [Google Scholar]
- 123.Kumar S et al. Impact of lenalidomide therapy on stem cell mobilization and engraftment post-peripheral blood stem cell transplantation in patients with newly diagnosed myeloma. Leukemia 21, 2035–2042 (2007). [DOI] [PubMed] [Google Scholar]
- 124.Mazumder A et al. Effect of lenalidomide therapy on mobilization of peripheral blood stem cells in previously untreated multiple myeloma patients. Leukemia 22, 1280–1281 (2007). [DOI] [PubMed] [Google Scholar]
- 125.Micallef INM et al. Plerixafor (Mozobil) for stem cell mobilization in patients with multiple myeloma previously treated with lenalidomide. Bone Marrow Transpl 46, 350–355 (2011). [DOI] [PubMed] [Google Scholar]
- 126.Musto P et al. Second primary malignancies in multiple myeloma: an overview and IMWG consensus. Ann Oncol 28, 228–245 (2017). [DOI] [PubMed] [Google Scholar]
- 127.Jones JR et al. Second malignancies in the context of lenalidomide treatment: an analysis of 2732 myeloma patients enrolled to the Myeloma XI trial. Blood Cancer J 6, e506–e506 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.CARRIER M, GAL GL, TAY J, WU C & LEE AY Rates of venous thromboembolism in multiple myeloma patients undergoing immunomodulatory therapy with thalidomide or lenalidomide: a systematic review and meta-analysis: Venous thromboembolism in multiple myeloma. J Thromb Haemost 9, 653–663 (2011). [DOI] [PubMed] [Google Scholar]
- 129.Chen C et al. Expanded safety experience with lenalidomide plus dexamethasone in relapsed or refractory multiple myeloma. Brit J Haematol 146, 164–170 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lenalidomide and Venous Thrombosis in Multiple Myeloma. New Engl J Medicine 354, 2079–2080 (2006). [PubMed] [Google Scholar]
- 131.Scarpace S et al. Arterial thrombosis in four patients treated with thalidomide. Leukemia Lymphoma 46, 239–242 (2005). [DOI] [PubMed] [Google Scholar]
- 132.Palumbo A et al. Prevention of thalidomide- and lenalidomide-associated thrombosis in myeloma. Leukemia 22, 414–423 (2008). [DOI] [PubMed] [Google Scholar]
- 133.Kaushal V, Kaushal GP, Melkaveri SN & Mehta P Thalidomide protects endothelial cells from doxorubicin-induced apoptosis but alters cell morphology. J Thromb Haemost 2, 327–334 (2004). [DOI] [PubMed] [Google Scholar]
- 134.Nardone B et al. Risk of Rash Associated With Lenalidomide in Cancer Patients: A Systematic Review of the Literature and Meta-analysis. Clin Lymphoma Myeloma Leukemia 13, 424–429 (2013). [DOI] [PubMed] [Google Scholar]
- 135.Tinsley SM, Kurtin SE & Ridgeway JA Practical Management of Lenalidomide-Related Rash. Clin Lymphoma Myeloma Leukemia 15, S64–S69 (2015). [DOI] [PubMed] [Google Scholar]
- 136.Lee MJ et al. Lenalidomide desensitization for delayed hypersensitivity reactions in 5 patients with multiple myeloma. Brit J Haematol 167, 127–131 (2014). [DOI] [PubMed] [Google Scholar]
- 137.Pawlyn C et al. Lenalidomide-induced diarrhea in patients with myeloma is caused by bile acid malabsorption that responds to treatment. Blood 124, 2467–2468 (2014). [DOI] [PubMed] [Google Scholar]
- 138.Fisher SL & Phillips AJ Targeted protein degradation and the enzymology of degraders. Curr Opin Chem Biol 44, 47–55 (2018). [DOI] [PubMed] [Google Scholar]
- 139.Lai AC & Crews CM Induced protein degradation: an emerging drug discovery paradigm. Nat Rev Drug Discov 16, 101–114 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Burslem GM & Crews CM Proteolysis-Targeting Chimeras as Therapeutics and Tools for Biological Discovery. Cell (2020) doi: 10.1016/j.cell.2019.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bondeson DP et al. Catalytic in vivo protein knockdown by small-molecule PROTACs. Nat Chem Biol 11, 611–617 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhu YX et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 118, 4771–4779 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Sievers QL, Gasser JA, Cowley GS, Fischer ES & Ebert BL Genome-wide screen identifies cullin-RING ligase machinery required for lenalidomide-dependent CRL4CRBN activity. Blood 132, 1293–1303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Patil A, Manzano M & Gottwein E Genome-wide CRISPR screens reveal genetic mediators of cereblon modulator toxicity in primary effusion lymphoma. Blood Adv 3, 2105–2117 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Tateno S et al. Genome-wide screening reveals a role for subcellular localization of CRBN in the anti-myeloma activity of pomalidomide. Sci Rep-uk 10, 4012 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kortüm KM et al. Targeted sequencing of refractory myeloma reveals a high incidence of mutations in CRBN and Ras pathway genes. Blood 128, 1226–1233 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Thakurta A et al. Absence of mutations in cereblon (CRBN) and DNA damage-binding protein 1 (DDB1) genes and significance for IMiD therapy. Leukemia 28, 1129–1131 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Gooding S et al. Multiple Cereblon genetic changes associate with acquired resistance to Lenalidomide or Pomalidomide in Multiple Myeloma. Blood (2020) doi: 10.1182/blood.2020007081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Huang S-Y et al. Expression of cereblon protein assessed by immunohistochemicalstaining in myeloma cells is associated with superior response of thalidomide- and lenalidomide-based treatment, but not bortezomib-based treatment, in patients with multiple myeloma. Ann Hematol 93, 1371–1380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Franssen LE et al. Cereblon loss and up-regulation of c-Myc are associated with lenalidomide resistance in multiple myeloma patients. Haematologica 103, e368–e371 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Heintel D et al. High expression of cereblon ( CRBN ) is associated with improved clinical response in patients with multiple myeloma treated with lenalidomide and dexamethasone. Brit J Haematol 161, 695–700 (2013). [DOI] [PubMed] [Google Scholar]
- 152.Jonasova A et al. High level of full-length cereblon mRNA in lower risk myelodysplastic syndrome with isolated 5q deletion is implicated in the efficacy of lenalidomide. Eur J Haematol 95, 27–34 (2014). [DOI] [PubMed] [Google Scholar]
- 153.Schuster SR et al. The clinical significance of cereblon expression in multiple myeloma. Leukemia Res 38, 23–8 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Pourabdollah M et al. High IKZF1/3 protein expression is a favorable prognostic factor for survival of relapsed/refractory multiple myeloma patients treated with lenalidomide. J Hematol Oncol 9, 123 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Dimopoulos K et al. Expression of CRBN, IKZF1, and IKZF3 does not predict lenalidomide sensitivity and mutations in the cereblon pathway are infrequent in multiple myeloma. Leukemia Lymphoma 60, 1–9 (2018). [DOI] [PubMed] [Google Scholar]
- 156.Gandhi AK et al. Measuring cereblon as a biomarker of response or resistance to lenalidomide and pomalidomide requires use of standardized reagents and understanding of gene complexity. Brit J Haematol 164, 233–44 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Zhu YX, Kortuem KM & Stewart AK Molecular mechanism of action of immune-modulatory drugs thalidomide, lenalidomide and pomalidomide in multiple myeloma. Leukemia Lymphoma 54, 683–687 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Krönke J et al. IKZF1 expression is a prognostic marker in newly diagnosed standard-risk multiple myeloma treated with lenalidomide and intensive chemotherapy: a study of the German Myeloma Study Group (DSMM). Leukemia 31, 1363–1367 (2017). [DOI] [PubMed] [Google Scholar]
- 159.Reichermeier KM et al. PIKES Analysis Reveals Response to Degraders and Key Regulatory Mechanisms of the CRL4 Network. Mol Cell 77, 1092–1106.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 160.Järås M et al. Csnk1a1 inhibition has p53-dependent therapeutic efficacy in acute myeloid leukemia. J Exp Medicine 211, 605–12 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Scharenberg C et al. Progression in patients with low- and intermediate-1-risk del(5q) myelodysplastic syndromes is predicted by a limited subset of mutations. Haematologica 102, 498–508 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Lodé L et al. Emergence and evolution of TP53 mutations are key features of disease progression in myelodysplastic patients with lower-risk del(5q) treated with lenalidomide. Haematologica 103, e143–e146 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Hoofnagle AN, Becker JO, Wener MH & Heinecke JW Quantification of Thyroglobulin, a Low-Abundance Serum Protein, by Immunoaffinity Peptide Enrichment and Tandem Mass Spectrometry. Clin Chem 54, 1796–1804 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Zhang B et al. Clinical potential of mass spectrometry-based proteogenomics. Nat Rev Clin Oncol 16, 256–268 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Donovan KA et al. Thalidomide promotes degradation of SALL4, a transcription factor implicated in Duane Radial Ray syndrome. Elife 7, e38430 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.An J et al. pSILAC mass spectrometry reveals ZFP91 as IMiD-dependent substrate of the CRL4CRBN ubiquitin ligase. Nat Commun 8, 15398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Petzold G, Fischer ES & Thomä NH Structural basis of lenalidomide-induced CK1α degradation by the CRL4CRBN ubiquitin ligase. Nature 532, 127–130 (2016). [DOI] [PubMed] [Google Scholar]
- 168.Najafabadi HS et al. C2H2 zinc finger proteins greatly expand the human regulatory lexicon. Nat Biotechnol 33, 555–562 (2015). [DOI] [PubMed] [Google Scholar]
- 169.Higgins JJ, Pucilowska J, Lombardi RQ & Rooney JP A mutation in a novel ATP-dependent Lon protease gene in a kindred with mild mental retardation. Neurology 63, 1927–1931 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Eichner R et al. Immunomodulatory drugs disrupt the cereblon–CD147–MCT1 axis to exert antitumor activity and teratogenicity. Nat Med 22, 735–743 (2016). [DOI] [PubMed] [Google Scholar]
- 171.Lee KM et al. Disruption of the cereblon gene enhances hepatic AMPK activity and prevents high-fat diet-induced obesity and insulin resistance in mice. Diabetes 62, 1855–64 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Nguyen TV et al. Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell 61, 809–820 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Mita M et al. Phase I Study of E7820, an Oral Inhibitor of Integrin α-2 Expression with Antiangiogenic Properties, in Patients with Advanced Malignancies. Clin Cancer Res 17, 193–200 (2011). [DOI] [PubMed] [Google Scholar]
- 174.Simon GR et al. A phase I study of tasisulam sodium (LY573636 sodium), a novel anticancer compound in patients with refractory solid tumors. Cancer Chemoth Pharm 68, 1233–1241 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Rigas JR et al. Phase I clinical and pharmacological study of chloroquinoxaline sulfonamide. Cancer Res 52, 6619–23 (1992). [PubMed] [Google Scholar]
- 176.Punt CJA et al. Phase I and pharmacokinetic study of E7070, a novel sulfonamide, given at a daily times five schedule in patients with solid tumors. A study by the EORTC-Early Clinical Studies Group (ECSG). Ann Oncol 12, 1289–1293 (2001). [DOI] [PubMed] [Google Scholar]
- 177.Han T et al. Anticancer sulfonamides target splicing by inducing RBM39 degradation via recruitment to DCAF15. Sci New York N Y 356, eaal3755 (2017). [DOI] [PubMed] [Google Scholar]
- 178.Uehara T et al. Selective degradation of splicing factor CAPERα by anticancer sulfonamides. Nat Chem Biol 13, 675–680 (2017). [DOI] [PubMed] [Google Scholar]
- 179.Ting TC et al. Aryl Sulfonamides Degrade RBM39 and RBM23 by Recruitment to CRL4-DCAF15. Cell Reports 29, 1499–1510.e6 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Faust TB et al. Structural complementarity facilitates E7820-mediated degradation of RBM39 by DCAF15. Nat Chem Biol 16, 7–14 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Du X et al. Structural Basis and Kinetic Pathway of RBM39 Recruitment to DCAF15 by a Sulfonamide Molecular Glue E7820. Structure 27, 1625–1633.e3 (2019). [DOI] [PubMed] [Google Scholar]
- 182.Slabicki M et al. The CDK inhibitor CR8 acts as a molecular glue degrader that depletes cyclin K. Nature 1–5 (2020) doi: 10.1038/s41586-020-2374-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Mayor-Ruiz C et al. Rational discovery of molecular glue degraders via scalable chemical profiling. Nat Chem Biol 16, 1199–1207 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Lv L et al. Discovery of a molecular glue promoting CDK12-DDB1 interaction to trigger Cyclin K degradation. Elife 9, e59994 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nusse R & Clevers H Wnt/β-Catenin Signaling, Disease, and Emerging Therapeutic Modalities. Cell 169, 985–999 (2017). [DOI] [PubMed] [Google Scholar]
- 186.Simonetta KR et al. Prospective discovery of small molecule enhancers of an E3 ligase-substrate interaction. Nat Commun 10, 1402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Kerres N et al. Chemically Induced Degradation of the Oncogenic Transcription Factor BCL6. Cell Reports 20, 2860–2875 (2017). [DOI] [PubMed] [Google Scholar]
- 188.Slabicki M et al. Small-molecule-induced polymerization triggers degradation of BCL6. Nature 1–5 (2020) doi: 10.1038/s41586-020-2925-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Weber EW, Maus MV & Mackall CL The Emerging Landscape of Immune Cell Therapies. Cell 181, 46–62 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Neelapu SS et al. Chimeric antigen receptor T-cell therapy - assessment and management of toxicities. Nat Rev Clin Oncol 15, 47–62 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Roybal KT & Lim WA Synthetic Immunology: Hacking Immune Cells to Expand Their Therapeutic Capabilities. Annu Rev Immunol 35, 229–253 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Stasi AD et al. Inducible Apoptosis as a Safety Switch for Adoptive Cell Therapy. New Engl J Med 365, 1673–1683 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Chiocca EA et al. Regulatable interleukin-12 gene therapy in patients with recurrent high-grade glioma: Results of a phase 1 trial. Sci Transl Med 11, eaaw5680 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Verma R, Mohl D & Deshaies RJ Harnessing the Power of Proteolysis for Targeted Protein Inactivation. Mol Cell (2020) doi: 10.1016/j.molcel.2020.01.010. [DOI] [PubMed] [Google Scholar]
- 195.Banaszynski LA, Chen L, Maynard-Smith LA, Ooi AGL & Wandless TJ A Rapid, Reversible, and Tunable Method to Regulate Protein Function in Living Cells Using Synthetic Small Molecules. Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T & Kanemaki M An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat Methods 6, 917–922 (2009). [DOI] [PubMed] [Google Scholar]
- 197.Chung HK et al. Tunable and reversible drug control of protein production via a self-excising degron. Nat Chem Biol 11, 713–720 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Neklesa TK et al. Small-molecule hydrophobic tagging–induced degradation of HaloTag fusion proteins. Nat Chem Biol 7, 538–543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Buckley DL et al. HaloPROTACS: Use of Small Molecule PROTACs to Induce Degradation of HaloTag Fusion Proteins. Acs Chem Biol 10, 1831–7 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nabet B et al. Rapid and direct control of target protein levels with VHL-recruiting dTAG molecules. Biorxiv 2020.03.13.980946 (2020) doi: 10.1101/2020.03.13.980946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nabet B et al. The dTAG system for immediate and target-specific protein degradation. Nat Chem Biol 14, 431–441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Clackson T et al. Redesigning an FKBP-ligand interface to generate chemical dimerizers with novel specificity. Proc National Acad Sci 95, 10437–10442 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lee SM et al. A Chemical Switch System to Modulate Chimeric Antigen Receptor T Cell Activity through Proteolysis-Targeting Chimaera Technology. Acs Synth Biol (2020) doi: 10.1021/acssynbio.9b00476. [DOI] [PubMed] [Google Scholar]
- 204.Weber EW et al. Transient “rest” induces functional reinvigoration and epigenetic remodeling in exhausted CAR-T cells. Biorxiv 2020.01.26.920496 (2020) doi: 10.1101/2020.01.26.920496. [DOI] [Google Scholar]
- 205.Koduri V et al. Peptidic degron for IMiD-induced degradation of heterologous proteins. P Natl Acad Sci Usa 116, 2539–2544 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Carbonneau S et al. An IMiD-inducible degron provides reversible regulation for chimeric antigen receptor expression and activity. Cell Chem Biol (2020) doi: 10.1016/j.chembiol.2020.11.012. [DOI] [PubMed] [Google Scholar]
- 207.Jan M et al. Reversible ON- and OFF-switch chimeric antigen receptors controlled by lenalidomide. Sci Transl Med 13, eabb6295 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Colombo MP & Trinchieri G Interleukin-12 in anti-tumor immunity and immunotherapy. Cytokine Growth F R 13, 155–168 (2002). [DOI] [PubMed] [Google Scholar]
- 209.Varshavsky A The Ubiquitin System, an Immense Realm. Biochemistry-us 81, 167–176 (2012). [DOI] [PubMed] [Google Scholar]
- 210.Zheng N & Shabek N Ubiquitin Ligases: Structure, Function, and Regulation. Annu Rev Biochem 86, 129–157 (2017). [DOI] [PubMed] [Google Scholar]
- 211.Komander D & Rape M The Ubiquitin Code. Annu Rev Biochem 81, 203–229 (2012). [DOI] [PubMed] [Google Scholar]
- 212.Martinez-Høyer S. & Karsan A. Mechanisms of lenalidomide sensitivity and resistance. Exp Hematol (2020) doi: 10.1016/j.exphem.2020.09.196. [DOI] [PubMed] [Google Scholar]
- 213.Cortes M, Wong E, Koipally J & Georgopoulos K Control of lymphocyte development by the Ikaros gene family. Curr Opin Immunol 11, 167–171 (1999). [DOI] [PubMed] [Google Scholar]
- 214.Cortés M & Georgopoulos K Aiolos Is Required for the Generation of High Affinity Bone Marrow Plasma Cells Responsible for Long-Term Immunity. J Exp Medicine 199, 209–219 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Shaffer AL et al. IRF4 addiction in multiple myeloma. Nature 454, 226–231 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Gandhi R et al. Activation of the aryl hydrocarbon receptor induces human type 1 regulatory T cell–like and Foxp3+ regulatory T cells. Nat Immunol 11, 846–853 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Quintana FJ et al. Aiolos promotes TH17 differentiation by directly silencing Il2 expression. Nat Immunol 13, 770–777 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Elyada E et al. CKIα ablation highlights a critical role for p53 in invasiveness control. Nature 470, 409–413 (2011). [DOI] [PubMed] [Google Scholar]
- 219.Nijhawan D et al. Cancer vulnerabilities unveiled by genomic loss. Cell 150, 842–54 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Hoshino S et al. A human homologue of the yeast GST1 gene codes for a GTP-binding protein and is expressed in a proliferation-dependent manner in mammalian cells. Embo J 8, 3807–14 (1989). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Zhouravleva G et al. Termination of translation in eukaryotes is governed by two interacting polypeptide chain release factors, eRF1 and eRF3. Embo J 14, 4065–72 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Matyskiela ME et al. SALL4 mediates teratogenicity as a thalidomide-dependent cereblon substrate. Nat Chem Biol 14, 981–987 (2018). [DOI] [PubMed] [Google Scholar]
- 223.Knobloch J & Rüther U Shedding light on an old mystery: Thalidomide suppresses survival pathways to induce limb defects. Cell Cycle 7, 1121–1127 (2008). [DOI] [PubMed] [Google Scholar]
- 224.Coleman KG & Crews CM Proteolysis-Targeting Chimeras: Harnessing the Ubiquitin-Proteasome System to Induce Degradation of Specific Target Proteins. Annu Rev Cancer Biology 2, 41–58 (2018). [Google Scholar]
- 225.Gerry CJ & Schreiber SL Unifying principles of bifunctional, proximity-inducing small molecules. Nat Chem Biol 16, 369–378 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Stanton BZ, Chory EJ & Crabtree GR Chemically induced proximity in biology and medicine. Science 359, eaao5902 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Nowak RP et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat Chem Biol 14, 706–714 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Huang H-T et al. A Chemoproteomic Approach to Query the Degradable Kinome Using a Multi-kinase Degrader. Cell Chem Biol 25, 88–99.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Bondeson DP et al. Lessons in PROTAC Design from Selective Degradation with a Promiscuous Warhead. Cell Chem Biol 25, 78–87.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Dobrovolsky D et al. Bruton tyrosine kinase degradation as a therapeutic strategy for cancer. Blood 133, 952–961 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Buhimschi AD et al. Targeting the C481S Ibrutinib-Resistance Mutation in Bruton’s Tyrosine Kinase Using PROTAC-Mediated Degradation. Biochemistry-us 57, 3564–3575 (2018). [DOI] [PubMed] [Google Scholar]
- 232.Schapira M, Calabrese MF, Bullock AN & Crews CM Targeted protein degradation: expanding the toolbox. Nat Rev Drug Discov 18, 949–963 (2019). [DOI] [PubMed] [Google Scholar]
- 233.Yamazoe S et al. Heterobifunctional Molecules Induce Dephosphorylation of Kinases–A Proof of Concept Study. J Med Chem 63, 2807–2813 (2019). [DOI] [PubMed] [Google Scholar]
- 234.Banik SM et al. Lysosome-targeting chimaeras for degradation of extracellular proteins. Nature 584, 291–297 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Neklesa TK & Crews CM Greasy tags for protein removal. Nature 487, 308–309 (2012). [DOI] [PubMed] [Google Scholar]
- 236.Ma A et al. Discovery of a first-in-class EZH2 selective degrader. Nat Chem Biol 16, 214–222 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.