

Abstract
We investigated the product ion spectra of [M+Na]+ from diterpene diester species and low molecular mass metabolites analyzed by electrospray ionization (ESI). Mainly, the formation of protonated salt structures was proposed to explain the observed neutral losses of carboxylic acids. It also facilitates understanding sodium retention on product ions or on neutral losses. In addition, the occurrence of consecutive carboxylic acid losses is rather unexpected under resonant excitation conditions. Quantum calculation demonstrated that the exothermic character of such neutral losses can represent a relevant explanation. There is no doubt that the formation and role of the protonated salt structures will be helpful for a better understanding and software-assisted interpretation of tandem mass spectra from small molecules, especially in the ever-growing metabolomics field.
Introduction
The annotation of product ion spectra of [M+Na]+ precursor ions formed from diterpene diester species and low molecular mass metabolites under electrospray ionization (ESI) conditions featured in Ludwig et al.’s work lead us to further investigate the product ion spectra of [M+Na]+ precursors.1 Studying and understanding product ion spectra nowadays represents a major concern in the metabolomics field.2 Indeed, the development of efficient untargeted tandem mass spectrometry workflows (e.g., data independent acquisition, DIA)3–5 will generate huge amounts of MS/MS datasets, thus requiring the development of relevant “chemistry-compatible” cheminformatics tools to address metabolite annotation notably via MS/MS molecular networking.6–8 When combined with liquid chromatography, ESI is the most popular desorption/ionization technique for metabolomic studies,9 often competitively yielding both intact protonated and sodiated molecules for a given metabolite. In their recent paper, Ludwig et al.1 examined in great detail the annotation of the largely understudied but highly informative sodiated molecules [M+Na]+, with a special emphasis on their corresponding product ions observed by considering exclusively covalent bond cleavages (CBC). In particular, the authors interpreted and annotated these product ion spectra by homology to those derived from corresponding protonated species activated through collisions under resonant excitation conditions (collision-induced-dissociations or CID in an LTQ-Orbitrap instrument).1 For example, major fragment ions observed in the high-resolution CID spectrum of a particular sodiated diterpene diester denoted a m/z difference of 21.9819 (+/−5 ppm) resulting from the exchange of one Na+ by one H+ (“mNa-mH” mass difference). Such a mass difference points to sodium cation retention on the given product ions, while unshifted peaks were interpreted as resulting from sodiated neutral losses. This strategy was extended to hundreds of metabolites by mining polyfunctional compounds included in the NIST 17 spectral database such as lipids whose sodiated ions are known to exhibit particular fragmentation patterns.10
Discussion
The work of Ludwig et al. caught and retained our attention by its attempt to rationalize the formation of fragment ions generated by collisional dissociations of sodiated molecules, with the objective of implementing computational methods to automatically annotate MS and MS/MS spectra within spectral databases.1 Noteworthy, such rationalization of fragmentation mechanisms could be highly relevant for elucidation of unknown metabolite structure in biological extracts. However, the CBC mechanisms were considered independently of “chemical reality” i.e., without strict compliance to chemical rules. For instance, in the case of sodiated molecules, the authors assume that such precursor ions can dissociate through two main mechanisms: either by charge migration fragmentation (CMF) or by charge retention fragmentation (CRF),1 as initially suggested by Demarque et al.11 Although such a systematic description could represent an acceptable semi-empirical in silico approach, it might ignore important chemical rules while interpreting and illustrating dissociation of sodiated species.
In Ludwig et al. paper figure 2 shows sodium as a coordinated neutral metal in the CMF pathway,1 while one would expect a neutral salt entity. Indeed, one should assume that sodium-coordinated species appear strictly as charge solvated forms, thus yielding by CID, free Na+ cation.12 Actually, it is possible to propose mechanistic alternatives for rationalizing fragmentations of sodiated species (as well as protonated ones) following fundamental chemical rules,6–8 especially when considering the concept of protonated sodium salt entities.9 Scheme 1, adapted from Colsch et al.12 illustrates the fragmentation pathways of a sodiated lysoglycerophosphoethanolamine (LGPE, m/z 434) leading to the observation of complementary ions: one with sodium retention (m/z 164) and the other (m/z 271) resulting from the loss of a sodium containing neutral entity.
Figure 2.

(a) Structure of the studied [AiPr+H]+ and [AEt+H]+ ions; (b) exothermic dissociative pathway with intermediate structures from [AiPr+H]+ (violet stepwise pathway: the → B+ → Ccy+ sequence) and [AEt+H]+ (blue stepwise pathway: the → E+ → F+ → Gcy+ sequence) resulting in stabilization of the monoester Ccy+ and Gcy+ carbocation; in competition with the cation stabilizations, competitive processes allowing the second ester loss are added: from B+ (red stepwise pathway B+ → C+ → D+ sequence) and from F+ (green stepwise pathway: the F+ → H+ → I+ sequence). Correspondence between structures from Figure 2 and m/z values from Figure 1 (considering that the decatrienoic acid chain has been modified to propionic acid for simplification of the calculation procedure): [AiPr+H]+ and [AEt+H]+ corresponds to m/z 589 in Figure 1; B+, C+ and Ccy+, m/z 501; F+, Gcy+ and H+, m/z 423; I+, m/z 335.
Scheme 1:
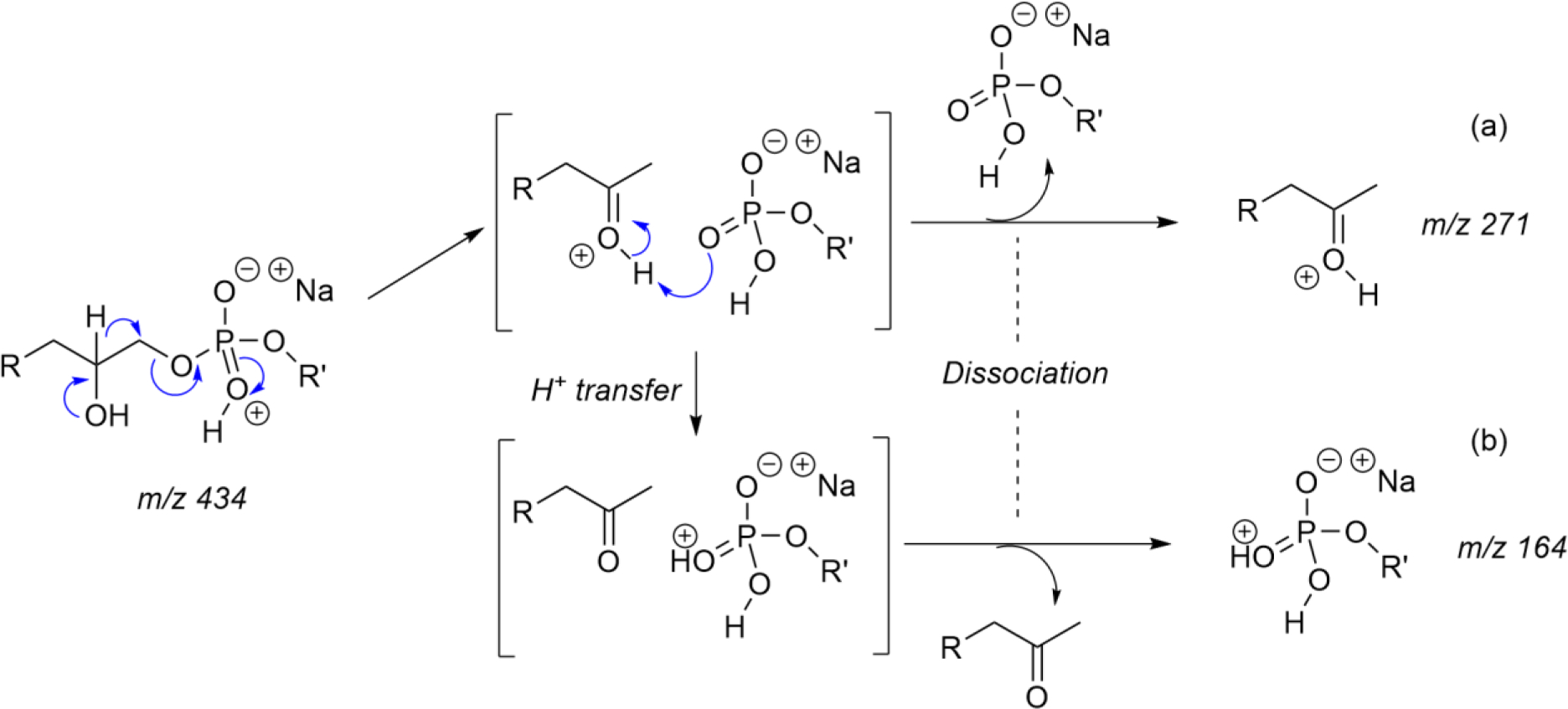
Two complementary ions formed by decomposition of the protonated sodium salt of a LGPE (m/z 434, R=C11H23) (a) ion resulting from the loss of the neutral phosphonate sodium salt (m/z 271); (b) protonated salt with retention of sodium (m/z 164).
It is necessary to consider chemically-compatible annotations of the desorbed sodiated molecules at the origin of CBCs.16 Indeed, two possible structures can be considered: (i) charge solvated forms (i.e., Na+ liganded by heteroatom(s) annotated as [M+Na]+) and (ii) protonated salts annotated as zwitterionic species [(M-H+Na)+H]+ with the mobile proton located at the heteroatoms.16 When submitted to CID, the former species would yield exclusively free Na+,10,12–14 while the protonated salt would dissociate directly or indirectly (by prototropy) by CBC(s) with salt retention either on product ions or attached to lost neutrals.15 Thus, the objective of the present letter is to propose alternative mechanisms to the CRF/CMF ones based solely on interpreting the chemistry of Na+ retention either in product ions or neutral losses occurring during fragmentation of sodiated molecules. By way of an example, the fragmentation mechanism of the sodiated diterpene di-ester from Figure 1 of Ludwig et al., (i.e., m/z 589 from 12β-O-[deca-2Z,4E,6E-trienoyl]13-isobutyroyloxy 4β-deoxyphorbol, Scheme 2, Figure 1 and Supporting Information Figure S1) has been reconsidered and discussed using density-functional theory (DFT) modeling. This will provide evidence of the validity of the proposed alternative mechanisms, which are exclusively based on protonated salt structures i.e., sodiated zwitterionic (ZW) species, thus focusing on the location of ion-ion interaction within these structures.
Figure 1.

Product ion spectrum of the sodiated diterpene diester (m/z 589.3116) acquired at a resolution of 30,000 using an LTQ-Orbitrap. Experimental conditions for resonant CID excitation: qz, 0.25; normalized collision energy (NCE), 30%; excitation duration, 10 msec; He buffer gas pressure, 5 × 10−5 mbar. Of note, Na+ cation cannot be detected under these conditions due to the low-mass-cut-off of the LTQ and the very high motion frequency limit of the FT/MS detection mode.
Scheme 2.
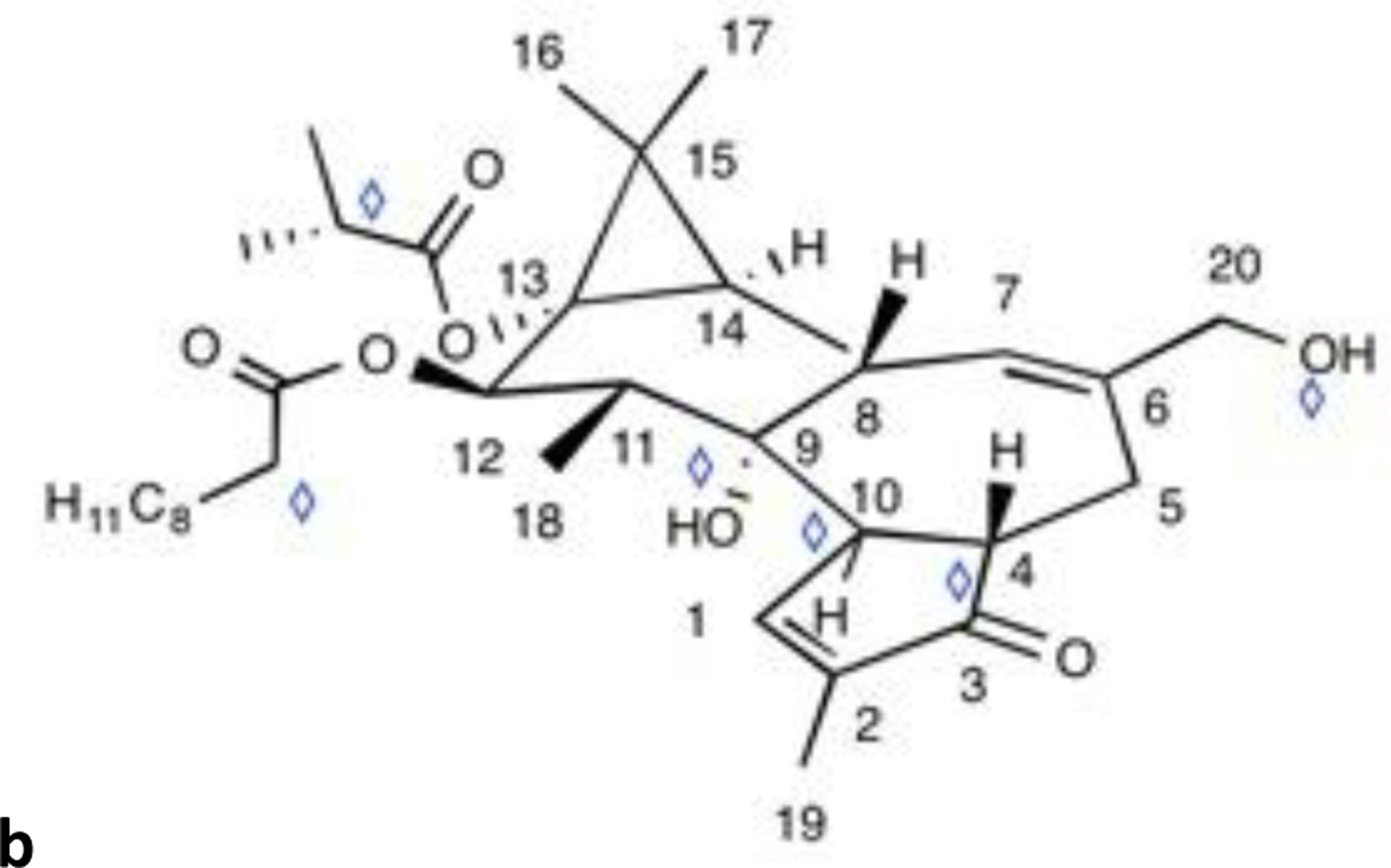
Structure of the studied diterpene di-ester 12β-O-[deca-2Z,4E,6E-trienoyl]13-iso-butyroyloxy 4β-deoxyphorbol (C34H46O7, Mw=566.3244 Da). The heteroatoms such as oxygen atoms are competitive protonation sites. Diamonds mark the possible deprotonation sites (enolizable and hydroxyl groups).
In ESI, the salt can be competitively located at diverse acidic sites within different ZW forms: (i) at the α (at C-4) and γ (at C-10) enolizable positions of the α-β unsaturated C-3 ketone, yielding the a and a’ forms, and (ii) at the α positions of the C-12 and C-13 esters giving the b and b’ forms (Supporting Information Schema S1). Deprotonation at the C-9 and C-20 hydroxy groups, thus yielding a ZW molecule can be ruled out due to its relative low gas phase acidity (compared to that of enolizable sites).17 The co-existence of tautomeric a and a’ forms of [(M-H+Na)+H]+ allows the mobilizable proton to migrate through prototropy at diverse positions on the oxygen atoms or on an unsaturated site. Especially, this mobile proton can be competitively localized on one of two ester groups at C-12 and C-13, independently of its C-4 or C-10 origin. It is expected that these protonated sites would promote dissociations into shared fragment ions corresponding to losses of the corresponding isobutyric and fatty acid neutrals. Finally, the salt site at C-3 (a and a’ forms) could be considered a spectator in these CBC processes.
Note that the reported CID spectrum (Figure 1) and that used by Ludwig et al.1 extracted from the MassIVE spectral database18 (Supporting Information Figure S1) have been recorded with the same instrument explaining their high degree of similarity. Under these CID conditions, the competitive losses of sodium salts of isobutyric and decatrienoic acids resulting in ions at m/z 479 and m/z 401, due to CMF, are not detected by contrast to the CRF-induced competitive losses of carboxylic acids. Indeed, the CID spectrum (Figure 1) mainly displays three abundant sodiated product ions resulting from: (i) competitive releases of isobutyric and decatrienoic acids from protonated esters at m/z 423 and m/z 501 and (ii) consecutive releases of the two carboxylic acids at m/z 335, which is rather unexpected under resonant conditions. Indeed, product ions being characterized by a larger motion frequency than that of precursor ions, cannot be excited in contrast to that occurring in non-resonant excitation with RF only multipole. Thus, the consecutive dissociations in the ion trap are somewhat hindered, except when the apparent energy threshold of the consecutive process is sufficiently weak compared to the residual internal energy of the first-generation product ions.
Organic chemistry mechanisms led us to the interpretation of the carboxylic acid releases from the protonated salt. However, the precursor ion absence at a normalized collision energy NCE = 30% suggests that these releases are significantly favored. The strained 3-membered ring proximity may assist carboxylic acid elimination (Supporting Information Scheme S2). However, it is more difficult to propose mechanisms to explain the consecutive losses of the isobutyric and decatrienoic acids. So, a pertinent pathway must be proposed to explain these consecutive processes.
Under low energy CID conditions, the sodiated molecules considered as protonated salt mainly fragment through CBC (Supporting Information Scheme S2) by losing the same neutrals that those released by the protonated molecules.1 Thus, it is reasonable to infer that the sodiated site remains a spectator of the major fragmentation processes, resulting in a better interpretation of these competitive and consecutive major losses. Hence to simplify, modeling of the protonated [M+H]+ molecules was performed. In addition, for reasons of computational time and the reasonable assumption that the length of the unsaturated C9H13- chain of the ester at C-12 would have little or no influence on the results, the modeling was carried out with the unchanged isobutyrate ester at C-13 and the modified long chain ester into the propionate ester at C-12 of the 6-membered ring of the diterpenic skeleton A (Figure 2). Modeling of the competitive/consecutive isobutyric and propionic acid losses has been performed at B3LYP/6-311G** level, the nature of minima identified by the nature of their frequencies. The values reported in the manuscript are Gibbs free energies in kJ/mol.
The protonated [AEt+H]+ propanoate ester is slightly more stable by 15.8 kJ/mol than the protonated isobutyrate [AiPr+H]+ ester. Their direct dissociations by the corresponding carboxylic acid loss are exothermic and give rise to formation of species B+ (from [AiPr+H]+, red pathway) and Gcy+ (from [AEt+H]+, blue pathway) with stabilization energies of 136.3 kJ/mol and 155.5 kJ/mol, respectively.
i). Protonation on the isobutyrate ester at C-13 (red pathway)
The ester bond cleavage is concomitant with the cyclopropane ring opening. From the isomeric product B+ ion, it was possible to quench the charge by exothermic cyclization in Ccy+ i.e., by −165.1 kJ/mol which represents a small transition state (TS[B->CCY]+) energy. This occurs in competition with the consecutive loss of EtCO2H related to the second ester bond cleavage. In a first step, a proton is transferred from C-8 to the ester carbonyl group at C-12 with an energy cost of 23.4 kJ/mol (i.e., TS[B->C]+), leading to the C+ ion with an exothermicity of −158 kJ/mol. In a second step, the release of the propanoic acid occurs almost spontaneously, producing the D+ product ion stabilized by 42.6 kJ/mol with respect to C+ (−200.6 kJ/mol).
ii). protonation of the propanoate ester (blue and green pathways)
In a similar manner, the C-12 propanoic acid release is associated to the cyclopropane ring opening and leads to the tertiary carbocation E+ intermediate via exothermic reaction (by −49.3 kJ/mol). The propionic acid interacts poorly with the fragment ion and easily yields F+. From the latter, two possible dissociative pathways occur. The most direct process consists of charge quenching by a nucleophilic attack of tertiary carbocation by the carbonyl oxygen atom of isobutyrate ester. It results in a 6-membered 1,3-dioxo ring Gcy+ (−155.5 kJ/mol) which is a very stable form likely not to further fragment. A possible competitive pathway (green pathway) is the migration of a mobile proton from C-16 or C-17 (acidic methyl groups due to the vicinal C-15 carbocation) to the carbonyl oxygen atom of the isobutyrate ester resulting in the intermediate H+ ion. This isomerization requiring a low TS[F->H]+ energy level (only 6.7 kJ/mol), is slightly exothermic by −12.1 kJ/mol. The H+ intermediate spontaneously yield the I+ product ion through an exothermic isobutyric acid release by −59.3 kJ/mol.
The proposed mechanisms based on classic organic chemistry (Supporting Information Scheme S3) for the first generation of product ions at m/z 501 and m/z 423 due to the competitive C3H7COOH and C9H13COOH losses from the m/z 589 sodiated diterpene diester are consistent with those predicted from the above calculations performed on the protonated molecules carrying shorter chain at C-12. First and foremost, these calculations offer mechanisms for the consecutive isobutyric acid releases from the product ion at m/z 423 due to the C9H13COOH neutral loss which is not easily described since it mainly involved a ring size reduction according to quasi concerted ring opening and reclosing (Supporting Information Scheme S3). Finally, the calculation provides us with a definitive explanation on why consecutive losses of carboxylic acids occur due to their exothermicity in the ion trap although only the precursor ion was submitted to resonant excitation.
In conclusion, we present a particular model to explain dissociation of sodiated species under low-energy conditions that is a chemically- rather than computationally-driven approach. The model proposed in this paper is indeed fully compliant with chemical reactivity and basic rules, and accentuates the necessity of zwitterionic species for explaining the fragment ions observed under low-energy decomposition of sodiated species. The basic premise is that the initial desorption of sodiated molecule yields protonated salts post-desolvation. Under low-energy CID conditions, bond cleavages are then promoted by the mobilizable proton bound to heteroatoms of the sodiated molecule, while the salt group remains spectator to the fragmentation processes. This implies sodium cation retention within salts either on product ions or within neutral losses. With this in mind, we re-investigated and re-interpretated the fragmentation mechanisms of the representative sodiated diterpene diester initially introduced by Ludwig et al. in their paper,1 and also made use of quantum calculations to demonstrate and confirm the premise is correct.
As to the supporting information Scheme 1 presents the proposed structures of protonated salt located at the C-3 ketone and the enolizable site at C-12. While Scheme S2 shows the proposed fragmentation mechanisms for the competitive losses from C-12 and/or C-13 positions and Scheme S3 points to the potential decompositions occurring during minimization of the rearrangement resulting in the isobutyric acid release and ring reduction (ion I+).
Supplementary Material
Acknowledgements
This research was supported in part by the Intramural Research Program of the National Institute on Drug Abuse, NIH.
References
- (1).Ludwig M; Broeckling CD; Dorrestein PC; Dührkop K; Schymanski EL; Böcker S; Nothias L-F Studying Charge Migration Fragmentation of Sodiated Precursor Ions in Collision-Induced Dissociation at the Library Scale. J. Am. Soc. Mass Spectrom 2021, 32 (1), 180–186. 10.1021/jasms.0c00240. [DOI] [PubMed] [Google Scholar]
- (2).Domingo-Almenara X; Montenegro-Burke JR; Benton HP; Siuzdak G Annotation: A Computational Solution for Streamlining Metabolomics Analysis. Anal. Chem 2018, 90 (1), 480–489. 10.1021/acs.analchem.7b03929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Wang Y; Feng R; Wang R; Yang F; Li P; Wan J-B Enhanced MS/MS Coverage for Metabolite Identification in LC-MS-Based Untargeted Metabolomics by Target-Directed Data Dependent Acquisition with Time-Staggered Precursor Ion List. Anal. Chim. Acta 2017, 992, 67–75. 10.1016/j.aca.2017.08.044. [DOI] [PubMed] [Google Scholar]
- (4).Bruderer T; Varesio E; Hidasi AO; Duchoslav E; Burton L; Bonner R; Hopfgartner G Metabolomic Spectral Libraries for Data-Independent SWATH Liquid Chromatography Mass Spectrometry Acquisition. Anal. Bioanal. Chem 2018, 410 (7), 1873–1884. 10.1007/s00216-018-0860-x. [DOI] [PubMed] [Google Scholar]
- (5).Barbier Saint Hilaire P; Rousseau K; Seyer A; Dechaumet S; Damont A; Junot C; Fenaille F Comparative Evaluation of Data Dependent and Data Independent Acquisition Workflows Implemented on an Orbitrap Fusion for Untargeted Metabolomics. Metabolites 2020, 10 (4), 158. 10.3390/metabo10040158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Blaženović I; Kind T; Ji J; Fiehn O Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8 (2), 31. 10.3390/metabo8020031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Tsugawa H Advances in Computational Metabolomics and Databases Deepen the Understanding of Metabolisms. Curr. Opin. Biotechnol 2018, 54, 10–17. 10.1016/j.copbio.2018.01.008. [DOI] [PubMed] [Google Scholar]
- (8).Narayanaswamy P; Teo G; Ow JR; Lau A; Kaldis P; Tate S; Choi H MetaboKit: A Comprehensive Data Extraction Tool for Untargeted Metabolomics. Mol. Omics 2020, 16 (5), 436–447. 10.1039/D0MO00030B. [DOI] [PubMed] [Google Scholar]
- (9).Junot C; Fenaille F; Colsch B; Bécher F High Resolution Mass Spectrometry Based Techniques at the Crossroads of Metabolic Pathways. Mass Spectrom. Rev 2014, 33 (6), 471–500. 10.1002/mas.21401. [DOI] [PubMed] [Google Scholar]
- (10).Colsch B; Fenaille F; Warnet A; Junot C; Tabet J-C Mechanisms Governing the Fragmentation of Glycerophospholipids Containing Choline and Ethanolamine Polar Head Groups. Eur. J. Mass Spectrom 2017, 23 (6), 427–444. 10.1177/1469066717731668. [DOI] [PubMed] [Google Scholar]
- (11).Demarque DP; Crotti AEM; Vessecchi R; Lopes JLC; Lopes NP Fragmentation Reactions Using Electrospray Ionization Mass Spectrometry: An Important Tool for the Structural Elucidation and Characterization of Synthetic and Natural Products. Nat. Prod. Rep 2016, 33 (3), 432–455. 10.1039/c5np00073d. [DOI] [PubMed] [Google Scholar]
- (12).Colsch B; Damont A; Junot C; Fenaille F; Tabet J-C Experimental Evidence That Electrospray-Produced Sodiated Lysophosphatidyl Ester Structures Exist Essentially as Protonated Salts. Eur. J. Mass Spectrom 2019, 25 (3), 333–338. 10.1177/1469066719838924. [DOI] [PubMed] [Google Scholar]
- (13).Attygalle AB; Chan C-C; Axe FU; Bolgar M Generation of Gas-Phase Sodiated Arenes Such as [(Na3(C6H4)+] from Benzene Dicarboxylate Salts. J. Mass Spectrom. JMS 2010, 45 (1), 72–81. 10.1002/jms.1690. [DOI] [PubMed] [Google Scholar]
- (14).Chan C-C; Axe FU; Bolgar M; Attygalle AB Reactivity of Gaseous Sodiated Ions Derived from Benzene Dicarboxylate Salts toward Residual Water in the Collision Gas. J. Mass Spectrom. JMS 2010, 45 (10), 1130–1138. 10.1002/jms.1792. [DOI] [PubMed] [Google Scholar]
- (15).Darii E; Alves S; Gimbert Y; Perret A; Tabet J-C Meaning and Consequence of the Coexistence of Competitive Hydrogen Bond/Salt Forms on the Dissociation Orientation of Non-Covalent Complexes. J. Chromatogr. B Analyt. Technol. Biomed. Life. Sci 2017, 1047, 45–58. 10.1016/j.jchromb.2016.09.038. [DOI] [PubMed] [Google Scholar]
- (16).Damont A; Olivier M-F; Warnet A; Lyan B; Pujos-Guillot E; Jamin EL; Debrauwer L; Bernillon S; Junot C; Tabet J-C; Fenaille F Proposal for a Chemically Consistent Way to Annotate Ions Arising from the Analysis of Reference Compounds under ESI Conditions: A Prerequisite to Proper Mass Spectral Database Constitution in Metabolomics. J. Mass Spectrom. JMS 2019, 54 (6), 567–582. 10.1002/jms.4372. [DOI] [PubMed] [Google Scholar]
- (17).Bartmess JE; Scott JA; McIver RT Scale of Acidities in the Gas Phase from Methanol to Phenol. J. Am. Chem. Soc 1979, 101 (20), 6046–6056. 10.1021/ja00514a030. [DOI] [Google Scholar]
- (18).Aron AT; Gentry EC; McPhail KL; Nothias L-F; Nothias-Esposito M; Bouslimani A; Petras D; Gauglitz JM; Sikora N; Vargas F; van der Hooft JJJ; Ernst M; Kang KB; Aceves CM; Caraballo-Rodríguez AM; Koester I; Weldon KC; Bertrand S; Roullier C; Sun K; Tehan RM; Boya P CA; Christian MH; Gutiérrez M; Ulloa AM; Tejeda Mora JA; Mojica-Flores R; Lakey-Beitia J; Vásquez-Chaves V; Zhang Y; Calderón AI; Tayler N; Keyzers RA; Tugizimana F; Ndlovu N; Aksenov AA; Jarmusch AK; Schmid R; Truman AW; Bandeira N; Wang M; Dorrestein PC Reproducible Molecular Networking of Untargeted Mass Spectrometry Data Using GNPS. Nat. Protoc 2020, 15 (6), 1954–1991. 10.1038/s41596-020-0317-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.