

Abstract
Antibiotic resistance has emerged as one of the biggest public health concerns all over the world. In an effort to combat bacterial infections, a series of imidazolidine-4-one derivatives with potent and broad-spectrum antibacterial activity and low rates of drug resistance were developed by mimicking the salient physiochemical features of host defense peptides. These small molecules displayed potent activity against both Gram-negative and Gram-positive bacteria including several multidrug-resistant bacteria strains. Meanwhile, time-kill kinetics and drug resistance studies suggested that the most potent compound 3 could not only eliminate the bacteria rapidly but also exhibit a low probability of drug resistance in MRSA over many passages. Further mechanistic studies suggested that 3 eradicated bacterial pathogens by disintegrating membranes of both Gram-negative and Gram-positive bacteria. Together with their small molecular weight and low production cost compared with HDPs, these imidazolidine-4-one compounds may be developed into a new generation of antibiotic therapeutics combating emergent drug resistance.
Graphical Abstract

INTRODUCTION
Infections caused by antibiotic-resistant bacteria in hospitals and the community continue to threaten the public health in the 21st century.1 It is estimated that 70,000 patients die due to antibacterial resistance every year.2 The alarmingly escalating resistance to conventional drugs developed by strains including methicillin-resistant Staphylococcus epidermidis (MRSE), methicillin-resistant Staphylococcus aureus (MRSA), Klebsiella pneumoniae (K. pneumoniae), and Escherichia coli (E. coli) continuously forces the exploration for new antibiotic agents.3 Antimicrobial peptides (AMPs), also known as host defense peptides (HDPs), are a part of the innate immune system, which protects a host from invading pathogens, and have attracted substantial interest in recent decades.4 These short cationic polypeptides appear in all species of complex life including plants, insects, crustaceans, mammals, and so on. For example, one of the most important HDPs, LL-37, consists of 37 amino acids. It is the sole member of the human cathelicidin family, and it has good antimicrobial activity.5 Interaction with bacterial membranes driven by hydrophobic and cationic groups in HDPs, which have no specific targets on cells, renders the emergence of resistance less probable than conventional antibiotics.6–8 Generally, a cationic charged domain and a proper ratio of hydrophobic residues are two common physical features of HDPs, endowing HDPs with considerable selectivity toward bacteria over mammalian cells. This is because the outer leaflet of mammalian cell membranes consists of zwitterionic lipids, while the outer leaflet of bacteria membranes is rich in negatively charged phospholipids. The cationic charge of HDPs promotes the selectivity to bacteria membranes over the mammalian cell membranes through electrostatic attraction, which makes HDPs have lower hemolysis risks and cytotoxicity.9,10 The hydrophobic patches facilitate association with the lipid layer of bacteria membranes through hydrophobic interaction.4 It should be noted that, although HDPs share this common feature of bacterial membrane activity, the detailed actions of HDPs on bacterial membranes could be diverse and complex. Several models were proposed, including molecular electroporation, barrel stave model, carpet model, and sinking raft model.11 Furthermore, membrane action is not the only mechanism of HDPs adopted to kill bacteria. Many of them are bacteria cell-permeable and have intracellular targets, which further helps them overcome the resistance through the combination of multiple bactericidal mechanims.12,13
Although some antibiotic peptides such as colistin, gramicidin, and daptomycin are being used in the clinic, their antibacterial mechanisms are different from those of HDPs, and as a result they generally exhibit a narrow spectrum of activities.14,15 The development of HDPs as potential antibiotics is still facing considerable challenges due to their intrinsic drawbacks, such as large size, expensive large-scale production, low bioavailability, susceptibility to proteolysis, toxicity, and so on.2,3,16,17 Significant effort has been devoted to countering these problems, one of which is to develop synthetic mimics of HDPs. Briefly, there are at least three kinds of synthetic mimics of HDPs: cationic polymers,18–21 peptidomimetic oligomers (include β-peptides,22 peptoids,23 oligoacyllysines,24 α-AA peptides,25 γ-AApeptides,26,27 and oligoureas28), or small molecules.3,17,29 Many of them have made great progress and success on clinical trials, e.g., brilacidin30,31 and ceragenins,32 delineating the promising future of HDPs.
Among different types, small molecules are of particular interest because they are more drug-like and easy to scale-up for practical applications. To continue our efforts to develop small-molecule HDP mimics to combat emerging antibiotic resistance,3,9,17,29 we decided to look into the derivatives of imidazolidine. The imidazolidine-4-one scaffold provides versatile opportunities for chemical modification, resulting in plenty of biological activities, including G-protein-coupled receptor (GPCR) antagonists,33 anticancer agents,34 and also antibacterial activity35,36 or antifungal functions.37,38 Among these applications, antibiotic activity is one of the most important research topics of imidazolidine derivatives.36,39 However, moderate antibiotic activity made them ineligible for clinical use. Herein, we introduced both cationic groups and hydrophobic groups onto the imidazolidine-4-one scaffold to mimic the amphipathic structures of HDPs through the strategy of dimerization, which had previously been demonstrated as a promising strategy to enhance the antibacterial activity.9,40
RESULTS AND DISCUSSION
Design and Synthesis of Mimics.
Previously, we designed and synthesized HDP-mimicking small molecules including bis-cyclic guanidines41 and dimeric lysine N-alkylamides,9 some of which demonstrated strong and broad-spectrum activity against a panel of Gram-negative and Gram-positive strains. In those scaffolds, the benzene ring and alkyl chains act as hydrophobic groups and guanidines and amino groups are cationic groups. Here, we discovered that bisimidazolidines could be conveniently obtained via the dimerization and cyclization of the γ-substituted-N-acylated-N-aminoethyl peptides (γ-AApeptide) building blocks (Scheme 1). It consists of one pair of symmetric hydrophobic groups and one pair of cationic groups. The hydrophobicity of compounds could be easily adjusted by conjugating different alkyl chlorides in the step of synthesizing the c intermediate (Scheme 1).
Scheme 1.

General Scheme for Synthesis of Dimeric Imidazolidine-4-one Derivatives 1–11
Synthesis of these molecules was quite straightforward. Briefly, building block a was coupled with benzene-1,4-diamine using 1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3-oxid hexafluorophosphate (HATU) as the coupling agent. After the Fmoc group was removed by diethylamine, alkyl chloride was added to react with the terminal primary amine to obtain c. Paraformaldehyde and K2CO3 were used to form the imidazolidine-4-one ring according to the method introduced previously.42 After HPLC purification, the Cbz protecting group of d was removed using Pd/C under a H2 atmosphere to give the final products. A series of molecules (1–11) bearing different hydrophobic and hydrophilic groups were synthesized using a similar method. The scheme of synthesizing compounds 12 and 13 is shown in the Experimental Section.
Antibacterial Activity and Selectivity.
After obtaining these compounds, their antibacterial activity was tested against a panel of Gram-positive and Gram-negative strains, including methicillin-resistant S. aureus (MRSA), methicillin-resistant S. epidermidis (MRSE), K. pneumoniae, and E. coli (Table 1). Interestingly, the antibacterial activity of these compounds demonstrates a decent relationship with hydrophobicity of the side chains. As shown in Table 1, 1 and 2 has no activity against any tested bacterial strains due to the deficient hydrophobicity. When the number of carbons in alkyl chains increased to 10 (compound 3), the activity improved dramatically, with MICs less than 6 μg/mL for all four bacterial strains. Increasing the alkyl tail length to 12 carbons led to compound 4, which exhibited the slightly better activity against Gram-positive strains MRSA (1 μg/mL) and MRSE (2 μg/mL). However, it was not active toward Gram-negative bacteria E. coli and K. pneumoniae. When the length of alkyl tails increased continuously to 14 and 16 (compounds 5 and 6, respectively), no antibacterial activity was observed. This suggested that carbons in the alkyl tails should not be over 12 when designing these imidazolidine derivatives. Obviously, antibacterial activity of compounds was relevant to their hydrophobicity, which could be estimated by Clog P values. Compounds 1–6 had gradually increasing Clog P values. When this value reached around 8.5, the activity appeared to be the best. However, hydrophobicity is not the only decisive factor, as the Clog P of compound 7 was also around 8.5 but it failed to show any bacteria inhibiting activity up to 25 μg/mL. We speculated that long alkyl tails are easier than bulky groups to insert into the lipid bilayer of bacterial membranes. The activity of compounds 12 and 13 also followed this rule. As expected, the replacement of 4-aminobutyl groups with methylphenyl groups (compound 10) abolished its activity due to the lack of cationic charge for initial static interaction with negatively charged bacterial membranes. Compound 9 was obtained by switching the position of the hydrophobic alkyl tails with hydrophilic amine groups. Unfortunately, it failed to show any activity. To examine whether the imidazolidine-4-one rings contribute to the antibacterial activity or not, compound 11 was designed and tested. Although 11 still displayed potent antibacterial activity against Gram-positive bacteria including both MRSA and MRSE, it was not active toward Gram-negative bacteria. This indicated that the imidazolidine-4 one ring was helpful in improving antimicrobial activity.
Table 1.
Structures of All Bis-cyclic Imidazolidine Derivatives and Their Antibacterial Activity
| Structure | MIC (μg/mL) | Hemolysis (HC50, μg/mL) | Selectivity (HC50/MIC of MRSA) | Clog P | ||||
|---|---|---|---|---|---|---|---|---|
| # | MRSA | MRSE | E. coli | K. pneumoniae | ||||
| 1 |

|
>25 | >25 | >25 | >25 | >250 | - | 4.51 |
| 2 |

|
>25 | >25 | >25 | >25 | >250 | - | 6.63 |
| 3 |

|
2 | 5 | 5 | 5 | >250 | >125 | 8.74 |
| 4 |
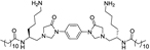
|
1 | 2 | >25 | >25 | >250 | >250 | 10.86 |
| 5 |
|
>25 | >25 | >25 | >25 | >250 | - | 12.97 |
| 6 |

|
>25 | >25 | >25 | >25 | >250 | - | 15.09 |
| 7 |
|
>25 | >25 | >25 | >25 | >250 | - | 8.91 |
| 8 |
|
>25 | >25 | >25 | >25 | >250 | - | 6.07 |
| 9 |
|
>25 | >25 | >25 | >25 | >250 | - | 4.05 |
| 10 |
|
>25 | >25 | >25 | >25 | >250 | - | 12.48 |
| 11 |
|
2 | 2 | >25 | >25 | >250 | >125 | 6.50 |
| 12 |

|
1 | 5 | >25 | 5 | 125 | 125 | 7.06 |
| 13 |
|
1 | 2 | >25 | >25 | 250 | 250 | 7.23 |
| 14 | Ciprofloxacin | 0.15 | 0.15 | 0.07 | 0.3 | - | - | |
All measurements were repeated three times in duplicate.
Methicillin-resistant S. aureus (MRSA) (ATCC 33591), Methicillin-resistant S. epidermidis (MRSE) (RP62A), E. coli (ATCC 25922), and K. pneumoniae (ATCC 13383). The compound with the most potent activity is shown in blue color.
To assess the selectivity of the compounds against mammalian blood cells, we subsequently studied the hemolytic activity of these compounds on mice blood cells. A higher HC50 value indicates higher selectivity and safer application to mammals. As shown in Table 1, the HC50 values of most structures have exceeded the testing range (up to 250 μg/mL), which was much higher than MICs, suggesting that these compounds have excellent selectivity toward bacteria. Interestingly, compounds 12 and 13 show lower HC50 values, which are 125 and 250 μg/mL, respectively. This somehow may suggest that guanidine groups are likely to induce more hemolysis than amino groups. It is notable that the HC50 of compound 3 was >125-fold higher than its MIC for MRSA. Following that, we tested the cytotoxicity of compound 3 against HeLa cells (Figure 1). The result shows that the IC50 of compound 3 was 74.83 μg/mL, >38-fold selectivity toward MRSA, which further confirmed its excellent selectivity and therapeutic potential.
Figure 1.

MTT assay of compound 3 against HeLa cells.
Salt Sensitivity.
Physiological conditions for medical applications are usually with high salt concentration.43 Therefore, investigation of the effectiveness of cations on the antibacterial activity against four strains was conducted. As shown in Table 2, monovalent (Na+, K+) and divalent cations (2 mM Ca2+) were added into tryptic soybean broth (TSB) medium. 1 × PBS was applied to make TSB medium with Na+ and K+. The result indicated that the addition of these cations had a slight influence on the antibacterial activity of compound 3, which made it promising to be applied in vivo.
Table 2.
Effects of Physiological Salts on the Antibacterial Activities of Compound 3
| MIC (μg/mL) |
||||
|---|---|---|---|---|
| cations | MRSA | MRSE | E. coli | K. pneumoniae |
| Na+, K+ | 4 | 7 | 5 | 7 |
| Ca2+ | 2 | 2 | 7 | 5 |
Bactericidal Time-Kill Kinetics.
It is well known that HDPs could kill bacteria rapidly.44 Since 3 was designed to mimic the mode of action of HDPs, we conducted time-kill studies to explore the kinetics of compound 3 on killing MRSA and E. coli at a range of concentrations. As shown in Figure 2, 3 could thoroughly eliminate MRSA within 60 min at concentrations of 24 and 48 μg/mL. Only 30 min was required at the concentrations of 24 and 48 μg/mL for eradicating E. coli, which was faster than MRSA. This observation simply showed that compound 3 was effective on killing these bacteria.
Figure 2.

Time-kill curves of 3 against (a) E. coli and (b) MRSA. Negative controls were untreated bacteria. The killing activity was monitored for 2 h. (a) Drug concentrations were 12, 24, and 48 μg/mL. (b) Drug concentrations were 6, 12, 24, and 48 μg/mL.
Mechanistic Studies.
Fluorescence Microscopy.
Since our compounds were designed based on the principle of HDPs, we speculated that the same antibacterial mechanism of HDPs was involved here. First, we applied dyes 4',6-diamidino-2-phenylindole (DAPI) and propidium iodide (PI) to conduct the fluorescence microscopy assay. As it is known, DAPI is generally used to stain cells irrespective to their viability while PI could only stain the dead or injured cells. MRSA and E. coli were treated with 3 and then incubated with PI and DAPI. As shown in Figure 3, negative controls without the treatment of 3 only have blue fluorescence, suggesting the intact membrane of MRSA and E. coli. By contrast, when both bacteria strains were treated with 3 for 2 hours, blue and red fluorescence were all observed, demonstrating that compound 3 has disintegrated the membranes.
Figure 3.

Fluorescence micrographs of E. coli and MRSA treated with 3 at 2 × MIC for 2 h: (a1-a4) MRSA; (a1) control, no treatment, DAPI channel; (a2) control, no treatment, PI channel; (a3) 3-treated MRSA, DAPI channel; (a4) 3-treated MRSA, PI channel,; (b1-b4) E. coli; (b1) control, no treatment, DAPI channel; (b2) control, no treatment, PI channel; (b3) 3-treated E. coli, DAPI channel; (b4) 3-treated E. coli, PI channel. All images were acquired using a 10×/22 eyepiece lens and a 100×/1.25 objective lens. Scale bar = 10 μm.
Membrane Depolarization Study.
To further characterize the antibacterial mechanism of molecule 3, we conducted a membrane depolarization study using DiSC3(5) as a potentiometric probe. DiSC3(5) could accumulate into the living bacteria membrane, leading to a weak fluorescence intensity due to self-quenching. However, after the integrity of the membrane is interrupted, the fluorescence intensity would increase sharply due to the release of the fluorophore. As shown in Figure 4, when E. coli was treated with compound 3 at concentrations of 0.5 × MIC, 1 × MIC, 2 × MIC, and 4 × MIC after incubating 30 min. Different amounts of the fluorescence intensity increase in a concentration-dependent manner were observed, while the negative control kept decreasing. A decreasing trend of negative control may be because the dye kept accumulating into bacteria cells. 1% Triton X-100 was applied as a positive control. This result demonstrated that the bacterial membranes were disrupted by compound 3.
Figure 4.

Membrane depolarization of compound 3 against E. coli using DiSC3(5) as the dye. The negative control was bacteria without drug treatment. Positive control was bacteria treated with 1% Triton X-100.
Inner Membrane (IM) Permeabilizaton.
Propidium iodide (PI) is a dye that will fluoresces upon binding to the cellular DNA.35,45 An increase in normalized fluorescence will be detected once the bacterial membrane is compromised when being incubated with the PI dye. Thus, we applied this assay to confirm the inner membrane interruption ability of compound 3. As shown in Figure 5, the cell membranes of both E. coli and MRSA were permeabilized fast by compound 3 at a 2-fold MIC value, while negative controls had no change. The increasing intensity of MRSA was lower than E. coli. This difference is correlated with the result of the time-kill study, which exhibited that E. coli was being killed faster than MRSA.
Figure 5.

Inner membrane permeabilization ability of compound 3 against MRSA and E. coli.
TEM Study.
Transmission electron microscopy (TEM) is a direct visualization method to observe the cell membrane morphology. The effect of 3 on the MRSA and E. coli membrane was observed by this technology (Figure 6). In Figure 6A,B, the membrane of bacteria without drug treatment was intact. After being treated with 3 (Figure 6C,D), the number of bacteria was less and their membrane was all dispersed, implying the membrane disruptive ability of compound 3 was strong.
Figure 6.

Transmission electron micrographs of cell membrane damage. (A) MRSA cells without antibacterial treatment; (B) E. coli cells without antibacterial treatment; (C) MRSA cells after being treated with 3 at 6 μg/mL; (D) E. coli cells after being treated with 3 at 12 μg/mL.
Drug Resistance Study.
The low propensity to induce drug resistance is one of the most appealing features of HDPs. Therefore, we conducted the drug resistance study of compound 3 against MRSA. After bacteria were incubated in 13 passages, the MIC of 3 remained almost unchanged, which suggested that 3 had the minimum propensity to induce bacterial resistance. In contrast, norfloxacin, which belongs to the fluoroquinolone family of antibiotics, was shown to induce drug resistance against MRSA rapidly, exhibiting a more than 22-fold increase in the MIC (Figure 7).
Figure 7.

Fold increase in MIC of compound 3 and norfloxacin toward MRSA.
Biofilm Prevention.
The National Institutes of Health (NIH) revealed that among all microbial and chronic infections, 65 and 80%, respectively, are associated with biofilm formation.46 Biofilm formation is involved in a large part of bacterial infections, and the biofilm matrix polymers make the natural and chemical agents hard to adequately attack and destroy infectious biofilm populations.47,48 Thus, new agents preventing biofilm formation are needed to reverse this situation. We evaluated the ability of compound 3 to inhibit the biofilm formation of E. coli. As shown in Figure 8, more than 55% biofilm formation of E. coli could be inhibited at a concentration of 3 μg/mL. Ciprofloxacin was included as the positive control here.
Figure 8.

Biological activity of compound 3 and ciprofloxacin on E. coli biofilm formation.
CONCLUSIONS
In summary, a series of bis-cyclic imidazolidine-4-one derivatives were rationally designed and synthesized. Among them, the lead compound 3 was discovered, which displayed strong activity with low toxicity and rapid action against both Gram-positive and Gram-negative strains including MRSA, MRSE, E. coli, and K. pneumoniae. Mechanistic studies indicated that these compounds kill bacteria through disruption of the cell membranes, analogous to that of antibacterial HDPs. Furthermore, the antibiotic activity of 3 was unchanged during 13 passages and the biofilm could also be inhibited at low drug concentration. Therefore, our design of bis-cyclic imidazolidine derivatives would shed light on developing new potent small-molecule antibacterial agents to fight against drug resistance. Further optimization of activity and in vivo studies with compound 3 is undergoing in our lab.
EXPERIMENTAL SECTION
General Information.
Solvents and other reagents were purchased from either Sigma-Aldrich or Fisher Scientific and were used without further purification. All reactions were monitored by thin-layer chromatography. Visualization was accomplished using a UV (254 nm) lamp. Column chromatography was carried out with silica gel (200–300 mesh). The final products were purified on a Waters Breeze 2 HPLC system and lyophilized on a Labconco lyophilizer. The purity of the compounds was determined to be >95% by analytical HPLC (1 mL/min flow, 5–100% linear gradient of solvent B (0.1% TFA in acetonitrile) in A (0.1% TFA in water) over 40 min). The NMR spectra were obtained on a Varian Inova 400 instrument. High-resolution mass spectra of compounds were identified using an Agilent Technologies 6540 UHD accurate-mass Q-TOF LC/MS spectrometer.
General Synthesis of Compounds 1–11.
Starting structure a and phenylalanine building block ((S)-N-(2-((((9H-fluoren-9-yl)-methoxy)carbonyl)amino)-3-phenylpropyl)-N-((allyloxy)carbonyl)-glycine) were synthesized in a method reported before.49,50 The synthesis of compound 3 is shown below. Other compounds were prepared using a similar synthesis method. To a solution of a (350 mg, 0.54 mmol) and N,N-diisopropylethylamine (187 μL, 1.08 mmol) in DMF (15 mL), HATU (413 mg, 1.08 mmol) was added. Then, p-phenylenediamine (29.3 mg, 0.27 mmol) was added into the solution. After being stirred at room temperature for 2 h, the mixture was acidified with 1 M HCl (50 mL). Ethyl acetate (15 mL × 3) was used to extract the product. The combined EA solution was washed with brine three times, dried over Na2SO4, and filtered. After the solution was removed using a rotavapor, the residue was purified by column chromatography (hexane/ethyl acetate = 1:1) to afford the b (315 mg, yield of 85%). b (315 mg, 0.23 mmol) was added to CH3CN/ ethylenediamine (1:1) solution and allowed to stir for 30 min. The solvent was evaporated, and then cold ether was added to precipitate the product, which was dissolved in CH2Cl2 (20 mL) and used directly without further purification. To this solution was added diisopropylethylamine (402 μL, 2.3 mmol) followed by decanoyl chloride (220 μL, 1.15 mmol), which was added dropwise. After stirring for 2 h, the solution was washed with 1 M HCl (15 mL × 1) and brine and dried with Na2SO2 to afford crude c, which was treated with 1:1 CH2Cl2/TFA at room temperature for 1 h. The solution was then removed using the rotavapor. The residue (30 mg, 0.026 mmol) was dissolved in 15 mL of MeOH solution, and then paraformaldehyde (3.1 mg, 0.104 mmol) and K2CO3 (14.4 mg, 0.10 mmol) were added. The mixtures were heated to reflux for 5 h. MeOH was removed by vacuum, and the residue was washed with 1 M HCl and brine and dried over Na2SO4 to provide d, which was subjected to hydrogenation in the presence of 10% wet Pd/C under a hydrogen atmosphere for 2 h in MeOH. After the reaction is completed, Pd/C was filtered and MeOH was evaporated. The residue was purified by HPLC to afford compound 3 (28 mg, yield of 16%) as a white solid with 99% purity.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))-bis(6-aminohexane-1,2-diyl))dihexan amide (1).
The title compound was prepared from 260 mg (0.40 mmol) of a, 21.8 mg (0.2 mmol) of p-phenylenediamine, and 137 μL (1.0 mmol) of hexanoyl chloride according to the general procedure affording 35 mg of product 1 in 26% yield as a white solid with 97% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.66 (s, 4H), δ 7.60 (m, 6H), δ 4.54–4.60 (dd, J = 16 Hz, 4H), δ 3.86 (s, 2H), δ 3.37–3.47 (q, J = 40 Hz, 4H), δ 2.73–2.75 (m, 4H), δ 2.58–2.60 (m, 4H), δ 2.03–2.07 (t, J = 16 Hz, 4H), δ 1.45–1.41 (m, 10H), δ 1.23–1.35 (m, 14H), 0.79–0.82 (t, J = 12 Hz, 6H). HRMS (ESI) C36H62N8O4 [M+ H]+ calcd = 671.4972. Found = 671.4930.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(6-aminohexane-1,2-diyl))dioctan amide (2).
The title compound was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 130 μL (0.8 mmol) of octanoyl chloride according to the general procedure affording 21 mg of product 2 in 19% yield as a white solid with 95% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.64 (s, 4H), δ 7.55 (s, 6H), δ 4.52–4.56 (d, J = 16 Hz, 4H), δ 3.34–3.41 (m, 6H), δ 2.73 (s, 4H), δ 2.57 (s, 4H), δ 2.01–2.24 (t, J = 12 Hz, 4H), δ 1.45 (m, 10H), δ 1.18–1.28 (m, 22H), δ 0.78 (m, 6H). HRMS (ESI) C40H70N8O4 [M+ H]+ calcd = 727.5598. Found = 727.5599.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(6-aminohexane-1,2-diyl))bis(decanamide) (3).
The synthesis of compound 3 is shown above as an example in the general procedure. The product (28 mg) was obtained in 16% yield as a white solid with 99% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.63 (s, 4H), δ 7.58 (s, 2H), δ 7.55 (s, 4H), δ 4.52–4.56 (d, J = 16 Hz, 4H), δ 3.82 (s, 2H), δ 3.37 (s, 4H), δ 2.71–2.72 (d, J = 4 Hz, 4H), δ 2.56 (s, 4H), δ 2.01–2.04 (t, J = 12 Hz, 4H), δ 1.45–1.49 (m, 12H), δ 1.05–1.32 (m, 28H), δ 0.78–0.81 (t, J = 12 Hz,6H). HRMS (ESI) C44H78N8O4 [M+ H]+ calcd = 783.6224. Found =783.6242.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(6-aminohexane-1,2-diyl))didodecanamide (4).
The title compound was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 175 μL (0.8 mmol) of dodecanoyl chloride according to the general procedure affording 28 mg of product 4 in 21% yield as a white solid with 99% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.63 (s, 4H), δ 7.55–7.60 (m, 6H), δ 4.52–4.57 (d, J = 20 Hz, 4H), δ 3.83 (s, 4H), δ 3.37–3.41(m, 4H), δ 2.71–2.73 (d, J = 8 Hz, 4H), δ 2.58 (s, 4H), δ 2.01–2.05 (m, 4H), δ 1.45–1.47 (m, 10H), δ 1.26–1.30 (m, 36H), δ 0.80–0.83 (t, J = 12 Hz, 6H). HRMS (ESI) C48H86N8O4 [M + H]+ calcd = 839.6850. Found [M + 2H]/2 = 420.3471.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(6-aminohexane-1,2-diyl))ditetradecanamide (5).
Compound 5 was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 197.4 mg (0.8 mmol) of tetradecanoyl chloride according to the general procedure affording 26 mg of product 5 in 18.7% yield as a white solid with 99% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.70 (s, 4H), δ 7.58–7.60 (d, J = 8 Hz, 2H), δ 7.55 (s, 4H), δ 4.52–4.57 (d, J = 20 Hz, 4H), δ 3.33–3.41(m, 6H), δ 2.69–2.74 (q, J = 20 Hz, 4H), δ 2.58 (s, 4H), δ 2.00–2.04 (t, J = 16, 4H), δ 1.44–1.48 (m, 10H), δ 1.15–1.17 (m, 46H), δ 0.79–0.82 (t, J = 12 Hz, 6H). HRMS (ESI) C52H94N8O4 [M + H]+ calcd = 895.7476. Found [M + H] = 895.7477.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(6-aminohexane-1,2-diyl))dipalmitamide (6).
The title compound was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 220 mg (0.8 mmol) of palmitoyl chloride according to the general procedure affording 30 mg of product 6 in 20.4% yield as a white solid with 99% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.68 (s, 4H), δ 7.58–7.60 (d, J = 8 Hz, 2H), δ 7.55 (s, 4H), δ 4.51–4.61 (dd, J = 40 Hz, 4H), δ 3.34–3.46 (m, 6H), δ 2.71–2.74 (d, J = 12 Hz, 4H), δ 2.59 (s, 4H), δ 2.00–2.04 (t, J = 16, 4H), δ 1.44–1.49 (m, 10H), δ 1.15–1.18 (m, 54H), δ 0.79–0.83 (t, J = 16 Hz, 6H). HRMS (ESI) C56H102N8O4 [M + H]+ calcd = 951.8102 Found [M + H] = 951.8089.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))-bis(6-aminohexane-1,2-diyl))bis(2-((3S,5S,7S)-adamantan-1-yl)- acetamide) (7).
The title compound was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 170 μL (0.8 mmol) of 2-((3R,5R,7R)-adamantan-1-yl)acetyl chloride according to the general procedure affording 16 mg of product 7 in 12% yield as a white solid with 98% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.62 (s, 4H), δ 7.54 (s, 4H), δ 7.43–7.45 (d, J = 8 Hz, 2H), δ 4.49–4.59 (dd, J = 36 Hz, 4H), δ 3.85 (s, 2H), δ 3.32–3.48 (dd, J = 64 Hz, 4H), δ 2.72–2.73 (d, J = 4 Hz, 4H), δ 2.56 (s, 4H), δ 1.76–1.82 (m, 10H), δ 1.51–1.55 (m, 32H), δ 1.29–1.34 (m, 4H). HRMS (ESI) C48H74N8O4 [M+ H]+ calcd = 827.5911. Found [M + H]+ = 827.5872.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))-bis(6-aminohexane-1,2-diyl))bis(2-(naphthalen-2-yl)acetamide) (8).
The title compound was prepared from 200 mg (0.31 mmol) of a, 17 mg (0.16 mmol) of p-phenylenediamine, and 16.4 mg (0.8 mmol) of 2-(naphthalen-2-yl)acetyl chloride according to the general procedure affording 15 mg of product 8 in 11.9% yield as a white solid with 97% purity. 1H NMR (400 MHz, DMSO-d6) δ 8.01–8.03 (d, J = 8 Hz, 2H), δ 7.73–7.80 (m, 7H), δ 7.46 (s, 4H), δ 7.45 (s, 4H), δ 7.36–7.42 (m, 7H), δ 4.49–4.55 (dd, J = 24 Hz, 4H), δ 3.85 (s, 2H), δ 3.57–3.58(m, 4H), δ 3.36–3.40 (m, 4H), δ 2.63–2.71 (m, 8H), δ 1.45–1.54 (m, 6H), δ 1.20–1.36 (m, 6H). HRMS (ESI) C48H58N8O4 [M + H]+ calcd = 811.4659. Found [M + H] = 811.4653.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(3-phenylpropane-1,2-diyl))bis(4-aminobutanamide) (9).
The title compound was prepared from 200 mg (0.38 mmol) of phenylalanine building block, 17 mg (0.19 mmol) of p-phenylenediamine, and 251 mg (0.98 mmol) of benzyl (4-chloro-4-oxobutyl)-carbamate according to the general procedure affording 21 mg of product 9 in 11.8% yield as a white solid with 99.9% purity. 1H NMR (400 MHz, DMSO-d6) δ 7.83–7.85 (d, J = 8 Hz, 2H), δ 7.67 (s, 4H), δ 7.57 (s, 4H), δ 7.24–7.26 (m, 5H), δ 7.15–7.19 (m, 5H), δ 4.57–4.61 (dd, J = 16 Hz, 4H), δ 4.07–4.09 (m, 2H), δ 3.44–3.46 (m, 4H), δ 2.83–2.84 (d, J = 4 Hz, 2H), δ 2.47–2.68 (m, 10H), δ 2.04–2.08 (m, 4H), δ 1.61–1.64 (m, 4H). HRMS (ESI) C38H50N8O4 [M + H]+ calcd = 683.4033. Found [M + H]+ = 683.4028.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))- bis(3-phenylpropane-1,2-diyl))bis(decanamide) (10).
The title compound was prepared from 200 mg (0.38 mmol) of phenylalanine building block 20 mg (0.19 mmol) of p-phenylenediamine, and 170 μL (0.95 mmol) of decanoic acid according to the general procedure affording 39 mg of product 10 in 25% yield as a white solid with 99% purity.1H NMR(400 MHz, CDCl3) δ 7.52 (s, 4H), δ 7.30–7.33 (m, 5H), δ 7.17–7.21 (m, 5H), δ 5.51–5.53 (d, J = 8 Hz, 2H), δ 4.63–4.66 (d, J = 12 Hz, 2H), δ 4.57–4.58 (d, J = 4 Hz, 2H), δ 4.30–4.35 (q, J = 20 Hz, 2H), δ 3.54 (s, 4H), δ 2.92–2.93 (d, J = 4 Hz, 4H), δ 2.13–2.16 (t, J = 12 Hz, 4H), δ 2.12–2.16 (m, 4H), δ 1.53–1.57 (t, J = 16 Hz, 4H), δ 1.19–1.28 (m, 24H), δ 0.85–0.88 (t, J = 12 Hz, 6H). HRMS (ESI) C50H72N6O4 [M + 2H]/2+ calcd = 411.2886. Found [M + 2H]/2+ = 411.2889.
N,N'-((2S,2'S)-(((1,4-Phenylenebis(azanediyl))bis(2-oxoethane-2,1-diyl))bis(azanediyl))bis(6-aminohexane-1,2-diyl))bis- (decanamide) (11).
Intermediate c was prepared from 200 mg (0.38 mmol) of a, 20 mg (0.19 mmol) of p-phenylenediamine, and 170 μL (0.95 mmol) of decanoic acid according to the general procedure. Then, a mixture of DCM/trifluoroacetic acid (1:1) was applied to cleave the Boc protecting group. After being purified with HPLC, Pd/ C in a H2 atmosphere was applied to deprotect the Cbz group. Compound 11 (29 mg) in 20% yield as a white solid with 99% purity was obtained. 1H NMR (400 MHz, DMSO-d6) δ 10.54 (s, 2H), δ 8.91 (s, 1H), δ 8.78 (s, 1H), δ 7.73–7.78 (m, 4H), δ 7.53 (s, 4H), δ 4.01 (s, 2H), δ 3.88 (m, 4H), δ 3.05 (m, 2H), δ 2.92 (m, 2H), δ 2.73 (m, 4H), δ 2.07–2.10 (t, J = 12 Hz, 4H), δ 1.438–1.48 (m, 10H), δ 1.17–1.21 (m, 24H), δ 0.80–0.83 (t, J = 12 Hz, 6H). HRMS (ESI) C38H70N8O4 [M + H]+ calcd = 759.6224. Found [M + H]+ = 759.6231.
Synthesis of Compound 12.
Compound 3 (17 mg, 0.022 mmol), 17 μL (0.13 mmol) of triethylamine, and 13 mg (0.046 mmol) of 1,3-bis(tert-butoxycarbonyl)-2-methyl-2-thiopseudourea were added into a 25 mL flask with 7 mL of DMF at 0 °C(Scheme 2). After stirring for 10 min, 12 mg (0.046 mmol) of HgCl2 was added into the above solution. The reaction was stopped after 2 h. Ethyl acetate (50 mL) was used to dilute the reaction solution, and then washed with 50 mL of water (2×) and 50 mL of brine (2×). Then, after being dried over Na2SO4 and evaporation of ethyl acetate in vacuo, DCM/TFA (1:1) was used to cleave the Boc protecting group. HPLC was used to purify the final product. The product (16 mg) was obtained in 84% yield as a white solid with 97% purity. Compound 14 was obtained using exactly the same method in 88% yield as a white solid, 99% purity.
Scheme 2.

General Synthesis Method of Compounds 12 and 13
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))-bis(6-guanidinohexane-1,2-diyl))bis(decanamide) (12).
1H NMR (400 MHz, DMSO-d6) δ 7.51–7.60 (m, 8H), δ 4.52–4.58 (dd, J =4 Hz, 4H), δ 4.00 (m, 6H), δ 3.84 (m, 2H), δ 3.34–3.45 (q, J = 44 Hz, 4H), δ 3.0–3.04 (q, J = 16 Hz, 4H), δ 2.58 (m, 4H), δ 2.01–2.06 (t, J = 20 Hz, 4H), δ 2.44 (m, 12H), δ 1.16 (m, 28H), δ 0.78–0.82 (t, J = 16 Hz, 6H). HRMS (ESI) C46H82N12O4 [M + H]+ calcd = 867.6660. Found [M + H]+ = 867.6648.
N,N'-((2S,2'S)-(1,4-Phenylenebis(4-oxoimidazolidine-3,1-diyl))-bis(6-guanidinohexane-1,2-diyl))bis(2-((3S,5S,7S)-adamantan-1-yl)acetamide) (13).
1H NMR (400 MHz, DMSO-d6) δ 7.52 (s, 4H), δ 7.41–7.43 (m, 4H), δ 4.55–4.56 (d, J = 4 Hz, 2H), δ 4.47–4.48 (d, J = 4 Hz, 2H), δ 3.69 (m, 6H), δ 3.42–3.45 (d, J = 12 Hz, 2H), δ 3.30–3.33 (d, J = 12 Hz, 2H), δ 2.99–3.03 (q, J = 16 Hz, 4H), δ 2.53–2.58 (m, 4H), δ 1.73–1.79 (m, 10H), δ 1.40–1.55 (m, 32H), δ 1.19–1.31 (m, 6H). HRMS (ESI) C50H78N12O4 [M + H]+ calcd = 911.6347. Found [M + H]+ = 911.6334.
Minimum Inhibitory Concentrations (MICs) against Bacteria.
The antimicrobial assay of the compounds was conducted on the following four bacteria strains: K. pneumoniae (ATCC 13383), E. coli (ATCC 25922), methicillin-resistant S. aureus (MRSA, ATCC 33591), and methicillin-resistant S. epidermidis (MRSE, RP62A). The procedures were followed as reported previously.51 Compound samples were diluted with TSB medium from 5 mg/mL DMSO solution to 100 μg/mL. First, bacteria strains were incubated in TSB for 16 h. Then, 100 μL of suspension was transferred to 4 mL of fresh medium and incubated for another 6 h to reach the log phase stage. After that, bacteria were diluted to reach 106 colony-forming units per milliliter (CFU/mL) according to the OD600 value. Different drug concentrations were obtained by series dilution in TSB. The highest drug concentration was 25 μg/mL. The MICs were determined as the lowest concentration that completely inhibits the bacteria growth. All measurements were repeated at least three times in duplicate each time.
Hemolytic Assays.
Compounds at a concentration of 1 mg/mL were 2-fold serially diluted in PBS, and 50 μL of each was added to 96-well plates. Fresh red blood cells of mice were collected by centrifugation at a speed of 500–700g for 10 min. Following that, PBS was used to wash the red blood cells with the same method three times. After addition of 20-fold volume PBS to washed red blood cells, 50 μL of the resulting solution was added to each vial containing the serially diluted compound and incubated at 37 °C for 1 h. 96-well plates were centrifuged at 3500 rpm for 10 min, and then 30 μL of the suspension was taken out and transferred to another 96-well plate containing 100 μL of PBS in each vial. The absorbance of the mixture was read at 540 nm using a Biotek Synergy HT plate reader. The negative control was the PBS buffer not treated with the drug, and the positive control was the PBS buffer treated with 2% Triton. The hemolysis activity was calculated by the formula % hemolysis = [(Abssample - AbsPBS)/(AbsTriton - AbsPBS)] × 100. The experiment was repeated at least three times in duplicate each time.
MTT Assays.
HeLa cells in good condition were seeded into a 96-well plate at a concentration of 5 × 103 cells/well in 100 μL of complete growth medium. After 24 h of attachment at 37 °C and 5% CO2, the growth medium was removed and 100 μL of growth medium containing various concentrations of compound 3 was applied. Then, the cells were incubated at 37 °C and 5% CO2. Cell viability was monitored after 24 h using a cell counting Kit-8 (CCK-8). For each well, 10 μL of CCK-8 solution was added. Absorbance at 490 nm of the whole solution was measured after 1 h in the incubator. Experiments were performed in triplicate and repeated three times.
Salt Sensitivity Assay.
MIC values were tested by the same method as mentioned before, except for the bacteria incubating medium. 1 × PBS (250 mL) was used to dissolve 7.5 g of TSB powder under high-temperature and high-pressure sterilization conditions to obtain the medium with Na+ and K+. CaCl2 TSB medium (2 mM) was obtained by adding CaCl2 into the TSB medium, which was made of DI water and TSB powder.
Time-Kill Study.
Time–kill kinetics studies of compound 3 were performed to determine the antibacterial efficiency. After being incubated in 4 mL of TSB medium overnight at 37 °C while shaking, 100 μL of bacterial solution was transferred into 4 mL of fresh medium and incubated for 6 h in the same condition to obtain the mid-log phase bacteria. MRSA or E. coli suspension (106 CFU/mL) was made and then incubated with different concentrations of 3 at 37 °C for 10, 30, 60, and 120 min. The MRSA and drug mixture were diluted 102-fold and then spread on the TSB agar plates. E. coli and the drug mixture were diluted 104-fold. These plates were incubated at 37 °C for 20 h. The numbers of colonies on each plate were counted and plotted against the incubation time. The experiment was repeated three times.
Fluorescence Microscopy.
Fluorescent dyes propidium iodide (PI) and 4',6'-diamidino-2-phenylindole dihydrochloride (DAPI) can be used to distinguish the viable cells from the dead cells. 106 CFU/ mL bacteria solution was diluted 100 times and mixed with compound 3 at a concentration of 2 × MIC. Then, the mixture was incubated for 2 h and centrifuged at 4000g for 15 min at 4 °C to harvest the cells. After washing with PBS three times, PI (5 μg/mL) and DAPI (10 μg/mL) were used to dye cells for 15 min. Controls were carried out without compounds. The bacterial cells were then visualized and analyzed by employing a Zeiss Axio imager Z1 optical microscope with an oil-immersion objective. The experiment was repeated three times in duplicate each time.
Membrane Depolarization Study.
Mid-log phase E. coli bacteria solution was obtained as above. Then, cells were collected by centrifugation and washed with 5 mM HEPES and 5 mM glucose. HEPES(5 mM)/5 mM glucose/100 mM KCl (1:1:1) was used to resuspend bacteria cells until the OD value at 600 nm was around 0.05. Following that, 192 μL of bacterial suspension and 8 μL of 10 μM DiSC3(5) were added into a 96-well plate. The fluorescence of the suspension was monitored at room temperature for 30 min at an excitation wavelength of 622 nm and emission wavelength of 670 nm. After 30 min, compound 3 was added into the wells and the increased potential was monitored. The concentration of compound 3 in each well should be 0.5 × MIC, 1 × MIC, 2 × MIC, and 4 × MIC. Triton X-100 (1%) was applied as the positive control. The experiment was repeated three times in duplicate each time.
Inner Membrane (IM) Permeabilization.
The experiment followed the method reported before.52 Mid-log phase MRSA and E. coli bacteria solution was obtained as above. Then, cells were collected by centrifugation at a speed of 1000g. HEPES(5 mM)/5 mM glucose (1:1) solution was used to wash the bacteria three times and suspend them to 106 CFU/mL. Bacteria solution (150 μL) and 50 μL of 10 μM PI was mixed in 96-well plates. Fluorescence was measured for about 8 min at 2 min intervals at an excitation wavelength of 535 nm and emission wavelength of 617 nm. Then, compound 3 was added and continuously read for 12 min. The negative control with no drug treatment was added the identical amount of water. The experiment was repeated three times.
Drug Resistance Study.
The first MIC value was measured following the same method as the MIC assay mentioned before. Then, the well suspended with bacteria after the last clear well was employed to make the bacterial suspension (approximately 106 CFU/mL) for the next experiment to measure new MIC until it was repeated for 13 passages.
Biofilm Prevention Study.
E. coli was first incubated at 37 °C overnight in TSB medium. Then, 100 μL of bacterial solution was transferred into 4 mL of fresh medium and incubated for 6 h under the same condition to obtain the mid-log phase bacteria. After that, compound 3, bacteria, and medium were all added into a 96-well plate for the MIC assay. After incubating for 2 days, suspended E. coli was dumped and washed with DI water. Crystal violet solution (200 μL, 0.1%) was added into each well to stain for 15 min. Then, the biomass was washed with DI water until water became colorless. After drying, 200 μL of 30% acetic acid solution was added into each vial and left to stand for 15 min. The solution in each vial was mixed, and then 125 μL of solution was transferred into another 96-well plate. The OD value was read at 595 nm. Relative biofilm biomass values were normalized by the biomass value of the control (no addition of compounds). Experiments were conducted in triplicate, and the data are presented as mean ± STDEV.
TEM Study.
MRSA and E. coli (106 CFU/mL) were obtained as shown in MIC studies. Then, both strains were mixed with compound 3 at a concentration of 2 × MIC for 2 h at 37 °C. The bacteria cells were first collected by centrifugation and then washed with PBS three times. Following that, the bacteria were resuspended in deionized water. Control bacterial samples were obtained without adding compound 3. Prepared samples were dropped on the surface of grids. Extra solution was absorbed slightly by filter paper. The grids were dried in a vacuum oven at a temperature of 50 °C for 30 s. TEM images were obtained on a FEI Morgagni 268D TEM with an Olympus MegaView III camera on the microscope. The microscope uses Analysis software to run the camera. The microscope was operated at 60 kV.
Supplementary Material
ACKNOWLEDGMENTS
This work was generously supported by NSF 1708500 (J.C.), NIH 5R01AI149852 (J.C.), and NIH 9R01AI152416–06 (J.C.). We also want to thank Tao Xin for his valuable discussion.
ABBREVIATION USED
- HDPs
host defense peptides
- MRSE
Staphylococcus epidermidis
- K. pneumoniae
Klebsiella pneumoniae
- E. coli
Escherichia coli
- AMPs
antimicrobial peptides
- α-AA peptides
α-substituted-N-acylated-N-aminoethyl peptides
- γ-AA peptides
γ-substituted-N-acylated-N-aminoethyl peptides
- HATU
1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]-pyridinium-3-oxide hexafluorophosphate
- Cbz
carboxybenzyl
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- DAPI
4',6-diamidino-2-phenylindole
- PI
propidium iodide
- DiSC3(5)
3,3'-dipropylthiadicarbocyanine iodide
- NIH
National Institutes of Health
- TEM
transmission electron microscopy
- TEA
triethylamine
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.0c00171.
Characterization and purities of compounds (PDF)
Molecular formula strings and some data (CSV)
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.jmedchem.0c00171
Contributor Information
Minghui Wang, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Ruixuan Gao, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Mengmeng Zheng, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Peng Sang, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
Chunpu Li, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States; Department of Medical Oncology, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
En Zhang, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States; School of Pharmaceutical Sciences, Institute of Drug Discovery and Development, Key Laboratory of Advanced Pharmaceutical Technology (Ministry of Education), Zhengzhou University, Zhengzhou 450001, PR China.
Qi Li, Department of Medical Oncology, Shuguang Hospital, Shanghai University of Traditional Chinese Medicine, Shanghai 201203, China.
Jianfeng Cai, Department of Chemistry, University of South Florida, Tampa, Florida 33620, United States.
REFERENCES
- (1).Boucher HW; Talbot GH; Bradley JS; Edwards JE; Gilbert D; Rice LB; Scheld M; Spellberg B; Bartlett J Bad Bugs, No Drugs: No ESKAPE! An Update from the Infectious Diseases Society of America. Clin. Infect. Dis. 2009, 48,1–12. [DOI] [PubMed] [Google Scholar]
- (2).Ghosh C; Sarkar P; Issa R; Haldar J Alternatives to Conventional Antibiotics in the Era of Antimicrobial Resistance. Trends Microbiol. 2019, 27, 323–338. [DOI] [PubMed] [Google Scholar]
- (3).Teng P; Huo D; Nimmagadda A; Wu J; She F; Su M; Lin X; Yan J; Cao A; Xi C; Hu Y; Cai J Small Antimicrobial Agents Based on Acylated Reduced Amide Scaffold. J. Med. Chem. 2016, 59, 7877–7887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Nguyen LT; Haney EF; Vogel HJ The Expanding Scope of Antimicrobial Peptide Structures and Their Modes of Action. Trends Biotechnol. 2011, 29, 464–472. [DOI] [PubMed] [Google Scholar]
- (5).Kahlenberg JM; Kaplan MJ Little Peptide, Big Effects: The Role of LL-37 in Inflammation and Autoimmune Disease. J. Immunol. 2013, 191, 4895–4901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Zasloff M Antimicrobial Peptides of Multicellular Organisms. Nature 2002, 415, 389. [DOI] [PubMed] [Google Scholar]
- (7).Yeaman MR; Yount NY Mechanisms of Antimicrobial Peptide Action and Resistance. Pharmacol. Rev. 2003, 55, 27–55. [DOI] [PubMed] [Google Scholar]
- (8).Hancock REW; Sahl H-G Antimicrobial and Host-defense Peptides as New Anti-Infective Therapeutic Strategies. Nat. Biotechnol. 2006, 24, 1551. [DOI] [PubMed] [Google Scholar]
- (9).Niu Y; Wang M; Cao Y; Nimmagadda A; Hu J; Wu Y; Cai J; Ye X-S Rational Design of Dimeric Lysine N-Alkylamides as Potent and Broad-Spectrum Antibacterial Agents. J. Med. Chem. 2018, 61, 2865–2874. [DOI] [PubMed] [Google Scholar]
- (10).Mercer DK; O’Neil DA Peptides As The Next Generation of Anti-infectives. Future Med. Chem. 2013, 5, 315–337. [DOI] [PubMed] [Google Scholar]
- (11).Sengupta D; Leontiadou H; Mark AE; Marrink S-J Toroidal Pores Formed by Antimicrobial Peptides Show Significant Disorder. Biochim. Biophys. Acta, Biomembr. 2008, 1778, 2308–2317. [DOI] [PubMed] [Google Scholar]
- (12).Brown KL; Hancock REW Cationic Host Defense (Antimicrobial) Peptides. Curr. Opin. Immunol. 2006, 18, 24–30. [DOI] [PubMed] [Google Scholar]
- (13).Tyers M; Wright GD Drug Combinations: A Strategy to Extend The Life of Antibiotics in The 21st Century. Nat. Rev. Microbiol. 2019, 17, 141–155. [DOI] [PubMed] [Google Scholar]
- (14).Wang R; van Dorp L; Shaw LP; Bradley P; Wang Q; Wang X; Jin L; Zhang Q; Liu Y; Rieux A; Dorai-Schneiders T; Weinert LA; Iqbal Z; Didelot X; Wang H; Balloux F The Global Distribution and Spread of The Mobilized Colistin Resistance Gene Mcr-1. Nat. Commun. 2018, 9, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wenzel M; Rautenbach M; Vosloo JA; Siersma T; Aisenbrey CHM; Zaitseva E; Laubscher WE; van Rensburg W; Behrends JC; Bechinger B; Hamoen LW The Multifaceted Antibacterial Mechanisms of the Pioneering Peptide Antibiotics Tyrocidine and Gramicidin S. MBio 2018, 9, No. e00802–08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Choi S; Isaacs A; Clements D; Liu D; Kim H; Scott RW; Winkler JD; DeGrado WF De Novo Design and In Vivo Activity of Conformationally Restrained Antimicrobial Arylamide Foldamers. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 6968–6973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Su M; Xia D; Teng P; Nimmagadda A; Zhang C; Odom T; Cao A; Hu Y; Cai J Membrane-Active Hydantoin Derivatives as Antibiotic Agents. J. Med. Chem. 2017, 60, 8456–8465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Zhu T; Xu S; Rahman A; Dogdibegovic E; Yang P; Pageni P; Kabir MP; Zhou X-D; Tang C Cationic Metallo-Polyelectrolytes for Robust Alkaline Anion-Exchange Membranes. Angew. Chem. 2018, 57, 2388–2392. [DOI] [PubMed] [Google Scholar]
- (19).Nimmagadda A; Liu X; Teng P; Su M; Li Y; Qiao Q; Khadka NK; Sun X; Pan J; Xu H; Li Q; Cai J Polycarbonates with Potent and Selective Antimicrobial Activity toward Gram-Positive Bacteria. Biomacromolecules 2017, 18, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Venkataraman S; Hedrick JL; Ong ZY; Yang C; Ee PLR; Hammond PT; Yang YY The Effects of Polymeric Nanostructure Shape on Drug Delivery. Adv. Drug Delivery Rev. 2011, 63, 1228–1246. [DOI] [PubMed] [Google Scholar]
- (21).Song Z; Tan Z; Cheng J Recent Advances and Future Perspectives of Synthetic Polypeptides from N-Carboxyanhydrides. Macromolecules 2019, 52, 8521–8539. [Google Scholar]
- (22).Liu D; DeGrado WF De Novo Design, Synthesis, and Characterization of Antimicrobial β-Peptides. J. Am. Chem. Soc. 2001, 123, 7553–7559. [DOI] [PubMed] [Google Scholar]
- (23).Czyzewski AM; Jenssen H; Fjell CD; Waldbrook M; Chongsiriwatana NP; Yuen E; Hancock REW; Barron AE In Vivo, In Vitro, and In Silico Characterization of Peptoids as Antimicrobial Agents. PLoS One 2016, 11, No. e0135961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Jammal J; Zaknoon F; Kaneti G; Goldberg K; Mor A Sensitization of Gram-Negative Bacteria to Rifampin and OAK Combinations. Sci. Rep. 2015, 5, 9216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sang P; Shi Y; Teng P; Cao A; Xu H; Li Q; Cai J Antimicrobial AApeptides. Curr. Top. Med. Chem. 2017, 17, 1266–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Shi Y; Patel NA; Cai J Discovery of A Macrocyclic γ-AApeptide Binding to lncRNA GAS5 And its Therapeutic Implication in Type 2 Diabetes. Future Med. Chem. 2019, 11, 2233–2235. [DOI] [PubMed] [Google Scholar]
- (27).Nimmagadda A; Shi Y; Cai J γ-AApeptides As A New Strategy for Therapeutic Development. Curr. Med. Chem. 2019, 26, 2313–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Teyssiéres E; Corre J-P; Antunes S; Rougeot C; Dugave C; Jouvion G; Claudon P; Mikaty G; Douat C; Goossens PL; Guichard G Proteolytically Stable Foldamer Mimics of Host-Defense Peptides with Protective Activities in a Murine Model of Bacterial Infection. J. Med. Chem. 2016, 59, 8221–8232. [DOI] [PubMed] [Google Scholar]
- (29).Teng P; Nimmagadda A; Su M; Hong Y; Shen N; Li C; Tsai L-Y; Cao J; Li Q; Cai J Novel Bis-cyclic Guanidines As Potent Membrane-active Antibacterial Agents with Therapeutic Potential. Chem. Commun. 2017, 53, 11948–11951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tew GN; Scott RW; Klein ML; DeGrado WF De Novo Design of Antimicrobial Polymers, Foldamers, and Small Molecules: From Discovery to Practical Applications. Acc. Chem. Res. 2010, 43, 30–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Mensa B; Howell GL; Scott R; DeGrado WF Comparative Mechanistic Studies of Brilacidin, Daptomycin, and the Antimicrobial Peptide LL16. Antimicrob. Agents Chemother. 2014, 58, 5136–5145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lai X-Z; Feng Y; Pollard J; Chin JN; Rybak MJ; Bucki R; Epand RF; Epand RM; Savage PB Ceragenins: Cholic Acid-Based Mimics of Antimicrobial Peptides. Acc. Chem. Res. 2008, 41, 1233–1240. [DOI] [PubMed] [Google Scholar]
- (33).Handzlik J; Szymańska E; Nędza K; Kubacka M; Siwek A; Mogilski S; Handzlik J; Filipek B; Kieć-Kononowicz K Pharmacophore Models Based Studies on The Affinity and Selectivity Toward 5-HT1A with Reference to α1-adrenergic Receptors Among Arylpiperazine Derivatives of Phenytoin. Bioorg. Med. Chem. 2011, 19, 1349–1360. [DOI] [PubMed] [Google Scholar]
- (34).Subtel’na I; Atamanyuk D; Szymańska E; Kieć-Kononowicz K; Zimenkovsky B; Vasylenko O; Gzella A; Lesyk R Synthesis of 5-arylidene-2-amino-4-azolones and Evaluation of Their Anticancer Activity. Bioorg. Med. Chem. 2010, 18, 5090–5102. [DOI] [PubMed] [Google Scholar]
- (35).Kieć-Kononowicz K; Szymańska E; Motyl M; Holzer W; Białecka A; Kasprowicz A Synthesis, Spectral and Antimicrobial Properties of 5-chloroarylidene Aromatic Derivatives of Imidazoline4-one. Pharmazie 1998, 53, 680–684. [PubMed] [Google Scholar]
- (36).Matys A; Podlewska S; Witek K; Witek J; Bojarski AJ; Schabikowski J; Otrębska-Machaj E; Latacz G; Szymańska E; Kieć-Kononowicz K; Molnar J; Amaral L; Handzlik J Imidazolidine-4-one Derivatives in the Search for Novel Chemosensitizers of Staphylococcus Aureus MRSA: Synthesis, Biological Evaluation and Molecular Modeling Studies. Eur. J. Med. Chem. 2015, 101, 313–325. [DOI] [PubMed] [Google Scholar]
- (37).Szymańska E; Kieć-Kononowicz K; Białecka A; Kasprowicz A Antimicrobial Activity of 5-arylidene Aromatic Derivatives of Hydantoin. Farmaco 2002, 57, 39–44. [DOI] [PubMed] [Google Scholar]
- (38).Karolak-Wojciechowska J; Szymańska E; Fruziński A; Kieć Kononowicz K Crystallographic Studies of (Z) and (E) Isomers of 2-amino-(2-chlorobenzylidene)-1-methy-lH-imidazol-4(5H)-one. J. Mol. Struct. 2010, 966, 14–17. [Google Scholar]
- (39).Qian L; Sun G Durable and Regenerable Antimicrobial Textiles: Synthesis and Applications of 3-methylol-2,2,5,5-tetramethyl-imidazolidin-4-one (MTMIO). J. Appl. Polym. Sci. 2003, 89, 2418–2425. [Google Scholar]
- (40).Lorenzon EN; Piccoli JP; Santos-Filho NA; Cilli EM Dimerization of Antimicrobial Peptides: A Promising Strategy to Enhance Antimicrobial Peptide Activity. Protein Pept. Lett. 2019, 26, 98–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Li C; Teng P; Peng Z; Sang P; Sun X; Cai J Bis-Cyclic Guanidines as a Novel Class of Compounds Potent against Clostridium difficile. ChemMedChem 2018, 13, 1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Knight BJ; Stache EE; Ferreira EM Complementary Stereochemical Outcomes in Proline-Based Self-Regenerations of Stereocenters. Org. Lett. 2014, 16, 432–435. [DOI] [PubMed] [Google Scholar]
- (43).Ma Z; Yang J; Han J; Gao L; Liu H; Lu Z; Zhao H; Bie X Insights into the Antimicrobial Activity and Cytotoxicity of Engineered α-Helical Peptide Amphiphiles. J. Med. Chem. 2016, 59, 10946–10962. [DOI] [PubMed] [Google Scholar]
- (44).Ge Y; MacDonald DL; Holroyd KJ; Thornsberry C; Wexler H; Zasloff M In Vitro Antibacterial Properties of Pexiganan, An Analog of Magainin. Antimicroh. Agentb Chemother. 1999, 43, 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Hoque J; Konai MM; Gonuguntla S; Manjunath GB; Samaddar S; Yarlagadda V; Haidar J lMembrane Active Small Molecules Show Selective Broad Spectrum Antibacterial Activity with No Detectable Resistance and Eradicate Biofilms. J. Med. Chem. 2015, 58, 5486–5500. [DOI] [PubMed] [Google Scholar]
- (46).Jamal M; Ahmad W; Andleeb S; J alii F; Imran M; Nawaz MA; Hussain T; Ali M; Rafiq M; Kamil MA Bacterial Biofilm and Associated Infections. J. Chin.Med. Assoc. 2018, 81, 7–11. [DOI] [PubMed] [Google Scholar]
- (47).Davies D Understanding Biofilm Resistance to Antibacterial Agents. Nat. Rev. Drug Discovery 2003, 2, 114. [DOI] [PubMed] [Google Scholar]
- (48).Arciola CR; Campoccia D; Montanaro L Implant Infections: Adhesion, Biofilm Formation and Immune Evasion. Nat. Rev. Microbiol. 2018, 16, 397–409. [DOI] [PubMed] [Google Scholar]
- (49).Niu Y; Padhee S; Wu H; Bai G; Qiao Q; Hu Y; Harrington L; Burda WN; Shaw LN; Cao C; Cai J Lipo-γ-AApeptides as a New Class of Potent and Broad-Spectrum Antimicrobial Agents. J. Med. Chem. 2012, 55, 4003–4009. [DOI] [PubMed] [Google Scholar]
- (50).Teng P; Shi Y; Sang P; Cai J γ-AApeptides as a New Class of Peptidomimetics. Chem. – Eur. J. 2016, 22, 5458–5466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Isaksson J; Brandsdal BO; Engqvist M; Flaten GE; Svendsen JSM; Stensen W A Synthetic Antimicrobial Peptidomimetic (LTX 109): Stereochemical Impact on Membrane Disruption. J. Med. Chem. 2011, 54, 5786–5795. [DOI] [PubMed] [Google Scholar]
- (52).Konai MM; Ghosh C; Yarlagadda V; Samaddar S; Haidar J Membrane Active Phenylalanine Conjugated Lipophilic Norspermidine Derivatives with Selective Antibacterial Activity. J. Med. Chem. 2014, 57, 9409–9423. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.