

ABSTRACT
Stimulator of interferon genes (STING) pathway activation predicts the effectiveness of targeting the PD-1/PD-L1 axis in lung cancer. Active IFN-γ signaling is a common feature in tumors that respond to PD-1/PD-L1 blockade. The connection between IFN-γ and STING signaling in cancer cells has not been documented. We showed that IFN-γ caused DNA damage and the accumulation of cytosolic dsDNA, leading to the activation of the cGAS- and IFI16-dependent STING pathway in lung adenocarcinoma cells. IFN-γ-induced iNOS expression and nitric oxide production were responsible for DNA damage and STING activation. Additional etoposide treatment enhanced IFN-γ-induced IFN-β and CCL5 expression. Tumor-infiltrating T cells stimulated with a combination of anti-CD3 and anti-PD-1 antibodies caused STING activation and increased IFN-β and CCL5 expression in lung adenocarcinoma. These effects were abrogated by the addition of an IFN-γ neutralizing antibody. Our results suggest that the activation of tumor-infiltrating T cells could alter the tumor microenvironment via the IFN-γ-mediated activation of STING signaling in cancer cells.
KEYWORDS: IFN-γ, STING, DNA damage, lung adenocarcinoma, T cell activation
Introduction
Targeting the programmed cell death-1 (PD-1) or programmed cell death ligand 1 (PD-L1) axis alone or in combination with chemotherapy has shown remarkable antitumor effects and has become the standard of care for advanced non-small-cell lung cancer (NSCLC) patients, leading to improved survival.1,2 However, a large proportion of NSCLC patients derive limited or no benefits from treatment with PD-1/PD-L1 inhibitors.3 In recent years, numerous investigations have focused on identifying biomarkers for the prediction of patient responses to anti-PD-1/PD-L1 therapy.4,5 Della Corte et al. reported that the activation of the stimulator of interferon genes (STING) pathway predicts the effectiveness of cancer responding to immunotherapy. Moreover, STING activation is associated with increased levels of intrinsic DNA damage and biomarkers of an active immune microenvironment in NSCLC.6,7
Cellular DNA is under constant threat from exogenous and endogenous damage. DNA damage can be detected as a danger-associated molecular pattern using cytosolic and endosomal sensors and adaptor molecules, leading to the production of type I interferon, proinflammatory cytokines, and chemokines.8 Cyclic GMP-AMP synthase (cGAS) is an important cytosolic nucleic acid sensor of double-stranded DNA.9,10 cGAS activation generates cyclic dinucleotide cyclic GMP-AMP (cGAMP), which in turn triggers the STING signaling pathway and induces IFN-β expression.11 In addition to cGAS, interferon-inducible factor-16 (IFI16) can recognize cytosolic double-stranded DNA, single-stranded DNA, and damaged nuclear DNA.12–14 Activation of the STING pathway in human macrophages and keratinocytes requires the cooperation of cGAS and IFI16.15,16
The STING pathway may be involved in the induction of the antitumor immune response in the context of DNA damage.17–19 DNA-damaging agents are widely used in clinical oncology. Exposure to ionizing radiation (IR) and chemotherapeutic agents causes DNA double-strand breaks (DSBs). Etoposide-induced DNA damage can trigger NF-κB-dependent transcription by activating the cGAS-independent STING pathway.20 Ionizing radiation-induced antitumor immunity depends on STING activation and IFN-β induction.21 However, the association between cancer immunotherapy and the induction of cellular DNA damage in cancer cells and the underlying mechanisms have not been fully elucidated. Emerging experimental and clinical evidence suggests that the DNA damage and repair landscape is also associated with cancer responses to immune-directed therapies.7
Substantial evidence shows that active IFN-γ signaling is a common feature in tumors that respond to immune checkpoint blockade.22 The loss of IFN-γ pathway genes in tumor cells reduces the likelihood of a response to immunotherapy.23,24 The precise contributions of IFN-γ to immunotherapy, however, remain to be defined. The cellular effects of IFN-γ have been described, including the activation of cellular immunity and the induction of cell cycle arrest and cell death in human cancer cells.25–28 Recent studies using state-of-the-art technology have shown that IFN-γ secreted from tumor antigen-stimulated T cells can diffuse extensively to alter the tumor microenvironment in a distant area. These findings imply that activated tumor-infiltrating CD8+ T cells modulate the behavior of remote tumor cells through IFN-γ.29,30 IFN-γ alone or in combination with other inflammatory cytokines, including IL-1β and TNF-α, induces DNA damage in HeLa and cholangiocarcinoma cells.31,32 Whether IFN-γ-induced DNA damage triggers STING activation in cancer cells has not been documented.
In this study, we show that IFN-γ induced nuclear DNA damage and cytosolic dsDNA formation in lung adenocarcinoma cells via the upregulation of inducible nitric oxide synthase (iNOS) expression and nitric oxide (NO) production. IFN-γ-induced DNA damage activated the STING pathway. The DNA sensors cGAS and IFI16 are involved in STING activation induced by IFN-γ. Our results suggest that IFN-γ can shape the tumor immune microenvironment and exert antitumor effects by activating the STING pathway in cancer cells.
Materials and methods
Cell lines and cell culture
Human lung adenocarcinoma A549 (ATCC Cat# CRL-7909), HCC827 (KCLB Cat# 70827), H2444 (ATCC Cat# CRL-5945), H2030 (ATCC Cat# CRL-5914), and H1975 (ATCC Cat# CRL-5908) cell lines were obtained from Cobioer Biosciences (Nanjing, China). Short tandem repeat (STR) analyses were performed as follows: the analyses of the A549, HCC827, and H1975 cell lines were performed in 2017, and the analyses of the H2444 and H2030 cell lines were performed in 2019. A549, H2444, and H2030 cells harbor mutations in KRAS, and H1975 and HCC827 cells harbor EGFR mutations. The A549 cells were cultured in F12K medium containing 10% fetal bovine serum (Gibco, Grand Island, NY, USA), 100 U/ml penicillin and 0.1 mg/ml streptomycin (HyClone, Logan, NT, USA). The other cell lines were cultured in RPMI 1640 (HyClone) containing 10% fetal bovine serum supplemented with 100 U/ml penicillin and 0.1 mg/ml streptomycin. The cells were maintained at 37°C in a humidified incubator with 5% CO2. All cell lines were confirmed to be Mycoplasma-negative (Biothrive Sci. & Tech. Ltd., Shanghai, China), and the cell lines were used within 3 months after resuscitation.
Ex vivo culture of patient-derived lung cancer explants
Fresh tissues were obtained from patients undergoing pulmonary resection before radiation or chemotherapy at the Department of Thoracic Surgery, Tongji Hospital. Ex vivo culture was performed as previously described.33 Briefly, the tumor tissues were dissected into 1 mm3 cubes and placed on a gelatin sponge (Hushida, Jiangxi, China) in RPMI-1640 media supplemented with 10% heat-inactivated FBS. Then, the indicated amounts of IFN-γ or anti-CD3 mAb, anti-PD-1 Ab, and anti-IFN-γ Ab were added to the cultures. The tissues were cultured at 37°C for 48 h and collected for RNA and protein extraction. This study was performed following the Declaration of Helsinki. The use of human tissue samples was approved by the Institutional Ethics Committee of Huazhong University of Science and Technology.
Antibodies and reagents
All antibodies and reagents are listed in Table S1.
Activation of T lymphocytes in vitro
Human peripheral blood mononuclear cells (PBMCs) were isolated from EDTA-K2 anticoagulated blood using Ficoll-Paque medium (GE Healthcare, Uppsala, Sweden) and density gradient centrifugation. PBMCs (2 × 106 cells/well, 6-well plate) were cultured alone or treated with 1 µg/mL CD3 mAb (BD Bioscience, San Jose, CA, USA). Two days later, the supernatants were collected and cultured with tumor cells. Alternatively, CD3 T lymphocytes were isolated from the PBMCs using magnetically labeled CD3 MicroBeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Subsequently, purified CD3 T cells (2 × 106 cells/well, 6-well plate) were stimulated with 1 µg/mL CD3 mAb and 1 µg/mL CD28 mAb (R&D Systems, Minneapolis, MN, USA). The supernatants were collected for the culturing of tumor cells.
Detection of IFN-γ expression by enzyme-linked immunosorbent assay (ELISA)
PBMCs or purified T cells were stimulated with or without anti-CD3/CD28 mAbs for 24 h ~ 72 h. The supernatants were collected using centrifugation. The levels of IFN-γ in the supernatants were quantified using a commercial ELISA kit (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
Cell viability
Cell viability was measured using a CCK-8 kit (Dojindo Molecular Laboratories, Kumamoto, Japan). A549 cells were seeded (3,000 cells/100 μL/well) in 96-well plates and cultured overnight before being exposed to the indicated treatment. The absorbance was measured at 450 nm using a microplate spectrophotometer (TECAN, Baldwin Park, CA, USA).
Nitric oxide detection
The quantification of nitric oxide in cells after exposure to IFN-γ was performed using a commercial kit obtained from Beyotime Biotechnology (Beyotime Biotechnology, Shanghai, China) according to the manufacturer’s instructions.
siRNA transfection
The siRNAs targeting JAK1, JAK2, cGAS, IFI16, and STING were synthesized by RiboBio (Guangzhou, China). The targeted sequences are shown in Table S2. Transient transfection was used to deliver the siRNAs. Briefly, the siRNAs (50 nM) and Lipofectamine 3000 (Invitrogen) were gently premixed in medium without serum according to the manufacturer’s guidelines. The transfection mixture was added to the culture plate, and subsequently, cell suspensions were seeded into the culture plates and maintained for 24 ~ 72 h. The efficacy of the target gene knockdown was verified by an immunoblot analysis.
RNA isolation and quantitative real-time PCR analysis
Total RNA was extracted using TRIzol reagent (Takara Bio Inc., Shiga, Japan). The mRNA was reverse transcribed into cDNA using an RT reagent kit (Vazyme, Nanjing, China). The target gene mRNA expression was quantified using real-time polymerase chain reaction (PCR) with a SYBR Green Master Mix Kit (Vazyme) and Applied Biosystems (Vilnius, Lithuania). Template-free negative controls were included, and all reactions were assayed in triplicate. We used ACTB as an internal control and determined the relative expression of the target genes according to the 2−ΔΔCt method. The primers were obtained from TsingKe Biology Technology (Wuhan, China). The sequences of the primers are shown in Table S2.
Histone extraction
The cells were harvested and resuspended in Triton Extraction Buffer (TEB: PBS containing 0.5% Triton X-100 and 2 mM PMSF) at a cell density of 107 cells/ml. Then, the cells were lysed on ice for 10 min with gentle stirring and centrifuged at 400 g for 10 min at 4°C. The cell pellet was resuspended in 0.2 N HCl at a density of 4 × 107 cells/mL and incubated at 4°C overnight. The samples were centrifuged at 6,500 g for 10 min at 4°C. The supernatant was used for immunoblot analysis after neutralization with 2 M NaOH.
Western blot analysis
The cells were lysed using RIPA lysis buffer (50 mM Tris pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, and 0.1% SDS). The proteins were resolved using SDS–PAGE and transferred to Immobilon®-P membranes (Merck Millipore, Darmstadt, Germany). The blots were developed using an enhanced chemiluminescent detection system (Tanon 5200, Shanghai, China).
Immunoprecipitation
Cells were lysed using a Nuclear and Cytoplasmic Protein Extraction kit (Beyotime Biotechnology, Shanghai, China). The cytoplasmic and nuclear fractions were obtained according to the manufacturer’s instructions. The cytoplasmic fractions were used for the immunoprecipitation assay. A portion of each cytoplasmic fraction was retained as an input control. The remaining cytoplasmic fractions were incubated with 2 μg of primary antibodies per 500 μg of total protein overnight at 4°C, followed by the addition of 50 µL of protein A/G beads (Thermo Fisher) and incubation for 1 h at room temperature. The beads were washed three times with wash buffer, and the bound proteins were eluted by boiling in sample buffer for 10 min.
Immunofluorescence analysis
The cells were seeded on poly-L-lysine-coated coverslips and treated with the indicated reagents (IFN-γ) for various times. Subsequently, the cells were fixed with 4% paraformaldehyde (Servicebio, Wuhan, China) for 15 min at room temperature and then permeabilized with 0.3% Triton X-100 (Standard Reagent, Hyderabad, India) in phosphate-buffered saline (PBS) for 10 min. After fixation and permeabilization, the cells were blocked with 1% bovine serum albumin (BSA) and 0.3% Triton X-100 in PBS for 60 min at room temperature. Then, the cells were incubated with primary antibodies at 4°C overnight, followed by incubation with fluorescently conjugated secondary antibodies. Finally, all cells were stained with DAPI (Servicebio, Wuhan, China). The cells were visualized under a wide-field fluorescence microscope (Carl Zeiss, Baden-Württemberg, Germany). The number of γ-H2AX-positive puncta was quantified using ImageJ. We analyzed three randomly selected images from each culture condition.
Immunofluorescence confocal microscopy
The cells were embedded with coverslips and treated with or without IFN-γ for 24 h. Subsequently, the cells were fixed with 4% paraformaldehyde and permeabilized with 0.05% Tween 20 and 0.005% Triton-X for 10 min. After blocking with 1% BSA, the cells were incubated with specific primary antibodies at 4°C overnight, washed with PBS, and incubated with fluorescently conjugated secondary antibodies. Finally, the nuclei were stained with DAPI for 15 min. The stained cells were visualized and captured by confocal microscopy (Zeiss LSM 780). The colocalized proportion of cGAS and dsDNA in three visual fields with more than 50 cells was further analyzed by ImageJ software and calculated by Manders’ coefficients.
Detection of cytosolic DNA
Cells (10,000 ~ 20,000 cells/well) were seeded on 12 mm diameter coverslips in 0.5 mL of culture medium. The cells were treated with IFN-γ for 24 h, fixed with 4% paraformaldehyde for 10 min at room temperature, and washed with PBS. Then, the cells were permeabilized using PBS buffer containing 0.05% Tween 20 and 0.005% Triton-X for 10 min at room temperature. The slides were washed three times with PBS, and nonspecific binding was blocked with PBS containing 1% BSA for 60 min at room temperature. Then, the slides were incubated with antibodies against dsDNA (1:800), cGAS (1:400), and γ-H2AX (1:200) overnight at 4°C. The slides were incubated with diluted secondary antibodies (1:200, 1% BSA) and DAPI. The number of cytosolic dsDNA-positive cells was quantified using ImageJ. We analyzed three randomly selected images, and the number of counted cells is indicated in the related figures.
Comet assay
A comet assay was performed using an OxiSelectTM Comet Assay Kit according to the manufacturer’s instructions (cat# STA-350, CELL BIOLABS, San Diego, CA). In brief, 75 µL of Comet Agarose per well was added to the OxiSelect™ Comet Slide to create a Base Layer. The samples were prepared using 75 µL of Comet Agarose per well with 1 × 104 cells/mL. The slides were kept at 4°C in the dark for 15 min, and then, the slides were soaked with prechilled lysis buffer (~25 mL/slide) for 60 min. The slides were immersed in alkaline solution for 30 min at 4°C in the dark and subjected to TBE electrophoresis (30 V, 10 mA) for 15 min. The slides were washed with prechilled DI H2O three times and replaced with cold 70% ethanol for 5 minutes. After the slides were completely air dried, 100 µL/well of diluted Vista Green DNA Dye was added, and the slides were incubated at room temperature for 15 min. Finally, the slides were viewed by epifluorescence microscopy using a FITC filter. DNA damage was measured in Olive Tail Moments (OTM) using CometScore software (casplab_1.2.3b2, https://casplab.com/download).
Flow cytometry
Cultured cells (5 × 105 cells/sample) were harvested and fixed with eBioscience IC Fixation Buffer (eBioscience, Waltham, Massachusetts, USA), and subsequently, the cells were permeabilized using permeabilization buffer (Invitrogen, USA) and stained with antibodies against IFN-β and iNOS at 4°C for 30 min. A flow cytometric analysis was performed using an Attune N x T Acoustic cytometer (Thermo Fisher Scientific, Waltham, Massachusetts, USA). The data were analyzed using FlowJo software v10 (BD Biosciences).
Staining of dead cells
A549 cells were stained with 7-AAD for 30 min at 4°C in the dark and subjected to flow cytometric analysis. Unstained cells were used as a staining control.
Detection of cGAMP
cGAMP detection was performed as previously described.15 Briefly, cells (1 ~ 2 x 107/sample) were lysed in cold 80% methanol. Cell debris was removed by centrifugation, and the samples were subjected to three rounds of butanol: water extraction. After the samples were mixed well, nitrogen evaporation was performed until the methanol was completely volatilized. Then, the sample was completely dissolved in 400 μl of ammonium acetate solution filtered with a 0.22 μm membrane and subjected to LC–MS analysis.
Statistical analysis
The data shown in the bar graphs are presented as the mean ± SD unless indicated otherwise. The RT–PCR and ELISA results are presented as the mean of triplicate samples, and the error bars represent the SD. Unpaired two-sided Student’s t-tests were used for comparisons between two groups. Multiple t-test analyses with the Holm-Sidak method were used for comparisons among multiple groups. GraphPad Prism V.8.0 (GraphPad Software) was used for the graphing and statistical analysis. The differences were considered statistically significant at p < .05.
Results
IFN-γ triggers the DNA damage response in human lung adenocarcinoma cells
First, to determine whether IFN-γ signaling triggers DNA damage, we assessed the expression level of phosphorylated histone H2AX (Ser139), which is an early marker of DNA damage, in lung cancer cells in response to IFN-γ. Western blot analysis showed that A549 and HCC827 cells treated with IFN-γ exhibited an increase in H2AX phosphorylation (Figure 1a). γ-H2AX foci analysis is a reliable and exquisitely sensitive monitor of double-strand break formation and repair. IFN-γ treatment led to a significant increase in the number of γ-H2AX foci per cell compared to that in mock-treated cells (Figure 1b).
Figure 1.
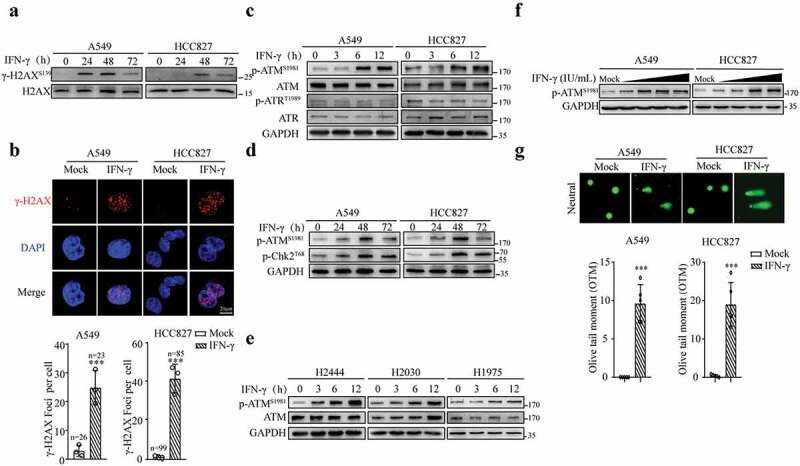
IFN-γ triggers DNA damage in lung adenocarcinoma cells. (a) Representative immunoblots showing phosphorylated H2AX and total H2AX expression in A549 and HCC827 cells treated with or without IFN-γ (100 IU/mL) for the indicated time intervals. The experiments were repeated at least four times. (b) A549 and HCC827 cells were treated with or without IFN-γ for 24 h. γ-H2AΧ foci were assessed using fluorescence microscopy (scale bar, 20 μm). DNA was visualized with DAPI. The data are expressed as the mean ± SD and are replicates of a representative experiment. Quantification of γ-H2ΑΧ foci per cell in each group was performed in the indicated number of cells (n); ***p < .001. (c-e) Whole cell lysates from IFN-γ-treated or untreated cells were subjected to immunoblotting using antibodies against p-ATM, p-ATR, p-Chk2, total ATM and ATR, and GAPDH was used as a loading control. (f) Whole-cell lysates from A549 and HCC827 cells treated with IFN-γ at concentrations of 100, 250, 500, and 1000 IU/mL were subjected to immunoblotting. (g) A549 and HCC827 cells were treated as described in B and subjected to a comet analysis. The olive tail moment (OTM) was used as an index of DNA damage. OTM is defined as the fraction of tail DNA multiplied by the distance between the centers of gravity for DNA in the head and tail. At least 50 cells per treatment were analyzed. The data are expressed as the mean ± SD and are replicates of a representative experiment. The same experiments were repeated at least twice.
ATM and ATR are two essential regulators involved in the cellular response to DNA damage.34 As shown in Figure 1c, ATM phosphorylation at Ser 1981 was increased in A549 and HCC827 cells as early as 6 h after IFN-γ treatment. IFN-γ did not affect the phosphorylation of ATR at Thr 1989. Notably, radiation treatment increased ATR phosphorylation at Thr 1989 (Fig. S1A). IFN-γ stimulation also led to a significant increase in the phosphorylation of the ATM target protein kinase Chk2 on Thr 68, confirming ATM activation by IFN-γ (Figure 1d). In addition to A549 and HCC827 cells, IFN-γ could induce ATM activation in other lung cancer cells, including H2444, H2030, and H1975 cells (Figure 1e). To determine whether ATM phosphorylation in response to IFN-γ is dose-dependent, we used gradient amounts of IFN-γ to stimulate A549 cells. The lower amount of IFN-γ (10 IU/mL) had a limited effect on the induction of ATM phosphorylation (Figure 1f).
To assess DNA damage in detail, we performed a comet assay in cells receiving IFN-γ or radiation treatment. The comet tail moment under neutral conditions reflects DNA breaks. As expected, comet tail moments were increased in the radiation-treated cells (Fig. S1B). The results of the comet assay showed that compared to the untreated cells, comet tail moments were significantly increased in the cells treated with IFN-γ (Figure 1g). Thus, our results demonstrate that IFN-γϒis capable of inducing DNA breaks in tumor cells.
Increased iNOS expression and NO production are responsible for the DNA damage induced by IFN-γ
Several studies have proposed that IFN-γ alone or mainly in combination with other inflammatory cytokines can increase the levels of ROS in cancer cells,32,35 primary hepatocytes,36 and macrophages.37 ROS can contribute to DNA damage. We assessed whether IFN-γ increased the level of ROS in lung cancer cells. We found that IFN-γ only increased the ROS levels in the A549 cells but not in the HCC827 and H2030 cells, and in contrast, IFN-γ reduced the ROS levels in HCC827 and H2030 cells (Fig. S2A). Notably, the baseline ROS levels in HCC827 and H2030 cells were much higher than those in A549 cells. Our data indicate that the IFN-γ-ROS axis may not be responsible for the IFN-γ-induced DNA damage in these cells.
Nitric oxide has also been documented to induce DNA damage in cancer cells.38–41 Chan et al. reported that IFN-γ upregulates inducible nitric oxide synthase (iNOS) expression in macrophages.42 Therefore, we investigated the role of nitric oxide in the induction of DNA damage by IFN-γ in lung cancer cells. The A549 and HCC827 cells treated with IFN-γ exhibited significant increases in iNOS expression at the mRNA (Figures 2a) and protein Figures 2(b,c) levels. We also observed that compared to the mock-treated cells, the production of nitric oxide (NO) was increased in the IFN-γ-treated cells (Figures 2d).
Figure 2.
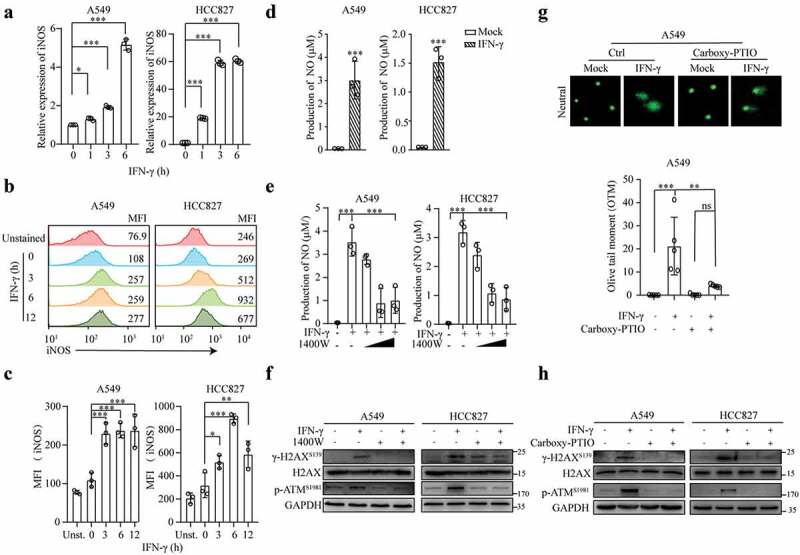
IFN-γ-induced iNOS expression and NO production are responsible for DNA damage. (a-c) A549 and HCC827 cells were treated with IFN-γ (100 IU/mL) for the indicated time intervals. qRT–PCR was used to measure the mRNA expression of iNOS (a). The relative iNOS expression was normalized to β-actin expression. The data represent the mean ± SD of each sample in triplicate. The experiments were repeated at least twice. Representative flow cytometry plots showing intracellular iNOS expression (b). The data are expressed as the mean ± SEM of 3 independent experiments (c). (d and e) Cell lysates from mock- or IFN-γ-treated cells (d) in the presence or absence of the iNOS inhibitor 1400 W (e) were subjected to NO production measurement using the Griess reagent method. The data represent the mean ± SD of each sample in triplicate. (f) Representative immunoblots showing phosphorylated ATM and H2AX expression in A549 and HCC827 cells treated with or without IFN-γ (100 IU/mL) in the presence or absence of 1400 W (500 μM) for 24 h. The experiments were repeated at least twice. (g) A549 cells were treated with or without IFN-ϒ in the presence or absence of the NO scavenger carboxy-PTIO (100 nM) for 24 h and subjected to a comet assay. (h) A549 cells were treated as described in G. Cell lysates were subjected to immunoblot analyses.
To confirm that iNOS induction is mediated by IFN-γ signaling, we knocked down JAK1 and JAK2 using siRNA in A549 and HCC827 cells, and the resultant cells were stimulated with IFN-γ. JAK2 knockdown abolished the IFN-γ-induced upregulation of iNOS, while JAK1 knockdown had a similar effect but to a lesser extent (Fig. S2B). Our data show the involvement of IFN-γ-mediated JAK1/JAK2 signaling in the induction of iNOS.
Subsequently, to determine whether IFN-γ-induced NO triggers DNA damage, we used the iNOS inhibitor 1400 W. The IFN-γ-induced production of NO in cancer cells was significantly reduced in the cells cultured with 250 μM and 500 μM 1400 W (Figure 2e). Moreover, culturing cells with IFN-γ in the presence of 1400 W abrogated the IFN-γ-mediated phosphorylation of H2AX and ATM (Figure 2f). To further confirm whether NO is responsible for IFN-γ-induced DNA damage, we used the NO scavenger carboxy-PTIO to culture cells in the presence of IFN-γ. The removal of NO inhibited the IFN-γ-induced DNA damage (Figure 2g) and phosphorylation of H2AX and ATM (Figure 2h). Notably, the amount of 1400 W and NO scavenger used at the given concentration did not significantly affect the cell viability (Figs. S2C and D). Our results demonstrate that the IFN-γ-induced upregulation of iNOS expression and NO production is responsible for the induction of DNA damage in tumor cells.
IFN-γ-induced DNA damage activates the STING pathway
The DNA damage response is tightly connected to inflammatory gene expression. Several studies have shown that the cellular DNA damage caused by chemotherapy and radiation can activate the STING pathway in cancer cells and result in the upregulation of inflammatory gene expression.20,43 Because we demonstrated that IFN-γ could induce DNA damage, we sought to determine whether the STING pathway is subsequently activated. We first evaluated whether the STING pathway is functional in A549 cells by transfecting the cells with herring testes (HT) DNA. As shown in Figures 3(a,b), A549 cells transfected with HT-DNA exhibited upregulated phosphorylation of TBK1, IRF3, and p65 and enhanced transcription of the IFN-β coding gene IFNB1, which is the most important target gene of the STING pathway. Our data indicate that the STING pathway can be activated by exogenous DNA in A549 cells.
Figure 3.
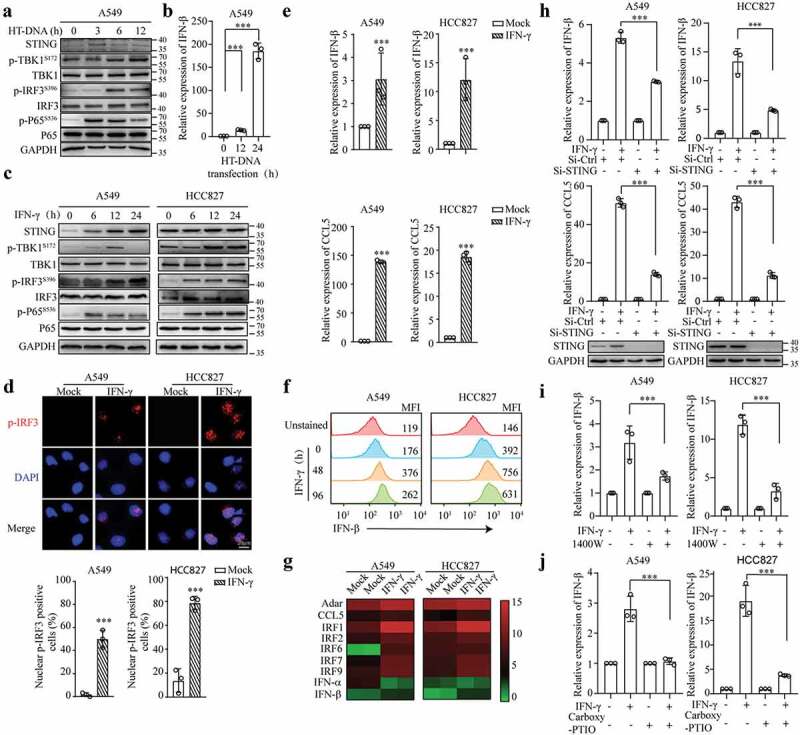
IFN-γ-induced DNA damage triggers STING activation in lung cancer cells. (a and b) A549 cells were transfected with HT-DNA (1 μg/mL). Immunoblots showing STING, p-TBK1, TBK1, p-IRF3, IRF3, p-p65, p65, and GAPDH expression (a). qRT–PCR was used to measure the mRNA expression of IFN-β (b). (c) Whole-cell lysates from A549 and HCC827 cells treated with or without IFN-γ (100 IU/mL) were subjected to immunoblotting. (d) A549 and HCC827 cells were treated with IFN-γ for 24 h. The expression and translocation of p-IRF3 were assessed using fluorescence microscopy (scale bar, 20 μm). DNA was visualized with DAPI. The number of nuclear p-IRF3-positive cells in 3 independent fields containing at least 80 cells in each group. *** p < .001. (e) qRT–PCR was used to measure the mRNA expression of IFN-βand CCL5 in A549 and HCC827 cells treated as described in D. (f) A549 and HCC827 cells were treated with IFN-γ for 48 h and 96 h. Representative flow cytometric plots showing IFN-β expression. The expression levels of IFN-β were analyzed by the mean fluorescence intensity (MFI). The experiments were repeated at least twice. (g) qRT–PCR was used to analyze the expression of STING-related genes in A549 and HCC827 cells treated as described in D. (h) A549 and HCC827 cells transfected with siRNA-Ctrl or siRNA-STING were treated with IFN-γ for 24 h. qRT–PCR was used to measure the mRNA expression of IFN-β and CCL5. (i and j) A549 and HCC827 cells were treated with IFN-γ in the presence or absence of 1400 W (500 μM) (i) or the NO scavenger carboxy-PTIO (100 nM) (j) for 24 h. IFN-β expression was analyzed by qRT–PCR. The data represent the mean ± SD of each sample in triplicate; *** p < .001.
Then, we investigated the effect of IFN-γ on STING signaling in A549 and HCC827 cells. Western blot analysis showed that the levels of phosphorylated TBK1, IRF3, and p65 were significantly increased in the IFN-γ–treated A549 and HCC827 cells compared to those in the untreated cells (Figure 3c). Notably, IFN-γ increased STING expression in A549 cells and HCC827 cells to a lesser extent. The basal expression level of STING in HCC827 cells was higher than that in A549 cells. IFN-γ-induced STING expression was STAT1-dependent (Fig. S3A). The significant increase in the translocation of p-IRF3 into the nucleus in the cells upon the IFN-γ treatment further confirmed the activation of the STING pathway (Figure 3d).
The qRT–PCR and flow cytometry results showed that IFN-β expression was increased in the cells treated with IFN-γ Figures 3(e,f). In addition to IFN-β, IFN-γ significantly enhanced the transcription of interferon-stimulated genes, including Adar, CCL5, and IRF1/2/7/9,which have been reported to be involved in the STING pathway44 (Figure 3g). Furthermore, knocking down STING by transfection with siRNA-STING led to a significant reduction in the IFN-γ-induced upregulation of IFN-β and CCL5 expression (Figure 3h), suggesting that STING is required for IFN-γ-induced IFN-βand CCL5 expression. STING activation in certain cell types triggers cell death.45,46 Interestingly, the downregulation of STING slightly increased cell death; however, additional IFN-γ did not increase the death of STING-knockdown cells under 24 h of culture conditions (Fig. S3B), suggesting that a lack of STING might affect the responsiveness of tumor cells to IFN-γ-mediated cytotoxic effects.
To determine the association between IFN-γ-induced DNA damage and STING activation, cells were treated with IFN-γ in the presence of the iNOS inhibitor 1400 W. As shown in Figure S3C, 1400 W reduced IFN-γ-induced phosphorylation of IRF3. Furthermore, the IFN-γ-induced upregulation of IFN-β expression was significantly decreased in the presence of 1400 W (Figure 3i) or the NO scavenger carboxy-PTIO (Figure 3j). Altogether, our data show that IFN-γ-induced DNA damage activates the STING pathway in lung cancer cells.
cGAS is required for IFN-γ-mediated activation of the STING pathway
We speculated whether cGAS is required for the IFN-γ-induced activation of STING signaling. IFN-γ did not significantly affect cGAS expression at the mRNA and protein levels in A549 and HCC827 cells (Fig. S4A and Figure 4a). Subsequently, we examined the cellular localization of cGAS. As shown in Figures 4(b,C), cGAS was mainly expressed in the cytoplasm in A549 and HCC827 cells. IFN-γ stimulation resulted in cGAS aggregation in the cytoplasm. As shown in Figure 4d, the knockdown of cGAS using siRNA significantly reduced IFN-γ-mediated IRF3 phosphorylation and translocation into the nucleus, consequently inhibiting IFN-β and CCL5 expression Figures 4(e,f). Similar to STING depletion, cGAS downregulation increased cell death. The cells lacking cGAS expression were resistant to IFN-γ-mediated cytotoxic effects (Fig. S4B). Our results demonstrate that cGAS is required for IFN-γ-mediated STING activation.
Figure 4.
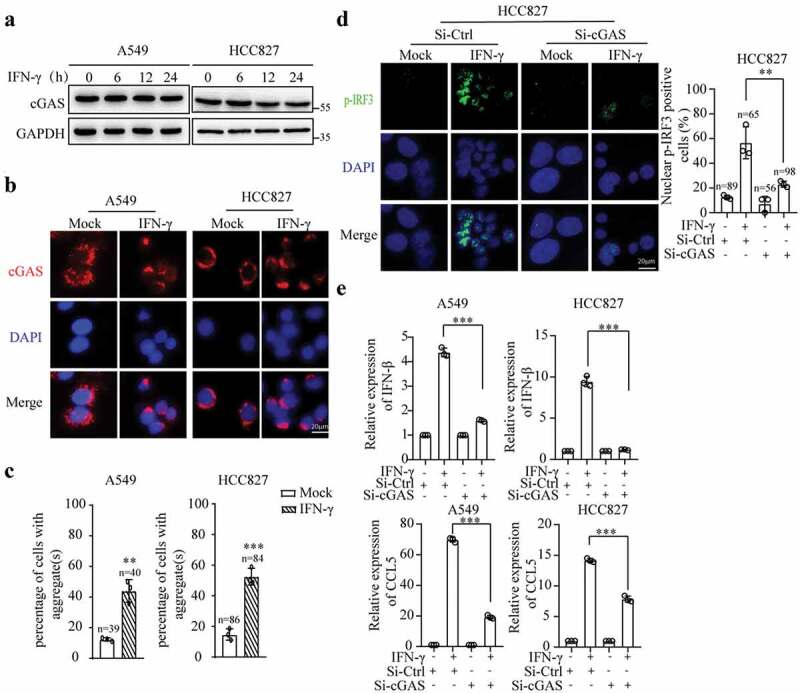
cGAS is required for IFN-γ-induced STING activation. (a) Immunoblots showing cGAS expression in A549 and HCC827 cells treated with or without IFN-γ (100 IU/mL). (b) A549 and HCC827 cells were treated with IFN-γ for 12 h. The cellular localization of cGAS was assessed using fluorescence microscopy (scale bar, 20 μm). DNA was visualized with DAPI. (c) The number of cells with aggregate(s) larger than 8 μm was quantified by ImageJ. The data are expressed as the mean ± SD and are replicates of a representative experiment; *** p < .001. (d and e) A549 and HCC827 cells were transfected with siRNA-Ctrl or siRNA-cGAS for 48 h, and subsequently, the cells were treated with IFN-γ (100 IU/mL) for 24 h. The expression and translocation of p-IRF3 were assessed (d). Nuclear pIRF3-positive cells were quantified in 3 independent fields containing the indicated number of cells in each group. qRT–PCR was used to measure the mRNA expression of IFN-β and CCL5(e). The data represent the mean ± SD of each sample in triplicate. The experiments were repeated at least twice; ** p < .01, *** p < .001.
IFN-γstimulation induces the presence of cytosolic dsDNA
Emerging evidence shows that irradiation-induced double-strand DNA breaks involving the formation of micronuclei ultimately release DNA into the cytoplasm, which can be recognized by cGAS.43,47 To determine whether IFN-γ-induced DNA damage results in the presence of cytosolic dsDNA, we used an anti-dsDNA antibody to detect dsDNA in the cytoplasm. In the immunofluorescence analysis, limited cytosolic dsDNA was observed in the A549 and HCC827 cells. IFN-γ stimulation significantly increased the number of cytosolic dsDNA-positive cells (Figure 5a). Interestingly, the addition of 1400 W reduced the number of cytosolic dsDNA-positive cells after IFN-γ treatment (Figure 5b), indicating that IFN-γ-induced DNA damage is responsible for the presence of cytosolic dsDNA.
Figure 5.
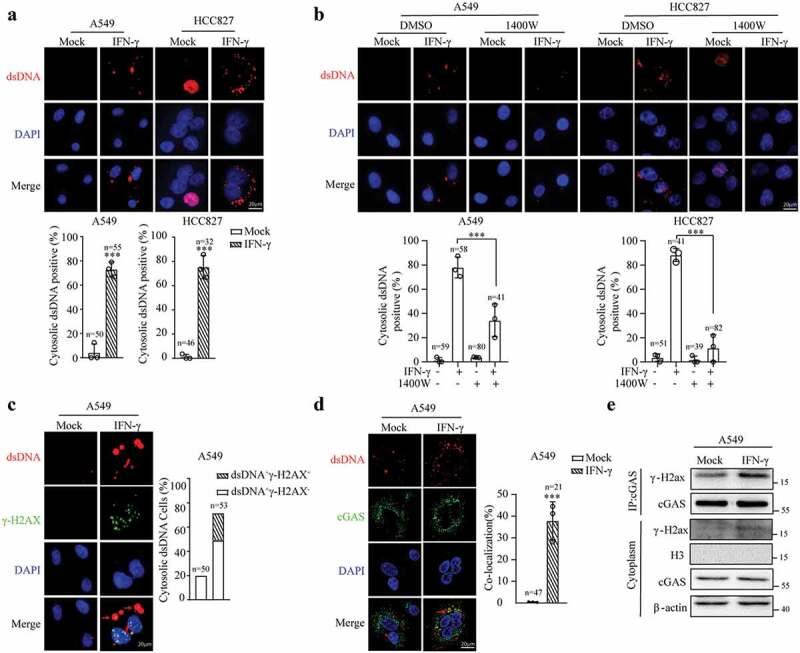
IFN-γ treatment results in the accumulation of cytosolic dsDNA. (a and b) A549 and HCC827 cells were treated with IFN-γ (100 IU/mL) for 24 h (a) or in the presence or absence of 1400 W (500 μM) (b). Subsequently, the cells were stained with an antibody against dsDNA. Cytosolic dsDNA was assessed using immunofluorescence. Nuclear DNA was visualized with DAPI. The data are expressed as the mean ± SD of replicates in a representative experiment. The number of cytosolic dsDNA-positive cells in 3 independent fields containing the indicated number of cells. The experiments were repeated twice; *** p < .001. (c and d) A549 cells were treated with IFN-γ (100 IU/mL) for 24 h and immunostained with antibodies against cytosolic dsDNA, γ-H2AX (c) or cGAS (d). The bar graphs represent the percentage of cytosolic dsDNA-positive cells relative to the total number of cells in 3 randomly selected fields containing at least 50 cells in each group. Cytosolic dsDNA was classified as dsDNA+γ-H2AX− and dsDNA+γ-H2AX+ (C). The colocalization of cGAS and dsDNA was analyzed by confocal microscopy. The degree of colocalization of cGAS and dsDNA was quantified by Manders’ colocalization coefficients (MCCs); *** p < .001. (e) Cytoplasmic fractions of IFN-γ-or mock-treated A549 cells were subjected to immunoprecipitation with an antibody against cGAS. Immunoblots showing γ-H2AX associated with cGAS. The experiments were repeated at least twice.
We used γ-H2AX staining to confirm the presence of damaged cytosolic dsDNA. γ-H2AX foci were prominently observed in the nucleus in the IFN-γ-treated cells, and approximately 31.5% of cytosolic dsDNA was γ-H2AX-positive in the IFN-γ-treated cells (Figure 5c). Subsequently, we examined whether cytosolic dsDNA is associated with cGAS. Immunofluorescence confocal microscopic analysis showed that a significant fraction of dsDNA colocalized with cGAS in the cells treated with IFN-γ (Figure 5d). The immunoprecipitation analysis of the cytoplasmic fraction of cell lysates using anti-cGAS antibodies confirmed that cGAS was associated with γ-H2AX (Figure 5e). Our results reveal that the presence of dsDNA in the cytosol could be the underlying mechanism by which IFN-γ induces cGAS activation.
IFI16 is involved in IFN-γ-mediated activation of the STING pathway
Several studies have shown that in human monocytes and keratinocytes, STING activation by cGAS requires the cooperation of IFI16, which shuttles between the nucleus and cytosol.15,16,48 IFN-γ upregulated IFI16 expression at the mRNA (Figure 6a) and protein levels (Figure 6b). To determine whether IFI16 plays a role in the IFN-γ-induced activation of the STING pathway, we used transient transfection with small interfering RNA (siRNA) to knock down IFI16. We found that IFI16 knockdown significantly reduced IFN-γ-induced IRF3 phosphorylation (Figure 6c) and the nuclear translocation of IRF3 (Figure 6d). The downregulation of IFI16 also significantly decreased IFN-γ-induced IFN-β expression (Figure 6e). These data suggest that IFI16 is involved in the IFN-γ-induced activation of the STING pathway.
Figure 6.
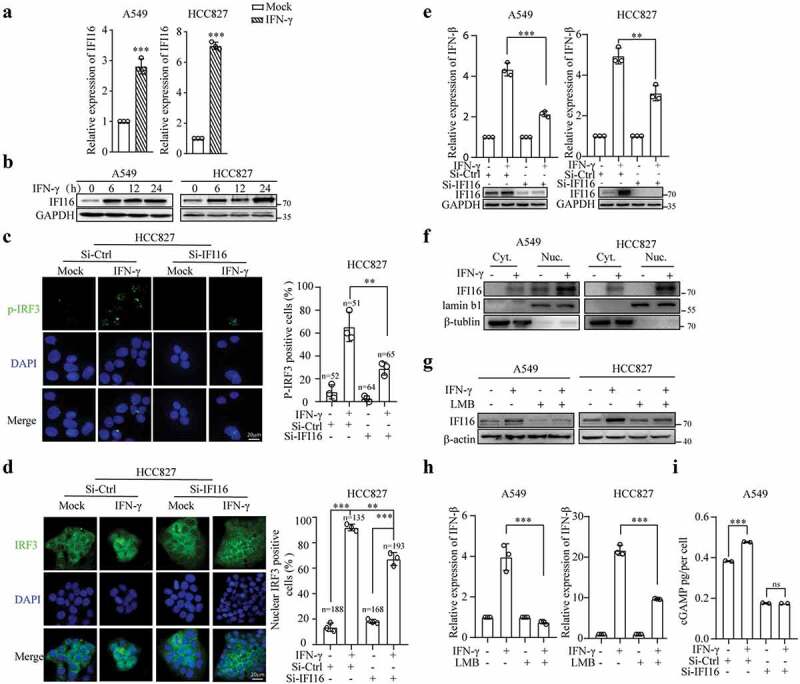
Upregulation and translocation of IFI16 mediated by IFN-γ is required for STING activation. (a) qRT–PCR analysis of IFI16 expression in A549 and HCC827 cells treated with IFN-γ (100 IU/mL) for 12 h and (b) immunoblots showing IFI16 expression in A549 and HCC827 cells treated with IFN-γ for the indicated time intervals. (c-e) HCC827 cells were transfected with siRNA-Ctrl or siRNA-IFI16 for 48 h, and subsequently, the cells were treated with IFN-γ for 24 h. The p-IRF3-positive cells (c) and nuclear IRF3-positive cells (d) were assessed; ***P < .001. qRT–PCR was used to measure the mRNA expression of IFN-βϒ(e). The data represent the mean ± SD of each sample in triplicate. The experiments were repeated at least twice. (f) A cytoplasmic nuclear separation kit was used to extract the cytoplasmic and nuclear fractions of A549 and HCC827 cells treated with or without IFN-γ (100 IU/mL) for 24 h. Immunoblot analysis of IFI16 expression in the extracted cytoplasmic and nuclear fractions. (g and h) Cells were pretreated with LMB (20 nM) or an equal volume of ethanol for 1 h, and subsequently, the cells were treated with IFN-γ (100 IU/mL) or mock treatment for 24 h. Cytoplasmic IFI16 expression levels were analyzed by immunoblotting (g), and qRT–PCR was used to assess IFN-β expression (h). The data represent the mean ± SD of each sample in triplicate. The experiments were repeated at least two times. (i) A549 cells were transfected with siRNA-Ctrl or siRNA-IFI16 for 48 h, and subsequently, the cells were treated with IFN-γ (100 IU/mL) for 8 h. The level of cGAMP was measured by LC–MS. The data represent the mean ± SD of each sample in duplicate, *** p < .001.
To further understand how IFI16 is involved in the IFN-γ-mediated activation of STING signaling, we first examined the cellular localization of IFI16 at a steady state and in response to IFN-γ stimulation in A549 and HCC827 cells. Immunoblotting of cytoplasmic and nuclear fractions was carried out to determine the level and distribution of the IFI16 protein. A549 and HCC827 cells exhibited high levels of IFI16 in the nuclear fraction and very low to almost undetectable levels of IFI16 in the cytoplasmic fraction. IFN-γ stimulation increased the expression level of IFI16 in the nuclear fraction, and interestingly, cytoplasmic IFI16 was also enhanced (Figure 6f). Subsequently, to determine whether the cytoplasmic translocation of IFI16 in response to IFN-γ stimulation is a critical step in activating the STING pathway, we used leptomycin B (LMB) to inhibit CRM1-mediated nuclear export49 and examined cytosolic IFI16 expression. As shown in Figure 6g, LMB prevented the translocation of IFI16 from the nucleus to the cytoplasm in response to IFN-γ. Moreover, LMB significantly inhibited IFN-γ-induced IFN-β expression (Figure 6h).
We conducted a cGAS assay, which directly measures cyclic GAMP-AMP (cGAMP) production, to determine whether IFI16 affects the enzymatic activity of cGAS. IFN-γ stimulation significantly increased cGAMP levels. The knockdown of IFI16 significantly reduced the basal level of cGAMP and IFN-γ-mediated cGAMP production (Figure 6i).
Etoposide enhances the IFN-γ-induced upregulation of IFN-β and CCL5 expression
Subsequently, we investigated whether the combination of etoposide and IFN-γ enhances STING activation. As shown in Figure S5A, etoposide had a significant dose-dependent cytotoxic effect on A549 cells. We first examined whether 25 μM etoposide induced DNA damage and STING activation in lung adenocarcinoma cells. As expected, etoposide induced the upregulation of H2AX and ATM phosphorylation Figures 7(a,b), and similar to IFN-γ stimulation, etoposide induced cytosolic dsDNA formation (Fig. S5B). As shown in Figure 7c, etoposide increased TBK1 and IRF3 phosphorylation. Consequently, IFN-β and CCL5 expression was upregulated (Figure 7d).
Figure 7.
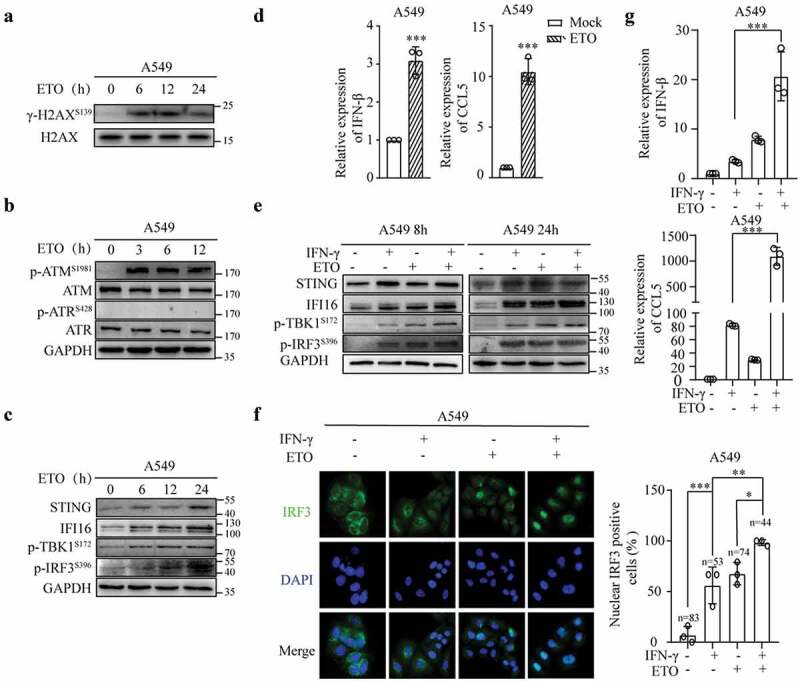
Etoposide enhances IFN-γ-induced IFN and CCL5 expression. (a) Immunoblots showing phosphorylated H2AX and total H2AX expression in A549 cells treated with ETO (25 μM). (b) Whole-cell lysates of A549 cells treated with ETO (25 μM) were subjected to an immunoblot analysis of p-ATM, p-ATR, total ATM, and total ATR expression. GAPDH was used as a loading control. (c) Immunoblots showing STING pathway-associated molecules expressed in A549 cells treated with ETO (25 μM). (d) qRT–PCR analysis of IFN-β and CCL5 expression in A549 cells treated with ETO (25 μM) for 24 h. (e) A549 cells were treated with IFN-γ and ETO alone or IFN-γ in combination with ETO for 8 h and 24 h. Whole-cell lysates were subjected to immunoblot analyses of STING, IFI16, p-TBK1, and p-IRF3, and GAPDH was used as a loading control. (f) A549 cells were treated as described in E for 8 h. Nuclear IRF3-positive cells were quantified in 3 independent fields containing the indicated number of cells in each group. (g) qRT–PCR was used to analyze IFN-β and CCL5 expression. The data represent the mean ± SD of each sample in triplicate.
Then, we speculated that the combination of etoposide and IFN-γ could further increase the activation of the STING pathway. As shown in Figure S5C, the combination of etoposide and IFN-γ significantly increased cell death after 48 h but not after 24 h of culture. Etoposide in combination with IFN-γ increased TBK1 phosphorylation as early as 8 h of culture compared to the cells cultured with IFN-γ alone (Figure 7e). The nuclear translocation of IRF3 was also increased in cells treated with etoposide and IFN-γ compared to that in the cells treated with IFN-γ or etoposide alone (Figure 7f). The combination of etoposide and IFN-γ enhanced IFN-β and CCL5 expression compared to that in the cells treated with etoposide or IFN-γ alone (Figure 7g).
Activated T cells induce DNA damage and activate the STING pathway in tumor cells through the secretion of IFN-γ
IFN-γ is a key effector molecule of cellular immunity and is secreted by activated T lymphocytes. Therefore, we examined whether T cell activation induces DNA damage and ATM activation in cancer cells and whether this effect is IFN-γ-dependent. Peripheral blood mononuclear cells (PBMCs) were stimulated with anti-CD3 mAb for 48 h. A significant amount of IFN-γ was produced (Figure 8a). As shown in Figure 8b, A549 and HCC827 cells cultured with supernatants from CD3-activated PBMCs exhibited increases in the phosphorylation of H2AX and ATM. The addition of an IFN-γ neutralizing antibody to the cultures prevented the phosphorylation of H2AX and ATM, indicating that IFN-γ produced by CD3-activated PBMCs is responsible for the induction of DNA damage and ATM activation. Notably, the supernatants from the PBMCs stimulated with anti-CD3 mAb for only 24 h with lower levels of IFN-γ were also able to induce the phosphorylation of H2AX and ATM Figure 8(c,d).
Figure 8.
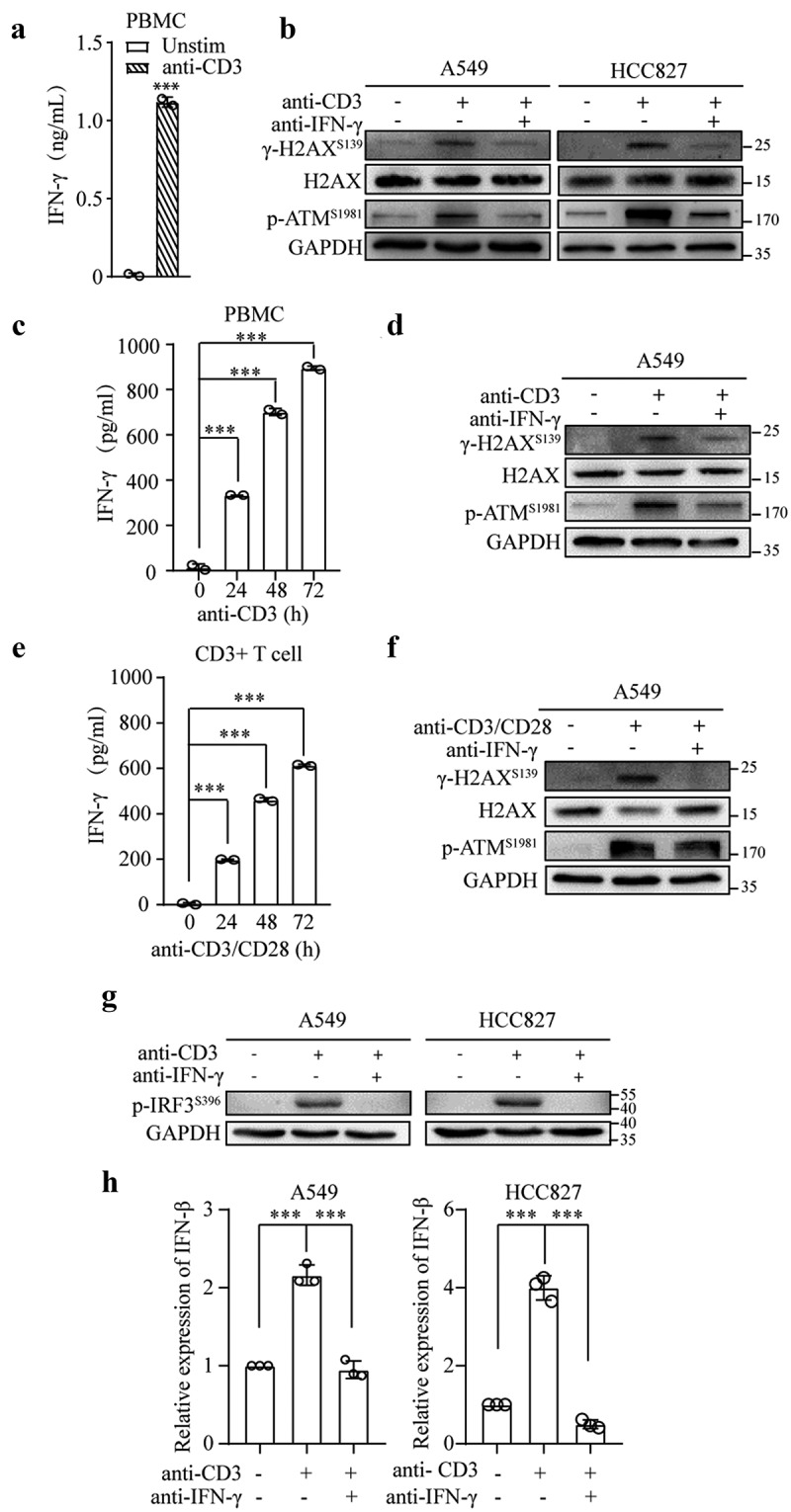
T cell activation triggers the STING pathway in tumor cells by the production of IFN-γ. (a and b) PBMCs (1 × 106 cells/mL) were stimulated with anti-CD3 mAb (1 μg/mL) in the presence or absence of an anti-IFN-γ antibody (10 μg/mL) for 48 h. Subsequently, the culture supernatants were collected. The secretion of IFN-γ was assessed by ELISA (a). The mean ± SD of triplicate cultures is shown; *** p < .001. The collected supernatants were cultured with A549 or HCC827 cells for 24 h. Immunoblots showing phosphorylated H2AX and ATM, total H2AX, and GAPDH expression (b). (c and d) PBMCs were treated as described in A for the indicated time intervals. The secretion of IFN-γ was assessed by ELISA (c). Supernatants obtained after 24 h of stimulation of PBMCs were cultured with A549 cells. Immunoblots showing phosphorylated H2AX and ATM, total H2AX, and GAPDH expression (d). (e and f) CD3+ T cells (1 × 106 cells/mL) isolated from PBMCs were stimulated with anti-CD3 mAb (1 μg/mL) and anti-CD28 mAb (1 μg/mL) in the presence or absence of anti-IFN-γ antibody (10 μg/mL) for 24–72 h. ELISA was used to measure IFN-ϒ in the culture supernatants (e). The mean ± SD of triplicate cultures is shown. The supernatants collected from 72 h of stimulation of T cells were used to culture A549 cells for 24 h. Immunoblots showing phosphorylated H2AX and ATM, total H2AX, and GAPDH expression (f). (g and h) Culture supernatants collected after 48 h of stimulation of PBMCs (1 x 106 cells/ml) with anti-CD3 mAb (1 μg/mL) in the presence or absence of IFN-γ neutralizing antibody (10 μg/mL) were cultured with A549 and HCC827 cells. Whole-cell lysates were subjected to an immunoblot analysis of phosphorylated IRF3 expression (g), and qRT–PCR was used to assess IFN-β expression (h).
To further confirm that IFN-γ produced by activated T cells is responsible for the induction of DNA damage, we isolated CD3 T cells from PBMCs and stimulated these cells with anti-CD3 and anti-CD28 mAbs. As shown in Figure 8e, IFN-γ was secreted by activated T cells. The supernatants from 72 h of T cell stimulation cultured with A549 cells resulted in an increase in the phosphorylation of H2AX and ATM. The neutralization of IFN-γ significantly reduced H2AX and ATM phosphorylation (figure 8f).
Subsequently, we investigated whether the activation of T cells could directly affect the STING pathway in A549 and HCC827 cells. PBMCs were stimulated with anti-CD3 mAb in the presence or absence of an IFN-γ neutralizing antibody for 48 h. The supernatants were cultured with A549 and HCC827 cells. As shown in Figures 8g and h, the supernatants collected from the anti-CD3 mAb-stimulated PBMCs significantly increased the phosphorylation of IRF3 and IFN-β expression. Interestingly, these effects were abrogated in the presence of an IFN-γ neutralizing antibody. Our results demonstrate that T cell activation can stimulate the STING pathway in tumor cells via the production of IFN-γ.
Tumor-infiltrating T cells stimulated with αCD3 and αPD-1 activate STING signaling in human lung adenocarcinoma
To further validate our in vitro findings in cell lines, we used human-derived lung adenocarcinoma to investigate the influence of IFN-γ on the STING pathway. As shown in Figures S6A and B, additional IFN-γ significantly increased the phosphorylation of IRF3 and IFN-β expression in human-derived lung adenocarcinoma.
Subsequently, we examined whether the stimulation of tumor-infiltrating T cells with an anti-CD3 mAb alone or in combination with an anti-PD-1 antibody could directly activate STING signaling in lung adenocarcinoma. As shown in Figure 9a, the stimulation of tumor-infiltrating T cells with anti-CD3 and anti-PD-1 antibodies significantly increased STAT1 phosphorylation. An additional IFN-γ neutralizing antibody abolished STAT1 phosphorylation, indicating that tumor-infiltrating T cells were activated and produced IFN-γ, subsequently activating IFN-γ signaling. Moreover, the stimulation of tumor-infiltrating T cells with anti-CD3 mAb resulted in the increased phosphorylation of H2AX and ATM, indicating that the activation of tumor-infiltrating T cells induces a DNA damage response. Neutralizing IFN-γ prevented the activated T cell-mediated phosphorylation of H2AX and ATM (Figure 9b). In the same experimental setting, we observed that the stimulation of tumor specimens with anti-CD3 and anti-PD-1 increased the phosphorylation of IRF3 (Figure 9c) and IFN-βand CCL5 expression (Figures 9(d,e) and Fig. S6C). These effects were abrogated in the presence of an IFN-γ neutralizing antibody Figures 9(d,e). These results indicate that the activation of tumor-infiltrating T cells could induce IFN-γ-dependent STING activation.
Figure 9.
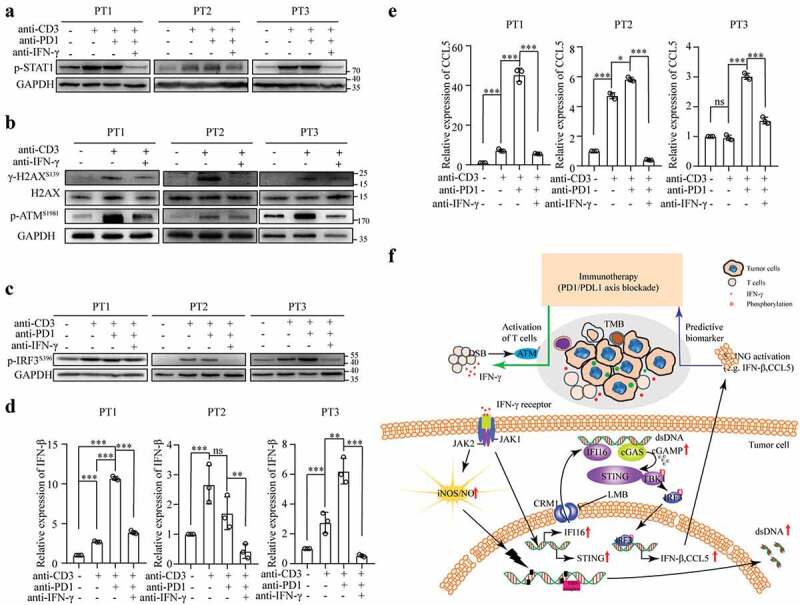
Activation of tumor-infiltrating T cells triggers STING activation in human lung adenocarcinoma. (a-e) Fresh lung tumor tissues were directly cultured with anti-CD3 mAb alone or a combination of anti-CD3 mAb (1 μg/mL) and anti-PD-1 antibody (10 μg/mL) in the presence or absence of IFN-γ neutralizing antibody for 48 h. The tissue lysates were subjected to an immunoblot analysis to determine phosphorylated STAT1 (a), H2AΧ, ATM (b), and IRF3 (c). RNA was extracted from the tissues and used to analyze IFN-β (d) and CCL5 (e) expression. (f) The connection between active IFN-γ signaling and STING activation in lung cancer cells is illustrated.
Overall, both the IFN-γ and STING pathways are predictive biomarkers of tumors that respond to PD-1/PD-L1 blockade-based immunotherapy in NSCLC. We showed that IFN-γ produced by activated T lymphocytes could cause DNA damage, leading to STING activation in lung adenocarcinoma cells. Our study establishes a connection between activated T cells and the STING pathway in cancer cells via IFN-γ (figure 9f).
Discussion
DNA damage and cellular responses to DNA damage are important determinants of cancer outcomes following radiation therapy, chemotherapy, and immune-directed therapies.7,50 Several studies have shown how radiation therapy and chemotherapy induce DNA damage and repair and revealed the biological consequences of the cellular response to DNA damage.50 The role of IFN-γ, which is the most important mediator involved in immune-directed therapies,51,52 in the induction of DNA damage in cancer cells and the subsequent cellular response to DNA damage have not been fully investigated. Hubackova et al. reported that IFN-γ could induce TGF-β/Smad signaling-dependent DNA damage and senescence in HeLa cells.32 IFN-γ did not activate TGF-β/Smad signaling in lung cancer cells (data not shown); instead, we showed that IFN-γ upregulated iNOS expression and resulted in the production of NO in lung adenocarcinoma cells. Inhibiting NO production resulted in the abrogation of IFN-γ-induced DNA damage. NO is the precursor of the highly reactive nitrogen species peroxynitrite (ONOO-), which is a required factor in oxidative DNA damage. High levels of iNOS-dependent DNA damage could cause DNA double-strand breaks and genomic instability.53
The DNA damage response plays a crucial role in determining tumor adjuvanticity through the production of adjuvant molecules, notably type I interferon and proinflammatory cytokines.54 DNA damage can activate innate immune responses by activating the cGAS-STING pathway.55,56 cGAS can detect endogenous nucleus-derived DNA in cancer cells in the form of ruptured micronuclei or cytosolic dsDNA9,10 and is activated by DNA in a length-dependent manner.57 Interestingly, in the nucleus, the reactivity of cGAS against self-DNA is inhibited by binding the nucleosome acidic patch.58,59 Our studies showed that IFN-γ treatment resulted in the accumulation of dsDNA in the cytosol. Moreover, a significant fraction of cytosolic dsDNA colocalized with cGAS in the IFN-γ-treated cells but not the untreated cells. The knockdown of cGAS abrogated the IFN-γ-mediated upregulation of IFN-β and CCL5 expression, suggesting that IFN-γ-induced STING activation is cGAS-dependent. However, the characteristics of IFN-γ-induced cytosolic dsDNA and the precise interaction among IFN-γ-induced cytosolic dsDNA, cGAS, and STING remain unclear and warrant further investigation.
In addition to cGAS, IFI16 is also involved in STING pathway activation.16 Almine and colleagues elegantly demonstrated that in human keratinocytes, both IFI16 and cGAS are required for the full activation of the STING pathway in response to exogenous DNA and DNA viruses.15 IFI16 is required for DNA sensing in macrophages by promoting the production and function of cGAMP.16 IFI16 has also been reported to interact with STING to promote STING phosphorylation and translocation.15 In our study, we found that IFI16 was required for IFN-γ-mediated STING activation. Upon IFN-γ stimulation, IFI16 translocates from the nucleus to the cytosol. We did not observe an association between IFI16 and STING or cGAS (data not shown). Although the knockdown of IFI16 reduced the enzymatic activity of cGAS, the underlying mechanisms remain largely unclear.
Notably, STING activation can often be suppressed by genetic mutations or direct epigenetic silencing of STING. Kitajima and coworkers reported that STING expression is silenced in KRAS-LKB1(KL) mutant lung cancers. LKB1 loss results in epigenetic level suppression of STING expression.60 A recent study reported that inhibitors of MEK and PARP induce DNA damage and STING signaling in KRAS mutant cells but not A549 cells.61 In our study, we used A549 cells, which harbor mutations in KRAS and LKB1. As expected, we found that STING expression in A549 cells was much lower than that in HCC827 cells. Interestingly, STING expression was increased in both the A549 and HCC827 cells in response to IFN-γ treatment in a STAT1-dependent manner. Our findings provide a rationale for further developing combination therapy to effectively improve cGAS-STING activity in lung cancer cells with intrinsically low STING expression.
Using an in vitro assay of lung cancer cell lines and recombinant IFN-γ, for the first time, we demonstrated the role of IFN-γ in the induction of DNA damage and the cellular response to DNA damage in a stepwise manner. More importantly, we found that the activation of T cells in PBMCs or tumors by anti-CD3 mAb could induce DNA damage and trigger STING signaling in cancer cells. These effects were abolished by IFN-γ neutralization. Activation of the STING pathway leads to IFN production and T cell priming and may play a promising role in cancer immunotherapy. However, to date, precisely how activated STING signaling affects the tumor immune microenvironment remains unclear and warrants further investigation. Our results suggest that IFN-γ could alter the tumor microenvironment and is connected to innate immunity via the induction of DNA damage and activation of STING signaling in cancer cells. These findings expand our understanding of IFN-γ-mediated signaling in cancer cells.
Supplementary Material
Funding Statement
This work was supported by the National Natural Science Foundation of China (Grant numbers 81874168, 82173103, and 81672808or L.L.).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data and materials availability:
All data are available in the main text.
Author contributions:
Conceptualization: HX, YC, LL, XF
Methodology: HX, BW, SH, BH, CF, LL
Investigation: HX, YX, ZY, BW, SH, LL
Visualization: HX, YX, LL
Funding acquisition: LL
Project administration: LL, XF
Supervision: LL, XF
Writing-original draft: HX, LL
Writing-review & editing: HX, YX, ZY, BW, SH, YC, LL
Supplementary material
Supplemental data for this article can be accessed on the publisher’s website
References
- 1.Mathew M, Enzler T, Shu CA, Rizvi N.. Rizvi combining chemotherapy with PD-1 blockade in NSCLC. Pharmacol Ther. 2018;186:130–16. doi: 10.1016/j.pharmthera.2018.01.003. [DOI] [PubMed] [Google Scholar]
- 2.Reck M, Papadimitrakopoulou VA, Cappuzzo F, Jotte R, Mok TSK, Sandler A, Waterkamp D, Coleman S, Asakawa T, Socinski M, et al. Phase III clinical trials in chemotherapy-naive patients with advanced NSCLC assessing the combination of atezolizumab and chemotherapy. Annals Oncology. 2016;27:vi451. doi: 10.1093/annonc/mdw383.94. [DOI] [Google Scholar]
- 3.Herzberg B, Campo MJ, Gainor J. Immune checkpoint inhibitors in non-small cell lung cancer. Oncologist. 2017;22(1):81–88. doi: 10.1634/theoncologist.2016-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishino M, Ramaiya NH, Hatabu H, Hodi F. Monitoring immune-checkpoint blockade: response evaluation and biomarker development. Nat Rev Clin Oncol. 2017;14(11):655–668. doi: 10.1038/nrclinonc.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Niu M, Yi M, Li N, Luo S, Wu K. Predictive biomarkers of anti-PD-1/PD-L1 therapy in NSCLC. Exp Hematol Oncol. 2021;10(1). doi: 10.1186/s40164-021-00211-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Della Corte CM, Sen T, Gay CM, Ramkumar K, Diao L, Cardnell RJ, Rodriguez BL, Stewart CA, Papadimitrakopoulou VA, Gibson L, et al. STING pathway expression identifies NSCLC with an immune-responsive phenotype. J Thoracic Oncology. 2020;15(5):777–791. doi: 10.1016/j.jtho.2020.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mouw KW, Goldberg MS, Konstantinopoulos PA, D A. D’Andrea DNA damage and repair biomarkers of immunotherapy response. Cancer Discov. 2017;7(7):675–693. doi: 10.1158/2159-8290.CD-17-0226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paludan SR. Activation and regulation of DNA-driven immune responses. Micro and Mole Bio Rev. 2015;79(2):225–241. doi: 10.1128/MMBR.00061-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao P, Ascano M, Wu Y, Barchet W, Gaffney B, Zillinger T, Serganov A, Liu Y, Jones R, Hartmann G, et al. Cyclic G(2 ‘,5 ‘) pA(3 ‘,5 ‘)p is the metazoan second messenger produced by DNA-Activated Cyclic GMP-AMP synthase. Cell. 2013;153(5):1094–1107. doi: 10.1016/j.cell.2013.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun L, Wu J, Du F, Chen X, Chen Z. Cyclic GMP-AMP synthase is a cytosoLIC DNA sensor that activates the type I interferon pathway. Science. 2013;339(6121):786–791. doi: 10.1126/science.1232458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ablasser A, Goldeck M, Cavlar T, Deimling T, Witte G, Röhl I, Hopfner K-P, Ludwig J, Hornung V. cGAS produces a 2 ‘-5 ‘-linked cyclic dinucleotide second messenger that activates STING. Nature. 2013;498(7454):380-+. doi: 10.1038/nature12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jin T, Perry A, Jiang J, Smith P, Curry J, Unterholzner L, Jiang Z, Horvath G, Rathinam V, Johnstone R, et al. Structures of the HIN domain: DNA complexes reveal ligand binding and activation mechanisms of the AIM2 inflammasome and IFI16 receptor. Immunity. 2012;36(4):561–571. doi: 10.1016/j.immuni.2012.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jakobsen MR, Bak RO, Andersen A, Berg RK, Jensen SB, Jin T, Laustsen A, Hansen K, Ostergaard L, Fitzgerald KA, et al. IFI16 senses DNA forms of the lentiviral replication cycle and controls HIV-1 replication. Proceedings of the National Academy of Sciences of the United States of America,110: E4571–E4580,2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Morrone SR, Wang T, Constantoulakis LM, Hooy RM, Delannoy MJ, Sohn J. Cooperative assembly of IFI16 filaments on dsDNA provides insights into host defense strategy. Proceedings of the National Academy of Sciences of the United States of America,111: E62–E71,2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Almine JF, O’Hare CAJ, Dunphy G, Haga IR, Naik RJ, Atrih A, Connolly DJ, Taylor J, Kelsall IR, Bowie AG, et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat Commun. 2017;8(1). doi: 10.1038/ncomms14392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jonsson KL, Laustsen A, Krapp C, Skipper KA, Thavachelvam K, Hotter D, Egedal JH, Kjolby M, Mohammadi P, Prabakaran T, et al. IFI16 is required for DNA sensing in human macrophages by promoting production and function of cGAMP. Nat Commun. 2017;8(1). doi: 10.1038/ncomms14391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ho SSW, Zhang WL, Tan N, Khatoo M, Suter M, Tripathi S, Cheung FG, Lim W, Tan P, Ngeow J, et al. The DNA structure-specific endonuclease MUS81 mediates DNA sensor STING-dependent host rejection of prostate cancer cells. Immunity. 2016;44(5):1177–1189. doi: 10.1016/j.immuni.2016.04.010. [DOI] [PubMed] [Google Scholar]
- 18.Lu C, Paschall AV, Shi H, Savage N, Waller JL, Sabbatini ME, Oberlies NH, Pearce C, Liu K. The MLL1-H3K4me3 axis-mediated PD-L1 expression and pancreatic cancer immune evasion. JNCI-J Natl Cancer Inst. 2017;109(6). doi: 10.1093/jnci/djw283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Takashima K, Takeda Y, Oshiumi H, Shime H, Okabe M, Ikawa M, Matsumoto M, Seya T. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. Biochem Biophys Res Commun. 2016;478(4):1764–1771. doi: 10.1016/j.bbrc.2016.09.021. [DOI] [PubMed] [Google Scholar]
- 20.Dunphy G, Flannery SM, Almine JF, Connolly DJ, Paulus C, Jønsson KL, Jakobsen MR, Nevels MM, Bowie AG, Unterholzner L, et al. Non-canonical activation of the DNA sensing adaptor STING by ATM and IFI16 mediates NF-kappa B signaling after nuclear DNA damage. Mol Cell. 2018;71(5):745-+. doi: 10.1016/j.molcel.2018.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng L, Liang H, Xu M, Yang X, Burnette B, Arina A, Li X-D, Mauceri H, Beckett M, Darga T, et al. STING-dependent cytosolic DNA sensing promotes radiation-induced type I interferon-dependent antitumor immunity in immunogenic tumors. Immunity. 2014;41(5):843–852. doi: 10.1016/j.immuni.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest. 2017;127(8):2930–2940. doi: 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. Loss of IFN-gamma pathway genes in tumor cells as a mechanism of resistance to anti-CTLA-4 therapy. Cell. 2016;167(2):397-+. doi: 10.1016/j.cell.2016.08.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shin DS, Zaretsky JM, Escuin-Ordinas H, Garcia-Diaz A, Hu-Lieskovan S, Kalbasi A, Grasso CS, Hugo W, Sandoval S, Torrejon DY, et al. Primary resistance to PD-1 blockade mediated by JAK1/2 mutations. Cancer Discov. 2017;7(2):188–201. doi: 10.1158/2159-8290.CD-16-1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schoenborn JR, and Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. In: Alt FW, et al., editors. Advances in immunology. Vol. 96; 2007. p. 41–101.Alt FW, et al. ed .2007. :. [DOI] [PubMed] [Google Scholar]
- 26.Burke F, East N, Upton C, Patel K, Balkwill F. Interferon gamma induces cell cycle arrest and apoptosis in a model of ovarian cancer: enhancement of effect by batimastat. Eur J Cancer. 1997;33(7):1114–1121. doi: 10.1016/S0959-8049(97)88065-3. [DOI] [PubMed] [Google Scholar]
- 27.Shin E-C, Ahn JM, Kim CH, Choi Y, Ahn YS, Kim H, Kim SJ, Park JH. IFN-gamma induces cell death in human hepatoma cells through a trail/death receptor-mediated apoptotic pathway. Int J Cancer. 2001;93(2):262–268. doi: 10.1002/ijc.1310. [DOI] [PubMed] [Google Scholar]
- 28.Fang C, Weng T, Hu S, Yuan Z, Xiong H, Huang B, Cai Y, Li L, Fu X. IFN-gamma-induced ER stress impairs autophagy and triggers apoptosis in lung cancer cells. Oncoimmunology. 2021;10(1). doi: 10.1080/2162402X.2021.1962591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hoekstra ME, Bornes L, Dijkgraaf FE, Philips D, Pardieck IN, Toebes M, Thommen DS, van Rheenen J, Schumacher TNM. Long-distance modulation of bystander tumor cells by CD8(+) T-cell-secreted IFN-gamma. Nat Cancer. 2020;1(3):291-+. doi: 10.1038/s43018-020-0036-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thibaut R, Bost P, Milo I, Cazaux M, Lemaître F, Garcia Z, Amit I, Breart B, Cornuot C, Schwikowski B, et al. Bystander IFN-gamma activity promotes widespread and sustained cytokine signaling altering the tumor microenvironment. Nat Cancer. 2020;1(3):302-+. doi: 10.1038/s43018-020-0038-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jaiswal M, LaRusso NF, Burgart LJ, J G. Gores inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res. 2000;60:184–190. [PubMed] [Google Scholar]
- 32.Hubackova S, Kucerova A, Michlits G, Kyjacova L, Reinis M, Korolov O, Bartek J, Hodny Z. IFN gamma induces oxidative stress, DNA damage and tumor cell senescence via TGF beta/SMAD signaling-dependent induction of Nox4 and suppression of ANT2. Oncogene. 2016;35(10):1236–1249. doi: 10.1038/onc.2015.162. [DOI] [PubMed] [Google Scholar]
- 33.Ravindranathan P, Lee T-K, Yang L, Centenera MM, Butler L, Tilley WD, Hsieh J-T, Ahn J-M, Raj GV. Peptidomimetic targeting of critical androgen receptor-coregulator interactions in prostate cancer. Nat Commun. 2013;4(1). doi: 10.1038/ncomms2912. [DOI] [PubMed] [Google Scholar]
- 34.Blackford AN, Jackson ATM SP. ATR, and DNA-PK: The trinity at the heart of the DNA damage response. Mol Cell. 2017;66(6):801–817. doi: 10.1016/j.molcel.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 35.Wang Y, Yu X, Song H, Feng D, Jiang Y, Wu S, Geng J. The STAT-ROS cycle extends IFN-induced cancer cell apoptosis. Int J Oncol. 2018;52(1):305–313. doi: 10.3892/ijo.2017.4196. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe Y, Suzuki O, Haruyama T, Akaike T. Interferon-gamma induces reactive oxygen species and endoplasmic reticulum stress at the hepatic apoptosis. J Cell Biochem. 2003;89(2):244–253. doi: 10.1002/jcb.10501. [DOI] [PubMed] [Google Scholar]
- 37.Sonoda J, Laganière J, Mehl IR, Barish GD, Chong L-W, Li X, Scheffler IE, Mock DC, Bataille AR, Robert F, et al. Nuclear receptor ERR alpha and coactivator PGC-1 beta are effectors of IFN-gamma-induced host defense. Genes Dev. 2007;21(15):1909–1920. doi: 10.1101/gad.1553007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nguyen T, Brunson D, Crespi CL, Penman BW, Wishnok JS, Tannenbaum SR. DNA damage and mutation in human cells exposed to nitric oxide in vitro. Proceedings of the National Academy of Sciences of the United States of America.1992;89:3030–3034, [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kiziltepe T, Hideshima T, Ishitsuka K, Ocio EM, Raje N, Catley L, Li C-Q, Trudel LJ, Yasui H, Vallet S, et al. JS-K, a GST-activated nitric oxide generator, induces DNA double-strand breaks, activates DNA damage response pathways, and induces apoptosis in vitro and in vivo in human multiple myeloma cells. Blood. 2007;110(2):709–718. doi: 10.1182/blood-2006-10-052845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei L, Gravitt PE, Song H, Maldonado AM, Ozbun MA. Nitric oxide induces early viral transcription coincident with increased DNA damage and mutation rates in human papillomavirus-infected cells. Cancer Res. 2009;69(11):4878–4884. doi: 10.1158/0008-5472.CAN-08-4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C, Gong G, Sheh A, Muthupalani S, Bryant EM, Puglisi DA, Holcombe H, Conaway EA, Parry NAP, Bakthavatchalu V, et al. Interleukin-22 drives nitric oxide-dependent DNA damage and dysplasia in a murine model of colitis-associated cancer. Mucosal Immunol. 2017;10(6):1504–1517. doi: 10.1038/mi.2017.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chan ED, H DW. IFN-γ + LPS induction of iNOS is modulated by ERK, JNK/SAPK, and p38 mapk in a mouse macrophage cell line. American J Physiology-Cell Physiology. 2001;280(3):C441–C450. doi: 10.1152/ajpcell.2001.280.3.C441. [DOI] [PubMed] [Google Scholar]
- 43.Harding SM, Benci JL, Irianto J, Discher DE, Minn AJ, Greenberg RA. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature. 2017;548(7668):466-+. doi: 10.1038/nature23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ishikawa H, Barber G. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–U674. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Liu S, Guan W. STING signaling promotes apoptosis, necrosis, and cell death: An overview and update. Mediators Inflamm. 2018;2018:1–4. doi: 10.1155/2018/1202797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Murthy AMV, Robinson N, Kumar S. Crosstalk between cGAS-STING signaling and cell death. Cell Death Differ. 2020;27(11):2989–3003. doi: 10.1038/s41418-020-00624-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackenzie KJ, Carroll P, Martin C-A, Murina O, Fluteau A, Simpson DJ, Olova N, Sutcliffe H, Rainger JK, Leitch A, et al. cGAS surveillance of micronuclei links genome instability to innate immunity. Nature. 2017;548(7668):461-+. doi: 10.1038/nature23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li T, Diner BA, Chen J, and M I. Cristea acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:10558–10563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sun H, Huang Y, Mei S, Xu F, Liu X, Zhao F, Yin L, Zhang D, Wei L, Wu C, et al. A nuclear export signal is required for cGAS to sense cytosolic DNA. Cell Rep. 2021;34(1):108586. doi: 10.1016/j.celrep.2020.108586. [DOI] [PubMed] [Google Scholar]
- 50.Goldstein M, Kastan MB. The DNA damage response: implications for tumor responses to radiation and chemotherapy. Annu Rev. Med. 2015;66:129–143. [DOI] [PubMed] [Google Scholar]
- 51.Peng W, Liu C, Xu C, Lou Y, Chen J, Yang Y, Yagita H, Overwijk WW, Lizée G, Radvanyi L, et al. PD-1 blockade enhances T-cell migration to tumors by elevating IFN-gamma inducible chemokines. Cancer Res. 2012;72(20):5209–5218. doi: 10.1158/0008-5472.CAN-12-1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Das R, et al. Combination therapy with anti-CTLA-4 and anti-PD-1 leads to distinct immunologic changes in vivo. J Immunology. 2015;194(3):950–959. doi: 10.4049/jimmunol.1401686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murata M, Thanan R, Ma N, Kawanishi S. Role of nitrative and oxidative DNA damage in inflammation-related carcinogenesis. J Biomed Biotechnol. 2012;2012:1–11. doi: 10.1155/2012/623019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chabanon RM, Rouanne M, Lord CJ, Soria J-C, Pasero P, Postel-Vinay S. Targeting the DNA damage response in immuno-oncology: developments and opportunities. Nat Rev Cancer. 2021;21(11):701–717. doi: 10.1038/s41568-021-00386-6. [DOI] [PubMed] [Google Scholar]
- 55.Reislander T, Groelly FJ, Tarsounas M. DNA Damage and Cancer Immunotherapy: a STING in the tale. Mol Cell. 2020;80(1):21–28. doi: 10.1016/j.molcel.2020.07.026. [DOI] [PubMed] [Google Scholar]
- 56.Li C, Liu W, Wang F, Hayashi T, Mizuno K, Hattori S, Fujisaki H, Ikejima T. DNA damage-triggered activation of cGAS-STING pathway induces apoptosis in human keratinocyte HaCaT cells. Mol Immunol. 2021;131:180–190. doi: 10.1016/j.molimm.2020.12.037. [DOI] [PubMed] [Google Scholar]
- 57.Luecke S, Holleufer A, Christensen MH, Jonsson KL, Boni GA, Sorensen LK, Johannsen M, Jakobsen MR, Hartmann R, Paludan SR. cGAS is activated by DNA in a length-dependent manner. EMBO Rep. 2017;18(10):1707–1715. doi: 10.15252/embr.201744017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pathare GR, Decout A, Glück S, Cavadini S, Makasheva K, Hovius R, Kempf G, Weiss J, Kozicka Z, Guey B, et al. Structural mechanism of cGAS inhibition by the nucleosome. Nature. 2020;587(7835):668-+. doi: 10.1038/s41586-020-2750-6. [DOI] [PubMed] [Google Scholar]
- 59.Khan SU, Singh M, Valavoor S, Khan MU, Lone AN, Khan MZ, Khan MS, Mani P, Kapadia SR, Michos ED, et al. Dual antiplatelet therapy after percutaneous coronary intervention and drug-eluting stents a systematic review and network meta-analysis. Circulation. 2020;142(15):1425–1436. doi: 10.1161/CIRCULATIONAHA.120.046308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kitapma S, Ivanova E, Guo S, Yoshida R, Campisi M, Sundararaman SK, Tange S, Mitsuishi Y, Thai C, Masudaa S, et al. Suppression of STING associated with LKB1 loss in KRAS-driven lung cancer. Cancer Discov. 2019;9(1):34–45. doi: 10.1158/2159-8290.CD-18-0689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang B, Li X, Fu Y, Guo E, Ye Y, Li F, Liu S, Xiao R, Liu C, Lu F, et al. MEK inhibition remodels the immune Landscape Of Mutant KRAS tumors to overcome resistance to PARP and immune checkpoint inhibitors. Cancer Res. 2021;81(10):2714–2729. doi: 10.1158/0008-5472.CAN-20-2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.