

SUMMARY
Intrahepatic cholangiocarcinoma (ICC) contains abundant myofibroblasts derived from hepatic stellate cells (HSCs) through an activation process mediated by TGF-β. To determine the role of programmed death-ligand 1 (PD-L1) in myofibroblastic activation of HSCs, we disrupted PD-L1 of HSCs by shRNA or anti-PD-L1 antibody. We find that PD-L1, produced by HSCs, is required for HSC activation by stabilizing TGF-β receptors I (TβRI) and II (TβRII). While the extracellular domain of PD-L1 (amino acids 19–238) targets TβRII protein to the plasma membrane and protects it from lysosomal degradation, a C-terminal 260-RLRKGR-265 motif on PD-L1 protects TβRI mRNA from degradation by the RNA exosome complex. PD-L1 is required for HSC expression of tumor-promoting factors, and targeting HSC PD-L1 by shRNA or Cre/loxP recombination suppresses HSC activation and ICC growth in mice. Thus, myofibroblast PD-L1 can modulate the tumor microenvironment and tumor growth by a mechanism independent of immune suppression.
Graphical Abstract
In brief
Sun et al. find that PD-L1, originally thought to be expressed by cancer cells and immune cells, is expressed by the myofibroblasts of liver cancer and that the myofibroblast PD-L1 can modulate the hepatic tumor microenvironment and cholangiocarcinoma growth by a mechanism independent of the PD-L1/PD-1-mediated immune suppression.
INTRODUCTION
Intrahepatic cholangiocarcinoma (ICC) is the second most common lethal liver malignancy originated from the biliary epithelium with limited treatment options. ICC development and progression are determined by genetic and non-genetic factors in cancer cells and also by those in the hepatic microenvironment (Fingas et al., 2011; Cadamuro et al., 2013; Zhang et al., 2020). ICC is surrounded by a dense desmoplastic stroma with the myofibroblasts as a major cellular component, and the reciprocal crosstalk between cancer cells and the myofibroblasts influences ICC growth, metastasis, immunosuppression, and chemo-resistance (Cadamuro et al., 2019; Fingas et al., 2011). The myofibroblasts within the hepatic tumor microenvironment are mainly derived from hepatic stellate cells (HSCs) through an activation process mediated by TGF-β (Kang et al., 2011, 2015; Liu et al., 2013). Elucidating the mechanism of HSC activation may lead to new targets to suppress the hepatic tumor microenvironment and ICC.
Programmed death-ligand 1 (PD-L1, also named CD274 or B7-H1) is an immune checkpoint protein modulating cancer immune evasion by interacting with its receptor, programmed cell death protein 1 (PD-1, also called PDCD1 or CD279), on the cell surface of B or T cells (Finger et al., 1997; Shi et al., 2013; Thibult et al., 2013; Dong et al., 1999). PD-L1’s binding to PD-1 leads to T cell apoptosis and inhibited T cell proliferation and cytotoxic activity, contributing to immune escape of cancer (Dong et al., 1999, 2002). Pharmacologic compounds targeting the PD-1/PD-L1 axis have been developed and approved by FDA for the treatment of various forms of cancer (Jelinek et al., 2018; Han et al., 2020). PD-L1 is detected in cancer cells and immune cells, including tumor-associated macrophages (Loeuillard et al., 2020). In addition to immune modulation, PD-L1 was found in cancer cells where it regulates cell apoptosis, glucose metabolism, and autophagy through activating tumor-intrinsic signals (Azuma et al., 2008; Chang et al., 2015). PD-L1 in the nucleus regulates a cohesion complex that ensures proper cohesion and segregation of sister chromatids for genomic stability maintenance (Yu et al., 2020). In addition, PD-L1 binds to mRNA of a panel of DNA damage-related genes to protect them from RNA exosome-mediated degradation (Tu et al., 2019). Although activated HSC/myofibroblasts are a major component of the pro-tumor microenvironment of the liver, the role of PD-L1 in HSCs, particularly in the HSC activation process mediated by TGF-β, remains uninvestigated.
TGF-β induces HSCs to express α-smooth muscle actin (αSMA), fibronectin, and type I collagen, markers of myofibroblastic activation of HSCs (Chen et al., 2020; Liu et al., 2020). In addition, it promotes HSCs to form actin-based stress fibers, a characteristic of the myofibroblasts. We first used these TGF-β signaling readouts to test whether PD-L1 regulates myofibroblastic activation of HSCs induced by TGF-β and found that targeting PD-L1 of HSCs suppresses the protein level of TGF-β receptors I (TβRI) and II (TβRII), phosphorylation of SMAD3, and myofibroblastic activation of HSCs induced by TGF-β1. Since HSCs do not express PD-1 receptor, these effects of PD-L1 are independent of the PD-L1/PD-1 signaling axis. Interestingly, PD-L1 regulates TβRI and TβRII by two distinct mechanisms; PD-L1 protects TβRII protein from lysosome-mediated degradation, whereas PD-L1 protects TβRI mRNA from degradation by the RNA exosome complex. RNA sequencing revealed that TGF-β1 promotes HSCs to express a panel of tumor-promoting paracrine factors in a PD-L1-dependent manner; and, functionally, targeting PD-L1 of HSCs impairs the tumor-promoting effect of HSCs in vitro and in subcutaneous and orthotopic ICC implantation mouse models. Thus, PD-L1 of HSCs represents a target for suppressing the hepatic tumor microenvironment and ICC.
RESULTS
Targeting PD-L1 suppresses myofibroblastic activation of HSCs induced by TGF-β1
We first created PD-L1 knockdown HSCs and used the cells to test if PD-L1 influenced myofibroblastic activation of HSCs induced by TGF-β1. Lentiviruses encoding PD-L1 shRNA-1 or PD-L1 shRNA-2 were used to transduce HSCs to knockdown PD-L1, and lentiviruses encoding non-targeting shRNA (NT shRNA) were used as the control. Cells were then stimulated with TGF-β1 (5 ng/mL) and collected for western blot (WB) and immunofluorescence (IF) for stellate cell activation markers, αSMA, fibronectin, and type I collagen. WB revealed that TGF-β1 stimulation led to increased expression of αSMA, fibronectin, and collagen 1 protein in control cells, and this effect of TGF-β1 was abolished in PD-L1 knockdown HSCs (p < 0.05, Figures 1A and S1A). IF for αSMA confirmed that, while more than 50% of control HSCs were activated into myofibroblasts by TGF-β1, less than 20% of PD-L1 knockdown HSCs were activated under a same condition (p < 0.001, Figures 1B and S1B). We have previously showed that anti-PD-L1 antibody (clone H1A, generated in Dr. Haidong Dong’s laboratory) targets amino acids (aa) 20–32 of the PD-L1 extracellular domain and blocks the interaction between PD-L1 and CKLF-like MARVEL transmembrane domain-containing protein 6 (CMTM6), leading to lysosomal degradation of PD-L1 in cancer cells (Tu et al., 2019). Therefore, HSCs pre-incubated with H1A (40 μg/mL) for 6 h were co-stimulated with TGF-β1 (5 ng/mL) to test whether HSC activation was influenced by H1A. Isotype-matched control IgG was used as the control. Consistent with the data of PD-L1 knockdown, H1A reduced PD-L1 protein level and myofibroblastic activation of HSCs induced by TGF-β1 (p < 0.05, Figures 1C and 1D). Thus, PD-L1 of HSCs is required for TGF-β1-mediated activation of HSCs into myofibroblasts in vitro.
Figure 1. Targeting PD-L1 protein suppresses myofibroblastic activation of HSCs induced by TGF-β1 by reducing TGF-β receptor I (TβRI) and II (TβRII) protein levels.

(A) Primary human HSCs, transduced with either control shRNA (NT shRNA) lentiviruses or PD-L1 shRNA-1 lentiviruses, were stimulated without or with TGF-β1 (5 ng/mL) for 24 h and collected for western blot (WB) for expression of HSC activation markers. The blot was re-probed for Hsc70 for protein loading control and densitometry data are shown on the right. TGF-β1-stimulated upregulation of a-smooth muscle actin (αSMA), fibronectin, and collagen 1A1 was reduced by PD-L1 knockdown in HSCs. *p < 0.05, ***p < 0.001 by ANOVA, n = 3 repeats.
(B) HSCs described in (A) were collected for immunofluorescence (IF) for αSMA. The percentage of activated-HSC/myofibroblasts was higher in TGF-β1-stimulated control HSCs than TGF-β1-stimulated PD-L1 knockdown HSCs. ***p < 0.001 by ANOVA, n = 5 randomly selected microscopic fields per group, each containing 50–100 cells. Scale bar, 100 μm.
(C and D) HSCs pre-incubated with non-immune IgG (control) or anti-PD-L1 antibody (clone H1A, 40 μg/mL) for 6 h were co-stimulated without orwith TGF-β1 (5 ng/mL) for 24 h. Cells were collected for WB and IF for HSC activation markers. Targeting PD-L1 protein by H1A suppressed myofibroblastic activation of HSCs induces by TGF-β1. *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA, n = 3 repeats for WB, and for IF n = 5 microscopic fields per group, each containing 50–100 cells. Scale bar, 100 μm.
(E) Control and PD-L1 knockdown HSCs were stimulated without or with TGF-β1 (5 ng/mL) for 30 min and collected for WB for phosphorylation of SMAD3 (P-SMAD3). The blot was re-probed for GAPDH for protein loading control. TGF-β1-induced P-SMAD3 was reduced by PD-L1 knockdown in HSCs. **p < 0.01 by ANOVA, n = 3 repeats.
(F) PD-L1 of HSCs was knocked down by three different shRNAs and cells were collected for WB. Both TβRI and TβRII protein level of HSCs were reduced by PD-L1 knockdown. **p < 0.01, ***p < 0.001 by ANOVA, n = 3 repeats.
(G and H) PD-L1 knockdown HSCs or H1A-incubated HSCs were incubated with lysosomal inhibitors (bafilomycin [BAF, 10 nM] or E64d [10 μg/mL] + Pepstatin A [PepA, 10 μg/mL]) or proteasomal inhibitor MG132 (25 μM) and collected for WB for TβRI and TβRII. The TβRII protein level in PD-L1-deficient cells was rescued by the lysosomal inhibitors, but not by the proteasomal inhibitor. The PD-L1 protein in H1A antibody-incubated HSCs was also rescued by the lysosomal inhibitors. The TβRI protein level was not rescued by either inhibitor. *p < 0.05, **p < 0.01, ***p < 0.001 by ANOVA, n = 3. All data are represented as mean ± SEM.
Targeting PD-L1 reduces TβRI and TβRII protein levels of HSCs through distinct mechanisms
To understand how PD-L1 deficiency suppressed HSC activation, we performed WB for phosphorylation of SMAD3 (P-SMAD3) induced by TGF-β1, an early signaling event of the TGF-β1 signaling pathway. Two different PD-L1 shRNAs and H1A consistently reduced the P-SMAD3 level induced by TGF-β1 in HSCs (p < 0.01, Figures 1E, S1C, and S1D), suggesting that targeting PD-L1 may inhibit TGF-β1 signaling at the TGF-β receptor level. WB for TβRI and TβRII was thus performed, which revealed that PD-L1 knockdown, by three different shRNAs, consistently reduced TβRI and TβRII protein levels of HSCs (p < 0.01, Figure 1F). Incubation of the cells with the inhibitors targeting lysosomes (bafilomycin [10 nM] or E64d [10 μg/mL] + pepstatin A [10 μg/mL]) or proteasomes (MG132, 25 μM) demonstrated that the lysosomal inhibitors, but not the proteasomal inhibitor MG132, prevented TβRII from downregulation by PD-L1 deficiency (p < 0.05, Figures 1G and 1H). In contrast, neither prevented TβRI from downregulation. Consistent with the fact that the H1A antibody induces lysosomal degradation of PD-L1 (Tu et al., 2019), the lysosomal inhibitors also prevented PD-L1 downregulation by H1A (p < 0.01, Figure 1H). Interestingly, quantitative real-time RT-PCR (qRT-PCR) revealed that TβRI mRNA levels were reduced, whereas TβRII mRNA levels were elevated by PD-L1 knockdown in HSCs (p < 0.05, Figure 2A). These data suggest distinct mechanisms by which PD-L1 regulates TβRI and TβRII in HSCs.
Figure 2. PD-L1 knockdown leads to ubiquitination and lysosomal degradation of TβRII.

(A) Control and PD-L1 knockdown HSCs were collected for real-time quantitative RT-PCR (qRT-PCR) for TβRI and TβRII mRNA. Each was detected by five different primer pairs by qPCR. PD-L1 knockdown in HSCs reduced TβRI mRNA level (left), but not TβRII mRNA level (right). *p < 0.05, **p < 0.01 by ANOVA, n = 3.
(B) Cycloheximide (40 μg/mL) was added in cell culture to inhibit protein translation and cells were collected for WB for endogenous TβRII at different time points. Time-dependent downregulation of endogenous TβRII protein was accelerated in PD-L1 knockdown HSCs compared with control HSCs. *p < 0.05 by ANOVA, n = 4.
(C) HSCs expressing TβRII-HA fusion protein were transduced with NT shRNA or PD-L1 shRNA lentiviruses and incubated with either DMSO (control) or bafilomycin (10 nM). Cells were then collected for immunoprecipitation (IP) with anti-HA followed by WB for ubiquitin (Ub) to detected ubiquitination of TβRII-HA. In both DMSO- and bafilomycin-incubated groups, TβRII-HA ubiquitination level was higher in PD-L1 knockdown HSCs than in control HSCs. Data are representative of multiple repeats with similar results.
(D) HSCs expressing TβRII-HA fusion protein were transduced with control or PD-L1 shRNA lentiviruses. Serum-starved cells were then stimulated with TGF-β1 and collected at different time points, 0, 15, and 45 min, for double IF for HA (green) and LAMP-1 (marker of lysosomes, red). The rate of TβRII-HA/LAMP-1 co-localization (arrows, yellow) was time-dependently increased by TGF-β1 in control HSCs, but not in PD-L1 knockdown HSCs. The rate of TβRII-HA/LAMP-1 co-localization was higher in PD-L1 knockdown HSCs than in control HSCs at the basal condition. ***p < 0.001 by ANOVA, n > 10 cells per group. Scale bar, 20 μm. All data are represented as mean ± SEM except data in (C).
To rule out the off-target effect of PD-L1 shRNA on TβRI and TβRII of HSCs, primary HSCs were isolated from mice in which the cd274 gene, encoding murine PD-L1, is flanked by two loxP sites (Sage et al., 2018) and Cre adenoviruses (AdCre) were used to delete the cd274 gene in vitro. HSCs transduced with GFP adenoviruses (AdGFP) were used as the control. This Cre/loxP-mediated gene knockout approach generated consistent data; PD-L1 deletion reduced TβRI and TβRII protein levels of murine HSCs (p < 0.01), and also reduced TβRI mRNA (p < 0.05) but not TβRII mRNA levels (Figure S2A). Thus, PD-L1 may regulate TβRII at the protein level and TβRI at the mRNA level in HSCs; this hypothesis was tested by the experiments below.
Targeting PD-L1 leads to ubiquitination, lysosomal targeting, and degradation of TβRII
To support that PD-L1 regulates TβRII protein, we first assessed the stability of TβRII protein in control and PD-L1 knockdown HSCs. Cycloheximide (40 μg/mL) was added to HSC culture to block protein synthesis and cells were collected at different time points for WB to quantitate endogenous TβRII protein levels. Chasing TβRII protein in the presence of cycloheximide demonstrated that the stability of TβRII in HSCs was reduced by PD-L1 knockdown; the half-life of TβRII was 82 min in control HSCs and shortened to 49 min in PD-L1 knockdown HSCs (p < 0.05, Figure 2B). Thus, targeting PD-L1 reduces the stability of TβRII protein in HSCs.
Since targeting of TβRII for degradation requires tagging TβRII protein with ubiquitin (Liu et al., 2013; Chen et al., 2020), we performed immunoprecipitation (IP) to test whether PD-L1 knockdown promoted TβRII ubiquitination in HSCs. All commercial anti-TβRII antibodies are poor for IP, so we generated HSCs expressing TβRII-HA fusion protein by retroviral transduction to test the hypothesis (Liu et al., 2013; Chen et al., 2020). Anti-HA antibody was used to pull down TβRII-HA from HSC lysates and WB for ubiquitin was followed to quantitate TβRII tagged by ubiquitin. As expected, TβRII ubiquitination level in control cells was low due to degradation of TβRII and it was elevated by blocking lysosomal function by bafilomycin (Figure 2C). Importantly, TβRII ubiquitination levels were markedly higher in PD-L1 knockdown HSCs than in control HSCs for both DMSO- and bafilomycin-incubated cells (Figure 2C). These data suggest that PD-L1 knockdown indeed promotes ubiquitination of TβRII in HSCs.
We next analyzed lysosomal targeting of TβRII induced by TGF-β1 in control and PD-L1-deficient HSCs. Control and PD-L1 knockdown HSCs, expressing TβRII-HA, were stimulated with TGF-β1 (5 ng/mL) for different times (0, 15, and 45 min) and collected for double IF for HA (green) and lysosomal-associated membrane protein 1 (LAMP-1, marker of late endosome/lysosomes) (red) (Liu et al., 2013; Chen et al., 2020). TGF-β1 stimulation led to a time-dependent increase of TβRII-HA/LAMP-1 co-localization in control HSCs (arrows, yellow) (p < 0.001, Figure 2D), but not in PD-L1 knockdown HSCs. At the basal level, the rate of TβRII-HA/LAMP-1 co-localization was higher in PD-L1 knockdown HSCs than in control HSCs (p < 0.001, Figure 2D), supporting that PD-L1 knockdown promotes the sorting of TβRII-HA to the lysosomes. At the basal condition, HSCs incubated with the H1A antibody (40 μg/mL) also displayed a higher rate of TβRII-HA/LAMP-1 co-localization than control HSCs, which was further enhanced by bafilomycin (p < 0.05, Figure S2B). Taken together, these data support that targeting PD-L1 leads to ubiquitination of TβRII and subsequent lysosomal targeting and degradation of TβRII in HSCs.
The PD-L1 extracellular domain interacts with TβRII to increase TβRII protein stability
We next performed co-immunoprecipitation (coIP) to test the hypothesis that PD-L1 binds to TβRII to prevent it from lysosomal targeting. Anti-HA was used to pull down TβRII-HA from HSC lysates and WB was followed to detect co-precipitated PD-L1. PD-L1 and TβRII-HA were co-precipitated from HSC lysates by anti-HA antibody, suggesting that both formed a complex in HSCs (Figure 3A, upper). HSCs expressing FLAG-tagged TβRI (TβRI-FLAG) and HA-tagged PD-L1 (HA-PD-L1) were collected for coIP using anti-FLAG antibody, which revealed no co-precipitation of the two proteins (Figure 3A, lower). Thus, PD-L1 forms a complex with TβRII protein, but not TβRI protein, in HSCs.
Figure 3. The extracellular domain of PD-L1 binds to TβRII to protect it from degradation.

(A) Upper: HSCs expressing TβRII-HA fusion protein were subjected to IP using anti-HA and co-precipitated PD-L1 was detected by WB. PD-L1 and TβRII-HA were co-precipitated by anti-HA. Lower: HSCs expressing TβRI-FLAG and HA-PD-L1 fusion proteins were subjected to coIP using anti-FLAG. HA-PD-L1 and TβRI-FLAG were not co-precipitated by anti-FLAG. Data are representative of multiple repeats with similar results.
(B) HSCs expressing TβRII-HA fusion protein were transduced with retroviruses encoding full-length PD-L1 (FL), the extracellular domain of PD-L1 (Ex), or the C-terminal portion of PD-L1 (the transmembrane domain + cytoplasmic domain [T + C]). Cells were collected for coIP using anti-FLAG followed by WB for HA to detect PD-L1/TβRII-HA binding. TβRII-HA was co-precipitated with PD-L1 FL or PD-L1 Ex by anti-FLAG.
(C) HSCs co-expressing TβRII-HA and FLAG-PD-L1 FL were collected for double IF for HA (red) and FLAG (green). TβRII-HA and FLAG-PD-L1 FL co-localized at the plasma membrane (arrows) and in the endosomes (arrowheads) of a HSC. Cell nuclei were stained by DAPI. Scale bar, 20 μm.
(D) FLAG-tagged PD-L1 domains were introduced into PD-L1 knockdown HSCs and cells were collected for WB for TβRII. PD-L1 FL and PD-L1 Ex rescued the TβRII protein level of PD-L1 knockdown HSCs. **p < 0.01 by ANOVA, n = 3.
(E) Control and PD-L1 knockdown HSCs, stimulated without or with TGF-β1 (5 ng/mL) for 6 h, were collected for biotinylation of cell surface protein followed by streptavidin agarose pull down to quantitate TβRII at the plasma membrane of HSCs. TGF-β1 stimulation reduced plasma membrane TβRII and at the basal condition PD-L1 knockdown reduced the plasma membrane TβRII level. **p < 0.01, ***p < 0.001 by ANOVA, n = 3. PM, plasma membrane. Data in (D and E) are represented as mean ± SEM.
Human PD-L1 possesses three domains: an extracellular domain (aa 19–238), a transmembrane domain (aa 239–259), and a cytoplasmic domain (aa 260–290). To investigate which domain binds to TβRII, FLAG-tagged PD-L1 subdomains (Tu et al., 2019) were introduced into HSCs by viral transduction so that HSCs expressing the PD-L1 extracellular domain (PD-L1 Ex) or the C-terminal portion of PD-L1 (PD-L1 transmembrane domain + cytoplasmic domain; PD-L1 T + C) were obtained. HSCs expressing full-length PD-L1 (PD-L1 FL) were used as the control. Anti-FLAG antibody was used to pull down PD-L1 from cell lysates and WB for HA was followed to quantitate associated TβRII-HA, which showed co-precipitation of TβRII-HA with PD-L1 FL or PD-L1 Ex (Figure 3B). Double IF performed with HSCs co-expressing TβRII-HA and FLAG-PD-L1 FL revealed co-localization of the two proteins at the plasma membrane (yellow, arrows) and in the endosomes (arrowheads) (Figure 3C). In regard to the PD-L1 Ex mutant, it was not targeted to the plasma membrane due to the lack of the transmembrane domain but it still co-localized with TβRII-HA in the cytoplasm of HSCs (arrows, Figure S3A). GST pull-down assay confirmed a direct binding between TβRII and the PD-L1 extracellular domains (Figure S3B). Functionally, when the PD-L1 subdomains were expressed in HSCs, PD-L1 FL and PD-L1 Ex, but not PD-L1 T + C, prevented TβRII protein from downregulation by PD-L1 knockdown (p < 0.01, Figure 3D). To investigate whether PD-L1 influences plasma membrane localization of TβRII in HSCs, we performed a biotinylation assay and found that the plasma membrane TβRII level was significantly reduced in PD-L1 knockdown HSCs than in control HSCs (p < 0.01, Figure 3E). It was downregulated drastically by TGF-β1 in both control and PD-L1 knockdown HSCs (p < 0.001, Figure 3E), consistent with the notion that TβRII undergoes endocytosis in response to TGF-β1 stimulation. An in vitro binding assay performed with TβRII and biotinylated TGF-β1 revealed that pre-incubation of TβRII with the PD-L1 extracellular domain did not influence TβRII/TGF-β1 binding (Figure S3C). Taken together, the PD-L1 extracellular domain binds to TβRII at both the transporting vesicles and plasma membrane of HSCs to protect TβRII from lysosomal targeting and degradation.
Knockdown of PD-L1 reduces TβRI mRNA stability of HSCs
In cancer cells, PD-L1 interacts with RNA and enhances the stability of mRNA of genes related to DNA damage responses (Tu et al., 2019). Since TβRI mRNA was reduced by PD-L1 knockdown in HSCs, we next tested whether the stability of TβRI mRNA was impacted by PD-L1 knockdown. Actinomycin D (5 μg/mL), a transcription inhibitor, was added into HSC culture to block gene transcription and cells were collected at different time points for qRT-PCR for TβRI mRNA. In control HSCs, TβRI mRNA was not stable and it was downregulated in a time-dependent manner (Figure 4A). However, it was downregulated much faster in PD-L1 knockdown HSCs than in control HSCs (p < 0.05, Figure 4A), supporting that PD-L1 knockdown indeed reduced TβRI mRNA stability in HSCs.
Figure 4. The C-terminal portion of PD-L1 protects TβRI mRNA from degradation.

(A) Actinomycin D (5 μg/mL) was added in cell culture to block gene transcription and cells were collected for qRT-PCR for TβRI mRNA at different time points. Knockdown of PD-L1 by two different shRNAs consistently accelerated degradation of TβRI mRNA in HSCs. *p < 0.05, **p < 0.01 by ANOVA, n = 3.
(B) Upper: anti-PD-L1 was used for RNA immunoprecipitation (RIP) to pull down PD-L1 protein and co-precipitated RNA was quantitated by qRT-PCR. Non-immune IgG was used as the control. TβRI mRNA co-immunoprecipitated with PD-L1 protein. ***p < 0.001 by ANOVA, n = 3. Lower: fluorescence in situ hybridization for TβRI mRNA (red) and IF for FLAG-PD-L1 FL protein were performed on the same cells. TβRI mRNA and FLAG-PD-L1 FL protein co-localized in the cytoplasm of a HSC (yellow, arrows). Scale bar, 20 μm.
(C) FLAG-tagged PD-L1 domains were introduced into PD-L1 knockdown HSCs and cells were collected for RIP using anti-FLAG. TβRI mRNA co-precipitated with PD-L1 FL or PD-L1 C + T by RIP. ***p < 0.001 by ANOVA, n = 3.
(D and E) FLAG-tagged PD-L1 domains were introduced into PD-L1 knockdown HSCs by retroviral transduction and cells were collected for WB (D) and qRT-PCR (E) for TβRI. PD-L1 FL and PD-L1 C + T rescued TβRI mRNA and TβRI protein of PD-L1 knockdown HSCs. **p < 0.01, ***p < 0.001 by ANOVA, n = 3.
(F) FLAG-tagged PD-L1 domains were introduced into PD-L1 knockdown HSCs followed by stimulation without or with TGF-β1 (5 ng/mL) overnight (upper) or 30 min (lower). Cells were collected for WB for HSC myofibroblatic activation markers or P-SMAD3. PD-L1 FL, but not the PD-L1 Ex and PD-L1 T + C mutants, rescued the impaired TGF-β signaling and myofibroblastic activation of PD-L1 knockdown HSCs. *p < 0.05, **p < 0.01, ****p < 0.0001 by ANOVA, n = 3. All data are represented as mean ± SEM.
The cytoplasmic domain of PD-L1 binds to and stabilizes TβRI mRNA
We next performed RNA immunoprecipitation (RIP) to test whether PD-L1 interacted with TβRI mRNA in HSCs. Anti-PD-L1 antibody was used to pull down PD-L1 from cell lysates and qRT-PCR was used to quantitate co-precipitated RNA. TβRI mRNA, but not TβRII mRNA, was detected in the precipitates, supporting that PD-L1 binds to TβRI mRNA, but not TβRII mRNA (p < 0.001, Figure 4B, upper and Figure S4A, left). RIP sequencing data also revealed that PD-L1 binds to TβRI mRNA in MDA-MB-231 breast cancer cells (GEO: GSE128613) (Figure S4A, right). HSCs expressing FLAG-PD-L1 FL were collected for co-detection of FLAG-tagged PD-L1 by immunofluorescence staining and TβRI mRNA by fluorescence in situ hybridization. Fluorescence confocal microscopy indeed revealed that FLAG-PD-L1 FL protein (green) and TβRI mRNA (red) co-localized in the cytoplasm of HSCs (arrows, Figure 4B, lower).
To investigate how PD-L1 binds to TβRI mRNA, HSCs expressing FLAG-tagged PD-L1 subdomains were collected for RIP assay. Anti-FLAG was used to pull down PD-L1 and qRT-PCR was used to detect co-precipitated TβRI mRNA. TβRI mRNA was co-precipitated with PD-L1 FL or PD-L1 T + C by anti-FLAG, suggesting that the PD-L1 cytoplasmic domain (aa 260–290) interacted with TβRI mRNA in HSCs (p < 0.05, Figure 4C). Consistently, PD-L1 FL and PD-L1 T + C, but not PD-L1 Ex, prevented TβRI mRNA and protein from downregulation by PD-L1 knockdown (p < 0.05, Figures 4D and 4E). We next performed WB to study the influence of the PD-L1 mutants on the TGF-β signaling of HSCs. The impaired TGF-β signaling and myofibroblastic activation of HSCs were rescued by PD-L1 FL, but not by either PD-L1 truncation mutant, consistent with the notion that the TGF-β signaling in cells requires the participation of both TβRI and TβRII receptors (Figure 4F).
The PD-L1 cytoplasmic domain protects TβRI mRNA from degradation by the RNA exosome complex
To investigate how PD-L1 protects TβRI mRNA stability, we focused on the RNA exosome complex, a multiprotein complex related to RNA decay. Exosome component 10 (EXOSC10), harboring 3′-5′ exoribonuclease activity, is a catalytic component and exosome component 4 (EXOSC4) is a non-catalytic structure component of the RNA exosome complex (Tu et al., 2019). To test whether TβRI mRNA is degraded by the exosome complex, EXOSC10 or EXOSC4 of HSCs were knocked down by shRNA lentiviruses. qRT-PCR revealed that TβRI mRNA level was increased by knockdown of EXOSC10 or EXOSC4 and WB confirmed that TβRI protein level was higher in EXOSC10 knockdown HSCs or EXOSC4 knockdown HSCs compared with control HSCs (p < 0.05, Figures 5A and S4B). In addition, the TβRI mRNA level, reduced by PD-L1 knockdown in HSCs, was rescued by EXOSC10 shRNA or EXOSC4 shRNA (p < 0.01, Figure 5B, left and Figure S4C). RNA stability assay revealed that PD-L1 knockdown accelerated the degradation of TβRI mRNA in HSCs and that this phenotype was abrogated by knockdown of both PD-L1 and EXOSC10 (p < 0.05, Figure 5B, right). Thus, the RNA exosome complex is responsible for TβRI mRNA decay in control and PD-L1 knockdown HSCs.
Figure 5. A RLRKGR motif on PD-L1 protects TβRI mRNA from degradation by competing with the RNA exosome complex.

(A) EXOSC10 was knocked down by two different shRNAs and the cells were collected for qRT-PCR (upper) and WB (lower) for TβRI. Knockdown of EXOSC10 led to increases of TβRI mRNA and protein level of HSCs. *p < 0.05, **p < 0.01 by ANOVA, n = 3.
(B) Left: control HSCs, HSCs with PD-L1 knockdown, EXOSC10 knockdown, or knockdown of both were collected for qRT-PCR and WB for TβRI. EXOSC10 knockdown rescued TβRI mRNA and TβRI protein of PD-L1 knockdown HSCs. ***p < 0.001 by ANOVA, n = 3. Right: the stability of TβRI mRNA was assessed in the presence of Actinomycin D. PD-L1 knockdown accelerated the degradation of TβRI mRNA in HSCs and this effect was abrogated by knockdown of EXOSC10. *p < 0.05 by ANOVA, n = 3.
(C) RIP assay revealed that knockdown of PD-L1 led to increased binding of TβRI mRNA to EXOSC10 in HSCs. ***p < 0.001 by ANOVA, n = 3.
(D) RIP assay revealed that PD-L1 FL and PD-L1 T + C competed off EXOSC10/TβRI mRNA binding promoted by PD-L1 knockdown in HSCs. ***p < 0.001 by ANOVA, n = 3.
(E) Left: the RLRKGR motif on PD-L1 T + C was changed to ALAAGA (PD-L1 T + C [4A]) or ALAAGR (PD-L1 T + C [3A]). RIP assay revealed that both mutants abrogated PD-L1 T + C/TβRI mRNA binding in HSCs. ***p < 0.001 by ANOVA, n = 5. Right: qRT-PCR and WB revealed that both mutants failed to rescue TβRI mRNA and TβRI protein of PD-L1 knockdown HSCs compared with wild-type PD-L1 T + C. ***p < 0.001 by ANOVA, n = 4.
(F) RIP assay showed that PD-L1 T + C competed off EXOSC10/TβRI mRNA binding in PD-L1 knockdown HSCs, and this effect was abrogated by either mutant. **p < 0.01, ***p < 0.001 by ANOVA, n = 3. All data are represented as mean ± SEM.
To test whether PD-L1 protects TβRI mRNA stability by competing off EXOSC10/TβRI mRNA binding, PD-L1 knockdown HSCs and EXOSC10 knockdown HSCs were collected for RIP. Our RIP data demonstrated that PD-L1 knockdown led to increased EXOSC10/TβRI mRNA binding and, reversely, that EXOSC10 knockdown led to increased PD-L1/TβRI mRNA binding in HSCs (p < 0.001, Figure 5C p < 0.01, Figure S4D). In addition, the increased EXOSC10/TβRI mRNA binding in HSCs, induced by PD-L1 knockdown, was competed off by the PD-L1 T + C mutant or PD-L1 FL (p < 0.001, Figure 5D). Thus, the PD-L1 cytoplasmic domain protects TβRI mRNA from degradation by binding to TβRI mRNA and competing off RNA exosome/TβRI mRNA binding in HSCs.
A RLRKGR motif on the PD-L1 cytoplasmic domain binds to and stabilizes TβRI mRNA in HSCs
PD-L1 prefers a “GVAGAW” sequence as its binding site on RNA (where V denotes A, G, or C, and W denotes A or U) (Tu et al., 2019) and the 3′ UTR of TβRI mRNA contains eight GVAGAW motifs. However, it is unknown which amino acid(s) on PD-L1 bind to TβRI mRNA. We noticed that the PD-L1 cytoplasmic domain is enriched with positively charged amino acids arginine (R) and lysine (K), which have been predicted as the preferred binding residues for the negatively charged RNA phosphates (Kruger et al., 2018). To test whether the R and K residues mediate PD-L1/TβRI mRNA binding, we mutated 260-RLRKGR-265 on PD-L1 to ALAAGA or ALAAGR by site-directed mutagenesis to generate PD-L1 T + C (4A) and PD-L1 T + C (3A) mutants (Figure 5E, left). RIP data revealed that, while PD-L1 T + C bound to TβRI mRNA in HSCs, this binding was abrogated for either mutant (p < 0.001, Figure 5E, left). Functionally, PD-L1 T + C rescued the TβRI mRNA level reduced by PD-L1 knockdown in HSCs, whereas either mutant did not (p < 0.001, Figure 5E, right). Moreover, PD-L1 T + C competed off the increased EXOSC10/TβRI mRNA binding induced by PD-L1 knockdown in HSCs and this effect was abrogated for either mutant (p < 0.01, Figure 5F). Together, these data support that PD-L1 binds to and stabilizes TβRI mRNA through an RLRKGR motif at its cytoplasmic domain.
Targeting PD-L1 blocks HSCs from producing tumor-promoting factors
We have previously demonstrated that TGF-β1 stimulated HSCs to release paracrine tumor-promoting factors (Liu et al., 2013, 2020; Chen et al., 2020; Wang et al., 2019). To test whether PD-L1 influences HSC production of tumor-promoting factors, we collected control and PD-L1-deficient HSCs for RNA sequencing analysis. Our data (GEO: GSE167173) revealed that 2,410 genes were turned on for transcription by TGF-β1 in control HSCs and that 2,824 genes were PD-L1 dependent as their transcripts were downregulated by PD-L1 knockdown (Figure S5A, p < 0.05). These two gene sets were overlapping, leading to the identification of 1,163 genes as PD-L1-dependent TGF-β1-inducible targets (Figures S5A and S5B). As analyzed by Ingenuity Pathway Analysis, the 1,163 genes are involved in numerous signaling pathways, such as HSC activation, actin cytoskeleton signaling, integrin signaling, and molecular mechanisms of cancer (Figure S5A, lower). A set of genes (32 genes) related to HSC activation are shown in Figures S6A and S6B (p < 0.05). The RNA sequencing data of TGFBR1, TGFBR2, CD274, ACTA2, FN1, and COL1A1, are consistent with our WB or qRT-PCR results (Figures 1A, 2A, 6A, left). The transcript of PD-1 was undetectable by RNA sequencing. A gene set encoding paracrine tumor-promoting factors was also identified as PD-L1-dependent TGF-β1 targets (p < 0.05, Figure 6A, right). WB confirmed that, in response to TGF-β1, HSCs generated and released tumor-promoting factors, such as CTGF, FGF2, IGF1, IGFBP3, and thrombospondin-2, in a PD-L1-dependent manner (p < 0.05, Figures 6B and S7A).
Figure 6. Targeting PD-L1 suppresses the ICC-promoting effect of HSCs in vitro and in a subcutaneous ICC/HSC co-implantation mouse model.

(A) Control and PD-L1 knockdown HSCs, without or with TGF-β1 stimulation (5 ng/mL) for 24 h, were subjected to RNA sequencing and the data were analyzed using the Morpheus tool. The heatmaps depict the mRNA levels of genes related to HSC activation (left) and the tumor-promoting effect of HSCs (right). The scale bar represents the minimum expression value (blue) to the maximum value (red).
(B) Cell culture media collected from HSCs, stimulated without or with TGF-β1 (5 ng/mL) for 24 h, were subjected to WB for tumor-promoting factors. TGF-β1 promoted HSCs to produce and release tumor-promoting factors and this effect was abrogated by knockdown of PD-L1 in HSCs. **p < 0.01, ***p < 0.001 by ANOVA, n = 5.
(C) HuCCT1 cells tagged by firefly luciferase were mixed with HSCs and they were co-injected into nude mice subcutaneously. HuCCT1 chemiluminescence was quantitated by in vivo tumor imaging using a Xenogen IVIS 200 machine and Living Image software. Quantitative HuCCT1 chemiluminescence data revealed that control HSCs promoted the implantation of HuCCT1 cells in mice and that this effect of HSCs was reduced by knockdown of PD-L1 of HSCs. *p < 0.05 by ANOVA, n = 12 per group.
(D) Tumor size was measured using a caliper at different days, and tumor growth curves were generated. Control HSCs promoted HuCCT1 growth in mice and this effect of HSCs was reduced by knockdown of PD-L1 of HSCs. *p < 0.05 by ANOVA, n = 7 per group.
(E) The lysates of HuCCT1 tumors were subjected to WB, which revealed that the levels of αSMA and HSC-derived tumor-promoting factors were all reduced in tumors arising from HuCCT1/PD-L1 knockdown HSC co-injections than in tumors arising from control co-injections. *p < 0.05, **p < 0.01 by ANOVA, n = 5, 6.
(F) αSMA IF showed that the myofibroblast densities were lower in tumors arising from HuCCT1/PD-L1 knockdown HSC co-injections than in tumors arising from control co-injections. *p < 0.05 by ANOVA, n = 7, 7. Scale bar, 50 μm. All data are represented as mean ± SEM except those in (A).
Targeting PD-L1 suppresses the tumor-promoting effect of HSCs in vitro and in a subcutaneous ICC implantation mouse model
To evaluate PD-L1 expression in the myofibroblasts of ICC, a CyTOF antibody panel was used to stain human cholangiocarcinoma sections for multiplex mass cytometry and a Hyperion Imaging System was used for data acquisition and analysis (Loeuillard et al., 2020). Using imaging mass cytometry, we identified PD-L1-positive myofibroblasts in ICC patients (white arrows, Figure S7B). Based on this, we tested the role of HSC PD-L1 for ICC using in vitro and in vivo studies. Conditioned medium (CM) was collected from HSCs and its role for HuCCT1 human cholangiocarcinoma cell migration and proliferation was assessed by Transwell migration and MTS assay. As shown in Figures S7C and S7D, the CM of HSCs promoted HuCCT1 migration and proliferation in vitro compared with the basal culture medium. Importantly, the CM of PD-L1 knockdown HSCs was less effective at promoting HuCCT1 migration (p < 0.01, Figure S7C) and proliferation (p < 0.05, Figure S7D, left) than that of control HSCs. The CM of HSCs incubated with H1A antibody was also less effective at promoting HuCCT1 proliferation than that of control HSCs (p < 0.05, Figure S7D, right). Thus, targeting PD-L1 suppresses the tumor-promoting effect of HSCs in vitro.
We next used tumor/HSC co-injection to translate our in vitro findings into the in vivo tumor microenvironment. Nude mice do not have cytotoxic T cells, allowing us to assess the role of HSC PD-L1 for ICC without the influence the immune system. HuCCT1 cells (0.5 × 106) were mixed with control or PD-L1 knockdown HSCs (0.5 × 106) in vitro and they were co-injected into mice subcutaneously. HuCCT1 cells were tagged by firefly luciferase by lentiviral transduction before co-injection, so their implantation in mice was assessed by in vivo imaging of firefly luciferase (Tu et al., 2015; Liu et al., 2013). As revealed by in vivo tumor imaging, co-injection of HuCCT1 with either control or PD-L1 knockdown HSCs promoted the implantation of HuCCT1 cells in mice compared with the HuCCT1 alone injection group (Figure 6C). However, PD-L1 knockdown HSCs were less effective at promoting HuCCT1 implantation in mice than control HSCs (p < 0.05, Figure 6C). Tumor size was measured using a caliper and tumor growth curves also demonstrated that, while both control and PD-L1 knockdown HSCs promoted HuCCT1 growth in mice, PD-L1 knockdown HSCs were less effective at promoting HuCCT1 growth than control HSCs (p < 0.05, Figure 6D). αSMA IF and WB revealed that the myofibroblast densities were lower in tumors arising from HuCCT1/HSC-PD-L1 shRNA co-injections than in tumors arising from control co-injections (p < 0.05, Figures 6E and 6F). Consistently, the levels of HSC-derived paracrine factors, such as CTGF, IGFBP3 and thrombospondin-2, were all reduced in tumors arising from HuCCT1/HSC-PD-L1 shRNA co-injections than in tumors arising from control co-injections (p < 0.05, Figure 6E). Since the H1A antibody reduces PD-L1 of HSCs, HSCs incubated with the H1A antibody (40 μg/mL) for 24 h were collected for HuCCT1/HSC co-injections and HSCs incubated with control IgG or H1A + PD-L1 (aa 19–238) were used for control co-injections. This co-injection protocol also generated data supporting that targeting of PD-L1 of HSCs suppresses myofibroblastic activation of HSCs and ICC-promoting effects of HSCs in mice (Figure S8).
Cre/loxP-mediated cd274/PD-L1 deletion in activated HSC/myofibroblasts suppresses ICC growth in mice
To determine the pathophysiological relevance of HSC PD-L1 for ICC, we set up a crossbreeding between a cd274/PD-L1 floxed mutant mouse line (Sage et al., 2018) and a collagen1A1-Cre transgenic mouse line (Dou et al., 2018; Wang et al., 2019) in order to delete the cd274 gene in activated HSC/myofibroblasts of the hepatic tumor microenvironment. PD-L1+/+Cre mice (control) and matched PD-L1F/FCre mice were selected as recipients for orthotopic implantation of SB murine ICC cells (Loeuillard et al., 2020; Liu et al., 2013). Four weeks later, we found that ICC orthotopic implantation led to smaller SB tumors in PD-L1F/FCre mice compared with PD-L1+/+Cre mice (p < 0.05, Figure 7A). As revealed by IF and WB, the average αSMA IF density and αSMA protein level were lower in SB tumors of PD-L1F/FCre mice than in SB tumors of PD-L1+/+Cre mice (p < 0.05, Figures 7B and 7C). The protein levels of HSC-derived tumor-promoting factors, CTGF, IGFBP3, and thrombospondin-2, were also lower in SB tumors of PD-L1F/FCre mice (p < 0.05, Figure 7C). Double IF for αSMA and PD-L1 revealed that the average PD-L1 expression level was much reduced in PD-L1F/FCre myofibroblasts (arrowheads) than in PD-L1+/+Cre myofibroblasts (arrows) (p < 0.0001, Figure 7D). Together, these in vivo data support that targeting of HSC PD-L1 inhibits myofibroblastic activation of HSCs and ICC growth in mice.
Figure 7. Cre/loxP-mediated cd274/PD-L1 deletion in activated-HSC/myofibroblasts suppresses ICC growth in mice.

(A) SB murine ICC cells were implanted into the livers of PD-L1+/+Cre (control) and PD-L1F/FCre mice by portal vein injection. SB implantation led to smaller tumors in PD-L1F/FCre mice than in PD-L1+/+Cre mice. *p < 0.05 by t test, n = 6, 7.
(B) Isolated SB ICC tumors were subjected to IF for αSMA. The average αSMA IF density was reduced in SB tumors of PD-L1F/FCre mice than in SB tumors of PD-L1+/+Cre mice. *p < 0.05 by t test, n = 4 tumors. Scale bar: 50 μm.
(C) WB performed with tumor lysates revealed that the protein levels of αSMA, CTGF, IGFBP3, and thrombospondin-2 were all reduced in SB tumors of PD-L1F/FCre mice than in those of PD-L1+/+Cre mice. *p < 0.05, **p < 0.01 by t test, n = 5, 6.
(D) SB tumors were subjected to double IF for αSMA and PD-L1. The average PD-L1 expression level was much lower in PD-L1F/FCre myofibroblasts (arrowheads) than in PD-L1+/+Cre myofibroblasts (arrows). ****p < 0.0001 by t test, n = 23, 17. Scale bar, 50 μm.
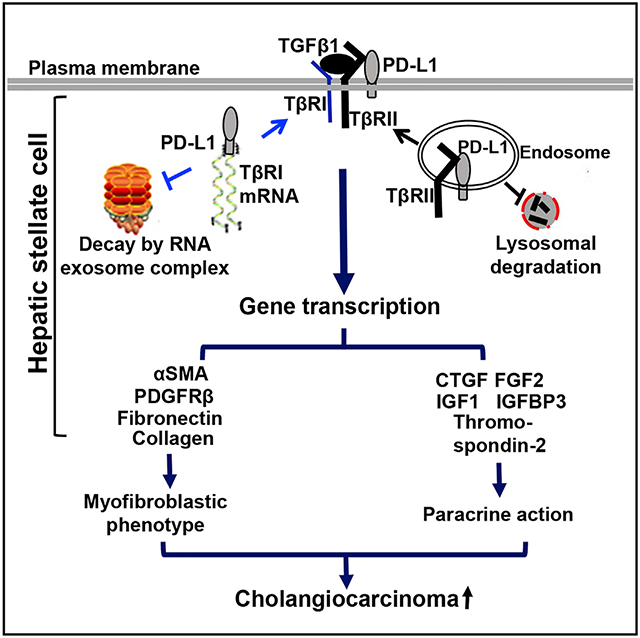
(E) A schematic presentation of this study demonstrating two distinct mechanisms by which PD-L1 protects TβRI mRNA and TβRII protein of HSCs. Together, PD-L1 stabilizes TGF-β receptors and promotes HSC myofibroblastic activation. PM, plasma membrane. All data are represented as mean ± SD.
DISCUSSION
Inhibitors targeting the PD-L1/PD-1 immune checkpoint proteins have been shown to unleash host antitumor immune responses and induce durable responses in the clinic against a growing list of solid tumors and various B cell lymphomas (Goodman et al., 2017). They are therefore considered the most promising drugs for cancers and currently under investigation for advance ICC in multiple clinical trials. However, the role of PD-L1 in the myofibroblasts of ICC remains undetermined. In this regard, our study demonstrates that PD-L1 is expressed in activated-HSC/myofibroblasts where it regulates TβRII protein levels and TβRI mRNA levels and promotes activation of HSCs into tumor-promoting myofibroblasts (Figure 7E). Targeting HSC PD-L1 by shRNA or Cre/loxP recombination suppresses the tumor-promoting effect of HSCs in subcutaneous and orthotopic ICC implantation mouse models.
Current studies in the field of immunotherapy research mainly focus on PD-L1/PD-1-mediated interactions between cancer cells and immune cells, and the contribution of cancer-associated fibroblasts (CAFs) to cancer immune evasion has been overlooked. Our data generated from the myofibroblastic activation of HSCs, relevant for the CAFs of liver cancer, support that the CAFs may be an important source of PD-L1 for various types of cancer and a significant contributor to cancer immune escape in general. Interestingly, we found that TGF-β1 differentially regulates PD-L1 expression in HuCCT1 cancer cells and HSCs; TGF-β1 stimulation (5 ng/mL, 24 h) promotes PD-L1 expression in HSCs (Figures 1A, 1C, and 6A), whereas it reduces PD-L1 expression in HuCCT1 cells (Figure S9A). These data indicate that gene transcription, posttranscriptional modifications, and intracellular trafficking of PD-L1 are regulated by cell-type-specific mechanisms. Our RNA sequencing data also reveal that TGF-β1 promotes HSCs to express and release TGF-β1 and TGF-β2, which are potent immunosuppressants (Figure 6A). Together, these data highlight the role of activated-HSC/myofibroblasts in the immunosuppression of cancer in the liver. Understanding the myofibroblast/immune cell crosstalk may lead to novel strategies to target cancer immune evasion and overcome drug resistance of cancer.
An interesting aspect of this study is the identification of two distinct mechanisms by which PD-L1 regulates TβRI and TβRII in the myofibroblasts. The TGF-β signaling events induced within a cell include the formation of a TβRI/TβRII complex at the plasma membrane, endocytosis of the receptors, phosphorylation of SMADs in the cytoplasm, nuclear translocation of SMADs, and gene transcription in the nucleus (Chen, 2009; Shi and Massague, 2003; Wrana et al., 1994). Following receptor endocytosis, a fraction of the TβRI/TβRII receptor complexes is sorted from the endosomes to lysosomes for degradation and another is sorted back to the plasma membrane for re-use (Mitchell et al., 2004; Massague and Kelly, 1986; Hayes et al., 2002). TGF-β induces downregulation of the receptors and Smurf1/2 are the E3 ubiquitin ligases targeting the receptors for ubiquitination and degradation (Kavsak et al., 2000; Ebisawa et al., 2001). In contrast to this simple model, Koli and Arteaga (1997) reported that 35S-labeled TβRII in mink lung epithelial cells is rapidly downregulated under TGF-β stimulation with a half-life of approximately 45 min, whereas 35S-labeled TβRI has a longer half-life (approximately 12 h) and is not considerably affected by TGF-β stimulation, suggesting that TβRI and TβRII are regulated by distinct mechanisms. Here, we show that the PD-L1 extracellular domain binds to the TβRII protein to protect it from lysosomal degradation, whereas a C-terminal RLRKGR motif on PD-L1 binds to TβRI mRNA to protect it from the RNA exosome complex-mediated degradation. To date, this is the only study focusing on the stability of TβRI mRNA and interrogating how TβRI mRNA is stabilized by PD-L1 and downregulated by the RNA exosome complex.
Our H1A anti-PD-L1 antibody targets aa 20–32 of the PD-L1 extracellular domain (Tu et al., 2019). The fate of PD-L1 in cancer cells is determined by a competitive binding between PD-L1, CMTM6, and H1A antibody. While PD-L1/CMTM6 binding leads to plasma membrane localization of PD-L1 and protects PD-L1 from lysosomal degradation (Burr et al., 2017; Mezzadra et al., 2017), H1A competes off PD-L1/CMTM6 binding, resulting in PD-L1 lysosomal targeting and degradation (Tu et al., 2019). Here, we show that H1A promotes lysosomal targeting and degradation of TβRII as well. We also collected HSCs incubated with therapeutic anti-PD-L1 antibodies, atezolizumab (10 μg/mL) or durvalumab (20 μg/mL), for 16 h for coIP to determine TβRII/PD-L1 binding and HSCs incubated with isotype-matched control IgG as the control. Interestingly, the two antibodies, blocking PD-L1/PD-1 binding, led to increased TβRII and PD-L1 protein levels, as well as increased PD-L1/TβRII binding in HSCs (Figure S9B), suggesting that they may lock the PD-L1/TβRII protein complexes at the plasma membrane to prevent them from endocytosis, lysosomal sorting, and degradation. Since receptor endocytosis is required for TGF-β signaling, we can predict that these antibodies may inhibit myofibroblastic activation of HSCs mediated by TGF-β.
More than 6,500 RNAs are downregulated by PD-L1 knockdown in cancer cells (Tu et al., 2019). Our RNA sequencing data revealed that, in TGF-β-stimulated HSCs, 2,824 RNAs are downregulated upon PD-L1 knockdown, supporting that PD-L1 indeed regulates RNA stability globally in HSCs (Figure S5A). The reason why only 1,163 out of 2,410 TGF-β1 target genes are affected by PD-L1 knockdown could be: (1) HSCs with 100% of PD-L1 knockdown did not survive and HSCs expressing a low level of PD-L1 and maintaining a low level of TGF-β signal were in fact collected for RNA sequencing. In these cells, the low level of TGF-β signal led to expression of the 1,247 TGF-β1 targets at a comparable level of control cells. (2) Since HSCs incubated with TGF-β1 for 24 h were collected for RNA sequencing, some of the TGF-β1 targets identified may be direct targets of TGF-β1 and some may not be. In this case, the 1,163 genes may be direct targets of the TGF-β/SMAD pathway and the 1,247 genes may be indirect targets regulated by the cytokines stimulated by TGF-β1 or by the non-canonical TGF-β1 signaling pathways.
Ingenuity Pathway Analysis revealed that the 1,163 PD-L1-dependent TGF-β targets are involved in multiple signaling pathways, including the TGF-β signaling, IGF signaling, FAK signaling, integrin signaling, actin cytoskeleton signaling, and molecular mechanisms of cancer. We also found that, under TGF-β1 stimulation, HSCs express and release paracrine factors, such as CTGF, IGF1, FGF2, IGFBP3, and thrombospondin-2, in a PD-L1-dependent manner (Figures 6B and S7A). Clinical data showed that IGFBP3 is overexpressed in certain cancers, including esophageal squamous cell carcinoma (ESCC) and grade IV glioma, and that its high expression is associated with poor postsurgical prognosis of ESCC patients (Natsuizaka et al., 2014) and glioma patients (Chen et al., 2019). Similarly, plasma thrombospondin-2 level is regarded to be a candidate diagnosis biomarker for early non-small cell lung cancer, pancreatic cancer, and cholangiocarcinoma of patients (Jiang et al., 2019; Le Large et al., 2020; Kim et al., 2017). Taken together, HSC PD-L1 can modulate tumor growth by regulating the release of paracrine factors, independent of the PD-L1/PD-1-mediated immune suppression.
In summary, HSCs are a source of PD-L1, and HSC PD-L1 binds to and stabilizes TβRII protein and TβRI mRNA thereby promoting TGF-β-stimulated activation of HSCs into myofibroblasts. Targeting PD-L1 blocks activated-HSC/myofibroblasts from producing tumor-promoting factors thereby suppressing their tumor-promoting effect in vitro and in mice. PD-L1 in the activated-HSC/myofibroblasts represents a plausible target for suppressing HSC activation, the hepatic tumor microenvironment, and ICC.
Limitations of the study
HSCs are a precursor of CAFs of liver cancer. The CAFs are a major cellular component of the tumor microenvironment that reciprocally crosstalk with various types of cells within the tumors, including endothelial cells and immune cells, in addition to cancer cells. This study only focuses on the role of the myofibroblast PD-L1 for cancer cells, and its role in endothelial function and tumor angiogenesis, as well as its contribution to the immunosuppressive tumor microenvironment, which has not been explored. Further studies toward these directions may help us uncover novel mechanisms and therapeutic targets to advance our understanding of the tumor microenvironment and improve anti-cancer efficacy of PD-L1/PD-1 immunotherapy.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to lead contact Ningling Kang (nkang@umn.edu).
Materials availability
The constructs generated in this study will be made available from the lead contact upon request.
Data and code availability
The RNA sequencing data for control and PD-L1 knockdown cells have been deposited to Gene Expression Omnibus and are publicly available as of the date of publication (GEO: GSE167173). Original Western blot and microscopy data generated in this study will be made available and shared by the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cells
Primary human hepatic stellate cells (HSCs) with passage <9 and murine HSCs with passage 1 were used for the experiments. The cells, including the primary human and murine HSCs, HuCCT1 human ICC cells, and SB murine ICC cells, were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL), and streptomycin (100 g/mL), at 37°C in 5% CO2. They were routinely monitored for mycoplasma infection and free of infection during the experiments. The procedure for murine HSC isolation was approved by the Institutional Animal Care and Use Committee (IACUC) of University of Minnesota. Both male and female mice, older than 4 months, were used for in situ liver perfusion for HSC isolation and the cells were pooled together for the experiments.
Mice
Breeding of the PD-L1 floxed mutant mouse line to the collagen1A1-Cre transgenic mouse line has been approved by IACUC of University of Minnesota; ICC implantation into mice has been approved by IACUC of University of Minnesota and Mayo Clinic. Seven-week-old nude mice (NCI Athymic NCr-nu/nu Mice) were purchased from the Charles River Laboratories and housed in the specific-pathogen-free facility. All mice were cohoused together (4 mice/cage) in the climate controlled and 12-h light/dark cycling conditions. Since the collagen1A1-Cre transgene is only transmitted to male mice, 8-week-old male PD-L1+/+Cre mice and PD-L1F/FCre mice were used as the recipients for orthotopic ICC implantation.
Clinical samples
Multiplex mass-cytometry performed with human ICC samples has been approved by the Mayo Clinic Institutional Review Board (no. 707–03). Informed consent was obtained from the patients and the samples were de-identified for patient privacy protection. Since we only did a descriptive study confirming PD-L1 expression in the myofibroblasts of patient ICC, the information of the patients (sex, gender, age and others) was not included and analyzed.
METHOD DETAILS
Inducing HSC activation by TGFβ
To induce myofibroblastic activation of primary human HSCs, HSCs serum-starved overnight were incubated with TGF-β1 (5 ng/mL) for 24 h and collected for Western blot analysis (WB) for the markers of HSC activation, such as alpha-smooth muscle actin (αSMA). fibronectin, and type 1 collagen. To study TGF-β1-induced SMAD3 phosphorylation, serum-starved HSCs were stimulated with TGF-β1 for 30 min and collected for WB.
Plasmids and site-directed mutagenesis
The retroviral constructs encoding full length PD-L1 or a PD-L1 domain with FLAG tag at the N-terminus were created by Dr. Zhenkun Lou’s laboratory (Tu et al., 2019). To create PD-L1 T + C (4A) and PD-L1 T + C (3A) mutants, Q5 Site-Directed Mutagenesis kit was purchased from New England Biolabs (E0554 Ipswich, MA) and PCR primers were designed so that 260-RLRKGR-265 on PD-L1 was replaced with 260-ALAAGA-265 or 260-ALAAGR-265. The retroviral constructs TβRII-HA and TβRI-FLAG with the tag at the C-terminus were created previously (Liu et al., 2013).
Viral packaging and transduction of cells
Packaging of retroviruses and lentiviruses were done as we described previously (Kang et al., 2010; Liu et al., 2013, 2020; Chen et al., 2020). In brief, plasmids encoding viral elements were co-transfected into HEK-293 T cells by following the manufacturer’s protocol of Effectene® Transfection Reagent. Virus-containing supernatants were harvested 48 and 72 h after plasmid transfection. Transduction of HSCs was conducted by incubating the cells with virus-containing supernatant overnight (1:1 dilution with complete DMEM, supplemented with 8 μg/mL polybrene). HSCs collected 72 h later were used for determining gene expression or knockdown.
Immunofluorescence staining (IF)
Cultured HSCs or murine tumor cryosections (7 μm in thickness) were used for IF staining, as we described (Chen et al., 2020; Liu et al., 2020; Wang et al., 2019). In brief, HSCs were first fixed with 4% paraformaldehyde for 10 min followed by permeabilization by 0.5% Triton X-100 for 3 min. After rinsing the samples with 1 x PBS, 10% goat serum was added to block non-specific antibody binding sites and a primary antibody, fluorescent secondary antibody, and DAPI were added to label the target protein and cell nuclei. Signals were visualized and captured with a Zeiss LSM 510 confocal microscope (Cal Zeiss AG, Jena, Germany) (Chen et al., 2020; Liu et al., 2020; Wang et al., 2019). To determine TβRII-HA/LAMP-1 co-localization in HSCs, HSCs serum-starved overnight were incubated with TGFβ1 (5 ng/mL) and collected at different time points, 0, 15, and 45 min, for IF. Image acquisition and analysis of TβRII-HA/LAMP-1 co-localization were done as we previously described (Liu et al., 2013, 2020).
Western Blot analysis (WB)
Cultured cells or tumor samples were lysed with RIPA buffer supplemented with protease inhibitors and a phosphatase inhibitor cocktail. After protein quantification, 5–50 μg of total proteins per sample were loaded into an SDS-PAGE gel for electrophoresis followed by protein transfer onto a nitrocellulose membrane (10600002, GE Healthcare Life Sciences). The membrane was then incubated with a primary antibody and secondary antibody conjugated with horseradish peroxidase (HRP). Signals were detected with HRP chemiluminescent substrates and densitometry was done by using the ImageJ software (NIH) (Wang et al., 2019; Dou et al., 2018).
TβRII degradation assay
To assess the stability of TβRII protein in control and HSCs with PD-L1 knockdown, HSCs were transduced with lentiviruses encoding control shRNA or PD-L1 shRNA. HSCs were then incubated with cycloheximide (40 μg/mL) to block protein synthesis followed by cell collection for WB for endogenous TβRII at different time points (0, 30, 60, 90, 120, 240 min). Densitometry was conducted by using the ImageJ software and TβRII degradation curves were generated with the GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA) (Liu et al., 2013, 2014).
Biotinylation for plasma membrane TβRII
To quantitate TβRII at the plasma membrane, control and PD-L1 knockdown HSCs in 60 mm dishes were serum-starved overnight followed by incubation with TGF-β1 (5 ng/mL) for 6 h. Cell were then incubated with EZ-Link Sulfo-NHS-Biotin for 30 min at 4°C to label cell surface proteins. After removing free biotins, the RIPA lysis buffer was used to lyse the cells and streptavidin agarose beads were used to pull down biotinylated proteins (Tu et al., 2015; Liu et al., 2020). TβRII precipitated (cell surface TβRII) was quantitated by WB using anti-TβRII (Liu et al., 2014; Tu et al., 2015). HRP-conjugated streptavidin was used to detect total biotinylated proteins, which was used as the control (Chen et al., 2020; Liu et al., 2020).
Co-immunoprecipitation (coIP)
To investigate the interactions between PD-L1 and TGF-β receptors in HSCs, HSCs expressing TβRII-HA were transduced by viruses encoding FLAG-tagged full-length PD-L1 or a PD-L1 domain and cells were lysed with IP buffer containing 0.5% NP40. The lysates containing equal volumes and equal amounts of proteins were incubated with 1–5 μg anti-FLAG and 30 μL slurry of Protein G Sepharose overnight to capture FLAG and its associated proteins. On the following day, the beads were precipitated by centrifugation and TβRII-HA in the precipitates was quantitated by WB using anti-HA antibody (Liu et al., 2013, 2014; Tu et al., 2015). Reversely, anti-HA antibody and agarose beads were used to pull down TβRII-HA and WB was used to detect co-precipitated PD-L1.
Real-time RT-PCR (qRT-PCR)
QRT-PCR was performed with RNA recovered from immunoprecipitation or extracted from HSCs. RNA was quantitated by absorbance at 260 nm, and 500 ng RNA was reverse transcribed by using the SuperScript III kit. QPCR was conducted by using SYBR Green Supermix according to the manufacturers’ protocols. GAPDH was used as a control and the results were calculated by the 2−ΔΔCt method. The primers used for qPCR are in (Table S1).
RNA immunoprecipitation assay (RIP)
RIP was performed with the EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit according to the manufacturer’s instructions. In brief, RIP lysis buffer supplemented with a protease inhibitor cocktail and RNase inhibitors was used to lyse the cells and cell lysates were incubated with a specific antibody (such as anti-PD-L1, anti-FLAG, or anti-EXOSC10) and protein A/G magnetic beads overnight at 4°C. On the following day, the protein/RNA complexes on the beads were precipitated by magnetic force and RNA eluted was detected by qRT-PCR(Tu et al., 2019).
GST pull down assay
The pGEX6P1-GST and pGEX6P1-GST-TβRII bacterial expression vectors were constructed in our lab previously and the recombinant proteins were generated and purified from E. Coli (BL21 DE3) using Glutathione Sepharose 4B beads, as we described (Liu et al., 2013). The PD-L1 extracellular domain recombinant protein and biotinylated TGF-β1 were purchased from commercial vendors. In vitro binding assay was performed by incubating two or three proteins in 200 μL binding buffer (25 mM Tris-HCl pH7.6, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1% NP-40, 0.1 mM PMSF, protease inhibitors) for 4 h at 4°C followed by centrifugation to precipitate GST-TβRII and bound proteins. Pulled down PD-L1 was detected by WB using anti-PD-L1 (ab205921 Abcam) and biotinylated TGF-β1 was detected by WB using streptavidin conjugated with HRP. Lastly, GST and GST-TβRII used for GST pull down assay were visualized by Ponceau S staining.
RNA sequencing and data analysis
To identify transcriptional targets of TGF-β1 and those are also modulated by PD-L1, 4 groups of HSCs were used for RNA sequencing, HSCs transduced with control shRNA, HSCs transduced with control shRNA with TGF-β1 stimulation (5 ng/mL) for 24 h, HSCs transduced with PD-L1 shRNA, and HSCs transduced with PD-L1 shRNA with TGF-β1 stimulation (5 ng/mL) for 24 h. Each group contained 3 independent cell samples (biological triplicates). RNA samples were isolated by using the RNeasy Plus Mini Kit (QIAGEN). RNA sequencing and bioinformatics analysis were performed by the University of Minnesota Genomic Center, as we previously described (Dou et al., 2018; Chen et al., 2020). In brief, after samples were checked for quantity by using fluorimetric RiboGreen assay and quality by capillary electrophoresis, RNA samples with higher than 500 ng were converted to Illumina sequencing libraries using Illumina’s Truseq Stranded mRNA Sample Preparation Kit. Truseq libraries were then subjected to cluster using Illumina cBot instrument and sequencing by HiSeq2500. De-multiplexed FASTQ files containing the raw reads for each gene were generated by using the Illumina Real Time Analysis (RTA) software and Illumina’s CASAVA software 1.8.2. Human genome (hg19) was used as the reference. Data were analyzed by the EdgeR package so genes differentially expressed were identified based on the analyzing criteria (fold change >2 and FDR < 0.05). Differentially expressed genes were then analyzed with Venn diagrams (https://bioinfogp.cnb.csic.es/tools/venny/) and Ingenuity Pathway Analysis (Qiagen). Heatmaps were generated by using the Heatmapper (Babicki et al., 2016) and the Morpheus tools (https://software.broadinstitute.org/morpheus/) with raw read counts as input (Dou et al., 2018). Raw counts were also used as input for gene set enrichment analysis (GSEA) which was performed using default settings (1000 permutations) and for a maximum size of sets of 500 (Subramanian et al., 2005).
MTS cell proliferation assay
Conditioned media (CMs) were collected from control HSCs, HSCs with PD-L1 knockdown, or HSCs incubated with H1A antibody (40 μg/mL) and they were used as stimulants for HuCCT1 cells proliferation in vitro. 5,000 HuCCT1 cells suspended in 100 μL of CM were seeded into each well of a 96-well plate and cells were collected 24, 48, 72 and 96 h later for CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit (Promega), according to the manufacturer recommended protocol (Liu et al., 2013; Chen et al., 2020).
Transwell cell migration assay
CMs collected from HSCs were used as stimulants for HuCCT1 Transwell migration assay. The assay was set up by plating 5,000 HuCCT1 cells into an upper chamber and the CM into the lower chamber of a Transwell apparatus (6.5 mm Transwell® with 8.0 μm Pore Polycarbonate Membrane Insert) (Corning Incorporated). After incubation at 37°C for 4 h, cells migrated into the lower chamber and attached to the lower surface of the membrane were stained with DAPI. DAPI-stained cell nuclei were visualized and counted under a fluorescence microscopy (Liu et al., 2013).
Mass-cytometry and cancer/HSC co-injection
Multiplex mass-cytometry with human ICC sections was done with a CyTOF antibody panel as we previously described (Loeuillard et al., 2020). To test whether PD-L1 depletion in HSCs influences ICC growth in mice, 0.5 × 106 HuCCT1 human ICC cells were mixed with 0.5 × 106 HSCs with or without PD-L1 knockdown in vitro followed by co-injection of them into the lower flank of a nude mouse subcutaneously (Liu et al., 2013; Wang et al., 2019). HuCCT1 cells were tagged by firefly luciferase by lentiviral transduction, so their implantation in mice was monitored by in vivo imaging with a Xenogen IVIS 200 and the Living Image software (Caliper Life Sciences) (Liu et al., 2013; Tu et al., 2015). Tumor sizes were measured by a caliper at different times and tumor volumes were calculated by an equation: V (tumor volume: mm3) = 0.5 × [W (width: mm)]2 × L (longer diameter: mm) (Liu et al., 2013; Tu et al., 2015). To investigate the effect of anti-PD-L1 (H1A) on myofibroblastic activation of HSCs in vivo and HuCCT1 growth, HSCs pre-incubated with H1A (40 μg/mL) for 24 h in culture were collected for HuCCT1/HSC co-injections. HSCs pre-incubated with H1A (40 μg/mL) and PD-L1 Ex (R & D Systems, 10 μg/mL) or HSCs pre-incubated with control IgG were collected for co-injections as control groups.
HSC isolation and orthotopic ICC implantation
The cd274/PD-L1 floxed mutant mouse line was generated previously (Sage et al., 2018) and it was cross-bred to a collagen1A1-Cre transgenic mouse line (Dou et al., 2018; Wang et al., 2019). A PCR-based genotyping protocol was used to identify mouse offspring. Primary HSCs were isolated from PD-L1F/F mice, as we described (Dou et al., 2018), and they were transduced with adenoviruses encoding GFP or Cre (MOI = 100). Mice with the following genotypes, PD-L1+/+Cre and PD-L1F/FCre, were selected for SB murine ICC orthotopic implantation (Loeuillard et al., 2020), which was done by injecting the cells into the portal vein of the mice (0.75 × 106 cells per mouse), as we previously did (Liu et al., 2013; Dou et al., 2018). Mice were sacrificed 28 days later and SB tumors were isolated, measured, and stored for IF and WB.
Co-detection of TβRI mRNA and PD-L1 protein
The co-detection was performed with HSCs expressing FLAG-PD-L1 FL by using the RNAscope 2.5 High Definition Red Assay, RNA-Protein Co-Detection Ancillary Kit, and RNAscope Probe-Hs-TGFBR1 according to the manufacturer’s protocol (Advanced Cell Diagnostics). In brief, after the amplification steps for the RNA probe detection, cells were blocked with the co-detection blocking solution and stained with anti-FLAG (M2) and Alexa Fluor 488 secondary antibody diluted in co-detection antibody diluent for IF staining. Following DAPI staining of the cell nucleus, the cells were mounted and imaged using Zeiss fluorescence confocal microscope.
QUANTIFICATION AND STATISTICAL ANALYSIS
Information of data analysis for the experiments is shown in the figure legends and the Results section. Data are expressed as either mean ± SD or mean ± SEM. Statistical analysis was done by using the Prism 6 software. For experiments with 2 groups, data were analyzed by unpaired Student’s t-test; for experiments with more than 2 groups, data were analyzed by ANOVA (analysis of variance) followed by a post hoc test. p <0.05 is considered statistically significant. For WB data with cultured cells, n represents independent repeats; for IF with cultured cells, n represents the number of cells or number of confocal images analyzed; for animal studies, n represents number of animals. RNA sequencing data were analyzed by the EdgeR package so genes differentially expressed were identified based on the analyzing criteria (fold change >2 and FDR < 0.05). Ingenuity Pathway Analysis (Qiagen) and Venn diagrams (https://bioinfogp.cnb.csic.es/tools/venny/) were used to analyze genes identified and Heatmapper and the Morpheus tools (https://software.broadinstitute.org/morpheus/) were used to generate heatmaps.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| PD-L1 (RIP, IP and IF; WB for mouse PD-L1) | Cell Signaling Technology | Cat#13684; RRID:AB_2687655 |
| PD-L1 (WB for human PD-L1) | Abcam | Cat#ab213524; RRID:AB_2857903 |
| PD-L1 (WB for human PD-L1) | Abcam | Cat#ab205921; RRID:AB_2687878 |
| TβRII (WB) | Abcam | Cat#ab184948; RRID:AB_2818975 |
| TβRI (WB) | Abcam | Cat#ab31013; RRID:AB_778352 |
| αSMA (IF and WB) | Abcam | Cat#ab5694; RRID:AB_2223021 |
| Fibronectin (WB) | Abcam | Cat#ab2413; RRID:AB_2262874 |
| Type 1 collagen | Southern Biotech | Cat#1310–01; RRID:AB_2753206 |
| pSMAD3 (S423/425) (C25A9) | Cell Signaling Technology | Cat#9520; RRID:AB_2193207 |
| SMAD3 | Cell Signaling Technology | Cat#9523; RRID:AB_2193182 |
| HA (12CA5) (WB, IF, IP) | Roche Diagnostics | Cat#11583816001; RRID:AB_514505 |
| HA (Chip grade) (IP) | Abcam | Cat#ab9110; RRID:AB_307019 |
| HA (IF) | Santa Cruz Biotechnology, Inc. | Cat#sc-7392; RRID:AB_627809 |
| HA (IF) | Cell Signaling Technology | Cat#3724; RRID:AB_1549585 |
| GAPDH | Invitrogen | Cat#AM4300; RRID:AB_2536381 |
| LAMP-1 (H4A3) | Santa Cruz Biotechnology, Inc. | Cat#sc-20011; RRID:AB_626853 |
| EXOSC10 (WB) | Abcam | Cat#ab50558; RRID:AB_869937 |
| EXOSC10 (RIP) | Abcam | Cat#ab264343 |
| EXOSC4/RRP41 | Abcam | Cat#ab137250 |
| Ubiquitin (P4D1) | Cell Signaling Technology | Cat#3936; RRID:AB_331292 |
| CTGF | Abcam | Cat#ab209780 |
| IGF-1 (H-9) | Santa Cruz Biotechnology, Inc. | Cat#sc-518040 |
| FGF2 (C-2) | Santa Cruz Biotechnology, Inc. | Cat#sc-74412; RRID:AB_1122854 |
| FLAG (M2) | MilliporeSigma | Cat#F1804; RRID:AB_262044 |
| Thrombospondin-2 | Abcam | Cat#ab112543; RRID:AB_10863103 |
| IGFBP3 | Abcam | Cat#ab193910 |
| Control IgG (DA1E) (IP) | Cell Signaling Technology | Cat#3900; RRID:AB_1550038 |
| PD-L1 (clone H1A) | Dr. Haidong Dong’s lab | (Liu et al., 2016) |
| Atezolizumab | MedChemExpress | Cat#HY-P9904 |
| Durvalumab | MedChemExpress | Cat#HY-P9919 |
| Control human IgG | Thermo Fisher Scientific | Cat#02-7102;RRID:AB_2532958 |
| Bacterial and virus strains | ||
| DH5α competent cells | Thermo Fisher Scientific | Cat#18265017 |
| BL21(DE3) Competent Cells | Thermo Fisher Scientific | Cat#EC0114 |
| Biological samples | ||
| Human ICC patient samples | Mayo Clinic | (Loeuillard et al., 2020) |
| Chemicals, peptides, and recombinant proteins | ||
| TGF-β1 | R & D Systems | Cat#7754-BH |
| Biotinylated TGF-β1 | Reprokine Research Immunity | Cat#RKP01137B |
| HRP-streptavidin | Thermo Fisher Scientific | Cat#N100 |
| PD-L1 extracellular domain recombinant protein | R & D Systems | Cat#9049-B7 |
| MG132 | Cayman Chemical Company | Cat#10012628 |
| E64d | Tocris | Cat#4545 |
| Pepstatin A | MilliporeSigma | Cat#51648 |
| Bafilomycin | Tocris | Cat#1334 |
| Protease inhibitor cocktail | Thermo Fisher Scientific | Cat#88266 |
| Phosphatase inhibitor cocktail | Thermo Fisher Scientific | Cat#78428 |
| RIPA buffer | Cell Signaling Technology | Cat#9806 |
| HRP chemiluminescent substrate | Santa Cruz Biotechnology, Inc. | Cat#sc-2048 |
| RNase inhibitors | MilliporeSigma | Cat#3335399001 |
| DAPI | Thermo Fisher Scientific | Cat#D1306 |
| EZ-Link Sulfo-NHS-Biotin | Thermo Fisher Scientific | Cat#21217 |
| Streptavidin agarose beads | Millipore | Cat#S1638 |
| Transwell® | Corning Incorporated | Cat#3422 |
| HRP-conjugated streptavidin | Thermo Fisher Scientific | Cat#434323 |
| Protein G Sepharose beads | GE Healthcare | Cat#GE17-0618-01 |
| RNeasy Plus Mini Kit | QIAGEN | Cat#74134 |
| Glutathione Sepharose 4B beads | GE Healthcare | GE17-0756-01 |
| Critical commercial assays | ||
| SuperScript III kit | Invitrogen | Cat#18080–051 |
| SYBR Green Supermix | Bio-Rad | Cat#1725120 |
| EZ-Magna RIP™ RNA-Binding Protein Immunoprecipitation Kit | MilliporeSigma | Cat#17–701 |
| CellTiter 96 Aqueous Non-Radioactive Cell Proliferation Assay kit | Promega | Cat#G5421 |
| RNAscope 2.5 High Definition Red Assay | Advanced Cell Diagnostics | Cat#322350 |
| RNA-Protein Co-Detection Ancillary Kit | Advanced Cell Diagnostics | Cat#323180 |
| RNAscope Probe-Hs-TGFBR1 | Advanced Cell Diagnostics | Cat#431041 |
| Q5 Site-Directed Mutagenesis Kit | New England Biolabs | Cat#E0554 |
| Effectene® Transfection Reagent | Qiagen | Cat#301425 |
| Deposited data | ||
| RNA sequencing data (TGFβ1-stimulated control and PD-L1 knockdown cells) | This paper | GEO: GSE167173 |
| Experimental models: Cell lines | ||
| Human primary hepatic stellate cells | ScienCell Research Laboratories | Cat#5300 |
| HuCCT1 | Dr. Gregory Gores’s lab | (Razumilava et al., 2014; Fingas et al., 2011, 2013) |
| SB murine ICC cells | Dr. Gregory Gores’s lab | (Loeuillard et al., 2020; Diggs et al., 2021) |
| Experimental models: Organisms/strains | ||
| Nude mice (NCI Athymic NCr-nu/nu Mice) | Charles River Laboratories | Cat#553 |
| Cd274/PD-L1 floxed mutant mouse line | Dr. Arlene H. Sharpe’s lab | (Sage et al., 2018) |
| Collagen1A1-Cre transgenic mouse line | Dr. Tatiana Kisseleva’s lab | (Dou et al., 2018; Wang et al., 2019) |
| Oligonucleotides | ||
| PD-L1 shRNA-1 | MilliporeSigma | Cat#TRCN0000056916 |
| PD-L1 shRNA-2 | MilliporeSigma | Cat#TRCN0000056914 |
| PD-L1 shRNA-3 | MilliporeSigma | Cat#TRCN0000056915 |
| EXOSC10 shRNA-1 | MilliporeSigma | Cat#TRCN0000006340 |
| EXOSC10 shRNA-2 | MilliporeSigma | Cat#TRCN0000006341 |
| EXOSC4 shRNA-1 | MilliporeSigma | Cat#TRCN0000051364 |
| EXOSC4 shRNA-2 | MilliporeSigma | Cat#TRCN0000051365 |
| Non-Targeting shRNA control | MilliporeSigma | Cat#SHC202 |
| Primers for qRT-PCR are in Table S1 | ||
| Recombinant DNA | ||
| PLVX3-full-length PD-L1 | Dr. Zhenkun Lou’s lab | (Tu et al., 2019) |
| PLVX3-extracellular domain of PD-L1 | Dr. Zhenkun Lou’s lab | (Tu et al., 2019) |
| PLVX3-cytoplasmic domain of PD-L1 | Dr. Zhenkun Lou’s lab | (Tu et al., 2019) |
| PLVX3-cytoplasmic domain of PD-L1(4A) | This paper | N/A |
| PLVX3-cytoplasmic domain of PD-L1(3A) | This paper | N/A |
| pMD.2G | Dr. Zhenkun Lou’s lab | (Tu et al., 2019) |
| psPAX2 | Addgene | Cat#12260 |
| pMMP-TβRII-HA | Dr. Ningling Kang’s lab | (Liu et al., 2013) |
| pMMP-TβRI-FLAG | Dr. Ningling Kang’s lab | (Liu et al., 2013) |
| pGEX6P1-GST | Dr. Ningling Kang’s lab | (Liu et al., 2013) |
| pGEX6P1-GST-TβRII | Dr. Ningling Kang’s lab | (Liu et al., 2013) |
| Software and algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 6.0 | GraphPad Software | http://www.graphpad.com/ |
| Xenogen IVIS 200 and the Living Image software | Caliper Life Sciences | http://www.caliperls.com/assets/011/6716.pdf |
| Ingenuity Pathway Analysis | Qiagen | https://digitalinsights.qiagen.com/products-overview/discovery-insights-portfolio/analysis-and-visualization/qiagen-ipa/ |
| Venny 2.1.0 | By Juan Carlos Oliveros | https://bioinfogp.cnb.csic.es/tools/venny/ |
| Morpheus | Broad Institute | https://software.broadinstitute.org/morpheus/ |
Highlights.
Hepatic stellate cells, the precursor of cancer-associated fibroblasts, express PD-L1
PD-L1 protects TGF-β receptor II protein and TGF-β receptor I mRNA from degradation
PD-L1 promotes activation of hepatic stellate cells into myofibroblasts
Targeting of myofibroblast PD-L1 selectively suppresses liver cancer in mice
ACKNOWLEDGMENTS
NIH grants R01CA160069 R01CA187027 to N.K., R37AA021171 to V.H.S., the Cell Biology Core of the Mayo Clinic Center for Cell Signaling in Gastroenterology (P30DK084567), and the Mayo Clinic Hepatobiliary SPORE (P50 CA210964) Developmental Research Program. L.S. is funded by the China Scholarship Council.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
INCLUSION AND DIVERSITY
One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. We worked to ensure that the study questionnaires were prepared in an inclusive way. We worked to ensure diversity in experimental samples through the selection of the cell lines.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2022.110349.
REFERENCES
- Azuma T, Yao S, Zhu G, Flies AS, Flies SJ, and Chen L (2008). B7-H1 is a ubiquitous antiapoptotic receptor on cancer cells. Blood 111, 3635–3643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, and Wishart DS (2016). Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 44, W147–W153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, et al. (2017). CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature 549, 101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadamuro M, Brivio S, Mertens J, Vismara M, Moncsek A, Milani C, Fingas C, Cristina Malerba M, Nardo G, Dall’Olmo L, et al. (2019). Platelet-derived growth factor-D enables liver myofibroblasts to promote tumor lymphangiogenesis in cholangiocarcinoma. J. Hepatol 70, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadamuro M, Nardo G, Indraccolo S, Dall’Olmo L, Sambado L, Moserle L, Franceschet I, Colledan M, Massani M, Stecca T, et al. (2013). Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 58, 1042–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, Chen Q, Gindin M, Gubin MM, Van DerWindt GJ, et al. (2015). Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell 162, 1229–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Chen PY, Lin YY, Feng LY, Chen SH, Chen CY, Huang YC, Huang CY, Jung SM, Chen LY, and Wei KC (2019). Suppression of tumor growth via IGFBP3 depletion as a potential treatment in glioma. J. Neurosurg 132, 168–179. [DOI] [PubMed] [Google Scholar]
- Chen Y, Li Q, Tu K, Wang Y, Wang X, Liu D, Chen C, Liu D, Yang R, Qiu W, and Kang N (2020). Focal adhesion kinase promotes hepatic stellate cell activation by regulating plasma membrane localization of TGFbeta receptor 2. Hepatol. Commun 4, 268–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YG (2009). Endocytic regulation of TGF-beta signaling. Cell Res. 19, 58–70. [DOI] [PubMed] [Google Scholar]
- Diggs LP, Ruf B, Ma C, Heinrich B, Cui L, Zhang Q, Mcvey JC, Wabitsch S, Heinrich S, Rosato U, et al. (2021). CD40-mediated immune cell activation enhances response to anti-PD-1 in murine intrahepatic cholangiocarcinoma. J. Hepatol 74, 1145–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. (2002). Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat. Med 8, 793–800. [DOI] [PubMed] [Google Scholar]
- Dong H, Zhu G, Tamada K, and Chen L (1999). B7-H1, a third member of the B7 family, co-stimulates T-cell proliferation and interleukin-10 secretion. Nat. Med 5, 1365–1369. [DOI] [PubMed] [Google Scholar]
- Dou C, Liu Z, Tu K, Zhang H, Chen C, Yaqoob U, Wang Y, Wen J, Van Deursen J, Sicard D, et al. (2018). P300 acetyltransferase mediates stiffness-induced activation of hepatic stellate cells into tumor-promoting myofibroblasts. Gastroenterology 154, 2209–2221 e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, and Miyazono K (2001). Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J. Biol. Chem 276, 12477–12480. [DOI] [PubMed] [Google Scholar]
- Fingas CD, Bronk SF, Werneburg NW, Mott JL, Guicciardi ME, Cazanave SC, Mertens JC, Sirica AE, and Gores GJ (2011). Myofibroblast-derived PDGF-BB promotes Hedgehog survival signaling in cholangiocarcinoma cells. Hepatology 54, 2076–2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fingas CD, Mertens JC, Razumilava N, Sydor S, Bronk SF, Christensen JD, Rizvi SH, Canbay A, Treckmann JW, Paul A, et al. (2013). Polo-like kinase 2 is a mediator of hedgehog survival signaling in cholangiocarcinoma. Hepatology 58, 1362–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finger LR, Pu J, Wasserman R, Vibhakar R, Louie E, Hardy RR, Burrows PD, and Billips LG (1997). The human PD-1 gene: complete cDNA, genomic organization, and developmentally regulated expression in B cell progenitors. Gene 197, 177–187. [DOI] [PubMed] [Google Scholar]
- Goodman A, Patel SP, and Kurzrock R (2017). PD-1-PD-L1 immune-checkpoint blockade in B-cell lymphomas. Nat. Rev. Clin. Oncol 14, 203–220. [DOI] [PubMed] [Google Scholar]
- Han Y, Liu D, and Li L (2020). PD-1/PD-L1 pathway: current researches in cancer. Am. J. Cancer Res 10, 727–742. [PMC free article] [PubMed] [Google Scholar]
- Hayes S, Chawla A, and Corvera S (2002). TGF beta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J. Cell Biol 158, 1239–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelinek T, Paiva B, and Hajek R (2018). Update on PD-1/PD-L1 inhibitors in multiple myeloma. Front. Immunol 9, 2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang YM, Yu DL, Hou GX, Jiang JL, Zhou Q, and Xu XF (2019). Serum thrombospondin-2 is a candidate diagnosis biomarker for early non-small-cell lung cancer. Biosci. Rep 39, BSR20190476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang N, Gores GJ, and Shah VH (2011). Hepatic stellate cells: partners in crime for liver metastases? Hepatology 54, 707–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang N, Shah VH, and Urrutia R (2015). Membrane-to-nucleus signals and epigenetic mechanisms for myofibroblastic activation and desmoplastic stroma: potential therapeutic targets for liver metastasis? Mol. Cancer Res 13, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang N, Yaqoob U, Geng Z, Bloch K, Liu C, Gomez T, Billadeau D, and Shah V (2010). Focal adhesion assembly in myofibroblasts fosters a microenvironment that promotes tumor growth. Am. J. Pathol 177, 1888–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, and Wrana JL (2000). Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol. Cell 6, 1365–1375. [DOI] [PubMed] [Google Scholar]
- Kim J, Bamlet WR, Oberg AL, Chaffee KG, Donahue G, Cao XJ, Chari S, Garcia BA, Petersen GM, and Zaret KS (2017). Detection of early pancreatic ductal adenocarcinoma with thrombospondin-2 and CA19-9 blood markers. Sci. Transl. Med 9, eaah5583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koli KM, and Arteaga CL (1997). Processing of the transforming growth factor beta type I and II receptors. Biosynthesis and ligand-induced regulation. J. Biol. Chem 272, 6423–6427. [DOI] [PubMed] [Google Scholar]
- Kruger DM, Neubacher S, and Grossmann TN (2018). Protein-RNA interactions: structural characteristics and hotspot amino acids. RNA 24, 1457–1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Large TYS, Meijer LL, Paleckyte R, Boyd LNC, Kok B, Wurdinger T, Schelfhorst T, Piersma SR, Pham TV, Van Grieken NCT, et al. (2020). Combined expression of plasma thrombospondin-2 and CA19-9 for diagnosis of pancreatic cancer and distal cholangiocarcinoma: a proteome approach. Oncologist. 25, e634–e643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Billadeau DD, Abdelhakim H, Leof E, Kaibuchi K, Bernabeu C, Bloom GS, Yang L, Boardman L, Shah VH, and Kang N (2013). IQGAP1 suppresses TbetaRII-mediated myofibroblastic activation and metastatic growth in liver. J. Clin. Invest 123, 1138–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Li J, Xiang X, Guo L, Tu K, Liu Q, Shah VH, and Kang N (2014). PDGF receptor alpha promotes TGF-beta signaling in hepatic stellate cells via transcriptional and posttranscriptional regulation of TGF-beta receptors. Am. J. Physiol. Gastrointest. Liver Physiol 307, G749–G759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Fu X, Wang Y, Wang X, Wang H, Wen J, and Kang N (2020). Protein diaphanous homolog 1 (Diaph1) promotes myofibroblastic activation of hepatic stellate cells by regulating Rab5a activity and TGFbeta receptor endocytosis. FASEB J. 34, 7345–7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Wu X, Cao S, Harrington SM, Yin P, Mansfield AS, and Dong H (2016). B7-H1 antibodies lose antitumor activity due to activation of p38 MAPK that leads to apoptosis of tumor-reactive CD8(+) T cells. Sci. Rep 6, 36722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeuillard E, Yang J, Buckarma E, Wang J, Liu Y, Conboy C, Pavelko KD, Li Y, O’Brien D, Wang C, et al. (2020). Targeting tumor-associated macrophages and granulocytic myeloid-derived suppressor cells augments PD-1 blockade in cholangiocarcinoma. J. Clin. Invest 130, 5380–5396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massague J, and Kelly B (1986). Internalization of transforming growth factor-beta and its receptor in BALB/c 3T3 fibroblasts. J. Cell Physiol 128, 216–222. [DOI] [PubMed] [Google Scholar]
- Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, De Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, et al. (2017). Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 549, 106–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell H, Choudhury A, Pagano RE, and Leof EB (2004). Ligand-dependent and -independent transforming growth factor-beta receptor recycling regulated by clathrin-mediated endocytosis and Rab11. Mol. Biol. Cell 15, 4166–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natsuizaka M, Kinugasa H, Kagawa S, Whelan KA, Naganuma S, Subramanian H, Chang S, Nakagawa KJ, Rustgi NL, Kita Y, et al. (2014). IGFBP3 promotes esophageal cancer growth by suppressing oxidative stress in hypoxic tumor microenvironment. Am. J. Cancer Res 4, 29–41. [PMC free article] [PubMed] [Google Scholar]
- Razumilava N, Gradilone SA, Smoot RL, Mertens JC, Bronk SF, Sirica AE, and Gores GJ (2014). Non-canonical Hedgehog signaling contributes to chemotaxis in cholangiocarcinoma. J. Hepatol 60, 599–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage PT, Schildberg FA, Sobel RA, Kuchroo VK, Freeman GJ, and Sharpe AH (2018). Dendritic cell PD-L1 limits autoimmunity and follicular T cell differentiation and function. J. Immunol 200, 2592–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Chen S, Yang L, and Li Y (2013). The role of PD-1 and PD-L1 in T-cell immune suppression in patients with hematological malignancies. J. Hematol. Oncol 6, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, and Massague J (2003). Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 113, 685–700. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U S A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibult ML, Mamessier E, Gertner-Dardenne J, Pastor S, Just-Landi S, Xerri L, Chetaille B, and Olive D (2013). PD-1 is a novel regulator of human B-cell activation. Int. Immunol 25, 129–137. [DOI] [PubMed] [Google Scholar]
- Tu K, Li J, Verma VK, Liu C, Billadeau DD, Lamprecht G, Xiang X, Guo L, Dhanasekaran R, Roberts LR, et al. (2015). Vasodilator-stimulated phosphoprotein promotes activation of hepatic stellate cells by regulating Rab11-dependent plasma membrane targeting of transforming growth factor beta receptors. Hepatology 61, 361–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu X, Qin B, Zhang Y, Zhang C, Kahila M, Nowsheen S, Yin P, Yuan J, Pei H, Li H, et al. (2019). PD-L1 (B7-H1) competes with the RNA exosome to regulate the DNA damage response and can be targeted to sensitize to radiation or chemotherapy. Mol. Cell 74, 1215–1226 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Tu K, Liu D, Guo L, Chen Y, Li Q, Maiers JL, Liu Z, Shah VH, Dou C, et al. (2019). p300 acetyltransferase is a cytoplasm-to-nucleus shuttle for SMAD2/3 and TAZ nuclear transport in transforming growth factor beta-stimulated hepatic stellate cells. Hepatology 70, 1409–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrana JL, Attisano L, Wieser R, Ventura F, and Massague J (1994). Mechanism of activation of the TGF-beta receptor. Nature 370, 341–347. [DOI] [PubMed] [Google Scholar]
- Yu J, Qin B, Moyer AM, Nowsheen S, Tu X, Dong H, Boughey JC, Goetz MP, Weinshilboum R, Lou Z, and Wang L (2020). Regulation of sister chromatid cohesion by nuclear PD-L1. Cell Res 30, 590–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Yang H, Wan L, Wang Z, Wang H, Ge C, Liu Y, Hao Y, Zhang D, Shi G, et al. (2020). Single-cell transcriptomic architecture and intercellular crosstalk of human intrahepatic cholangiocarcinoma. J. Hepatol 73, 1118–1130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA sequencing data for control and PD-L1 knockdown cells have been deposited to Gene Expression Omnibus and are publicly available as of the date of publication (GEO: GSE167173). Original Western blot and microscopy data generated in this study will be made available and shared by the lead contact upon request.
This study does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.