

Abstract
Myricetin, a flavonoid found in fruits and vegetables, is known to have antioxidant and anticancer effects. However, the anticancer effects of myricetin on SK-BR-3 human breast cancer cells have not been elucidated. In the present study, the anticancer effects of myricetin were confirmed in human breast cancer SK-BR-3 cells. As the concentration of myricetin increased, the cell viability decreased. DAPI (4′,6-diamidino-2-phenylindole) and Annexin V/PI staining also revealed a significant increase in apoptotic bodies and apoptosis. Western blot analysis was performed to confirm the myricetin-induced expression of apoptosis-related proteins. The levels of cleaved PARP and Bax proteins were increased, and that of Bcl-2 was decreased. The levels of proteins in the mitogen-activated protein kinase (MAPK) pathway were examined to confirm the mechanism of myricetin-induced apoptosis, and it was found that the expression levels of phosphorylated c-Jun N-terminal kinase (p-JNK) and phosphorylated mitogen-activated protein kinases (p-p38) were increased, whereas that of phosphorylated extracellular-regulated kinase (p-ERK) was decreased. It was also demonstrated that myricetin induced autophagy by promoting autophagy-related proteins such as microtubule-associated protein 1A/1B-light chain 3 (LC 3) and beclin 1. In addition, 3-methyladenine (3-MA) was used to evaluate the association between cell viability and autophagy in cells treated with myricetin. The results showed that simultaneous treatment with 3-MA and myricetin promoted the apoptosis of breast cancer cells. Furthermore, treatment with a JNK inhibitor reduced cell viability, promoted Bax expression, and reduced the expression of p-JNK, Bcl-2, and LC 3-II/I. These results suggest that myricetin induces apoptosis via the MAPK pathway and regulates JNK-mediated autophagy in SK-BR-3 cells. In conclusion, myricetin shows potential as a natural anticancer agent in SK-BR-3 cells.
Keywords: myricetin, breast cancer, apoptosis, autophagy, MAPK pathway
Introduction
Breast cancer is the second most common cancer in the world, affecting 1.9 million individuals and causing 601,000 deaths annually (1). Patients with breast cancer show rapid tumor progression and metastasis (2), and treatment strategies for such patients include surgical resection, radiotherapy, targeted therapies, and chemotherapy (3). However, these treatment methods affect not only the cancer cells, but also normal cells, and can cause adverse outcomes such as resistance, hypertension, venous and arterial thromboembolism, and metabolic disorders, resulting in poor prognosis (4). Anticancer treatments using natural products can prevent or minimize the risk of side effects, compared to chemotherapy, and such treatment can exert numerous anticancer activities. Therefore, there is a need for research on anticancer therapy based on natural products (5).
Flavonoids are the most common group of plant polyphenols, and can be largely divided into flavones, flavonols, flavanones, flavanonols, flavanols or catechins, anthocyanins and chalcones (6). Among the flavonols, myricetin is included. Myricetin (3,3′,4′,5,5′,7-hexahydroxyflavone, C15H10O8) is abundantly found in bayberry, nuts, red wine, and green tea (7-9). Previous studies have demonstrated the anti-oxidant, antiviral, antibacterial, and anticancer effects of myricetin (10-12). Myricetin has been found to exhibit anticancer effects against pancreatic (13), liver (14), prostate (15), thyroid (16) and breast cancer (17-19) in vitro. In particular, myricetin targets the mitochondrial apoptosis pathway to inhibit cellular proliferation and induce apoptosis (20). However, the anticancer effects of myricetin in regards to breast cancer SK-BR-3 cells have not been evaluated. Additionally, there is a lack of studies on the mechanism of apoptosis caused by myricetin and its relation to autophagy.
Apoptosis is one of many key anticancer mechanisms involving programmed cell death, and is characterized by cell contraction, nuclear condensation, and blebbing of the cell membrane. Apoptosis is an essential process, and abnormal apoptosis may cause mutated cells to progress into tumor cells (21). Apoptosis involves two pathways: extrinsic and intrinsic. The mitochondria play a key role in the intrinsic pathway, and it is also affected by Bax (a pro-apoptotic protein) and Bcl-2 (an anti-apoptotic protein). DNA damage and cellular stress lead to a relative increase in the expression of the Bax protein, and this increases the permeability of the outer mitochondrial membrane. During this process, cytochrome c is released from the intermembrane space of the mitochondria. Cytochrome c creates the caspase complex, inhibits Bcl-2, and causes a caspase chain reaction (22) that fragments the PARP protein. The fragmented PARP protein binds to the ends of DNA and becomes activated, following which NAD and ATP are depleted in the cells, causing apoptosis (23,24).
The mitogen-activated protein kinase (MAPK) pathway is involved in apoptosis via its regulation of pro-apoptotic and anti-apoptotic proteins through various mechanisms. The main proteins involved in the MAPK pathway include extracellular-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 mitogen-activated protein kinases (p38). ERKs are activated via the stimulation of growth factors that cause cell differentiation and proliferation. ERKs inhibit apoptosis by promoting anti-apoptotic proteins and suppressing pro-apoptotic proteins (25,26). In addition, JNK and p38 are activated by stress, and are involved in maintaining the balance between cell survival and death. These proteins promote pro-apoptotic proteins and suppress anti-apoptotic proteins to stimulate apoptosis (27,28). Thus, these proteins regulate the MAPK pathway and are fundamental for the induction of apoptosis.
Autophagy is commonly known to inhibit apoptosis and inhibit the activity of caspases; however, excessive degradation of the cellular cytoplasm through autophagy may lead to apoptosis and cellular death (29). When the Bcl-2/beclin 1 complex is dissociated, beclin1 recruits autophagic proteins to initiate autophagy (30,31). These autophagic proteins form a double membrane to become autophagosomes that combine with lysosomes and degrade old organelles and proteins. In this process, phycoerythrin (PE) combines with 1A/1B-light chain 3 (LC 3)-I to form LC 3-II, which can bind to phagophores. Thus, LC 3 and beclin1 are used as marker proteins that indicate the activation of autophagy (32).
Therefore, in the present study, inhibition of the cell proliferation of SK-BR-3 cells, in which the anticancer efficacy of myricetin is unclear, was confirmed, and whether inhibition of viability is induced through apoptosis was further investigated. In addition, the pathway through which apoptosis occurs was identified, and the association between apoptosis and autophagy in SK-BR-3 cells treated with myricetin was examined, which has not been studied previously.
Materials and methods
Reagents and antibodies
Myricetin (Fig. 1A), purity ≥96.0% used in this experiment, was purchased from Sigma-Aldrich/Merck KGaA. RPMI-1640 medium for cell culturing was purchased from Welgene, and fetal bovine serum (FBS) and penicillin/streptomycin were purchased from Gibco BRL/Thermo Fisher Scientific, Inc. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 4′,6-diamidino-2-phenylindole (DAPI), and acridine orange were purchased from Sigma-Aldrich/Merck KGaA, and the fluorescein isothiocyanate (FITC) Annexin V apoptosis detection kit was purchased from BD Pharmingen. Cell lysis buffer was purchased from Invitrogen/Thermo Fisher Scientific, Inc. Polyadenosine diphosphate-ribose polymerase (PARP, rabbit, 1:1,000, #9542), Bcl-2 associated X (Bax, rabbit, 1:1,000, #2772), B cell lymphoma 2 (Bcl-2, rabbit, 1:1,000, #4223), microtubule-associated protein 1A/1B-light chain 3 (LC 3, rabbit, 1:1,000, #4108), beclin 1 (rabbit, 1:1,000, #3738), total c-Jun N-terminal kinase (t-JNK, rabbit, 1:1,000, #9252), phosphorylated c-Jun N-terminal kinase (p-JNK, rabbit, 1:1,000, #4668), total extracellular-regulated kinase (t-ERK, rabbit, 1:1,000, #9102), phosphorylated extracellular-regulated kinase (p-ERK, rabbit, 1:1,000, #9101), total mitogen-activated protein kinases (t-p38, rabbit, 1:1,000, #9212), phosphorylated mitogen-activated protein kinases (p-p38; rabbit, 1:1,000, #9211), and rabbit IgG (rabbit, 1:1,000, #7074) antibodies were purchased from Cell Signaling Technology (CST), and β-actin (mouse, 1:1,000, sc-47778) and mouse IgG (mouse, 1:1,000, sc-516102) antibodies were purchased from Santa Cruz Biotechnology Inc. JNK inhibitor (SP600125) was purchased from Sigma-Aldrich/Merck KGaA and 3-MA was purchased form MedChemExpress.
Figure 1.
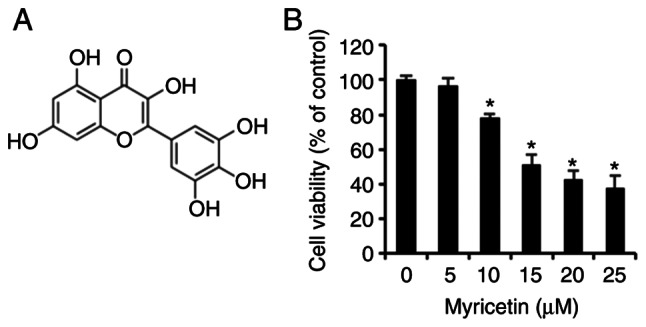
Myricetin inhibits cell viability in SK-BR-3 cells. (A) The chemical structure of myricetin. (B) Cell viability (as measured by an MTT assay) of SK-BR-3 cells treated with 0, 5, 10, 15, 20, and 25 µM of myricetin for 24 h. The control group (0 µM) was treated with the same amount of DMSO. Data are presented as mean and standard deviation (SD) for three samples. *P<0.05 vs. control group. MTT, 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide; DMSO, dimethyl sulfoxide.
Cell culture
Her2-positive SK-BR-3 breast cancer cells were purchased from the Korean Cell Line Bank (KCLB, Seoul, Korea). The cells were cultured in a 75 cm2 flask using RPMI-1640 medium containing 5% FBS and 1% penicillin/streptomycin at 37.5% in a CO2 incubator. The cells were passaged when they were 70% confluent in the 75 cm2 flask.
MTT assay
The SK-BR-3 cells were plated in a 96-well plate at a density of 4×104 cells/ml and placed in an incubator. After 24 h, the cells were treated for 24 h with the following concentrations of myricetin: 0, 5, 10, 15, 20, and 25 µM. Following this, the cells were incubated with 40 µl of MTT solution at 1 mg/ml for 90 min. The MTT solution was removed, and the cells were treated with 100 µl of dimethyl sulfoxide (DMSO). Absorbance (optical density) was measured at 595 nm using an ELISA reader (Bio-Rad Laboratories) and Microplate Manager® 6 software (Bio-Rad Laboratories) to calculate the viability of the cells.
DAPI staining
The SK-BR-3 cells were plated in a 60-mm dish at a density of 2×105 cells/ml and placed in an incubator for 24 h. The cells were treated with 0, 10, and 20 µM of myricetin for 24 h. The medium was removed, and the cells were washed three times with PBS. The cells were fixed in 4% paraformaldehyde for 15 min and washed three times with PBS. DAPI was added to the cells and the reaction was allowed to occur for 1 min under dark conditions. The nuclear morphology of the reacted cells was observed with a fluorescence microscope (Zeiss AG, magnification, ×200). Apoptotic cells were counted as DAPI-positive cells/total cells in three random fields.
Acridine orange staining
The SK-BR-3 cells were plated in a 60-mm dish at a density of 2×105 cells/ml and placed in an incubator for 24 h. The cells were treated with 0, 10, and 20 µM of myricetin for 24 h. The medium was removed, and the cells were washed with PBS. Subsequently, the cells were fixed in 4% paraformaldehyde for 15 min and washed twice with PBS. The cells were stained with acridine orange and observed under a fluorescence microscope (Zeiss AG, magnification, ×200).
Annexin V/PI staining
The Annexin V apoptosis detection kit was used for quantitative analysis of apoptosis. The SK-BR-3 cells were cultured in a 75 cm2 flask and treated with 0, 10, and 20 µM of myricetin for 24 h. The cells were trypsinized with trypsin-EDTA and centrifuged (300 × g, 5 min) into a pellet, following which 1X binding buffer was used to re-suspend the cells to a concentration of 1×106 cells/ml. The cells were reacted with FICT-conjugated Annexin V and PE-conjugated propidium iodide (PI) for 15 min, and analyzed using a FACSCalibur™ flow cytometer (BD Biosciences).
Western blot analysis
The SK-BR-3 cells were cultured in a 75 cm2 flask and treated with 0, 10, and 20 µM of myricetin for 24 h. The cells were trypsinized and centrifuged (300 × g, 5 min) into a pellet, and a cell lysis buffer (Invitrogen/Thermo Fisher Scientific, Inc.) was used to the separate proteins. The cells were centrifuged at 18,000 × g for 5 min, and the supernatant was collected. The proteins were quantified using the Bradford protein assay (Bio-Rad Laboratories) and separated by size using a 12% SDS-PAGE gel. The proteins were transferred to a nitrocellulose membrane (Bio-Rad Laboratories) and blocked using 5% skim milk. The primary antibody was reacted overnight at 4°C. The secondary antibody was reacted at room temperature for 2 h, and the proteins were reacted with ECL detection reagents (Pierce/Thermo Fisher Scientific, Inc.). Multiple proteins were tested on one membrane via stripping. Protein expression was measured using the Image J Launcher (provided by NCBI).
Statistical analysis
The results are expressed as mean ± standard deviation. Statistical analyses were performed using a Student's t test and using a one-way ANOVA followed by Dunnett's or Tukey's test, when more than two conditions were compared. Values were considered statistically significant at P<0.05.
Results
Myricetin inhibits the viability of SK-BR-3 cells
An MTT assay was conducted to determine the effects of myricetin on the cell viability of 3 human breast cancer SK-BR-cells. The SK-BR-3 cells treated with 0, 5, 10, 15, 20, and 25 µM of myricetin for 24 h exhibited viabilities of 96.5% at 5 µM, 78.1% at 10 µM, 51.4% at 15 µM, 42.5% at 20 µM, and 37.9% at 25 µM. Thus the SK-BR-3 cells showed a dose-dependent decrease in viability compared to that of the control group (Fig. 1B).
Myricetin induces apoptosis in SK-BR-3 cells
To investigate whether the decrease in the viability of SK-BR-3 cells was mediated by apoptosis, the cells were stained with DAPI to assess morphological changes. The cells were treated with 0, 10, and 20 µM of myricetin for 24 h and stained with DAPI. Cells in the myricetin-treated group showed the presence of apoptotic bodies such as chromatin condensation, and nuclear fragmentation. The percentage of apoptotic cells was 1.59% in the control group, 6.65% in the 10 µM myricetin group, and 12.78% in the 20 µM myricetin group, indicating a dose-dependent increase in the numbers of apoptotic bodies (Fig. 2A and C). To analyze the percentage of apoptosis after DAPI staining, the cells were stained with Annexin V/PI and analyzed using flow cytometry. The ratio between cells in early and late apoptosis was 9.8% in the control group, 12.5% in the 10 µM myricetin group, and 38.5% in the 20 µM myricetin group. This indicated that there was significant dose-dependent increase in the ratio between cells in early and late apoptosis (Fig. 2B and D). To measure the expression levels of PARP, Bax, and Bcl-2 proteins (apoptosis-regulatory proteins), SK-BR-3 cells were treated with myricetin at concentrations of 0, 10, and 20 µM for 24 h, and the proteins were used in a western blot analysis. In the groups treated with 10 and 20 µM myricetin, the expression levels of cleaved-PARP and Bax (pro-apoptotic protein) were increased, whereas that of Bcl-2 (anti-apoptotic protein) was decreased compared to the expression levels in the control group (Fig. 3A).
Figure 2.

Myricetin induces apoptotic cell death in SK-BR-3 cells. (A) Fluorescence microscopic images of SK-BR-3 cells treated with myricetin (10 and 20 µM for 24 h) and stained with DAPI. The arrows indicate chromatin condensation in SK-BR-3 cells. Scale bar, 10 µm. (B) Flow cytometry results of SK-BR-3 cells treated with myricetin (10 and 20 µM for 24 h) and were stained with Annexin V/PI. (C) The bar graph represents the apoptotic bodies (% of total cells). (D) Total cell population (%) in groups treated with 0, 10, and 20 µM of myricetin. The control group (0 µM) was treated with the same amount of DMSO. Data are displayed as the mean ± SD (n=3). *P<0.05 vs. control group. DAPI, 4′, 6-diamidino-2-phenylindole; PI, propidium iodide; DMSO, dimethyl sulfoxide.
Figure 3.

Myricetin-induced apoptosis occurs through the MAPK pathway in SK-BR-3 cells. (A) Western blot analysis of expression levels of cleaved PARP, Bax, and Bcl-2 in SK-BR-3 cells after myricetin (10, 20 µM) treatment. (B) Western blot analysis of SK-BR-3 cells treated with myricetin (10 and 20 µM for 24 h) to measure the levels of ERK, p-ERK, JNK, p-JNK, p38, and p-p38 proteins. The control group (0 µM) was treated with the same amount of DMSO, and β-actin was used as a loading control. Data are displayed as the mean ± SD (n=3). *P<0.05 vs. control group. PARP, polyadenosine diphosphate-ribose polymerase; Bax, Bcl-2 associated X; Bcl-2, B cell lymphoma 2; MAPK, mitogen-activated protein kinase; ERK, extracellular-regulated kinase; JNK, c-Jun N-terminal kinase; p38, p38 mitogen-activated protein kinases; DMSO, dimethyl sulfoxide.
Myricetin modulates the MAPK pathway to induce apoptosis
The results of the previous experiments indicated that myricetin induces apoptosis in breast cancer SK-BR-3 cells. To investigate the underlying mechanism, the expression levels of the ERK, JNK, and p38 proteins in the MAPK pathway were assessed through western blot analysis. Compared to the control group, the groups treated with 10 and 20 µM myricetin showed a dose-dependent increase in p-JNK and p-38 levels. In contrast, the levels of t-JNK and t-p38 levels were decreased. Compared to the expression levels in the control group, p-ERK expression was decreased and t-ERK expression was increased in the groups treated with 10 and 20 µM myricetin (Fig. 3B). Compared to the control group, the myricetin treatments showed reduced expression of p-ERK/t-ERK. In particular, treatment with 20 µM myricetin led to a significant decrease in p-ERK/t-ERK expression. In contrast, p-JNK/t-JNK expression was significantly higher in the myricetin (10 and 20 µM) treatment groups than in the control. p-p38/t-p38 expression was also significantly higher in the 20 µM myricetin treatment group than in the control.
Myricetin induces protective autophagy
Vacuoles, characteristic features typical of autophagy, were observed in the myricetin-treated SK-BR-3 cells (Fig. 4A). To determine whether myricetin induces autophagy in SK-BR-3 cells, acidic vesicular organelles (AVOs) were identified through acridine orange staining. A small number of AVOs were observed in the control group; however, the number of AVOs increased as the concentration of myricetin increased in the experimental cells (Fig. 4B).
Figure 4.

Myricetin-induced morphological changes in SK-BR-3 cells. (A) Morphological changes (in particular, autophagic vacuoles) observed under a fluorescence microscope in SK-BR-3 cells treated with 10 and 20 µM of myricetin for 24 h. (B) Fluorescence microscopic images of SK-BR-3 cells stained with acridine orange to detect AVOs. The cytoplasm and the nucleus are stained fluorescent green, and the AVOs are stained fluorescent red. Scale bar, 10 µm. AVOs, acidic vesicular organelles.
Western blot analysis was conducted to assess the p-mTOR, beclin 1 and LC 3 proteins, which are related to autophagy. Compared to the control group, the groups treated with 10 and 20 µM myricetin showed increased expression of beclin1 and LC 3-II/I (Fig. 5A). Conversely, p-mTOR/t-mTOR expression was significantly decreased. Additionally, 3-MA (an inhibitor of autophagy) was used to evaluate the association between autophagy and myricetin-induced apoptosis. First, the MTT assay was conducted to assess cell viability after the inhibition of myricetin-induced autophagy. The MTT assay included the following groups: the control group, 3-MA (1 mM, 1 h) pre-treatment group, myricetin (MYR) (10 µM, 24 h) treatment group, and 3-MA (1 mM, 1 h) pre-treatment with myricetin (10 µM, 24 h) treatment group. The cell viability in these groups was as follows: 98.0% in the 3-MA (1 mM, 1 h) pre-treatment group, 78.5% in the myricetin (10 µM, 24 h) treatment group, and 70.4% in the 3-MA (1 mM, 1 h) pretreatment and myricetin (10 µM, 24 h) treatment group. Cell viability was significantly lower in the 3-MA (1 mM, 1 h) pre-treatment with myricetin (10 µM, 24 h) treatment group than in the myricetin (10 µM, 24 h) treatment group (Fig. 5B). Based on these observations, proteins were collected from each group, and the Bax, Bcl-2, and LC 3 proteins were evaluated through western blot analysis. Compared to the control group, the myricetin (10 µM, 24 h) treatment group showed increased levels of Bax and LC 3-II/I and decreased levels of Bcl-2. The 3-MA (1 mM, 1 h) pre-treatment with myricetin (10 µM, 24 h) treatment group showed increased levels of Bax and reduced levels of Bcl-2 and LC 3-II/I compared to these levels in the myricetin (10 µM, 24 h) treatment group (Fig. 5C and D).
Figure 5.

Myricetin causes cytoprotective autophagy in SK-BR-3 cells. (A) Western blot analysis of p-mTOR, beclin 1 and LC 3-II/I expression in SK-BR-3 cells after treatment with myricetin (10 and 20 µM). (B) Cell viability (as measured by an MTT assay) of SK-BR-3 cells pretreated with 1 mM 3-MA (autophagy inhibitor) for 1 h, followed by treatment with myricetin (10 µM for 24 h). (C) Western blot analysis of Bax, Bcl-2, and LC 3-II/I expression in SK-BR-3 cells. (D) Densitometric quantification of the bands in C. The control group (0 µM) was treated with the same amount of DMSO, and β-actin was used as a loading control. Data are displayed as the mean ± SD (n=3). *P<0.05 vs. control group; #P<0.05 vs. myricetin treatment group. MYR, myricetin; LC 3, microtubule-associated protein 1A/1B-light chain 3; MTT, 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide; 3-MA, 3-methyladenine; Bax, Bcl-2 associated X; Bcl-2, B cell lymphoma 2; DMSO, dimethyl sulfoxide.
Myricetin regulates autophagy through the activation of JNK
The JNK inhibitor, SP600125, was used to assess the effects of myricetin-activated JNK on cell viability. This assay included the following groups: the control group, SP600125 (5 µM, 1 h) pre-treatment group, myricetin (MYR) (10 µM, 24 h) treatment group, and SP600125 (5 µM, 1 h) pre-treatment and myricetin (10 µM, 24 h) treatment group. The cell viabilities were as follows: 99.1% in the SP600125 (5 µM, 1 h) pre-treatment group, 79.9% in the myricetin (10 µM, 24 h) treatment group, and 73.7% in the SP600125 (5 µM, 1 h) pre-treatment and myricetin (10 µM, 24 h) treatment group. The SP600125 (5 µM, 1 h) pre-treatment and myricetin (10 µM, 24 h) treatment group showed significantly reduced cell viability compared to that in the myricetin (10 µM, 24 h) treatment group (Fig. 6A). The expression levels of p-JNK, Bax, Bcl-2, and LC 3, were assessed through western blot analysis. The myricetin (10 µM, 24 h) treatment group showed results similar to that in the previous experiment. In the SP600125 pre-treatment and myricetin (10 µM, 24 h) treatment group, the levels of p-JNK/t-JNK, Bcl-2, and LC 3-II/I were lower, whereas that of Bax was higher than those in the myricetin (10 µM, 24 h) treatment group (Fig. 6B and C).
Figure 6.

Myricetin-induced autophagy is regulated by JNK in SK-BR-3 cell. (A) Cell viability (as measured by an MTT assay) of SK-BR-3 cells pretreated with SP600125 (a JNK inhibitor; 5 µM for 1 h) followed by treatment with myricetin (MYR) (10 µM for 24 h). (B) Western blot analysis of p-JNK, Bax, Bcl-2, and LC 3-II/I expression in SK-BR-3 cells. (C) Densitometric quantification of the bands in B. The control group (0 µM) was treated with the same amount of DMSO, and β-actin was used as a loading control. Data are displayed as the mean ± SD (n=3). *P<0.05 vs. control group; #P<0.05 vs. myricetin treatment group. MTT, 3-(4,5-dimethylthiazol-2-yl)-2.5-diphenyltetrazolium bromide; JNK, c-Jun N-terminal kinase; Bax, Bcl-2 associated X; Bcl-2, B cell lymphoma 2; LC 3, microtubule-associated protein 1A/1B-light chain 3; DMSO, dimethyl sulfoxide.
Discussion
Although breast cancer can be treated through various surgical and therapeutic means, these treatments have known side effects. However, anticancer treatments using natural products can reduce the risk of side effects, compared to chemotherapy, and such treatments can exert many anti-cancer activities. Therefore, anticancer treatments using natural substances have been increasingly endorsed (5). In the present study, we assessed whether myricetin induces apoptosis and autophagy in breast cancer SK-BR-3 cells and investigated the correlation between myricetin-induced apoptosis and autophagy.
The experiment was performed after establishing a non-toxic concentration in normal cells (13,33). In the MTT assay, the viability of SK-BR-3 cells was significantly decreased after treatment with myricetin in a dose-dependent manner. According to Phillips et al (13), when pancreatic cancer cells were treated with myricetin, cell viability decreased significantly in a concentration-dependent manner, from 12.5 µM myricetin. Similarly, in the present experiment, significant results were observed following 10 µM myricetin treatments, and cell viability was decreased in a concentration-dependent manner. Therefore, myricetin is thought to decrease the viability of SK-BR-3 cells.
Apoptotic bodies are formed by the condensation of the nucleus and chromatin, and are a key feature of apoptosis. Apoptotic bodies can be observed under the microscope through DAPI staining (34). Therefore, the SK-BR-3 cells were treated with myricetin, stained with DAPI, and observed under a fluorescence microscope. The myricetin treatment groups showed reduced cell viability compared to that in the control group, and the myricetin-treated cells showed apoptotic bodies. Compared to the control group, both the myricetin-treated groups had significantly higher numbers of apoptotic cells. According to Jo et al (16), when myricetin was used to treat thyroid cancer cells, apoptotic bodies increased in a concentration-dependent manner. Similarly, in the SK-BR-3 cells, the number of apoptotic bodies was increased in the myricetin-treated group in a concentration-dependent manner. Cells undergoing apoptosis exhibit distinct changes to their morphology, including the surface exposure of phosphatidylserine which is usually present inside the phospholipid bilayer of the cell membrane. Apoptosis can be divided into early and late stages depending on the severity of cell membrane damage (35). To assess these changes, the cells were stained with Annexin V/PI, and cellular apoptosis was quantitated through flow cytometry. The myricetin treatment groups had higher numbers of cells in early and late apoptosis than those in the control group. These results suggest that the myricetin-induced decrease in cell viability is mediated by apoptosis.
Furthermore, our results confirmed the myricetin-induced expression of apoptosis-related proteins. Apoptosis is mainly regulated by the Bcl-2 family of proteins, Bcl-2/Bcl-XL (anti-apoptotic) and Bax and Bak (pro-apoptotic), and the relative ratio of these proteins may inhibit or promote apoptosis (36). DNA damage and cellular stress leads to a relative increase in the expression of the pro-apoptotic Bax protein, which increases the permeability of the outer mitochondrial membrane. This causes the release of cytochrome c, which inhibits the anti-apoptotic Bcl-2 protein and induces a caspase chain reaction. Subsequently, the PARP protein is fragmented, and NAD and ATP are depleted in cells, resulting in apoptosis (22-24). In this study, myricetin increased the levels of the pro-apoptotic Bax protein, cleaved PARP, and reduced the levels of the anti-apoptotic Bcl-2 protein. These results suggest that myricetin induces apoptosis in SK-BR-3 cells by regulating the levels of PARP, Bax, and Bcl-2.
The MAPK pathway regulates the pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins, and is involved in apoptosis. The downstream molecules of the MAPK pathway include the ERK1/2, JNK, and p38 proteins. The activation of ERK promotes Bcl-2 and suppresses Bax, thus inhibiting apoptosis (25,26), and JNK and p38 play important roles in balancing cell death and survival. Activated JNK and p38 increase Bax expression and decrease Bcl-2 expression to promote apoptosis (27,28). These regulatory mechanisms of proteins in the MAPK pathway are fundamental for the induction of apoptosis. According to Innajak et al (37), when goniothalamin was used to treat SK-BR-3 cells, apoptosis was induced by decreasing p-ERK, while increasing the expression of p-JNK and p-p38 proteins. Similarly, when SK-BR-3 cells were treated with myricetin, the same results were obtained. This suggests that myricetin treatments induce apoptosis in SK-BR-3 breast cancer cells through the MAPK pathway which inhibits ERK and activates JNK and p38.
SK-BR-3 cells treated with myricetin showed vacuoles, one of the hallmarks of autophagy. Autophagy is commonly known to inhibit apoptosis and suppress the activity of caspases; however, excessive degradation of the cellular cytoplasm through autophagy may lead to apoptosis and cellular death (29). Acidic vesicular organelles (AVOs) are characteristic of cells in autophagy, and can be identified through the accumulation of acridine orange in the acidic compartments (38). Here, the SK-BR-3 cells were stained with acridine orange to examine whether myricetin induces autophagy. Although cells in the control group had a small number of AVOs, the myricetin (10, 20 µM) treatment groups showed a high number of AVOs.
Several proteins are activated in the process of autophagy. At initial stages, the Bcl-2/beclin 1 complex is dissociated, following which beclin 1 recruits autophagic proteins to initiate autophagy (30,31). The autophagic proteins form a double membrane to form autophagosomes that then combine with lysosomes. The resulting autolysosome degrades old cellular organelles and proteins (32). mTOR is an important protein that inhibits autophagy. Therefore, a decrease in p-mTOR affects the induction of autophagy (39). In this process, LC 3 combines with phagophores. Through our western blot analysis, we observed the expression of p-mTOR, beclin 1 and LC 3, which confirmed the presence of autophagy-related proteins in myricetin-treated SK-BR-3 cells. The myricetin (10, 20 µM) treatment groups showed significantly higher levels of the beclin 1 protein than the control group. Similarly, the levels of LC 3-II/I were significantly increased in the myricetin (20 µM) treatment group. Taken together, the morphological features of autophagy (such as increased numbers of vacuoles and AVOs) and the increased levels of beclin 1 (a marker of autophagy) and LC 3-II/I suggest that myricetin induces autophagy in SK-BR-3 cells.
To assess the effects of myricetin-induced autophagy on cell viability, we conducted an MTT assay using 3-MA, an inhibitor of autophagy. Zhu et al (40) observed a tendency of decreased cell viability in 3-MA treatments. Similarly, in this experiment, when myricetin and 3-MA were applied in combination, cell viability showed a tendency to decrease further. Moreover, as a result of western blotting, the pro-apoptosis protein Bax increased and the anti-apoptosis protein Bcl-2 decreased. Accordingly, these findings indicate that the inhibition of autophagy induces apoptosis and reduces cell viability, suggesting that myricetin-induced autophagy exerts protective effects on SK-BR-3 cells.
To examine the effects of the JNK protein on cell viability in the previous experiment, we conducted an MTT assay using SP600125, a JNK inhibitor. Studies by Yu et al (41) and Chen et al (42) demonstrated that JNK inhibition leads to a further decrease in cell viability, and our experiments showed similar results. To confirm that the reduced cell viability after the inhibition of JNK was related to autophagy, we evaluated the expression levels of LC 3 and apoptosis-related proteins through western blot analysis. Yu et al (41) reported that SP600125 pre-treatment followed by treatment with a natural compound resulted in a lower level of LC 3-II/I expression than in the natural compound treatment group. Similarly, in our study, SP600125 (20 µM, 1 h) pre-treatment followed by myricetin (10 µM, 24 h) treatment led to reduced expression levels of the LC 3-II/I protein compared to that in the myricetin (10 µM, 24 h) treatment group. Additionally, Bcl-2/Bax expression levels were also reduced. These finding suggest that autophagy is mediated by JNK, as previously reported by Chen et al (42). JNK degrades the Bcl-2/beclin 1 complex, thus allowing beclin 1 to initiate autophagy. This leads to the increased expression of the LC 3 protein and promotes autophagy (43). These results indicate that myricetin induces apoptosis via the JNK pathway in SK-BR-3 breast cancer cells, and that autophagy is regulated by the JNK pathway. Furthermore, myricetin-induced autophagy also had a protective role in SK-BR-3 cells. Therefore, JNK inhibition suppressed autophagy, which protected the cells and led to increased apoptosis.
In conclusion, myricetin significantly inhibited the viability of breast cancer SK-BR-3 cells (in a dose-dependent manner) by promoting apoptosis. Western blot analysis showed that myricetin increased the expression of the pro-apoptotic PARP and Bax proteins, and reduced the expression of the anti-apoptotic Bcl-2 protein. In addition, the myricetin-activated MAPK pathway induced apoptosis in SK-BR-cells. The expression levels of autophagic proteins (beclin 1 and LC 3-II/I) were also increased, indicating that myricetin induced autophagy in SK-BR-3 cells. An MTT assay with the 3-MA inhibitor revealed that myricetin-induced autophagy had a protective effect in cells. Moreover, the JNK protein was shown to play an important role in regulating autophagy (Fig. 7). Taken together, these results suggest that myricetin has anticancer efficacy against breast cancer SK-BR-3 cells and that inhibition of autophagy increases anticancer efficacy through apoptosis. However, additional studies are required to examine whether myricetin induces autophagy as well as apoptosis in vivo and to examine the relationship between the two mechanisms.
Figure 7.

Schematic of myricetin-induced apoptosis and autophagy in SK-BR-3 cells. Myricetin-induced apoptosis through the intrinsic pathway by regulated the MAPK pathway, and autophagy through JNK signaling in SK-BR-3 cells. MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase.
Acknowledgments
Not applicable.
Funding Statement
This study was supported by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology (2021R1A2C1010912).
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Authors' contributions
SHH, CC and JYJ designed the study. SHH, JHL and JSW performed the experiments and GHJ, SHJ and EJH performed the reagent preparation and analyzed the data. SKK and BKP developed the methodology. SHH wrote the manuscript and YSP, BSK and JYJ revised the manuscript. All authors read and approved the manuscript and agree to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work, in particular the data, are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare no competing interest.
References
- 1.Global Burden of Disease Cancer Collaboration. Fitzmaurice C, Abate D, Abbasi N, Abbastabar H, Abd-Allah F, Abdel-Rahman O, Abdelalim A, Abdoli A, Abdollahpour I, et al. Global, regional, and national cancer incidence, mortality, years of life lost, years lived with disability, and disability-adjusted life-years for 29 cancer groups, 1990 to 2017: A systematic analysis for the global burden of disease study. JAMA Oncol. 2019;5:1749–1768. doi: 10.1001/jamaoncol.2019.2996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: Epidemiology and etiology. Cell Biochem Biophys. 2015;72:333–338. doi: 10.1007/s12013-014-0459-6. [DOI] [PubMed] [Google Scholar]
- 3.Brown LC, Mutter RW, Halyard MY. Benefits, risks, and safety of external beam radiation therapy for breast cancer. Int J Womens Health. 2015;7:449–458. doi: 10.2147/IJWH.S55552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seltzer JH, Gintant G, Amiri-Kordestani L, Singer J, Koplowitz LP, Moslehi JJ, Barac A, Yu AF. Assessing cardiac safety in oncology drug development. Am Heart J. 2019;214:125–133. doi: 10.1016/j.ahj.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mitra S, Dash R. Natural products for the management and prevention of breast cancer. Evid Based Complement Alternat Med. 2018;2018:8324696. doi: 10.1155/2018/8324696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Panche AN, Diwan AD, Chandra SR. Flavonoids: An overview. J Nutr Sci. 2016;5:e47. doi: 10.1017/jns.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song X, Tan L, Wang M, Ren C, Guo C, Yang B, Ren Y, Cao Z, Li Y, Pei J. Myricetin: A review of the most recent research. Biomed Pharmacother. 2021;134:111017. doi: 10.1016/j.biopha.2020.111017. [DOI] [PubMed] [Google Scholar]
- 8.Ross JA, Kasum CM. Dietary flavonoids: Bioavailability, metabolic effects, and safety. Annu Rev Nutr. 2002;22:19–34. doi: 10.1146/annurev.nutr.22.111401.144957. [DOI] [PubMed] [Google Scholar]
- 9.Devi KP, Rajavel T, Habtemariam S, Nabavi SF, Nabavi SM. Molecular mechanisms underlying anticancer effects of myricetin. Life Sci. 2015;142:19–25. doi: 10.1016/j.lfs.2015.10.004. [DOI] [PubMed] [Google Scholar]
- 10.Masuda T, Miura Y, Inai M, Masuda A. Enhancing effect of a cysteinyl thiol on the antioxidant activity of flavonoids and identification of the antioxidative thiol adducts of myricetin. Biosci Biotechnol Biochem. 2013;77:1753–1758. doi: 10.1271/bbb.130315. [DOI] [PubMed] [Google Scholar]
- 11.Zheng AW, Chen YQ, Zhao LQ, Feng JG. Myricetin induces apoptosis and enhances chemosensitivity in ovarian cancer cells. Oncol Lett. 2017;13:4974–4978. doi: 10.3892/ol.2017.6031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ong KC, Khoo HE. Biological effects of myricetin. Gen Pharmacol. 1997;29:121–126. doi: 10.1016/S0306-3623(96)00421-1. [DOI] [PubMed] [Google Scholar]
- 13.Phillips PA, Sangwan V, Borja-Cacho D, Dudeja V, Vickers SM, Saluja AK. Myricetin induces pancreatic cancer cell death via the induction of apoptosis and inhibition of the phosphatidylinositol 3-kinase (PI3K) signaling pathway. Cancer Lett. 2011;308:181–188. doi: 10.1016/j.canlet.2011.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li M, Chen J, Yu X, Xu S, Li D, Zheng Q, Yin Y. Myricetin suppresses the propagation of hepatocellular carcinoma via down-regulating expression of YAP. Cells. 2019;8:358. doi: 10.3390/cells8040358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ye C, Zhang C, Huang H, Yang B, Xiao G, Kong D, Tian Q, Song Q, Song Y, Tan H, et al. The natural compound myricetin effectively represses the malignant progression of prostate cancer by inhibiting PIM1 and disrupting the PIM1/CXCR4 interaction. Cell Physiol Biochem. 2018;48:1230–1244. doi: 10.1159/000492009. [DOI] [PubMed] [Google Scholar]
- 16.Jo S, Ha TK, Han SH, Kim ME, Jung I, Lee HW, Bae SK, Lee JS. Myricetin induces apoptosis of human anaplastic thyroid cancer cells via mitochondria dysfunction. Anticancer Res. 2017;37:1705–1710. doi: 10.21873/anticanres.11502. [DOI] [PubMed] [Google Scholar]
- 17.Jiao D, Zhang XD. Myricetin suppresses p21-activated kinase 1 in human breast cancer MCF-7 cells through downstream signaling of the β-catenin pathway. Oncol Rep. 2016;36:342–348. doi: 10.3892/or.2016.4777. [DOI] [PubMed] [Google Scholar]
- 18.Soleimani M, Sajedi N. Myricetin apoptotic effects on T47D breast cancer cells is a P53-independent approach. Asian Pac J Cancer Prev. 2020;21:3697–3704. doi: 10.31557/APJCP.2020.21.12.3697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Knickle A, Fernando W, Greenshields AL, Rupasinghe HPV, Hoskin DW. Myricetin-induced apoptosis of triple-negative breast cancer cells is mediated by the iron-dependent generation of reactive oxygen species from hydrogen peroxide. Food Chem Toxicol. 2018;118:154–167. doi: 10.1016/j.fct.2018.05.005. [DOI] [PubMed] [Google Scholar]
- 20.Morales P, Haza AI. Selective apoptotic effects of piceatannol and myricetin in human cancer cells. J Appl Toxicol. 2012;32:986–993. doi: 10.1002/jat.1725. [DOI] [PubMed] [Google Scholar]
- 21.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 22.Elumalai P, Gunadharini DN, Senthilkumar K, Banudevi S, Arunkumar R, Benson CS, Sharmila G, Arunakaran J. Induction of apoptosis in human breast cancer cells by nimbolide through extrinsic and intrinsic pathway. Toxicol Lett. 2012;215:131–142. doi: 10.1016/j.toxlet.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 23.Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S, Smulson M. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932–22940. doi: 10.1074/jbc.274.33.22932. [DOI] [PubMed] [Google Scholar]
- 24.Murphy KM, Ranganathan V, Farnsworth ML, Kavallaris M, Lock RB. Bcl-2 inhibits Bax translocation from cytosol to mitochondria during drug-induced apoptosis of human tumor cells. Cell Death Differ. 2000;7:102–111. doi: 10.1038/sj.cdd.4400597. [DOI] [PubMed] [Google Scholar]
- 25.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Naturey. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 26.Lu Z, Xu S. ERK1/2 MAP kinases in cell survival and apoptosis. IUBMB Life. 2006;58:621–631. doi: 10.1080/15216540600957438. [DOI] [PubMed] [Google Scholar]
- 27.Dhanasekaran DN, Reddy EP. JNK signaling in apoptosis. Oncogene. 2008;27:6245–6251. doi: 10.1038/onc.2008.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cuenda A, Rousseau S. p38 MAP-kinases pathway regulation, function and role in human diseases. Biochim Biophys Acta. 2007;1773:1358–1375. doi: 10.1016/j.bbamcr.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 29.Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT, Liu B, Bao JK. Programmed cell death pathways in cancer: A review of apoptosis, autophagy and programmed necrosis. Cell Prolif. 2012;45:487–498. doi: 10.1111/j.1365-2184.2012.00845.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.He C, Levine B. The beclin 1 interactome. Curr Opin Cell Biol. 2010;22:140–149. doi: 10.1016/j.ceb.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Oberstein A, Jeffrey PD, Shi Y. Crystal structure of the Bcl-XL-beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem. 2007;282:13123–13132. doi: 10.1074/jbc.M700492200. [DOI] [PubMed] [Google Scholar]
- 32.Parzych KR, Klionsky DJ. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid Redox Signal. 2014;20:460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Chen AY, Ye X, Li B, Rojanasakul Y, Rankin GO, Chen YC. Myricetin inhibits proliferation of cisplatin-resistant cancer cells through a p53-dependent apoptotic pathway. Int J Oncol. 2015;47:1494–1502. doi: 10.3892/ijo.2015.3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cummings BS, Wills LP, Schnellmann RG. Measurement of cell death in Mammalian cells. Curr Protoc Pharmacol. 2012;56:12.8.1–12.8.24. doi: 10.1002/0471141755.ph1208s56. [DOI] [PubMed] [Google Scholar]
- 35.Yoo GS, Lee JM, Lee CH, Jang JB, Lee KS. Study of apoptosis by scirpi tuber in Hela cell and MCF-7 cell. J Korean Obstet Gynecol. 2011;24:1–13. [Google Scholar]
- 36.Kale J, Osterlund EJ, Andrews DW. BCL-2 family proteins: Changing partners in the dance towards death. Cell Death Differ. 2017;25:65–80. doi: 10.1038/cdd.2017.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Innajak S, Mahabusrakum W, Watanapokasin R. Goniothalamin induces apoptosis associated with autophagy activation through MAPK signaling in SK-BR-3 cells. Oncol Rep. 2016;35:2851–2858. doi: 10.3892/or.2016.4655. [DOI] [PubMed] [Google Scholar]
- 38.Traganos F, Darzynkiewicz Z. Lysosomal proton pump activity: Supravital cell staining with acridine orange differentiates leukocyte subpopulations. Methods Cell Biol. 1994;41:185–194. doi: 10.1016/S0091-679X(08)61717-3. [DOI] [PubMed] [Google Scholar]
- 39.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584:1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu ML, Zhang PM, Jiang M, Yu SW, Wang L. Myricetin induces apoptosis and autophagy by inhibiting PI3K/Akt/mTOR signalling in human colon cancer cells. BMC Complement Med Ther. 2020;20:209. doi: 10.1186/s12906-020-02965-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yu H, Wu CL, Wang X, Ban Q, Quan C, Liu M, Dong H, Li J, Kim GY, Choi YH, et al. SP600125 enhances C-2-induced cell death by the switch from autophagy to apoptosis in bladder cancer cells. J Exp Clin Cancer Res. 2019;38:448. doi: 10.1186/s13046-019-1467-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen S, Dobrovolsky VN, Liu F, Wu Y, Zhang Z, Mei N, Guo L. The role of autophagy in usnic acid-induced toxicity in hepatic cells. Toxicol Sci. 2014;142:33–44. doi: 10.1093/toxsci/kfu154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jin HO, Hong SE, Park JA, Chang YH, Hong YJ, Park IC, Lee JK. Inhibition of JNK-mediated autophagy enhances NSCLC cell sensitivity to mTORC1/2 inhibitors. Sci Rep. 2016;6:28945. doi: 10.1038/srep28945. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.